

Abstract

A series of novel anticancer hydrazinotriazolothiadiazine-based derivatives were designed based on the structure–activity relationship of the previously reported anticancer triazolothiadiazines. These derivatives were synthesized and biologically screened against full NCI-60 cancer cell lines revealing compound 5l with a potential antiproliferative effect. 5l was screened over 16 kinases to study its cytotoxic mechanism which showed to inhibit glycogen synthase kinase-3 β (GSK-3β) with IC50 equal to 0.883 μM and 14-fold selectivity over CDK2. Also, 5l increased active caspase-3 levels, induced cell cycle arrest at the G2-M phase, and increased the percentage of Annexin V-fluorescein isothiocyanate-positive apoptotic cells in PC-3 prostate cancer-treated cells. Molecular docking and dynamics were performed to predict the binding mode of 5l in the GSK-3β ATP binding site. 5l can be utilized as a starting scaffold for developing potential GSK-3β inhibitors.
1. Introduction
Cancer is among the most common causes of mortality worldwide and the second leading death cause in the United States.1 According to cancer statistics reported in 2020 by the American Cancer Society, prostate cancer is the leading cancer type with estimated new cases and the second with estimated cancer deaths in the US in 2020.1 Despite continuing efforts for the development of anticancer agents, the current cancer therapeutic modalities are still limited because of accompanied side effects and development of drug resistance.2 Thus, identifying novel effective therapies for various types of cancers is a continuing focus of medical research.
In the last two decades, the basis of cancer evolution has been tackled on the molecular basis, allowing the characterization of a plethora of key cellular components involved in carcinogenesis. Among these cellular components are protein kinases, which can trigger tumorigenesis when mutated or overly expressed. Protein kinases play a pivotal role in nearly all cellular functions through controlling metabolism, transcription, cell division, movement, survival, signaling, and programmed cell death.3 They transfer a γ phosphate group from an ATP molecule to substrate proteins by catalyzing the reaction of adding a phosphate group into a nucleophilic amino acid in the presence of an Mg2+ ion.4 In general, they fall, according to the amino acid being phosphorylated, into four main groups, tyrosine kinases (TKs), serine/threonine kinases (STKs),3 dual specificity kinases, which phosphorylate both serine/threonine and tyrosine amino acids,5 and histidine kinases.6 Kinases, together with phosphatases, control over the reversible protein phosphorylation process, in which any unbalance leads to various disease states.3,7,8 Impaired kinase activity results in uncontrolled cell proliferation, which is common in cancer.9 Because of its involvement in many diseases including cancer, this enzyme family has become one of the most important drug targets in the 21st century.8
Glycogen synthase kinase-3 (GSK-3), an enzyme that was reported in late 1970s, was known to regulate glycogen synthase; hence, its name was driven.10−12 Later on, it was found to modulate several other cellular activities by phosphorylating over 100 protein targets making it the busiest kinase.13 GSK-3 is a poly functional STKs that exists in two isoforms, GSK-3α and GSK-3β, which share a high degree of similarity within their active kinase domain.11 GSK-3β is implicated in many age-related disorders such as diabetes,14 Alzheimer’s disease,15 bipolar disorder,16 inflammation,17 cardiovascular diseases,18 and cancer.19 The role of GSK-3β in cancer is paradoxical as it is reported to serve as either a tumor suppressor or tumor promoter.20 It is reported to be a tumor suppressor in breast,21−23 lung,24,25 oral,26,27 and skin28,29 cancers, and tumor promoter in prostate,30 colorectal,31,32 glioblastoma multiforme,33,34 renal,35 pancreas,36−39 leukemia,40,41 and hepatocellular carcinoma,42,43 through inhibition of autophagy mediated by activation of mammalian target of rapamycin complex-1 (mTORC1) activity.44
GSK-3β is upregulated in androgen receptor (AR)-dependent prostate cancer.30 Inhibition of GSK-3β has been found to suppress prostate cancer cells proliferation in vitro45−49 and tumor growth in xenograft in vivo.50 Inhibitors of GSK-3β that have reached clinical trials are summarized in Figure 1A.51 Among the reported inhibitors, it was found that many possess fused 5–6-membered heterocyclic ring systems52,53 (I–III, Figure 1B).
Figure 1.

(A) Representative GSK-3β inhibitors under clinical trials, (B) GSK-3 inhibitors having 5–6-fused ring system (I–III) including our discovered triazolothiadiazine (5l).
Triazolothiadiazine-based scaffolds possess versatile medicinal uses54 such as antimicrobial,55 anti-inflammatory,56 antitubercular,57 and anticancer agents.58−72 Triazolothiadiazine-based scaffolds (Figure 1B) bear some common structural features with the 5–6-membered fused ring system GSK3B inhibitors.
In this work, structure–activity relationship (SAR) of triazolothiadiazine compounds with reported anticancer activity is generated to give insights into the design of our novel hydrazinotriazolothiadiazine kinase inhibitor analogues. Thus, a series of disubstituted hydrazinotriazolothiadiazine analogues were synthesized and biologically assessed on NCI-60 cell lines. Among the tested compounds, 5l was found to possess potential GSK-3β inhibitory activity through target fishing and kinase screening.
2. Results and Discussion
2.1. Rational Design Based on SAR of Reported Anticancer Triazolothiadiazines
By looking at the reported anticancer triazolothiadiazines, general structural features can be concluded (Figure 2). The fused 5–6-membered ring system is essential for activity. The unfused 5-membered ring systems were found to be inactive.58−60 The 3-Mercapto-oxadiazole (IV, Figure 2)60 and the 3-mercapto-4-amino-(1,2,4)-triazole (V, Figure 2)58−60 are reported to be inactive. Unsaturation of the thiadiazine ring appears to be important for activity, as the saturated derivative was reported to be inactive (VI, Figure 2).61 Aromatic substitution at position 3 of the triazole ring is connected through a spacer with varied length of zero,62−66 one,58,59 two,67 or three58,61,68−70 atoms. The 3-atom spacer showed better activity. Aromatic substitution at 6-position of the triazolothiadiazine ring showed superior activity over the aliphatic counterparts. Halogeno-substituted aryls, especially with chlorine,61−63,65−68,70 were better with regard to activity than other electron-donating groups, as aliphatic substitution demolished the compounds’ anticancer activity.68 Substitution at position 7 of the triazolothiadiazine ring can be important for activity, but not essential;71 in addition, it can be utilized for blocking metabolic oxidation of thioether.61
Figure 2.

General SAR for triazolothiadiazine analogues possessing anticancer/antitumor activity.
In our design, we kept the fused 5–6 triazolothiadiazine unsaturated ring system which is essential for activity. In position 3 of the triazolothiadiazine, we kept the three-atom spacer represented by an arylidene hydrazone and varied the aryl moiety with substituents of diverse steric, electronic, and polar properties. In position 6 of the triazolothiadiazine, a phenyl or 4-chloro-phenyl was kept.
2.2. Chemistry
The synthesis of target compounds 5 is shown in Scheme 1. The synthesis of compound 5a was attempted as a one-pot reaction of equimolar amounts of purpald 1, 2-bromoacetophenone 2a and benzaldehyde 4a in refluxing absolute ethanol and few drops of glacial acetic acid as reported by Thirupaiah and Vedula;72 however, inspite of the consumption of the starting materials as monitored by thin-layer chromatography (TLC), the target compound was not attained with appropriate degree of purity and was obtained in poor yields.
Scheme 1.

Reagents and conditions: (a) HCl, EtOH, H2O, reflux 1 h, 70–80%; (b) EtOH, reflux 2 h, 85–92%.
The lack of efficient methodology for synthesis of compounds 5 prompted us to do the reaction in a stepwise fashion through the stepwise synthesis of the hydrazino intermediate 3 followed by reaction with the corresponding aldehyde. This would allow us to have higher purity and yields of the final compounds. As, to the best of our knowledge, there was no literature precedence for the synthesis of intermediate 3, it was decided to develop synthetic methodology toward these compounds taking into account that the nucleophilicity of sulfhydryl, amino, and hydrazino groups are very close. Attempts to react 1 and 2a in refluxing ethanol in the absence of base, presence of NaOH, Na2CO3, or acetic acid resulted in a mixture of products that were difficult to purify. Dickinson and Jacobsen73 selectively reacted benzaldehyde at the amino group of purpald in very good yield by dissolving purpald in 1 M HCl and adding benzaldehyde dissolved in ethanol over 10 min, and the reaction was left stirring for further 10 min. Applying the same methodology using phenacylbromide successfully afforded the desired product 3a in very good yield (80%). The same reaction conditions also gave 3b in 79% yield. With intermediate 3 in hand, we proceeded toward the synthesis of target compounds 5 which were easily prepared in excellent yields (83.1–91.5%) through the reaction of the appropriate aldehyde and intermediates 3.
Refluxing a mixture of 3 and an aldehyde 4 in ethanol while monitoring the reaction progress by TLC effectively yielded the desired conversion in 2 h. The reaction mixture was then cooled to room temperature and quenched with Na2CO3 solution. The crude precipitate formed was filtered, washed with water, dried, and recrystallized from appropriate organic solvent.
The Fourier transform infrared (FTIR) spectra of the hydrochloride salts of compounds 3a and 3b showed broad peaks in the 3500–2500 cm–1. This peak disappeared after the coupling of 3a and 3b with aldehyde.
2.3. Biological Evaluation
2.3.1. In Vitro Antiproliferative Screening against NCI-60 Cell Lines
All twenty synthesized compounds (5a–t) were screened by the National Cancer Institute (NCI), NIH, Bethesda, Maryland, USA (https://dtp.cancer.gov/compsub/)74 under the Developmental Therapeutic Program (DTP) against NCI-60 human tumor cell lines. Compounds screened with respective NCI codes NSC-809289, NSC-809281, NSC-809280, NSC-809286, NSC-809284, NSC-809285, NSC-809288, NSC-809282, NSC-809287, NSC-809283, NSC-809290, NSC-806976, NSC-806975, NSC-806979, NSC-806978, NSC-809292, NSC-809294, NSC-809291, NSC-809293, and NSC-806977 were tested for determination of percent inhibition of the full NCI-60 cell panel at a concentration of 10 μM. The compounds showed moderate inhibitory activity except for compound 5l which showed the highest percent inhibition over the 60 cell lines with mean growth inhibition percentage of 45.44% (Figure 3) (Figure S1).
Figure 3.

Heat map depicting the growth inhibition percentage of the compounds 5a–t (y-axis) at concentration of 10 μM against the NCI-60 cell lines (x-axis).
The series of the compounds with p-choloro substitution (5k–t) is more active than the unsubstituted series (5a–j), an observation consistent with our derived SAR of the reported triazolothiadiazine anticancer agents (Figure 2). This observation is consistent over the whole series except for the compounds with biphenyl hydrazone compounds, where the compound with the unsubstituted phenyl ring at position 6 (5f) is more active than the p-cholro substituted congenere (5p).
Concerning the phenyl ring of the hydrazinyl substitution at position 3 of the triazolothiadiazine ring, its unsubstitution (5a and 5k), its substitution with p-phenyl (5f and 5p), or replacement with 2-furyl (5h and 5r) or 3-thiophenyl (5i and 5s) rings rendered the compounds inactive as concluded from their mean percent inhibition, which are the least when compared to the rest of the series (5b–e, 5g, 5j, 5l–o, 5q, and 5t). On the other hand, substitution of the phenyl ring generally enhances anticancer activity, especially with p-CF3 (5l) which dramatically increased the activity especially in RXF 393, UO-31, MCF7, and PC-3 cell lines.
5l showed 83.63, 96.48, and 79.34% inhibition on NSCLC HOP-62, HOP-92, and NCI-H226 cell lines, respectively. Moreover, it showed 79.74 and 124.92% inhibition on CNS SNB-19 and SNB-75 cell lines, respectively. Also, it showed 83.65, 82.61, 80.14, 96.96, and 91.7% inhibition on melanoma MALME-3M, ovarian OVCAR-4, ovarian OVCAR-8, renal RXF 393, and breast HS 578T cell lines, respectively. On renal cancer cell lines, its inhibition percentage was 72.4 and 71.64% on 786-0 and ACHN cell lines, respectively (Figure 4).
Figure 4.

Percent inhibition of compound (5l) against 22 cell lines from the NCI-60 cell lines that exhibit the percent inhibition range from 50 to 125% (red: >90%, blue: 80–90%, green: 70–80%, black: <70%).
2.3.2. Ligand-Based Target Fishing and Kinase Screening
In pursuit of identifying a possible molecular kinase target(s) for the prepared series of compounds, the ligand-based target fishing (LBTF) approach was adopted (Figure 5), implementing 3D similarity searching between a query compound (5l) and a target-annotated compound database, binding database (www.bindingdb.org) (downloaded on June 6th 2018, 1.4 M compounds).75−77 The FastROCS shape comparison software toolkit78 was used for the 3D similarity searching. The target fishing results are shown in Table T1 in the Supporting Information. Among the targets of the 500 compounds most similar to 5l, 21 kinases were retrieved. Of the 21 resulting kinases, the following 8 kinases were discarded: (i) adenosine kinase and (ii) leucine-rich repeat serine/threonine-protein kinase 2 (no relation to cancer), (iii) serine/threonine-protein kinase PknB (PknB) (not present in human genome), (iv) serine/threonine-protein kinase PLK1 (PLK1), (v) dual-specificity protein kinase CLK3 (CLK3), and (vi) vascular endothelial growth factor receptor 3 (VEGFR3) (low activity of the resulting compounds, with IC50 > 10 μM), (vii) dual specificity protein kinase CLK4 (CLK4), and (viii) casein kinase I (CK1) (only one inhibitor retrieved from the target fishing). GSK-3 alpha (GSK-3α) and serine/threonine-protein kinase pim-3 (PIM3) were considered; to account for the isoform selectivity of the designed compounds within the kinase family, and vascular endothelial growth factor receptor 2 (VEGFR2) was considered in order to test the antiangiogenic effect of the designed compounds, resulting in 16 kinases: cyclin-dependent kinase 1 (CDK1), cyclin-dependent kinase 2 (CDK2), epidermal growth factor receptor (EGFR), fibroblast growth factor receptor 1 (FGFR1), GSK-3α, GSK-3 beta (GSK-3β), insulin-like growth factor 1 receptor (IGF1R), VEGFR2, mitogen-activated protein kinase 8 (MAPK8), MAP kinase-interacting serine/threonine-protein kinase 1 (MKNK1), serine/threonine-protein kinase pim-1 (PIM1), serine/threonine-protein kinase pim-2 (PIM2), serine/threonine-protein kinase pim-2 (PIM3), phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha isoform (PIK3C2A), MAP3K7-binding protein 1 (TAK1/TAB1), and hepatocyte growth factor receptor (MET).
Figure 5.

Computational LBTF workflow.
Compound 5l was screened on 16 kinases to investigate for the possible mechanism underlying its anticancer activity. The 16 kinases are distributed through the kinase families as follows: (i) the CAMK group (MKNK1 and PIM1/2/3), (ii) the CMGC group (CDK1/2, GSK3α/β and MAPK8), (iii) the TK group (EGFR, FGFR1, IGF1R, VEGFR2 and MET), (iv) lipid kinase (PI3K-C2α), and (v) the STE group (MAP3K7). Compound 5l was screened over the kinases to determine percent inhibition at 10 μM concentration. GSK-3β was determined to have the highest percent inhibition with 71% (Figure 6). The screening results showed 5l selectivity toward GSK-3β kinase over the rest of the kinases especially CDK1 and CDK2, which have high sequence similarity with GSK-3β,53 with 7 and 14 folds, respectively.
Figure 6.

Results of kinase screening of compound 5l on the 16 kinases represented in kinase percent inhibition at a concentration of 10 μM. The experiments were conducted in duplicates.
IC50 of 5l on GSK-3β was then measured to be 0.883 ± 0.0294 μM. The AR-A014418 GSK-3β inhibitor79 was used as a reference with measured IC50 = 0.370 ± 0.0111 μM.
2.3.3. Effect of 5l on Caspase-3 Level
Caspase-3 (active) level measurement and cell cycle analysis were performed on PC-3 prostate cancer cells, which were chosen because of the reported correlation between GSK-3β inhibition and prostate cancer cells suppressed proliferation in vitro45−49 and suppressed tumor growth in xenograft in vivo.50 GSK-3β inhibition induces apoptosis in cancer cell lines,80 so that the active caspase-3 level was evaluated in PC-3 cells in order to confirm the cytotoxic activity of 5l on PC-3 cells and compared to its levels in control untreated PC-3 cells. As shown in Figure 7, caspase-3 was 7.63-fold higher in 5l-treated PC-3 cells compared to control. These results further confirm the apoptotic potential of 5l.
Figure 7.

Caspase-3 (active) levels measured in both 5l-treated and untreated cells. Values are presented as means ± S.D. from three independent experiments performed in triplicates. *p < 0.05 significant from control untreated PC-3 cells using the Student’s t-test.
2.3.4. Effect of 5l on Cell Cycle
Cell cycle analysis showed that 5l caused cell cycle arrest at the G2-M phase, as shown in Figure 8. The number of 5l-treated cells were 2.1- and 12.9-fold higher in the G2 and pre-G1 phases, compared to control, respectively. In addition, 5l caused 10.5- and 63.7-fold increase in early and late apoptotic cells, respectively, as compared to control. Moreover, 5l treatment caused a 2-fold increase in necrotic cells compared to control. These results, together with NCI results, strongly suggest that compound 5l has an antiproliferative effect on PC-3 cells.
Figure 8.

PC-3 cell cycle analysis results. (a) Histograms representing the cell cycle phases in 5l-treated cells compared to control untreated cells. (b) Annexin V-FITC and PI-double stained untreated vs 5l-treated PC-3 cells analyzed with flowcytometry. Percentages of living (bottom left), early apoptotic (bottom right), necrotic (top left), and late apoptotic (top right) cells are shown. (c) Stacked histogram representing cells percentages of each cell cycle phase in 5l-treated PC-3 cells compared to control.
2.4. Molecular Modeling
2.4.1. Molecular Docking
The molecular docking experiment was conducted in order to predict the binding mode of 5l inside the GSK-3β ATP binding site. The GSK-3β protein structure co-crystallized with AR-A014418, the compound used as a reference in GSK-3β IC50 determination (PDB ID: 1Q5K),79 was used as protein. Docking was carried out using FRED and HYBRID modules of OpenEye software.81−83 Both applications used the same search algorithm and scoring function; however, HYBRID utilizes co-crystallized ligand pose information to improve binding pose prediction, unlike FRED which docks ligands blindly. First, docking validation for both modules was done through the pose retrieval experiment by docking AR-A014418 in the binding site and measuring root-mean-square deviation (rmsd) between the co-crystallized and docked poses. rmsd was measured to be 8.357 Å and 0.599 Å, for FRED and HYBRID, respectively, which gives HYBRID better credibility in pose prediction over FRED. The docked pose predicted by HYBRID reproduces the same interactions that the co-crystallized pose possesses [hydrogen bonds with valine 135 (Val135) and proline 136 (Pro136) amino acids of the hinge region], while the FRED-predicted pose failed to reproduce such interactions showing a great deviation from the co-crystallized pose (Figure 9).
Figure 9.
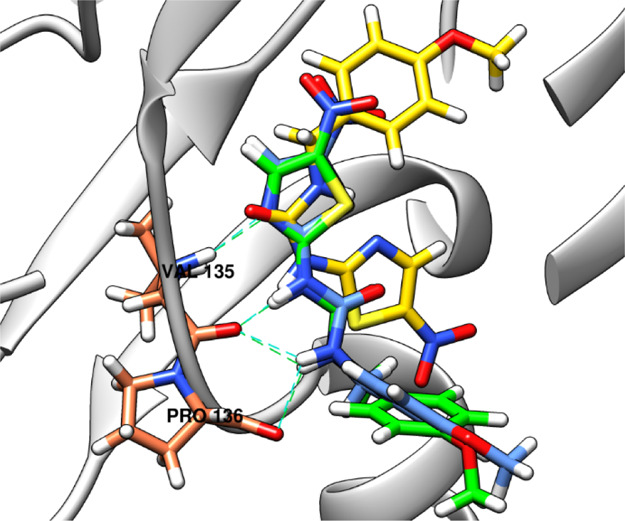
Docking validation results. HYBRID-predicted docked pose of AR-A014418 (blue) is superimposed with the co-crystallized AR-A014418 (green) with rmsd = 0.599 Å, while the docked pose predicted by FRED (yellow) shows a great deviation with rmsd = 8.357 Å. Hydrogen bonds of co-crystallized AR-A014418 (green dashed lines) with Val135 and Pro136 are conserved by HYBRID docked pose (cyan dashed lines), while the FRED docked pose shows no hydrogen bonds with the protein amino acids. Valine 135 and proline 136 amino acids of the hinge region are depicted in coral, while the protein ribbon in gray colors.
Docking of 5l with both FRED and HYBRID was used to predict the binding mode of 5l inside the ATP binding pocket of GSK-3β. The binding pose predicted by HYBRID, referred to as 5l_HYBRID throughout the text, was similar to the reported pose of GSK-3 inhibitors with pyrazolpyridine and pyrazolopyridazine scaffolds similar to triazolothiadiazines52,53 (Figure 10). They are reported to have two hydrogen bonds with Val135, where N2 of the pyrazole acts a hydrogen bond acceptor and NH at position 3 acts as a hydrogen bond donor and one hydrogen bond with aspartate (Asp133), where NH at position 1 of the pyrazole acts a hydrogen bond donor. Lacking the hydrogen bond donor NH at position 1 of triazolothiadiazine makes it lose the hydrogen bond with Asp133, while having hydrogen bond acceptor N2 of triazole and hydrogen bond donor NH at position 3 makes it keep the two hydrogen bonds with Val135. In addition, the 4-cholorophenyl is placed in the back pocket of the ATP binding site and the 4-CF3 phenyl hydrazone is positioned toward the solvent accessible region, similar to the reported pyrazolopyridazine GSK-3 inhibitor (Figure 10). On the other hand, the binding pose predicted by FRED, referred to as 5l_FRED throughout the text, is flipped with regard to the pose reported by pyrazolpyridines and pyrazolopyridazines GSK-3 inhibitors (Figure 10). The 4-chlorophenyl is positioned toward the solvent accessible region, and the 4-CF3 phenyl hydrazone is placed in the back pocket of the ATP binding site. It is also predicted to make a hydrogen bond with Val135 through the hydrogen bond donor NH at position 3.
Figure 10.
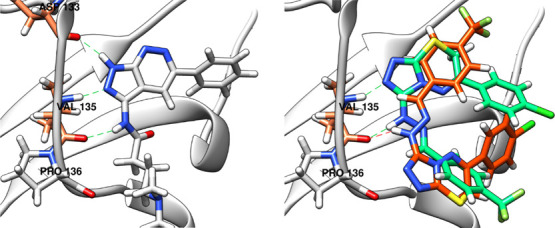
Left: Reported binding mode of a pyrazolopyridazine GSK-3 inhibitor (gray) with three hydrogen bonds with Asp133 and Val135 (green dashed lines). Right: Binding mode of 5l predicted by FRED (5l_FRED, orange), showing one hydrogen bond formed with Val135 (orange dashed line), and HYBRID (5l_HYBRID, green), showing two hydrogen bonds formed with Val135 (green dashed lines).
2.4.2. Molecular Dynamics
Molecular dynamics (MD) simulations were conducted to test the stability of 5l binding modes predicted by both FRED (5l_FRED) and HYBRID (5l_HYBRID) docking. AR-A014418 was also subjected for MD simulations to account for its binding mode and protein dynamics as a reference, resulting in a total of three MD runs. MD was carried out for 20 ns each, at NPT ensemble (constant number of particles, pressure, and temperature), for minimization and equilibration purposes.
The rmsd, a measure of time-dependent structure stability, is measured for protein backbone atoms (Cα, C, and N) for the three MD runs (Figure 11). The rmsd reached stability after 1 ns with no prominent fluctuation throughout the three MD runs. The average rmsd was 1.60 ± 0.21, 1.69 ± 0.26, and 1.53 ± 0.22 Å (mean ± SD) for AR-A014418, 5l_HYBRID, and 5l_FRED proteins, respectively. These results give an indication of proteins stability during the three runs. Protein residue conformational changes and stability are measured by per residue root-mean-square fluctuation (RMSF) (Figure 11). All amino acid residues of the binding site have RMSF < 1 Å, which indicates a relatively stable binding with AR-A014418, 5l_HYBRID and 5l_FRED and minimal local conformational changes in the binding site. Protein compactness and equilibrium conformation were assessed by measuring the protein radius of gyration (Rg) throughout the simulation time (Figure 11). Rg values show consistency within the three experiments, which is an indication of protein conformational stability throughout the simulation time. As shown in Figure 11, Rg values are 44.7 ± 0.12, 44.7 ± 0.12, and 44.7 ± 0.12 Å (mean ± SD) for AR-A014418, 5l_HYBRID, and 5l_FRED proteins, respectively. The time stability of Rg confirms our knowledge that major conformational changes need micro-to-milli second simulation time period to emerge, on the contrary to the nanosecond time scale, in which major protein conformational changes rarely occur.
Figure 11.

Top: Protein rmsd of the three MD runs showing stability after 1 ns and minimal fluctuation throughout the simulation time. Middle: Per residue RMSF of the three MD runs showing RMSF < 1 Å for the binding site residues (colored rectangles) with low RMSF in the hinge region residues (red rectangle). Bottom: Protein radius of gyration (Rg) of the three MD runs showing compactness of the protein throughout the simulation time with no major conformational changes. AR-A014418, 5l_HYBRID, and 5l_FRED proteins are depicted in green, blue, and yellow colors, respectively.
Ligand rmsd analysis throughout the time of simulation showed AR-A014418 to have the least deviation from the co-crystallized pose, with an rmsd of 1.46 ± 0.34 Å (mean ± SD). Comparing 5l_HYBRID and 5l_FRED ligands rmsd analysis revealed a greater deviation of 5l_FRED than that of 5l_HYBRID with an average rmsd of 2.08 ± 0.46 and 2.71 ± 0.44 Å (mean ± SD) for 5l_HYBRID and 5l_FRED, respectively (Figure 12). This indicates that 5l_FRED abandoned its initial pose, depicted in Figure 10, to another distant binding mode. However, 5l_HYBRID had more stable binding mode indicated by its lower rmsd and bidentate-hydrogen-bond anchorage with the Val135 hinge region amino acid (Figure 12).
Figure 12.

rmsd of the ligand during the three MD runs: AR-A014418 (green), 5l_HYBRID docked pose (blue), and 5l_FRED docked pose (yellow). The 5l_FRED docked pose shows the highest deviation which indicates the instability of this binding pose.
Hydrogen bonding analysis between the ligands and their proteins was carried out in order to investigate the hydrogen bonding contribution to the overall binding interactions. AR-A014418, 5l_HYBRID, and 5l_FRED had 0.67 ± 0.57, 0.64 ± 0.56, and 0.59 ± 0.52 Å (mean ± SD) hydrogen bonds through the 20 ns simulation time. The lower average number of 5l_FRED hydrogen bonds with its protein indicates its weaker binding compared to the number of hydrogen bonds of AR-A014418 and 5l_HYBRID with their proteins that showed comparable binding stability (Figure 13). The main hydrogen bonding interaction is shown to be with the carbonyl and amine moieties of the Val135 residue. This is depicted in Figure 13 in which hydrogen bonds with the ligands and their proteins are compared with their hydrogen bonds with the Val135 amino acid. Both AR-A014418 and 5l_HYBRID maintain their hydrogen bonding interactions with Val135. On the other hand, 5l_FRED lost its hydrogen bonding interactions with Val135 in the first few picoseconds of the simulation. The one hydrogen bond kept by 5l_FRED is attributed to the Pro136 hydrogen bond acceptor carbonyl moiety.
Figure 13.

Number of hydrogen bonds formed during the simulation time between the ligands and the protein (left), and the ligands and Valine 135 (right).
Finally, comparing rmsd of docked poses of 5l_HYBRID and 5l_FRED makes the HYBRID-predicted docking pose more acceptable. The lower rmsd of 5l_HYBRID and its higher number of hydrogen bonds throughout the MD simulation assures 5l_HYBRID pose stability. These results together suggest 5l_HYBRID to be the binding mode adopted by 5l inside the GSK-3β protein.
3. Conclusions
5l has been discovered to be a novel GSK-3β inhibitor with antiproliferative activity on the PC-3 prostate cancer cell line through NCI-60 cancer cell line screening and LBTF. This study has demonstrated the apoptotic effect of 5l on PC-3 cells as shown by its ability to increase active caspase-3 levels and arrest cell cycle at G2-M phase. A combination of molecular docking and dynamics simulations was used in predicting the binding mode of 5l, which adopted that of reported similar pyrazolopyridazine GSK-3 inhibitors. 5l can be used as a scaffold for further development of GSK-3β inhibitors.
4. Experimental Section
4.1. Chemistry
4.1.1. General
Starting materials and reagents were purchased from Sigma-Aldrich (USA) or Alfa Aesar Organics and used without further purification. Solvents were purchased from Fisher Scientific or Sigma-Aldrich and used without further purification. Reactions were monitored by analytical TLC, performed on silica gel 60 F254 packed on aluminum sheets, purchased from Merck, with visualization under UV light (254 nm). Melting points were recorded on Stuart Scientific apparatus and were uncorrected. 1HNMR spectra were recorded in δ scale given in ppm on a Bruker 400 MHz spectrophotometer and referred to trimethylsilyl at Cairo University. 13CNMR spectra were recorded in δ scale given in ppm on a Bruker 101 MHz spectrophotometer at Cairo University. FT-IR spectra were recorded on a Thermo Scientific Nicolet iS10 spectrometer at Ain Shams University. Electron ionization (EI)–mass spectroscopy (MS) spectra were recorded on a Thermo Scientific ISOLT mass spectrometer at the Regional Center for Mycology and Biotechnology, Al-Azhar University. Elemental analyses were performed on a Thermo Scientific Flash 2000 elemental analyzer at the Regional Center for Mycology and Biotechnology, Al-Azhar University.
4.1.2. Synthesis
4.1.2.1. General Procedure for the Preparation of 3a,b
A solution of 4-amino-5-hydrazinyl-4H-1,2,4-triazole-3-thiol (purpald) (1, 1 equiv, 30.1 mmol, 4.4 g) in 1 M HCl (60 mL) was heated till complete dissolution. Then, the respective 2-bromo-4′-substituted acetophen-1-one (2, 0.9 equiv, 27.09 mmol) dissolved in hot absolute methanol (60 mL) was added to 4-amino-5-hydrazineyl-4H-1,2,4-triazole-3-thiol (purpald) HCl solution dropwise over 15 min. The reaction mixture was heated under reflux for 45 min. The reaction mixture was then diluted with 20 mL water portionwise while hot and then left to crystallize at room temperature for 3 h and in the freezer overnight. The reaction mixture was then filtered, washed with minimal amount of water (10 mL), and dried to afford the titled compounds 3a–b. The compound is purified by crystallization form aqueous ethanol to yield pure crystals.
4.1.2.2. 2-(6-Phenyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-3-yl)hydrazin-1-ium Chloride (3a)
The titled compound was separated as yellowish white crystals (6.13 g, 80%); mp 205–206 °C; 1H-NMR (400 MHz, DMSO-d6): δ 4.34 (s, 2H), 7.52–7.62 (m, 3H), 8.00 (d, J = 7.3 Hz, 2H), 10.18 (br s, 1H, NH, D2O exchangeable). 13C-NMR (100 MHz, DMSO-d6): δ 23.2, 128.1, 129.4, 132.7, 133.3, 139.9, 151.3, 156.1. FTIR (KBr, cm–1) ṽ: 3727, 3482, 3380, 3310, 3261, 3205, 3004, 2910, 2790, 2742, 1734, 1630, 1546, 1448, 1415, 1287, 1228, 1194, 1163, 1062, 1049, 1025, 1007, 935, 910, 852, 828, 772, 741, 693, 675, 627, 603, 562, 499. EI-MS: (Mwt: 282.75): m/z (% rel. int.), 282.34 [M+, (16.15%)], 75.11 (100%); Anal. Calcd for C10H11ClN6S: C, 42.48; H, 3.92; N, 29.72. Found: C, 42.71; H, 3.85; N, 29.89.
4.1.2.3. 2-(6-(4-Chlorophenyl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-3-yl)hydrazin-1-ium Chloride (3b)
The titled compound was separated as yellowish white crystals (6.78 g, 79%); mp 184–185 °C; 1H-NMR (400 MHz, DMSO-d6): δ 4.48 (s, 2H), 7.67 (d, J = 8.7 Hz, 2H), 8.13 (d, J = 8.7 Hz, 2H), 9.95 (br s, 1H, NH). FTIR (KBr, cm–1) ṽ: 3726, 3410, 3339, 3250, 3130, 3080, 2987, 2921, 2671, 1711, 1590, 1543, 1494, 1415, 1396, 1332, 1278, 1222, 1194, 1089, 1043, 1010, 933, 906, 844, 752, 709, 640, 571, 503, 471, 457, 419. EI-MS: (Mwt: 317.19): m/z (% rel. int.), 319.61 [M+ + 2, (27.33%)], 317.14 [M+, (34.92%)], 253.44 (100%); Anal. Calcd for C10H10Cl2N6S: C, 37.87; H, 3.18; N, 26.50. Found: C, 38.11; H, 3.27; N, 26.78.
4.1.2.4. General Procedure for the Preparation of 5a–t
A solution of the 2-(6-(4-substituted)phenyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-3-yl)hydrazin-1-ium chloride (3a,b) (1 equiv, 3.5 mmol) in 50% ethanol (30 mL) was stirred under reflux until dissolution. Then, the freshly prepared solution of aldehyde (4, 0.9 equiv, 3.15 mmol) in ethanol (5 mL) was added to the reaction mixture. The reaction mixture was maintained under reflux for 2 h and quenched with 5 mL saturated Na2CO3 solution. The crude product was isolated by hot filtration, followed by crystallization from 50% aqueous dimethylformamide (DMF). The product was then dried under vacuum in a vacuum oven overnight.
4.1.2.5. (E)-3-(2-Benzylidenehydrazinyl)-6-phenyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine (5a)
The titled compound was crystallized from aqueous DMF as yellow crystals (0.92 g, 87.3%); mp 220–223 °C; 1H NMR (400 MHz, DMSO-d6): δ 10.92 (s, 1H, NH, D2O exchangeable), 8.47 (s, 1H, CH), 8.18–8.07 (m, 2H, Ar H), 7.72–7.64 (m, 2H, Ar H), 7.65–7.54 (m, 3H, Ar H), 7.48–7.34 (m, 3H, Ar H), 4.41 (s, 2H, CH2). 13C NMR (101 MHz, DMSO-d6): δ 154.38, 151.39, 144.60, 137.56, 135.18, 133.90, 132.21, 129.75, 129.37, 129.24, 128.01, 126.88, 40.33. FTIR (KBr, cm–1) ṽ: 3140, 3060, 3010, 2966, 1732, 1716, 1610, 1590, 1526, 1473, 1445, 1392, 1328, 1307, 1392, 1328, 1307, 1293, 1227, 1174, 1113, 1057, 1013, 945, 894, 834, 760, 688, 615, 599, 507, 471, 458. EI-MS: (Mwt: 334.10): m/z (% rel. int.), 334.00, [M+, (20.89%)], 254.75 (100%); Anal. Calcd for C17H14N6S: C, 61.06; H, 4.22; N, 25.13; S, 9.59. Found: C, 60.87; H, 4.38; N, 25.39; S, 9.67.
4.1.2.6. (E)-6-Phenyl-3-(2-(4-(trifluoromethyl)benzylidene)hydrazinyl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine (5b)
The titled compound was crystallized from aqueous DMF as yellow crystals (1.13 g, 89.2%); mp 207–215 °C; 1H NMR (400 MHz, DMSO-d6): δ 11.24 (s, 1H, NH, D2O exchangeable), 8.55 (s, 1H, CH), 8.12 (s, J = 8 Hz, 2H, Ar H), 7.89 (d, J = 7.9 Hz, 2H, Ar H), 7.78 (d, J = 8.1 Hz, 2H, Ar H), 7.65–7.53 (m, 3H, Ar H), 4.42 (s, 2H, CH2). 13C NMR (101 MHz, DMSO-d6): δ 154.48, 151.09, 142.53, 139.33, 137.74, 133.96, 132.17, 129.33, 128.04, 127.30, 126.09, 126.05, 123.30, 40.10. FTIR (KBr, cm–1) ṽ: 3622, 3529, 3124, 3020, 2912, 2858, 2827, 2638, 1620, 1597, 1535, 1481, 1446, 1396, 1327, 1230, 1103, 1068, 1014, 987, 925, 898, 840, 821, 759, 732, 686, 594. EI-MS: (Mwt: 402.08): m/z (% rel. int.), 402.24 [M+, (17.75%)], 57.47 (100%); Anal. Calcd for C18H13F3N6S: C, 53.73; H, 3.26; F, 14.16; N, 20.89; S, 7.97. Found: C, 53.91; H, 3.48; N, 21.23; S, 8.19.
4.1.2.7. (E)-3-(2-(3-Methoxybenzylidene)hydrazinyl)-6-phenyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine (5c)
The titled compound was crystallized from aqueous DMF as off-white crystals (1 g, 87.1%); mp 212–216 °C; 1H NMR (400 MHz, DMSO-d6): δ 10.97 (s, 1H, NH, D2O exchangeable), 8.43 (s, 1H, CH), 8.12 (d, J = 7.3 Hz, 2H, Ar H), 7.65–7.53 (m, 3H, Ar H), 7.35 (t, J = 7.9 Hz, 1H, Ar H), 7.29–7.20 (m, 2H, Ar H), 6.96 (dd, J = 7.9, 2.9 Hz, 1H, Ar H), 4.41 (s, 2H, CH2), 3.79 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6): δ 159.99, 154.33, 151.31, 144.28, 137.51, 136.77, 134.00, 132.15, 130.32, 129.35, 128.03, 119.84, 115.81, 110.97, 55.56, 40.11. FTIR (KBr, cm–1) ṽ: 3167, 3001, 2970, 2920, 2835, 1610, 1581, 1535, 1269, 1114, 686. EI-MS: (Mwt: 364.11): m/z (% rel. int.), 364.67 [M+, (11.56%)], 51.49 (100%); Anal. Calcd for C18H16N6OS: C, 59.33; H, 4.43; N, 23.06; O, 4.39; S, 8.80. Found: C, 59.60; H, 4.56; N, 22.94; S, 8.87.
4.1.2.8. (E)-3-(2-(4-Hydroxy-3-methoxybenzylidene)hydrazinyl)-6-phenyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine (5d)
The titled compound was crystallized from aqueous DMF as yellow crystals (1.07 g, 89.2%); mp 210–213 °C; 1H NMR (400 MHz, DMSO-d6): δ 10.69 (s, 1H, NH, D2O exchangeable), 9.42 (s, 1H, OH, D2O exchangeable), 8.35 (s, 1H, CH), 8.16–8.09 (m, 2H, Ar H), 7.71–7.52 (m, 3H, Ar H), 7.27 (d, J = 1.9 Hz, 1H, Ar H), 7.03 (dd, J = 8.2, 1.9 Hz, 1H, Ar H), 6.83 (d, J = 8.1 Hz, 1H, Ar H), 4.40 (s, 2H, CH2), 3.80 (s, 3H, OCH3). 13C NMR (101 MHz, DMSO-d6): δ 154.18, 151.61, 148.62, 148.48, 145.25, 137.29, 133.97, 132.14, 129.35, 127.98, 126.76, 121.70, 115.81, 108.95, 55.95, 40.38. FTIR (KBr, cm–1) ṽ: 3619, 3566, 3136, 3005, 2966, 2935, 1636, 1609, 1513, 1445, 1463, 1431, 1402, 1349, 1340, 1328, 1291, 1272, 1254, 1196, 1167, 1122, 1099, 1062, 1032, 860, 822, 759, 690, 623, 605, 593. EI-MS: (Mwt: 380.1): m/z (% rel. int.), 380.73 [M+, (28.87%)], 364.40 (100%); Anal. Calcd for C18H16N6O2S: C, 56.83; H, 4.24; N, 22.09; O, 8.41; S, 8.43. Found: C, 57.06; H, 4.38; N, 22.31; S, 8.29.
4.1.2.9. (E)-6-Phenyl-3-(2-(4-(dimethylamino)benzylidene)hydrazinyl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine (5e)
The titled compound was crystallized from aqueous DMF as yellowish brown crystals (1.03 g, 86.6%); mp 216–220 °C; 1H NMR (400 MHz, DMSO-d6): δ 10.55 (s, 1H, NH, D2O exchangeable), 8.33 (s, 1H, CH), 8.21–8.06 (m, 2H, Ar H), 7.69–7.54 (m, 3H, Ar H), 7.50 (d, J = 8.5 Hz, 2H, Ar H), 6.75 (d, J = 8.8 Hz, 2H, Ar H), 4.38 (s, 2H, CH2), 2.96 (s, 6H, 2CH3). 13C NMR (101 MHz, DMSO-d6): δ 153.85, 151.76, 151.45, 145.74, 136.96, 134.06, 132.04, 129.31, 128.16, 127.98, 122.82, 112.35, 40.53. FTIR (KBr, cm–1) ṽ: 3178, 3124, 3093, 3066, 2993, 2889, 2858, 2804, 1597, 1581, 1527, 1512, 1473, 1446, 1365, 1303, 1234, 1180, 1095, 1056, 945, 802, 748, 694, 524. EI-MS: (Mwt: 377.14): m/z (% rel. int.), 377.13 [M+, (27.01%)], 160.53 (100%); Anal. Calcd for C19H19N7S: C, 60.46; H, 5.07; N, 25.98; S, 8.49. Found: C, 60.75; H, 5.11; N, 26.23; S, 8.61.
4.1.2.10. (E)-3-(2-([1,1′-Biphenyl]-4-ylmethylene)hydrazinyl)-6-phenyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine (5f)
The titled compound was crystallized from aqueous DMF as dark yellow crystals (1.13 g, 87.3%); mp 218–220 °C; 1H NMR (400 MHz, DMSO-d6): δ 10.99 (s, 1H, NH, D2O exchangeable), 8.52 (s, 1H, CH), 8.15 (d, J = 8.0, 2H, Ar H), 7.81–7.70 (m, 6H, Ar H), 7.65–7.54 (m, 3H, Ar H), 7.49 (t, J = 7.6 Hz, 2H, Ar H), 7.43–7.36 (m, 1H, Ar H), 4.42 (s, 2H, CH2). 13C NMR (101 MHz, DMSO-d6): δ 154.26, 151.34, 143.96, 141.12, 139.93, 137.43, 134.51, 134.05, 132.13, 129.46, 129.34, 128.17, 128.05, 127.43, 127.05, 40.62, 40.46. FTIR (KBr, cm–1) ṽ: 3566, 3447, 3156, 3053, 3206, 2993, 2900, 1610, 1590, 1556, 1484, 1353, 1291, 1098, 1075, 902, 840, 723, 618. EI-MS: (Mwt: 410.13): m/z (% rel. int.), 410.13 [M+, (31.43%)], 248.40 (100%); Anal. Calcd for C23H18N6S: C, 67.30; H, 4.42; N, 20.47; S, 7.81. Found: C, 67.53; H, 4.59; N, 20.39; S, 7.98.
4.1.2.11. (E)-3-(2-(4-(Benzyloxy)benzylidene)hydrazinyl)-6-phenyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine (5g)
The titled compound was crystallized from aqueous DMF as yellow crystals (1.2 g, 86.4%); mp 213–215 °C; 1H NMR (400 MHz, DMSO-d6): δ 10.75 (s, 1H, NH, D2O exchangeable), 8.41 (s, 1H, CH), 8.13 (d, J = 7.0 Hz, 2H, Ar H), 7.73–7.51 (m, 5H, Ar H), 7.48 (d, J = 7.0 Hz, 2H, Ar H), 7.45–7.38 (m, 2H, Ar H), 7.38–7.31 (m, 1H, Ar H), 7.09 (d, J = 8.4 Hz, 2H, Ar H), 5.16 (s, 2H, OCH2), 4.40 (s, 2H, CH2). 13C NMR (101 MHz, DMSO-d6): δ 159.70, 144.55, 137.32, 137.25, 133.97, 132.13, 129.35, 128.93, 128.43, 128.38, 128.18, 128.11, 127.98, 115.57, 69.78, 40.40. FTIR (KBr, cm–1) ṽ: 3446, 3160, 3060, 3010, 2914, 2868, 1605, 1508, 1474, 1380, 1247, 1170, 1007, 954, 921, 898, 833, 764, 751, 696, 622, 601, 531, 517, 472, 435. EI-MS: (Mwt: 440.14): m/z (% rel. int.), 440.87 [M+, (42.51%)], 399.26 (100%); Anal. Calcd for C24H20N6OS: C, 65.44; H, 4.58; N, 19.08; O, 3.63; S, 7.28. Found: C, 65.31; H, 4.75; N, 19.36; S, 7.09.
4.1.2.12. (E)-3-(2-(Furan-2-ylmethylene)hydrazinyl)-6-phenyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine (5h)
The titled compound was crystallized from aqueous DMF as buff crystals (0.85 g, 83.1%); mp 190–196 °C; 1H NMR (400 MHz, DMSO-d6): δ 10.90 (s, 1H, NH, D2O exchangeable), 8.44 (s, 1H, CH), 8.19–8.08 (m, 2H, Ar H), 7.80 (d, J = 1.9 Hz, 1H, furan H), 7.65–7.52 (m, 3H, Ar H), 6.80 (d, J = 3.4 Hz, 1H, furan H), 6.62 (dd, J = 3.4, 1.8 Hz, 1H, furan H), 4.40 (s, 2H, CH2). 13C NMR (101 MHz, DMSO-d6): δ 154.36, 151.16, 150.37, 144.72, 137.40, 134.77, 133.97, 132.16, 129.34, 128.06, 112.48, 111.90, 40.11. FTIR (KBr, cm–1) ṽ: 3525, 3421, 3143, 3097, 3059, 3012, 2958, 2900, 2804, 1589, 1465, 1327, 1111, 1060, 1014, 933, 732, 686, 590. EI-MS: (Mwt: 324.07): m/z (% rel. int.), 324.60 [M+, (22.81%)], 256.75 (100%); Anal. Calcd for C15H12N6OS: C, 55.54; H, 3.73; N, 25.91; O, 4.93; S, 9.88. Found: C, 55.79; H, 3.81; N, 26.14; S, 9.97.
4.1.2.13. (E)-6-Phenyl-3-(2-(thiophen-3-ylmethylene)hydrazinyl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine (5i)
The titled compound was crystallized from aqueous DMF as buff crystals (0.94 g, 87.6%); mp 210–212 °C; 1H NMR (400 MHz, DMSO-d6): δ 10.79 (s, 1H, NH, D2O exchangeable), 8.55 (s, 1H, CH), 8.12 (d, J = 7.2 Hz, 2H, Ar H), 7.80 (d, J = 3.4 Hz, 1H, thiophene H), 7.65–7.53 (m, 4H, Ar and thiophene H), 7.46 (d, J = 5.1 Hz, 1H, thiophene H), 4.40 (s, 2H, CH2). 13C NMR (101 MHz, DMSO-d6): δ 154.19, 151.33, 140.50, 138.46, 137.26, 134.05, 132.11, 129.32, 128.05, 127.86, 126.51, 124.91, 40.63. FTIR (KBr, cm–1) ṽ: 3503, 3148, 3057, 2950, 2905, 1680, 1590, 1534, 1473, 1384, 1332, 1309, 1288, 1224, 1242, 1164, 1108, 1075, 871, 821, 780, 755, 686, 643, 628, 600, 558, 472. EI-MS: (Mwt: 340.05): m/z (% rel. int.), 340.72 [M+, (36.74%)], 103.33 (100%); Anal. Calcd for C15H12N6S2: C, 52.92; H, 3.55; N, 24.69; S, 18.84. Found: C, 53.17; H, 3.71; N, 24.95; S, 18.97.
4.1.2.14. (E)-3-(2-((1H-Indol-3-yl)methylene)hydrazinyl)-6-phenyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine (5j)
The titled compound was crystallized from aqueous DMF as yellow crystals (1.06 g, 89%); mp 206–212 °C; 1H NMR (400 MHz, DMSO-d6): δ 11.45 (s, 1H, NH, indole NH′, D2O exchangeable), 10.45 (s, 1H, NH, D2O exchangeable), 8.58 (s, 1H, CH), 8.28 (d, J = 7.9 Hz, 1H, indole H), 8.20–8.10 (m, 2H, Ar H), 7.72 (d, J = 2.7 Hz, 1H, indole H), 7.58 (qd, J = 8.7, 7.8, 3.6 Hz, 3H, Ar H), 7.42 (dt, J = 8.0, 1.0 Hz, 1H, indole H), 7.18 (ddd, J = 8.2, 7.0, 1.3 Hz, 1H, indole H), 7.04 (ddd, J = 8.0, 7.1, 1.1 Hz, 1H, indole H), 4.40 (s, 2H, CH2). 13C NMR (101 MHz, DMSO-d6): δ 162.86, 153.94, 152.06, 142.23, 137.40, 136.94, 134.14, 132.03, 129.33, 129.11, 128.01, 124.76, 122.91, 122.54, 120.53, 112.44, 112.11, 40.50, 39.88, 39.67, 39.46, 39.25. FTIR (KBr, cm–1) ṽ: 3170, 3151, 3109, 3055, 2927, 2881, 2738, 3692, 2611, 2569, 2499, 2314, 1662, 1597, 1581, 1469, 1446, 1384, 1099, 752, 666. EI-MS: (Mwt: 373.11): m/z (% rel. int.), 373.86 [M+, (17.93%)], 160.29 (100%); Anal. Calcd for C19H15N7S: C, 61.11; H, 4.05; N, 26.26; S, 8.59. Found: C, C, 61.38; H, 4.16; N, 26.44; S, 8.70.
4.1.2.15. (E)-3-(2-Benzylidenehydrazinyl)-6-(4-chlorophenyl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine (5k)
The titled compound was crystallized from aqueous DMF as yellow crystals (1.06 g, 91.2%); mp 216–221 °C; 1H NMR (400 MHz, DMSO-d6): δ 10.97 (s, 1H, NH, D2O exchangeable), 8.46 (s, 1H, CH), 8.15 (d, J = 8.2 Hz, 2H, Ar H), 7.76–7.57 (m, 4H, Ar H), 7.53–7.31 (m, 3H, Ar H), 4.41 (s, 2H, CH2). 13C NMR (101 MHz, DMSO-d6): δ 153.13, 151.47, 144.77, 137.32, 136.99, 135.28, 132.79, 129.79, 129.69, 129.41, 129.21, 126.89, 40.25. FTIR (KBr, cm–1) ṽ: 3432, 3144, 3053, 2974, 2917, 1619, 1590, 1486, 1445, 1409, 1325, 1089, 1071, 1008, 986, 885, 808, 756, 699, 649, 564, 529, 517, 474. EI-MS: (Mwt: 368.06): m/z (% rel. int.), 368.54 [M+, (19.15%)], 61.98 (100%); Anal. Calcd for C17H13ClN6S: C, 55.36; H, 3.55; Cl, 9.61; N, 22.79; S, 8.69. Found: C, 55.49; H, 3.79; N, 22.63; S, 8.75.
4.1.2.16. (E)-6-(4-Chlorophenyl)-3-(2-(4-(trifluoromethyl)benzylidene)hydrazineyl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine (5l)
The titled compound was crystallized from aqueous DMF as yellow crystals (1.23 g, 89.3%); mp 226–230 °C; 1H NMR (400 MHz, DMSO-d6): δ 11.24 (s, 1H, NH, D2O exchangeable), 8.54 (s, 1H, CH), 8.15 (d, J = 8.4 Hz, 2H, Ar H), 7.90 (d, J = 8.1 Hz, 2H, Ar H), 7.79 (d, J = 8.2 Hz, 2H, Ar H), 7.66 (d, J = 8.6 Hz, 2H, Ar H), 4.41 (s, 2H, CH2). 13C NMR (101 MHz, DMSO-d6): δ 153.28, 151.30, 143.15, 139.27, 137.63, 137.04, 132.68, 129.75, 129.47, 129.38, 127.34, 125.99, 123.26, 40.13. FTIR (KBr, cm–1) ṽ: 3622, 3091, 3027, 2906, 1751, 1602, 1591, 1508, 1479, 1326, 1152, 1092, 1015, 850, 806, 771, 595, 458. EI-MS: (Mwt: 436.04): m/z (% rel. int.), 436.40 [M+, (27.86%)], 181.02 (100%); Anal. Calcd for C18H12ClF3N6S: C, 49.49; H, 2.77; Cl, 8.12; F, 13.05; N, 19.24; S, 7.34. Found: C, 49.36; H, 2.96; N, 19.08; S, 7.38.
4.1.2.17. (E)-6-(4-Chlorophenyl)-3-(2-(3-methoxybenzylidene)hydrazinyl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine (5m)
The titled compound was crystallized from aqueous DMF as yellow crystals (1.15 g, 91.5%); mp 212–216 °C; 1H NMR (400 MHz, DMSO-d6): δ 10.99 (s, 1H, NH, D2O exchangeable), 8.45 (s, 1H, CH), 8.15 (d, J = 8.2 Hz, 2H, Ar H), 7.66 (d, J = 8.7 Hz, 2H, Ar H), 7.35 (t, J = 7.9 Hz, 1H, Ar H), 7.24 (d, J = 7.7 Hz, 2H, Ar H), 7.01–6.92 (m, 1H, Ar H), 4.40 (s, 2H, CH2), 3.79 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6): δ 159.96, 153.16, 151.44, 144.77, 137.40, 137.01, 136.73, 132.78, 130.32, 129.77, 129.41, 119.95, 115.87, 110.99, 55.56, 40.17. FTIR (KBr, cm–1) ṽ: 3626, 3161, 3074, 2998, 2970, 2920, 2876, 2831, 1590, 1556, 1473, 1451, 1353, 1281, 1198, 1092, 1079, 1006, 919, 840, 807, 792, 699, 584, 473. EI-MS: (Mwt: 398.07): m/z (% rel. int.), 398.47 [M+, (29.65%)], 235.56 (100%); Anal. Calcd for C18H15ClN6OS: C, 54.20; H, 3.79; Cl, 8.89; N, 21.07; O, 4.01; S, 8.04. Found: C, 54.37; H, 4.02; N, 21.30; S, 8.17.
4.1.2.18. (E)-6-(4-Chlorophenyl)-3-(2-(4-hydroxy-3-methoxybenzylidene)hydrazinyl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine (5n)
The titled compound was crystallized from aqueous DMF as yellow crystals (1.1 g, 84.2%); mp 218–220 °C; 1H NMR (400 MHz, DMSO-d6): δ 10.72 (s, 1H, NH, D2O exchangeable), 9.43 (s, 1H, OH, D2O exchangeable), 8.36 (s, 1H, CH), 8.15 (d, J = 8.7 Hz, 2H, Ar H), 7.65 (d, J = 8.6 Hz, 2H, Ar H), 7.27 (d, J = 1.9 Hz, 1H, Ar H), 7.03 (dd, J = 8.2, 1.9 Hz, 1H, Ar H), 6.83 (d, J = 8.1 Hz, 1H, Ar H), 4.39 (s, 2H, CH2), 3.80 (s, 3H, OCH3). 13C NMR (101 MHz, DMSO-d6): δ 152.97, 151.48, 148.68, 148.47, 145.21, 136.96, 136.95, 132.90, 129.82, 129.40, 126.79, 121.56, 115.89, 108.97, 55.93, 40.41. FTIR (KBr, cm–1) ṽ: 3404, 3180, 3038, 2996, 2922, 2736, 2599, 1640, 1617, 1555, 1522, 1480, 1428, 1408, 1341, 1317, 1288, 1260, 1200, 1107, 1064, 1035, 849, 781, 746, 655, 575, 468. EI-MS: (Mwt: 414.06): m/z (% rel. int.), 414.03 [M+, (49.90%)], 164.56 (100%); Anal. Calcd for C18H15ClN6O2S: C, 52.11; H, 3.64; Cl, 8.54; N, 20.26; O, 7.71; S, 7.73. Found: C, 51.97; H, 3.88; N, 20.59; S, 7.82.
4.1.2.19. (E)-6-(4-Chlorophenyl)-3-(2-(4-(dimethylamino)benzylidene)hydrazinyl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine (5o)
The titled compound was crystallized from aqueous DMF as yellow crystals (1.13 g, 87.2%); mp 209–212 °C; 1H NMR (400 MHz, DMSO-d6): δ 10.54 (s, 1H, NH, D2O exchangeable), 8.32 (s, 1H, CH), 8.16 (d, J = 8.6 Hz, 2H, Ar H), 7.66 (d, J = 8.6 Hz, 2H, Ar H), 7.50 (d, J = 8.7 Hz, 2H, Ar H), 6.76 (d, J = 8.8 Hz, 2H, Ar H), 4.38 (s, 2H, CH2), 2.97 (s, 6H, 2CH3). 13C NMR (101 MHz, DMSO-d6): δ 152.81, 151.63, 151.48, 145.69, 136.91, 136.72, 132.93, 129.82, 129.40, 128.10, 122.82, 112.39, 40.63, 40.21. FTIR (KBr, cm–1) ṽ: 3223, 3066, 2918, 1651, 1583, 1490, 1474, 1366, 1345, 1305, 1183, 1092, 1054, 947, 751, 727, 696, 581, 519, 472. EI-MS: (Mwt: 411.10): m/z (% rel. int.), 411.13 [M+, (10.64%)], 126.14 (100%); Anal. Calcd for C19H18ClN7S: C, 55.40; H, 4.40; Cl, 8.61; N, 23.80; S, 7.78. Found: C, 55.73; H, 4.29; N, 24.11; S, 7.69.
4.1.2.20. (E)-3-(2-([1,1′-Biphenyl]-4-ylmethylene)hydrazinyl)-6-(4-chlorophenyl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine (5p)
The titled compound was crystallized from aqueous DMF as yellow crystals (1.21 g, 86.3%); mp 220–223 °C; 1H NMR (400 MHz, DMSO-d6): δ 11.01 (s, 1H, NH, D2O exchangeable), 8.53 (s, 1H, CH), 8.17 (d, J = 8.7 Hz, 2H, Ar H), 7.81–7.75 (m, 4H, Ar H), 7.77–7.69 (m, 2H, Ar H), 7.67 (d, J = 8.6 Hz, 2H, Ar H), 7.49 (dd, J = 8.5, 6.8 Hz, 2H, Ar H), 7.39 (t, J = 7.4 Hz, 1H, Ar H), 4.41 (s, 2H, CH2). 13C NMR (101 MHz, DMSO-d6): δ 153.19, 151.27, 144.04, 141.15, 139.92, 137.25, 137.02, 134.48, 132.87, 129.86, 129.46, 129.43, 128.19, 127.46, 127.40, 127.06, 40.63. FTIR (KBr, cm–1) ṽ: 3626, 3151, 3086, 3018, 2984, 2894, 1610, 1590, 1474, 1397, 1312, 1292, 1096, 1087, 1008, 983, 969, 847, 838, 812, 763, 744, 636, 617, 514, 487. EI-MS: (Mwt: 444.09): m/z (% rel. int.), 444.03 [M+, (26.11%)], 144.96 (100%); Anal. Calcd for C23H17ClN6S: C, 62.09; H, 3.85; Cl, 7.97; N, 18.89; S, 7.21. Found: C, 62.38; H, 4.03; N, 19.12; S, 7.08.
4.1.2.21. (E)-3-(2-(4-(Benzyloxy)benzylidene)hydrazinyl)-6-(4-chlorophenyl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine (5q)
The titled compound was crystallized from aqueous DMF as yellow crystals (1.35 g, 90.2%); mp 216–220 °C; 1H NMR (400 MHz, DMSO-d6): δ 10.79 (s, 1H, NH, D2O exchangeable), 8.42 (s, 1H, CH), 8.16 (d, J = 8.6 Hz, 2H, Ar H), 7.71–7.58 (m, 4H, Ar H), 7.47 (d, J = 7.1 Hz, 2H, Ar H), 7.45–7.37 (m, 2H, Ar H), 7.37–7.31 (m, 1H, Ar H), 7.09 (d, J = 8.7 Hz, 2H, Ar H), 5.16 (s, 2H, OCH2), 4.39 (s, 2H, CH2). 13C NMR (101 MHz, DMSO-d6): δ 159.74, 152.99, 151.44, 144.49, 137.31, 137.01, 136.97, 132.89, 129.83, 129.40, 128.91, 128.35, 128.32, 128.19, 128.16, 115.60, 69.80, 40.64. FTIR (KBr, cm–1) ṽ: 3626, 3216, 3032, 2913, 1654, 1603, 1584, 1471, 1353, 1310, 1246, 1170, 1101, 1006, 972, 858, 832, 745, 714, 653, 625, 534, 458. EI-MS: (Mwt: 474.10): m/z (% rel. int.), 474.73 [M+, (22.01%)], 311.27 (100%); Anal. Calcd for C24H19ClN6OS: C, 60.69; H, 4.03; Cl, 7.46; N, 17.69; O, 3.37; S, 6.75. Found: C, 60.97; H, 4.21; N, 17.85; S, 6.89.
4.1.2.22. (E)-6-(4-Chlorophenyl)-3-(2-(furan-2-ylmethylene)hydrazinyl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine (5r)
The titled compound was crystallized from aqueous DMF as dark yellow crystals (1 g, 88.5%); mp 206–212 °C; 1H NMR (400 MHz, DMSO-d6): δ 10.93 (s, 1H, NH, D2O exchangeable), 8.44 (s, 1H, CH), 8.15 (d, J = 8.6 Hz, 2H, Ar H), 7.80 (d, J = 1.8 Hz, 1H, furan H), 7.66 (d, J = 8.6 Hz, 2H, Ar H), 6.80 (d, J = 3.4 Hz, 1H, furan H), 6.62 (dd, J = 3.4, 1.8 Hz, 1H, furan H), 4.40 (s, 2H, CH2). 13C NMR (101 MHz, DMSO-d6): δ 153.24, 151.15, 150.26, 144.73, 137.31, 137.03, 134.88, 132.74, 129.80, 129.40, 112.49, 112.04, 40.40. FTIR (KBr, cm–1) ṽ: 3447, 3135, 3070, 2990, 2956, 2883, 2809, 1600, 1591, 1568, 1470, 1338, 1223, 1105, 1078, 935, 884, 851, 757, 594, 565, 472. EI-MS: (Mwt: 358.04): m/z (% rel. int.), 358.18 [M+, (48.55%)], 354.59 (100%); Anal. Calcd for C15H11ClN6OS: C, 50.21; H, 3.09; Cl, 9.88; N, 23.42; O, 4.46; S, 8.94. Found: C, 50.49; H, 3.21; N, 23.70; S, 8.86.
4.1.2.23. (E)-6-(4-Chlorophenyl)-3-(2-(thiophen-3-ylmethylene)hydrazineyl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine (5s)
The titled compound was crystallized from aqueous DMF as yellow crystals (1.03 g, 87.2%); mp 218–221 °C; 1H NMR (400 MHz, DMSO-d6): δ 10.80 (s, 1H, NH, D2O exchangeable), 8.55 (s, 1H, CH), 8.16 (d, J = 8.3 Hz, 2H, Ar H), 7.81 (t, J = 2.7 Hz, 1H, thiophene H), 7.66 (d, J = 8.6 Hz, 2H, Ar H), 7.62 (dd, J = 5.3, 2.9 Hz, 1H, thiophene H), 7.45 (d, J = 4.8 Hz, 1H, thiophene H), 4.39 (s, 2H, CH2). 13C NMR (101 MHz, DMSO-d6): δ 152.93, 151.54, 140.64, 138.58, 137.02, 136.92, 132.92, 129.81, 129.41, 127.80, 126.38, 124.98, 40.21. FTIR (KBr, cm–1) ṽ: 3626, 3448, 3097, 2915, 1734, 1651, 1623, 1560, 1485, 1408, 1315, 1238, 1107, 1088, 1070, 949, 810, 769, 784, 726, 689, 565, 444. EI-MS: (Mwt: 374.01): m/z (% rel. int.), 374.95 [M+, (16.22%)], 45.07 (100%); Anal. Calcd for C15H11ClN6S2: C, 48.06; H, 2.96; Cl, 9.46; N, 22.42; S, 17.10. Found: C, 48.32; H, 3.09; N, 22.71; S, 17.04.
4.1.2.24. (E)-3-(2-((1H-Indol-3-yl)methylene)hydrazineyl)-6-(4-chlorophenyl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine (5t)
The titled compound was crystallized from aqueous DMF as dark yellow crystals (1.13 g, 87.9%); mp 206–212 °C; 1H NMR (400 MHz, DMSO-d6): δ 11.46 (s, 1H, NH1″, D2O exchangeable), 10.49 (s, 1H, NH, D2O exchangeable), 8.58 (s, 1H, CH), 8.32–8.24 (m, 1H, indole H), 8.16 (d, J = 8.6 Hz, 2H, Ar H), 7.73 (d, J = 2.6 Hz, 1H, indole H), 7.65 (d, J = 8.6 Hz, 2H, Ar H), 7.43 (d, J = 8.1 Hz, 1H, indole H), 7.18 (ddd, J = 8.1, 7.0, 1.3 Hz, 1H, indole H), 7.05 (t, J = 7.2 Hz, 1H, indole H), 4.40 (s, 2H, CH2). 13C NMR (101 MHz, DMSO-d6): δ 152.77, 152.00, 142.26, 137.43, 136.85, 136.68, 133.02, 129.82, 129.39, 129.17, 124.78, 122.89, 122.54, 120.49, 112.46, 112.11, 40.62. FTIR (KBr, cm–1) ṽ: 3626, 3421, 3170, 3010, 2922, 1651, 1508, 1479, 1390, 1339, 1249, 1091, 1057, 814, 751, 608, 518, 472. EI-MS: (Mwt: 407.07): m/z (% rel. int.), 407.80 [M+, (51.57%)], 116.79 (100%); Anal. Calcd for C19H14ClN7S: C, 55.95; H, 3.46; Cl, 8.69; N, 24.04; S, 7.86. Found: C, 56.12; H, 3.75; N, 24.27; S, 7.90.
4.2. Biological Evaluation
4.2.1. In Vitro Antiproliferative Activity against NCI-60 Cell Lines
The synthesized compounds were screened for their cytotoxic activity against 60 cell lines by the National Cancer Institute (NCI), NIH, Bethesda, Maryland, USA74 under the Developmental Therapeutics Program (DTP) (https://dtp.cancer.gov/compsub/) for NCI-60 human tumor cell lines screen. The operation of this screen utilizes 60 different human tumor cell lines, representing leukemia, NSCLC, colon, CNS, ovarian, renal, prostate, breast cancers, and melanoma. The compounds are initially screened at 10 μM concentration for measuring the cell growth percent. The data of the single dose treatment are represented as mean graph (Figure S1).
4.2.2. In Vitro Kinase Screening
Compound 5l was screened against the 16 kinases for kinase inhibition percent determination in Thermo Fisher Scientific corporation, Madison, WI, USA (https://www.thermofisher.com/eg/en/home.html), under the SelecScreen program. The assay of the compounds is carried out according to the manufacturer’s protocol with the ATP concentration at the km (Michaelis constant) of each kinase, except for MAPK8, where ATP concentration was 100 μM.
4.2.3. In Vitro GSK-3β IC50 Determination
The IC50 of 5l against GSK-3β was determined using the Kinase-Glo Max Assay kit (Promega#V6071) of BPS Biosciences corporation, San Diego, CA, USA (www.bpsbioscience.com) (catalog #79700). The following concentrations were prepared for the test compounds: 10, 1, 0.1, and 0.01 μM. The reaction mixture and the test compounds concentrations were prepared according to the manufacturer’s protocol. The Kinase-Glo (Promega) was used as a detection reagent, and luminescence was measured using a Bio Tek Synergy 2 microplate reader. Measures of the control and sample were carried out in duplicates. The reference GSK-3β inhibitor used was AR-A014418.79
4.2.4. Annexin V/PI Staining Apoptosis Assay
Apoptosis and cell cycle analysis studies were performed at Cell Culture Unit, Vacsera, Cairo, Egypt. The PC-3 cells were treated with 5l at the IC50 concentration (0.883 μM) for 48 h and cultured in six well plates. Annexin V-FITC Apoptosis Detection Kit (BioVision, CA, USA) was used to stain the cells with 5 μL propidium iodide (PI) and 5 μL Annexin V-FITC after being fixed for 0.5 h in cold ethanol. The FACScan flow cytometer (Beckman Coulter, CA, USA) was used for the cells’ analysis with a 15 min period of dark and room temperature treatment. Apoptosis was evaluated in terms of the fluorescein isothiocyanate (FITC)-positive cells.
4.2.5. Cell Cycle Analysis
PC-3 cells were treated with 0.883 μM of compound 5l. 5l-treated PC-3 cells were trypsinized, washed with phosphate buffered saline (PBS), resuspended in cold methanol, and kept overnight at 4 °C, 48 h after treatment. The cells were then resuspended in sodium citrate buffer with RNase and incubated at 37 °C for 30 min. The cells were then centrifugated and collected to be resuspended in PBS and filtered. FACSCalibur Flow Cytometer (BD Biosciences, San Jose, CA, USA) was then used for cell cycle analysis.
4.2.6. Caspase-3 Determination in PC-3 Cells Treated with 5l
Caspase-3 (active) levels were measured in the cell lysates using the human caspase-3 ELISA kit obtained from Invitrogen (catalog #KHO1091, CA, USA), according to the manufacturer’s instructions.
4.2.7. Statistical Analysis
Statistical analysis was performed using the Student’s t-test for unpaired data. The values were acquired from three experiments conducted independently in triplicates, and data were represented as mean ± standard deviation (S.D.). GraphPad Prism, version 5.01 (GraphPad Software, CA, USA), was used for data analysis and figures generation. p < 0.05 was set as the statistical significance level.
4.3. Molecular Modeling
4.3.1. Ligand-Based Target Fishing
LBTF was carried out using 3D similarity searching between a query compound and a target-annotated compound database. Compound 5l, the most active compound in NCI-60 cell line screening, was used as a query, while the binding database (www.bindingdb.org) (downloaded on June 6th 2018, 1.4 M compounds) was used as the database.75−77 First, a multiconformer file of the binding database was generated using in-house multithreaded OMEGA python script written using the OMEGA Toolkit.84 3D similarity was then calculated between the query molecule and every conformer in the BindingDB multiconformer file. The FastROCS shape comparison software toolkit,78 the GPU version of ROCS (Rapid Overlay of Chemical Structures) software,85 was used for the 3D similarity searching. ROCS target fishing ability was validated in a target fishing protocol for the determination of polypharmacology of selected drugs.86 The similarity measurement in FastROCS depends on two criteria, shape similarity, and color similarity. According to ROCS philosophy, shape is the shape of the molecule described using a Gaussian function which describes the molecule’s volume. Color is the functional group of the molecule. Each similarity is measured by the Tanimoto coefficient which has a value of [0, 1]; 0 means totally dissimilar, and 1 means totally similar (the same molecule). Combining both shape and color Tanimoto coefficients gives the Tanimoto combo coefficient with a value of [0, 2]. The results were ranked with the Tanimoto combo in a descending order.
4.3.2. Molecular Docking
The ligands were sketched using MarvinSketch and energy-minimized using OpenBabel software implementing the steepest descent algorithm for a convergence criterion of 10–6 kcal/mol/Å and 10,000 iterations using MMFF94s (Merck molecular force field static variant) for energy evaluation at each step.
The GSK-3β protein structure (PDB ID: 1Q5K) was downloaded from the RCSB Protein Data Bank (http://www.rcsb.org/).87,88 Protein structure was prepared using Make Receptor utility of the OEDOCKING 3.4.0.2 module of OpenEye software.81 The box of search area was determined around AR-A014418 co-crystallized ligand (PDB ID: TMU) with the default dimensions, followed by detection of shape potential of the binding site. Ligands were then prepared by multiconformer file generation using OMEGA 3.1.2.2 OpenEye software.89,90 Docking was carried out by FRED and HYBRID applications of the OEDOCKING 3.4.0.2 module of OpenEye software.81−83 DockRMSD was used to measure docking rmsd.91 All protein–ligand figures generation was carried out using UCSF Chimera visualization software.92
4.3.3. Molecular Dynamics
Simulation system preparation was carried out using VMD 1.9.3 software.93 The protein structure was first cleared of any nonprotein atoms (water, ions, or ligands); then, the topology file for the protein was generated using the psfgen 1.6.4 package implemented through VMD. The topology file of the ligands was generated by SwissParam web server.94 The protein–ligand complex was constructed manually by incorporating both topology files, which is then solvated in the TIP3P water model, forming a box of 14 Å margin in every direction of the protein atoms using the VMD solvate 1.7 package, followed by charge neutralization with Na+Cl– ions using VMD autoionize 1.4 package, ending up with systems of 72,000 atoms, approximately. MD calculations were carried out using NAMD 2.13 software.95 100,000 minimization steps were carried out using the coupled conjugate gradient and line search algorithms of NAMD, followed by the 20 ns NPT equilibration phase. Protein parameters were provided by the CHARMM36 force field96−98 (July 2017 release), while ligand parameters were generated using SwissParam. The dynamics configuration file was set to neglect 1–2 and 1–3 atom interactions, and fully consider 1–4 interactions. Pair list distance for calculating van der Waals and electrostatic interactions were set to 13.5 Å and restarted every integration cycle, while calculated within 12, with 10 Å switch distance for function smoothing. Velocity and position were calculated using the velocity Verlet integration method, while electrostatics were calculated using the particle mesh Ewald sum method,99 all with a time step of 2 fs. The frequency of calculating the nonbonded and electrostatic interactions was 2 fs (1 time step). Langevin dynamics was used for keeping the system temperature constant at 310 K (37 °C) with 1 ps– damping coefficient applied only to heavy atoms. The Langevin piston with 200 fs oscillation period and 50 fs damping period was used to keep the pressure constant at 1 atm (1.01325 bar), coupled with the Nose–Hoover method100 to control barostat fluctuations. The dynamics simulation analysis was carried out using ProDy python package101,102 and VMD rmsd trajectory tool. All analysis charts were constructed using Matplotlib 3.0.2 python plotting library.103
Acknowledgments
This work was supported by The Center for Drug Research and Development (CDRD), Faculty of Pharmacy, The British University in Egypt. We gratefully acknowledge the support of OpenEye Scientific Software Inc. for offering an academic license. We also gratefully acknowledge the support of NVIDIA Corporation with the donation of the Titan Xp GPU used for molecular dynamics calculations. The authors acknowledge Mohamed S. A. Elsayed for fruitful discussions about NCI screening results and Mai M. Shaalan and Ahmed Aladawy for technical support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c01829.
Copies of NMR spectra for compounds 5a–t, kinase inhibitors retrieved from target fishing, and NCI screening mean graph example (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Siegel R. L.; Miller K. D.; Jemal A. Cancer Statistics, 2020. Ca-Cancer J. Clin. 2020, 70, 7–30. 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- Szakács G.; Paterson J. K.; Ludwig J. A.; Booth-Genthe C.; Gottesman M. M. Targeting Multidrug Resistance in Cancer. Nat. Rev. Drug Discovery 2006, 5, 219–234. 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- Manning G.; Whyte D. B.; Martinez R.; Hunter T.; Sudarsanam S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Roskoski R. A Historical Overview of Protein Kinases and Their Targeted Small Molecule Inhibitors. Pharmacol. Res. 2015, 100, 1–23. 10.1016/j.phrs.2015.07.010. [DOI] [PubMed] [Google Scholar]
- Dhanasekaran N.; Reddy E. P. Signaling by Dual Specificity Kinases. Oncogene 1998, 17, 1447–1455. 10.1038/sj.onc.1202251. [DOI] [PubMed] [Google Scholar]
- Besant P. G.; Tan E.; Attwood P. V. Mammalian Protein Histidine Kinases. int. J. Biochem. Cell Biol. 2003, 35, 297–309. 10.1016/S1357-2725(02)00257-1. [DOI] [PubMed] [Google Scholar]
- Alonso A.; Sasin J.; Bottini N.; Friedberg I.; Friedberg I.; Osterman A.; Godzik A.; Hunter T.; Dixon J.; Mustelin T. Protein Tyrosine Phosphatases in the Human Genome. Cell 2004, 117, 699–711. 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- Cohen P. Protein Kinases — the Major Drug Targets of the Twenty-First Century?. Nat. Rev. Drug Discovery 2002, 1, 309–315. 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- Shchemelinin I.; Sefc L.; Necas E. Protein Kinases, Their Function and Implication in Cancer and Other Diseases. Folia Biol. 2006, 52, 81–100. [PubMed] [Google Scholar]
- Embi N.; Rylatt D. B.; Cohen P. Glycogen Synthase Kinase-3 from Rabbit Skeletal Muscle. Separation from Cyclic-AMP-Dependent Protein Kinase and Phosphorylase Kinase. Eur. J. Biochem. 1980, 107, 519–527. [PubMed] [Google Scholar]
- Woodgett J. R. Molecular Cloning and Expression of Glycogen Synthase Kinase-3/Factor A. EMBO J. 1990, 9, 2431–2438. 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeflich K. P.; Luo J.; Rubie E. A.; Tsao M.-S.; Jin O.; Woodgett J. R. Requirement for Glycogen Synthase Kinase-3beta in Cell Survival and NF-KappaB Activation. Nature 2000, 406, 86–90. 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- Beurel E.; Grieco S. F.; Jope R. S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. 10.1016/j.pharmthera.2014.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame S.; Zheleva D. Targeting Glycogen Synthase Kinase-3 in Insulin Signalling. Expert Opin. Ther. Targets 2006, 10, 429–444. 10.1517/14728222.10.3.429. [DOI] [PubMed] [Google Scholar]
- Martinez A.; Perez D. I. GSK-3 Inhibitors: A Ray of Hope for the Treatment of Alzheimer’s Disease?. J. Alzheimer’s Dis. 2008, 15, 181–191. 10.3233/jad-2008-15204. [DOI] [PubMed] [Google Scholar]
- Klein P. S.; Melton D. A. A Molecular Mechanism for the Effect of Lithium on Development. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 8455–8459. 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jope R. S.; Yuskaitis C. J.; Beurel E. Glycogen Synthase Kinase-3 (GSK3): Inflammation, Diseases, and Therapeutics. Neurochem. Res. 2007, 32, 577–595. 10.1007/s11064-006-9128-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H.; Woodgett J.; Maamari M.; Force T. Targeting GSK-3 Family Members in the Heart: A Very Sharp Double-Edged Sword. J. Mol. Cell. Cardiol. 2011, 51, 607–613. 10.1016/j.yjmcc.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin I.; Eturi A.; De Souza A.; Pamarthy S.; Tavora F.; Giles F. J.; Carneiro B. A. Glycogen Synthase Kinase-3 Beta Inhibitors as Novel Cancer Treatments and Modulators of Antitumor Immune Responses. Cancer Biol. Ther. 2019, 20, 1047–1056. 10.1080/15384047.2019.1595283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walz A.; Ugolkov A.; Chandra S.; Kozikowski A.; Carneiro B. A.; O’Halloran T. V.; Giles F. J.; Billadeau D. D.; Mazar A. P. Molecular Pathways: Revisiting Glycogen Synthase Kinase-3β as a Target for the Treatment of Cancer. Clin. Cancer Res. 2017, 23, 1891–1897. 10.1158/1078-0432.CCR-15-2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farago M.; Dominguez I.; Landesman-Bollag E.; Xu X.; Rosner A.; Cardiff R. D.; Seldin D. C. Kinase-Inactive Glycogen Synthase Kinase 3beta Promotes Wnt Signaling and Mammary Tumorigenesis. Cancer Res. 2005, 65, 5792–5801. 10.1158/0008-5472.CAN-05-1021. [DOI] [PubMed] [Google Scholar]
- Tan Z.; Zheng H.; Liu X.; Zhang W.; Zhu J.; Wu G.; Cao L.; Song J.; Wu S.; Song L.; et al. MicroRNA-1229 Overexpression Promotes Cell Proliferation and Tumorigenicity and Activates Wnt/β-Catenin Signaling in Breast Cancer. Oncotarget 2016, 7, 24076–24087. 10.18632/oncotarget.8119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H.; Li L.; Yang S.; Wang D.; Zhong S.; Zhao J.; Tang J. MicroRNA-29a Contributes to Drug-Resistance of Breast Cancer Cells to Adriamycin through PTEN/AKT/GSK3β Signaling Pathway. Gene 2016, 593, 84–90. 10.1016/j.gene.2016.08.016. [DOI] [PubMed] [Google Scholar]
- Zheng H.; Saito H.; Masuda S.; Yang X.; Takano Y. Phosphorylated GSK3beta-Ser9 and EGFR Are Good Prognostic Factors for Lung Carcinomas. Anticancer Res. 2007, 27, 3561–3569. [PubMed] [Google Scholar]
- Lin G.; Liu B.; Meng Z.; Liu Y.; Li X.; Wu X.; Zhou Q.; Xu K. MiR-26a Enhances Invasive Capacity by Suppressing GSK3β in Human Lung Cancer Cells. Exp. Cell Res. 2017, 352, 364–374. 10.1016/j.yexcr.2017.02.033. [DOI] [PubMed] [Google Scholar]
- Mishra R.; Nagini S.; Rana A. Expression and Inactivation of Glycogen Synthase Kinase 3 Alpha/ Beta and Their Association with the Expression of Cyclin D1 and P53 in Oral Squamous Cell Carcinoma Progression. Mol. Cancer 2015, 14, 20. 10.1186/s12943-015-0300-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He K.; Tong D.; Zhang S.; Cai D.; Wang L.; Yang Y.; Gao L.; Chang S. E.; Guo B.; Song T.; et al. MiRNA-99b-3p Functions as a Potential Tumor Suppressor by Targeting Glycogen Synthase Kinase-3β in Oral Squamous Cell Carcinoma Tca-8113 Cells. int. J. Oncol. 2015, 47, 1528–1536. 10.3892/ijo.2015.3135. [DOI] [PubMed] [Google Scholar]
- Ma C.; Wang J.; Gao Y.; Gao T.-W.; Chen G.; Bower K. A.; Odetallah M.; Ding M.; Ke Z.; Luo J. The Role of Glycogen Synthase Kinase 3beta in the Transformation of Epidermal Cells. Cancer Res. 2007, 67, 7756–7764. 10.1158/0008-5472.CAN-06-4665. [DOI] [PubMed] [Google Scholar]
- Qiu H.-J.; Lu X.-H.; Yang S.-S.; Weng C.-Y.; Zhang E.-K.; Chen F.-C. MiR-769 Promoted Cell Proliferation in Human Melanoma by Suppressing GSK3B Expression. Biomed. Pharmacother. 2016, 82, 117–123. 10.1016/j.biopha.2016.04.052. [DOI] [PubMed] [Google Scholar]
- Li B.; Thrasher J. B.; Terranova P. Glycogen Synthase Kinase-3: A Potential Preventive Target for Prostate Cancer Management. Urol. Oncol. 2015, 33, 456–463. 10.1016/j.urolonc.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakoori A.; Mai W.; Miyashita K.; Yasumoto K.; Takahashi Y.; Ooi A.; Kawakami K.; Minamoto T. Inhibition of GSK-3 Beta Activity Attenuates Proliferation of Human Colon Cancer Cells in Rodents. Cancer Sci. 2007, 98, 1388–1393. 10.1111/j.1349-7006.2007.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T.; Lai Q.; Wang S.; Cai J.; Xiao Z.; Deng D.; He L.; Jiao H.; Ye Y.; Liang L.; et al. MicroRNA-224 Sustains Wnt/β-Catenin Signaling and Promotes Aggressive Phenotype of Colorectal Cancer. J. Exp. Clin. Cancer Res. 2016, 35, 21. 10.1186/s13046-016-0287-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao S.; Yang Z.; Lv R.; Zhao J.; Wu M.; Liao Y.; Liu Q. MiR-135b Contributes to the Radioresistance by Targeting GSK3β in Human Glioblastoma Multiforme Cells. PLoS One 2014, 9, e108810 10.1371/journal.pone.0108810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagini S.; Sophia J.; Mishra R. Glycogen Synthase Kinases: Moonlighting Proteins with Theranostic Potential in Cancer. Semin. Cancer Biol. 2019, 56, 25–36. 10.1016/j.semcancer.2017.12.010. [DOI] [PubMed] [Google Scholar]
- Pal K.; Cao Y.; Gaisina I. N.; Bhattacharya S.; Dutta S. K.; Wang E.; Gunosewoyo H.; Kozikowski A. P.; Billadeau D. D.; Mukhopadhyay D. Inhibition of GSK-3 Induces Differentiation and Impaired Glucose Metabolism in Renal Cancer. Mol. Cancer Ther. 2014, 13, 285–296. 10.1158/1535-7163.MCT-13-0681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcea G.; Manson M.; Neal C.; Pattenden C.; Sutton C.; Dennison A.; Berry D. Glycogen Synthase Kinase-3 Beta; a New Target in Pancreatic Cancer?. Curr. Cancer Drug Targets 2007, 7, 209–215. 10.2174/156800907780618266. [DOI] [PubMed] [Google Scholar]
- Marchand B.; Arsenault D.; Raymond-Fleury A.; Boisvert F.-M.; Boucher M.-J. Glycogen Synthase Kinase-3 (GSK3) Inhibition Induces Prosurvival Autophagic Signals in Human Pancreatic Cancer Cells. J. Biol. Chem. 2015, 290, 5592–5605. 10.1074/jbc.M114.616714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W.; Li Y.; Gou S.; Xiong J.; Wu H.; Wang C.; Yan H.; Liu T. MiR-744 Increases Tumorigenicity of Pancreatic Cancer by Activating Wnt/β-Catenin Pathway. Oncotarget 2015, 6, 37557–37569. 10.18632/oncotarget.5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.-w.; Liu G.-h.; Liu Y.-q.; Zhao H.-c.; Yang Z.; Zhao C.-l.; Zhang X.-f.; Ye H. Over-Expression of MicroRNA-940 Promotes Cell Proliferation by Targeting GSK3β and SFRP1 in Human Pancreatic Carcinoma. Biomed. Pharmacother. 2016, 83, 593–601. 10.1016/j.biopha.2016.06.057. [DOI] [PubMed] [Google Scholar]
- Abrahamsson A. E.; Geron I.; Gotlib J.; Dao K.-H. T.; Barroga C. F.; Newton I. G.; Giles F. J.; Durocher J.; Creusot R. S.; Karimi M.; et al. Glycogen Synthase Kinase 3beta Missplicing Contributes to Leukemia Stem Cell Generation. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 3925–3929. 10.1073/pnas.0900189106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F.; Zhang L.; van Laar T.; van Dam H.; Ten Dijke P. GSK3β Inactivation Induces Apoptosis of Leukemia Cells by Repressing the Function of C-Myb. Mol. Biol. Cell 2011, 22, 3533–3540. 10.1091/mbc.E11-06-0483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park N. R.; Cha J. H.; Jang J. W.; Bae S. H.; Jang B.; Kim J.-H.; Hur W.; Choi J. Y.; Yoon S. K. Synergistic Effects of CD44 and TGF-Β1 through AKT/GSK-3β/β-Catenin Signaling during Epithelial-Mesenchymal Transition in Liver Cancer Cells. Biochem. Biophys. Res. Commun. 2016, 477, 568–574. 10.1016/j.bbrc.2016.06.077. [DOI] [PubMed] [Google Scholar]
- Zhuang L.; Wang X.; Wang Z.; Ma X.; Han B.; Zou H.; Wu Z.; Dong S.; Qu Z.; Zang Y.; et al. MicroRNA-23b Functions as an Oncogene and Activates AKT/GSK3β/β-Catenin Signaling by Targeting ST7L in Hepatocellular Carcinoma. Cell Death Discovery 2017, 8, e2804 10.1038/cddis.2017.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azoulay-Alfaguter I.; Elya R.; Avrahami L.; Katz A.; Eldar-Finkelman H. Combined Regulation of MTORC1 and Lysosomal Acidification by GSK-3 Suppresses Autophagy and Contributes to Cancer Cell Growth. Oncogene 2015, 34, 4613–4623. 10.1038/onc.2014.390. [DOI] [PubMed] [Google Scholar]
- Liao X.; Thrasher J. B.; Holzbeierlein J.; Stanley S.; Li B. Glycogen Synthase Kinase-3beta Activity Is Required for Androgen-Stimulated Gene Expression in Prostate Cancer. Endocrinology 2004, 145, 2941–2949. 10.1210/en.2003-1519. [DOI] [PubMed] [Google Scholar]
- Mazor M.; Kawano Y.; Zhu H.; Waxman J.; Kypta R. M. Inhibition of Glycogen Synthase Kinase-3 Represses Androgen Receptor Activity and Prostate Cancer Cell Growth. Oncogene 2004, 23, 7882–7892. 10.1038/sj.onc.1208068. [DOI] [PubMed] [Google Scholar]
- Sun A.; Shanmugam I.; Song J.; Terranova P. F.; Thrasher J. B.; Li B. Lithium Suppresses Cell Proliferation by Interrupting E2F-DNA Interaction and Subsequently Reducing S-Phase Gene Expression in Prostate Cancer. Prostate 2007, 67, 976–988. 10.1002/pros.20586. [DOI] [PubMed] [Google Scholar]
- Li R.; Erdamar S.; Dai H.; Sayeeduddin M.; Frolov A.; Wheeler T. M.; Ayala G. E. Cytoplasmic Accumulation of Glycogen Synthase Kinase-3beta Is Associated with Aggressive Clinicopathological Features in Human Prostate Cancer. Anticancer Res. 2009, 29, 2077–2081. [PubMed] [Google Scholar]
- Schütz S. V.; Schrader A. J.; Zengerling F.; Genze F.; Cronauer M. V.; Schrader M. Inhibition of Glycogen Synthase Kinase-3β Counteracts Ligand-Independent Activity of the Androgen Receptor in Castration Resistant Prostate Cancer. PLoS One 2011, 6, e25341 10.1371/journal.pone.0025341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q.; Yang J.; Han S.; Liu J.; Holzbeierlein J.; Thrasher J. B.; Li B. Suppression of Glycogen Synthase Kinase 3 Activity Reduces Tumor Growth of Prostate Cancer in Vivo. Prostate 2011, 71, 835–845. 10.1002/pros.21300. [DOI] [PubMed] [Google Scholar]
- Xu M.; Wang S. L.; Zhu L.; Wu P. Y.; Dai W. B.; Rakesh K. P. Structure-Activity Relationship (SAR) Studies of Synthetic Glycogen Synthase Kinase-3β Inhibitors: A Critical Review. Eur. J. Med. Chem. 2019, 164, 448–470. 10.1016/j.ejmech.2018.12.073. [DOI] [PubMed] [Google Scholar]
- Witherington J.; Bordas V.; Garland S. L.; Hickey D. M. B.; Ife R. J.; Liddle J.; Saunders M.; Smith D. G.; Ward R. W. 5-Aryl-Pyrazolo[3,4-b]Pyridines: Potent Inhibitors of Glycogen Synthase Kinase-3 (GSK-3). Bioorg. Med. Chem. Lett. 2003, 13, 1577–1580. 10.1016/S0960-894X(03)00134-3. [DOI] [PubMed] [Google Scholar]
- Witherington J.; Bordas V.; Haigh D.; Hickey D. M. B.; Ife R. J.; Rawlings A. D.; Slingsby B. P.; Smith D. G.; Ward R. W. 5-Aryl-Pyrazolo[3,4-b]Pyridazines: Potent Inhibitors of Glycogen Synthase Kinase-3 (GSK-3). Bioorg. Med. Chem. Lett. 2003, 13, 1581–1584. 10.1016/s0960-894x(03)00135-5. [DOI] [PubMed] [Google Scholar]
- Khan I.; Ibrar A.; Abbas N. Triazolothiadiazoles and Triazolothiadiazines-Biologically Attractive Scaffolds. Eur. J. Med. Chem. 2013, 63, 854–868. 10.1016/j.ejmech.2013.01.060. [DOI] [PubMed] [Google Scholar]
- Kaplancıklı Z. A.; Turan-Zitouni G.; Özdemir A.; Revial G. New Triazole and Triazolothiadiazine Derivatives as Possible Antimicrobial Agents. Eur. J. Med. Chem. 2008, 43, 155–159. 10.1016/j.ejmech.2007.03.019. [DOI] [PubMed] [Google Scholar]
- Aytaç S. P.; Tozkoparan B.; Kaynak F. B.; Aktay G.; Göktaş Ö.; Ünüvar S. Synthesis of 3,6-Disubstituted 7H-1,2,4-Triazolo[3,4-b]-1,3,4-Thiadiazines as Novel Analgesic/Anti-Inflammatory Compounds. Eur. J. Med. Chem. 2009, 44, 4528–4538. 10.1016/j.ejmech.2009.06.026. [DOI] [PubMed] [Google Scholar]
- Suresh Kumar G. V.; Rajendra Prasad Y.; Mallikarjuna B. P.; Chandrashekar S. M. Synthesis and Pharmacological Evaluation of Clubbed Isopropylthiazole Derived Triazolothiadiazoles, Triazolothiadiazines and Mannich Bases as Potential Antimicrobial and Antitubercular Agents. Eur. J. Med. Chem. 2010, 45, 5120–5129. 10.1016/j.ejmech.2010.08.023. [DOI] [PubMed] [Google Scholar]
- Puthiyapurayil P.; Poojary B.; Buridipad S. K. Synthesis, Characterization and Biological Evaluation of a Novel Series of 1,2,4-Triazolo-[3,4-b]-1,3,4-Thiadiazines Containing an Amide Linkage. J. Heterocycl. Chem. 2014, 51, E55–E67. 10.1002/jhet.1766. [DOI] [Google Scholar]
- Aytaç P. S.; Durmaz I.; Houston D. R.; Çetin-Atalay R.; Tozkoparan B. Novel Triazolothiadiazines Act as Potent Anticancer Agents in Liver Cancer Cells through Akt and ASK-1 Proteins. Bioorg. Med. Chem. 2016, 24, 858–872. 10.1016/j.bmc.2016.01.013. [DOI] [PubMed] [Google Scholar]
- Ahmad A.; Varshney H.; Rauf A.; Sherwani A.; Owais M. Synthesis and Anticancer Activity of Long Chain Substituted 1,3,4-Oxadiazol-2-Thione, 1,2,4-Triazol-3-Thione and 1,2,4-Triazolo[3,4-b]-1,3,4-Thiadiazine Derivatives. Arabian J. Chem. 2017, 10, S3347–S3357. 10.1016/j.arabjc.2014.01.015. [DOI] [Google Scholar]
- LaPorte M. G.; Wang Z.; Colombo R.; Garzan A.; Peshkov V. A.; Liang M.; Johnston P. A.; Schurdak M. E.; Sen M.; Camarco D. P.; et al. Optimization of Pyrazole-Containing 1,2,4-Triazolo-[3,4-b]Thiadiazines, a New Class of STAT3 Pathway Inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 3581–3585. 10.1016/j.bmcl.2016.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat K. S.; Poojary B.; Prasad D. J.; Naik P.; Holla B. S. Synthesis and Antitumor Activity Studies of Some New Fused 1,2,4-Triazole Derivatives Carrying 2,4-Dichloro-5-Fluorophenyl Moiety. Eur. J. Med. Chem. 2009, 44, 5066–5070. 10.1016/j.ejmech.2009.09.010. [DOI] [PubMed] [Google Scholar]
- Kamel M. M.; Megally Abdo N. Y. Synthesis of Novel 1,2,4-Triazoles, Triazolothiadiazines and Triazolothiadiazoles as Potential Anticancer Agents. Eur. J. Med. Chem. 2014, 86, 75–80. 10.1016/j.ejmech.2014.08.047. [DOI] [PubMed] [Google Scholar]
- Khan I.; Zaib S.; Ibrar A.; Rama N. H.; Simpson J.; Iqbal J. Synthesis, Crystal Structure and Biological Evaluation of Some Novel 1,2,4-Triazolo[3,4-b]-1,3,4-Thiadiazoles and 1,2,4-Triazolo[3,4-b]-1,3,4-Thiadiazines. Eur. J. Med. Chem. 2014, 78, 167–177. 10.1016/j.ejmech.2014.03.046. [DOI] [PubMed] [Google Scholar]
- Khan I.; Ibrar A.; Zaib S.; Ahmad S.; Furtmann N.; Hameed S.; Simpson J.; Bajorath J.; Iqbal J. Active Compounds from a Diverse Library of Triazolothiadiazole and Triazolothiadiazine Scaffolds: Synthesis, Crystal Structure Determination, Cytotoxicity, Cholinesterase Inhibitory Activity, and Binding Mode Analysis. Bioorg. Med. Chem. 2014, 22, 6163–6173. 10.1016/j.bmc.2014.08.026. [DOI] [PubMed] [Google Scholar]
- Ansari M.; Shokrzadeh M.; Karima S.; Rajaei S.; Hashemi S. M.; Mirzaei H.; Fallah M.; Emami S. Design, Synthesis and Biological Evaluation of Flexible and Rigid Analogues of 4H-1,2,4-Triazoles Bearing 3,4,5-Trimethoxyphenyl Moiety as New Antiproliferative Agents. Bioorg. Chem. 2019, 93, 103300. 10.1016/j.bioorg.2019.103300. [DOI] [PubMed] [Google Scholar]
- Shivarama Holla B.; Sooryanarayana Rao B.; Sarojini B. K.; Akberali P. M.; Suchetha Kumari N. Synthesis and Studies on Some New Fluorine Containing Triazolothiadiazines as Possible Antibacterial, Antifungal and Anticancer Agents. Eur. J. Med. Chem. 2006, 41, 657–663. 10.1016/j.ejmech.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Husain A.; Rashid M.; Shaharyar M.; Siddiqui A. A.; Mishra R. Benzimidazole Clubbed with Triazolo-Thiadiazoles and Triazolo-Thiadiazines: New Anticancer Agents. Eur. J. Med. Chem. 2013, 62, 785–798. 10.1016/j.ejmech.2012.07.011. [DOI] [PubMed] [Google Scholar]
- Rashid M.; Husain A.; Mishra R.; Karim S.; Khan S.; Ahmad M.; Al-wabel N.; Husain A.; Ahmad A.; Khan S. A. Design and Synthesis of Benzimidazoles Containing Substituted Oxadiazole, Thiadiazole and Triazolo-Thiadiazines as a Source of New Anticancer Agents. Arabian J. Chem. 2019, 12, 3202. 10.1016/j.arabjc.2015.08.019. [DOI] [Google Scholar]
- Arandkar V.; Vedula R. R. A Facile One-Pot Expeditious Synthesis of Triazolothiadiazines and Anticancer Activity. Phosphorus, Sulfur Silicon Relat. Elem. 2019, 194, 533–539. 10.1080/10426507.2018.1542394. [DOI] [Google Scholar]
- Farghaly T. A. E.-R.; Abdallah M. A.; Mahmoud H. K. Synthesis of Novel 1,2,4-Triazoles and Triazolo-Thiadiazines as Anticancer Agents. Turk. J. Chem. 2015, 39, 955–969. 10.3906/kim-1504-13. [DOI] [Google Scholar]
- Thirupaiah B.; Vedula R. R. Novel One-Pot Multicomponent Synthesis of Substituted 2,3-Dihydro-2-(6-(4-Hydroxy-6-Methyl-2-Oxo-2H-Pyran-3-Yl)-7H-[1,2,4]Triazolo[3,4-b][1,3,4]Thiadiazin-3-Yl)Phthalazine-1,4-Diones and Substituted 3-[3-(N′-Benzylidene-Hydrazino)-7H-[1,2,4] Triazolo[3,4-. Synth. Commun. 2014, 44, 513–519. 10.1080/00397911.2013.817584. [DOI] [Google Scholar]
- Dickinson R. G.; Jacobsen N. W. 6-Substituted s-Triazolo[4,3-b]-s-Tetrazine-3-Thiols: A Sensitive and Specific Test for Aldehydes. J. Chem. Soc., Perkin Trans. 1 1975, 975–979. 10.1039/P19750000975. [DOI] [Google Scholar]
- Shoemaker R. H. The NCI60 Human Tumour Cell Line Anticancer Drug Screen. Nat. Rev. Cancer 2006, 6, 813–823. 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- Chen X.; Liu M.; Gilson M. A Web-Accessible Molecular Recognition Database. Comb. Chem. High Throughput Screening 2001, 4, 719–725. 10.2174/1386207013330670. [DOI] [PubMed] [Google Scholar]
- Liu T.; Lin Y.; Wen X.; Jorissen R. N.; Gilson M. K. BindingDB: A Web-Accessible Database of Experimentally Determined Protein–Ligand Binding Affinities. Nucleic Acids Res. 2007, 35, D198–D201. 10.1093/nar/gkl999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilson M. K.; Liu T.; Baitaluk M.; Nicola G.; Hwang L.; Chong J. BindingDB in 2015: A Public Database for Medicinal Chemistry, Computational Chemistry and Systems Pharmacology. Nucleic Acids Res. 2015, 44, D1045–D1053. 10.1093/nar/gkv1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FastROCS Toolkit 2017. Oct 2; OpenEye Scientific Software: Santa Fe, NM. http://www.eyesopen.com.
- Bhat R.; Xue Y.; Berg S.; Hellberg S.; Ormö M.; Nilsson Y.; Radesäter A.-C.; Jerning E.; Markgren P.-O.; Borgegård T.; et al. Structural Insights and Biological Effects of Glycogen Synthase Kinase 3-Specific Inhibitor AR-A014418. J. Biol. Chem. 2003, 278, 45937–45945. 10.1074/jbc.M306268200. [DOI] [PubMed] [Google Scholar]
- Yoshino Y.; Ishioka C. Inhibition of Glycogen Synthase Kinase-3 Beta Induces Apoptosis and Mitotic Catastrophe by Disrupting Centrosome Regulation in Cancer Cells. Sci. Rep. 2015, 5, 1–14. 10.1038/srep13249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OEDOCKING 3.4.0.2; OpenEye Scientific Software: Santa Fe, NM. http://www.eyesopen.com.
- McGann M. FRED Pose Prediction and Virtual Screening Accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. 10.1021/ci100436p. [DOI] [PubMed] [Google Scholar]
- McGann M. FRED and HYBRID Docking Performance on Standardized Datasets. J. Comput.-Aided Mol. Des. 2012, 26, 897–906. 10.1007/s10822-012-9584-8. [DOI] [PubMed] [Google Scholar]
- Omega Toolkit 2017. Oct 2; OpenEye Scientific Software: Santa Fe, NM. http://www.eyesopen.com.
- ROCS 3.3.2.2; OpenEye Scientific Software: Santa Fe, NM. http://www.eyesopen.com.
- AbdulHameed M. D. M.; Chaudhury S.; Singh N.; Sun H.; Wallqvist A.; Tawa G. J. Exploring Polypharmacology Using a ROCS-Based Target Fishing Approach. J. Chem. Inf. Model. 2012, 52, 492–505. 10.1021/ci2003544. [DOI] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burley S. K.; Berman H. M.; Bhikadiya C.; Bi C.; Chen L.; Di Costanzo L.; Christie C.; Dalenberg K.; Duarte J. M.; Dutta S.; et al. RCSB Protein Data Bank: Biological Macromolecular Structures Enabling Research and Education in Fundamental Biology, Biomedicine, Biotechnology and Energy. Nucleic Acids Res. 2019, 47, D464–D474. 10.1093/nar/gky1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OMEGA 3.1.2.2; OpenEye Scientific Software: Santa Fe, NM. http://www.eyesopen.com.
- Hawkins P. C. D.; Skillman A. G.; Warren G. L.; Ellingson B. A.; Stahl M. T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell E. W.; Zhang Y. DockRMSD: An Open-Source Tool for Atom Mapping and RMSD Calculation of Symmetric Molecules through Graph Isomorphism. J. Cheminf. 2019, 11, 40. 10.1186/s13321-019-0362-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera - A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Zoete V.; Cuendet M. A.; Grosdidier A.; Michielin O. SwissParam: A Fast Force Field Generation Tool for Small Organic Molecules. J. Comput. Chem. 2011, 32, 2359–2368. 10.1002/jcc.21816. [DOI] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kalé L.; Schulten K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell A. D.; Bashford D.; Bellott M.; Dunbrack R. L.; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- Mackerell A. D. Jr.; Feig M.; Brooks C. L. III Extending the Treatment of Backbone Energetics in Protein Force Fields: Limitations of Gas-Phase Quantum Mechanics in Reproducing Protein Conformational Distributions in Molecular Dynamics Simulations. J. Comput. Chem. 2004, 25, 1400–1415. 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- Best R. B.; Zhu X.; Shim J.; Lopes P. E. M.; Mittal J.; Feig M.; MacKerell A. D. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain Χ1 and Χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. 10.1063/1.470117. [DOI] [Google Scholar]
- Evans D. J.; Holian B. L. The Nose-Hoover Thermostat. J. Chem. Phys. 1985, 83, 4069–4074. 10.1063/1.449071. [DOI] [Google Scholar]
- Bakan A.; Meireles L. M.; Bahar I. ProDy: Protein Dynamics Inferred from Theory and Experiments. Bioinformatics 2011, 27, 1575–1577. 10.1093/bioinformatics/btr168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakan A.; Dutta A.; Mao W.; Liu Y.; Chennubhotla C.; Lezon T. R.; Bahar I. Evol and ProDy for Bridging Protein Sequence Evolution and Structural Dynamics. Bioinformatics 2014, 30, 2681–2683. 10.1093/bioinformatics/btu336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter J. D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. 10.1109/MCSE.2007.55. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.