

A supramolecular platform has been demonstrated to be a powerful tool for screening efficient targeted gene delivery vehicles.
Abstract
It requires multistep synthesis and conjugation processes to incorporate multifunctionalities into a polyplex gene vehicle to overcome numerous hurdles during gene delivery. Here, we describe a supramolecular platform to precisely control, screen, and optimize molecular architectures of siRNA targeted delivery vehicles, which is based on rationally designed host-guest complexation between a β-cyclodextrin–based cationic host polymer and a library of guest polymers with various PEG shape and size, and various density of ligands. The host polymer is responsible to load/unload siRNA, while the guest polymer is responsible to shield the vehicles from nonspecific cellular uptake, to prolong their circulation time, and to target tumor cells. A series of precisely controlled molecular architectures through a simple assembly process allow for a rapid optimization of siRNA delivery vehicles in vitro and in vivo for therapeutic siRNA-Bcl2 delivery and tumor therapy, indicating the platform is a powerful screening tool for targeted gene delivery vehicles.
INTRODUCTION
RNA interference (RNAi) has emerged as a promising gene therapy strategy for human diseases through silencing harmful genes by complementary small interfering RNA (siRNA) in target cells (1, 2). The main obstacle for RNAi-based therapy is the safe and efficient delivery of siRNA to target cells (3, 4). While cationic polymers forming polyplex nanoparticles (PNPs) are a class of promising siRNA delivery vehicles, there remains a major challenge to incorporate multiple functional features into a single vehicle system to overcome numerous hurdles during the gene delivery, which usually requires tedious multistep chemical synthesis and conjugation processes (5, 6). The multiple functions required for an efficient siRNA delivery system may include siRNA protection, serum stability in blood, overcoming nonspecific cellular binding/uptake and clearance by the reticuloendothelial systems (RES), low cytotoxicity, specific targeting ability to cells of interest, and efficient cytoplasmic release (7, 8).
For example, positively charged PNP vehicles show strong nonspecific binding to all types of cells, because of the abundant anionic phospholipid phosphatidylserines on cell surfaces (9), and also interact with serum components, resulting in short circulation time and toxic effects. While surface shielding by PEGylation of PNPs can improve serum stability, reduce nonspecific cellular binding/uptake and clearance by RES, and prolong in vivo circulation, it also compromises the cellular uptake by target cells (10–12). Conjugation of ligands onto PNPs is an appealing strategy to achieve targeted delivery mediated by ligands interacting with receptors on target cells, maximize therapeutic effects, avoid nonspecific silencing in bystander cells, and reduce the amount of siRNA required (13, 14). Therefore, it is desirable to have both PEGylation shielding and ligand-based targeted delivery property in a single PNP vehicle system (15).
Often, the overall delivery efficiency will be cooperatively affected by the molecular architectural factors, such as size, number, density, and spatial organization of both surface shielding polymers and targeting ligands (16). To achieve an efficient targeted delivery, it is important to precisely control the molecular architectures of the vehicles and readily screen the vehicles to optimize the delivery systems for optimal therapeutic effects (17, 18). In other words, before construction of the ligand-based targeting system, it is necessary to optimize the surface shielding by screening out the desired architectures of polyethylene glycol (PEG) on the PNP vehicles, such as the size, length, shape, and density of the PEG (12), as well as to maximize the ligand-receptor interaction by screening out the desired architectures of ligands attached to the distal ends of PEG on the PNP vehicles. For a given cell type, the interactions between ligands and receptors are collectively influenced by the number, density, and spatial organization of the ligands on vehicles (19, 20). Receptors usually exist as “cluster” on cell membranes; thus, multivalent ligands on vehicles would be beneficial for more efficient ligand-receptor interaction (21, 22). Until now, optimal parameters related to surface shielding and ligand-receptor interaction for achieving maximized efficiency of siRNA targeted delivery are not clear in many studies due to lack of convenient screening platforms (17, 18).
Here, we report a supramolecular platform for precisely controlling and optimizing the molecular architectures of an siRNA targeted delivery vehicle, which is developed on the basis of host-guest supramolecular self-assembly processes (Fig. 1). Supramolecular host-guest chemistry has offered a powerful and convenient approach for constructing complicated nanostructures self-assembled from individually tunable and relatively simple molecular building blocks (23–26). In this study, an siRNA vehicle is constructed by two basic modular components: a host polymer based on β-cyclodextrin (β-CD) and a guest polymer functionalized by the adamantyl (Ad) group. Specifically, the host is a star-shaped cationic polymer with multiple arms of poly(2-dimethylaminoethyl methacrylate) (pDMAEMA) linked to a β-CD core with bioreducible disulfide bonds (β-CD–SS–pDMAEMA, and further shortened to CD-SS-P), which has been proven as a safe and efficient DNA carrier (25). The guest is a series of Ad-functionalized PEG polymers with linear or comb-like structures and with various numbers and densities of folic acid (FA) as targeting ligand on the distal ends of PEG. By taking advantage of the β-CD cavity that can strongly form host-guest complex with Ad moiety (with an association constant of Ka ≈ 105 M−1 at room temperature) (27), the host and the guest integrate into a complex polymeric vehicle that can condense siRNA with its pDMAEMA segments, forming core-shell PNPs with siRNA loaded at the core and PEG and ligands as the shell. By switching the host and guest in different combinations, a series of siRNA delivery vehicles could be simply obtained with precisely controlled molecular architectures for screening their functional features in a rapid and convenient way, to optimize the vehicles for the maximized therapeutic effects.
Fig. 1. Schematic illustration of the engineering platform for controlling, screening, and optimizing molecular architectures of siRNA targeted delivery vehicles.

Supramolecular siRNA delivery vehicles are formed by host-guest complexation between CD-SS-pDMAEMA host polymer and Ad-PEG or Ad-PEG-FA guest polymers, which subsequently condense siRNA to form core-shell PNPs. The host polymer is responsible to condense and load siRNA, to assist endosomal escape induced by the “proton sponge” effect, and to unload siRNA in the cytoplasm in the target tumor cells, while the guest polymer is responsible to shield the vehicles from nonspecific cellular uptake, to prolong the circulation time, and to actively target the tumor cells. By switching the host and guest in different combinations, the molecular architectures of the PNPs are precisely controlled, which are subsequently evaluated in vitro in cell cultures to screen out the optimized PNPs, followed by further in vivo validation for targeted delivery of therapeutic siRNA-Bcl2 in a mouse tumor model.
In this study, to obtain the optimal architectural parameters of the host-guest complex vehicles for efficient PEG shielding and FA-based ligand targeting, as well as for the maximized overall siRNA delivery efficiency, we first screened the PEG architectures that are linear and comb shaped and of different lengths and sizes. After the PEG architectures were optimized, the PEGs were conjugated with FA at their distal ends with different numbers and densities. We chose FA because it is a well-characterized ligand (28) targeting to the folate receptor (FR), which is overexpressed in numerous cancer cells (29). Now a series of host-guest PNPs with the shells of PEG and conjugated FA ligand of different architectures and densities were obtained and evaluated in both FR-positive and FR-negative cells. The optimized PNPs for targeted delivery of therapeutic siRNA for cancer therapy were further evaluated in mice tumor models. The in vivo data showed good correlation with the in vitro data, indicating that the supramolecular platform is a useful screening tool for targeted gene delivery vehicles.
RESULTS AND DISCUSSION
Host polymer for siRNA condensation and cytoplasmic delivery
The host-guest screening system is composed of the CD-SS-P host polymer for siRNA condensation and endosomal escape, and switchable guest polymers for PNP shielding and targeting. The redox-sensitive CD-SS-P with multiple arms of pDMAEMA linked to the β-CD core by disulfide bonds was synthesized using the method in our previous report (25). The control polymer CD-P, where multiple arms of pDMAEMA were linked to the β-CD core without disulfide bonds, was synthesized too (refer to the Supplementary Materials). The 1H nuclear magnetic resonance (NMR) spectrum of CD-SS-P is shown in Fig. 2A. The β-CD core was grafted with four arms of pDMAEMA. The average degree of polymerization of each pDMAEMA arm was 12. A single peak was observed in the gel permeation chromatography (GPC) elution curve of CD-SS-P (Fig. 2B). The molecular weight was determined to be 7.8 kDa with PDI (polydispersity index) of 1.15. The CD-SS-P was able to completely condense siRNA at an N/P ratio of 3 and above (Fig. 2C). We selected the redox-sensitive CD-SS-P as the host polymer component for the screening platform for the following reasons: (i) The pDMAEMA arms can condense siRNA efficiently and assist endosomal escape of PNPs induced by the “proton sponge” effect; (ii) the β-CD core can allow host-guest complexation of different guest polymers (PEG with different architectures and ligand density) to form the PNP shells; (iii) the disulfide bonds can be cleaved by the intracellular glutathione to readily release siRNA into cytoplasm; and (iv) the redox-sensitive biodegradability of CD-SS-P makes the polymer very low cytotoxic (25). To confirm that CD-SS-P can deliver siRNA into cells and readily release siRNA in cytoplasm, both CD-SS-P and CD-P were labeled by rhodamin-B-isothiocyanate (RITC) via the hydroxyl groups of β-CD and used to deliver siRNA labeled by fluorescein isothiocyanate (FITC) in cultured KB cells (Fig. 2D). The spatial confocal microscopic images showed that green dots (FITC-siRNA) and red dots (RITC-CD-SS-P) were observed separately after internalization (1, 2, and 24 hours after incubation), indicating that most of the siRNA was released from the CD-SS-P carrier (Fig. 2D, upper panel). However, the control CD-P without disulfide linkages failed to release the siRNA in cytosol, because most of the dots were yellow, which are the colocalized FITC-siRNA and RITC-CD-P carrier (Fig. 2D, bottom panel).
Fig. 2. Characterization of CD-SS-P host polymer and its properties for siRNA condensation and cytoplasmic delivery.
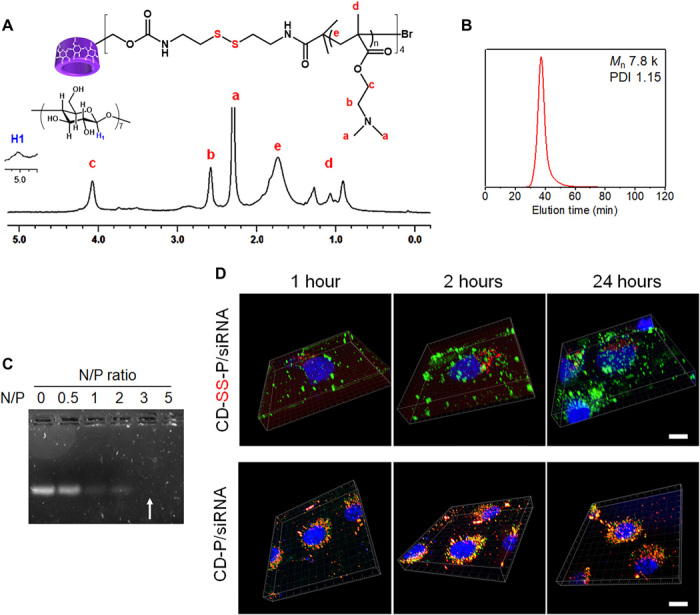
(A) 1H NMR spectrum of CD-SS-P in D2O. (B) GPC curve of CD-SS-P host polymer. Phosphate-buffered saline (PBS) (0.5×) was used as eluent. (C) siRNA condensation by CD-SS-P evaluated by gel retardation assay. (D) Three-dimensional (z-stack) confocal microscopic images showing intracellular distribution of rhodamin-B-isothiocyanate (RITC)–labeled polymer vehicles (red) and fluorescein isothiocyanate (FITC)–labeled siRNA (green) in KB cells at 1, 2, and 24 hours after transfection. RITC dye was labeled to hydroxyl groups of β-CD of CD-SS-P and CD-P. Cell nucleus was stained by Hoechst 33342 (blue). Scale bars, 10 μm.
Screening of PEG architectures and ligand densities
To obtain an efficient siRNA targeted delivery system, nonspecific binding to cells must be first minimized, as the positively charged gene vehicle tends to bind to the surfaces of all cells (30, 31). PEGylation shielding is the predominant method to minimize the nonspecific binding. Once minimal nonspecific binding is achieved with proper architectures of PEG, the number and density of the ligands at the distal ends of PEG can be optimized. To this end, we assembled the CD-SS-P host polymer with a series of Ad-PEG guest polymers having linear and comb-shaped structures and different sizes, without or with distal ligands (Fig. 3, A and B), to screen and find out the best combinations for the most efficient siRNA targeted delivery.
Fig. 3. Screening and optimizing of PEG architectures and ligand densities for efficient siRNA targeted delivery.

(A) Linear and comb-shaped Ad-PEG host polymers are used to screen the architectures (length, size, and shape) of PEG shell for minimized nonspecific binding. (B) Ad-PEG with distal FA ligands are used to screen and optimize the architectures and overall effects of nonspecific binding and ligand-induced specific binding. (C) Particle sizes and zeta potentials of siRNA-loaded PNPs formed by H/G carriers with various linear and comb-shaped Ad-PEG guests. Host polymer CD-SS-P without a guest polymer (H) was used as a control. PNPs were prepared at N/P ratio of 20 in PBS. (D) Mean fluorescence intensity (MFI) of KB and A549 cells at 4 hours after transfection with FITC-siRNA–loaded PNPs formed by various H/G and H carriers, measured by flow cytometry. **P < 0.01, when compared with H. (E) Fluorescence intensity of KB cells at 1 hour after incubation on ice with FITC-siRNA–loaded H/G targeted carriers, plotted against siRNA concentration, after subtracting that of corresponding H/G without FA. (F) Plots of fluorescence intensity of KB cells versus time during the dissociation of the bound PNPs from the surfaces of the cells. The KB cells were preincubated to equilibrium with PNPs formed by FITC-siRNA–loaded H/G targeted carriers, followed by reincubation in PBS. For (C) to (F), all data were presented in means ± SD (n = 3). a.u., arbitrary units.
First, linear Ad-PEG (PL) with Mn of 4k, 8k, 12k, and 20k (PL4k, PL8k, PL12k, and PL20k), and comb-shaped Ad-PEG (PC) with Mn of 4k, 8k, 12k, and 20k (PC4k, PC8k, PC 12k, and PC20k) were synthesized (fig. S1) and screened as shielding polymers for the siRNA PNPs for achieving the minimized nonspecific binding to nontarget cells. By combining the CD-SS-P host and the above Ad-PEG guest polymers, a series of host-guest siRNA gene carriers (shortened as H/G followed by the name of the guest used in a parenthesis) were obtained. The particle size and zeta potential of the siRNA-loaded PNPs formed by the H/G carriers were measured (Fig. 3C). For both linear and comb-shaped Ad-PEG guests, with the increase in PEG molecular weight, the PNP particle size increased from around 75 to 230 nm, whereas the zeta potential decreased from around 32 to below 10 mV. With similar molecular weights, comb-shaped Ad-PEG was more efficient to reduce the zeta potential than linear Ad-PEG. The H/G(PL20k), H/G(PC8k), H/G(PC12k), and H/G(PC20k) carriers gave very low zeta potentials (<10 mV) at an N/P ratio of 20, while their particle size remained within a suitable range for gene delivery (<250 nm). Gene delivery PNPs with low zeta potentials (<10 mV) may have minimal nonspecific binding and prolonged in vivo circulation (32).
To evaluate the H/G carriers for nonspecific binding, FITC-siRNA–loaded PNPs formed by the H/G carriers were cultured with two cell lines, the KB (FR overexpressed, FR+) and A549 (FR deficient, FR−) cells. The cell-associated mean fluorescence intensity (MFI) at 4 hours after transfection was measured to determine the shielding effects of the PEG shells with different sizes and architectures (Fig. 3D). The CD-SS-P host polymer (H) alone without PEG shielding showed very high MFI values in both cell lines, indicating a very high nonspecific binding. H/G(PL20k), H/G(PC8k), H/G(PC12k), and H/G(PC20k) exhibited significantly lower MFI values than other carriers in both cell lines. Linear PEG with longer lengths showed a stronger shielding effect than those with shorter ones. As compared with linear PEG, comb-shaped PEG with similar molecular weight displayed greater shielding power. However, it is noted that longer PEG led to larger particle sizes (Fig. 3C), which may affect the pharmacokinetics of the PNPs (19). On the basis of the results, for linear Ad-PEG, PL20k showed the best effect on PNP shielding. For comb-shaped Ad-PEG, PC8k, PC12k, and PC20k showed the best effect on PNP shielding, whereas PC8k gave smaller particle size than PC12k and PC20k. Therefore, the linear PL20k and the comb-shaped PC8k were chosen for further screening and optimizing of the overall effects of nonspecific binding and ligand-induced siRNA targeted delivery.
Next, FA as the targeting ligand was conjugated to the distal end(s) of PL20k and PC8k. For PL20k, there was only one –OH end, so one FA ligand was conjugated to give PL20k-FA1 (PL20kF1) (fig. S1). For PC8k, there were multiple –OH ends, so 1, 3, 6, 9, and 12 FA ligands were conjugated, to give a series of PC8k-FAn (n = 1, 3, 6, 9, and 12) (PC8kFn) (Fig. 3B). The CD-SS-P host and the six guests (one PL20kF1 and five PC8kFn) were used to form six H/G self-assembled siRNA targeted delivery carriers, and FR+ KB cells were used to evaluate the siRNA-loaded H/G carriers for their specific binding to the FRs on the cells, to screen and find out the optimized combinations of the PEG architecture and the FA ligand density of the H/G carriers.
Binding affinity and dissociation kinetics are two key parameters to determine the specific binding of the H/G carriers to the surface of the KB cells (31, 33). The binding affinity of H/G targeted delivery carriers to KB cells was analyzed by comparing the specific binding and nonspecific binding. FITC-siRNA–loaded H/G carriers with FA ligand (specific binding) and corresponding H/G carriers without ligand (nonspecific binding) were incubated with KB cells respectively on ice for 1 hour, and the fluorescence intensity of the cells was measured after washing out the unbound PNPs. The difference of the fluorescence intensity of the cells treated by H/G with FA and without FA, which represents the net specific binging, was plotted against the siRNA concentration (Fig. 3E). Among six H/G targeted delivery carrier systems, the fluorescence of H/G(PC8kF6) increased much more than other carriers with an increase in siRNA concentration (i.e., PNP concentration), indicating that its PNPs had the strongest specific binding to KB cells. The measured EC50 (the concentration leading to 50% binding) for H/G(PC8kF6) was the lowest among all formulations. The EC50 value was approximately 10-fold lower than those of the monovalent ligand formulations H/G(PL20kF1) and H/G(PC8kF1).
For investigation of the dissociation kinetics, a dissociation binding experiment was conducted. After incubation of FITC-siRNA–loaded PNPs (2000 μmol/ml) in the KB cells for 4 hours, the cells were washed to allow the bound PNPs to dissociate from the surfaces of the cells, while the fluorescence intensity of the cells with remaining bound PNPs were measured and plotted against time (Fig. 3F). The curves showed that among six H/G targeted carriers, the longest dissociation time was needed for the H/G(PC8kF6) delivery system, which is in line with the result that the PNPs formed by H/G(PC8kF6) had the strongest specific binding to KB cells.
It was reported that FR exists as “receptor clusters” in the membrane of KB cells (20, 22, 34, 35). Our results showed that carriers with low FA densities failed to achieve a high binding avidity between the carriers and the cell surfaces. For example, H/G(PL20kF1) and H/G(PC8kF1) only had weak monovalent interaction with the receptor clusters. The binding avidity increased with the ligand density, because higher ligand density allowed multivalent interaction between the carriers and the receptor clusters. When FA density increased beyond optimal values, the binding avidity decreased. Overcrowding may possibly prevent ligands from optimal orientation and spatial arrangement for efficient binding (19). Also, competitions may occur between multiple ligands for a single receptor, which tends to limit the access of ligands to receptors. Several recent publications also shared similar observations with ours (19, 20, 30, 31).
The above results indicate that, for the specific case of KB cells, the intermediate FA ligand density together with the size and architecture of the comb-shaped PEG in the H/G(PC8kF6) carrier led to the optimal specific binding to the target KB cells, whereas the nonspecific binding to nontarget cells was efficiently minimized. It was also confirmed that the H/G(PC8kF6)-based PNPs showed undetectable cytotoxicity at an N/P ratio up to 50 (fig. S2A) and excellent capacity against protein adsorption (fig. S2B). Thus, the formulation based on H/G(PC8kF6) was identified to be the optimized H/G targeted delivery carrier for the following siRNA delivery and gene silencing studies.
Targeted delivery of siRNA to FR+ KB cells by H/G(PC8kF6)
To demonstrate whether the strong binding between the H/G(PC8kF6) carrier and KB cell could lead to efficient FR-mediated endocytosis and delivery of the payload siRNA, the cellular uptake of FITC-siRNA–loaded H/G(PC8kF6) by both KB (FR+) and A549 (FR−) cells were evaluated. First, to validate the use of KB and A549 cells, FR expression levels of the two cell lines were investigated by immunoblotting analysis (Fig. 4A). KB cells showed high expression of α-FR (KB, I), whereas α-FR expression for A549 cells was negligible (A549, I). Expression of α-FR in KB cells decreased after the cells were knocked down by Lipofectamine 2k/siRNA-α-FR (KB, II). Next, it was tested whether H/G(PC8kF6) could selectively deliver siRNA to FR+ KB cells. Both KB and A549 cells were incubated with FITC-siRNA–loaded PNPs for 4 hours and then stained with α-FR antibody. The flow cytometry results revealed that uptake of FITC-siRNA delivered by H/G(PC8kF6) was very efficient in FR+ KB cells where FR expression was also very high, but less efficient in FR− A549 cells where FR expression was also low (Fig. 4B). However, no selectivity was observed for FITC-siRNA delivery by nontargeted carrier H alone. Figure 4C further confirmed that the cellular uptake of H/G(PC8kF6)-based PNPs was directed by FR-mediated endocytosis. Depression of FR expression (line II) and competitive blocking of FR by free FA (line III) reduced the cellular uptake in KB cells, in comparison with KB cells without any pretreatment (line IV). Confocal microscopic observations further confirmed that the H/G(PC8kF6)-based PNPs selectively targeted and efficiently delivered FITC-siRNA to KB cells, but showed negligible nonspecific cellular update by A549 cells (Fig. 4D). However, the nontargeted PNPs formed by H alone delivered FITC-siRNA to both KB and A549 cells.
Fig. 4. Cellular uptake of PNPs formed by FITC-siRNA and H or H/G(PC8kF6) in FR+ KB and FR− A549 cells.

(A) Western blot analysis of FR (α-FR) expression in KB and A549 cells. I, Cells were not pretreated. II, FR receptors of the cells were silenced by pretreating cells with Lipofectamine 2k/siRNA-α-FR. The β-actin expression was used as a control. (B) Flow cytometry analysis showing the relationships between cellular uptake of FITC-siRNA–loaded PNPs and the expressions of FR on cells of KB and A549 cells. Cells were first treated with FITC-loaded PNPs for 4 hours and then fixed and stained using anti-rabbit α-FR as primary antibody and Alexa Fluor 546–labeled goat anti-rabbit antibody as secondary antibody. (C) Flow cytometric histograms showing the effect of FR receptor silencing and blocking of FR on cellular uptake of PNPs. I, Blank cells without pretreatment. II, The FR receptors of the cells were knocked down by pretreating cells with Lipofectamine 2k/siRNA-α-FR and then transfected with PNPs for 4 hours. III, Cells were first blocked with 600 μM free FA for 4 hours and then transfected with PNPs for 4 hours. IV, Cells directly transfected with PNPs for 4 hours without pretreatment. MFIs of KB and A549 under various conditions and treatment were quantitatively analyzed. Data were presented as means ± SD (n = 3). (D) Confocal laser scanning microscopic analysis of the cellular uptake of PNPs in FR+ KB cells and FR− A549 cells. Green, FITC-labeled siRNA. Blue, nucleus stained with DAPI. Scale bars, 20 μm.
GFP gene silencing by siRNA-loaded H/G(PC8kF6)
The silencing effect and targeted delivery of siRNA–green fluorescent protein (GFP) by H/G(PC8kF6) were evaluated in both KB-GFP (FR+) and A549-GFP (FR−) cells, which stably express enhanced green fluorescent protein (EGFP). Flow cytometry histograms showing the GFP expression levels and the GFP silencing efficiency of the cells after delivery of siRNA-GFP are presented in fig. S3 (A and B). The nonspecific delivery of siRNA-GFP by H alone reduced GFP protein expression in both FR+ KB-GFP and FR− A549-GFP cells. However, H/G(PC8kF6) specifically delivered siRNA-GFP to FR+ KB-GFP cells. When siRNA-GFP (500 pmol) was delivered by H/G(PC8kF6), up to 88% of GFP expression was silenced in FR+ KB-GFP cells, whereas only 10% of GFP expression was silenced in FR− A549-GFP cells. siRNA-GFP delivered by H/G(PC8kF6) also reduced mRNA levels selectively in FR+ KB-GFP cells (fig. S3C). The reduction in GFP mRNA indicates that siRNA delivered by H/G(PC8kF6) silenced target gene expression via mRNA degradation. The reduction in both GFP protein and GFP mRNA induced by H/G(PC8kF6)/siRNA-GFP in KB-GFP cells was in a dose-dependent manner. The results indicate that the H/G(PC8kF6)/siRNA-GFP PNPs only specifically silenced GFP expression in target cells and did not silence the gene expression in nontarget cells. In some cases, targeted and PEGylated vectors may not reach the activity of the uncoated positively charged vectors (36). However, in this study, silencing mediated by H/G(PC8kF6) was remarkably stronger than that mediated by the control polymer H in KB cells. As this silencing experiment was performed under serum protein conditions, the superb shielding property of H/G(PC8kF6) could maintain particle stability and active endocytosis, while high protein absorption of H led to particle aggregation, which compromised the cellular internalization.
Suppression of Bcl-2 gene expression by siRNA-loaded H/G(PC8kF6)
The siRNA targeted delivery by H/G(PC8kF6) carrier was further evaluated for delivering therapeutic Bcl-2 siRNA (siRNA-Bcl2). H, H/G(PC8k), and commercial product Lipofectamine were used as controls. Expression of Bcl-2 in the mRNA and protein levels and apoptosis of cells were investigated. The Bcl-2 gene is an important cell apoptosis regulator, which is overexpressed in cancer cells, preventing apoptosis of cancer cells (37). Inhibition of Bcl-2 expression is a strategy to recover apoptotic pathway of cancer cell. The Bcl-2 gene down-regulation was measured by both mRNA and protein levels at 48 hours after transfection. Figure 5A shows the relative Bcl-2 mRNA expression levels in cells analyzed by reverse transcription quantitative polymerase chain reaction (RT-qPCR). When siRNA-Bcl2 was transfected by the H/G(PC8kF6) carrier, the Bcl-2 mRNA expression was suppressed to 20% in KB cells but remained at a high level of 83% in A549, indicating that the H/G(PC8kF6) carrier selectively delivered siRNA to the target KB cells with high efficiency, while minimized the siRNA delivery to the nontarget A549 cells. When siRNA-Bcl2 was transfected by the nontargeted carriers H and Lipofectamine 2k (commercial reagent), both KB and A549 cells received Bcl-2 gene down-regulation to a medium level. The two nontargeted carriers delivered siRNA to both cells with medium efficiency. Similar down-regulation trends of Bcl-2 protein level were observed by Western blot (Fig. 5B). To further verify the downstream effects of the delivery of Bcl-2 siRNA, annexin V–FITC and propidium iodide (PI) costaining was applied to the cells, and flow cytometry analyses were conducted to quantify the cell apoptosis (Fig. 5C). Clearly, H/G(PC8kF6) treatment resulted in the highest level of cell apoptosis in FR+ KB cells among all the tested groups, while negligible apoptosis level was observed for H/G(PC8kF6)-treated FR− A549 cells (Fig. 5D). The results indicate that Bcl-2 siRNA delivered by H/G(PC8kF6) could specifically silence the Bcl-2 expression in FR+ KB cells and induce selective cell apoptosis while minimizing cell apoptosis in nontarget cells.
Fig. 5. Delivery of Bcl-2 siRNA.
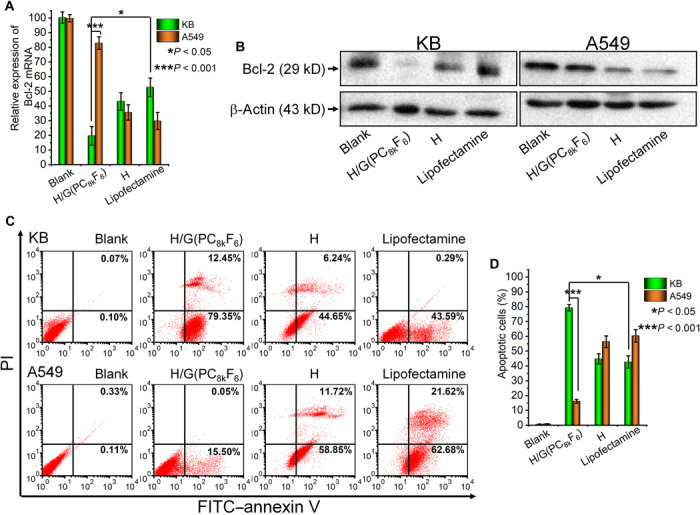
(A) Down-regulation of Bcl-2 mRNA levels as quantified by RT-qPCR (n = 3). The relative level of mRNA expression was calculated over the untreated control (blank). (B) Suppression of Bcl-2 protein expression levels as evaluated by Western blot. β-Actin was used as negative controls. (C) Representative scatter dot-plot of apoptotic cells obtained by FITC–annexin V/PI costaining assay in flow cytometry experiments. (D) Quantitative analysis of the percentage of apoptotic cells using annexin V–FITC/PI costaining assay (n = 3) by flow cytometry.
In vivo therapeutic efficacy of siRNA-Bcl2 targeted delivery in KB xenograft tumor model
To further prove the optimized H/G(PC8kF6) carrier could function efficiently in vivo as a shielding and targeted delivery system, the siRNA-Bcl2–loaded H/G(PC8kF6) PNPs were evaluated in a mouse KB xenograft tumor model. Phosphate-buffered saline (PBS), H, and H/G(PC8k) treatment groups were also tested as controls. Anticancer efficacy and safety of PNPs were evaluated by tumor volume changes, body weight change, survival rate, hematological and urinal test, tumor histological analysis, and immunofluorescence of Bcl-2. Male BALB/c nude mice were inoculated with KB cells in the right flank. When the tumors reached 50 to 100 mm3 on day 10 after the inoculation, PNPs loaded with siRNA-Bcl2 at a dose of 1000 pmol/kg were injected via the tail vein every 2 days until day 20 after the first injection (Fig. 6A). Tumor growth and body weight of the mice were measured every 2 days. A pronounced inhibition in tumor growth was observed for the H/G(PC8kF6) group (Fig. 6B). From days 4 to 20, the tumors for the control groups increased for 9- to 22-fold in volume, whereas the growth of tumors for the H/G(PC8kF6) group was significantly suppressed. The tumor volume of the H/G(PC8k) group was suppressed to ~0.14-fold of PBS group, which is more efficient than other Bcl-2–siRNA targeted delivery systems reported (38). It must be noted that the antitumor capacity of H/G(PC8k) was also significantly improved although it was not as good as H/G(PC8kF6), probably due to the stability of particles, which would lead to long in vivo circulation and kill cancer via enhanced permeability and retention (EPR) effect. It was beneficial from the optimized length and architecture of PEG as corona of polyplexes. In addition to long circulation and passive targeting, active targeting of H/G(PC8kF6) through receptor-ligand interaction further improved the anticancer efficiency by tumor retention. The body weight was steady or slightly increased for the H/G(PC8kF6) group, showing that the PNPs were safe, while other groups also maintained the body weight during the 21-day treatment period (Fig. 6C). Images of the KB xenograft tumors of the mice at days 0 and 10 and day 20 (or experimental end point) are shown in fig. S4. Figure 6D shows the images of the tumors from various treatment groups at the experimental end point. A 100% survival rate up to 21 days after treatment was observed for the KB tumor–bearing mice treated with PNPs of H/G(PC8kF6), as compared with 35 to 85% survival rates for those treated by other control formulations (Fig. 6E). The ex vivo histological analyses further confirmed that the formulations showed no acute toxicity in the liver, kidney, heart, spleen, and lung (fig. S5). The hematology results indicate that all measured factors were within normal ranges (39), suggesting that no inflammatory reaction was associated with the treatments for all groups, and no liver injury was found (fig. S6A). Urine samples were examined and confirmed that kidney functions were not affected by the PNP treatment (fig. S6B).
Fig. 6. Antitumor efficacy of siRNA-Bcl2–loaded H/G(PC8kF6) PNPs against KB tumors and whole-body and ex vivo fluorescence imaging of KB tumor–bearing mice injected with PNPs loaded with Cy5-siRNA.

(A) Timeline of treatment of BALB/c nude mice. Ten days after subcutaneous inoculation of KB cells, male BALB/c nude mice were injected with PNPs loaded with siRNA-Bcl2 via tail vein injection every 2 days. All survival mice were euthanized at day 21. (B) The KB tumor growth curves of mice treated with PNPs loaded with siRNA-Bcl2 (dosage, 1000 pmol/kg) based on H/G(PC8kF6) and other control groups [PBS and siRNA-Bcl2–loaded PNPs based on PEI 25k, H, and H/G(PC8k)]. One injection every 2 days until day 20 (6 mice per group). Data are presented as means ± SD; six mice per group. Data of the H/G(PC8kF6) and H/G(PC8k) groups were compared using one-tailed t test. *P < 0.05, **P < 0.01, and ***P < 0.001. (C) Body weight change of KB tumor–bearing mice during 21-day anticancer treatment. Body weight percentage (%) = Body weight (Dayn)/Body weight (Day0) × 100. Data of the H/G(PC8kF6) and H/G(PC8k) groups were compared using one-tailed t test. *P < 0.05, **P < 0.01. (D) Images of the tumors harvested from the mice at day 21 after the initial treatment (or at the end point of the experiment). (E) Survival curves of KB tumor–bearing mice in response to treatment via tail vein injection with PNPs loaded with siRNA-Bcl2. (F) In vivo fluorescence imaging of KB tumor–bearing mice assessing the biodistribution, tumorous targeting, and retention of PNPs. Mice were imaged at different time points (2, 8, 24, and 36 hours) using IVIS live imaging system. Red circles indicate tumor areas. (G) Fluorescence intensity of Cy5-siRNA in tumor area at various time points. The tumor accumulation was calculated based on the quantification of Cy5 intensity in red circles. Photo credit: H.B., Zhejiang University.
Hematoxylin and eosin (H&E) examination of the tumor sections revealed that the tumors treated with siRNA-Bcl2–loaded H/G(PC8kF6) had extensive granulation and showed evidence of necrosis (fig. S7A, upper panel). TUNEL (terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling) staining showed extensive apoptosis in the tumors of the H/G(PC8kF6) group (fig. S7B, lower panel). In addition, immunofluorescence staining for Bcl-2 showed that green fluorescence signals of the H/G(PC8kF6) group was much weaker than those of the other control groups (fig. S7B). The results indicate that the H/G(PC8kF6) group significantly decreased Bcl-2 expression, which benefited the apoptosis of tumor cells.
Whole-body and ex vivo fluorescence imaging
Qualitative nanoparticle biodistribution was studied to evaluate the tumorous targeting and retention of PNPs of H/G(PC8kF6) loaded with Cy5-labeled siRNA (Cy5-siRNA). PBS, H, and H/G(PC8k) were used as control groups. Whole-body fluorescence distribution and organ distribution (tumor, liver, kidney, heart, and lung) of nanoparticles at various time points were evaluated. KB tumor–bearing mice were intravenously injected with the PNPs via the tail vein at a single dose of 1000 pmol/kg of Cy5-siRNA, followed by imaging at different time points through measuring fluorescence signal to capture the whole-body distribution pattern (Fig. 6F). The fluorescence signals of siRNA in the H/G(PC8kF6) group started to accumulate early at 2 hours in the tumor area (red circle) and showed the strongest signal at 24 hours (Fig. 6F). The MFI of Cy5-siRNA in the tumor area for the H/G(PC8kF6) group was significantly higher than those of all other control groups at 2, 8, and 24 hours (fig. S7C). The results indicate that H/G(PC8kF6) PNPs exhibited prolonged circulation and tumor accumulation properties. Because of the limited penetration depth of the light source through the tissue in the whole-body imaging, a different set of mice were euthanized, and their tumors and major organs (liver, kidney, heart, and lung) were collected for ex vivo imaging (Fig. 6G). In fig. S7 (D to G), quantitative fluorescence intensity analyses confirmed that the H/G(PC8k) and H/G(PC8kF6) groups, both with appropriate PEG shielding, showed much lower accumulation in the liver and kidney at the first 24 hours, as compared with the polyethylenimine (PEI) and H groups (both without PEG shell). The excised tumors showed a stronger signal at 2, 8, and 24 hours after injection for H/G(PC8kF6) PNPs, as compared with all the other control groups, indicating that the optimized PC8kF6 shell could enhance the tumor accumulation and retention of the PNPs. It is noted that H/G(PC8kF6) also accumulated in the lung at the 8-hour time point. It was probably due to the lung tissue also expressing some level of FRs (40). Both the therapeutic efficacy and biodistribution results strongly support that H/G(PC8kF6) exhibited long blood circulation and efficient tumor accumulation properties and could successfully deliver siRNA-Bcl2 to tumor cells to silence Bcl-2 protein expression and induce tumor cell apoptosis, resulting in efficient tumor growth suppression.
CONCLUSIONS
We have successfully demonstrated a supramolecular platform that can precisely control, screen, and optimize the molecular architectures of siRNA targeted delivery vehicles, which is developed on the basis of host-guest complexation between a β-CD–based cationic host polymer and an Ad-functionalized PEG guest polymer. The β-CD–Ad host-guest complexation integrates the host polymer and the guest polymer, while the host polymer is responsible to condense and load siRNA and to unload siRNA in the cytoplasm in the target tumor cells, and the guest polymer is responsible to shield the vehicles from nonspecific cellular uptake, to prolong the circulation time, and to actively target the tumor cells. A library of guest polymers with PEG of different shapes and sizes and various density of ligands allows for simple “assembly” of a series of siRNA vehicles with precisely controlled molecular architectures and for a rapid in vitro screening out of the optimized siRNA delivery vehicle, which is validated in vivo for efficient targeted delivery of therapeutic siRNA-Bcl2 into tumor cell in mice animal model, to efficiently suppress the growth of the tumor. The platform can be further adapted and applied for delivery of microRNA (miRNA) and DNA and other therapeutics, such as small-molecule anticancer drugs and anti-inflammatory drugs, or for codelivery of genes and drugs. Moreover, if a reasonably large library of host and guest polymers is developed, then the in vitro screening can be carried out using patient-derived cancer cells, and the optimized delivery vehicles can be a unique formulation for efficient patient-specific cancer treatment. Therefore, the strategy may be promising for potential personalized and precision cancer therapy applications.
MATERIALS AND METHODS
Polymer synthesis
Star-shaped pDMAEMA with β-CD core and disulfide linkers (CD-SS-P, host) as host polymer was synthesized using copper (I)–mediated atom transfer radical polymerization (ATRP) based on our previous report (25). Two types of Ad-terminated PEG with various molecular weights as guest polymers, linear PEG (PL) and comb-shaped p(PEGMA) (PC), were synthesized. The synthesis methods are described in the Supplementary Materials. Folate was conjugated to Ad-PEG and Ad-p(PEGMA) ends using carbodiimide chemistry, which is described in the Supplementary Materials in detail.
Polyplex formation
Polyplexes for cell binding, cellular uptake, knockdown, and endocytosis pathway investigations were prepared as follows, unless otherwise indicated: 20 μl of siRNA ore FITC-labeled siRNA (concentration, 20 μmol/liter) and the calculated amount of polymer at an N/P ratio of 20 in PBS were mixed by vortexing and incubated for 30 min at room temperature to form siRNA-loaded nanoparticles.
Particle size and zeta potential measurements
The size and zeta potential of siRNA-loaded PNPs were measured by a Malvern Zetasizer Nano ZS. PNPs containing 40 pmol of siRNA were prepared in 1 ml of PBS before measurement.
Cell line and cell cultures
FR-overexpressed human nasopharynx carcinoma cells (KB, FR positive, FR+) [American Type Culture Collection (ATCC)] and FR-deficient human adenocarcinomic cells (A549, FR negative, FR−) (ATCC) were used in our experiments. KB-GFP and A549-GFP, which stably express GFP, were constructed by Cell Biolabs Inc. RPMI 1640 medium, no FA, was from Life Technologies, supplemented with 10% fetal bovine serum (FBS). All cells were cultured in the folic-free media for at least 7 days before use. The expression of FRs in KB and A549 cells was determined by Western blot using α-FR goat polyclonal immunoglobulin G (IgG) (Santa Cruz Biotechnology) as the primary antibody and horseradish peroxidase (HRP)–goat anti-rabbit (Life Technologies) as the secondary antibody.
Determination of specific cell binding
For determination of the binding avidity of the targeted delivery systems, saturation binding experiment was performed (31, 33). FITC-siRNA–loaded H/G(PL20kFA1) and H/G(PC8kFAn) carriers at N/P ratios of 20 with different siRNA concentrations (from 10 to 250 μmol/liter) were incubated with FR+ KB cells on ice for 1 hour. Following incubations, cells were washed with cold PBS and collected for FITC fluorescence analysis. In the meantime, the nonspecific bindings of the corresponding nontargeted delivery systems were measured using FITC-siRNA–loaded H/G(PL20k) and H/G(PC8k) carriers without the targeting FA ligands, at N/P ratios of 20 with different siRNA concentrations (from 0.5 to 2000 μmol/liter). The degree of specific binding of the targeted delivery systems was determined by analyzing the total and nonspecific binding.
For determination of dissociation kinetics, a dissociation binding experiment was conducted. Generally, FR+ KB cells were first incubated with a high concentration of FITC-siRNA–loaded targeted PNPs (concentration of siRNA was 2000 μmol/liter) on ice for 4 hours to allow equilibrium. After that, cells were collected by centrifugation and resuspended in cold PBS. These cell suspensions were constantly stirred, spun down, washed, and tested at indicated time points to determine remaining bound FITC-siRNA–loaded PNPs on cells. The fluorescence intensities were measured by a spectrofluorometer (Tecan, Infinite 200 PRO) using FITC settings.
Cellular uptake
FR+ KB or FR− A549 cells were seeded at a density of 8 × 104 cells per well in 24-well plates. Cell culture medium was replaced with fresh FA free medium after cell confluence reached 70%. Transfection reagents containing 20 pmol of FITC-labeled siRNA (N/P ratio of 20) were added to each well, followed by incubation at 37°C for 2 hours. Subsequently, the cells were then trypsinized, collected, and fixed with 4% paraformaldehyde (PFA) in PBS and then flow cytometric analysis was performed (Beckman Coulter). Cellular uptake was analyzed by detection of the emission of FITC at 530 nm with excitation at 495 nm. FCS Express 4 Flow Research Edition software was used to analyze the results and obtain MFI values. For competitive inhibition of cellular uptake by free FAs, cells were preincubated with 300 μM FAs for 1 hour, and the transfection medium also contained 300 μM FAs. For the cellular uptake after the α-FR proteins were down-regulated, both KB and A549 cells were first transfected by Lipofectamine 2000/siRNA–α-FR complexes, and cellular uptake measurements (same procedures as above) were conducted 24 hours after FR knock down. Western blots of whole-cell lysates were performed to verify the siRNA-mediated silencing of α-FR.
For confocal microscopic observations, both KB and A549 cells were grown on glass coverslips to ~50 to 70% confluency. Cells were incubated with FITC-siRNA–loaded polyplexes for 2 hours at 37°C, and thereafter fixed with 4% of PFA for 30 min. The cells were then washed, and the nucleus was stained with DAPI (4′,6-diamidino-2-phenylindole) (Sigma-Aldrich) at a concentration of 2 μg/ml for 2 min. Cellular uptake was analyzed by detection of the emission of FITC at 530 nm with excitation at 485 nm.
Western blotting
After transfection with siRNA-Bcl2–loaded PNP, cells were lysed with 50 μl of RIPA (radioimmunoprecipitation assay) lysis buffer (Thermo Fisher Scientific) containing protease inhibitor. Samples were mixed with Western blot loading buffer and β-mercaptoethanol and boiled for 5 min at 95°C. The proteins were separated on a 12% SDS–polyacrylamide gel electrophoresis gel. The proteins were transferred to a 0.2-μm polyvinylidene difluoride membrane by wet transfer cell (Bio-Rad). After electrophoresis, the membranes were blocked for 30 min with PBS-Tween containing 5% bovine serum albumin at room temperature. The Bcl-2 protein was detected by incubating the membranes with the primary antibody, an anti-rabbit Bcl-2 (1:500; Santa Cruz), for 1 hour at room temperature, followed with HRP-labeled goat anti-rabbit IgG (1:5000 dilution; Life Technology) as the secondary antibody for 1 hour at room temperature. Proteins were detected using enhanced chemiluminescence (Pierce). Anti-β-actin (Cell Signal) was used as a control for the protein loading.
Cell apoptosis analysis
Apoptosis was analyzed by flow cytometry with the FITC Annexin V/Dead Cell Apoptosis Kit (Invitrogen) according to the manufacturer’s protocol. Briefly, KB or A549 cells (8 × 104 per well) were seeded in 24-well plates and collected 24 hours after transfection. The cells were trypsinized, washed with cold PBS, and resuspended in 1× binding buffer (100 μl). To measure the apoptotic cell number, the cells were double stained with PI and annexin V solution, incubated for 15 min on ice, and resuspended in 1× PBS buffer (200 μl). The samples (10,000 cells for each sample) were then analyzed on a FACSCalibur flow cytometry with an analysis software (Beckman Coulter).
Intracellular gene silencing efficiency
The siRNA-induced silencing abilities of carriers were evaluated in both an FR-overexpressing KB-GFP cell line and an FR-deficient A549-GFP cell line. Generally, 150,000 KB-GFP cells or A549-GFP cells were seeded into six-well plates. When 50 to 70% confluence was reached, the cell culture media were replaced with fresh serum-free transfection media containing siRNA-GFP (5′-AAGUGGUAGAAGCCGUAGCdTdT-3′ antisense, Integrated DNA Technologies)–loaded PNPs at various doses of siRNA per well. After 4-hour incubations, the media were replaced with fresh complete cell growth media, and the GFP expression of cells was detected 24 hours after transfection. Flow cytometry was performed on a FACSCalibur (Beckman Coulter) to determine the GFP expression of every sample. The negative siRNA (containing a scrambled sequence that will not lead to the specific degradation of any known cellular mRNA) (antisense sequence: rArCrGrUrGrArCrArCrGrUrUrCrGrGrArGrArATT; sense sequence: rUrUrCrUrCrCrGrArArCrGrUrGrUrCrArCrGrUTT; Integrated DNA Technologies) were also performed as negative control.
To detect the mRNA transcription level of GFP after silencing, semiquantitative RT-PCR was performed. Transfection procedures were the same as described above. Twenty-four hours after knockdown, total RNA extractions were carried out with the RNeasy Mini Kit (Qiagen). First-strand cDNA was synthesized using 0.5 mg of total RNA, SuperScript reverse transcriptase, and oligo(dT) primer (Invitrogen). RT-PCR was performed with 30 cycles. The intensities of PCR-generated fragments were analyzed by the GBOX gel doc system (Syngene).
In vitro delivery of Bcl-2 siRNA
Both KB and A549 cells were transfected with PNPs containing Bcl-2 siRNA (sense: rGrUrArCrArUrCrCrArUrUrArUrArArGrCrUrGTT; antisense: rCrArGrCrUrUrArUrArArUrGrGrArUrGrUrArCTT; Integrated DNA Technologies). The transfection procedures were same as that of siRNA-GFP transfection. To measure the down-regulation effect of siRNA-Bcl2 transfection, quantitative RT-qPCR was performed. The Bcl-2 protein expression level after silencing was also determined by Western blotting. The downstream effect of silencing cell apoptosis was analyzed by FITC Annexin V/Dead Cell Apoptosis Kit (Invitrogen) by flow cytometry.
RT-qPCR analysis
At 48 hours after transfection, total RNA was extracted from the cells via Qiagen RNEasy Mini Kit. Subsequently, the RNA was transcribed reversely with oligo(dT) (Invitrogen) based on the manufacturer’s instruction. qPCR was conducted on 1.0 μl of cDNA with the SYBR Green PCR Kit (Qiagen) following the protocol provided by the manufacturer. The Bcl-2 primers used for the qPCR experiments were as follows: forward, 5′-TGTCCCTTTGACCTTGTTTCT-3′; reverse, 5′-TCATTTGCCATCTGGATTTT-3′. The actin primers used were as follows: forward, 5′-TACCTCATGAAGATCCTCACC-3′; reverse, 5′-TTTCGTGGATGCCACAGGAC-3′. The amplification conditions were as follows: denaturation at 95°C for 30 s for 40 cycles, annealing at 60°C for 20 s, and extension at 72°C for 20 s with an iCycler (Bio-Rad).
In vivo delivery of Bcl-2 siRNA
Animal care and handing procedures were in accordance with the guidelines approved by the ethics committee of Zhejiang University. All animal experimental protocols were approved by the Zhejiang University Animal Care and Use Committee (ZJU20190018). KB cells were purchased from ATCC and have passed the conventional tests of cell line quality control methods (e.g., morphology, isoenzymes, and mycoplasma) and were cultured in Dulbecco’s minimum essential medium containing 10% FBS at 37°C. Male BALB/c nude mice (6 to 8 weeks old, ~20 g body weight) were purchased from Zhejiang Academy of Medical Sciences and maintained in a pathogen-free environment. In vivo therapeutic efficacy of siRNA-Bcl2–loaded H/G(PC8kF6) PNPs was investigated in the KB xenograft tumor model. Male nude mice were injected subcutaneously in the right flank region with 200 μl of cell suspension containing 2 × 106 KB cells. When tumors reached 50 to 100 mm3 (~10th day after injection), mice were randomly divided into five groups with six mice in each group. PBS (100 μl) and various nanoparticles [PEI 25k, H, H/G(PC8k), and H/G(PC8kF6)] loaded with siRNA-Bcl2 (1000 pmol/kg) were injected via the tail vein every 2 days (d0, d2, d4, d6, d8, d10, d12, d14, d16, d18, and d20). Tumor growth and body weight of mice were measured every 2 days. Tumor size was calculated using the formula (tumor length) × (tumor width)2/2. The harvested tumor specimens and major organs (liver, kidney, heart, and spleen) were fixed in 4% neutral buffered PFA and embedded in paraffin. The paraffin-embedded tumors and organs were further cut into slices at a thickness of 10 μm for the H&E staining. The paraffin-embedded tumors were further sliced for TUNEL staining and immunofluorescence staining of Bcl-2 protein. Blood was drawn from mice, and urine sample was collected before sacrifice.
In vivo biodistribution
To investigate the biodistribution of H/G(PC8kF6) PNPs, KB tumor–bearing mice (tumor volume of 200 to 300 mm3) were anesthetized, and the formulations with Cy5-labeled siRNA (1000 pmol/kg, total volume of 100 μl) (GenePharma, China) were administrated via tail vein injection. To monitor the distribution of PNPs, in vivo imaging was conducted on IVIS spectrum (Caliper Life Sciences, US) at 2, 8, 24, and 36 hours after injection (n = 2). Afterward, the mice were euthanized, and the tumors and major organs (heart, lung, liver, and kidney) were collected for ex vivo imaging. The radiant efficiency of the tissues was calculated using Image 3.1 software.
Statistical analysis
Statistical analysis was performed using variance tests [analysis of variance (ANOVA)]. Data sets were compared using two-tailed, unpaired t test. P < 0.05 was regarded as statistically significant.
Supplementary Material
Acknowledgments
Funding: This work was supported by Singapore’s Ministry of Education Academic Research Fund nos. R-397-000-188-112, R-397-000-267-114, and R-397-000-296-114. Author contributions: Y.W. and J.L. conceived the study. Y.W., H.B., G.T., and J.L. designed the experiments. Y.W., J.Z., and X.S. performed the experiments. H.B. and G.T. conducted the animal model experiments. Y.W. and J.L. wrote the manuscript. J.L. supervised the project. All authors discussed the results and commented on the manuscript. Y.W. and H.B. contributed equally to this work. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/31/eabc2148/DC1
REFERENCES AND NOTES
- 1.Kim D. H., Rossi J. J., Strategies for silencing human disease using RNA interference. Nat. Rev. Genet. 8, 173–184 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Kim H. J., Kim A., Miyata K., Kataoka K., Recent progress in development of siRNA delivery vehicles for cancer therapy. Adv. Drug Deliv. Rev. 104, 61–77 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Stevenson M., Therapeutic potential of RNA interference. N. Engl. J. Med. 351, 1772–1777 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Blanco E., Shen H., Ferrari M., Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 33, 941–951 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aliabadi H. M., Landry B., Sun C., Tang T., Uludağ H., Supramolecular assemblies in functional siRNA delivery: Where do we stand? Biomaterials 33, 2546–2569 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Ulkoski D., Bak A., Wilson J. T., Krishnamurthy V. R., Recent advances in polymeric materials for the delivery of RNA therapeutics. Expert Opin. Drug Deliv. 16, 1149–1167 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Whitehead K. A., Langer R., Anderson D. G., Knocking down barriers: Advances in siRNA delivery. Nat. Rev. Drug Discov. 8, 129–138 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tatiparti K., Sau S., Kashaw S. K., Iyer A. K., siRNA delivery strategies: A comprehensive review of recent developments. Nanomaterials (Basel) 7, 77 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yeung T., Gilbert G. E., Shi J., Silvius J., Kapus A., Grinstein S., Membrane phosphatidylserine regulates surface charge and protein localization. Science 319, 210–213 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Miteva M., Kirkbride K. C., Kilchrist K. V., Werfel T. A., Li H., Nelson C. E., Gupta M. K., Giorgio T. D., Duvall C. L., Tuning PEGylation of mixed micelles to overcome intracellular and systemic siRNA delivery barriers. Biomaterials 38, 97–107 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fang Y., Xue J., Gao S., Lu A., Yang D., Jiang H., He Y., Shi K., Cleavable PEGylation: A strategy for overcoming the "PEG dilemma" in efficient drug delivery. Drug Deliv. 24, 22–32 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ozer I., Tomak A., Zareie H. M., Baran Y., Bulmus V., Effect of molecular architecture on cell interactions and stealth properties of PEG. Biomacromolecules 18, 2699–2710 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Ashley C. E., Carnes E. C., Philips G. K., Padilla D., Durfee P. N., Brown P. A., Hanna T. N., Liu J., Phillips B., Carter M. B., Carroll N. J., Jiang X., Dunphy D. R., Willman C. L., Petsev D. N., Evans D. G., Parikh A. N., Chackerian B., Wharton W., Peabody D. S., Brinker C. J., The targeted delivery of multicomponent cargos to cancer cells by nanoporous particle-supported lipid bilayers. Nat. Mater. 10, 389–397 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song E., Zhu P., Lee S.-K., Chowdhury D., Kussman S., Dykxhoorn D. M., Feng Y., Palliser D., Weiner D. B., Shankar P., Marasco W. A., Lieberman J., Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat. Biotechnol. 23, 709–717 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Mickler F. M., Vachutinsky Y., Oba M., Miyata K., Nishiyama N., Kataoka K., Bräuchle C., Ruthardt N., Effect of integrin targeting and PEG shielding on polyplex micelle internalization studied by live-cell imaging. J. Control. Release 156, 364–373 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Sasayama Y., Hasegawa M., Taguchi E., Kubota K., Kuboyama T., Naoi T., Yabuuchi H., Shimai N., Asano M., Tokunaga A., Ishii T., Enokizono J., In vivo activation of PEGylated long circulating lipid nanoparticle to achieve efficient siRNA delivery and target gene knock down in solid tumors. J. Control. Release 311-312, 245–256 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Lueckheide M., Vieregg J. R., Bologna A. J., Leon L., Tirrell M. V., Structure–property relationships of oligonucleotide polyelectrolyte complex micelles. Nano Lett. 18, 7111–7117 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Angell C., Xie S., Zhang L., Chen Y., DNA nanotechnology for precise control over drug delivery and gene therapy. Small 12, 1117–1132 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Elias D. R., Poloukhtine A., Popik V., Tsourkas A., Effect of ligand density, receptor density, and nanoparticle size on cell targeting. Nanomedicine 9, 194–201 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song X., Li R., Deng H., Li Y., Cui Y., Zhang H., Dai W., He B., Zheng Y., Wang X., Zhang Q., Receptor mediated transcytosis in biological barrier: The influence of receptor character and their ligand density on the transmembrane pathway of active-targeting nanocarriers. Biomaterials 180, 78–90 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Bray D., Levin M. D., Morton-Firth C. J., Receptor clustering as a cellular mechanism to control sensitivity. Nature 393, 85–88 (1998). [DOI] [PubMed] [Google Scholar]
- 22.Smart E. J., Mineo C., Anderson R. G. W., Clustered folate receptors deliver 5-methyltetrahydrofolate to cytoplasm of MA104 cells. J. Cell Biol. 134, 1169–1177 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song X., Zhu J.-l., Wen Y., Zhao F., Zhang Z., Li J., Thermoresponsive supramolecular micellar drug delivery system based on star-linear pseudo-block polymer consisting of β-cyclodextrin-poly(N-isopropylacrylamide) and adamantyl-poly(ethylene glycol). J. Colloid Interface Sci. 490, 372–379 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Zhu J.-l., Liu K. L., Wen Y., Song X., Li J., Host–guest interaction induced supramolecular amphiphilic star architecture and uniform nanovesicle formation for anticancer drug delivery. Nanoscale 8, 1332–1337 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Wen Y., Zhang Z., Li J., Highly efficient multifunctional supramolecular gene carrier system self-assembled from redox-sensitive and zwitterionic polymer blocks. Adv. Funct. Mater. 24, 3874–3884 (2014). [Google Scholar]
- 26.Zhao F., Yin H., Li J., Supramolecular self-assembly forming a multifunctional synergistic system for targeted co-delivery of gene and drug. Biomaterials 35, 1050–1062 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Harries D., Rau D. C., Parsegian V. A., Solutes probe hydration in specific association of cyclodextrin and adamantane. J. Am. Chem. Soc. 127, 2184–2190 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Chen C., Ke J., Zhou X. E., Yi W., Brunzelle J. S., Li J., Yong E.-L., Xu H. E., Melcher K., Structural basis for molecular recognition of folic acid by folate receptors. Nature 500, 486–489 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kelemen L. E., The role of folate receptor α in cancer development, progression and treatment: Cause, consequence or innocent bystander? Int. J. Cancer 119, 243–250 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Hak S., Helgesen E., Hektoen H. L., Huuse E. M., Jarzyna P. A., Mulder W. J. M., Haraldseth O., de Lange Davies C., The effect of nanoparticle polyethylene glycol surface density on ligand-directed tumor targeting studied in vivo by dual modality imaging. ACS Nano 6, 5648–5658 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poon Z., Chen S., Engler A. C., Lee H.-i., Atas E., von Maltzahn G., Bhatia S. N., Hammond P. T., Ligand-clustered "patchy" nanoparticles for modulated cellular uptake and in vivo tumor targeting. Angew Chem. Int. Ed. Engl. 49, 7266–7270 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He C., Hu Y., Yin L., Tang C., Yin C., Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials 31, 3657–3666 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Hong S., Leroueil P. R., Majoros I. J., Orr B. G., Baker J. R. Jr., Banaszak Holl M. M., The binding avidity of a nanoparticle-based multivalent targeted drug delivery platform. Chem. Biol. 14, 107–115 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Rothberg K. G., Ying Y. S., Kolhouse J. F., Kamen B. A., Anderson R. G. W., The glycophospholipid-linked folate receptor internalizes folate without entering the clathrin-coated pit endocytic pathway. J. Cell Biol. 110, 637–649 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Merzel R. L., Frey C., Chen J., Garn R., van Dongen M., Dougherty C. A., Kandaluru A. K., Low P. S., Marsh E. N. G., Banaszak Holl M. M., Conjugation dependent interaction of folic acid with folate binding protein. Bioconjug. Chem. 28, 2350–2360 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Meyer M., Wagner E., pH-responsive shielding of non-viral gene vectors. Expert Opin. Drug Deliv. 3, 563–571 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Shen M., Gong F., Pang P., Zhu K., Meng X., Wu C., Wang J., Shan H., Shuai X., An MRI-visible non-viral vector for targeted Bcl-2 siRNA delivery to neuroblastoma. Int. J. Nanomedicine 7, 3319–3332 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun W., Liu X.-Y., Ma L.-L., Lu Z.-L., Tumor targeting gene vector for visual tracking of Bcl-2 siRNA transfection and anti-tumor therapy. ACS Appl. Mater. Interfaces 12, 10193–10201 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Zaias J., Mineau M., Cray C., Yoon D., Altman N. H., Reference values for serum proteins of common laboratory rodent strains. J. Am. Assoc. Lab. Anim. Sci. 48, 387–390 (2009). [PMC free article] [PubMed] [Google Scholar]
- 40.Parker N., Turk M. J., Westrick E., Lewis J. D., Low P. S., Leamon C. P., Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay. Anal. Biochem. 338, 284–293 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Zhang Z.-X., Liu K. L., Li J., A thermoresponsive hydrogel formed from a star-star supramolecular architecture. Angew. Chem. Int. Ed. Engl. 52, 6180–6184 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/31/eabc2148/DC1