

Abstract
Background
Mutations in the FBXO7 gene can cause a rare chromosomal recessive neurodegenerative disease, Parkinsonian‐pyramidal syndrome (PPS). Patients with this syndrome mainly show early‐onset Parkinson's syndrome. Here, we present a Chinese family with infantile‐onset PPS caused by FBXO7 mutations.
Methods
The clinical phenotypes and medical records of the proband and his family members were collected. The proband, his sibling, and his parents underwent whole‐exome sequencing (WES) by next‐generation sequencing.
Results
The proband and his sibling had a typical PPS phenotype with onset during infancy. WES identified compound heterozygous variants in the FBXO7 gene, including a nonsense mutation, p. Trp134*, and a splicing mutation, IVS5‐1G > A, which were shared by both siblings and inherited from each of the parents. These variants have not been reported in literatures or databases. According to the American College of Medical Genetics and Genomics guidelines, the p. Trp134* and IVS5‐1G > A mutations were classified as pathogenic variants.
Conclusions
We report a case of siblings in a Chinese family with infantile‐onset PPS caused by FBXO7 gene mutations determined by WES. These findings will contribute to the in‐depth study of the pathogenesis of PPS among patients with FBXO7 gene mutations.
Keywords: FBXO7, PARK15, Parkinsonian‐pyramidal syndrome, whole‐exome sequencing
1. INTRODUCTION
Mutations in the FBXO7 gene, which encodes F‐box protein 7, can cause a rare autosomal recessive neurodegenerative disease—Parkinson's disease‐15 (PARK15). 1 This disease is also known as Parkinsonian‐pyramidal syndrome (PPS) or pallido‐pyramidal disease. Patients with this disease mainly show early‐onset Parkinson's syndrome accompanied by pyramidal disturbances and often exhibit psychomotor retardation, eyelid apraxia, chorea, and other atypical symptoms. 2
As of now, more than 20 cases of FBXO7 gene mutations in families with early‐onset Parkinson's syndrome have been reported internationally, and partial mutation types have been identified. 1 , 3 , 4 , 5 , 6 , 7 , 8 , 9 Homozygous or compound heterozygous mutations of the FBXO7 in previous report have been shown to cause juvenile‐onset Parkinsonism or early‐onset Parkinsonism with different age of onset. 6 Wei et al 4 reported the first Chinese case of juvenile‐onset Parkinsonism caused by compound heterozygous mutations of the FBXO7 gene. The patient was a 32‐year‐old man who began experiencing a progressive involuntary tremor at 16 years of age. As to why the onset age tends to range from the teens to the 20s 1 , 3 , 4 , 5 , 6 , 7 , 8 , 9 is unknown. The genetic background, especially that of consanguineous families, may be a strong contributor to age of onset.
In this study, we reported siblings in a Chinese family with infantile‐onset PPS and identified compound heterozygous mutations in FBXO7 gene by whole‐exome sequencing (WES). The family's analysis suggests that mutations in the FBXO7 gene may cause PARK15 to develop in infancy.
2. MATERIALS AND METHODS
2.1. Subjects
In this family, a couple with a normal phenotype gave birth to two affected children (Figure 1). The proband was a male, 6‐year‐old, first child with a term birth and showed no notable abnormalities at birth. At about 6 months of age, the proband had increased muscle tension, could not sit independently, and was diagnosed with "cerebral palsy," but rehabilitation treatment did not produce satisfactory results. Now, the proband could not walk independently, had increased muscle tone and limb stiffness, could not speak, and had intellectual developmental delays. His parents were healthy and gave birth to another boy in 2017. When this sibling was 6 months old, he showed increased muscle tension and poor cognitive development. He died at the age of 2 years from a severe lung infection. Neither the proband nor the younger sibling showed any abnormality in genetic metabolism screening by tandem mass spectrometry, karyotype analysis, chromosome microarray analysis (CMA) with CytoScan 750K (Affymetrix Inc.) arrays, routine magnetic resonance imaging (MRI) (Figure 2), and electroencephalo‐graph (EEG). This research was approved by the Ethics Committee of Gansu Province Maternal and Child Health Care Hospital. Written informed consent was obtained from all persons who participated in the study along with parental consent for children's participation in this research.
FIGURE 1.
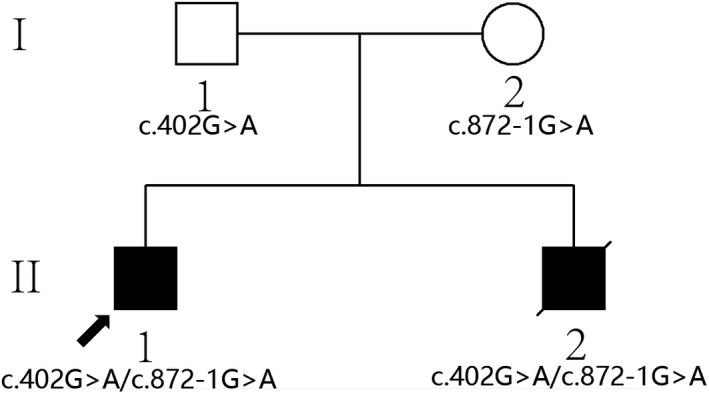
The pedigree of the Chinese family with FBXO7 mutations
FIGURE 2.
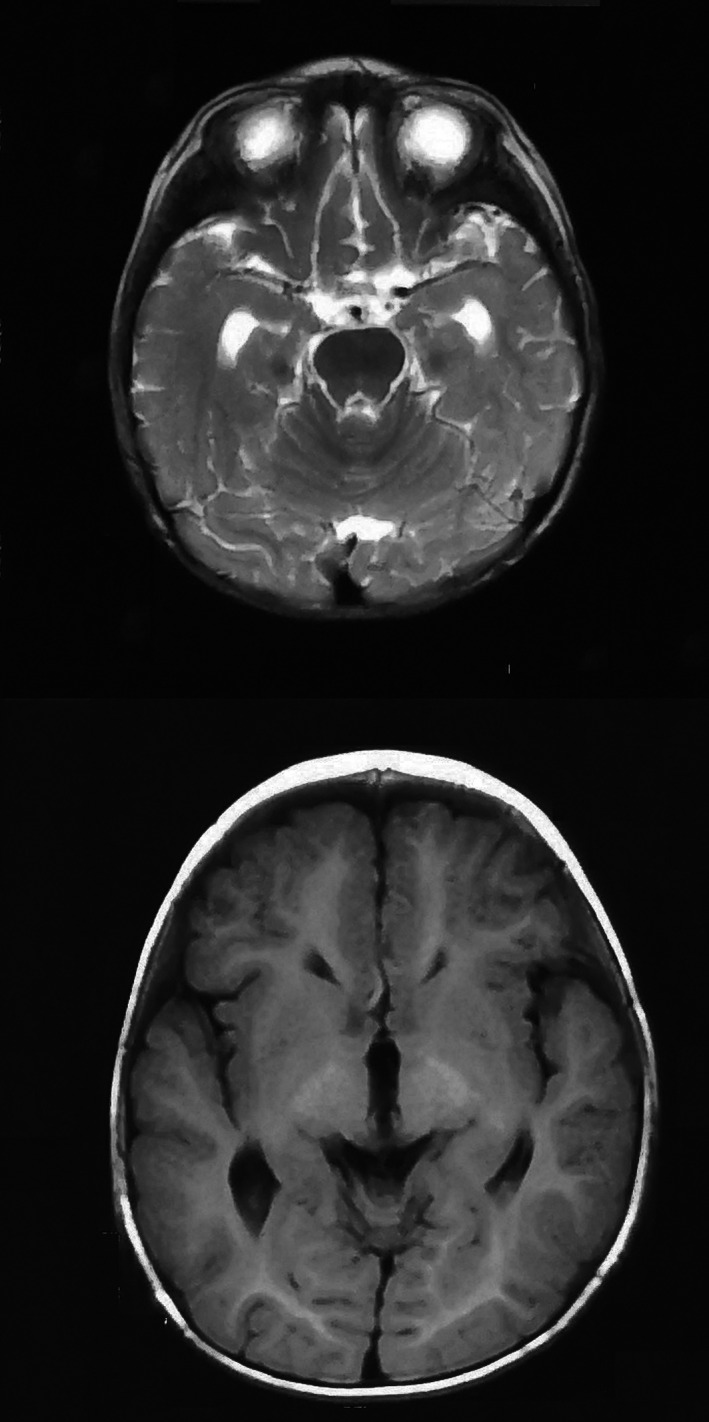
The magnetic resonance image of II 1
2.2. DNA extraction and WES
Peripheral venous blood (5 mL) was collected from the proband, his parents, and his younger sibling. A QIAamp DNA Blood Mini Kit (QIAGEN) was used to extract genomic DNA from the whole blood, and a Qubit 2.0 (Invitrogen) was used for nucleic acid quantification. Whole‐exome sequencing (WES) was performed on Hiseq 2000 sequencer (Illumina). SureSelect Human All Exon V5 kit (Agilent), Hiseq PE Cluster kit V4, and Hiseq SBS kit V4 (Illumina) were used for whole‐exome capture, library construction, and high‐throughput sequencing, respectively.
2.3. Bioinformatics analysis
Sequencing fragments were mapped with hg19 reference genome using BWA software (version 0.7.9a). PCR duplications were removed by Picard software (version 1.115). GATK (Genome Analysis Tool Kit, version 3.2) was employed to call variants; the processing included base quality score recalibration (BQSR), InDel realignment, duplicate removal, insertions/deletions (indels), and single‐nucleotide variants’ (SNVs) detection. Variants were annotated with VEP software (Variant Effect Predictor, Ensembl 73). Mutations in the functional coding region and splice site, which mainly involved loss of function mutations, missense mutations and non‐frameshift InDel mutations, were included in next step analysis. Loss of function mutations referred to nonsense, frameshift, and splicing site variants. Three major databases containing pathogenic or likely pathogenic variants, ClinVar, OMIM (Online Mendelian Inheritance in Man), and HGMD (Human Gene Mutation Database), were used to further screening. Multiple bioinformatics tools 10 , 11 , 12 were used to predict the functions of missense variants and annotate noncoding regulatory sequences. Each variant was classified according to the standards and guidelines for the interpretation of sequence variants published by the American College of Medical Genetics and Genomics. 13 DNA variants were described using HGVS nomenclature . 14
2.4. Sanger sequencing
Candidate disease–causing gene mutations were determined by bioinformatics analysis, and then, primers were designed to amplify the sequences around these sites. PCR products were purified by ethanol precipitation and sequenced on an ABI3500 sequencer (Applied Biosystems) using the BigDye 3.1 Sequencing Kit (Applied Biosystems).
3. RESULTS
The WES of the proband generated 10.9 G data, resulting in an average sequencing depth of 98× for target capture region. 92.99% of targeted bases were covered at ≥10×. The output data of WES of the family members met the requirements for the next step bioinformation analysis.
With the use of bioinformatics methods, compound heterozygosity variants in FBXO7 gene (NM_012179.3) were identified: c.402G > A and c.872‐1G > A, which were shared by the proband and the younger sibling. c.402G > A was inherited from their father, and c.872‐1G > A was inherited from their mother. The result was confirmed by Sanger sequencing in the family (Figure 3).
FIGURE 3.
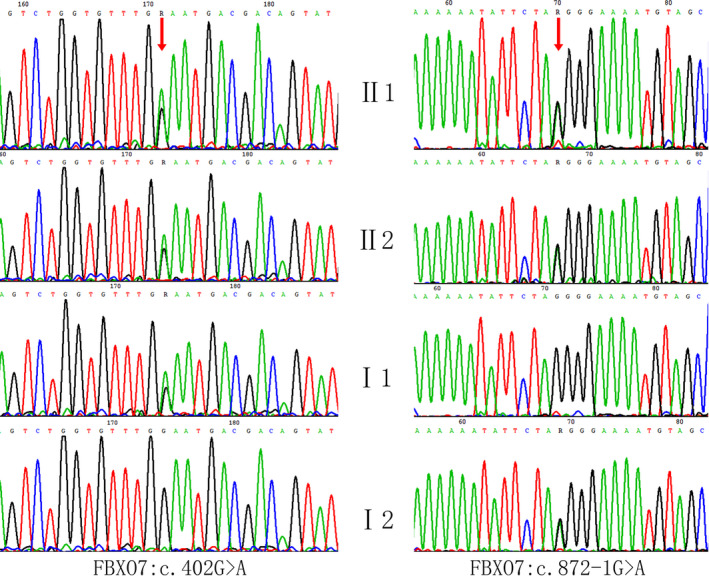
Sanger sequencing chromatograms of FBXO7 mutations in the family
These two variants were not reported in previous literatures. They were also absent from 1000 Genomes Project, the Exome Aggregation Consortium (ExAC), and dbSNP database and not identified in the variants database, ClinVar, OMIM, and HGMD. The c.402G > A variant transformed the tryptophan codon (UGG) into a termination codon (UGA), producing a truncated protein (p.Trp134*) and resulting in loss of function of FBXO7. The c.872‐1G > A variant was a splice site mutation, located in position −1 of intron 5, causing the abnormal splicing of exon 6 and production of aberrant proteins. These variants have not been reported in the literatures or databases. According to standards and guidelines of ACMG for the interpretation of sequence variants, the novel nonsense variant c.402G > A was weighted as very strong evidence of pathogenicity (PVS1), moderate evidence of pathogenicity (PM2 and PM4), and supporting evidence of pathogenicity (PP1) and splicing variant c. 872‐1G > A was weighted as PVS1, PM2, PM3, and PP1; based on the pathogenic criteria, these two variants were classified as pathogenic.
4. DISCUSSION
PARK15 is an extremely rare neurological disease. Beside Parkinsonism symptoms, patients also exhibit pyramidal tract signs, comprised of spasticity, tendon hyperreflexia, and positive pathologic reflex. In 1954, Davision 2 first reported five patients from three families (four of whom were from two inbred families) with typical tremor paralysis and pyramidal tract signs. In 2008, Shojaee 3 reported a case of strephenopodia deformity in an Iranian family, and 500 k SNP microarrays were used for genome‐wide linkage analysis. Setting an autosomal recessive genetic background, the mutation was found located in the 22q region, and then, a homozygous missense mutation (R378G) in exon 7 of FBXO7 gene was identified in that area. In 2009, Di Fonzo 1 reported an autosomal recessive homozygous truncation mutation of the FBXO7 gene (R498X) in two patients from an Italian family and found compound heterozygous mutations of the FBXO7 gene in another Dutch family (IVS7 + 1G > T, T22M); the disease was then officially named PARK15.
PARK15 is an autosomal recessive genetic disorder, and patients develop pyramidal tract symptoms such as spasticity, scissors gait, tendon hyperreflexia, and positive pathological characteristics during adolescence. After several years, extrapyramidal symptoms such as rigidity, tremor, and bradykinesia are presented but in a relatively slow progression, which could be better improved by dopamine treatment. Previous reports have shown that the patients with FBXO7 gene mutations exhibit variable age of onset, ranging from children and adolescents, to adults; the minimum age of onset is reported to be 10 years. 8 In this study, both of the two patients experienced onset during infancy, with manifestations of severe dystonia and spasm. The proband showed no obvious responses to rehabilitation treatment on motor function and still could not walk independently at the age of 6 years. He walked with a scissor gait with assistance, showing obvious damage to the extrapyramidal system; however, the proband and his sibling had no significant static tremor. Wei et al 4 performed phenotype analysis in 17 reported patients with FBXO7 mutations, indicating that all patients manifested myotonia and motor development retardation, 64.7% of patients had static or postural tremor, 87.5% of patients suffered postural instability, 64.7% and 75% of patients presented with pyramidal tract symptoms and tendon hyperreflexia, respectively. The majority of patients (88.2%) responded well to dopamine treatment, but some side effects were also observed. In this study, except for the lack of obvious static or postural tremor, the proband and his sibling shared similar phenotypes to those reported in the literature. The proband had been tried to treat with levodopa (with a starting dose of 50 mg/d) but had since shown little benefit with this drug, so it was discontinued, which further indicates the phenotype diversity of PPS patients and provides additional references for clinical diagnosis.
Human FBXO7 is encoded by two transcripts, among which, transcript 1 encodes 522 amino acid residues, and transcript 2 encodes 443 amino acid residues. To date, several mutations of FBXO7 that cause the PARK15 phenotype have been reported, including the missense mutations T22M, 1 L34R, 1 , 6 and R378G, 1 , 3 the nonsense mutation R498X, 1 , 7 , 8 , 9 and the splicing site mutation IVS7 + 1G>T. 1 L34R and T22M are located in the UbR domain, R378G is located in the F‐box domain, and R498X is in the PRR domain. In 2018, Wei et al 1 first reported a case of PARK15 in a Chinese patient due to complex heterozygous mutations of the FBXO7 gene, namely the missense mutation N51S and the nonsense mutation E470X. In this study, the two siblings were found to carry complex heterozygous mutations that were inherited from their parents, including the nonsense mutation p.Trp134* and the splicing site mutation IVS5‐1G > A, both of which seriously affected the structure of the FBXO7 gene, resulting in the loss of function. The patients had developed severe clinical phenotypes during infancy, which might be correlated with the types of mutations.
Previous studies have shown that the FBXO7 protein is located in mitochondria and involved in mitochondrial stability through interactions with Parkin and PINK1. FBXO7 dysfunction decreases the function of the ubiquitin‐proteasome system, resulting in neuron degeneration. 1 , 15 Recent studies have shown that FBXO7 is a stress reactive protein and can be upregulated by cellular stress. 16 Wild‐type FBXO7 protects cells from stress. However, the Parkinsonism‐associated mutant FBXO7 exacerbates the neurotoxin‐induced cell death and inhibits mitochondrial phagocytosis, which allows selective removal of damaged mitochondria and is an important process for mitochondrial quality control. 15 , 16 At present, the pathogenesis of Parkinsonism arising from FBXO7 mutations still keeps unclear. The phenotypic heterogeneity of patients may be related to the function alterations of FBXO7 different structural domains caused by its mutations. Studies on patients’ mutations and phenotypes will help to elucidate the pathogenesis and provide a basis for the treatment.
ACKNOWLEDGMENTS
The authors are thankful to the patient and the family members participating in the study. Genetic analysis was supported by National Research Institute for Family Planning. This work was supported by the National Key Research and Development Program of China (2016YFC1000307 and 2018YFC1002201).
Jin X, An L, Hao S, et al. Compound heterozygous variants of the FBXO7 gene resulting in infantile‐onset Parkinsonian‐pyramidal syndrome in siblings of a Chinese family. J Clin Lab Anal. 2020;34:e23324 10.1002/jcla.23324
Contributor Information
Yousheng Yan, Email: yys_521@126.com.
Xu Ma, Email: genetic@263.net.cn.
REFERENCES
- 1. Di Fonzo A, Dekker MC, Montagna P, et al. FBXO7 mutations cause autosomal recessive, early‐onset parkinsonian‐pyramidal syndrome. Neurology. 2009;72(3):240‐245. [DOI] [PubMed] [Google Scholar]
- 2. Davison C. Pallido‐pyramidal disease. J Neuropathol Exp Neurol. 1954;13:50‐59. [DOI] [PubMed] [Google Scholar]
- 3. Shojaee S, Sina F, Banihosseini SS, et al. Genome‐wide linkage analysis of a Parkinsonian‐pyramidal syndrome pedigree by 500 K SNP arrays. Am J Hum Genet. 2008;82:1375‐1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wei L, Ding L, Li H, et al. Juvenile‐onset parkinsonism with pyramidal signs due to compound heterozygous mutations in the F‐Box only protein 7 gene. Parkinsonism Relat Disord. 2018;47:76‐79. [DOI] [PubMed] [Google Scholar]
- 5. Conedera S, Apaydin H, Li Y, et al. FBXO7 mutations in Parkinson's disease and multiple system atrophy. Neurobiol Aging. 2016;40(92): 192.e1‐192.e5. [DOI] [PubMed] [Google Scholar]
- 6. Lohmann E, Coquel AS, Honore A, et al. A new F‐box protein 7 gene mutation causing typical Parkinson's disease. Mov Disord. 2015;30:1130‐1133. [DOI] [PubMed] [Google Scholar]
- 7. Gunduz A, Eken AG, Bilgic B, et al. FBXO7‐R498X mutation: phenotypic variability from chorea to early onset parkinsonism within a family. Parkinsonism Relat Disord. 2014;20:1253‐1256. [DOI] [PubMed] [Google Scholar]
- 8. Yalcin‐Cakmakli G, Olgiati S, Quadri M, et al. A new Turkish family with homozygous FBXO7 truncating mutation and juvenile atypical parkinsonism. Parkinsonism Relat Disord. 2014;20:1248‐1252. [DOI] [PubMed] [Google Scholar]
- 9. Paisan‐Ruiz C, Guevara R, Federoff M, et al. Early‐onset L‐dopa‐responsive parkinsonism with pyramidal signs due to ATP13A2, PLA2G6, FBXO7 and spatacsin mutations. Mov Disord. 2010;25:1791‐1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kumar P, Henikoff S. Ng PC Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073‐1081. [DOI] [PubMed] [Google Scholar]
- 11. Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen‐2. Curr Protoc Hum Genet. 2013;76:7.20.1‐7.20.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ramani R, Krumholz K, Huang YF, et al. PhastWeb: a web interface for evolutionary conservation scoring of multiple sequence alignments using phastCons and phyloP. Bioinformatics. 2019;35(13):2320‐2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17(5):405‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. den Dunnen JT. Sequence variant descriptions: HGVS nomenclature and mutalyzer. Curr Protoc Hum Genet. 2016;90(7):1‐7. [DOI] [PubMed] [Google Scholar]
- 15. Zhou ZD, Sathiyamoorthy S, Angeles DC, Tan EK. Linking F‐box protein 7 and parkin to neuronal degeneration in Parkinson's disease (PD). Mol Brain. 2016;9:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou ZD, Xie SP, Sathiyamoorthy S, et al. F‐box protein 7 mutations promote protein aggregation in mitochondria and inhibit mitophagy. Hum Mol Genet. 2015;24(22):6314‐6330. [DOI] [PubMed] [Google Scholar]