

To realize the potential of MSCs, bioengineering is needed to boost potency and control heterogeneity.
Abstract
More than 1050 clinical trials are registered at FDA.gov that explore multipotent mesenchymal stromal cells (MSCs) for nearly every clinical application imaginable, including neurodegenerative and cardiac disorders, perianal fistulas, graft-versus-host disease, COVID-19, and cancer. Several companies have or are in the process of commercializing MSC-based therapies. However, most of the clinical-stage MSC therapies have been unable to meet primary efficacy end points. The innate therapeutic functions of MSCs administered to humans are not as robust as demonstrated in preclinical studies, and in general, the translation of cell-based therapy is impaired by a myriad of steps that introduce heterogeneity. In this review, we discuss the major clinical challenges with MSC therapies, the details of these challenges, and the potential bioengineering approaches that leverage the unique biology of MSCs to overcome the challenges and achieve more potent and versatile therapies.
THE LANDSCAPE OF MSC THERAPIES
Multipotent mesenchymal stromal cells (MSCs) have been extensively investigated as a cell therapy, showing promise in treating an array of diseases by restoring organ homeostasis in inflamed, injured, or diseased tissues. Bone marrow–derived MSCs (BM-MSCs) were first described by Friedenstein et al. (1) in the late 1970s and continue to be the most commonly studied MSC source in preclinical and clinical studies. MSCs can also be easily isolated from multiple tissues including adipose tissue (AT), umbilical cord (UC), Wharton’s jelly, and the placenta (2). While initial therapeutic efforts were based on their multipotency, the discovery of their immunomodulatory and trophic properties motivated harnessing MSCs as a treatment for neurodegenerative and inflammatory diseases. To this end, MSCs have been investigated as a treatment for graft-versus-host disease (GvHD), multiple sclerosis (MS), Crohn’s disease (CD), amyotrophic lateral sclerosis (ALS), myocardial infarction (MI), and acute respiratory distress syndrome (ARDS), among others (Table 1) (3–5). MSCs are generally distinct from other cell therapies as their therapeutic effect not only is dictated by cell-cell contact but also may include a so-called hit-and-run mechanism. Here, paracrine effectors from their secretome, including soluble cytokines, growth factors, hormones, and miRNA, are transferred to target cells such as immune cells and cells of damaged tissues through secretion, the uptake of biologics-loaded submicrometer extracellular vesicles (EVs), and immune-mediated phagocytosis (6–9), which can lead to long-term effects. In line with this, many studies have shown that secreted biologics and MSC-derived EVs containing biologically active molecules (such as proteins, lipids, and nucleic acids) retain the biological activity of parental MSCs and demonstrate a similar therapeutic effect in selected animal models (10). Because the properties of secreted biologics and MSC-derived EVs have been thoroughly reviewed elsewhere (11–13), the current article focuses on MSC therapies.
Table 1. Representative indications of MSCs in clinical trials.
MPC, mesenchymal precursor cells; UC, umbilical cord; BM, bone marrow; AT, adipose tissue; Y, yes; N, no; TBD, to be determined; N/A, not available.
|
General indication |
Clinical indication |
Cell source |
Administration route |
Clinical efficacy (Y/N) |
Engineered (Y/N) |
Year started |
Phase | Status | Trial number | |
| Autoimmune disease |
Rheumatoid arthritis |
Allogeneic | MPC | Systemic | Y | N | 2013 | 2 | Complete | NCT1851070 |
| Systematic lupus erythematous |
Allogeneic | UC | Systemic | Promising | N | 2017 | 1 | Complete | NCT03171194 | |
| Cancer | Advanced gastrointestinal cancer |
Autologous | BM | Systemic | N | Y | 2013 | 1/2 | Terminated | NCT02008539 |
| Metastases solid tumors |
Autologous | BM | Systemic | Unknown | Y | 2013 | 1/2 | Complete | NCT01844661 | |
| Cardiac disorders | Acute mycardial infarction |
Allogeneic | BM | Systemic | Y | N | 2009 | 2 | Complete | NCT00877903 |
| Chronic heart failure |
Allogeneic | MPC-BM | Local | Y | N | 2014 | 3 | Ongoing, not recruiting |
NCT02032004 | |
| Class 2 or 3 heart failure |
Autologous | BM | Local | Y | N | 2008 | 2/3 | Complete | NCT008102328 | |
| Ischemic stroke | Allogeneic | BM | Systemic | N | N | 2011 | 2 | Complete | NCT01436487 | |
| GvHD | Chronic GvHD | Allogeneic | BM | Local | Unknown | N | 2012 | 2/3 | Unknown | NCT01526850 |
| Grade B to D acute GvHD |
Allogeneic | BM | Systemic | Y | N | 2006 | 3 | Complete | NCT00366145 | |
| Grade B to D acute GvHD |
Allogeneic | BM | Systemic | Y | N | 2015 | 3 | Complete | NCT02336230 | |
| IBD | Crohn’s disease | Allogeneic | AT | Local | Y | N | 2012 | 3 | Complete | NCT01541579 |
| Crohn’s disease | Allogeneic | BM | Systemic | TBD | N | 2007 | 3 | Ongoing, not recruiting |
NCT00482092 | |
| Ulcerative colitis | Allogeneic | BM | Systemic | N | N | 2010 | 2 | Complete | NCT01240915 | |
| Acute kidney injury |
Allogeneic | BM | Systemic | N | N | 2012 | 2 | Terminated | NCT01602328 | |
| Kidney disorders | Diabetic nephropathy |
Allogeneic | MPC-BM | Systemic | Y | N | 2013 | 1/2 | Complete | NCT01843387 |
| Liver/kidney failure |
Allogeneic | BM | Systemic | N | N | 2011 | 1/2 | Complete | NCT01429038 | |
| Renal transplantation |
Autologous | BM | Systemic | Y | N | 2008 | N/A | Complete | NCT00658073 | |
| Neurodegenerative disease |
Alzheimer’s disease |
Allogeneic | UC | Systemic | Unknown | N | 2012 | 1/2 | Ongoing, not recruiting |
NCT01547689 |
| ALS | Autologous | BM | Local | Y | Y | 2013 | 2 | Complete | NCT02017912 | |
| Chronic progressive MS |
Autologous | BM | Local | TBD | Y | 2019 | 2 | Recruiting | NCT03799718 | |
| Degenerative disc disease |
Allogeneic | MPC | Local | Y | N | 2011 | 2 | Complete | NCT01290367 | |
| MS | Autologous | BM | Systemic | TBD | N | 2014 | 2 | Ongoing, not recruiting |
NCT02239393 | |
| Respiratory disorders |
ARDS | Allogeneic | BM | Systemic | N/A | N | 2013 | 1 | Complete | NCT01775774 |
| Chronic obstructive pulmonary disease |
Allogeneic | BM | Systemic | N | N | 2008 | 2 | Complete | NCT00683722 | |
| Lung adenocarcinoma |
Allogeneic | UC | Systemic | TBD | Y | 2017 | 1/2 | Recruiting | NCT03298763 | |
| Skin disorder | Respiratory distress syndrome, adult |
Autologous | BM | Systemic | Unknown | N | 2014 | 2 | Unknown | NCT02112500 |
| Psoriasis vulgaris | Allogeneic | UC | Systemic | Y | N | 2015 | 1/2 | Unknown | NCT02491658 |
With more than 300 completed clinical trials using MSCs as of 2020, there is a wealth of information available to better understand what dictates their success and failure when investigated in humans. The TiGenix/Takeda phase 3 clinical trial that studied the use of MSCs for complex perianal fistulas in CD is arguably the most successful late-stage MSC trial to date (NCT01541579). In this study, adult CD patients with treatment-refractory, draining, complex perianal fistulas were enrolled in a randomized, double-blind, placebo-controlled phase 3 trial and treated with either a single intralesional injection of 120 million allogeneic AT-MSCs (Alofisel) or saline (14). At the primary end point of 24 weeks, combined remission was significantly higher in patients treated with Alofisel (~50% in treated group versus ~34% in placebo group). Greater incidences of remission in the Alofisel treatment group persisted in a subsequent 52-week follow-up (~56% in treated group versus ~38% in placebo group), demonstrating the potential of MSCs to substantially improve the standard-of-care in chronic illnesses like CD. As complex perianal fistulas refractory to conventional medical treatment strategies often require surgery with suboptimal outcomes, the success of this trial validated Alofisel as a novel therapeutic for addressing an unmet clinical need. As a result, Alofisel was granted both orphan drug status and central marketing authorization approval for CD by the European Medicines Agency (EMA), becoming the first allogeneic stem cell therapy to do so in the European Union. This designation enabled Alofisel to be processed through an expedited regulatory path, and Alofisel was approved in Europe for the treatment of complex perianal fistulas refractory to CD in 2018. The cost-effectiveness of Alofisel compared to standard of care will ultimately dictate its successful integration as a viable treatment for eligible patients. Alofisel has thus far been approved by the EMA for use in <10,000 patients under an orphan medicinal product designation (15). Although the mechanism of action in human patients is not well elucidated, results from preclinical studies of Alofisel indicate that induction of indoleamine 2,3-dioxygenase (IDO) in the presence of inflammatory factors such as interferon-γ (IFN-γ) is critical for the therapeutic effect of MSCs. This is because the enzymatic activity of IDO can inhibit T cell function and proliferation and increase the number of regulatory T cells, leading to an increase in anti-inflammatory cytokines [e.g., interleukin-10 (IL-10)] and decrease in pro-inflammatory cytokines [e.g., IFN-γ and tumor necrosis factor–α (TNF-α)] (14).
In addition to Alofisel, there are 10 globally approved MSC therapies including Prochymal (Osiris, approved in Canada and New Zealand), Temcell HS (JCR Pharmaceuticals, approved in Japan), Cartistem (Medipost, approved in South Korea), and Cellgram-AMI (FCB-Pharmicell, approved in South Korea) (16). The major approved indications are GvHD, CD, ALS, and MI (Table 2).
Table 2. MSC products that have received regulatory approval.
| Name | MSC type | Indication | Country of approval (year) | Company |
| Alofisel | Human AT-MSC | Complex perianal fistulas in CD | Europe (2018) | TiGenix NV/Takeda |
| Prochymal (remestemcel-L) |
Human BM-MSC | GvHD | Canada (2012) New Zealand (2012) |
Osiris Therapeutics Inc./ Mesoblast Ltd. |
| Temcell HS Inj | Human BM-MSC | GvHD | Japan (2015) | JCR Pharmaceuticals |
| Queencell | Human AT-MSC | Subcutaneous tissue defects | South Korea (2010) | Anterogen Co. Ltd. |
| Cupistem | Human AT-MSC | Crohn’s fistula | South Korea (2012) | Anterogen Co. Ltd |
| Neuronata-R | Human BM-MSC | Amytrophic lateral sclerosis | South Korea (2014) | Corestem Inc. |
| Cartistem | Human UC-MSC | Knee articular cartilage defects | South Korea (2012) | Medipost Co. Ltd. |
| Stemirac | Human BM-MSC | Spinal cord injury | Japan (2018) | Nipro Corp. |
| Stempeucel | Human BM-MSC | Critical limb ischemia | India (2016) | Stempeutics Research PVT |
| Cellgram-AMI | Human BM-MSC | Acute MI | South Korea (2011) | Pharmicell Co. Ltd. |
Several companies including Mesoblast, Athersys, Pluristem, Stempeutics, Cynata, and others are repurposing their MSC products for new indications. For example, Mesoblast has recently investigated the use of remestemcel-L to treat coronavirus disease 2019 (COVID-19) patients with moderate to severe ARDS, which is the major cause of death. The survival rate was 83% in ventilator-dependent COVID-19 patients when treated with two intravenous infusions of remestemcel-L. By comparison, the survival rate was only 12% in ventilator-dependent COVID-19 patients receiving standard of care during the same period (17). The capability of remestemcel-L to down-regulate pro-inflammatory cytokines and increase anti-inflammatory cytokines is believed to be the key mediator of the promising therapeutic efficacy for COVID-19 patients (18).
Despite the success of these therapeutics, most of the MSC therapies either have had no success in late-stage clinical trials or did not progress beyond preclinical studies. While MSCs demonstrate an exceptional safety profile, they have generally been therapeutically ineffective in humans. There are several factors that likely contribute to their suboptimal clinical outcomes, including heterogeneity in the potency of the MSC product, variable biodistribution and pharmacokinetics with different administration routes, and a limited understanding of the impact the host response has on therapeutic efficacy after administration (19, 20).
In this review, we summarize major clinical challenges for MSC therapies. These challenges are divided into three different categories, including challenges resulting from the manufacturing of MSCs (Fig. 1A), from administration of MSCs (Fig. 1B), and from the recipients (Fig. 1C). Under each challenge, we discuss major factors leading to the heterogeneity of the clinical outcome. We then highlight several examples that leverage bioengineering solutions to address the clinical challenges arising from MSC product quality, administration, and host factors and conclude that bioengineering strategies can and should be used to develop more potent and predictable MSC therapies (21–23).
Fig. 1. Major factors affecting the heterogeneity and ultimately the clinical outcome of MSCs.

(A) Outlines the major variables associated with preparation of the MSC product. Donor variations such as the health status, genetics, gender, and age can affect the potency of MSCs (2). MSCs can be harvested from multiple different sources, such as bone marrow, AT, and UC, causing additional variations in potency (20). Furthermore, different methods of isolating cells (needle versus biopsy) from these tissues and obtaining cells (enzymatic dissociation versus mechanical dissociation) can affect the potency of MSCs (28). The culture conditions, including the medium composition, oxygen levels, confluence, culture surface, flasks/bioreactors, passage number, and cell surface modification, are also reported to affect potency/homing (26, 29). Last, cryopreservation and thaw/culture rescue protocols can affect the viability, function, and homing of MSCs (50, 52, 53). (B) Outlines the major variables associated with the administration of MSCs that can affect the therapeutic outcome. The administration route (local/systemic), injection site (dense/nondense tissue), injection device properties (needle size/geometry), injection/infusion buffer, and cell carrier materials can affect the residence time, viability, and homing of MSCs (81, 84). (C) Outlines the major factors associated with the MSC recipients that can affect the therapeutic outcome. Host cytotoxic responses against MSCs are shown to have strong correlations with the therapeutic outcome (58). The therapeutic outcome is also dependent on the host disease/severity, which can result in highly variable microenvironmental factors (inflammation status, hypoxia, and ECM) that shape the function of MSCs (151).
OVERCOMING CLINICAL CHALLENGES RESULTING FROM THE MANUFACTURING OF MSCs
While the current clinical successes of MSC therapies are encouraging, albeit limited, the predominating failures emphasize the difficulty in predicting immunomodulatory and regenerative effects within human trials. This unpredictability is partially rooted in defining meaningful critical quality attributes (CQAs) of the MSC product. The International Society of Cell and Gene Therapy (ISCT) initially defined human MSCs (hMSCs) according to three minimal phenotypic criteria based on their morphology, surface markers, and trilineage differentiation (24). These criteria were reflective of the MSC “stemness,” not their immunomodulatory and regenerative effects that dictate their therapeutic properties. In 2019, ISCT updated their criteria for defining MSCs to include (i) tissue origin and (ii) associated functional assays to define their relevant therapeutic mode of action (25). ISCT also called for a moratorium on referring to MSCs as “mesenchymal stem cells” in literature, unless rigorous evidence for stemness both in vitro and in vivo is presented. Furthermore, as MSC therapies have reached a critical mass in clinical trials, regulatory authorities have mandated new minimal CQAs for MSCs related to in vitro potency (e.g., suppression of T cell proliferation or IDO expression), in addition to the evaluation of identify, viability, purity, potency, proliferative capacity, genomic stability, and microbiological testing (26, 27). Measuring these CQAs is a unique challenge in the field of cell therapy, as MSCs, for example, unlike traditional chemical drugs whose structure and potency can be narrowly defined, are dynamic “living therapies.”
Impact of sourcing and manufacturing/storage on the functions of MSCs
Heterogeneity in the MSC product
Living MSC therapies are an inherently heterogeneous population of cells whose therapeutic gene and protein expression profiles vary with the characteristics of the donor, MSC tissue of origin (2, 20), isolation method (28), and in vitro preparation methods (e.g., cell culture protocol and scale-up) (26, 29). The extraordinary heterogeneity of MSC product introduced during the manufacturing process emphasizes the need for both meaningfully characterizing and ultimately controlling the therapeutic potency of the MSC product (Fig. 1A).
Standardizing therapeutic potency of the MSC product is crucial before beginning clinical trials (30). This need has prompted research efforts toward developing improved in vitro potency assays that can accurately correlate the CQAs of the MSC product with their therapeutic function (31–35). Currently, the most widely used potency assay for the MSC product is based on in vitro inhibition of T cell proliferation using activated CD4+ T cells (26, 36, 37). This measurement is believed to be more representative of potency compared to surrogate markers for immunomodulatory function (i.e., IDO expression or TNF-α receptor expression) as it provides a direct readout of bioactivity (26).
While each MSC product is completely different, other than useful observations, currently accessible data are insufficient to conclusively determine how tissue source, isolation method, and culture/scale-up during MSC product development can affect the therapeutic efficacy of MSCs (Fig. 1A). To highlight this point, we consider the phase 3 trial using remestemcel-L conducted for both adult and pediatric GvHD populations (NCT00366145) that was unable to meet primary clinical end points in a mixed-age patient population, despite demonstrating a positive impact on the liver and gut in earlier stages of the study (38). All remestemcel-L MSCs used in the phase 3 trial were derived from a single donor, requiring cells to be expanded to passages 3 and 4 during manufacturing to yield enough MSCs to treat all 240 participants. Conversely, a separate phase 2 trial on therapeutic MSCs conducted by the University Hospital Frankfurt avoided this failure for the same clinical indication in a similar patient population. In this study, pooled MSCs were used to treat 26 patients with GvHD using passage 1 and 2 MSCs from eight donors to yield sufficient quantities of MSCs (39). Results from this study demonstrated improved clinical efficacy in GvHD patients at the primary end point (day 28), with an overall response rate of 77%. While this comparison appears to illustrate the importance of MSC sourcing and manufacturing at the commercial scale (29, 40), differences highlighted here as well as subtle differences in the preparation of the MSC product make it challenging to draw conclusions.
While potency assays may improve product quality by excluding MSCs with low potential therapeutic efficacy, strategies are needed to instead generate high-quality MSCs in sufficient quantities for large clinical trials. Several biomaterial strategies have been explored to maintain more homogeneous MSCs during the expansion phase of MSC manufacturing. For example, Rao et al. (41) showed that expanding hMSCs on soft poly(ethylene glycol) hydrogel matrices was able to avoid reductions in cell surface marker expression and cytokine expression that was observed when hMSCs were serially passaged on polystyrene. Growing MSCs in three-dimensional (3D) culturing systems has also been beneficial in maintaining early-passage MSC phenotype during expansion (42, 43). At the clinical stage, efforts to overcome MSC product heterogeneity have been best exemplified by Cynata Therapeutics, who developed a strategy for obtaining highly homogeneous MSCs using induced pluripotent stem cells (iPSCs). Because iPSCs have an exponentially larger proliferation capacity without undergoing differentiation compared to MSCs, they can be easily expanded to generate large quantities of iPSCs and then differentiated into MSCs after expansion to yield commercial quantities of MSCs (iPSC-MSCs) with a low passage number (44). For example, upward of 1 × 1022 passage 1 MSCs can be produced from a single iPSC population, with similar potency to low-passage BM-MSCs harvested from donors as assessed by T cell suppression (44, 45). The iPSC-MSC approach serves as an excellent solution for scaling MSC manufacturing without sacrificing therapeutic potency through the passage and expansion of cells (46). One caveat is that the intrinsic self-renewal and pluripotency of iPSCs may also be responsible for tumorigenic potential (47). However, this appears to be de-risked by clinical studies, showing that iPSC-MSCs are safe and effective. For example, Cynata’s lead product CYP-001 was shown to be effective for the treatment of steroid-resistant GvHD in a phase 1 clinical trial (NCT02923375) without showing any sign of tumorigenesis. Another phase 2 clinical trial including 448 osteoarthritis patients is expected to commence in early 2020 (48). Other approaches have used CRISPR-Cas9 technology to create a reversibly immortalized BM-MSC line, which avoids the phenotypic changes that occur with continued passaging and expansion (49).
Cryopreservation and culture rescue
The loss of MSC potency following cryopreservation is another important challenge in the development of high-quality MSC products. This clinical obstacle may be best addressed by optimizing the handling of MSCs rather than engineering their physical and functional properties. The preparation of most MSC therapeutics involves expanding cells ex vivo, cryogenically banking them until needed, thawing the banked MSCs at the bedside, and administering them to the patient (29). MSC processing between thawing and administration varies widely between clinical trials, which can have significant implications on the therapeutic effect of MSCs once administered. For example, a phase 2 study assessing MSCs as a treatment against the chronic inflammatory disorder ARDS was unable to achieve a significant clinical improvement compared to control groups, despite promising results in phase 1 and a satisfactory safety profile (50). Retrospective analysis of the MSC doses found that the viability of the freshly thawed and washed MSCs ranged widely from 36 to 85%, despite no significant changes in viability between doses before cryopreservation. Furthermore, only MSCs with the highest viability (70 to 85%) were able to improve oxygenation in patients compared to the placebo (51). Fundamental studies on MSC cryopreservation have demonstrated that freshly thawed MSCs have stunted immunosuppressive capabilities, with a reduced capacity to suppress T cell proliferation (52). Cell damage following cryopreservation can also alter their post-infusion biodistribution, engraftment, and clearance kinetics (22, 53). Furthermore, thawed MSCs exhibit diminished structural integrity upon rewarming, as the freezing process disrupts the actin cytoskeleton (53). This abnormal membrane structure marks thawed MSCs as a target for activated T cells, expediting the onset of immune clearance and significantly diminishing the lifetime of intact MSCs in patients following infusion (54).
Some investigators have avoided the detrimental effects caused by cryopreservation by changing the way in which MSCs are handled before administration. For example, the successful phase 3 clinical trial on Alofisel for perianal fistulas “culture-rescued” MSCs after thaw, a process that involves the “recovery” of freshly thawed MSCs under cell culture for a period of at least 24 hours between thawing and infusion (14). The clinical success of MSCs handled in this manner suggests that the associated detrimental effects of thawing not only are reversible but also introduce a feasible strategy for improving MSC product quality (52, 54).
Bioengineering solutions to boost the functions of MSCs
Engineering MSCs to boost the innate functions
Clinical trials to date demonstrate that MSCs can be safely infused in high doses (55) and display promising responses in some clinical indications. Quality control protocols to standardize MSC product potency may help reduce the risk of clinical failure, but they are unlikely to resolve the problem completely, as the innate function of MSCs is not always therapeutically sufficient for disease treatment (51, 56). To maximize clinical potency while preserving ease of use, simple alternative bioengineering strategies should be explored that can boost the innate function of MSCs, independent of cryopreservation, passage number, and donor and tissue source. Furthermore, bioengineering can serve as a powerful platform for translating new insights gained from the fundamental understanding of MSC behavior after infusion into more effective therapies (Fig. 2).
Fig. 2. Bioengineering solutions to boost the functions of MSCs.
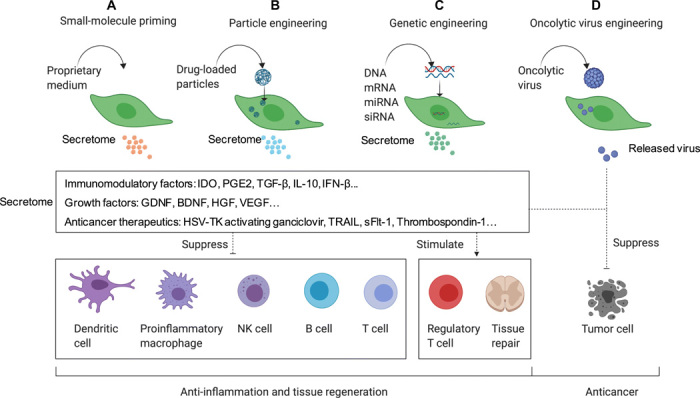
(A) Priming MSCs with small molecules is a simple and promising approach to induce the secretion of immunomodulatory and regenerative molecules, but the effect of small molecules only lasts a few hours to a few days. (B) MSCs can also be engineered with drug-loaded particles. These particles are intracellularly loaded into MSCs to sustain their immunosuppressive profile for an extended period of time, regardless of the source of MSCs, but particle preparation can increase the cost and complexity when compared to the use of free small molecules. (C) MSCs can be genetically engineered to overexpress a variety of different therapeutic molecules, including anti-inflammatory cytokines and growth factors, either to boost their innate functions or to overexpress other therapeutics and broaden their application to other diseases such as cancer. Viral vector–based genetic engineering typically has more efficient and durable gene expression but has some safety concerns because genes are integrated into the target cell genome. Nonviral vectors are safer, but the transfection efficiency is typically lower and gene expression is less durable. (D) OVs have also been used to engineer MSCs. MSCs function by shielding viruses to avoid immunogenicity and by releasing the virus in tumor tissue to kill tumor cells. One limitation is that regular OVs have only moderate infectivity, although this can be overcome by using certain viral variants with higher infectious capacity.
“Priming” MSCs with small molecules represents a simple strategy to exogenously boost their therapeutic function. Several “primed” neuroregenerative MSC products already have reached clinical investigation, with the most notable being Brainstorm Cell Therapeutics MSC product, NurOwn. NurOwn is a primed MSC product in which the innate regenerative capacity of MSCs is boosted using proprietary culture medium to express multiple neurotrophic factors (NTFs) including glial-derived neurotrophic factor (GDNF), brain-derived neurotrophic factor (BDNF), vascular endothelial growth factor (VEGF), and hepatocyte growth factor (HGF) (57, 58). When administered to patients with neurodegenerative diseases, NurOwn has been demonstrated to simultaneously deliver multiple NTFs, in addition to the immunomodulatory components innately secreted by MSCs (59). This combination has demonstrated impressive therapeutic efficacy in clinical trials, evidenced by a phase 2 clinical trial (NCT02017912) where ALS patients who received NurOwn demonstrated reduced ALS progression 24 months after infusion compared to controls. Brainstorm Cell Therapeutics is currently in the process of expanding the therapeutic scope of NurOwn, recruiting patients for a phase 2 clinical trial for MS (NCT03799718).
While small-molecule priming has utility, the effects only last several hours to a few days (60). With the emergence of techniques to improve the survival of transplanted cells, approaches to extend the exposure of small molecules to transplanted cells have also been developed. For example, loading MSCs with small-molecule encapsulating microparticles (MPs) has been used to boost the duration of product potency (61, 62). MPs comprise biocompatible materials that can be therapeutically tuned according to their composition, polymer molecular weight, extent of drug loading, and drug release. MSCs loaded with degradable MPs containing the steroid budesonide exhibited fourfold enhanced IDO activity in vitro compared to free budesonide-preconditioned MSCs and native MSCs. This led to a twofold improvement in the suppression of stimulated peripheral blood mononuclear cells (PBMCs) following IFN-γ stimulation (63).
MSCs can also be engineered to serve as a producer and carrier of biologics (Table 3). To produce the desired biologic within the MSC, viral DNA transduction and mRNA/DNA transfection are the most common approaches. For example, a study by Suresh et al. (64) reported that MSCs genetically engineered to express thioredoxin-1 (Trx1)—a powerful antioxidant, transcription factor, and growth factor regulator—improved cardiac function following MI in a rat model compared to unmodified MSCs. Although preclinical studies have shown promising results, it remains to be seen how these engineered MSCs may improve the therapeutic outcome in a clinical setting.
Table 3. Examples of bioengineered MSCs as living drug factories.
| Disease model | Cell source | Engineering approach | Expressed therapeutic | Reference |
| Neurodegenerative diseases |
Human BM-MSC | Priming | Neurotrophic factors (NTFs; include GDNF, BDNF, HGF, VEGF) |
Gothelf et al. (59) |
| Diabetes mellitus | Human AD-MSC | Priming | Insulin | Thakkar et al. (80) |
| Myocardial infarction | Mouse BM-MSC | Priming | Hypoxia-inducible factor 1, angiopoietin-1, VEGF, erythropoietin. |
Hu et al. (111) |
| Inflammatory diseases | Human BM-MSC | Microparticle (MP) delivery and priming |
Indoleamine-2,3-dioxygenase (IDO) |
Ankrum et al. (63) |
| Damaged tissue repair | Human AD-MSC | Biomaterial encapsulation | VEGF, HGF | Kim et al. (114) |
| Colitis | Human BM-MSC | Engineered hydrogel | IDO, programmed death ligand 1 (PD-L1), CCL8, CXCL9, CXCL10 |
García et al. (119) |
| Parkinson’s disease | Human BM-MSC | Genetic | Glial cell line–derived neurotrophic factor (GDNF) |
Hoban et al. (163) |
| Juvenile Huntington’s disease |
Human BM-MSC | Genetic | Brain-derived neurotrophic factor (BDNF) |
Deng et al. (164), Pollock et al. (165) |
| Parkinson’s disease | Human UC-MSC | Genetic | Hepatocyte growth factor (HGF) |
Liu et al. (166) |
| Multiple sclerosis | Human AT-MSC | Genetic | Interferon-β (IFN-β) | Marin-Banasco et al. (167) |
| Myocardial infarction | Rat BM-MSC | Genetic | Thioredoxin-1 (Trx-1) | Suresh et al. (64) |
| Osteoporosis | Porcine BM-MSC | Genetic | Bone morphogenic protein-6 gene |
Pelled et al. (168) |
| Osteoporosis | Mouse BM-MSC | Genetic | B cell–specific Moloney murine leukemia virus integration site 1 (Bmi1) |
Chen et al. (169) |
| Hepatic fibrosis/cirrhosis | Human BM-MSC | Genetic | Decorin | Jang et al. (170) |
Engineering MSCs to go beyond the innate functions
Bioengineering is a powerful approach for expanding the therapeutic scope of MSCs beyond their innate functions. This can be achieved by engineering MSCs to secrete either poorly expressed or non-native therapeutic proteins (Fig. 2). A key example of this approach is in the use of MSCs to generate anticancer therapeutics. Systemic drug toxicity is a pressing concern in chemotherapy and related cancer treatment (65). Unlike synthetic biomaterials such as nanoparticles, MSCs have intrinsic capability to temporarily evade the immune response and home to tumors (66). With these unique carrier features, engineered MSCs have been reimagined as anticancer “Trojan horses” that are able to safely deliver large doses of cancer-targeting biologics with a single MSC dose (Table 4). This approach was validated by Sasportas et al. (67), in which engineered MSC Trojan horses delivered TNF-related apoptosis-inducing ligand (TRAIL) to cancer cells, suppressing tumor growth in a highly malignant glioblastoma mouse model. MSCs have also been used to express other proteins, including herpes simplex virus–thymidine kinase (HSV-TK), enzymes for converting chemotherapy prodrugs into their active toxic compound (i.e., ganciclovir) (68, 69), soluble VEGF receptor-1, and thrombospondin-1, all of which have an antitumor effect (Table 4).
Table 4. Examples of bioengineered MSCs as anticancer Trojan horses.
| Disease model | Cell source | Expressed therapeutic | Reference |
| Pancreatic carcinoma | Murine BM-MSC | HSV-TK activating ganciclovir | Conrad et al. (171) |
| Hepatocellular carcinoma | Murine BM-MSC | HSV-TK activating ganciclovir | Niess et al. (69) |
| Pulmonary melanoma metastasis |
Rat BM-MSC | HSV-TK activating ganciclovir | Zhang et al. (21) |
| Glioblastoma | Human BM-MSC | HSV-TK activating ganciclovir + TRAIL secretion |
Shah (172) |
| Lewis lung cancer metastasis | Murine BM-MSC | Soluble vascular endothelial growth factor receptor-1 (sFlt-1) |
Hu et al. (173) |
| Glioblastoma | Human BM-MSC | Thrombospondin-1 | Choi et al. (174) |
| Metastatic breast cancer | Human BM-MSC | Cytosine deaminase under the control of the YAP/TAZ promoter activating 5-fluorocytosine |
Wu et al. (94) |
| Prostate cancer | Human BM-MSC | Thapsigargin-based prostate-specific antigen (PSA)–activated prodrug (G114) |
Levy et al. (61) |
| Glioblastoma | Human AT-MSC | Adenovirus expressing soluble hyaluronidase (ICOVIR17) |
Martinez-Quintanilla et al. (23) |
| Glioblastoma | Human BM-MSC | Oncolytic herpes simplex virus (oHSV) | Duebgen et al. (175) |
| Ovarian cancer | Human menstrual blood MSC | Oncolytic adenovirus | Alfano et al. (176) Moreno et al. (177) |
To avoid the off-target toxicity of the antitumor biologics produced by MSCs, MSCs have been engineered to only release their biologics in response to stimuli such as mechanical cues that are unique to the tumor’s physical characteristics, rather than release the biologics nonspecifically throughout the body (70). This was accomplished using a YAP/TAZ promoter, which is activated when the cells sense the stiff, collagen-rich matrix environment of solid tumors. YAP/TAZ-engineered MSCs targeted sites of high collagen deposition in the lungs of mice inoculated with metastatic Luc-RFP-231 breast cancer cells (70). By placing expression of the prodrug-activating enzyme cytosine deaminase under the control of the YAP/TAZ promoter, MSCs were able to locally activate the prodrug 5-fluorocytosine and reduce tumor burdens in collagen-rich areas of mice, without inducing drug-related systemic toxicity (70).
Oncolytic viruses (OVs) are a relatively recent development in cancer therapy, which uses viruses that are engineered to either directly lyse tumor cells or trigger antitumor immunity against cancerous cells (71). However, humoral immunity responses can quickly neutralize the efficacy of systemically injected OVs through various processes, including inactivation by complement proteins and immune-mediated phagocytosis (72, 73). To protect OVs from the recipient humoral immune system, cell-based carriers have been demonstrated as a useful approach for both producing OVs in vivo and delivering them to the tumor site (74). One limitation of using MSCs as OV carriers is that MSCs demonstrate only moderate infectivity when transduced (75). To overcome unsatisfactory infectivity, certain viral variants (i.e., Ad5/3 oncolytic adenovirus) can be used that are able to infect MSCs with higher viral loads (76).
Several MSC-based anticancer therapeutics have reached the clinical stage. For example, an ongoing phase 1/2 study of intraperitoneally administered AT-MSCs expressing both the oncolytic measles virus and a membrane-bound sodium iodine symporter is being investigated to enhance the measles virus’ therapeutic potency in patients with recurrent ovarian cancer (NCT02068794) (77). Another phase 1/2 trial (TREAT-ME1, NCT02008539), involving intravenously administered autologous MSCs engineered to express the tumor-specific HSV-TK gene, was also investigated for the treatment of gastrointestinal tumors (78). While the study was terminated in 2016 due to an insufficient number of patients meeting the eligibility criteria, investigators reported favorable safety and tolerability in patients who received the treatment (78). Last, TRAIL-releasing MSCs (MSC-TRAIL) are also being clinically investigated as a therapeutic against inoperable lung adenocarcinomas, and recruitment is currently underway for a phase 1/2 clinical trial (NCT03298763).
These examples demonstrate that engineered MSCs may provide a novel axis for developing a reproducible product, where quality control and potency hinge on the engineered behavior of the cells, rather than their comparatively unpredictable innate immunomodulatory properties. Although we focused on examples in anticancer therapeutics, there are many other instances of MSCs being engineered beyond their innate immunomodulatory and regenerative functions to treat various diseases (79). For example, MSCs have been preconditioned toward an insulin-secreting phenotype for the treatment of diabetes mellitus. Administration of these insulin-secreting MSCs in clinical trials has been shown to be safe and provide long-term control of hyperglycemia through a decreased exogenous insulin requirement and elevated levels of C-peptide, a molecule co-released with insulin from the pancreas (80). Regardless of the application, while engineered MSCs have the potential to better control therapeutic function compared to unaltered MSCs, ideally, the approach should be simple, robust, and amenable to large-scale cost-effective manufacturing.
OVERCOMING CLINICAL CHALLENGES FROM ADMINISTRATION
Just as living cell-based therapies pose unique challenges for meaningfully assessing in vitro potency, the behavior of MSCs, including the pharmacokinetic and biological properties of the infused MSCs, can be affected by the mode of administration (81–84). Specifically, factors such as the injection site, injection device properties, and properties of the carrier materials/buffer can affect the administration of cells (Fig. 1B). For example, different injection sites can cause variations in backpressure/reflux, and injection device properties (needle size/geometry) can cause variations in shear rate and shear stress during injection that are known to affect the viability of injected cells. The impact of these factors and potential solutions has been thoroughly reviewed elsewhere (84). In this section, we will summarize the various clinical challenges encountered with locally and systemically administered MSCs, and how these clinical findings can be leveraged to engineer more therapeutically consistent and effective MSC therapies.
Challenges associated with different administration routes
Local administration
Local administration is commonly used in clinical indications as it provides direct access to the disease site. As of 2018, 49% of registered MSC clinical trials use localized delivery (85). Most MSC therapies that have progressed to late-stage clinical trials have used local MSC administration (i.e., intrathecal, intralesional, and endocardial) within various clinical indications including lower back pain, perianal fistulas, and chronic heart failure (86). Local administration of MSCs is a more controlled delivery approach, making it easier to access the disease site that often results in better therapeutic responses (87). In ischemic stroke, for example, locally administering MSCs to the damaged site has shown to be more effective than intra-arterial and intravascular MSC injection in improving the neurological severity score (88). However, there are still clinical challenges associated with local administration that hinder therapeutic efficacy, primarily due to insufficient retention and survival of transplanted MSCs at the site of administration.
Insufficient retention and survival
Retention here is defined as the duration of localization of cells at the target site. The lack of retention following local administration has been attributed to multiple issues after administration, including cell death due to the hostile environment encountered at the disease site and poor engraftment into the tissue (89, 90). Although highly dependent on the clinical application, some cases have shown that less than 5% of administered cells remain at the site of injection in the hours following transplantation (91). For example, in a study using intracoronary injection of bone marrow stem cells in patients with MI, only 2.1% of radiolabeled stem cells remained at the site of injection after ~1 hour. In addition, most of the remaining signal was found primarily in the liver and spleen (92). This clinical study highlights that, despite injecting MSCs directly at the damaged tissue site, retention in these regions is still a major concern. Furthermore, the cells that are at the target site are often no longer viable due to immune-mediated damage and apoptosis (89, 91, 93). For instance, an in vivo rat study showed an undetectable level of viable MSCs 2 days following local injection at the site of MI (94). Moreover, limited diffusion of nutrients and oxygen can also affect the survival of cells following administration. Cells must be within ~200 μm of the nearest blood vessel for sufficient nutrients and oxygen, but it may take many days for vascularization to reach the cells, leading to cell death (95). Because nonviable cells have a reduced capacity to produce therapeutic biologics, this can compromise the potency of the MSCs. Together, these studies highlight that both the retention and survival of MSCs following local administration of MSCs must be enhanced to improve the therapeutic outcome.
Systemic administration
While local delivery of MSCs can help deliver paracrine factors directly to the diseased tissue, local administration is not a feasible option for many clinical indications, as more invasive injections can cause serious complications in many diseases (96). Alternatively, intravenous injection of MSCs is used, but the therapeutic utility has been limited due to insufficient homing to the target site. Furthermore, using systemic administration of MSCs has led to key challenges, namely, the instant blood-mediated inflammatory reaction (IBMIR) and insufficient residence time at the target site.
Instant blood-mediated inflammatory reaction
Elevated concentrations of procoagulants like tissue factor (TF) on the surface of MSC serve as a potent trigger for IBMIR, compromising cell engraftment, cell lifetime, and therapeutic potency (97–99). TF triggers the extrinsic pathway of coagulation, leading to thrombin generation, platelet activation, and fibrin cross-linking, which all contribute to adverse clinical outcomes. For instance, the intravenous infusion of allogeneic UC-MSCs into two patients with renal transplantation and chronic kidney disease triggered thrombosis in both patients and required emergency treatment to dissolve the resulting thromboemboli (98). Interestingly, both TF expression and the magnitude of IBMIR with MSC products used in clinical trials depend on the tissue and donor source as well as the passage number (100–102). In addition to triggering coagulation, IBMIR following MSC contact with human serum has been demonstrated to activate complement in all three classical, alternative, and lectin pathways (103). Complement and coagulation pathways are known to strongly interact in vivo, with cross-talk between the two paths leading to synergistic effects that enhance therapeutic MSC dysfunction and cytotoxicity (104). Consequently, the clinical safety of systemic MSC therapies in patients relies on the optimal control of IBMIR.
Insufficient residence time and homing
When MSCs are delivered systemically, a key factor for exerting maximal therapeutic benefit is their ability to remain in circulation for long enough to deliver therapeutic payloads to the damaged tissue. However, it is well known that intravenously administered MSCs are immediately concentrated in the lung capillaries and phagocytosed by monocytes within 24 hours (88, 105–107). This limits the MSC’s ability to deliver therapeutic payloads to the host environment via secreted paracrine factors to a short period of time following injection (106, 108) and limits cell homing to target tissues (i.e., bone marrow and nervous system). Entrapment of MSCs in the lung capillaries also increases susceptibility to immune clearance (83, 108). For example, clinical studies on intravenous administration of radiolabeled MSCs for MI showed a complete lack of MSC homing in the infarcted myocardium following intravascular injection (92, 109). In addition, a recent phase 1 clinical trial by Schweizer et al. (110) indicated that unmodified MSCs did not home to primary prostate tumors in adequate levels to warrant further development, although this may have been due to insufficient sensitivity for MSC detection. Alternatively, it is also possible that mouse tumors are not representative of human tumors, and therefore, promising preclinical results fail to be translated to human patients, highlighting the need to use large animal models or develop alternative models that can recapitulate key features of human diseases before the clinical study. Nevertheless, these clinical findings illustrate that systemically injected MSCs often fail to properly home to target tissues, making them insufficient at delivering therapeutic payloads to diseased sites.
Bioengineering approaches for improving MSC administration
Strategies to improve local administration
To improve the local administration of MSCs, multiple strategies have been investigated (Fig. 3). Among these strategies, priming MSCs in vitro is a simple approach. For example, hypoxic priming up-regulated expression of prosurvival factors such as hypoxia-inducible factor 1, which can help MSCs adapt to the disease site that is typically hypoxic. Consequently, hypoxia-primed MSCs exhibited ~40% less cell death on day 3 after intramyocardial injection compared with nonprimed MSCs in a rat model of MI, resulting in improved vascularization in the infarcted myocardium and better therapeutic efficacy (111). However, the effect of priming may not be preserved upon cryopreservation/thawing because the expression of prosurvival factors is highly dependent on the environment.
Fig. 3. Bioengineering solutions for improving administration of MSCs.
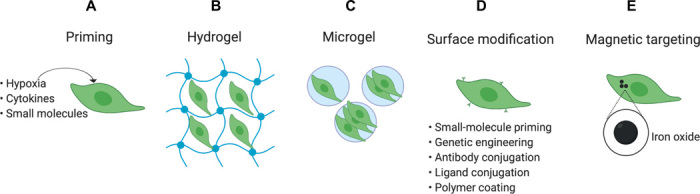
(A) Priming MSCs with hypoxia, inflammatory cytokines, and small molecules have been shown to improve the survival of MSCs, but the effect of priming may not be preserved upon cryopreservation/thawing. (B) Hydrogel is one of the most common biomaterials used to encapsulate MSCs and enhance their survival to several weeks following local administration, but the bulk size of hydrogel is only suitable for local administration, not for systemic administration. (C) Microgels containing one or several MSCs is another bioengineering solution to enhance the residence time and survival of MSCs. Unlike bulk hydrogel, which is only suitable for local administration, microgel can be suitable for both local and systemic injections. One potential limitation is that the physical barrier of the microgel may mask the receptors on MSCs that are important for their homing to diseased sites, although this may be addressed by using additional homing ligands on the microgel. (D) To improve the homing of MSCs to the target sites, the surface of MSCs can be modified with different homing ligands. This can be achieved through genetic engineering, antibody conjugation, or polymer coating of MSCs, but more work is required to achieve a critical mass of MSCs at the target site that can predictably modulate the biological signaling pathways. (E) MSCs can be engineered with intracellular iron oxide to efficiently direct MSCs to reach the target sites under guidance by an external magnetic field. Iron oxide also makes it possible to monitor the biodistribution of MSCs using magnetic resonance imaging, but more work is needed to understand whether the properties of iron oxide–engineered MSCs can be maintained during cryopreservation/thawing, which can cause leakage of iron oxide from MSCs.
Using biomaterials to encapsulate MSCs is another promising strategy to overcome challenges associated with local administration. For example, when a rat model of MI was treated with alginate-encapsulated human BM-MSCs or non-encapsulated MSCs, alginate encapsulation significantly increased the retention of MSCs at the infarction site and improved the therapeutic outcome with respect to increased microvasculature and decreased scar formation (112). While it was not clear in this study whether cell survival was also improved, biomaterial encapsulation has been demonstrated to prolong MSC survival by providing a mechanical barrier that helps both position cells at the target site and shield them from immune attack (91, 93). For example, in a separate rat MI model, immunohistology studies in vivo showed that MSC survival was sustained for up to 16 days following delivery of HGF-overexpressing MSCs in a synthetic peptide-based hydrogel compared to native MSCs that did not survive past day 2. This engineered MSC therapy demonstrated superior reduction in scar formation, accelerated angiogenesis, and increased ventricular wall thickness compared with native MSC (94). In another study, when alginate encapsulated MSCs were subcutaneously inoculated in mice with GvHD, a high percentage of encapsulated MSCs were alive 30 days after subcutaneous inoculation. Interestingly, subcutaneous inoculation of free MSCs also had a similar therapeutic effect in the same study, indicating that alginate encapsulation of MSCs may be not required in this specific condition. However, the lack of viability data for free MSCs makes this conclusion questionable, as free MSCs can survive for less than 30 days in the injection site; thus, the lack of difference may simply be because the GvHD animal model is relatively easy to treat, and it is hard to see the benefit of alginate encapsulation (113).
Compared with the above in vivo studies, where some key quantitative information is missing due to difficulty of extracting non-encapsulated MSCs, the difference between free MSCs and encapsulated MSCs has been more precisely characterized in vitro. For example, when MSCs were encapsulated in a fibrin-based scaffold, more than 95% of MSCs were viable for 14 days; in contrast, non-encapsulated MSCs had a viability of ~80.4 ± 10% on day 6 and 76.7 ± 3.6% on day 14 in vitro. Moreover, when encapsulated MSCs or non-encapsulated MSCs were added to the upper chamber of a Transwell system, whose lower chambers were filled with PBMCs stimulated with phytohemagglutin, only 1.8 ± 0.7% of encapsulated MSCs migrated out of the scaffolds, while 88.7 ± 8.1% of non-encapsulated MSCs migrated away from the initial site after 2 days. Consequently, encapsulated MSCs exhibited up to 10-fold higher local secretion of beneficial soluble factors, such as VEGF and HGF, in comparison to non-encapsulated cells, with the secretion highly dependent on cell number (114). Furthermore, hydrogels have been engineered more recently to boost MSCs potency in vitro. Tuning the in situ mechanical properties, such as pore size and stiffness, of 3D hydrogels that encapsulate injected MSCs has been shown to influence the relative cell proliferation, cell survival, and secretion of beneficial cytokines (115–117). For instance, Cai et al. (116) varied the amount of the thermoresponsive polymer poly(N-isopropylacrylamide) to alter the shear storage moduli of an encapsulating hydrogel. The best formulations showed more than twofold increases in cell proliferation on day 14 and up to threefold higher mRNA expression of relevant factors, such as angiopoietin and fibroblast growth factor-2, compared to formulations without the thermoresponsive polymer. Along with tuning mechanical properties, synthetic hydrogels with added linker molecules, including activated peptides and pro-inflammatory cytokines like IFN-γ, have also been explored as a technique to enhance MSC survival, persistence at the target site, and cytokine secretion (118, 119). For example, hydrogels engineered with the adhesive integrin-specific peptide GFOGER compared to the nonadhesive peptide GAOGER have been shown to significantly enhance the in vitro secretion of relevant cytokines, such as IL-8 and VEGF, and subsequently enhance MSC survival, engraftment, and bone repair in an in vivo mouse bone defect model (118).
While these studies demonstrated the impact of biomaterial encapsulation on the retention and survival of MSCs in vitro, additional work is still needed to further validate this in vivo. Nevertheless, promising results from the above preclinical studies using biomaterials to encapsulate MSCs have motivated their advancement to clinical trials. For example, Anterogen Ltd. is investigating the use of a hydrogel sheet containing allogeneic AT-MSCs in a multitude of diseases, including a phase 3 study on diabetic foot ulcers (NCT03370874), a phase 2 study on burn injury (NCT03183648), a phase 1/2 study on epidermolysis bullosa (NCT03183934), and phase 2 study on tendon injury (NCT03449082).
Strategies to improve systemic administration
Bioengineering strategies are being studied to address challenges from systemic administration related to both IBMIR and the insufficient residence time and homing of MSCs (Fig. 3). To attenuate IBMIR, Moll et al. (85) have recently advocated for the regular use of low-dose anticoagulants like heparin in the clinical setting. In addition, ABO antigens, complement, and coagulation factors that may be found in the AB plasma (ABP) used in MSC culture medium can amplify IBMIR in vitro (120). Replacing the ABP with a more defined, nonimmunogenic human serum albumin (HSA)–based supplement for MSC culture medium has been suggested as an approach to reduce the risk of IBMIR in MSC products. Recent clinical studies on GvHD showed an improved therapeutic effect when replacing ABP with HSA-supplemented medium and adding a low dose of heparin to MSC therapies (121, 122). This study highlights a simple, scalable, and clinically translatable technique for potentially improving the outcomes of many MSC therapies. In addition to the solutions discussed above, other bioengineering approaches have served as a useful platform in addressing MSC-mediated IBMIR. For instance, genetic engineering approaches (i.e., CRISPR-Cas9 or antisense RNA) to reduce expression of TF by MSCs, as well as engineering the MSCs with heparin cell surface coatings to prevent coagulation and complement properties, are bioengineering alternatives to systemic anticoagulation that have shown promise in preventing MSC-mediated IBMIR (97–99, 123). Furthermore, engineering MSCs to express blood regulatory molecules, such as CD46 and TF pathway inhibitor (85, 124), may be a beneficial strategy for suppressing IBMIR following systemic MSC administration. There are many promising bioengineering solutions to mitigate the effects of IBMIR. Translating these approaches into a clinical setting may provide a new tool for avoiding adverse IBMIR-related events following systemic MSC administration.
Independent of IBMIR, additional protective bioengineering strategies to increase MSC residence time and sufficiently deliver MSCs to target tissues have been developed in recent years. To enhance residence time, Mao et al. recently demonstrated a microencapsulation technique, in which individual MSCs were encapsulated in alginate-poly-d-lysine (PDL)-alginate (APA) microgels (particulate hydrogels with dimensions in the range of 30 to 50 μm). Using a single-cell microgel encapsulation approach has several distinct advantages compared to typical larger multicellular hydrogels for systemic administrations: The advantages include a reduced fibrotic capsule formation, a reduction in diffusion limitations that lead to hypoxic effects, and a higher surface area to volume ratio, which facilitates the release of biologics from encapsulated cells (125). Specifically, in the study by Mao et al., unlike a regular hydrogel that has a large volume and is not suitable for intravenous injection, the encapsulating microgel layer is on the order of 10 μm and can be easily injected intravenously. Furthermore, the encapsulating material did not interfere with the ability of MSCs to secrete therapeutic anti-inflammatory cytokines. Encapsulating MSCs into microgels significantly increased their residence time in vivo. Interestingly, encapsulating multiple cells per microgel had a half-life of >50 hours, whereas encapsulating a single cell per microgel had a half-life around 20 hours, although both were longer than unmodified MSCs that had a half-life of <2 hours in a mouse model (88, 108). Further analysis indicated that, compared with single-cell microgel encapsulates, multicellular microgel encapsulates had higher levels of collagen I and lower oxygen tension, both of which positively contributed to the prolonged residence time of encapsulated MSCs. These results indicate that controlling the cell number inside microgels is an important consideration to achieve the desired residence time of MSCs. Moreover, this improved in vivo residence time of APA-treated MSCs occurred despite the presence of innate and adaptive immune clearance mechanisms, leading to a significant improvement of therapeutic outcome in a bone marrow transplant model (108).
Engineering approaches that up-regulate ligands on MSCs have also improved homing by improving the interaction between MSCs and the inflamed endothelium or chemokines proximal to the disease site. Many studies have used various cocktails of soluble factors to increase the expression of CXCR4 and matrix metalloproteinases (MMPs), an approach that has yielded preclinical success in homing and disease recovery (126–130). For example, priming MSCs with valproic acid and lithium induced CXCR4 and MMP-9 up-regulation, which subsequently increased MSC homing, improved functional recovery, and reduced the infarct volume in the brain when tested in a rat model of cerebral ischemia (128). Alternatively, RNA- or DNA-based genetic engineering is a well-established approach that can be used to induce the continuous synthesis of homing ligands in vivo. For example, mRNA transfection was used to induce the expression of the homing ligands PSGL-1/SLeX as well as the anti-inflammatory cytokine IL-10 in MSCs (131). The engineered MSCs displayed increased homing to sites of inflammation/disease and showed an improved therapeutic impact in mouse models of skin inflammation (131) and experimental autoimmune encephalomyelitis (132). In addition, viral transduction protocols have been used to permanently overexpress homing factors in MSCs. Most notably, overexpression of CXCR4 increased the homing of engineered MSCs to the ischemic myocardium (133, 134), bone marrow (135, 136), and damaged intestinal mucosa (137) in preclinical models. Other bioengineering techniques have been investigated to chemically engineer the cell surface to improve MSC adhesion and homing, including hyaluronic acid coatings (138), enzymatic modifications (139, 140), binding of adhesion molecules (141–143), and the attachment of docking systems to bind homing antibodies such as PSGL-1 (22, 144).
Magnetically labeled MSCs have also been investigated as a means to direct cells to the target tissue with an external magnetic field (145–147). Iron oxide has no perceivable effects on MSC function in vitro and in vivo at treatment doses of ~23 pg per cell, and is deemed a safe material for MSC bioengineering (146, 148). Iron oxide–labeled MSCs demonstrated a 10-fold increase in retinal homing following intravenous infusion in a rat model, with magnetic MSCs better penetrating the inner and outer retina compared to nonmagnetic MSCs 1 week after injection (147). Furthermore, the improved homing of magnetic MSCs enhanced the overall expression of IL-10 in the retina, although this is believed to be largely due to increased endogenous expression of cytokines from retinal cells rather than from the MSCs directly (149).
Overall, there has been a vast amount of preclinical investigation into bioengineering strategies to improve the residence time and homing, but few have been translated into clinical trials. These bioengineering strategies may help to improve clinical outcomes for various diseases by addressing the challenges of insufficient MSC residence time and homing to diseased sites.
OVERCOMING CLINICAL CHALLENGES FROM THE HOST
Host factors affecting the therapeutic outcome of MSCs
Host cytotoxic responses against MSC
While the potency of the MSC product and the route of administration are critical parameters for the efficacy of MSC therapies in clinical trials, host factors are also an important consideration. Variations in the host cytotoxic response, inflammation status, and tissue microenvironment such as hypoxia and extracellular matrix (ECM) (stiffness) have been demonstrated as important factors in the efficacy of MSCs after administration (Fig. 1C) (58). This was most recently demonstrated in a study characterizing the PBMCs of patients who had variable therapeutic responses to intravenously administered MSCs (7). In this study, 16 patients with severe steroid-resistant grade 3 to 4 GvHD were treated with MSCs, and PBMCs were collected from each individual 24 hours after intravenous MSC administration. Subsequent analysis of the host PBMCs revealed that clinical responders to the MSC treatment elicited ex vivo cytotoxicity against MSCs that was almost fourfold higher than clinical nonresponders (7). This study demonstrated that the magnitude of immunosuppression in vivo is correlated to the recipient cytotoxic response against the infused MSCs.
Although the mechanisms underlying the role of the host response in a clinical setting are still unclear, several preclinical observations provide useful insight into its correlation with therapeutic outcome. In 2017, Galleu et al. (7) published an important study that identified a mechanism contributing to the immunosuppressive efficacy of intravenously administered MSCs that was not dependent on the typical CQAs of the cell product in isolation (i.e., IDO and TNF-α receptor expression). In a mouse model, it was observed that intravenously injected hMSCs underwent perforin-dependent apoptosis induced by the recipient immune system. The subsequent phagocytosis of apoptotic MSCs by recipient macrophages triggered the immune cells to produce additional IDO intracellularly, increasing the overall systemic IDO expression by ~2.5-fold. Furthermore, de Witte et al. (82) demonstrated in a mouse model that the phagocytosis of infused hMSCs by monocytes in the first 24 hours following injection triggered the immune cells to adopt an immunoregulatory phenotype. A separate study found that monocytes containing phagocytosed MSC debris then migrated to multiple tissues and established further immunotolerance in the adaptive immune system by promoting an immunoregulatory phenotype in lymphocytes (82). Together, these observations demonstrate that, in some clinical indications, stronger cytotoxic responses against MSCs improve the therapeutic effect of immunosuppressive MSCs by facilitating the adoption of a systemic immunoregulatory phenotype in the host. To this end, evaluating in vitro PBMC-mediated cytotoxicity using host immune cells may potentially be used as an indicator for success when recruiting patients for MSC treatment.
The importance of the host immune response on the efficacy of MSC therapy is also demonstrated in other diseases, such as cardiac ischemia (150). For example, a recent study has shown that the therapeutic benefit of cardiac stem cell therapy is not due to the production of new cardiomyocytes but through an acute sterile immune response mediated by host derived CCR2+ and CX3CR1+ macrophages. These macrophages resulted in the alteration of cardiac fibroblast activities, reduction in the ECM content, and functional improvement. Intracardiac injection of a chemical inducer, which induced a similar level of CCR2+ and CX3CR1+ macrophages locally, also provided functional improvement in the cardiac ischemic injury model.
Together, while the exact mechanism may vary depending on the disease and cell therapies used, the host immune responses play important roles in mediating the therapeutic benefit provided by cell-based therapies. Therefore, variations in the host immune response can also be responsible for the variability of cell therapies.
Host disease stage/severity
In addition to the cytotoxic response, host factors related to the stage of disease progression and disease microenvironment may also have implications in better predicting the clinical efficacy of MSC therapies. For example, many studies have suggested that early treatment is better than late treatment to achieve maximal therapeutic efficacy in certain indications. This has been best illustrated by a recent phase 2 study conducted by Athersys Inc. (NCT01436487), which investigated their allogeneic MSC product, Multistem, for treating ischemic stroke. The study was a clinical failure, showing no improvement in neurological outcome compared to placebo controls (151). However, retrospective analysis demonstrated that patients treated <36 hours following the onset of stroke had improved secondary outcomes compared to those treated between 36 and 48 hours. Hoping to leverage the temporal dependency of Multistem’s regenerative potency, the therapy will now be administered exclusively within the first 36 hours following stroke for their phase 3 study (NCT03545607).
At the clinical level, there has been limited progress in understanding the connection between therapeutic outcome and disease stage/severity, in part, due to difficulties in routinely sampling acutely ill patients (151). However, evidence from preclinical studies indicate that the microenvironment of the tissue surrounding the MSCs may be a contributing factor to the therapeutic efficacy in different disease stages. Inflammation, hypoxia, and the ECM in the disease site microenvironment are dynamic, and each parameter can influence MSC function in vivo (20). First, MSCs appear to be more potent in suppressing GvHD when inflammation is high and less potent when inflammation is low (20). Second, previous studies on environmental hypoxia have indicated that exposing MSCs to hypoxic conditions can induce various soluble bioactive molecules and enhance their angiogenic and regenerative potential (152). Third, in vitro studies on the impact of the ECM demonstrate that MSCs seeded on stiff surfaces have reduced suppression of allogeneic lymphocyte activation compared to MSCs seeded in softer, collagen-based 3D scaffolds (153). The stiffness of the ECM is positively correlated with the severity of fibrotic disease, which is highly variable between patients, thus likely contributing to mixed therapeutic outcomes (154). Moreover, along with the variations in inflammation, hypoxia, and the ECM, the increase in the number of damage-inflicting cells (T cells) with increasing disease severity may also influence the therapeutic effect of MSCs in vivo. For example, effector T cells in GvHD increase from nondetectable levels in the early disease stages to 90% of the total disease population in the late stage (155). Hence, the MSC doses (i.e., the number of infused MSCs) administered in late-stage GvHD become therapeutically insufficient, as they are substantially outnumbered by effector T cells. Together, these observations demonstrate that while cell engineering is able to boost the potency of MSCs, evaluating the status of recipients—especially the disease stage—will help optimize the dosing regimen and improve the clinical predictions of therapeutic response to MSC therapies.
Other host factors
In addition to the host immune response, other host factors may also affect the recruitment of MSCs to the disease site, which, in turn, can affect the therapeutic outcome for some clinical indications. For example, capillaries (10 to 15 μm in diameter) trap systemically infused MSCs (~20 μm in diameter) (83), compromising MSC homing to other organs. Moreover, MSC homing is also highly dependent on the secretion of chemokines, as the magnitude of chemokine secretion by the target organ in the host may be insufficient to recruit MSCs efficiently (156). Continued investigation of bioengineering strategies that can overcome these issues may represent a novel strategy to fully unleash the therapeutic potential of MSCs.
Bioengineering strategies relevant to host factors
Clinical and preclinical observations related to the role of the host in facilitating the MSC therapeutic response have inspired novel approaches to controlling these factors in a clinical setting. Patient stratification based on the host cytotoxic responses against MSCs or the disease severity/stage can be a simple strategy to help recruit patients who can likely benefit from MSC therapies (Fig. 4). This can be achieved by incubating MSCs with PBMCs from patients and testing the ability of PBMCs to induce MSC apoptosis in a cytotoxic assay (7). Priming the hosts to establish a microenvironment that can better use the therapeutic potential of MSCs represents another avenue to improve their potency and resulting response rate. For example, the water-soluble antioxidant vitamin C has the capacity to prevent oxidative stress and reduce damage to transplanted cells (157). In a spinal cord injury model, animals receiving intraperitoneal injection of vitamin C in combination with local injection of MSCs at the site of spinal cord injury had a better therapeutic outcome compared with MSCs or vitamin C alone (157). MSC recipients can also be primed with vasodilators such as sodium nitroprusside before systemic infusion of MSCs to circumvent MSC entrapment by the host capillaries (158). Preclinical studies have indicated that, following vasodilation, the accumulation of MSCs in the lungs was reduced by 15% and the recruitment of MSCs to the bone marrow was increased by 10 to 50% compared to untreated control hosts. Boosting the chemokine secretion by the target organ in the host has also been used to improve MSC homing (156). For example, preclinical studies have shown that, before systemic infusion of MSC, animals primed with irradiation had MSC homing to bone marrow increased by twofold compared to unirradiated controls. Irradiation of the animals increased the secretion of the chemoattractant SDF-1 at the damaged bone marrow site, which facilitated improved homing of MSCs in the irradiated population (156). Although host priming remains at the preclinical stage, continued investigation into this approach through irradiation, vasodilation, or other methods may represent a novel strategy to fully unleash the therapeutic potential of MSCs.
Fig. 4. Solutions relevant to host factors.
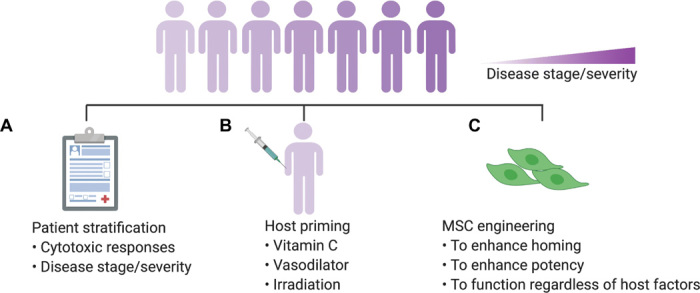
(A) Patient stratification based on the host cytotoxic responses against MSCs or the disease severity/stage can be used to recruit patients who can benefit from MSC therapies. (B) Priming the host with vitamin C can scavenge free radicals that compromise the potency of MSCs. Furthermore, priming the host with a vasodilator or irradiation can facilitate the homing of MSCs to the target sites, resulting in better therapeutic outcomes. (C) The identified host factors that affect the function of MSCs can guide the development of better MSCs, which can complement the host priming strategy and ultimately improve the therapeutic outcome. In particular, MSCs can be engineered to improve the homing to the target sites, engineered to maximum potency, or programmed to function regardless of the host environment. However, it is not clear yet if homing can be engineered to achieve a meaningful boost in efficacy in humans.
Last, an improved understanding of the role of the host environment in MSC function can also be used to guide novel approaches for MSC engineering. For example, priming MSCs with inflammatory cytokines or hypoxia represents an interesting bioengineering approach to boost their potency toward therapeutic applications (152, 159). It may also be useful to engineer the MSC secretome so that it functions independently from the surrounding microenvironment. For example, MSCs overexpressing anti-inflammatory cytokines such as IL-10 or IL-35 have been shown to improve the therapeutic effect compared with native MSCs (160, 161). By exogenously secreting anti-inflammatory factors in a pro-inflammatory environment, MSCs can more efficiently reduce inflammation and better achieve their desired function.
CONCLUSIONS
Although MSCs undoubtedly have immunomodulatory and regenerative therapeutic properties, attempting to apply MSCs “as is” without a clear target has proved to be unsuccessful in most clinical studies. Without a well-defined target, developing more effective “next-generation” MSC therapies will be limited. Specifically, a clear definition of the targets, ideally from the beginning of a project, is critical to guide the design of better MSC therapies. Thinking more about the target up front will help researchers to see how the baseline levels achieved by MSCs are typically suboptimal to activate target biology. This will also help provide a solid rationale to engineer MSCs to more efficiently act on these targets by secreting relevant factors and/or by interacting with the target cells through cell-cell contact and ultimately improve the therapeutic outcome.
Moreover, enormous challenges remain for MSC therapies—from the diverse origins of MSCs, the highly variable culture and cryopreservation conditions, the challenges associated with administration of MSCs, and the challenges of the host environment—that can also lead to unpredictable therapeutic outcomes. Continued exploration of engineering approaches that address these challenges should significantly improve the therapeutic efficacy for a broad range of clinical indications. In particular, boosting the potency of MSCs through engineering strategies such as small-molecule priming, MP engineering, and genetic modification (Fig. 2) provides a measurable property that can be examined throughout all stages of preclinical and clinical development, from well-defined potency assays and CQAs to therapeutic biomarkers in human clinical studies. It is also critical to ensure that these CQAs are preserved following cryopreservation and thawing at both the preclinical and clinical stages. Recent development of single-cell RNA sequencing will also enhance our understanding of MSC heterogeneity and phenotype shift during culture, which, in turn, may provide critical insights to improve the MSC manufacturing process (162). Coupled with other technologies such as iPSC-based MSCs and CRISPR-Cas9–based gene editing, there are many possibilities regarding what MSCs can functionally achieve. Furthermore, while local injection can position MSCs directly at the target site, the insufficient retention of MSCs can compromise the potency and duration of the therapeutic effect. Although strategies such as the use of biomaterials can help address these challenges, most studies have not decoupled cell survival from retention in vivo. Additional studies are needed to clarify this, which, in turn, may provide novel insights about the bottlenecks limiting the retention or survival of MSCs and guide the design of engineering approaches to develop better MSC therapies. For systemic administration, it is critical to properly engineer MSCs to modulate the IBMIR and improve the homing of infused MSCs (Fig. 3). Learning how the host factors impact function and delivery of MSCs will help both inform engineering strategies and inspire new approaches to prime the host (Fig. 4). One caveat is that preclinical mouse models often have limitations in recapitulating the key features of human diseases. Moreover, the infusion volume and number of cells used in mouse models are also very different from those in clinical patients. For example, when MSCs are intravenously injected into rodents, the dose is typically around 50 million/kg. However, the dose for intravenous injection in human patients is typically 1 million to 2 million/kg (19). Because the paracrine factors secreted from MSCs are dependent on cell number, different dosing can significantly affect the therapeutic outcome, assuming the mechanism of action is similar for different species. Consequentially, many promising preclinical results cannot be translated into clinical success. To obtain meaningful results, future studies should explore the use of large animal models that can better mimic the host disease conditions and dosing regimen in clinical settings.
Last, it should be noted that many of the failures in characterization, cell delivery, and thawing variability are limitations in process development, which is critical for ensuring that all procedures are robust and reliable and deliver the expected and intended outcomes in a repeatable manner. Process development is particularly important when large doses of MSCs need to be manufactured for clinical use. Preclinical work should shift to using MSCs manufactured under Good Manufacturing Practice (GMP) to facilitate clinical translation. Rather than just be hopeful that MSC efficacy will be preserved from preclinical to clinical studies (and during scale-up) and that the mechanism of action will include a relevant target with a robust response, engineering strategies can and should be used to engineer the mechanism of action with specific target biology in mind. While nature provides a basic therapeutic framework for MSC-based treatments, bioengineering tools will be the key to shatter translational barriers.
Acknowledgments
We would like to thank S. Aboulhouda and S. Aday for reading the manuscript and providing critical feedback. Funding: We are grateful for the funding support from U.S. NIH grants HL095722 (to J.M.K.) and NS107857 (to K.S.) and Department of Defense grant CA180698 (to K.S.). A grant (to J.M.K., A.A., A.H.A., and K.S.) from King Abdulaziz City for Science and Technology through the Center of Excellence for Biomedicine (CEBM) was also used to support this body of work. E.M.J.S. would also like to thank the Friedman Award for Scholars in Health, which funded her contribution. Author contributions: All authors contributed to the discussion, writing, and revising of the manuscript. Competing interests: During the drafting of this manuscript, J.M.K. has been a paid consultant and or equity holder for companies including Stempeutics, Sanofi, Celltex, LifeVaultBio, Takeda, Ligandal, Camden Partners, Stemgent, Biogen, Pancryos, Frequency Therapeutics, Quthero, and Mesoblast. K.S. has been a consultant for stem cell and oncology companies including Mesoblast, Cynata, and Merrimack. J.M.K. is also an inventor on a patent that was licensed to Mesoblast. J.M.K. holds equity in Frequency Therapeutics, a company that has licensed IP generated by J.M.K. that may benefit financially if the IP is further validated. The interests of J.M.K. were reviewed and are subject to a management plan overseen by his institutions in accordance with its conflict of interest policies. K.S. owns equity in and is a member of the board of directors of AMASA Therapeutics Inc., a company developing cell-based therapies for cancer. K.S.’s interests were reviewed and are managed by Brigham and Women’s Hospital and Partners HealthCare in accordance with their conflict of interest policies. D.B. is a consultant at AMASA Therapeutics Inc. J.M.K. and K.S. also have a multi-year collaborative grant from King Abdulaziz City for Science and Technology through the Center of Excellence for Biomedicine (CEBM) that involves training of Saudi students. The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper.
REFERENCES AND NOTES
- 1.Friedenstein A. J., Gorskaja J. F., Kulagina N. N., Fibroblast precursors in normal and irradiated mouse hematopoietic organs. Exp. Hematol. 4, 267–274 (1976). [PubMed] [Google Scholar]
- 2.Hass R., Kasper C., Bohm S., Jacobs R., Different populations and sources of human mesenchymal stem cells (MSC): A comparison of adult and neonatal tissue-derived MSC. Cell Commun. Signal 9, 12 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.A study of CYP-001 for the treatment of steroid-resistant acute graft versus host disease; https://clinicaltrials.gov/ct2/show/NCT02923375.
- 4.Prochymal® (human adult stem cells) intravenous infusion following acute myocardial infarction (AMI); https://clinicaltrials.gov/ct2/show/NCT00877903.
- 5.Safety and efficacy of intravenous autologous mesenchymal stem cells for MS: A phase 2 proof of concept study (MESCAMS); https://clinicaltrials.gov/ct2/show/NCT02239393.
- 6.Riazifar M., Pone E. J., Lotvall J., Zhao W., Stem cell extracellular vesicles: Extended messages of regeneration. Annu. Rev. Pharmacol. Toxicol. 57, 125–154 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galleu A., Riffo-Vasquez Y., Trento C., Lomas C., Dolcetti L., Cheung T. S., von Bonin M., Barbieri L., Halai K., Ward S., Weng L., Chakraverty R., Lombardi G., Watt F. M., Orchard K., Marks D. I., Apperley J., Bornhauser M., Walczak H., Bennett C., Dazzi F., Apoptosis in mesenchymal stromal cells induces in vivo recipient-mediated immunomodulation. Sci. Transl. Med. 9, eaam7828 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Prockop D. J., Concise review: Two negative feedback loops place mesenchymal stem/stromal cells at the center of early regulators of inflammation. Stem Cells 31, 2042–2046 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Xia X., Chan K. F., Wong G. T. Y., Wang P., Liu L., Yeung B. P. M., Ng E. K. W., Lau J. Y. W., Chiu P. W. Y., Mesenchymal stem cells promote healing of nonsteroidal anti-inflammatory drug-related peptic ulcer through paracrine actions in pigs. Sci. Transl. Med. 11, eaat7455 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Gomzikova M. O., James V., Rizvanov A. A., Therapeutic application of mesenchymal stem cells derived extracellular vesicles for immunomodulation. Front. Immunol. 10, 2663 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao T., Sun F., Liu J., Ding T., She J., Mao F., Xu W., Qian H., Yan Y., Emerging role of mesenchymal stem cell-derived exosomes in regenerative medicine. Curr. Stem Cell Res. Ther. 14, 482–494 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Baek G., Choi H., Kim Y., Lee H. C., Choi C., Mesenchymal stem cell-derived extracellular vesicles as therapeutics and as a drug delivery platform. Stem Cells Transl. Med. 8, 880–886 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rani S., Ryan A. E., Griffin M. D., Ritter T., Mesenchymal stem cell-derived extracellular vesicles: Toward cell-free therapeutic applications. Mol. Ther. 23, 812–823 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Panés J., García-Olmo D., Van Assche G., Colombel J. F., Reinisch W., Baumgart D. C., Dignass A., Nachury M., Ferrante M., Kazemi-Shirazi L., Grimaud J. C., de la Portilla F., Goldin E., Richard M. P., Leselbaum A., Danese S., Collaborators A. C. S. G., Expanded allogeneic adipose-derived mesenchymal stem cells (Cx601) for complex perianal fistulas in Crohn’s disease: A phase 3 randomised, double-blind controlled trial. Lancet 388, 1281–1290 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Detela G., Lodge A., EU regulatory pathways for ATMPs: Standard, accelerated and adaptive pathways to marketing authorisation. Mol. Ther. Methods Clin. Dev. 13, 205–232 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alliance for Regenerative Medicine; https://alliancerm.org/available-products/.
- 17.Mesoblast’s stem cell therapy shows 83% survival in ventilator-dependent COVID-19 patients; www.biospace.com/article/mesoblast-ltd-s-stem-cell-therapy-shows-83-percent-survival-in-covid-19-patients/.
- 18.Golchin A., Seyedjafari E., Ardeshirylajimi A., Mesenchymal stem cell therapy for COVID-19: Present or future. Stem Cell Rev. Rep. , 427–433 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galipeau J., Sensébé L., Mesenchymal stromal cells: Clinical challenges and therapeutic opportunities. Cell Stem Cell 22, 824–833 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi Y., Wang Y., Li Q., Liu K., Hou J., Shao C., Wang Y., Immunoregulatory mechanisms of mesenchymal stem and stromal cells in inflammatory diseases. Nat. Rev. Nephrol. 14, 493–507 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Zhang T. Y., Huang B., Yuan Z. Y., Hu Y. L., Tabata Y., Gao J. Q., Gene recombinant bone marrow mesenchymal stem cells as a tumor-targeted suicide gene delivery vehicle in pulmonary metastasis therapy using non-viral transfection. Nanomedicine 10, 257–267 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Ko I. K., Kean T. J., Dennis J. E., Targeting mesenchymal stem cells to activated endothelial cells. Biomaterials 30, 3702–3710 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinez-Quintanilla J., He D., Wakimoto H., Alemany R., Shah K., Encapsulated stem cells loaded with hyaluronidase-expressing oncolytic virus for brain tumor therapy. Mol. Ther. 23, 108–118 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dominici M., Le Blanc K., Mueller I., Slaper-Cortenbach I., Marini F. C., Krause D. S., Deans R. J., Keating A., Prockop D. J., Horwitz E. M., Minimal criteria for defining multipotent mesenchymal stromal cells. The international society for cellular therapy position statement. Cytotherapy 8, 315–317 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Viswanathan S., Shi Y., Galipeau J., Krampera M., Leblanc K., Martin I., Nolta J., Phinney D. G., Sensebe L., Mesenchymal stem versus stromal cells: International Society for Cell & Gene Therapy (ISCT(R)) Mesenchymal Stromal Cell committee position statement on nomenclature. Cytotherapy 21, 1019–1024 (2019). [DOI] [PubMed] [Google Scholar]
- 26.De Wolf C., Van De Bovenkamp M., Hoefnagel M., Regulatory perspective on in vitro potency assays for human mesenchymal stromal cells used in immunotherapy. Cytotherapy 19, 784–797 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Guadix J. A., Lopez-Beas J., Clares B., Soriano-Ruiz J. L., Zugaza J. L., Galvez-Martin P., Principal criteria for evaluating the quality, safety and efficacy of hMSC-based products in clinical practice: Current approaches and challenges. Pharmaceutics 11, 552 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Juneja S. C., Viswanathan S., Ganguly M., Veillette C., A simplified method for the aspiration of bone marrow from patients undergoing hip and knee joint replacement for isolating mesenchymal stem cells and in vitro chondrogenesis. Bone Marrow Res. 2016, 3152065 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yin J. Q., Zhu J., Ankrum J. A., Manufacturing of primed mesenchymal stromal cells for therapy. Nat. Biomed. Eng. 3, 90–104 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Trivedi A., Miyazawa B., Gibb S., Valanoski K., Vivona L., Lin M., Potter D., Stone M., Norris P. J., Murphy J., Smith S., Schreiber M., Pati S., Bone marrow donor selection and characterization of MSCs is critical for pre-clinical and clinical cell dose production. J. Transl. Med. 17, 128 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.François M., Romieu-Mourez R., Li M., Galipeau J., Human MSC suppression correlates with cytokine induction of indoleamine 2,3-dioxygenase and bystander M2 macrophage differentiation. Mol. Ther. 20, 187–195 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Ren G., Su J., Zhang L., Zhao X., Ling W., L’huillie A., Zhang J., Lu Y., Roberts A. I., Ji W., Zhang H., Rabson A. B., Shi Y., Species variation in the mechanisms of mesenchymal stem cell-mediated immunosuppression. Stem Cells 27, 1954–1962 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Zhukareva V., Obrocka M., Houle J. D., Fischer I., Neuhuber B., Secretion profile of human bone marrow stromal cells: Donor variability and response to inflammatory stimuli. Cytokine 50, 317–321 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Russell A. L., Lefavor R., Durand N., Glover L., Zubair A. C., Modifiers of mesenchymal stem cell quantity and quality. Transfusion 58, 1434–1440 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Chinnadurai R., Rajan D., Qayed M., Arafat D., Garcia M., Liu Y., Kugathasan S., Anderson L. J., Gibson G., Galipeau J., Potency analysis of mesenchymal stromal cells using a combinatorial assay matrix approach. Cell Rep. 22, 2504–2517 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salem B., Miner S., Hensel N. F., Battiwalla M., Keyvanfar K., Stroncek D. F., Gee A. P., Hanley P. J., Bollard C. M., Ito S., Barrett A. J., Quantitative activation suppression assay to evaluate human bone marrow–derived mesenchymal stromal cell potency. Cytotherapy 17, 1675–1686 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bloom D. D., Centanni J. M., Bhatia N., Emler C. A., Drier D., Leverson G. E., McKenna D. H., Gee A. P., Lindblad R., Hei D. J., Hematti P., A reproducible immunopotency assay to measure mesenchymal stromal cell–mediated T-cell suppression. Cytotherapy 17, 140–151 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin P. J., Uberti J. P., Soiffer R. J., Klingemann H., Waller E. K., Daly A. S., Herrmann R. P., Kebriaei P., Prochymal improves response rates in patients with steroid-refractory acute graft versus host disease (SR-GVHD) involving the liver and gut: Results of a randomized, placebo-controlled, multicenter phase III trial in GVHD. Biol. Blood Marrow Transplant. 16, S169–S170 (2010). [Google Scholar]
- 39.Kuçi Z., Bönig H., Kreyenberg H., Bunos M., Jauch A., Janssen J. W. G., Škifić M., Michel K., Eising B., Lucchini G., Bakhtiar S., Greil J., Lang P., Basu O., von Luettichau I., Schulz A., Sykora K.-W., Jarisch A., Soerensen J., Salzmann-Manrique E., Seifried E., Klingebiel T., Bader P., Kuçi S., Mesenchymal stromal cells from pooled mononuclear cells of multiple bone marrow donors as rescue therapy in pediatric severe steroid-refractory graft-versus-host disease: A multicenter survey. Haematologica 101, 985–994 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Galipeau J., The mesenchymal stromal cells dilemma—Does a negative phase III trial of random donor mesenchymal stromal cells in steroid-resistant graft-versus-host disease represent a death knell or a bump in the road? Cytotherapy 15, 2–8 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Rao V. V., Vu M. K., Ma H., Killaars A. R., Anseth K. S., Rescuing mesenchymal stem cell regenerative properties on hydrogel substrates post serial expansion. Bioeng. Transl. Med. 4, 51–60 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bunpetch V., Zhang Z. Y., Zhang X., Han S., Zongyou P., Wu H., Hong-Wei O., Strategies for MSC expansion and MSC-based microtissue for bone regeneration. Biomaterials 196, 67–79 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Ho S. S., Murphy K. C., Binder B. Y., Vissers C. B., Leach J. K., Increased survival and function of mesenchymal stem cell spheroids entrapped in instructive alginate hydrogels. Stem Cells Transl. Med. 5, 773–781 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ozay E. I., Vijayaraghavan J., Gonzalez-Perez G., Shanthalingam S., Sherman H. L., Garrigan D. T., Chandiran K., Torres J. A., Osborne B. A., Tew G. N., Slukvin I. I., Macdonald R. A., Kelly K., Minter L. M., Cymerus™ iPSC-MSCs significantly prolong survival in a pre-clinical, humanized mouse model of Graft-vs-host disease. Stem Cell Res. 35, 101401 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vodyanik M. A., Yu J., Zhang X., Tian S., Stewart R., Thomson J. A., Slukvin I. I., A mesoderm-derived precursor for mesenchymal stem and endothelial cells. Cell Stem Cell 7, 718–729 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cox G., Boxall S. A., Giannoudis P. V., Buckley C. T., Roshdy T., Churchman S. M., McGonagle D., Jones E., High abundance of CD271+ multipotential stromal cells (MSCs) in intramedullary cavities of long bones. Bone 50, 510–517 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee A. S., Tang C., Rao M. S., Weissman I. L., Wu J. C., Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat. Med. 19, 998 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cynata advances toward phase 2 clinical trial of Cymerus(TM) MSCs in patients with osteoarthritis; www.globenewswire.com/news-release/2019/10/04/1925275/0/en/Cynata-Advances-Toward-Phase-2-Clinical-Trial-of-Cymerus-TM-MSCs-in-Patients-with-Osteoarthritis.html.
- 49.Hu X., Li L., Yu X., Zhang R., Yan S., Zeng Z., Shu Y., Zhao C., Wu X., Lei J., Li Y., Zhang W., Yang C., Wu K., Wu Y., An L., Huang S., Ji X., Gong C., Yuan C., Zhang L., Liu W., Huang B., Feng Y., Zhang B., Haydon R. C., Luu H. H., Reid R. R., Lee M. J., Wolf J. M., Yu Z., He T.-C., CRISPR/Cas9-mediated reversibly immortalized mouse bone marrow stromal stem cells (BMSCs) retain multipotent features of mesenchymal stem cells (MSCs). Oncotarget 8, 111847–111865 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wilson J. G., Liu K. D., Zhuo H., Caballero L., McMillan M., Fang X., Cosgrove K., Vojnik R., Calfee C. S., Lee J. W., Rogers A. J., Levitt J., Wiener-Kronish J., Bajwa E. K., Leavitt A., McKenna D., Thompson B. T., Matthay M. A., Mesenchymal stem (stromal) cells for treatment of ARDS: A phase 1 clinical trial. Lancet Respir. Med. 3, 24–32 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matthay M. A., Calfee C. S., Zhuo H. J., Thompson B. T., Wilson J. G., Levitt J. E., Rogers A. J., Gotts J. E., Wiener-Kronish J. P., Bajwa E. K., Donahoe M. P., McVerry B. J., Ortiz L. A., Exline M., Christman J. W., Abbott J., Delucchi K. L., Caballero L., McMillan M., McKenna D. H., Liu K. D., Treatment with allogeneic mesenchymal stromal cells for moderate to severe acute respiratory distress syndrome (START study): A randomised phase 2a safety trial. Lancet Respir. Med. 7, 154–162 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Francois M., Copland I. B., Yuan S., Romieu-Mourez R., Waller E. K., Galipeau J., Cryopreserved mesenchymal stromal cells display impaired immunosuppressive properties as a result of heat-shock response and impaired interferon-gamma licensing. Cytotherapy 14, 147–152 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chinnadurai R., Garcia M. A., Sakurai Y., Lam W. A., Kirk A. D., Galipeau J., Copland I. B., Actin cytoskeletal disruption following cryopreservation alters the biodistribution of human mesenchymal stromal cells in vivo. Stem Cell Rep. 3, 60–72 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moll G., Alm J. J., Davies L. C., von Bahr L., Heldring N., Stenbeck-Funke L., Hamad O. A., Hinsch R., Ignatowicz L., Locke M., Lonnies H., Lambris J. D., Teramura Y., Nilsson-Ekdahl K., Nilsson B., Le Blanc K., Do cryopreserved mesenchymal stromal cells display impaired immunomodulatory and therapeutic properties? Stem Cells 32, 2430–2442 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lalu M. M., McIntyre L., Pugliese C., Fergusson D., Winston B. W., Marshall J. C., Granton J., Stewart D. J., Safety of cell therapy with mesenchymal stromal cells (SafeCell): A systematic review and meta-analysis of clinical trials. PLOS ONE 7, e47559 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bader P., Kuci Z., Bakhtiar S., Basu O., Bug G., Dennis M., Greil J., Barta A., Kallay K. M., Lang P., Lucchini G., Pol R., Schulz A., Sykora K. W., von Luettichau I., Herter-Sprie G., Uddin M. A., Jenkin P., Alsultan A., Buechner J., Stein J., Kelemen A., Jarisch A., Soerensen J., Salzmann-Manrique E., Hutter M., Schafer R., Seifried E., Klingebiel T., Bonig H., Kuci S., Effective treatment of steroid and therapy-refractory acute graft-versus-host disease with a novel mesenchymal stromal cell product (MSC-FFM). Bone Marrow Transplant. 53, 852–862 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gothelf Y., Abramov N., Harel A., Offen D., Safety of repeated transplantations of neurotrophic factors-secreting human mesenchymal stromal stem cells. Clin. Transl. Med. 3, 21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang Y., Chen X., Cao W., Shi Y., Plasticity of mesenchymal stem cells in immunomodulation: Pathological and therapeutic implications. Nat. Immunol. 15, 1009 (2014). [DOI] [PubMed] [Google Scholar]
- 59.Gothelf Y., Kaspi H., Abramov N., Aricha R., miRNA profiling of NurOwn (R): Mesenchymal stem cells secreting neurotrophic factors. Stem Cell. Res. Ther. 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ranganath S. H., Tong Z., Levy O., Martyn K., Karp J. M., Inamdar M. S., Controlled inhibition of the mesenchymal stromal cell pro-inflammatory secretome via microparticle engineering. Stem Cell Rep. 6, 926–939 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Levy O., Brennen W. N., Han E., Rosen D. M., Musabeyezu J., Safaee H., Ranganath S., Ngai J., Heinelt M., Milton Y., Wang H., Bhagchandani S. H., Joshi N., Bhowmick N., Denmeade S. R., Isaacs J. T., Karp J. M., A prodrug-doped cellular Trojan Horse for the potential treatment of prostate cancer. Biomaterials 91, 140–150 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ankrum J. A., Miranda O. R., Ng K. S., Sarkar D., Xu C., Karp J. M., Engineering cells with intracellular agent-loaded microparticles to control cell phenotype. Nat. Protoc. 9, 233–245 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ankrum J. A., Dastidar R. G., Ong J. F., Levy O., Karp J. M., Performance-enhanced mesenchymal stem cells via intracellular delivery of steroids. Sci. Rep. 4, 4645 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Suresh S. C., Selvaraju V., Thirunavukkarasu M., Goldman J. W., Husain A., Alexander Palesty J., Sanchez J. A., McFadden D. W., Maulik N., Thioredoxin-1 (Trx1) engineered mesenchymal stem cell therapy increased pro-angiogenic factors, reduced fibrosis and improved heart function in the infarcted rat myocardium. Int. J. Cardiol. 201, 517–528 (2015). [DOI] [PubMed] [Google Scholar]
- 65.Chabner B. A., Roberts T. G., Timeline—Chemotherapy and the war on cancer. Nat. Rev. Cancer 5, 65–72 (2005). [DOI] [PubMed] [Google Scholar]
- 66.Thanuja M. Y., Anupama C., Ranganath S. H., Bioengineered cellular and cell membrane-derived vehicles for actively targeted drug delivery: So near and yet so far. Adv. Drug Del. Rev. 132, 57–80 (2018). [DOI] [PubMed] [Google Scholar]
- 67.Sasportas L. S., Kasmieh R., Wakimoto H., Hingtgen S., van de Water J. A., Mohapatra G., Figueiredo J. L., Martuza R. L., Weissleder R., Shah K., Assessment of therapeutic efficacy and fate of engineered human mesenchymal stem cells for cancer therapy. Proc. Natl. Acad. Sci. U.S.A. 106, 4822–4827 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Matuskova M., Baranovicova L., Kozovska Z., Durinikova E., Pastorakova A., Hunakova L., Waczulikova I., Nencka R., Kucerova L., Intrinsic properties of tumour cells have a key impact on the bystander effect mediated by genetically engineered mesenchymal stromal cells. J. Gene Med. 14, 776–787 (2012). [DOI] [PubMed] [Google Scholar]
- 69.Niess H., Bao Q., Conrad C., Zischek C., Notohamiprodjo M., Schwab F., Schwarz B., Huss R., Jauch K. W., Nelson P. J., Bruns C. J., Selective targeting of genetically engineered mesenchymal stem cells to tumor stroma microenvironments using tissue-specific suicide gene expression suppresses growth of hepatocellular carcinoma. Ann. Surg. 254, 767–774 (2011). [DOI] [PubMed] [Google Scholar]
- 70.Liu L., Zhang S. X., Liao W., Farhoodi H. P., Wong C. W., Chen C. C., Segaliny A. I., Chacko J. V., Nguyen L. P., Lu M., Polovin G., Pone E. J., Downing T. L., Lawson D. A., Digman M. A., Zhao W., Mechanoresponsive stem cells to target cancer metastases through biophysical cues. Sci. Transl. Med. 9, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gujar S., Pol J. G., Kim Y., Lee P. W., Kroemer G., Antitumor benefits of antiviral immunity: An underappreciated aspect of oncolytic virotherapies. Trends Immunol. 39, 209–221 (2018). [DOI] [PubMed] [Google Scholar]
- 72.Wakimoto H., Ikeda K., Abe T., Ichikawa T., Hochberg F. H., Ezekowitz R. A. B., Pasternack M. S., Chiocca E. A., The complement response against an oncolytic virus is species-specific in its activation pathways. Mol. Ther. 5, 275–282 (2002). [DOI] [PubMed] [Google Scholar]
- 73.Ikeda K., Ichikawa T., Wakimoto H., Silver J. S., Deisboeck T. S., Finkelstein D., Harsh IV G. R., Louis D. N., Bartus R. T., Hochberg F. H., Chiocca E. A., Oncolytic virus therapy of multiple tumors in the brain requires suppression of innate and elicited antiviral responses. Nat. Med. 5, 881–887 (1999). [DOI] [PubMed] [Google Scholar]
- 74.Power A. T., Wang J., Falls T. J., Paterson J. M., Parato K. A., Lichty B. D., Stojdl D. F., Forsyth P. A. J., Atkins H., Bell J. C., Carrier cell-based delivery of an oncolytic virus circumvents antiviral immunity. Mol. Ther. 15, 123–130 (2007). [DOI] [PubMed] [Google Scholar]
- 75.Conget P. A., Minguell J. J., Adenoviral-mediated gene transfer into ex vivo expanded human bone marrow mesenchymal progenitor cells. Exp. Hematol. 28, 382–390 (2000). [DOI] [PubMed] [Google Scholar]
- 76.Hammer K., Kazcorowski A., Liu L., Behr M., Schemmer P., Herr I., Nettelbeck D. M., Engineered adenoviruses combine enhanced oncolysis with improved virus production by mesenchymal stromal carrier cells. Int. J. Cancer 137, 978–990 (2015). [DOI] [PubMed] [Google Scholar]
- 77.MV-NIS infected mesenchymal stem cells in treating patients with recurrent ovarian cancer; https://clinicaltrials.gov/ct2/show/NCT02068794.
- 78.Niess H., von Einem J. C., Thomas M. N., Michl M., Angele M. K., Huss R., Gunther C., Nelson P. J., Bruns C. J., Heinemann V., Treatment of advanced gastrointestinal tumors with genetically modified autologous mesenchymal stromal cells (TREAT-ME1): Study protocol of a phase I/II clinical trial. BMC Cancer 15, 237 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wei W., Huang Y., Li D., Gou H. F., Wang W., Improved therapeutic potential of MSCs by genetic modification. Gene Ther. 25, 538–547 (2018). [DOI] [PubMed] [Google Scholar]
- 80.Thakkar U. G., Trivedi H. L., Vanikar A. V., Dave S. D., Insulin-secreting adipose-derived mesenchymal stromal cells with bone marrow-derived hematopoietic stem cells from autologous and allogenic sources for type 1 diabetes mellitus. Cytotherapy 17, 940–947 (2015). [DOI] [PubMed] [Google Scholar]
- 81.Antunes M. A., Abreu S. C., Cruz F. F., Teixeira A. C., Lopes-Pacheco M., Bandeira E., Olsen P. C., Diaz B. L., Takyia C. M., Freitas I., Rocha N. N., Capelozzi V. L., Xisto D. G., Weiss D. J., Morales M. M., Rocco P. R. M., Effects of different mesenchymal stromal cell sources and delivery routes in experimental emphysema. Respir. Res. 15, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.de Witte S. F. H., Luk F., Sierra Parraga J. M., Gargesha M., Merino A., Korevaar S. S., Shankar A. S., O’Flynn L., Elliman S. J., Roy D., Betjes M. G. H., Newsome P. N., Baan C. C., Hoogduijn M. J., Immunomodulation by therapeutic mesenchymal stromal cells (MSC) is triggered through phagocytosis of MSC by monocytic cells. Stem Cells 36, 602–610 (2018). [DOI] [PubMed] [Google Scholar]
- 83.Eggenhofer E., Benseler V., Kroemer A., Popp F., Geissler E., Schlitt H., Baan C., Dahlke M., Hoogduijn M., Mesenchymal stem cells are short-lived and do not migrate beyond the lungs after intravenous infusion. Front. Immunol. 3, 297 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.O’Cearbhaill E. D., Ng K. S., Karp J. M., Emerging medical devices for minimally invasive cell therapy. Mayo Clin. Proc. 89, 259–273 (2014). [DOI] [PubMed] [Google Scholar]
- 85.Moll G., Ankrum J. A., Kamhieh-Milz J., Bieback K., Ringden O., Volk H. D., Geissler S., Reinke P., Intravascular mesenchymal stromal/stem cell therapy product diversification: Time for new clinical guidelines. Trends Mol. Med. 25, 149–163 (2019). [DOI] [PubMed] [Google Scholar]
- 86.Placebo-controlled study to evaluate rexlemestrocel-L alone or combined with hyaluronic acid in subjects with chronic low back pain (MSB-DR003); https://clinicaltrials.gov/ct2/show/NCT02412735.
- 87.Kanelidis A. J., Premer C., Lopez J., Balkan W., Hare J. M., Route of delivery modulates the efficacy of mesenchymal stem cell therapy for myocardial infarction a meta-analysis of preclinical studies and clinical trials. Circ. Res. 120, 1139 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ullah M., Liu D. D., Thakor A. S., Mesenchymal stromal cell homing: Mechanisms and strategies for improvement. Science 15, 421–438 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Madl C. M., Heilshorn S. C., Blau H. M., Bioengineering strategies to accelerate stem cell therapeutics. Nature 557, 335–342 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li L., Chen X., Wang W. E., Zeng C., How to improve the survival of transplanted mesenchymal stem cell in ischemic heart? Stem Cells Int. 2016, 9682757 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Burdick J. A., Mauck R. L., Gerecht S., To serve and protect: Hydrogels to improve stem cell-based therapies. Cell Stem Cell 18, 13–15 (2016). [DOI] [PubMed] [Google Scholar]
- 92.Hofmann M., Wollert K. C., Meyer G. P., Menke A., Arseniev L., Hertenstein B., Ganser A., Knapp W. H., Drexler H., Monitoring of bone marrow cell homing into the infarcted human myocardium. Circulation 111, 2198–2202 (2005). [DOI] [PubMed] [Google Scholar]
- 93.Fuhrmann T., Tam R. Y., Ballarin B., Coles B., Elliott Donaghue I., van der Kooy D., Nagy A., Tator C. H., Morshead C. M., Shoichet M. S., Injectable hydrogel promotes early survival of induced pluripotent stem cell-derived oligodendrocytes and attenuates longterm teratoma formation in a spinal cord injury model. Biomaterials 83, 23–36 (2016). [DOI] [PubMed] [Google Scholar]
- 94.Wu Z., Chen G., Zhang J., Hua Y., Li J., Liu B., Huang A., Li H., Chen M., Ou C., Treatment of myocardial infarction with gene-modified mesenchymal stem cells in a small molecular hydrogel. Sci. Rep. 7, 15826 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Muschler G. F., Nakamoto C., Griffith L. G., Engineering principles of clinical cell-based tissue engineering. J. Bone Joint Surg. Am. 86, 1541–1558 (2004). [DOI] [PubMed] [Google Scholar]
- 96.Gleeson B. M., Martin K., Ali M. T., Kumar A. H. S., Pillai M. G.-K., Kumar S. P. G., O’Sullivan J. F., Whelan D., Stocca A., Khider W., Barry F. P., O’Brien T., Caplice N. M., Bone marrow-derived mesenchymal stem cells have innate procoagulant activity and cause microvascular obstruction following intracoronary delivery: Amelioration by antithrombin therapy. Stem Cells 33, 2726–2737 (2015). [DOI] [PubMed] [Google Scholar]
- 97.Toma C., Wagner W. R., Bowry S., Schwartz A., Villanueva F., Fate of culture-expanded mesenchymal stem cells in the microvasculature: In vivo observations of cell kinetics. Circ. Res. 104, 398–402 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wu Z., Zhang S., Zhou L., Cai J., Tan J., Gao X., Zeng Z., Li D., Thromboembolism induced by umbilical cord mesenchymal stem cell infusion: A report of two cases and literature review. Transplant. Proc. 49, 1656–1658 (2017). [DOI] [PubMed] [Google Scholar]
- 99.Moll G., Ignatowicz L., Catar R., Luecht C., Sadeghi B., Hamad O., Jungebluth P., Dragun D., Schmidtchen A., Ringden O., Different procoagulant activity of therapeutic mesenchymal stromal cells derived from bone marrow and placental decidua. Stem Cells Dev. 24, 2269–2279 (2015). [DOI] [PubMed] [Google Scholar]
- 100.Oeller M., Laner-Plamberger S., Hochmann S., Ketterl N., Feichtner M., Brachtl G., Hochreiter A., Scharler C., Bieler L., Romanelli P., Couillard-Despres S., Russe E., Schallmoser K., Strunk D., Selection of tissue factor-deficient cell transplants as a novel strategy for improving hemocompatibility of human bone marrow stromal cells. Theranostics 8, 1421–1434 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Christy B. A., Herzig M. C., Montgomery R. K., Delavan C., Bynum J. A., Reddoch K. M., Cap A. P., Procoagulant activity of human mesenchymal stem cells. J. Trauma Acute Care Surg. 83, S164–S169 (2017). [DOI] [PubMed] [Google Scholar]
- 102.George M. J., Prabhakara K., Toledano-Furman N. E., Wang Y. W., Gill B. S., Wade C. E., Olson S. D., Cox C. S. Jr., Clinical cellular therapeutics accelerate clot formation. Stem Cells Transl. Med. 7, 731–739 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li Y., Lin F., Mesenchymal stem cells are injured by complement after their contact with serum. Blood 120, 3436–3443 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Markiewski M. M., Nilsson B., Ekdahl K. N., Mollnes T. E., Lambris J. D., Complement and coagulation: Strangers or partners in crime? Trends Immunol. 28, 184–192 (2007). [DOI] [PubMed] [Google Scholar]
- 105.Kraitchman D. L., Tatsumi M., Gilson W. D., Ishimori T., Kedziorek D., Walczak P., Segars W. P., Chen H. H., Fritzges D., Izbudak I., Young R. G., Marcelino M., Pittenger M. F., Solaiyappan M., Boston R. C., Tsui B. M., Wahl R. L., Bulte J. W., Dynamic imaging of allogeneic mesenchymal stem cells trafficking to myocardial infarction. Circulation 112, 1451–1461 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wagner B., Henschler R., Fate of intravenously injected mesenchymal stem cells and significance for clinical application. Adv. Biochem. Eng. Biotechnol. 130, 19–37 (2013). [DOI] [PubMed] [Google Scholar]
- 107.Scarfe L., Taylor A., Sharkey J., Harwood R., Barrow M., Comenge J., Beeken L., Astley C., Santeramo I., Hutchinson C., Ressel L., Smythe J., Austin E., Levy R., Rosseinsky M. J., Adams D. J., Poptani H., Park B. K., Murray P., Wilm B., Non-invasive imaging reveals conditions that impact distribution and persistence of cells after in vivo administration. Stem Cell. Res. Ther. 9, 332 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mao A. S., Ozkale B., Shah N. J., Vining K. H., Descombes T., Zhang L., Tringides C. M., Wong S. W., Shin J. W., Scadden D. T., Weitz D. A., Mooney D. J., Programmable microencapsulation for enhanced mesenchymal stem cell persistence and immunomodulation. Proc. Natl. Acad. Sci. U.S.A. 116, 15392–15397 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kang W. J., Kang H. J., Kim H. S., Chung J. K., Lee M. C., Lee D. S., Tissue distribution of 18F-FDG-labeled peripheral hematopoietic stem cells after intracoronary administration in patients with myocardial infarction. J. Nucl. Med. 47, 1295–1301 (2006). [PubMed] [Google Scholar]
- 110.Schweizer M. T., Wang H., Bivalacqua T. J., Partin A. W., Lim S. J., Chapman C., Abdallah R., Levy O., Bhowmick N. A., Karp J. M., De Marzo A., Isaacs J. T., Brennen W. N., Denmeade S. R., A phase i study to assess the safety and cancer-homing ability of allogeneic bone marrow-derived mesenchymal stem cells in men with localized prostate cancer. Stem Cells Transl. Med. 8, 441–449 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hu X., Yu S. P., Fraser J. L., Lu Z., Ogle M. E., Wang J. A., Wei L., Transplantation of hypoxia-preconditioned mesenchymal stem cells improves infarcted heart function via enhanced survival of implanted cells and angiogenesis. J. Thorac. Cardiovasc. Surg. 135, 799–808 (2008). [DOI] [PubMed] [Google Scholar]
- 112.Levit R. D., Landazuri N., Phelps E. A., Brown M. E., Garcia A. J., Davis M. E., Joseph G., Long R., Safley S. A., Suever J. D., Lyle A. N., Weber C. J., Taylor W. R., Cellular encapsulation enhances cardiac repair. J. Am. Heart Assoc. 2, e000367 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zanotti L., Sarukhan A., Dander E., Castor M., Cibella J., Soldani C., Trovato A. E., Ploia C., Luca G., Calvitti M., Mancuso F., Arato I., Golemac M., Jonjic N., Biondi A., Calafiore R., Locati M., D’Amico G., Viola A., Encapsulated mesenchymal stem cells for in vivo immunomodulation. Leukemia 27, 500–503 (2013). [DOI] [PubMed] [Google Scholar]
- 114.Kim I., Lee S. K., Yoon J. I., Kim D. E., Kim M., Ha H., Fibrin glue improves the therapeutic effect of MSCs by sustaining survival and paracrine function. Tissue Eng. Part A 19, 2373–2381 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Caldwell A. S., Rao V. V., Golden A. C., Anseth K. S., Porous bio-click microgel scaffolds control hMSC interactions and promote their secretory properties. Biomaterials 232, 119725 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cai L., Dewi R. E., Goldstone A. B., Cohen J. E., Steele A. N., Woo Y. J., Heilshorn S. C., Regulating stem cell secretome using injectable hydrogels with in situ network formation. Adv. Healthc. Mater. 5, 2758–2764 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Qazi T. H., Tytgat L., Dubruel P., Duda G. N., Vlierberghe S. V., Geissler S., Extrusion printed scaffolds with varying pore size as modulators of MSC angiogenic paracrine effects. ACS Biomater Sci. Eng. 5, 5348–5358 (2019). [DOI] [PubMed] [Google Scholar]
- 118.Clark A. Y., Martin K. E., Garcia J. R., Johnson C. T., Theriault H. S., Han W. M., Zhou D. W., Botchwey E. A., Garcia A. J., Integrin-specific hydrogels modulate transplanted human bone marrow-derived mesenchymal stem cell survival, engraftment, and reparative activities. Nat. Commun. 11, 114 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.García J. R., Quiros M., Han W. M., O’Leary M. N., Cox G. N., Nusrat A., García A. J., IFN-gamma-tethered hydrogels enhance mesenchymal stem cell-based immunomodulation and promote tissue repair. Biomaterials 220, 119403 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Moll G., Hult A., von Bahr L., Alm J. J., Heldring N., Hamad O. A., Stenbeck-Funke L., Larsson S., Teramura Y., Roelofs H., Nilsson B., Fibbe W. E., Olsson M. L., Le Blanc K., Do ABO blood group antigens hamper the therapeutic efficacy of mesenchymal stromal cells? PLOS ONE 9, e85040 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Baygan A., Aronsson-Kurttila W., Moretti G., Tibert B., Dahllof G., Klingspor L., Gustafsson B., Khoein B., Moll G., Hausmann C., Svahn B. M., Westgren M., Remberger M., Sadeghi B., Ringden O., Safety and side effects of using placenta-derived decidual stromal cells for graft-versus-host disease and hemorrhagic cystitis. Front. Immunol. 8, 795 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ringden O., Baygan A., Remberger M., Gustafsson B., Winiarski J., Khoein B., Moll G., Klingspor L., Westgren M., Sadeghi B., Placenta-derived decidua stromal cells for treatment of severe acute graft-versus-host disease. Stem Cells Transl. Med. 7, 325–331 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nilsson B., Korsgren O., Lambris J. D., Ekdahl K. N., Can cells and biomaterials in therapeutic medicine be shielded from innate immune recognition? Trends Immunol. 31, 32–38 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Soland M. A., Bego M., Colletti E., Zanjani E. D., Jeor S. S., Porada C. D., Almeida-Porada G., Mesenchymal stem cells engineered to inhibit complement-mediated damage. PLOS ONE 8, e60461 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kamperman T., Karperien M., Le Gac S., Leijten J., Single-cell microgels: Technology, challenges, and applications. Trends Biotechnol. 36, 850–865 (2018). [DOI] [PubMed] [Google Scholar]
- 126.Shi M., Li J., Liao L., Chen B., Li B., Chen L., Jia H., Zhao R. C., Regulation of CXCR4 expression in human mesenchymal stem cells by cytokine treatment: Role in homing efficiency in NOD/SCID mice. Haematologica 92, 897–904 (2007). [DOI] [PubMed] [Google Scholar]
- 127.Tsai L. K., Leng Y., Wang Z., Leeds P., Chuang D. M., The mood stabilizers valproic acid and lithium enhance mesenchymal stem cell migration via distinct mechanisms. Neuropsychopharmacology 35, 2225–2237 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tsai L.-K., Wang Z., Munasinghe J., Leng Y., Leeds P., Chuang D. M., Mesenchymal stem cells primed with valproate and lithium robustly migrate to infarcted regions and facilitate recovery in a stroke model. Stroke 42, 2932–2939 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yu Q., Chen L., You Y., Zou C., Zhang Y., Liu Q., Cheng F., Erythropoietin combined with granulocyte colonystimulating factor enhances MMP-2 expression in mesenchymal stem cells and promotes cell migration. Mol. Med. Report. 4, 31–36 (2011). [DOI] [PubMed] [Google Scholar]
- 130.Kim Y. S., Noh M. Y., Kim J. Y., Yu H. J., Kim K. S., Kim S. H., Koh S. H., Direct GSK-3beta inhibition enhances mesenchymal stromal cell migration by increasing expression of beta-PIX and CXCR4. Mol. Neurobiol. 47, 811–820 (2013). [DOI] [PubMed] [Google Scholar]
- 131.Levy O., Zhao W., Mortensen L. J., Leblanc S., Tsang K., Fu M., Phillips J. A., Sagar V., Anandakumaran P., Ngai J., Cui C. H., Eimon P., Angel M., Lin C. P., Yanik M. F., Karp J. M., mRNA-engineered mesenchymal stem cells for targeted delivery of interleukin-10 to sites of inflammation. Blood 122, e23–e32 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Liao W., Pham V., Liu L., Riazifar M., Pone E. J., Zhang S. X., Ma F., Lu M., Walsh C. M., Zhao W., Mesenchymal stem cells engineered to express selectin ligands and IL-10 exert enhanced therapeutic efficacy in murine experimental autoimmune encephalomyelitis. Biomaterials 77, 87–97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cheng Z., Ou L., Zhou X., Li F., Jia X., Zhang Y., Liu X., Li Y., Ward C. A., Melo L. G., Kong D., Targeted migration of mesenchymal stem cells modified with CXCR4 gene to infarcted myocardium improves cardiac performance. Mol. Ther. 16, 571–579 (2008). [DOI] [PubMed] [Google Scholar]
- 134.Zhang D., Fan G. C., Zhou X., Zhao T., Pasha Z., Xu M., Zhu Y., Ashraf M., Wang Y., Over-expression of CXCR4 on mesenchymal stem cells augments myoangiogenesis in the infarcted myocardium. J. Mol. Cell. Cardiol. 44, 281–292 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bobis-Wozowicz S., Miekus K., Wybieralska E., Jarocha D., Zawisz A., Madeja Z., Majka M., Genetically modified adipose tissue-derived mesenchymal stem cells overexpressing CXCR4 display increased motility, invasiveness, and homing to bone marrow of NOD/SCID mice. Exp. Hematol. 39, 686–696.e4 (2011). [DOI] [PubMed] [Google Scholar]
- 136.Chen W., Li M., Cheng H., Yan Z., Cao J., Pan B., Sang W., Wu Q., Zeng L., Li Z., Xu K., Overexpression of the mesenchymal stem cell Cxcr4 gene in irradiated mice increases the homing capacity of these cells. Cell Biochem. Biophys. 67, 1181–1191 (2013). [DOI] [PubMed] [Google Scholar]
- 137.Chen Z., Chen Q., Du H., Xu L., Wan J., Mesenchymal stem cells and CXC chemokine receptor 4 overexpression improved the therapeutic effect on colitis via mucosa repair. Exp. Ther. Med. 16, 821–829 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Corradetti B., Taraballi F., Martinez J. O., Minardi S., Basu N., Bauza G., Evangelopoulos M., Powell S., Corbo C., Tasciotti E., Hyaluronic acid coatings as a simple and efficient approach to improve MSC homing toward the site of inflammation. Sci. Rep. 7, 7991 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sackstein R., Merzaban J. S., Cain D. W., Dagia N. M., Spencer J. A., Lin C. P., Wohlgemuth R., Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat. Med. 14, 181–187 (2008). [DOI] [PubMed] [Google Scholar]
- 140.Abdi R., Moore R., Sakai S., Donnelly C. B., Mounayar M., Sackstein R., HCELL expression on murine msc licenses pancreatotropism and confers durable reversal of autoimmune diabetes in NOD mice. Stem Cells 33, 1523–1531 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sarkar D., Vemula P. K., Teo G. S., Spelke D., Karnik R., Weele Y., Karp J. M., Chemical engineering of mesenchymal stem cells to induce a cell rolling response. Bioconjug. Chem. 19, 2105–2109 (2008). [DOI] [PubMed] [Google Scholar]
- 142.Cheng H., Byrska-Bishop M., Zhang C. T., Kastrup C. J., Hwang N. S., Tai A. K., Lee W. W., Xu X., Nahrendorf M., Langer R., Anderson D. G., Stem cell membrane engineering for cell rolling using peptide conjugation and tuning of cell-selectin interaction kinetics. Biomaterials 33, 5004–5012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Xiao W., Green T. I. P., Liang X., Delint R. C., Perry G., Roberts M. S., Le Vay K., Back C. R., Ascione R., Wang H., Race P. R., Perriman A. W., Designer artificial membrane binding proteins to direct stem cells to the myocardium. Chem. Sci. 10, 7610–7618 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lo C. Y., Antonopoulos A., Dell A., Haslam S. M., Lee T., Neelamegham S., The use of surface immobilization of P-selectin glycoprotein ligand-1 on mesenchymal stem cells to facilitate selectin mediated cell tethering and rolling. Biomaterials 34, 8213–8222 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Arbab A. S., Jordan E. K., Wilson L. B., Yocum G. T., Lewis B. K., Frank J. A., In vivo trafficking and targeted delivery of magnetically labeled stem cells. Hum. Gene Ther. 15, 351–360 (2004). [DOI] [PubMed] [Google Scholar]
- 146.Song Y. S., Ku J. H., Monitoring transplanted human mesenchymal stem cells in rat and rabbit bladders using molecular magnetic resonance imaging. Neurourol.Urodyn. 26, 584–593 (2007). [DOI] [PubMed] [Google Scholar]
- 147.Yanai A., Hafeli U. O., Metcalfe A. L., Soema P., Addo L., Gregory-Evans C. Y., Po K., Shan X., Moritz O. L., Gregory-Evans K., Focused magnetic stem cell targeting to the retina using superparamagnetic iron oxide nanoparticles. Cell Transplant. 21, 1137–1148 (2012). [DOI] [PubMed] [Google Scholar]
- 148.Hsiao J. K., Tai M. F., Chu H. H., Chen S. T., Li H., Lai D. M., Hsieh S. T., Wang J. L., Liu H. M., Magnetic nanoparticle labeling of mesenchymal stem cells without transfection agent: Cellular behavior and capability of detection with clinical 1.5 T magnetic resonance at the single cell level. Magn. Reson. Med. 58, 717–724 (2007). [DOI] [PubMed] [Google Scholar]
- 149.Wang S., Lu B., Girman S., Duan J., McFarland T., Zhang Q.-s., Grompe M., Adamus G., Appukuttan B., Lund R., Non-invasive stem cell therapy in a rat model for retinal degeneration and vascular pathology. PLOS ONE 5, e9200 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Vagnozzi R. J., Maillet M., Sargent M. A., Khalil H., Johansen A. K. Z., Schwanekamp J. A., York A. J., Huang V., Nahrendorf M., Sadayappan S., Molkentin J. D., An acute immune response underlies the benefit of cardiac stem cell therapy. Nature 577, 405–409 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hess D. C., Wechsler L. R., Clark W. M., Savitz S. I., Ford G. A., Chiu D., Yavagal D. R., Uchino K., Liebeskind D. S., Auchus A. P., Sen S., Sila C. A., Vest J. D., Mays R. W., Safety and efficacy of multipotent adult progenitor cells in acute ischaemic stroke (MASTERS): A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol. 16, 360–368 (2017). [DOI] [PubMed] [Google Scholar]
- 152.Noronha N. C., Mizukami A., Caliari-Oliveira C., Cominal J. G., Rocha J. L. M., Covas D. T., Swiech K., Malmegrim K. C. R., Priming approaches to improve the efficacy of mesenchymal stromal cell-based therapies. Stem Cell. Res. Ther. 10, 131 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Yang J., Chen X., Yuan T., Yang X., Fan Y., Zhang X., Regulation of the secretion of immunoregulatory factors of mesenchymal stem cells (MSCs) by collagen-based scaffolds during chondrogenesis. Korean J. Couns. Psychother. 70, 983–991 (2017). [DOI] [PubMed] [Google Scholar]
- 154.Herrera J., Henke C. A., Bitterman P. B., Extracellular matrix as a driver of progressive fibrosis. J. Clin. Invest. 128, 45–53 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Tisato V., Naresh K., Girdlestone J., Navarrete C., Dazzi F., Mesenchymal stem cells of cord blood origin are effective at preventing but not treating graft-versus-host disease. Leukemia 21, 1992–1999 (2007). [DOI] [PubMed] [Google Scholar]
- 156.Kyriakou C., Rabin N., Pizzey A., Nathwani A., Yong K., Factors that influence short-term homing of human bone marrow-derived mesenchymal stem cells in a xenogeneic animal model. Haematologica 93, 1457–1465 (2008). [DOI] [PubMed] [Google Scholar]
- 157.Salem N., Salem M. Y., Elmaghrabi M. M., Elawady M. A., Elawady M. A., Sabry D., Shamaa A., Elkasapy A. H., Ibrhim N., Elamir A., Does vitamin C have the ability to augment the therapeutic effect of bone marrow-derived mesenchymal stem cells on spinal cord injury? Neural Regen. Res. 12, 2050–2058 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Gao J., Dennis J. E., Muzic R. F., Lundberg M., Caplan A. I., The dynamic in vivo distribution of bone marrow-derived mesenchymal stem cells after infusion. Cells Tissues Organs 169, 12–20 (2001). [DOI] [PubMed] [Google Scholar]
- 159.Szabo E., Fajka-Boja R., Kriston-Pal E., Hornung A., Makra I., Kudlik G., Uher F., Katona R. L., Monostori E., Czibula A., Licensing by inflammatory cytokines abolishes heterogeneity of immunosuppressive function of mesenchymal stem cell population. Stem Cells Dev. 24, 2171–2180 (2015). [DOI] [PubMed] [Google Scholar]
- 160.Nakajima M., Nito C., Sowa K., Suda S., Nishiyama Y., Nakamura-Takahashi A., Nitahara-Kasahara Y., Imagawa K., Hirato T., Ueda M., Kimura K., Okada T., Mesenchymal stem cells overexpressing interleukin-10 promote neuroprotection in experimental acute ischemic stroke. Mol. Ther. Methods Clin. Dev. 6, 102–111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Guo H., Li B., Wang W., Zhao N., Gao H., Mesenchymal stem cells overexpressing IL-35: A novel immunosuppressive strategy and therapeutic target for inducing transplant tolerance. Stem Cell. Res. Ther. 9, 254 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Huang Y., Li Q., Zhang K., Hu M., Wang Y., Du L., Lin L., Li S., Sorokin L., Melino G., Shi Y., Wang Y., Single cell transcriptomic analysis of human mesenchymal stem cells reveals limited heterogeneity. Cell Death Dis. 10, 368 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hoban D. B., Howard L., Dowd E., GDNF-secreting mesenchymal stem cells provide localized neuroprotection in an inflammation-driven rat model of Parkinson’s disease. Neuroscience 303, 402–411 (2015). [DOI] [PubMed] [Google Scholar]
- 164.Deng P., Torrest A., Pollock K., Dahlenburg H., Annett G., Nolta J. A., Fink K. D., Clinical trial perspective for adult and juvenile Huntington’s disease using genetically-engineered mesenchymal stem cells. Neural Regen. Res. 11, 702–705 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Pollock K., Dahlenburg H., Nelson H., Fink K. D., Cary W., Hendrix K., Annett G., Torrest A., Deng P., Gutierrez J., Nacey C., Pepper K., Kalomoiris S., McGee D. A. J. J., Gruenloh W., Fury B., Bauer G., Duffy A., Tempkin T., Wheelock V., Nolta J. A., Human mesenchymal stem cells genetically engineered to overexpress brain-derived neurotrophic factor improve outcomes in Huntington’s disease mouse models. Mol. Ther. 24, 965–977 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Liu X. S., Li J. F., Wang S. S., Wang Y. T., Zhang Y. Z., Yin H. L., Geng S., Gong H. C., Han B., Wang Y. L., Human umbilical cord mesenchymal stem cells infected with adenovirus expressing HGF promote regeneration of damaged neuron cells in a Parkinson’s disease model. Biomed. Res. Int. 2014, 909657 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Marin-Banasco C., Benabdellah K., Melero-Jerez C., Oliver B., Pinto-Medel M. J., Hurtado-Guerrero I., de Castro F., Clemente D., Fernandez O., Martin F., Leyva L., Suardiaz M., Gene therapy with mesenchymal stem cells expressing IFN-ss ameliorates neuroinflammation in experimental models of multiple sclerosis. Br. J. Pharmacol. 174, 238–253 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Pelled G., Sheyn D., Tawackoli W., Jun D. S., Koh Y., Su S., Cohn Yakubovich D., Kallai I., Antebi B., Da X., Gazit Z., Bae H., Gazit D., BMP6-engineered MSCs induce vertebral bone repair in a pig model: A pilot study. Stem Cells Int. 2016, 6530624 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Chen G., Zhang Y., Yu S., Sun W., Miao D., Bmi1 overexpression in mesenchymal stem cells exerts anti-aging and anti-osteoporosis effects by inactivating p16/p19 signaling and inhibiting oxidative stress. Stem Cells 0, 1–12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Jang Y. O., Cho M. Y., Yun C. O., Baik S. K., Park K. S., Cha S. K., Chang S. J., Kim M. Y., Lim Y. L., Kwon S. O., Effect of function-enhanced mesenchymal stem cells infected with decorin-expressing adenovirus on hepatic fibrosis. Stem Cells Transl. Med. 5, 1247–1256 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Conrad C., Husemann Y., Niess H., von Luettichau I., Huss R., Bauer C., Jauch K. W., Klein C. A., Bruns C., Nelson P. J., Linking transgene expression of engineered mesenchymal stem cells and angiopoietin-1-induced differentiation to target cancer angiogenesis. Ann. Surg. 253, 566–571 (2011). [DOI] [PubMed] [Google Scholar]
- 172.Shah K., Encapsulated stem cells for cancer therapy. Biomatter 3, e24278 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hu M., Yang J. L., Teng H., Jia Y. Q., Wang R., Zhang X. W., Wu Y., Luo Y., Chen X. C., Zhang R., Tian L., Zhao X., Wei Y. Q., Anti-angiogenesis therapy based on the bone marrow-derived stromal cells genetically engineered to express sFlt-1 in mouse tumor model. BMC Cancer 8, 306 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Choi S. H., Tamura K., Khajuria R. K., Bhere D., Nesterenko I., Lawler J., Shah K., Antiangiogenic variant of TSP-1 targets tumor cells in glioblastomas. Mol. Ther. 23, 235–243 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Duebgen M., Martinez-Quintanilla J., Tamura K., Hingtgen S., Redjal N., Wakimoto H., Shah K., Stem cells loaded with multimechanistic oncolytic herpes simplex virus variants for brain tumor therapy. J. Natl. Cancer Inst. 106, dju090 (2014). [DOI] [PubMed] [Google Scholar]
- 176.Alfano A. L., Nicola Candia A., Cuneo N., Guttlein L. N., Soderini A., Rotondaro C., Sganga L., Podhajcer O. L., Lopez M. V., Oncolytic adenovirus-loaded menstrual blood stem cells overcome the blockade of viral activity exerted by ovarian cancer ascites. Mol. Ther. Oncolytics 6, 31–44 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Moreno R., Rojas L. A., Villellas F. V., Soriano V. C., Garcia-Castro J., Fajardo C. A., Alemany R., Human menstrual blood-derived mesenchymal stem cells as potential cell carriers for oncolytic adenovirus. Stem Cells Int. 2017, 3615729 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]