

Dissecting the degradation of the Alzheimer’s disease–related protein Tau by the 20S proteasome at single-residue resolution.
Abstract
Intrinsically disordered proteins (IDPs) can be degraded in a ubiquitin-independent process by the 20S proteasome. Decline in 20S activity characterizes neurodegenerative diseases. Here, we examine 20S degradation of IDP tau, a protein that aggregates into insoluble deposits in Alzheimer’s disease. We show that cleavage of tau by the 20S proteasome is most efficient within the aggregation-prone repeat region of tau and generates both short, aggregation-deficient peptides and two long fragments containing residues 1 to 251 and 1 to 218. Phosphorylation of tau by the non-proline–directed Ca2+/calmodulin-dependent protein kinase II inhibits degradation by the 20S proteasome. Phosphorylation of tau by GSK3β, a major proline-directed tau kinase, modulates tau degradation kinetics in a residue-specific manner. The study provides detailed insights into the degradation products of tau generated by the 20S proteasome, the residue specificity of degradation, single-residue degradation kinetics, and their regulation by posttranslational modification.
INTRODUCTION
Intrinsically disordered proteins (IDPs) are abundant in the human proteome and are implicated as therapeutic targets in major human diseases (1). IDPs have amino acid sequences of low complexity and lack an ordered three-dimensional (3D) structure (1). This allows IDPs to dynamically bind to diverse interaction partners and thus influence many biological processes (1). The activity of IDPs is regulated by posttranslational modifications including phosphorylation and truncation (1, 2). Because of their structural instability, IDPs are particularly sensitive to proteolytic degradation (3–5).
Aggregation of IDPs into insoluble deposits is the hallmark of neurodegenerative diseases (3). Aggregates of the IDP tau are linked to the progression of Alzheimer’s disease (AD) and are found in other age-related disorders termed tauopathies (6). The longest tau isoform in the human central nervous system comprises 441 residues (7). The N-terminal ~150 residues of tau project away from the microtubule surface and are thus termed projection domain (8). The central part of the tau sequence is formed by pseudo-repeats, which bind to microtubules (8, 9) and are essential for pathogenic aggregation and folding into cross-β structure in tau amyloid fibrils (10, 11). Phosphorylated tau accumulates during the development of AD (6, 12).
The 20S proteasome forms the proteolytic core particle of the 26S proteasome holoenzyme (13). In contrast to the proteasomal degradation of most cellular proteins, IDPs can be degraded by the 20S proteasome in an ubiquitin- and adenosine triphosphate (ATP)–independent process without the necessity of the 19S regulatory particle (3–5). Soluble tau is degraded by the 20S proteasome (14, 15), while phosphorylation and aggregation of tau inhibit its turnover by the proteasome (2, 15–17). Decline of proteasomal activity and accumulation of tau have been linked to neurodegeneration (2, 18, 19): Decreased proteasomal activity results in tau accumulation, neurotoxicity, and cognitive dysfunction in cell and animal models of neurodegenerative disorders. Pharmacological activation of the 20S proteasome, direct administration of proteasome, or targeted proteasomal degradation of tau is therefore the focus of current therapeutic strategies targeting tauopathies (20, 21).
Here, we study the degradation of the IDP tau by the 20S proteasome through a residue-specific and quantitative approach that combines nuclear magnetic resonance (NMR) spectroscopy and mass spectrometry (MS). We provide detailed insights into the identity and properties of the proteasomal degradation products of tau, the single-residue degradation kinetics, and their specific regulation by phosphorylation in different tau domains/by different kinases.
RESULTS
The 20S proteasome processes tau
The 20S proteasome (20S) is a barrel-shaped complex comprised by two stacked heptameric β-rings that are sandwiched by two heptameric α-rings (Fig. 1A) (13). The proteolytic sites, which hydrolyze the peptide bonds of substrates, are located in the β subunits. IDPs thus traverse through the α-rings to reach the active sites in the interior of the 20S proteasome (Fig. 1B). To study degradation of the IDP tau, we recombinantly prepared 20S from Thermoplasma acidophilum, which contains only one type of α subunit and one type of β subunit. This 20S particle thus has 14 identical chymotrypsin-like active sites, which are positioned at equal distances around the β-rings (Fig. 1B). Electron microscopy (EM) showed intact barrel-shaped 20S complexes (Fig. 1C). The 441-residue isoform of tau (hTau40; also termed 2N4R tau; Fig. 1D) was also expressed in Escherichia coli.
Fig. 1. Degradation of the IDP tau by the 20S proteasome.
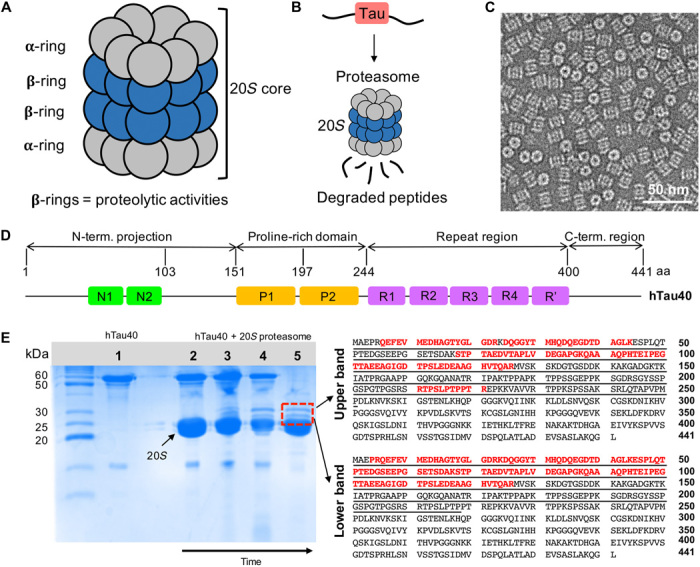
(A) Schematic representation depicting the architecture of the 20S proteasome (20S) comprising 28 subunits arranged in four heptameric rings (α7β7β7α7). (B) The proteolytic active sites of the 20S proteasome are located in its interior, thus enabling degradation of hTau40 into short peptides once it has entered the 20S core. (C) Negatively stained EM micrograph of the 20S proteasome. (D) Domain organization of full-length hTau40 composed of 441 amino acids (aa) (UniProt ID 10636-8). N1 and N2 are the two inserts in the N-terminal projection domain, P1 and P2 correspond to the two proline-rich regions, and R1 to R′ are five pseudo-repeats. (E) (Left) SDS-PAGE gel showing hTau40 (1) and the degradation of (2 to 5) hTau40 by the 20S proteasome over time. The samples were incubated at 37°C for 30 min (2), 90 min (3), and 150 min (4) and were subsequently put at 4°C for additional 48 hours (5). After 48 hours, two well-resolved bands at ~28 and ~30 kDa (red lined box) appeared. (Right) The amino acid sequences of the upper (~30 kDa) and lower bands were identified with in-gel analysis and marked in red. Both intermediates correspond to the N-terminal domain of hTau40.
Recombinant hTau40 was incubated with the 20S proteasome, and degradation was followed by SDS–polyacrylamide gel electrophoresis (PAGE) (Fig. 1E, left). After ~150 min, a clear decrease in the intensity of the hTau40 band at ~60 kDa was apparent (lane 4 in Fig. 1E). In addition, two bands running at ~30 and 28 kDa appeared. Analysis after 48 hours of incubation confirmed the presence of the two new bands, while the full-length protein was degraded to near completion (lane 5 in Fig. 1E).
N-terminal cleavage intermediates
The two intermediate bands were precisely and independently excised from the gel, subjected to in-gel digestion using trypsin, which specifically cleaves at the peptide bond C terminus of lysine or arginine residues, and analyzed using liquid chromatography (LC)–MS/MS. For both bands, the MS analysis confidently identified several peptides from the N-terminal domain (Fig. 1E, right). No peptides were identified in the region from 127 to 210, which contains multiple lysine and arginine residues such that trypsin digestion will produce too short sequences to be analyzed by LC-MS/MS. In the case of the upper band, the additional peptide RTPSLPTPPTR (residues 211 to 221 of hTau40) was identified (Fig. 1E, right).
We also separated the two long fragments using LC and detected their molecular weight by intact MS, giving masses of 25.782 and 22.257 kDa (fig. S1). Manual matching of the determined masses to N-terminal sequences of hTau40 showed that the long fragment contains residues 1 to 251, and the short one has residues 1 to 218. Previous studies showed that the upper band is recognized by the antibody Tau-5 (14), which binds to residues in the region from 218 to 225 (22).
Structural properties of N-terminal cleavage intermediate
To gain insight into the structural properties of the long tau fragments generated during 20S degradation, we recombinantly prepared a tau protein comprising residues 1 to 239 of hTau40. Tau(1–239) contains the full epitope for the Tau-5 antibody (residues 218 to 225) and has a length in between the two long N-terminal fragments. Particle size analysis by dynamic light scattering (fig. S2A) showed that both hTau40 and Tau(1–239) are more compact than the average size values for IDPs (fig. S2B) (23). hTau40, with an experimental size of 5.2 nm and an expected size for its number of residues of 5.5 nm, is 5% more compact than expected, while Tau(1–239) is 18% more compact than expected with 3.3 and 4 nm as experimental and expected sizes, respectively. Despite the stronger compaction of Tau(1–239), both proteins present the typical pattern of random coil conformation in circular dichroism spectra (fig. S2C).
Figure S2D shows the 1H-15N heteronuclear single-quantum coherence (HSQC) spectrum of 15N-labeled Tau(1–239). The backbone cross peaks are located in the region between 7.6 and 8.6 parts per million (ppm), which is characteristic for IDPs. When compared to hTau40, chemical shift perturbation was restricted to the most C-terminal residues of Tau(1–239) (fig. S2E), i.e., residues where Tau(1–239), but not hTau40, ends. Analysis of the secondary structure propensities using the chemical shifts of carbonyl and Cα (fig. S2F) furthermore showed that both hTau40 and Tau(1–239) are mainly random coil, in agreement with circular dichroism spectra (fig. S2C).
In addition, the single-residue analysis showed that Tau(1–239) contains elements of transient secondary structure: residues 116 to 119 with a tendency for helical structure and two short stretches (residues 150 to 152 and 225 to 230) with extended conformation. The same transiently structured regions were detected in hTau40 (fig. S2F). TALOS+ also identified four regions with preference for extended conformation (residues 275 to 279, 306 to 310, 337 to 339, and 392 to 399) and one with helical content (residues 431 to 437) in hTau40, in agreement with previous analysis (24). The presence of extended conformations in the repeat region has previously been suggested to be responsible for the observation that the repeat region of tau, which is not present in Tau(1–239), is less compact when compared to a pure random coil conformation. The combined data thus point to a compaction of the N-terminal cleavage intermediates of hTau40 (fig. S2, A and B).
Tau peptides generated by the 20S proteasome
To identify short tau peptides generated by 20S, we analyzed the released peptides in the supernatant after incubation of hTau40 and 20S using MS. The largest fraction of identified peptides was from hTau40’s pseudo-repeat region (Fig. 2, A and B). In addition, peptides from the C-terminal domain and the residue regions 2 to 13, 84 to 103, and 167 to 192 were detected but with very low responses in MS in the supernatant (Fig. 2C). The tau peptides and their cleavage sites identified by MS are generally in good agreement with the proteasomal cleavage sites predicted by NetChop 3.1 (Fig. 2B) (25).
Fig. 2. MS of tau peptides generated by the 20S proteasome.

(A) Domain organization of hTau40. (B) Amino acid sequence of hTau40 depicting in color [color code as in (A)] the 20S-generated peptides, which were identified by LC-MS/MS. The peptides underlined with black dots were also present in the in-solution sample but with low intensities. The slashes depict all identified cleavage sites. Cleavage sites predicted by the NetChop server are marked by arrows. The bar on top of the VQIVYK sequence indicates the ability of this sequence to form amyloid-like filaments (26). (C) (Left) Histogram representation of the peak area of 20S-generated tau peptides [color code as in (A)] identified by in-solution analysis. Insert depicting the sequences of the identified peptides and the cleavage sites (marked with slashes). (Right) Histogram representing the most intense peptides in the R3 region. A.U., arbitrary units. (D) ThT fluorescence during incubation of the peptide 309VYKPVDL315. The peptide (50, 100, and 150 μM) was incubated with heparin (peptide:heparin molar ratio of 4:1) in triplicates.
The peptide with the highest ion peak area was 309VYKPVDL315 (Fig. 2C, right). It partially overlaps with the hexapeptide sequence 306VQIVYK311 at the beginning of pseudo-repeat R3 (Fig. 2B).
20S-generated tau peptide does not form amyloid
The 306VQIVYK311 sequence is the most hydrophobic residue stretch of tau, is a major driving force for pathogenic tau aggregation, and can form amyloid-like filaments in isolation (26). We therefore tested whether the 20S-generated tau peptide 309VYKPVDL315 can aggregate into amyloid fibrils. To this end, the 309VYKPVDL315 peptide was incubated with heparin at a molar ratio of 4:1.
Figure 2D shows the results from thioflavin-T (ThT) fluorescence measurements of 309VYKPVDL315/heparin samples at three different peptide concentrations during incubation at 37°C for 6 days. For all of the samples, the background-corrected ThT intensity was very low and did not increase during incubation (Fig. 2D). No increase in ThT intensity was detected even when the peptide was incubated for 6 days in the absence of heparin (fig. S3). Because ThT fluorescence intensity increases upon binding to amyloid fibrils, the data show that the 20S-generated peptide 309VYKPVDL315 is not able/has a very low propensity to form amyloid fibrils.
20S degradation of tau at single-residue resolution
To gain insight into the kinetics of degradation of tau by the 20S proteasome and define its residue specificity, we used NMR spectroscopy. Figure 1A displays the 2D 1H-15N HSQC spectrum of 15N-labeled hTau40. The NMR spectrum was recorded at 5°C to attenuate the exchange of amide protons with solvent and thus exchange-induced NMR signal broadening. Comparison of the HSQC spectrum of hTau40 alone with the spectra recorded after 30 min and 66 hours (red) in the presence of 20S (hTau40:20S molar ratio of 4:1) showed that after 30 min, the spectrum of hTau40 was essentially unchanged (fig. S4), but after 66 hours, additional sharp cross peaks were present. Four of the newly appearing cross peaks overlapped with signals observed in a natural abundance 1H-15N HSQC spectrum of the 309VYKPVDL315 peptide, i.e., the peptide with the highest ion peak area in MS (fig. S5). The degradation-associated cross peaks were not observed for a separate sample, which additionally contained the proteasome inhibitor oprozomib (Fig. 3A, right spectrum).
Fig. 3. Kinetics of tau degradation by the 20S proteasome.

(A) Superposition of 2D 1H-15N HSQC spectra of hTau40 at 5°C in the presence of the 20S proteasome after 3 hours (black) and 66 hours (red) in the absence (left) and presence (right) of the proteasome inhibitor oprozomib. (B) (Top) Evolution of relative peak intensities, I(t)/I0, in 2D 1H-15N HSQC spectra of hTau40 in the presence of 20S with increasing incubation time at 5°C. I0 is the cross-peak intensity observed in the first HSQC. (Middle) Residue-specific rate constants of a first-order model of the 20S degradation kinetics of hTau40. Correlation coefficients for the fit to the first-order model are color-coded (color code bar to the right). Error bars represent SD. (Bottom) Evolution of relative peak intensities in 2D 1H-15N HSQC spectra of hTau40 in the presence of the 20S proteasome and the proteasome inhibitor oprozomib.
When tau is degraded by the proteasome into small peptides, the chemical environment of residues changes. To gain insights into the kinetics of 20S degradation, the intensity of IDP cross peaks at their location in the absence of 20S can be analyzed (27). Because a 1H-15N backbone correlation can be observed for every non-proline residue in the 2D 1H-15N HSQC, up to 397 (441 residues minus the C terminus and 43 prolines, and depending on signal overlap) sequence-specific probes for tau degradation are thus available.
The top panel in Fig. 3B displays the decrease of NMR signal intensities along the hTau40 sequence with increasing 20S incubation time. The fastest decrease occurred in the repeat domain. To derive residue-specific degradation rates, we fitted first-order decay kinetics via linear regression to the residue-specific intensity data. The highest rates occurred in repeat R3 and reached up to 0.015 hours−1 at 5°C (Fig. 3B, middle, and table S1). Fast degradation kinetics were also observed in the other pseudo-repeats, in agreement with similar sequence compositions. In addition, tau’s C terminus as well as residues ~220 to 250 at the end of the proline-rich region were rapidly affected by degradation.
Oprozomib predominantly inhibits the chymotrypsin-like activity of the 20S proteasome (28). Detailed analysis of the hTau40 spectra in the presence of both 20S and the small-molecule oprozomib showed that the cross peaks of residues in R2 and R3 decreased in intensity by up to 20% after 66 hours (Fig. 3B, bottom, and table S1). Thus, the 20S complex has residual proteolytic activity, which is not inhibited by oprozomib.
Phosphorylation of tau by CaMKII blocks 20S degradation
A large number of kinases can phosphorylate tau (29). These include proline-directed kinases [e.g., glycogen synthase kinase 3β (GSK3β) and cyclin-dependent kinase 5 (cdk5)] that phosphorylate proline-serine/threonine motifs, notably in the proline-rich region of tau, as well as non-proline–directed kinases [e.g., microtubule affinity-regulating kinase (MARK), protein kinase A (PKA), and Ca2+/calmodulin-dependent protein kinase II (CaMKII)], which phosphorylate the KXGS motifs in the pseudo-repeats. CaMKII phosphorylates tau at several sites (30) and colocalizes with neurofibrillary tangles (NFTs) in AD brains (31).
To gain insight into the influence of substrate phosphorylation on 20S degradation, we phosphorylated recombinant hTau40 with CaMKII in vitro. SDS-PAGE demonstrated an upfield shift in the hTau40 band, confirming successful phosphorylation (Fig. 4A). According to MS/MS analysis, CaMKII phosphorylates S131 and T135 in the projection domain, T212 and S214 in P2, S262 in R1, and S356 in R4 (30). 1H-15N NMR spectroscopy further showed that S214, S356, and S413 are fully phosphorylated in hTau40 (Fig. 4B). In addition, S262, S324, and S352 were found to be partially phosphorylated (Fig. 4B).
Fig. 4. Phosphorylation of tau by CaMKII blocks 20S degradation.

(A) SDS-PAGE gel demonstrating phosphorylation of hTau40 by CaMKII in the presence of calmodulin. (B) (Left) Enlarged region with phosphorylated residues taken from the first 2D 1H-15N HSQC recorded at 5°C for a total duration of 3 hours on CaMKII-phosphorylated hTau40 in the presence of 20S. On top, the location of the phosphorylated residues is marked by short black lines in the context of the domain diagram of hTau40. (Right) Superposition of the first 2D 1H-15N HSQC spectrum (black; total measurement time: 3 hours) of CaMKII-phosphorylated hTau40 in the presence of 20S with the spectrum completed after 66 hours (red). (C) Relative peak intensities in 2D 1H-15N HSQC spectra of CaMKII-phosphorylated hTau40 in the presence of the 20S proteasome with increasing time of incubation at 5°C (from red to blue).
We then incubated CaMKII-phosphorylated hTau40 with 20S proteasome at 5°C. Even after 66 hours, no degradation peaks were observed in the 1H-15N HSQC spectrum (Fig. 4B and fig. S6). In addition, hTau40 cross-peak intensities remained largely unaffected (Fig. 4C and fig. S6). Similarly, CaMKII phosphorylation of the tau construct K18, which only contains the repeat domain, attenuated its degradation by the 20S proteasome (fig. S7). Thus, phosphorylation of tau by CaMKII interferes with the degradation of tau by the 20S proteasome.
Impact of phosphorylation by GSK3β on tau degradation kinetics
GSK3β is ubiquitously expressed in mammalian tissue and has been implicated as a major tau kinase in AD (32). In vitro modification of hTau40 by GSK3β results in phosphorylation of S46, T175, T181, S202, T205, T212, T217, T231, S235, S396, S400, and S404 (33). NMR confirmed complete phosphorylation of S396, S400, and S404 (Fig. 5A). In contrast to CaMKII phosphorylation (Fig. 4), phosphorylation by GSK3β did not block proteasomal processing of hTau40 [Figs. 5A (red spectrum) and 6]. Analysis of cross-peak intensities at increasing 20S incubation times further showed that rapid degradation occurred in repeats R2 and R3 of hTau40 (Fig. 5B).
Fig. 5. Proteasomal degradation kinetics of GSK3β-phosphorylated tau.
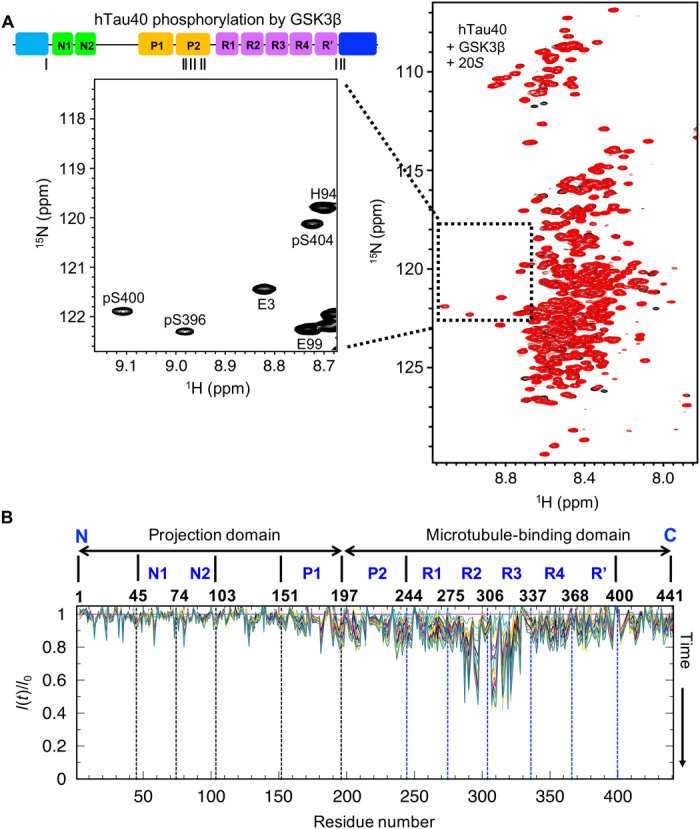
(A) (Left) Enlarged region with phosphorylated residues taken from the first 2D 1H-15N HSQC recorded at 5°C for a total duration of 3 hours on GSK3β-phosphorylated hTau40 in the presence of 20S. On top, the cartoon depicts the sites of phosphorylation of hTau40 by GSK3β. (Right) Superposition of the first 2D 1H-15N HSQC spectrum (black; total measurement time: 3 hours) of GSK3β-phosphorylated hTau40 in the presence of 20S with the spectrum completed after 66 hours (red). (B) Relative peak intensities in 2D 1H-15N HSQC spectra of GSK3β-phosphorylated hTau40 in the presence of the 20S proteasome with increasing time of incubation at 5°C (from red to blue).
Fig. 6. Phosphorylation-dependent degradation of the AD-related protein tau by the 20S proteasome.

(A and B) Per-residue rate constants for degradation of tau by the 20S proteasome. Residue-specific rate constants of a first-order model of the 20S degradation kinetics of hTau40 at 5°C (A, top; same as in Fig. 3B), in the presence of the inhibitor oprozomib (A, bottom), of hTau40 phosphorylated by CaMKII (B, top), and of hTau40 phosphorylated by GSK3β (B, bottom). Correlation coefficients for the fit to the first-order model are color-coded (color code bars to the right). Error bars represent SD. (C) Schematic representation illustrating the phosphorylation-dependent degradation of the AD-related protein tau by the 20S proteasome: Wild-type tau (hTau40) is degraded by the 20S proteasome starting from the pseudo-repeat region and the C-terminal domain, producing short peptides (blue, pink, and orange) from those regions, followed by degradation of the N-terminal domain, which generates two long N-terminal fragments. Depending on the sites of phosphorylation, 20S degradation of tau is inhibited (CaMKII; top) or attenuated (GSK3β; bottom). The color code of different hTau40 domains is described in Fig. 1.
Figure 6 (A and B) compares the residue-specific degradation rates of unmodified hTau40 in the presence of the 20S proteasome (Fig. 6A, top), unmodified hTau40 in the presence of 20S and the inhibitor oprozomib (Fig. 6A, bottom), CaMKII-phosphorylated hTau40 and 20S (Fig. 6B, top), and GSK3β-phosphorylated hTau40 and 20S (Fig. 6B, bottom, and table S1). As calculated from the time-dependent decrease in cross-peak intensities, GSK3β-phosphorylated hTau40 is most efficiently processed by the 20S proteasome in repeats R2 and R3. The phosphorylation of selected residues in tau’s C-terminal domain, however, blocks cleavage of peptide bonds in this region. In addition, the decay of NMR signals in the proline-rich region was strongly attenuated (Fig. 5B and fig. S4), in agreement with phosphorylation of T212, T217, T231, and S235 by GSK3β (33).
DISCUSSION
Within the cell, IDPs are constantly synthesized and degraded by the proteasome. Because they lack a globular structure, IDPs can directly be processed by the 20S proteasome without the need for previous ubiquitination and unfolding by the 26S proteasome (3–5, 34). In parallel, IDPs can be degraded in a ubiquitin-dependent manner by the 26S proteasome. Aggregates of IDPs cannot properly be degraded by the proteasome and are instead processed through autophagy (18, 19). In addition, tau aggregates might inhibit the activity of proteasomes and thereby contribute to neurodegeneration (2, 17, 18). Detailed insights into the processing of tau and other IDPs by the 20S proteasome may therefore be important for treating neurodegeneration and other human diseases (34).
Inhibition of the proteasome by small molecules results in increased amounts of tau in SH-SY5Y cells and rat brain (14, 35). In addition, the four-repeat isoform hTau43 (also termed 0NR4 tau) was shown to be degraded by the human 20S proteasome in vitro without previous ubiquitination (14). In agreement with the latter study, which used human 20S (14), we observed two relatively stable populations of long tau fragments from the N terminus when incubating hTau40 with the 20S proteasome from T. acidophilum (Fig. 1). To determine the identity of the two hTau40 fragments, we performed MS analysis and found that the long and short fragments contain residues 1 to 251 and 1 to 218, respectively (Fig. 1).
Proteasomes cleave their substrates to short peptides with mean lengths between 6 and 10 amino acids (4, 36). Longer (>50 amino acids) degradation intermediates are rarely detected, because the substrate is thought not to dissociate from the proteasome during the degradation process. The presence of two long truncated tau fragments during 20S degradation is therefore unexpected. The more than 200-residue-long tau fragments contain multiple, potential proteasomal cleavage sites (Fig. 2B). To investigate whether the generation of these fragments is the result of specific structural properties of the N-terminal domain of hTau40, we characterized this domain at a single-residue level by NMR spectroscopy. The analysis showed that Tau(1–239) is more compact than hTau40 (fig. S2). We speculate that the more compact structure might interfere with 20S cleavage of the N-terminal fragments.
The short ~6- to 10-residue tau peptides generated by the 20S proteasome can further be cleaved by other proteases (2). In parallel, they might itself contain activity, which is relevant for pathological processes. Consistent with this hypothesis, the six-residue tau peptide 306VQIVYK311 can form insoluble amyloid-like filaments in vitro (26). We therefore used MS to identify the tau peptides generated by 20S degradation (Fig. 2). From the large number of different 20S-generated peptides, the tau peptide with the highest ion peak area was 309VYKPVDL315. Consistent with the high abundance of the 309VYKPVDL315 peptide generated by 20S degradation, signals corresponding to this peptide were identified in the NMR spectra of degraded tau (fig. S4). The 309VYKPVDL315 peptide lacks the first three amino acids of the filament-forming 306VQIVYK311 sequence but has four additional N-terminal residues including the two hydrophobic residues V313 and L315. Despite an overall high hydrophobicity, however, the tau peptide 309VYKPVDL315 did not aggregate into amyloid-like filaments in the presence of the aggregation enhancer heparin (Fig. 2D). Notably, all of the other 20S-generated peptides in the region from 308 to 320 also contain residue P312, i.e., a proline with known β-strand–breaking property (Fig. 2C, right). Cleavage of tau by the 20S proteasome thus generates peptides that are unable to aggregate into amyloid-like filaments.
A wide range of assays have been developed to follow protein degradation. These assays often sample the degradation reaction at discrete time points using SDS-PAGE and antibody binding, autoradiography, protein staining, or Western blotting (37). In addition, proteasome activity can be analyzed through the measurement of fluorescence anisotropy of small-molecule dyes attached to substrate proteins. The identity of degradation products can furthermore be determined using MS. Here, we combined MS with NMR to (i) gain insight into the structural properties of the long degradation intermediates of tau identified by MS and (ii) quantify degradation kinetics in the IDP tau with single-residue and high temporal resolution. MS and NMR spectroscopy are thereby complementary, because MS enables large-scale identification of substrate fragments and peptides generated by proteasomal degradation, but cannot identify all released peptides, lacks single-residue resolution, and is limited in temporal resolution. NMR spectroscopy makes it possible to follow substrate degradation, while the reaction occurs in the test tube, and quantify degradation kinetics at high spatial/per-residue and temporal resolution. On the other hand, a high number of generated peptides and fragments complicate their identification by NMR especially for large IDPs, such as tau, which have many cross peaks. In addition, it has to be taken into account that the cleavage of a peptide bond can be “sensed” by residues that are several positions removed from the site of proteolysis (27). Because of the abovementioned aspects, we believe that the combination of MS and NMR will also be useful to investigate differences in the degradation pattern and substrate selectivity of 20S proteasomes from different organisms.
Using NMR spectroscopy, we found that the 20S degradation of many tau residues follows first-order decay kinetics (Fig. 3). The maximum degradation rate reached ~0.015 hours−1 at 5°C, which corresponds to a degradation half-time of ~46 hours. The reported half-life of tau in HT22 cells is 60 hours (15). The analysis further showed that the 20S proteasome from T. acidophilum preferentially cleaves tau in the pseudo-repeat region, with the fastest rates observed in repeat R3 (Fig. 3). Repeat R3 is part of the cross-β structure of heparin-induced tau fibrils (38). In addition, R3 is located in the core of paired helical filaments purified from the brains of patients with AD (10). The data suggest that the 20S proteasome preferentially degrades the regions of tau, which are important for pathogenic aggregation.
SDS-PAGE analysis, in combination with antibody binding, was used to suggest that the degradation of tau by the 20S proteasome is bidirectional (14), supporting degradation models in which 20S degradation has a preference for the free NH2 or COOH terminus of a substrate (39). In contrast, we find that the proteasome degradation of tau is most efficient in the repeat domain (followed by the C-terminal domain; Fig. 3). Our results are thus in agreement with reports showing that the 20S proteasome can initiate endoproteolytic cleavage at internal sites of IDPs (5). The efficient cleavage of the pseudo-repeat region also enables the generation of the two long fragments from the N terminus of tau (Fig. 1).
The strength of the quantitative, combined MS/NMR approach was further supported by the experiments, in which we studied the influence of phosphorylation of tau on its degradation by the 20S proteasome (Figs. 4 and 5). Tau molecules found in NFTs in the brains of patients with AD are hyperphosphorylated, and dysregulation of tau phosphorylation has been linked to neuronal toxicity (6). Consistent with the hypothesis that impaired proteasomal degradation results in tau accumulation, phosphomimetic tau variants were less efficiently degraded by the proteasome in autophagy-deficient mouse embryonic fibroblasts (16).
The quantitative NMR-based degradation analysis showed that phosphorylation of tau by the non-proline–directed serine/threonine kinase CaMKII inhibits degradation of tau by the 20S proteasome (Figs. 4 and 6). When the proteasome cannot degrade tau, autophagy becomes important, in agreement with the observation that autophagy is the primary route for clearing phosphorylated tau in neurons (16). However, using the same quantitative approach, we found that tau phosphorylated by GSK3β, which phosphorylates Pro-Ser/Thr epitopes seen in NFTs in AD (32), only blocks cleavage in certain regions but does not interfere with tau cleavage in the pseudo-repeats R2 and R3 (Figs. 5 and 6). The regions of tau, which are no longer cleaved such as the C-terminal domain and the proline-rich domain, contain residues phosphorylated by GSK3β (Fig. 5). While GSK3β does not phosphorylate residues in the repeat region, CaMKII phosphorylates S262, S324, S352, and S356 and blocks degradation by the 20S proteasome (Figs. 4 and 6). Phosphorylation of S262, S324, S352, and S356 therefore appears to play an important role in the inhibition of tau degradation by the 20S proteasome. S262, S324, S352, and S356 are also phosphorylated by microtubule-associated protein/MARKs, and their phosphorylation affects tau aggregation as well as microtubule binding of tau (40). Currently, the mechanism of impaired degradation of CaMKII-phosphorylated tau is unknown but could involve (i) an impaired/restricted entry through the 20S gate formed by the first 12 amino acids of the α subunit and (ii) a blocked interaction with the catalytic sites in the β subunit. Our study provides the basis to quantify with single-residue resolution the degradation of tau and other IDPs, their different isoforms, and posttranslationally modified variants and thus gain mechanistic insight into disease-associated accumulation of IDPs.
MATERIALS AND METHODS
Preparation of tau
Unlabeled and 15N-labeled Tau protein (hTau40, UniProt ID 10636-8, 441 residues) were expressed in E. coli strain BL21(DE3) from a pNG2 vector (a derivative of pET-3a, Merck-Novagen, Darmstadt) in the presence of an antibiotic. In case of unlabeled protein, cells were grown in 1 to 10 liters of LB and induced with 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside) at OD600 (optical density at 600 nm) of 0.6 to 0.8. To obtain 15N-labeled protein, cells were grown in LB until an OD600 of 0.6 to 0.8 was reached, then centrifuged at low speed, washed with M9 salts (Na2HPO4, KH2PO4, and NaCl), and resuspended in minimal medium M9 supplemented with 15NH4Cl as the only nitrogen source and induced with 0.5 mM IPTG. After induction, the bacterial cells were harvested by centrifugation, and the cell pellets were resuspended in lysis buffer [20 mM MES (pH 6.8), 1 mM EGTA, and 2 mM dithiothreitol (DTT)] complemented with protease inhibitor mixture, 0.2 mM MgCl2, lysozyme, and deoxyribonuclease (DNase) I. Subsequently, cells were disrupted with a French pressure cell press (in ice-cold conditions to avoid protein degradation). In the next step, NaCl was added to a final concentration of 500 mM and boiled for 20 min. Denaturated proteins were removed by ultracentrifugation at 4°C. The supernatant was dialyzed overnight at 4°C against dialysis buffer [20 mM MES (pH 6.8), 1 mM EDTA, 2 mM DTT, 0.1 mM phenylmethylsulfonyl fluoride (PMSF), and 50 mM NaCl] to remove salt. The following day, the sample was filtered and applied onto a previously equilibrated ion-exchange chromatography column, and the weakly bound proteins were washed out with buffer A (same as the dialysis buffer). Tau protein was eluted with a linear gradient of 60% final concentration of buffer B [20 mM MES (pH 6.8), 1 M NaCl, 1 mM EDTA, 2 mM DTT, and 0.1 mM PMSF]. Protein samples were concentrated by ultrafiltration (5 kDa Vivaspin from Sartorius) and purified by gel filtration chromatography. Last, the protein was dialyzed against 50 mM sodium phosphate (NaP) (pH 6.8).
Preparation of 20S proteasome
20S proteasomes from T. acidophilum were expressed from pRSETA containing the bicistronic gene including psmA and psmB. Transformed BL21 cells were induced with 0.1 mM IPTG and incubated for 18 hours at 37°C. Harvested cells were resuspended in 3 ml of lysis buffer (50 mM Na2HPO4 pH 8.0, 300 mM NaCl) per 1 g of cells and lysed with the French press. The lysate was incubated at 65°C for 15 min. Heat-denatured proteins were removed by centrifugation at 30,000g at 4°C. Polyethylenimine (0.1%, w/v) was added to the supernatant to precipitate contaminating nucleic acids. Precipitated nucleic acids were removed by centrifugation at 100,000g for 1 hour. The supernatant was subjected to differential precipitation with polyethylene glycol 400 (PEG; number signifies the mean molecular weight of the PEG polymer). PEG400 was added to a concentration of 20% (v/v) to the supernatant under stirring at 18°C and incubated for 30 min. Precipitated proteins were removed by centrifugation at 30,000g for 30 min at 4°C. The supernatant was then precipitated by raising the concentration of PEG400 to 40% (v/v). The precipitate of this step contained the 20S proteasomes and was recovered by centrifugation at 30,000g for 30 min at 4°C and resuspended in purification buffer (0.05 M BisTris pH 6.5, 0.05 M K(OAc), 0.01 M Mg(OAc)2, 0.01 M β-Glycerophosphate) containing 5% (w/v) sucrose, 10 mM DTT, and 0.01% (w/v) lauryl maltose neopentyl glycol (LMNG) on an orbital shaker at 18°C. The resuspended material was loaded on 10 to 30% (w/v) sucrose gradients in purification buffer containing 5 mM DTT, which are centrifuged at 284,000g for 16 hours at 4°C. Gradients were harvested in 400 μl of fractions. SDS-PAGE was used to identify fractions containing 20S proteasomes. Selected fractions were pooled and precipitated by the addition of 40% (v/v) PEG400. After centrifugation (30,000g, 20 min), the supernatant was removed and the precipitate was resuspended in purification buffer containing 5% (w/v) sucrose, 10 mM DTT, and 0.01% (w/v) LMNG. The resuspended material was loaded on linear 10 to 40% (w/v) sucrose gradients in purification buffer containing 5 mM DTT, which are centrifuged at 284,000g for 18 hours at 4°C. Fractions containing 20S proteasomes are yet again identified by SDS-PAGE, precipitated and concentrated by the addition of 40% PEG400, and resuspended in purification buffer containing 5% (w/v) sucrose and 5 mM DTT, yielding the final purified protein preparation at 26 mg/ml. Protein concentrations were determined by the Bradford assay (Bio-Rad, Munich, Germany) using bovine serum albumin (BSA) as a standard.
Negative-stain EM
For grid preparation, a protein stock solution (6 mg/ml) was diluted to 0.25 mg/ml with standard buffer without sucrose. Glutaraldehyde was added to the diluted protein solution to a concentration of 0.1% (v/v). After incubation for 2.5 min at room temperature, the reaction was quenched by the addition of 50 mM l-aspartate (pH 6.5). A continuous carbon foil was floated on the protein solution for 1 min at 4°C. A holey carbon copper grid was used to remove the continuous carbon foil from the protein solution. Excess liquid was removed with a tissue paper. Proteins were stained by floating the grid on a saturated uranyl formate solution for 1 min at 4°C. Remaining staining solution was removed with a tissue, and the grid was dried under ambient conditions. Negative-stain EM images were taken with a Philips CM200 microscope (160 kV). Images were acquired at a magnification of ×66,000. The pixel size corresponds to 3.34 Å per pixel. The TVIPS charge-coupled device camera was used to record the micrographs.
Phosphorylation of hTau40
hTau40 was phosphorylated by CaMKII (recombinant human CaMKII alpha protein from Abcam) and GSK3β [recombinant human GSK3 beta protein (active) from Abcam]. The reaction was performed by mixing 0.2 mM hTau40 with 0.02 mg/ml kinase, 2 mM DTT, 2 mM ATP, 1 mM PMSF, and 5 mM MgCl2 in 40 mM Hepes (pH 7.4). In case of CaMKII, we additionally used 2 μM calmodulin (bovine calmodulin, recombinant from Sigma), 1 mM CaCl2, and, in case of GSK3β, 2 mM EGTA. The samples were incubated at 30°C overnight and buffer-exchanged to 50 mM NaP (pH 6.8). Protein concentrations were determined by the Bradford assay using BSA as a standard.
SDS-PAGE electrophoresis
For detection of hTau40 degradation products/fragments generated by the 20S proteasome (hTau40:20S molar ratio of 3:1), we used a 18% separating gel [ddH2O, 30% acrylamide, 1.5 M tris (pH 8.8), 10% SDS, 10% ammonium persulfate (APS), and tetramethylethylenediamine (TEMED)] and a 4% stacking gel [ddH2O, 30% acrylamide, 1 M tris (pH 6.8), 10% SDS, 10% APS, and TEMED]. For validation of hTau40 phosphorylation, we used a 12% separating gel and a 4% stacking gel.
Intact MS
hTau40 was incubated with 20S proteasome for 150 min at 37°C and 1 day at 4°C. The resulting reaction sample was in 50 mM NaP (pH 6.8). The buffer was exchanged to MS compatible sample buffer using Amicon Ultra centrifugal filters with a molecular weight cutoff of 3000. The filter was first washed using water. The reaction sample and 300 μl of sample buffer [0.1% formic acid (FA)] were then added to the filter and centrifuged at 7500g for 30 min. After removing the buffer, 300 μl of sample buffer was added and centrifuged for 30 min. The buffer exchange was then repeated one more time. Last, the samples were diluted to 100 ng/μl for the following MS analysis.
The intact MS experiment was performed on Q Exactive HF-X2 (Thermo Fisher Scientific) coupled to a Dionex UltiMate 3000 UHPLC system (Thermo Fisher Scientific) equipped with a PepSwift Monolithic Trap Column [200 μm inside diameter (ID) × 5 mm] and a ProSwift RP-4H Monolithic Nano Column (100 μm ID × 25 cm). The flow rate was set to 1 μl/min. Mobile phase A and mobile phase B were 0.1% (v/v) FA and 80% (v/v) acetonitrile (ACN), 0.08% FA, respectively. The gradient started at 20% B and increased to 50% B in 33 min and then kept B constant at 90% for 4 min, followed by re-equilibration of the column with 5% B. MS spectra were acquired with the following settings: microscans, 1; resolution, 120,000; mass analyzer, Orbitrap; automatic gain control (AGC) target, 3 × 106; injection time, 100 ms; mass range, 450 to 2000 mass/charge ratio (m/z).
In-gel sample preparation
hTau40 samples were incubated with 20S proteasome (molar ratio of 3:1) for different times (30, 90, and 150 min at 37°C and, additionally, 48 hours at 4°C). The samples were then analyzed by SDS-PAGE electrophoresis as described above. The two fragments (around 25 to 30 kDa) were carefully cut from the gel and used for in-gel analysis. The in-gel digestion of the two bands was performed using trypsin (Promega) to the gels. In the next step, the extracted peptides were desalted by using stage tips. In the last step, the samples were dried (SpeedVac) and readied for further analysis.
In-solution sample preparation
hTau40 was incubated with 20S proteasome (molar ratio of 3:1) at 37°C for 3 hours before the analysis. The samples were precipitated by acetone and put at −30°C overnight. Then, the samples were centrifuged at 14,000g for 10 min, and the supernatant was collected and dried. In the next step, contaminates were removed by the sp3 method, followed by direct injection into the mass spectrometer.
LC-MS/MS analysis
In-gel–digested peptides were analyzed using an Orbitrap Fusion Tribrid (Thermo Fisher Scientific) instrument. In-solution samples were analyzed using Orbitrap Fusion Lumos (Thermo Fisher Scientific). Both instruments are coupled to a Dionex UltiMate 3000 UHPLC system (Thermo Fisher Scientific) equipped with an in-house–packed C18 column (ReproSil-Pur 120 C18-AQ, 1.9 μm pore size, 75 μm inner diameter, 30 cm length, Dr. Maisch GmbH). Both Orbitrap Fusions (Tribrid and Lumos) were operating in data-dependent mode for MS2. Dried samples were resuspended in 5% ACN, 0.1% FA. Samples were centrifuged for 10 min at 14,000g, and the supernatants were transferred to new sample tubes. In both cases, the flow rate was set to 300 nl/min. Mobile phase A and mobile phase B were 0.1% FA (v/v) and 80% ACN, 0.08% FA (v/v), respectively. The gradient in Orbitrap Fusion Tribrid (in-gel samples) started at 10% B and increased to 42% B in 43 min and then kept B constant at 90% for 6 min, followed by re-equilibration of the column with 5% B. MS1 spectra were acquired with the following settings: resolution, 120,000; mass analyzer, Orbitrap; mass range, 380 to 1500 m/z; injection time, 50 ms; AGC target, 4 × 105; S-Lens radio frequency (RF) levels, 60; charge state, +2 to +7; dynamic exclusion after n time, n = 1, dynamic exclusion duration = 60 s. MS2 parameters were as follows: first mass, 120; activation type, higher-energy collisional dissociation (HCD); collision energy, 35; Orbitrap resolution, 30,000; maximum injection time, 250 ms; AGC target, 100,000. The gradient in Orbitrap Fusion Lumos (in-solution samples) increased to 30% B in 42 min and further to 40% B in 4 min and then kept B constant at 90% for 6 min, followed by re-equilibration of the column with 5% B. MS1 spectra were acquired with the following settings: resolution, 120,000; mass analyzer, Orbitrap; mass range, 350 to 1600 m/z; injection time, 50 ms; AGC target, 5 × 105; S-Lens RF levels, 30; charge state, +2 to +7; dynamic exclusion after n time, n = 1, dynamic exclusion duration = 30 s. MS2 parameters were as follows: first mass, 120; activation type, HCD; collision energy, 30; Orbitrap resolution, 15,000; maximum injection time, 120 ms; AGC target, 100,000.
LC-MS/MS data processing
Thermo Proteome Discoverer (2.1.0.81) was used for database searching. In Proteome Discoverer, the Sequest HT, fixed value peptide spectrum match validator, and Precursor Ions Area Detector nodes were used. Parameters for database searching were as follows: the hTau40 protein sequence (P10636-8) was downloaded from Swiss-Prot. Mass tolerance for precursors and fragment ions was set as ±10 and ±20 ppm, respectively. Maximal missed cleavage was 4. Dynamic modifications were set as oxidation (M) and acetylation (protein N terminus). For in-gel samples, fixed modification was carbamidomethylation (C). Trypsin was used as the enzyme, and its specificity was set as semi-specific. For in-solution sample, no enzyme was set. For precursor ions area detector, mass precision was 2 ppm. Only the peptides that were identified with high confidence were used in this study. For in-solution samples, the peak area of precursors was used for quantification of the identified peptides.
Peptide aggregation experiment
The peptide VYKPVDL was synthesized as trifluoroacetic acid salts by GenScript, and the stock solution (1 mM) was made in 25 mM Hepes (pH 7.4). To test whether the peptide can aggregate into amyloid fibrils, we used 50, 100, and 150 μM of the peptide in 25 mM Hepes (pH 7.4). The stock solution of ThT (purchased from Sigma) was prepared in ddH2O, and for the binding assay, 50 μM was used. When heparin (~20 kDa, Roth) was added to the sample, the molar ratio of the peptide to heparin was 4:1. ThT fluorescence was then measured with excitation at 440 nm and emission at 482 nm at 37°C using a multimode microplate reader (Spark 20M, TECAN).
NMR spectroscopy
2D 1H-15N HSQC and 3D spectra (HNCO and HNCA) of hTau40 and Tau(1–239) were acquired at 5°C on a Bruker 800 MHz spectrometer equipped with triple-resonance 5-mm cryogenic probe. The protein concentration was 125 μM in 50 mM NaP buffer (pH 6.8), 5% D2O, 0.1% NaN3, and 50 μM dextran sulfate sodium. Spectra were processed with TopSpin 3.5 (Bruker) and analyzed using Sparky.
NMR degradation experiments with 20S proteasome involving hTau40, phosphorylated hTau40, and hTau40 in the presence of the proteasome inhibitor were acquired and processed as explained above. 2D 1H-15N HSQC spectra were recorded for 15N-labeled hTau40 and 20S proteasome in a molar ratio of 4:1 in 50 mM NaP buffer (pH 6.8) and 10% D2O. The dead time between mixing hTau40 and 20S proteasome and starting the first HSQC experiments was ~30 min.
To study the kinetics of the degradation of hTau40 by the 20S proteasome, 60-min HSQCs were measured every hour during the first 24 hours and then for 180-min HSQCs every 3 hours (for a total of 38 measurements) up to 66 hours. In case of the sample with the inhibitor as well as the phosphorylated samples, 180-min HSQCs were recorded every 3 hours for a total of 22 measurements (66 hours). For our control sample, we used the proteasome inhibitor oprozomib (ApexBio), which was incubated for 2 hours at 37°C in 250 molar excess before the experiment.
Peak intensities were extracted from a series of 1H-15N HSQC datasets at predetermined time intervals. After peak assignment with the software Sparky, the peak intensities were normalized with respect to the initial peak intensity for each residue, taking into account the duration of each HSQC. A residue was excluded from plotting and further analysis if a consecutively recorded peak intensity increased to more than 115% of the relative intensity of the preceding measurement. Such an increase in peak intensity when compared to the preceding measurement can arise from more favorable relaxation properties in the generated peptides when compared to full-length tau. In addition, peak overlap can potentially cause fluctuating intensities.
The peak intensities at all recorded times of the remaining (i.e., not excluded) residues were analyzed by fitting to first-order decay kinetics via linear regression of the data with respect to the analytic solution of the normalized first-order decay model. The fitted first-order decay reaction constants were plotted for all nonexcluded residues of hTau40. The statistical uncertainty in the determined degradation rates expressed in terms of SDs of fits was estimated as follows. For each sample, we randomly excluded five (in case of samples hTau40 + 20S in molar ratios of 4:1 and 4.5:1) or three (in case of samples hTau40 + 20S + inhibitor, hTau40 + CaMKII + 20S, and hTau40 + 20S + GSK3β) intensity profiles collected at the various time intervals from the fitting procedure and repeated this procedure 20 times. The selection was performed by randomly drawing five (three, respectively) numbers from a uniform distribution over all profiles measured at different time intervals, and the fitting procedure was carried out on each of these subsamples and each amino acid residue. From the 21 fits per residue obtained this way (20 “undersampled” plus 1 fit based on all measured profiles), we calculated the sample SD and depicted it as error bars. The plots depicting degradation rates were plotted as the full-data fit (declared here as the mean estimated value) plus/minus the SD. In addition, we determined the Pearson correlation coefficients ρ for all respective fits, which are encoded in the color. Fits with an incorrect sign of ρ (i.e., implying an incorrect/unphysical trend) were excluded from the plot.
Supplementary Material
Acknowledgments
We thank N. Rezaei-Ghaleh for help with NMR experiments and the Max Planck society for support. Funding: The financial support from the German Research Foundation (DFG) through the Emmy Noether Program “GO 2762/1-1” (to A.G.) is acknowledged. P.F. is supported by a Manfred-Eigen-Fellowship from the Max Planck Institute for Biophysical Chemistry. M.Z. was supported by the advanced grant “787679-LLPS-NMR” of the European Research Council. Author contributions: T.U.-G. performed tau phosphorylation, NMR experiments, and data analysis. P.F. and K.-T.P. performed MS and data analysis. A.I.d.O. analyzed Tau(1–239) and performed NMR experiments and K18 degradation. F.H. prepared 20S proteasome. A.G. performed NMR data analysis. M.-S.C.-O. prepared Tau(1–239). A.C. supervised 20S preparation. H.U. supervised MS. E.M. and M.Z. designed the study. The manuscript was written through contributions of all authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD015349. The chemical shifts of Tau(1–239) were deposited in the BMRB (identifier: 28065). All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/30/eaba3916/DC1
REFERENCES AND NOTES
- 1.Wright P. E., Dyson H. J., Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 16, 18–29 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Y., Garg S., Mandelkow E.-M., Mandelkow E., Proteolytic processing of tau. Biochem. Soc. Trans. 38, 955–961 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Opoku-Nsiah K. A., Gestwicki J. E., Aim for the core: Suitability of the ubiquitin-independent 20S proteasome as a drug target in neurodegeneration. Transl. Res. 198, 48–57 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kisselev A. F., Akopian T. N., Woo K. M., Goldberg A. L., The sizes of peptides generated from protein by mammalian 26 and 20 S proteasomes. Implications for understanding the degradative mechanism and antigen presentation. J. Biol. Chem. 274, 3363–3371 (1999). [DOI] [PubMed] [Google Scholar]
- 5.Liu C.-W., Corboy M. J., DeMartino G. N., Thomas P. J., Endoproteolytic activity of the proteasome. Science 299, 408–411 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Y., Mandelkow E., Tau in physiology and pathology. Nat. Rev. Neurosci. 17, 5–21 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Goedert M., Spillantini M. G., Jakes R., Rutherford D., Crowther R. A., Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 3, 519–526 (1989). [DOI] [PubMed] [Google Scholar]
- 8.Weingarten M. D., Lockwood A. H., Hwo S. Y., Kirschner M. W., A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. U.S.A. 72, 1858–1862 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gustke N., Trinczek B., Biernat J., Mandelkow E.-M., Mandelkow E., Domains of tau protein and interactions with microtubules. Biochemistry 33, 9511–9522 (1994). [DOI] [PubMed] [Google Scholar]
- 10.Fitzpatrick A. W. P., Falcon B., He S., Murzin A. G., Murshudov G., Garringer H. J., Crowther R. A., Ghetti B., Goedert M., Scheres S. H. W., Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547, 185–190 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crowther T., Goedert M., Wischik C. M., The repeat region of microtubule-associated protein tau forms part of the core of the paired helical filament of Alzheimer’s disease. Ann. Med. 21, 127–132 (1989). [DOI] [PubMed] [Google Scholar]
- 12.Ihara Y., Nukina N., Miura R., Ogawara M., Phosphorylated tau protein is integrated into paired helical filaments in Alzheimer’s disease. J. Biochem. 99, 1807–1810 (1986). [DOI] [PubMed] [Google Scholar]
- 13.Bochtler M., Ditzel L., Groll M., Hartmann C., Huber R., The proteasome. Annu. Rev. Biophys. Biomol. Struct. 28, 295–317 (1999). [DOI] [PubMed] [Google Scholar]
- 14.David D. C., Layfield R., Serpell L., Narain Y., Goedert M., Spillantini M. G., Proteasomal degradation of tau protein. J. Neurochem. 83, 176–185 (2002). [DOI] [PubMed] [Google Scholar]
- 15.Poppek D., Keck S., Ermak G., Jung T., Stolzing A., Ullrich O., Davies K. J. A., Grune T., Phosphorylation inhibits turnover of the tau protein by the proteasome: Influence of RCAN1 and oxidative stress. Biochem. J. 400, 511–520 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodriguez-Martin T., Cuchillo-Ibáñez I., Noble W., Nyenya F., Anderton B. H., Hanger D. P., Tau phosphorylation affects its axonal transport and degradation. Neurobiol. Aging 34, 2146–2157 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Myeku N., Clelland C. L., Emrani S., Kukushkin N. V., Yu W. H., Goldberg A. L., Duff K. E., Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat. Med. 22, 46–53 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng J., North B. J., Zhang T., Dai X., Tao K., Guo J., Wei W., The emerging roles of protein homeostasis-governing pathways in Alzheimer’s disease. Aging Cell 17, e12801 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boland B., Yu W. H., Corti O., Mollereau B., Henriques A., Bezard E., Pastores G. M., Rubinsztein D. C., Nixon R. A., Duchen M. R., Mallucci G. R., Kroemer G., Levine B., Eskelinen E. L., Mochel F., Spedding M., Louis C., Martin O. R., Millan M. J., Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 17, 660–688 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones C. L., Njomen E., Sjögren B., Dexheimer T. S., Tepe J. J., Small molecule enhancement of 20S proteasome activity targets intrinsically disordered proteins. ACS Chem. Biol. 12, 2240–2247 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Silva M. C., Ferguson F. M., Cai Q., Donovan K. A., Nandi G., Patnaik D., Zhang T., Huang H. T., Lucente D. E., Dickerson B. C., Mitchison T. J., Fischer E. S., Gray N. S., Haggarty S. J., Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models. eLife 8, e45457 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Porzig R., Singer D., Hoffmann R., Epitope mapping of mAbs AT8 and Tau5 directed against hyperphosphorylated regions of the human tau protein. Biochem. Biophys. Res. Commun. 358, 644–649 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Marsh J. A., Forman-Kay J. D., Sequence determinants of compaction in intrinsically disordered proteins. Biophys. J. 98, 2383–2390 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mukrasch M. D., Bibow S., Korukottu J., Jeganathan S., Biernat J., Griesinger C., Mandelkow E., Zweckstetter M., Structural polymorphism of 441-residue tau at single residue resolution. PLOS Biol. 7, e34 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nielsen M., Lundegaard C., Lund O., Keşmir C., The role of the proteasome in generating cytotoxic T-cell epitopes: Insights obtained from improved predictions of proteasomal cleavage. Immunogenetics 57, 33–41 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Sawaya M. R., Sambashivan S., Nelson R., Ivanova M. I., Sievers S. A., Apostol M. I., Thompson M. J., Balbirnie M., Wiltzius J. J. W., McFarlane H. T., Madsen A. Ø., Riekel C., Eisenberg D., Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 447, 453–457 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Sprangers R., Li X., Mao X., Rubinstein J. L., Schimmer A. D., Kay L. E., TROSY-based NMR evidence for a novel class of 20S proteasome inhibitors. Biochemistry 47, 6727–6734 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Chauhan D., Singh A. V., Aujay M., Kirk C. J., Bandi M., Ciccarelli B., Raje N., Richardson P., Anderson K. C., A novel orally active proteasome inhibitor ONX 0912 triggers in vitro and in vivo cytotoxicity in multiple myeloma. Blood 116, 4906–4915 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hanger D. P., Anderton B. H., Noble W., Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 15, 112–119 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Yoshimura Y., Ichinose T., Yamauchi T., Phosphorylation of tau protein to sites found in Alzheimer’s disease brain is catalyzed by Ca2+/calmodulin-dependent protein kinase II as demonstrated tandem mass spectrometry. Neurosci. Lett. 353, 185–188 (2003). [DOI] [PubMed] [Google Scholar]
- 31.McKee A. C., Kosik K. S., Kennedy M. B., Kowall N. W., Hippocampal neurons predisposed to neurofibrillary tangle formation are enriched in type II calcium/calmodulin-dependent protein kinase. J. Neuropathol. Exp. Neurol. 49, 49–63 (1990). [DOI] [PubMed] [Google Scholar]
- 32.Hanger D. P., Noble W., Functional implications of glycogen synthase kinase-3-mediated tau phosphorylation. Int. J. Alzheimers Dis. 2011, 352805 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reynolds C. H., Betts J. C., Blackstock W. P., Nebreda A. R., Anderton B. H., Phosphorylation sites on tau identified by nanoelectrospray mass spectrometry: Differences in vitro between the mitogen-activated protein kinases ERK2, c-Jun N-terminal kinase and P38, and glycogen synthase kinase-3beta. J. Neurochem. 74, 1587–1595 (2000). [DOI] [PubMed] [Google Scholar]
- 34.Guharoy M., Lazar T., Tompa P., Disordered substrates of the 20S proteasome link degradation with phase separation. Proteomics 18, e1800276 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Liu Y. H., Wei W., Yin J., Liu G. P., Wang Q., Cao F. Y., Wang J. Z., Proteasome inhibition increases tau accumulation independent of phosphorylation. Neurobiol. Aging 30, 1949–1961 (2009). [DOI] [PubMed] [Google Scholar]
- 36.Wenzel T., Eckerskorn C., Lottspeich F., Baumeister W., Existence of a molecular ruler in proteasomes suggested by analysis of degradation products. FEBS Lett. 349, 205–209 (1994). [DOI] [PubMed] [Google Scholar]
- 37.Li Y., Tomko R. J. Jr., Hochstrasser M., Proteasomes: Isolation and activity assays. Curr. Protoc. Cell Biol. 67, 3.43.1–3.43.20 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang W., Falcon B., Murzin A. G., Fan J., Crowther R. A., Goedert M., Scheres S. H., Heparin-induced tau filaments are polymorphic and differ from those in Alzheimer’s and Pick’s diseases. eLife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Navon A., Goldberg A. L., Proteins are unfolded on the surface of the ATPase ring before transport into the proteasome. Mol. Cell 8, 1339–1349 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Biernat J., Gustke N., Drewes G., Mandelkow E. M., Mandelkow E., Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: Distinction between PHF-like immunoreactivity and microtubule binding. Neuron 11, 153–163 (1993). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/30/eaba3916/DC1