

MRNIP interacts directly with the MRE11:RAD50 nuclease to protect nascent DNA from degradation and consequent genome instability.
Abstract
The remodeling of stalled replication forks to form four-way DNA junctions is an important component of the replication stress response. Nascent DNA at the regressed arms of these reversed forks is protected by RAD51 and the tumor suppressors BRCA1/2, and when this function is compromised, stalled forks undergo pathological MRE11-dependent degradation, leading to chromosomal instability. However, the mechanisms regulating MRE11 functions at reversed forks are currently unclear. Here, we identify the MRE11-binding protein MRNIP as a novel fork protection factor that directly binds to MRE11 and specifically represses its exonuclease activity. The loss of MRNIP results in impaired replication fork progression, MRE11 exonuclease–dependent degradation of reversed forks, persistence of underreplicated genomic regions, chemosensitivity, and chromosome instability. Our findings identify MRNIP as a novel regulator of MRE11 at reversed forks and provide evidence that regulation of specific MRE11 nuclease activities ensures protection of nascent DNA and thereby genome integrity.
INTRODUCTION
Accurate inheritance of genetic information following cell division depends on the fidelity of genomic DNA replication. This process is impaired by damage to or modification of the DNA template or by replisome progression through regions that are inherently difficult to replicate. Hence, organisms have evolved elaborate mechanisms to mitigate the risk of replisome collapse during replication stress (1). In the context of cancer treatment, many chemotherapies induce cancer cell death by modifying genomic DNA to induce replication fork stalling.
Mutations in the BRCA1 and BRCA2 tumor suppressor genes predispose individuals to breast, ovarian, and prostate cancers (2). Both BRCA1 and BRCA2 are crucial DNA repair factors that facilitate homologous recombination (HR)–mediated repair of DNA double-strand breaks (DSBs) (3, 4). A growing body of evidence demonstrates HR-independent genome maintenance functions for BRCA1/2 and several other core DNA repair proteins such as WRN in the protection of stalled replication forks (5–11). In response to genotoxic stresses, stalled replication forks remodel via rehybridization of nascent DNA to form four-way DNA intermediates (12), facilitating tolerance or bypass of replication impediments. It is now evident that BRCA1/2 and RAD51 act in concert to stabilize stalled forks (12) and promote replication stress resistance by preventing the inappropriate nucleolytic degradation of reversed forks by the nucleases MRE11 and EXO1 (8, 13, 14). Several recent publications have yielded further insight, revealing essential roles for the SMARCAL1 chromatin remodeler, the DNA translocase ZRANB3, and even the RAD51 itself in replication fork reversal (12, 14–16). Loss of function of an additional subset of fork protection factors such as CtIP (17), BOD1L (18), ABRO1 (19), and RIF1 (20, 21) results in fork degradation licensed by the nuclease DNA2.
Replication stress–induced genome instability is a major factor that drives cancer development and progression, and therefore, understanding the complex mechanisms at play during the replication stress response is an important goal. The MRE11 nuclease can act as either a DNA endonuclease or a 3′-5′ exonuclease, and multiple studies using the MRE11 inhibitor mirin implicate the exonuclease function of MRE11 in reversed fork degradation (4, 8). MRE11 is recruited to stalled forks by the enzyme PARP1 [poly(adenosine diphosphate)–ribose] polymerase 1] (22) in concert with the histone methyltransferase MLL3/4 and its binding partner, PTIP (14). However, the mechanisms governing the regulation of MRE11 activity at reversed forks are poorly understood, and before this study, there was no evidence that direct dysregulation of nuclease functions at reversed forks might cause pathological resection of nascent DNA and genome instability.
MRN-interacting protein (C5ORF45/MRNIP) was recently identified as a novel factor that promotes repair of radiation-induced lesions via functional interactions with the DSB-binding MRE11-RAD50-NBS1 (MRN) complex (23). Here, we have used a combination of iPOND (isolation of proteins on nascent DNA), nuclease assays, and CRISPR-Cas9 and small interfering RNA (siRNA)–based loss-of-function studies to identify MRNIP as a novel replication fork protection factor. MRNIP associates with nascent DNA and promotes replication fork progression, resistance to replication stress agents, and chromosome stability. MRNIP binds directly to MRE11 and prevents the MRE11 and DNA2-dependent degradation of reversed forks. MRNIP specifically represses MRE11 exonuclease but not endonuclease activity in vitro. The fork protection functions of MRNIP are associated with its ability to bind MRE11, because MRNIP truncations displaying reduced MRE11 interaction are unable to rescue fork degradation induced by MRNIP loss. In summary, our work identifies MRNIP as a novel replication fork protection factor that likely functions by repressing the MRE11 exonuclease, and furthers our understanding of the molecular functions underlying the complex processes involved in replication fork protection.
RESULTS
Deletion of MRNIP using CRISPR-Cas9 results in genome instability
The novel DNA repair factor MRNIP was identified in an siRNA-based screen for novel regulators of genome stability and characterized as a functional MRN complex interactor that promotes Ataxia Telangiectasia-Mutated (ATM) signaling in response to radiation-induced DNA damage (23). All published work on MRNIP to date has been based on studies using RNA interference (RNAi). We examined the consequences of total deletion of MRNIP using homology-directed repair (HDR)–directed CRISPR-Cas9 protocols. Cells were transfected with Cas9 and HDR constructs expressing MRNIP-directed guide RNAs (gRNAs), and then insertion-positive cells were selected with puromycin. Surviving clones were isolated, subcultured, and screened for homozygous MRNIP deletion and complete loss of the MRNIP protein by a combination of genomic polymerase chain reaction (PCR), quantitative reverse transcription PCR (qRT-PCR), and Western blot. We successfully generated three cancer cell lines lacking MRNIP in two different backgrounds, hereafter referred to as MRNIP-A (HeLa) and MRNIP-3 or MRNIP-7 (HCT116) (Fig. 1, A and B). All MRNIP knockout (KO) cell lines exhibited evidence of DNA damage accumulation evidenced by increased accumulation of 53BP1 and γH2AX-positive foci (Fig. 1, C to F) and increased neutral COMET score (Fig. 1G). MRNIP KO cells also displayed an increased frequency of radial chromosomes and chromosome fusions (Fig. 1H). Levels of DNA damage were fully rescued in cell lines derived from MRNIP KO backgrounds engineered to stably reexpress FLAG-tagged MRNIP (Fig. 1, E and F). The percentage of cells with damage foci was notably higher in MRNIP KO HCT116 relative to HeLa cells, and in both cases, the increase was approximately three times above basal levels. The ability to rescue these phenotypes by reintroduction of MRNIP demonstrates that the observed DNA damage in MRNIP KO lines is a direct consequence of MRNIP deletion and is not a side effect of spurious off-target Cas9 activity. Because HeLa cells lack functional p53, it is also clear that our observations are not due to selection for p53 loss of function, an effect recently observed in clones derived using CRISPR-Cas9 gene editing protocols (24).
Fig. 1. Genetic deletion of MRNIP using CRISPR-Cas9 results in genome instability.
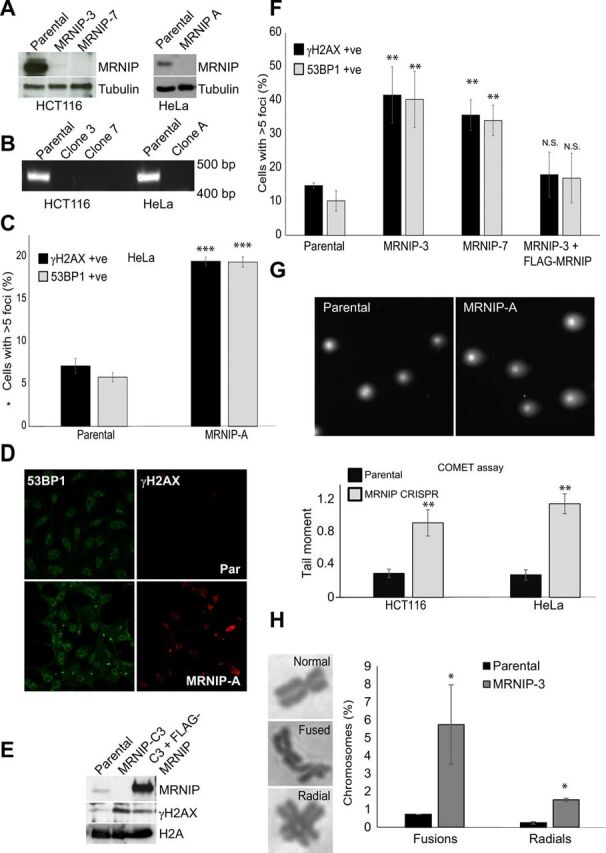
(A) Loss of MRNIP was confirmed by Western blotting of whole-cell extracts from parental HeLa and HCT116 cells and MRNIP KO CRISPR derivative lines. (B) Genomic DNA was isolated from lines used in (A), and PCR for the MRNIP locus was performed. (C and D) Parental HeLa and MRNIP KO CRISPR derivative lines were fixed and stained with antibodies recognizing γH2AX and 53BP1. Cells were counterstained with DAPI, and cells with more than five γH2AX foci were scored as positive. (E) Parental HeLa cells, MRNIP KO cells, and a derivative cell line stably reexpressing FLAG-MRNIP were lysed, and proteins were resolved by SDS-PAGE and probed with the indicated antibodies. (F) Parental HCT116 cells, two independent MRNIP KO cell lines (clones 3 and 7), and a cell line derived from clone 3 stably expressing FLAG-MRNIP were treated as in (C), and cells with more than five γH2AX foci were scored as positive. N.S., not significant. (G) Nuclei from parental HCT116 and HeLa and derivative MRNIP KO lines were isolated, then a neutral COMET assay was performed, and the results were plotted as tail moment. (H) Parental and MRNIP KO HCT116 cells were treated with colcemid for 3 hours, and then metaphase spreads were prepared and analyzed for chromosome abnormalities. The percentage of fused and radial chromosomes is displayed, along with representative images of abnormal chromosomes. Data represent the mean from three experimental repeats, and errors displayed represent SD (*P ≤ 0.05, **P ≤ 0.01, and ***P < 0.001 where indicated).
MRNIP associates with active forks to promote fork progression and resistance to replication stress agents
We also noted a marked increase in the percentage of cells with more than one 53BP1-containing OPT domain (figs. S1E and S3C), suggesting the persistence of underreplicated DNA from the previous cell cycle. Published work suggests that accumulation of replication intermediates underpins the damage phenotypes observed in MRN-deficient cells (25). On the basis of these data, we decided to assess survival of MRNIP KO cells following treatment with replication stress agents. CRISPR-mediated deletion of MRNIP sensitized both cell types to the ribonucleotide reductase inhibitor hydroxyurea (HU), the topoisomerase I inhibitor camptothecin (CPT), and the DNA cross-linker mitomycin C (Fig. 2, A and B, and fig. S1, A to C). MRNIP KO cells also exhibited significantly increased COMET tail moment following HU treatment (fig. S1D). Furthermore, MRNIP KO cells displayed an additional elevation in γH2AX and 53BP1 foci following treatment with HU or CPT at doses that caused minimal damage in parental cells (Fig. 2C). Collectively, these data suggest that MRNIP functions to maintain genome stability during replication stress.
Fig. 2. MRNIP associates with active forks to promote fork progression and resistance to replication stress agents.
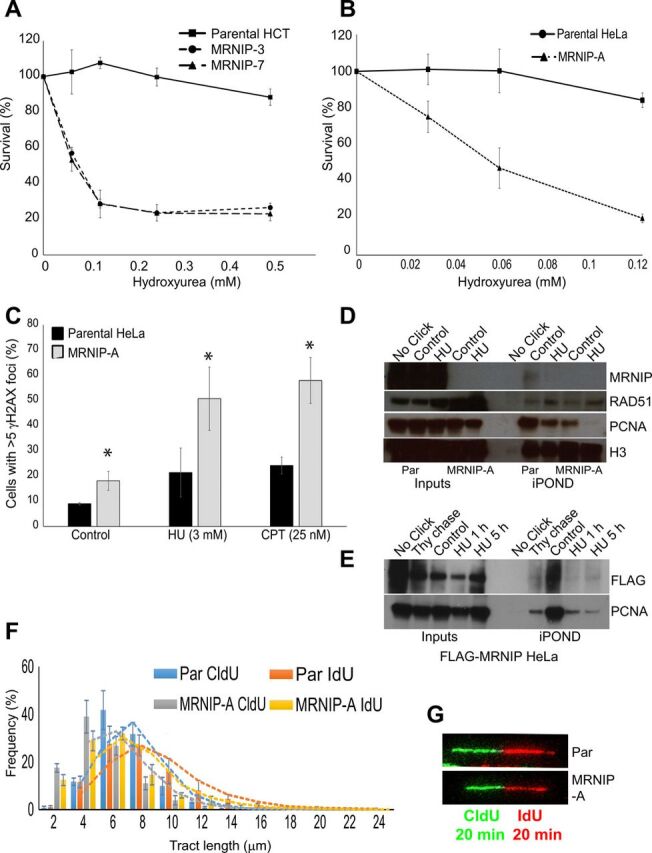
(A and B) Parental HeLa and HCT cells and MRNIP KO CRISPR derivative lines were treated with the indicated concentrations of HU or CPT. After 96 hours, an MTT assay was performed, and results were normalized to untreated controls. (C) Parental HeLa cells and MRNIP KO CRISPR derivative lines were fixed and stained with an antibody recognizing γH2AX. Cells were counterstained with DAPI, and those with more than five γH2AX foci were scored as positive. (D) Parental HeLa cells and MRNIP KO CRISPR derivative HeLa cells were incubated with EdU for 10 min in the presence or absence of 3 mM HU. The iPOND protocol was then performed, and eluates were resolved by SDS-PAGE and blotted with the indicated antibodies. The No-Click control represents a parental untreated sample, processed in the absence of biotin azide. (E) HeLa cells stably expressing FLAG-MRNIP were incubated with EdU for 10 min, then the medium was removed, and cells were incubated in medium containing 10 μM thymidine or 3 mM HU for the indicated times, before performing the iPOND protocol. The No-Click control represents the FLAG-WT untreated sample, processed in the absence of biotin azide. (F and G) Parental HeLa and HCT116 cells and MRNIP KO CRISPR derivative lines were labeled with CldU for 20 min and then IdU for 20 min. Green tracts represent CldU, and red tracts represent IdU staining—representative DNA fiber images are shown. Quantification of contiguous tract lengths from cells treated as in (C), displayed as a frequency value for each length. Data represent the mean from three experimental repeats, and the errors displayed represent SD (*P ≤ 0.05 where indicated).
On the basis of these findings, we sought to address the mechanism by which MRNIP prevents replication-associated DNA damage. To assess whether MRNIP associates with the replication fork, we carried out iPOND analyses (26). We identified endogenous MRNIP in iPOND eluates, but not in controls lacking the Click reagent or in eluates derived from cells lacking MRNIP (Fig. 2D), suggesting that MRNIP associates with the nascent DNA at the progressing fork. We also detected tagged MRNIP in iPOND eluates from HeLa cells stably overexpressing FLAG-MRNIP (Fig. 2E), and furthermore, thymidine chase resulted in loss of MRNIP in iPOND samples (Fig. 2E), providing additional evidence that MRNIP is a bona fide fork-associated protein. Addition of HU following 5-ethynyl-2’-deoxyuridine (EdU) labeling consistently reduced MRNIP association with the fork (Fig. 2D). Examination of proteins associated with nascent DNA in MRNIP KO cells after 1-hour treatment with HU revealed no alteration in RAD51 association with nascent DNA but markedly reduced levels of fork-associated proliferating cell nuclear antigen (PCNA) (Fig. 2D). Loss of PCNA in iPOND eluates has previously been attributed to fork collapse, suggesting that MRNIP is required to stabilize stalled replication forks (26, 27).
Therefore, we next sought to determine whether MRNIP promotes replication fork progression. We performed DNA fiber assays in growing MRNIP KO HeLa and HCT116 lines, in which nascent DNA was dually sequentially labeled with the nucleotide analogs CldU and IdU. In all clones tested, loss of MRNIP resulted in reduced replication fork progression, indicating the presence of replicative stress (Fig. 2, F and G, and fig. S2A) and suggesting that MRNIP has an important function at the progressing fork.
MRNIP prevents MRE11 and DNA2-dependent degradation of reversed forks
Given our findings, and the recent studies demonstrating a clear role for MRE11 in pathological resection of the regressed DNA arms of reversed replication forks (4, 9, 13, 14), we tested the integrity of nascent DNA at stalled forks in MRNIP KO cells via a DNA fiber assay involving sequential CldU and IdU labeling, followed by prolonged fork stalling with a high dose of HU. The ratio of IdU to CldU was used as a readout of the extent of nascent DNA degradation. As expected, parental HeLa and HCT116 cells maintained an IdU:CldU ratio of approximately 1 following HU treatment (Fig. 3, A to C). However, in both MRNIP-A KO HeLa and MRNIP-C3 KO HCT116 cells, the length of the second label was significantly reduced to approximately 0.6, suggestive of extensive nuclease-mediated degradation (Fig. 3, A to C). Pretreatment with the MRE11 exonuclease inhibitors PFM39 or Mirin or siRNA-mediated MRE11 depletion completely reversed replication fork degradation in HU-treated MRNIP KO cells (Fig. 3, A to E, and fig. S2C). Pathological fork degradation in MRNIP KO cells was also fully rescued by depletion or chemical inhibition of DNA2 (Fig. 3, D and E, and fig. S2B). In contrast, inhibition of the MRE11 endonuclease with either PFM01 or PFM03 did not significantly rescue degradation in MRNIP-deficient cells (Fig. 3F), while, as reported previously, inhibition of either the MRE11 exo- or endonuclease (but not DNA2 inhibition) rescued degradation in BRCA2-deficient cells (Fig. 3E and fig. S4D). These data demonstrate that MRNIP protects replication forks specifically from MRE11 exonuclease–dependent degradation in multiple cell types. We also observed proficient chromatin loading of MRE11 in MRNIP-deficient cells, in contrast to the role of MRNIP previously observed following ionizing radiation (fig. S4A), and examination of MRE11 localization with EdU-labeled nascent DNA via proximity ligation assay (PLA) revealed a significantly increased association in replication-stressed MRNIP-deficient cells (fig. S4, B and C). This is consistent with a model in which loss of MRNIP results in more persistent engagement of MRE11 with nascent DNA. In addition, replication fork degradation in MRNIP KO CRISPR-Cas9–generated cell lines was not an off-target effect or a secondary consequence of chronic DNA damage following extended periods of cell growth without MRNIP, as we observed fork degradation in additional cell lines including MCF7 breast cancer cells following transient depletion of MRNIP using two independent siRNA oligonucleotides (fig. S2D).
Fig. 3. MRNIP prevents MRE11 and DNA2-dependent degradation of reversed forks.
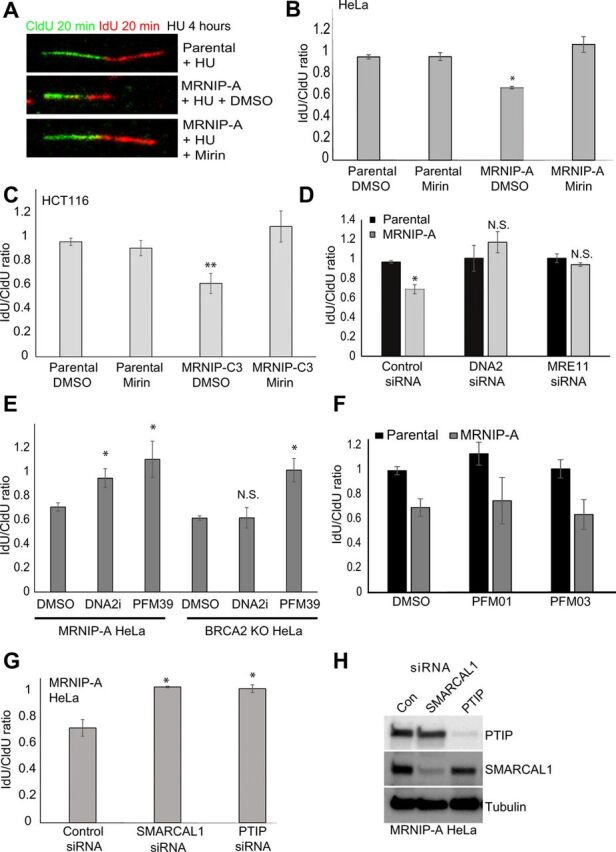
(A to C) Parental HeLa and HCT116 cells and MRNIP KO CRISPR derivative cell lines were labeled with CldU for 20 min and then IdU for 20 min before the addition of 4 mM HU in the presence or absence of 50 μM mirin for 5 hours (see Fig. 4A). Fork degradation was assessed via the IdU:CldU tract length ratio. A representative set of DNA fibers from each condition is displayed in (A). (D) Parental HeLa and a derivative MRNIP KO cell line were transfected with a nontargeting control siRNA or an siRNA targeting either MRE11 or DNA2. After 48 hours, cells were treated as in (A), and the IdU:CIdU tract length ratio for each condition is shown. (E) MRNIP-A KO or BRCA2 KO HeLa cells were labeled as in (A) and then cotreated with 4 mM HU and either DMSO, PFM39, or DNA2 inhibitor for 5 hours. Fork degradation was assessed via the IdU:CldU tract length ratio. (F and G) MRNIP KO HeLa cells were transfected with a nontargeting control siRNA or siRNAs targeting either PTIP or SMARCAL1. After 48 hours, cells were labeled with CldU for 20 min and then IdU for 20 min before the addition of 4 mM HU for 5 hours. Fork degradation was assessed via the IdU:CldU tract length ratio. (H) Cells were transfected as in (A) and then lysed, and the extracts were resolved by SDS-PAGE followed by blotting with the indicated antibodies. Data represent the mean from three experimental repeats, and the errors displayed represent SD (*P ≤ 0.05 and **P ≤ 0.01 where indicated).
To gather further evidence that nascent DNA degradation in MRNIP KO cells occurs at the regressed arms of reversed forks in an MRE11-dependent manner, we depleted the fork remodeling factor SMARCAL1 and the MRE11 recruitment factor PTIP individually in MRNIP KO cells. Both SMARCAL1 and PTIP knockdowns significantly rescued fork degradation associated with loss of MRNIP (Fig. 3, G and H). Furthermore, SMARCAL1 depletion rescued the increase in 53BP1 foci observed in MRNIP-depleted cells (fig. S3, A and B), suggesting that these DNA damage phenotypes are attributable to pathological events occurring at reversed forks. SMARCAL1 depletion resulted in increased γH2AX foci formation even in wild-type (WT) cells, and it was therefore difficult to fully interpret findings derived from this marker in SMARCAL1-depleted MRNIP KO cells. We therefore repeated the experiment in an additional cell line using depletion of the DNA translocase ZRANB3 to prevent fork reversal. Co-depletion of MRNIP and ZRANB3 resulted in a significant reduction in the proportion of cells with 53BP1 foci compared to MRNIP depletion alone, suggesting that events downstream of fork reversal lead to DNA damage in MRNIP KO cells (fig. S3, D and E). Similar findings were obtained in cells co-depleted of MRNIP and PTIP. SMARCAL1 and PTIP depletion also reversed the increased OPT domain expression observed in MRNIP-depleted cells (fig. S3C). Collectively, these findings suggest that MRE11 activity at reversed forks underpins the increase in DNA damage observed in MRNIP KO cells treated with replication stress agents.
Note that previous research identified a heterozygous deletion in MRE11 in the HCT116 cell line, which is thought to have a dominant-negative effect on MRN function (28, 29). However, our findings demonstrate that residual MRE11 exonuclease activity in HCT116 cells is sufficient to drive replication fork degradation, and we note again that we observe MRE11-dependent degradation in both HeLa and HCT116 MRNIP KO backgrounds. Furthermore, pathological degradation of nascent DNA and HU sensitivity in MRNIP-deficient cells was rescued fully by stable reexpression of FLAG-MRNIP in HeLa and HCT116 MRNIP KO cell lines (figs. S2, E and F, and S5, B and C), indicating that this phenotype is also not an off-target consequence of the CRISPR protocol.
MRNIP directly interacts with MRE11 and suppresses its exonuclease, but not endonuclease, activity
Given the established association between MRNIP and the MRN complex, we hypothesized that MRNIP might protect nascent DNA by modulating MRE11 nuclease functions. First, we produced recombinant myelin basic protein (MBP)–tagged MRNIP (rMRNIP) in bacteria to allow us to assess whether MRNIP interacts directly with the MRE11:RAD50 (MR) complex, because our previous association studies were limited to cell-based immunoprecipitation assays. Incubation of recombinant MR with MBP-MRNIP bound to amylose resin revealed a direct interaction between MBP-MRNIP and the MR complex (Fig. 4A). The MBP-only control sample did not pull down any detectable MR.
Fig. 4. MRNIP directly interacts with MRE11 and suppresses its exonuclease, but not endonuclease, activity.
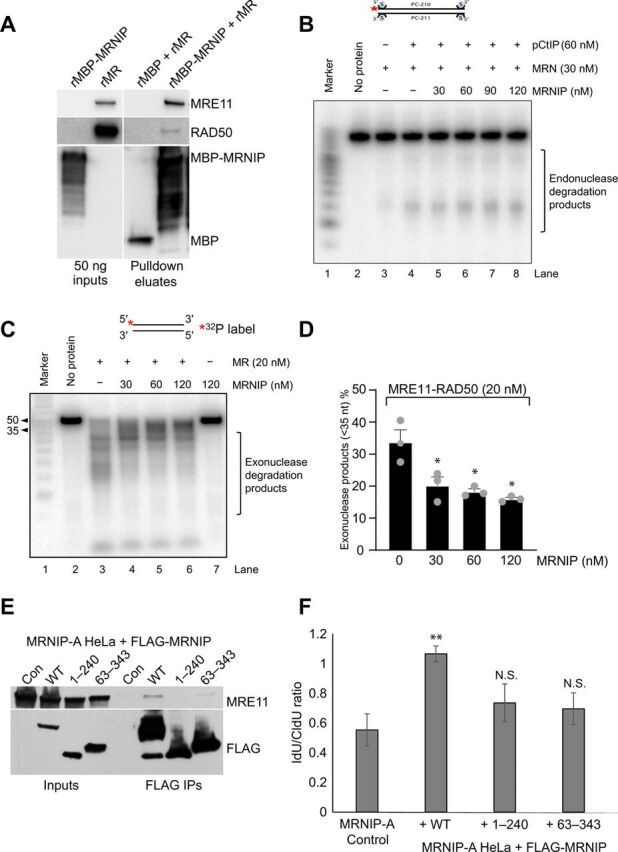
(A) Recombinant MBP or recombinant MBP-tagged MRNIP was bound to amylose resin and then incubated for 3 hours with recombinant MR complex. Unbound material was washed away, and bound proteins were eluted using 20 mM maltose. Samples were denatured and resolved via SDS-PAGE followed by blotting with the indicated antibodies. Input proteins (50 ng) are shown as a gauge of stochiometry. (B) Endonuclease assay with MRN (30 nM), pCtIP (60 nM), and various concentrations of MRNIP (30, 60, 90, and 120 nM) on a 5′ end-labeled 70–base pair (bp)–long dsDNA blocked at both ends with streptavidin. Red asterisk indicates the position of the labeling. (C) Exonuclease assay with MR (20 nM) and various concentrations of MRNIP (30, 60, and 120 nM) on a 5′ end-labeled 50-bp-long dsDNA. Red asterisk indicates the position of the labeling. (D) Quantitation of the assay shown in (C). nt, nucleotides. (E) MRNIP KO HeLa cells and derivative cell lines stably expressing FLAG-tagged WT MRNIP or N-terminally truncated (63–343) or C-terminally truncated (1–240) MRNIP were lysed, and FLAG immunoprecipitation was performed followed by elution with 3× FLAG peptide. Inputs and eluates were resolved by SDS-PAGE followed by Western blotting with the indicated antibodies. (F) Identical cell lines to (A) were plated and labeled with CldU for 20 min and then IdU for 20 min before the addition of 4 mM HU for 5 hours. Fork degradation was assessed via the IdU:CldU tract length ratio. Data represent the mean from three experimental repeats, and the errors displayed represent SD (*P ≤ 0.05 and **P ≤ 0.01 where indicated).
Spurred on by this finding, we next assessed the effect of MRNIP on the exonuclease activity of the MR complex against a double-stranded DNA (dsDNA) substrate and the endonuclease activity of the MRN complex against a dsDNA substrate with streptavidin-blocked ends (the latter in the presence and absence of a phosphorylated form of the endonuclease cofactor CtIP) (3). Titration of rMRNIP had no effect on the endonuclease activity under any condition tested (Fig. 4B and fig. S4E). Excitingly, however, we found that MRNIP specifically repressed the MRE11 exonuclease–mediated degradation of dsDNA (Fig. 4, C and D). This is consistent with our observation that degradation in MRNIP KO cells is reversible by inhibition of the exonuclease, but not the endonuclease, activity of MRE11.
MRNIP is a 37-kDa protein, predicted to have a small zinc finger–like N-terminal domain (Phyre2, SWISS-MODEL). The C terminus of the protein is predicted to include tracts of intrinsically disordered sequence, although it is possible that, on contact with a binding partner, this part of the polypeptide chain assumes a more ordered structure. Both zinc fingers and intrinsically disordered proteins can interact with nucleic acid–mimicking PAR chains, which are formed by PARP at replication forks and DNA breaks (22, 30). PARP1 has an established role in recruitment of MRE11 to stalled replication forks, and PARP is also thought to regulate MRE11 function at forks (31). Therefore, we initially generated FLAG-tagged MRNIP mutants lacking the C-terminal disordered region (1 to 240, ΔCT from now on) or the N-terminal region predicted to form a zinc finger (63 to 343, ΔNT from now on) in the GateWay system and used these constructs to derive stable cell lines expressing truncated proteins in the MRNIP KO HeLa background. FLAG immunoprecipitations in MRNIP KO cells expressing these mutants revealed that both the N terminus and the C terminus of MRNIP are important for MRNIP interaction with MRE11 (Fig. 4E). We note, however, that neither deletion causes a complete loss of MRE11 interaction, suggesting that MRNIP may contact MRE11 via multiple binding surfaces. Restoration of WT MRNIP expression in MRNIP KO cell lines consistently and fully rescued the fork degradation phenotype (Fig. 4F). However, neither the ΔNT or ΔCT truncation significantly rescued this phenotype (Fig. 4F), implying that the MRNIP-MRE11 interaction is important for fork protection.
Given the newly identified role of MRNIP in fork protection and the requirement for the MRNIP N-terminal domain, we hypothesized that MRNIP is co-recruited to forks with MRE11 via an interaction between the predicted NTD zinc finger motif and PAR chains. We therefore mutated all four cysteine residues (C4A mutant) predicted to coordinate a metal ion within the zinc finger–like structure (fig. S5, A and B). However, analysis of fork integrity in MRNIP null cells stably expressing FLAG-C4A MRNIP revealed that the predicted zinc finger motif of MRNIP is not required to mediate fork protection, implying that although this domain does form in vivo, it is not important for the function of MRNIP in replication fork protection (fig. S5B). Likewise, the S115A and Δ25 mutants of MRE11 were fully proficient in fork protection (fig. S5C), despite being involved in radioresistance (23). This is further evidence that MRNIP functions at forks and DSB breaks via very distinct mechanisms. Collectively, our data support a role for MRNIP as a novel fork-associated factor that protects newly formed DNA from pathological degradation, likely by repressing the exonuclease activity of MRE11.
DISCUSSION
The reversal of replication forks to form Holliday junction–like four-way DNA intermediates is now well established as an important response to diverse genotoxic stresses (12, 32). Our data indicate that the novel DNA repair factor MRNIP binds directly to MRE11, where it functions to protect reversed replication forks from pathological MRE11-dependent degradation, likely via repression of the MRE11 exonuclease. MRNIP loss sensitizes multiple cancer cell lines to replication stress agents and chemotherapies. We also present evidence that MRNIP loss results in elevated DNA damage and chromosomal instability.
Consistent with a protective role for MRNIP at reversed forks, we find that the degradation of nascent DNA and increased DNA damage observed in MRNIP KO cells are mitigated by SMARCAL1 depletion. We also provide several lines of evidence for MRE11 involvement in this process. First, small-molecule inhibitors of MRE11 exonuclease activity rescue degradation in MRNIP KO lines, and furthermore, depletion of MRE11 itself or the MRE11 fork recruitment factor PTIP also fully rescued the degradation phenotype.
Our confirmation that MRNIP could interact directly with the MR complex note is also in keeping with the immunoprecipitation procedures used to monitor MRE11-MRNIP association, which were all performed following extensive benzonase digestion, suggesting that the MRE11-MRNIP interaction is not mediated via indirect DNA binding. In addition, we also previously observed that expression of a nuclear localization sequence (NLS) mutant of MRNIP results in relocalization of the MRN complex to the cytosol (23), suggestive of direct interaction. It is possible that the association between MRNIP and nascent DNA is mediated via MRE11.
Depletion or chemical inhibition of DNA2 also completely rescued degradation in MRNIP KO cells. DNA2 is a 5′-3′ nuclease/helicase with important roles in DSB resection and Okazaki fragment maturation (33), which acts in concert with WRN in the degradation of reversed replication forks (34) even in cells proficient in RAD51 filament formation. This model suggests that DNA2 and MRE11 function in separate pathways of fork degradation (34). Recent studies have also demonstrated that both MRE11- and DNA2-dependent degradation can be modulated by depletion of RADX, a negative regulator of RAD51 (10, 11). Our findings constitute the first report of MRE11- and DNA2-dependent fork degradation—a scenario that might be unique to MRNIP-deficient cells. It is possible that dysregulation of MRE11 exonuclease activity in MRNIP-deficient cells results in unique intermediates that are dependent on the nuclease activity of DNA2 for continued processing. Given that MRE11 is a relatively slow exonuclease, it is possible that initial resection by MRE11 is a precursor to continued processing by more processive exonucleases such as DNA2. EXO1/MRE11 interplay at forks and breaks is well established (13, 35, 36). Degradation in MRNIP KO cells proceeds in a strictly MRE11 exonuclease–dependent manner, in contrast to BRCA2 KO cells where coordinated endo- and exonuclease activities drive degradation.
MRNIP is a relatively small protein with no obvious enzymatic functions. Nonetheless, both N- and C-terminal truncations of MRNIP exhibit reduced interaction with MRE11, and mutants lacking these regions cannot rescue fork degradation induced by MRNIP loss. MRNIP is predicted to contain a zinc finger–like N-terminal domain and an unstructured C-terminal domain. Both of these structural features are capable of binding to PAR chains at stalled forks (13) and DNA breaks (22, 30). This was intriguing, given the known role of PARP1 in recruitment of MRE11 to stalled replication forks. However, alanine substitution of all four predicted metal ion–coordinating cysteine residues did not compromise interaction with MRE11, and furthermore, the quadruple mutant protein retained full functionality in replication fork protection. This suggested that the MRNIP-MRE11 interaction is largely independent of PARP function, and this was confirmed by olaparib treatment (data not shown). Here, we also established that two MRNIP mutants that are unable to rescue radiosensitivity phenotypes are fully proficient in fork protection. This suggests that MRNIP is differently regulated in each scenario, in that both kinase inputs and key sequence features involved in promoting DSB repair have no apparent role at reversed forks. Hopefully, future investigations into the structure of MRNIP bound to the MR complex will prove revealing in this regard.
Last, a recent study has demonstrated a role for the yeast homolog of ATM (Tel1) in protecting replication forks from nuclease-mediated degradation (37). Given that MRE11 phosphorylation by ATM on Ser676 and Ser678 functions to repress resection at DSBs (38), and based on our previous observation that MRNIP promotes damage-induced ATM activation, it is tempting to speculate that MRNIP promotes MRE11 phosphorylation by ATM [or another phosphatidylinositol-3 kinase-related kinase (PIKK)] during replication stress to limit resection at reversed replication forks. This is the subject of further study in the Staples laboratory. In conclusion, we have identified MRNIP as a novel replication fork protection factor, which prevents nuclease-mediated degradation of stalled forks.
MATERIALS AND METHODS
Cell culture and CRISPR-Cas9 cell line generation
U2OS, MCF7, BRCA2 KO HeLa (a gift from the Moldovan Laboratory, Penn State), HeLa, and HCT116 Flp-In TREX cancer cells were maintained as adherent monolayers in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum at 37°C in an atmosphere of 5% CO2. Stable MRNIP KO CRISPR clones were created using Santa Cruz Biotechnology CRISPR-Cas9 and HDR plasmids (sc-412131-KO-2 and sc-412131-HDR-2), using the manufacturer’s protocol. In brief, cells were seeded in six-well plates and transfected with the relevant plasmids using Lipofectamine 2000. The following day, the cells were reseeded into 10-cm plates in the presence of puromycin (2 μg/ml). After a further 10 days, individual clones were picked, subcultured, and analyzed by genomic PCR, qRT-PCR, and Western blotting to confirm homozygous genetic deletion of MRNIP.
Stable cell line generation
Stable HeLa and HCT116 Flp-In cell lines expressing doxycycline-inducible FLAG-MRNIP (WT protein and relevant mutants) were generated via cotransfection with pObpA-Flp recombinase and either empty pDEST-Flag/FRT/TO or pDEST-Flag/FRT/TO-MRNIP according to the Flp-In manufacturer’s instructions (Invitrogen). Clones stably expressing FLAG-MRNIP were selected in medium containing blasticidin S (15 μg/ml) and hygromycin B (200 μg/ml) (Invitrogen). Expression was tested by addition of doxycycline (1 μg/ml) for 24 hours and Western blotting with anti-FLAG and anti-MRNIP antibodies.
RNAi transfections
MCF7, U2OS, HeLa, and HCT116 cells were transfected with 10 to 50 nM siRNA using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s instructions. Cells were collected, lysed, or fixed for analysis after 48 hours unless otherwise indicated.
Cell lysis and Western blotting
For the preparation of whole-cell extracts, cells were solubilized in lysis buffer [25 mM tris-HCl (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1 mM dithiothreitol (DTT), and 1 mM MgCl2] supplemented with benzonase (50 U/μl) (Novagen) and cOmplete protease inhibitors and PhosSTOP phosphatase inhibitors (Roche). Lysates were clarified by centrifugation at 16,000g for 15 min at 4°C. Gel electrophoresis was performed using 4 to 15% Mini-PROTEAN TGX precast gels (Bio-Rad). Briefly, samples were resolved in TGX running buffer and transferred to polyvinylidene difluoride (PVDF) membranes, which were then probed for the protein of interest using antibodies diluted in phosphate-buffered saline (PBS)–0.1% Tween 20 (Sigma-Aldrich) containing 5% Marvel.
Immunoprecipitation
To purify FLAG-tagged proteins, 1 mg of the whole-cell extract was incubated with 20 μl of M2-anti FLAG beads (Sigma-Aldrich) for 16 hours at 4°C. For immunoprecipitations using antibodies raised against endogenous proteins, 2 to 5 μg of antibody were incubated with the sample for 1 to 2 hours before addition to 20 μl of washed protein G beads (Santa Cruz Biotechnology) and incubation for 16 hours at 4°C. Beads were then pelleted and washed three times in 25× bed volume of the lysis buffer. The bound protein was eluted either by heating the beads at 95°C for 5 min with 2 × Lithium dodecyl sulphate (LDS) buffer (Invitrogen) or by incubation with 3 × FLAG peptide (100 ng/ml) (Sigma-Aldrich) according to the manufacturer’s instructions. Inputs represent 5% of the extract used for the immunoprecipitation.
Immunofluorescence
Cells were grown on glass coverslips in 24-well trays, transfected/treated as indicated, fixed with either methanol or 3% buffered paraformaldehyde for 10 min at room temperature, and permeabilized in PBS containing 0.5% Triton X-100 for 5 min at room temperature. Cells were incubated with primary antibody overnight in the cold room and detected using a secondary Alexa Fluor 488– or Alexa Fluor 594–conjugated goat anti-rabbit or anti-mouse immunoglobulin G fluorescent secondary (Invitrogen). Antibody dilutions and washes after incubations were performed in PBS. DNA was counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (1 μg/ml), and coverslips were mounted cell-side down in Shandon Immu-Mount medium (Thermo Fisher Scientific). Fluorescence microscopy was performed on a Zeiss LSM710 confocal microscope at ×40 or ×63 magnification. Images were captured and analyzed using Zen software (Zeiss).
Metaphase spreads
Cells were treated with 4 mM HU for 4 hours and released for 18 hours into fresh HU-free medium. Four hours before fixation, colcemid (100 ng/ml) (Sigma) was added to the cells, which were subsequently collected, washed in PBS, and resuspended in 5 ml of hypotonic solution (10 mM KCl) for 10 min at 37°C. Cells were fixed in cold fixation buffer (3:1 methanol/acetic acid). Cells were centrifuged, and the pellets were washed three times with cold fixative before spreading the chromosomes by dropping on cold slides. The slides were air-dried overnight and mounted with Antifade Mounting Medium containing DAPI (1 μg/ml). Fifty metaphases per sample were scored per sample in each experiment.
Survival assays
Cells were plated in six-well plates at a density of 100,000 cells per well and then transfected with the appropriate siRNAs if required. After 48 hours, cells were replated on 96-well plates at a density of 2000 cells per well, and the following day, these were treated as required, in quadruplicate. After a further 96 hours, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) reagent was added to the cells at a final concentration of 1 mg/ml, and these were incubated at 37°C for 3 hours. The medium was removed and replaced with 200 μl of dimethyl sulfoxide (DMSO) to solubilize the formazan product, and the absorbance of this product was assessed by quantifying optical density at 540 nm using a spectrophotometric microtiter plate reader. Results were normalized to untreated controls.
iPOND
iPOND was performed on HeLa cells exactly as described previously by Sirbu and colleagues (26, 27, 39). Briefly, newly synthesized DNA was labeled with 10 μM EdU for 10 min, cells were fixed in 1% formaldehyde and permeabilized, and the click reaction was performed using azide–polyethylene glycol biotin conjugate (Click Chemistry Tools). Cell preparations were then sonicated and EdU-labeled DNA was precipitated using streptavidin beads, before washing and elution in loading buffer containing 1 mM DTT.
DNA fiber assay
Cells were plated and transfected if appropriate, pulse-labeled with 25 μM CldU (Sigma-Aldrich) and 250 μM IdU (Sigma-Aldrich) for 20 min each, and then treated with HU (4 mM) for 5 hours if required. The cells were resuspended in PBS at 7.5 × 105 cells/ml. Then, 2.5 μl of cells was mixed with 7.5 μl of lysis buffer [200 mM tris (pH 7.5), 25 mM EDTA, and 0.5% SDS] on a clean slide (Thermo Fisher Scientific). After 7 min, the slides were tilted at 25° and then air-dried, before fixation in methanol/acetic acid (3:1). DNA fibers were denatured using 2.5 M HCl for 75 min and then washed extensively with 1× PBS before blocking in 1% bovine serum albumin (BSA)/PBS containing 0.2% Tween 20 for 1 hour. CldU- and IdU-labeled tracts were incubated with two anti-BrdU (5-bromo-2′-deoxyuridine) antibodies [one specific for CldU (Abcam) and the other for IdU (BD)], washed, and incubated with goat anti-mouse/rat Alexa Fluor 488 and Alexa Fluor 594 (Invitrogen). DNA fibers were visualized on a Zeiss LSM710 confocal microscope, and images were collected using Zen software and then analyzed with ImageJ.
Neutral COMET assays
Glass slides (Thermo Fisher Scientific) were prepared by adding 800 μl of molten normal melting point agarose (1%) to a slide followed by setting of the gel underneath a coverslip and overnight air drying. Following the appropriate treatment, exponentially growing cells were trypsinized and diluted to a density of 1 × 105 cells/ml. Low–melting point agarose (1% at 37°C) was then added to cells, mixed and added to a normal melting point agarose (NMPA)–coated slide, and covered by coverslip and left on ice for 2 min. Coverslips were then removed and placed in Coplin jars containing fresh neutral lysis buffer (2.5 M NaCl, 100 mM EDTA, 10 mM tris base, and 1% N-lauroylsarcosine; buffer pH was adjusted to 9.5)—cells were lysed for 1 hour at 4°C. Slides were then washed three times in fresh cold 1× tris-borate-EDTA (TBE) (10 min per wash), transferred to an electrophoresis tank, covered with cold electrophoresis buffer, and subjected to an electrophoretic current of 25 V for 25 min. Slides were then removed from the tank, washed with cold 1× PBS three times for 5 min each, and air-dried overnight. Slides were rehydrated in dH2O for 30 min, and SYBR Gold was added to each slide—excess stain was removed, and slides were air-dried overnight before analysis/storage.
In vitro MBP pulldowns
Amylose magnetic beads (NEB) (100 μl) were washed three times in wash buffer [50 mM tris (pH 7.4), 1 mM DTT, 10% glycerol, and 120 mM NaCl] and then split into two batches, and 5 μg of either MBP or MBP-MRNIP was bound to each set of beads via incubation in the cold room for 1 hour with rotation. Beads were then washed three times in lysis buffer and resuspended in 1 ml of wash buffer containing 1 μg of recombinant MR. This was incubated for 3 hours in the cold room with rotation, and then the beads were washed five times and bound proteins were eluted using 20 mM maltose.
Recombinant protein production
Recombinant MR was produced by the Cejka laboratory, via expression in Spodoptera frugiperda 9 (Sf9) cells, and purified as described previously (40).
Recombinant MBP-tagged MRNIP was produced as follows. Briefly, the MRNIP open reading frame (ORF) was amplified using the following primers: MRNIP-F, 5′-ATATGGATCCGCGTCGCTTCAGCGTTCTCGGG-3′; MRNIP-R, 5′-GCATCCATGGTCACACATCATCGAAGTCTTCC-3′. The PCR product was digested with Bam HI and Nco I and cloned into Bam HI and Nco I sites of pMALT-P-MBP (a gift from the Cejka laboratory) generating pMALT-P-MBP-MRNIP. MRNIP was expressed in Escherichia coli BL21(DE3)pLysS One Shot (Thermo Fisher Scientific) according to the manufacturer’s instructions. MBP-MRNIP was extracted from E. coli cell pellets with lysis buffer [50 mM tris-HCl (pH 7.5), 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM dithiothreitol (DTT), 10% glycerol, and 500 mM NaCl] and sonicated. MBP or MBP-MRNIP protein was bound to amylose resin (NEB) and eluted using 10 mM maltose. Purified MBP-MRNIP was loaded on a pre-equilibrated 16/600 HiLoad Superdex 75 pg column (GE Healthcare) using gel filtration buffer [50 mM tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 10% glycerol, and 1 mM DTT], and fractions were collected and then analyzed using SDS–polyacrylamide gel electrophoresis (PAGE) and Coomassie staining. Peak fractions were then pooled and concentrated.
DNA substrates and nuclease assays
The DNA substrates for the in vitro assays were used and radioactively labeled as described previously (41).The endonuclease assays with MRN, phosphorylated CtIP, and MRNIP were carried out in a 15-μl volume. They were assembled on ice in reaction buffer containing 25 mM tris-acetate (pH 7.5), 1 mM DTT, 5 mM magnesium acetate, 1 mM manganese acetate, 1 mM adenosine triphosphate (ATP), pyruvate kinase (80 U/ml) (Sigma-Aldrich), 1 mM phosphoenolpyruvate, BSA (0.25 mg/ml) (New England Biolabs), and 1 nM oligonucleotide–based DNA substrate (in molecules). Biotinylated DNA ends were blocked by adding 15 nM streptavidin and incubating the samples for 5 min at room temperature. The reaction was then returned on ice, where recombinant proteins were added. The reactions were incubated for 2 hours at 37°C and stopped by adding 0.5 μl of proteinase K (14 to 22 mg/ml) (Roche) and 0.5 μl of 0.5 M EDTA for 30 min at 50°C. The stopped reactions were mixed with an equal volume of loading dye [95% formamide, 20 mM EDTA, and bromophenol blue (1 mg/ml)] and boiled for 4 min at 95°C. The reaction products were separated by denaturing electrophoresis on 15% polyacrylamide gels (acrylamide:bisacrylamide, 19:1; Bio-Rad) containing 7 M urea. Radioactively labeled low–molecular weight marker (Affymetrix, J76410) was used where indicated. The samples were separated by electrophoresis in 1× TBE buffer (89 mM tris, 89 mM boric acid, and 2 mM EDTA), and the resolved gels were fixed in fixing solution (40% methanol, 10% acetic acid, and 5% glycerol) for 30 min at room temperature. The gels were then dried, exposed to storage phosphor screen, and scanned by a Typhoon imager (GE Healthcare). Quantitations were carried out using ImageJ software.
Differently from above, 5 nM oligonucleotide–based substrate (in molecules) was used for the exonuclease assays, and after addition of the proteins, reactions were incubated for 1 hour at 37°C. Stopping, reaction product separation, and analysis were performed as described for the endonuclease assays.
Proximity ligation assays
Cells were plated onto circular sterile coverslips in 24-well plates and grown for 24 hours. Cells were pretreated with 10 μM EdU for 20 min or pretreated with EdU for 20 min followed by a 2-hour treatment with 5 mM HU at 37°C. Cells were washed in 1× PBS before permeabilization (20 mM NaCl, 0.5% Triton X-100, 3 mM MgCl2, 300 mM sucrose, and 10 mM Pipes) for 5 min on ice. Cells were washed in 1× PBS before fixation in 4% paraformaldehyde in PBS for 10 min on ice. One further wash in 1× PBS was carried out before blocking in 5% BSA in PBS overnight at 4°C.
The click reaction (10 μM biotin azide, 2 mM CuSO4, and 10 mM sodium ascorbate in 1× PBS) was carried out on all samples pretreated with EdU as well as a “No-EdU” control that was included for each cell line. Briefly, cells were washed in 1× PBS before incubation for 1 hour at room temperature in either 200 μl of the Click reaction solution or the DMSO “No-Click” solution. Cells were washed in 1× PBS, and then the PLA assay (Sigma Duolink) was carried out according to the manufacturer’s instructions. Briefly, samples were incubated with rabbit anti-biotin (Bethyl, A150-109A: 1:3000) and mouse anti-MRE11 antibodies (Abcam, ab214: 1:100) in Duolink antibody dilution buffer at 37°C in a humidified chamber for 1 hour. Cells were then washed in Duolink buffer A and incubated with anti-rabbit Plus and anti-mouse minus PLA probes for 1 hour at 37°C. Ligase was then added and incubated for 30 min at 37°C. Cells were washed in buffer A, and then fluorescence signal was amplified by the addition of Duolink polymerase for 100 min at 37°C (in a dark humidified box). Cells were then washed in Duolink buffer B and finally in 0.01× buffer B. PLA foci were visualized on a Zeiss LSM710 confocal microscope, and images were captured using Zen software (Black edition).
Primers
The following primers were used: MRNIP Genomic Forward, GGTGGGAAGAGAAAAGCACTT; MRNIP Genomic Reverse, CAACACATGCTACAGAACCACTAG; MRNIP GW FOR 63-343, GGGGACAAGTTTGTACAAAAAAGCAGGCTTGCAGGGACAAGT; MRNIP GW REV STOP, GGGGACCACTTTGTACAAGAAAGCTGGGTTCACACATCATCATCGAAGTC; MRNIP FOR GW, GGGGACAAGTTTGTACAAAAAAGCAGGCTTGGCGTCGCTTCAGCGTTCTCGTTCAGAGC; MRNIP GW REV 1-240, GGGGACCACTTTGTACAAGAAAGCTGGGTTCATGAACTTTTTCTAGGTGGC; C3134AA FOR, AAGAGTGTCAAGTGGACAGCCAAAGCTGCTGGAGAGAAGCAGTCC; CC1215AA REV, TGCGCCTGGAAGAGGCGGGCGCTGCAGGCGCGTAGCACCCGAGAA; CC3134AAC REV, GGACTGCTTCTCTCCAGCAGCTTTGGCTGTCCACTTGACACTCTT; CC1215AA FOR, TTCTCGGGTGCTACGCGCCTGCAGCGCCCGCCTCTTCCAGGCGCA.
siRNA oligonucleotide sequences
All siRNAs were purchased from MWG Eurofins, high-performance liquid chromatography (HPLC)–purified, and synthesized with dTdT overhangs. The oligonucleotide sequences used are as follows: Mrnip-1, 5′-GCAAACAGCCUUCAUCCAA-3′; Mrnip-2, 5′-GUUAGGAGGGACAGGGUUC-3′; Smarcal1, 5′-GCUUUGACCUUCUUAGCAA-3′; Ptip, 5′-UGUUUGCAAUUGCGGAUUAUC-3′; and Zranb3, 5′-GAUCAGACAUCACACGAUU-3′.
List of antibodies
The following antibodies were used: Western blotting: MBP (Abcam, ab40390), MRE11 (GeneTex, 12D7), RAD50 (Bethyl, A300-184A), MRNIP (Abcam, C terminus, ab150917, 1:1000), actin (Abcam, ab1801), tubulin (Abcam, ab4074), SMARCAL1 (Santa Cruz Biotechnology, sc-376377), PTIP (Bethyl, A300-370A), RPA (Abcam, ab2175), γH2AX (Cell Signaling Technology, 9718), H2A (Abcam, ab18255), and FLAG-HRP (Sigma-Aldrich, A8592); immunofluorescence: 53BP1 (Abcam, ab21083, 1:2000) and γH2AX (Upstate, 1:1000).
Supplementary Material
Acknowledgments
We thank R. Cha, R. MacFarlane, and E. Hartsuiker for useful discussions. Funding: This research was funded by North West Cancer Research, UKRI and Cancer Research Wales grants to C.J.S., whose position is currently funded by a UKRI Future Leader Fellowship. MRNIP was initially identified while C.J.S. was funded by a Cancer Research UK Senior Fellowship awarded to S.J.C. Author contributions: C.J.S. and L.G.B. devised the experiments with input and advice from S.J.C. L.G.B. performed the majority of experiments including all CRISPR line generation and validation, iPOND, and DNA fiber and COMET assays, with associated analysis and data formatting. A.M.W. and E.A. contributed immunofluorescence studies and generation and validation of stable cell lines. I.C. and A.S. conducted nuclease assays. C.J.S. wrote the paper with contributions from all other authors and assisted with immunofluorescence and survival assays. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/28/eaba5974/DC1
REFERENCES AND NOTES
- 1.Zeman M. K., Cimprich K. A., Causes and consequences of replication stress. Nat. Cell Biol. 16, 2–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Apostolou P., Fostira F., Hereditary breast cancer: The era of new susceptibility genes. Biomed. Res. Int. 2013, 747318 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anand R., Ranjha L., Cannavo E., Cejka P., Phosphorylated CtIP functions as a co-factor of the MRE11-RAD50-NBS1 endonuclease in DNA end resection. Mol. Cell 64, 940–950 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Schlacher K., Christ N., Siaud N., Egashira A., Wu H., Jasin M., Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kolinjivadi A. M., Sannino V., de Antoni A., Técher H., Baldi G., Costanzo V., Moonlighting at replication forks—A new life for homologous recombination proteins BRCA1, BRCA2 and RAD51. FEBS Lett. 591, 1083–1100 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Fradet-Turcotte A., Sitz J., Grapton D., Orthwein A., BRCA2 functions: From DNA repair to replication fork stabilization. Endocr. Relat. Cancer 23, T1–T17 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Daza-Martin M., Starowicz K., Jamshad M., Tye S., Ronson G. E., MacKay H. L., Chauhan A. S., Walker A. K., Stone H. R., Beesley J. F. J., Coles J. L., Garvin A. J., Stewart G. S., McCorvie T. J., Zhang X., Densham R. M., Morris J. R., Isomerization of BRCA1–BARD1 promotes replication fork protection. Nature 571, 521–527 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Mijic S., Zellweger R., Chappidi N., Berti M., Jacobs K., Mutreja K., Ursich S., Chaudhuri A. R., Nussenzweig A., Janscak P., Lopes M., Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun. 8, 859 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iannascoli C., Palermo V., Murfuni I., Franchitto A., Pichierri P., The WRN exonuclease domain protects nascent strands from pathological MRE11/EXO1-dependent degradation. Nucleic Acids Res. 43, 9788–9803 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhat K. P., Krishnamoorthy A., Dungrawala H., Garcin E. B., Modesti M., Cortez D., RADX modulates RAD51 activity to control replication fork protection. Cell Rep. 24, 538–545 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dungrawala H., Bhat K. P., Meur R. L., Chazin W. J., Ding X., Sharan S. K., Wessel S. R., Sathe A. A., Zhao R., Cortez D., RADX promotes genome stability and modulates chemosensitivity by regulating RAD51 at replication forks. Mol. Cell 67, 374–386.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zellweger R., Dalcher D., Mutreja K., Berti M., Schmid J. A., Herrador R., Vindigni A., Lopes M., Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 208, 563–579 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lemaçon D., Jackson J., Quinet A., Brickner J. R., Li S., Yazinski S., You Z., Ira G., Zou L., Mosammaparast N., Vindigni A., MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun. 8, 860 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ray Chaudhuri A., Callen E., Ding X., Gogola E., Duarte A. A., Lee J.-E., Wong N., Lafarga V., Calvo J. A., Panzarino N. J., John S., Day A., Crespo A. V., Shen B., Starnes L. M., de Ruiter J. R., Daniel J. A., Konstantinopoulos P. A., Cortez D., Cantor S. B., Fernandez-Capetillo O., Ge K., Jonkers J., Rottenberg S., Sharan S. K., Nussenzweig A., Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535, 382–387 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kolinjivadi A. M., Sannino V., De Antoni A., Zadorozhny K., Kilkenny M., Técher H., Baldi G., Shen R., Ciccia A., Pellegrini L., Krejci L., Costanzo V., Smarcal1-mediated fork reversal triggers Mre11-dependent degradation of nascent DNA in the absence of Brca2 and stable Rad51 nucleofilaments. Mol. Cell 67, 867–881.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taglialatela A., Alvarez S., Leuzzi G., Sannino V., Ranjha L., Huang J.-W., Madubata C., Anand R., Levy B., Rabadan R., Cejka P., Costanzo V., Ciccia A., Restoration of replication fork stability in BRCA1- and BRCA2-deficient cells by inactivation of SNF2-family fork remodelers. Mol. Cell 68, 414–430.e8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Przetocka S., Porro A., Bolck H. A., Walker C., Lezaja A., Trenner A., von Aesch C., Himmels S.-F., D’Andrea A. D., Ceccaldi R., Altmeyer M., Sartori A. A., CtIP-mediated fork protection synergizes with BRCA1 to suppress genomic instability upon DNA replication stress. Mol. Cell 72, 568–582.e6 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Higgs M. R., Reynolds J. J., Winczura A., Blackford A. N., Borel V., Miller E. S., Zlatanou A., Nieminuszczy J., Ryan E. L., Davies N. J., Stankovic T., Boulton S. J., Niedzwiedz W., Stewart G. S., BOD1L is required to suppress deleterious resection of stressed replication forks. Mol. Cell 59, 462–477 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Xu S., Wu X., Wu L., Castillo A., Liu J., Atkinson E., Paul A., Su D., Schlacher K., Komatsu Y., You M. J., Wang B., Abro1 maintains genome stability and limits replication stress by protecting replication fork stability. Genes Dev. 31, 1469–1482 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garzón J., Ursich S., Lopes M., Hiraga S.-i., Donaldson A. D., Human RIF1-protein phosphatase 1 prevents degradation and breakage of nascent DNA on replication stalling. Cell Rep. 27, 2558–2566.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hiraga S.-i., Monerawela C., Katou Y., Shaw S., Clark K. R., Shirahige K., Donaldson A. D., Budding yeast Rif1 binds to replication origins and protects DNA at blocked replication forks. EMBO Rep. 19, e46222 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahel I., Ahel D., Matsusaka T., Clark A. J., Pines J., Boulton S. J., West S. C., Poly(ADP-ribose)-binding zinc finger motifs in DNA repair/checkpoint proteins. Nature 451, 81–85 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Staples C. J., Barone G., Myers K. N., Ganesh A., Gibbs-Seymour I., Patil A. A., Beveridge R. D., Daye C., Beniston R., Maslen S., Ahel I., Skehel J. M., Collis S. J., MRNIP/C5orf45 interacts with the MRN complex and contributes to the DNA damage response. Cell Rep. 16, 2565–2575 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haapaniemi E., Botla S., Persson J., Schmierer B., Taipale J., CRISPR–Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 24, 927–930 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Bruhn C., Zhou Z.-W., Ai H., Wang Z.-Q., The essential function of the MRN complex in the resolution of endogenous replication intermediates. Cell Rep. 6, 182–195 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Sirbu B. M., Couch F. B., Feigerle J. T., Bhaskara S., Hiebert S. W., Cortez D., Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 25, 1320–1327 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sirbu B. M., McDonald W. H., Dungrawala H., Badu-Nkansah A., Kavanaugh G. M., Chen Y., Tabb D. L., Cortez D., Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J. Biol. Chem. 288, 31458–31467 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takemura H., Rao V. A., Sordet O., Furuta T., Miao Z.-H., Meng L., Zhang H., Pommier Y., Defective Mre11-dependent activation of Chk2 by ataxia telangiectasia mutated in colorectal carcinoma cells in response to replication-dependent DNA double strand breaks. J. Biol. Chem. 281, 30814–30823 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Wen Q., Scorah J., Phear G., Rodgers G., Rodgers S., Meuth M., A mutant allele of MRE11 found in mismatch repair-deficient tumor cells suppresses the cellular response to DNA replication fork stress in a dominant negative manner. Mol. Biol. Cell 19, 1693–1705 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Altmeyer M., Neelsen K. J., Teloni F., Pozdnyakova I., Pellegrino S., Grøfte M., Rask M.-B. D., Streicher W., Jungmichel S., Nielsen M. L., Lukas J., Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP-ribose). Nat. Commun. 6, 8088 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bryant H. E., Petermann E., Schultz N., Jemth A.-S., Loseva O., Issaeva N., Johansson F., Fernandez S., Glynn P. M., Helleday T., PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 28, 2601–2615 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zellweger R., Lopes M., Dynamic architecture of eukaryotic DNA replication forks in vivo, visualized by electron microscopy. Methods Mol. Biol. 1672, 261–294 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Budd M. E., Campbell J. L., A yeast replicative helicase, Dna2 helicase, interacts with yeast FEN-1 nuclease in carrying out its essential function. Mol. Cell. Biol. 17, 2136–2142 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thangavel S., Berti M., Levikova M., Pinto C., Gomathinayagam S., Vujanovic M., Zellweger R., Moore H., Lee E. H., Hendrickson E. A., Cejka P., Stewart S., Lopes M., Vindigni A., DNA2 drives processing and restart of reversed replication forks in human cells. J. Cell Biol. 208, 545–562 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garcia V., Phelps S. E., Gray S., Neale M. J., Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature 479, 241–244 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoa N. N., Akagawa R., Yamasaki T., Hirota K., Sasa K., Natsume T., Kobayashi J., Sakuma T., Yamamoto T., Komatsu K., Kanemaki M. T., Pommier Y., Takeda S., Sasanuma H., Relative contribution of four nucleases, CtIP, Dna2, Exo1 and Mre11, to the initial step of DNA double-strand break repair by homologous recombination in both the chicken DT40 and human TK6 cell lines. Genes Cells 20, 1059–1076 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Menin L., Ursich S., Trovesi C., Zellweger R., Lopes M., Longhese M. P., Clerici M., Tel1/ATM prevents degradation of replication forks that reverse after topoisomerase poisoning. EMBO Rep. 19, e45535 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kijas A. W., Lim Y. C., Bolderson E., Cerosaletti K., Gatei M., Jakob B., Tobias F., Taucher-Scholz G., Gueven N., Oakley G., Concannon P., Wolvetang E., Khanna K. K., Wiesmüller L., Lavin M. F., ATM-dependent phosphorylation of MRE11 controls extent of resection during homology directed repair by signalling through Exonuclease 1. Nucleic Acids Res. 43, 8352–8367 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sirbu B. M., Couch F. B., Cortez D., Monitoring the spatiotemporal dynamics of proteins at replication forks and in assembled chromatin using isolation of proteins on nascent DNA. Nat. Protoc. 7, 594–605 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anand R., Pinto C., Cejka P., Methods to study DNA end resection I: Recombinant protein purification. Methods Enzymol. 600, 25–66 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Cannavo E., Cejka P., Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature 514, 122–125 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/28/eaba5974/DC1