

RACK7 recognizes H3.3G34R to suppress expression of MHC II complex components and their delivery pathway in pediatric GBM.
Abstract
Histone H3 point mutations have been identified in incurable pediatric brain cancers, but the mechanisms through which these mutations drive tumorigenesis are incompletely understood. Here, we provide evidence that RACK7 (ZMYND8) recognizes the histone H3.3 patient mutation (H3.3G34R) in vitro and in vivo. We show that RACK7 binding to H3.3G34R suppresses transcription of CIITA, which is the master regulator of MHC (major histocompatibility complex) class II molecules and genes involved in vesicular transport of MHC class II molecules to the cell surface, resulting in suppression of MHC class II molecule expression and transport. CRISPR-based knock-in correction of the H3.3G34R mutation in human pediatric glioblastoma (pGBM) cells significantly reduces overall RACK7 chromatin binding and derepresses the same set of genes as does knocking out RACK7 in the H3.3G34R pGBM cells. By demonstrating that H3.3G34R and RACK7 work together, our findings suggest a potential molecular mechanism by which H3.3G34R promotes cancer.
INTRODUCTION
Histone point mutations have been identified as possible driver mutations in pediatric gliomas, chondroblastomas, and giant cell tumors of bone (1–3). Specifically, mutation of lysine-27 (K) to methionine (M) on histone H3.3 or H3.1 is associated with pediatric diffuse intrinsic pontine gliomas (DIPGs) and glioblastomas (GBMs) arising in midline structures, while glycine–34 (G) to arginine (R) or valine (V) mutation on histone H3.3 are linked to GBM arising in the cerebral cortex of older adolescents and young adults (2, 3). Histone mutations have also been linked to giant cell tumor of bone and chondroblastoma (1). Besides histone H3, mutations were also identified in all four core histones in human cancers recently (4).
Overexpression of an H3K27M transgene causes a global decrease in H3K27me3, possibly by inhibiting the enzymatic activity of the Polycomb repressive complex 2 (PRC2) through an interaction with the catalytic subunit, EZH2 (5, 6). The K27M mutation cooperates with activated PDGFRA (platelet-derived growth factor receptor a), combined with p53 loss, to induce gliomagenesis (7–9). Another histone mutation, H3.3K36M, reduces H3K36 methylation, and recombinant H3.3K36M-containing nucleosomes inhibit the enzymatic activities of NSD2 and SETD2 (10, 11). The differentially expressed genes associated with H3K36 di- and trimethylation in the H3.3K36M cells are enriched in cancer pathways (11). The H3.3G34R/V transgenes have also been shown to reduce H3K36me3 levels on the same and nearby nucleosomes by inhibiting the activity of the H3K36 trimethyltransferase, SETD2 (5), but whether H3.3G34R/V is consistently associated with a reduced H3K36 methylation level in tumor samples remains unclear (2, 5). Unlike H3K27M, the role of H3.3G34R in cancer is less clear. For instance, expression of H3.3G34R in p53-deficient neural progenitor cells has no impact on proliferation when compared with H3.3K27M-expressing cells (9). By and large, the mechanisms by which the G34R mutation regulates chromatin and tumorigenesis are incompletely understood.
RACK7 (ZMYND8) was previously identified as a reader of the dual histone mark, H3K4me1/H3K14ac (12), and is related to BS69 (ZMYND11) that binds H3.3K36me3 (13, 14). RACK7 has been shown to repress transcription of metastasis-linked genes and has been proposed to have an antimetastasis function (12). RACK7 has also been shown to bind to active enhancers, where it suppresses overactivation of enhancers, which drive expression of genes involved in tumorigenesis (15). Here, we provide both in vitro and in vivo evidence demonstrating that RACK7 recognizes the H3.3G34R mutation. Correction of the H3.3G34R mutation and knocking out RACK7 in human pediatric H3.3G34R-containing pGBM cells both lead to up-regulation of MHC (major histocompatibility complex) class II immune response genes and vesicle pathway genes that facilitate transportation of MHC class II molecules, suggesting that G34R and RACK7 function together in cells, possibly to regulate MHC class II immune response.
MHC class II genes encode proteins that present antigenic peptides to CD4+ T cells to initiate and control immune responses (16, 17). Therefore, the expression of MHC class II genes is usually restricted to the antigen-presenting cells (APCs) such as dendritic cells, macrophages, and B cells (16). However, expression of MHC class II genes has also been detected in the CNS (central nervous system), including neural stem cells (18), microglia cells (19), and IFN-γ–induced astrocytes (20), raising the possibility that MHC II molecules may play a role in the CNS. In humans, MHC class II proteins have three classical molecules: human leukocyte antigen (HLA)–DR, HLA-DP, and HLA-DQ. The MHC class II molecule is a heterodimer consisting of an alpha and a beta chain assembled in the endoplasmic reticulum (ER) together with an invariant chain CD74 (aka Ii) (16). The MHC II–CD74 complex is transported from the ER, through the Golgi complex and trans-Golgi network, to the endosome/lysosome compartments (17, 21, 22), where CD74 is degraded, and peptides from endocytosed pathogens bind to MHC class II molecules with the help of HLA-DM and HLA-DO (22). HLA-DM and HLA-DO are two chaperones, which balance the antigen peptides binding to MHC class II molecules, making sure only strong peptide–MHC II bonds can form (23, 24). Last, MHC II–peptides containing vesicles fuse with the plasma membrane to activate T cells (22). Classical MHC class II genes, their chaperones, and CD74 are all under the control of the master transcription factor, CIITA (16), which we identify as a direct target of RACK7. Our findings suggest that RACK7 binds H3.3G34R and suppresses transcription of CIITA and vesicle pathway–related genes that are necessary for the activation of the MHC class II immune pathway. Given the established roles of the MHC class II genes in cancer, our findings may therefore be relevant to understanding the mechanism by which H3.3G34R regulates pediatric GBM.
RESULTS
RACK7 recognizes H3.3G34R mutation
RACK7 is a chromatin-binding protein with three tandemly arranged reader domains, including PHD (plant homeodomain), Bromo, and PWWP (Pro-Trp-Trp-Pro) domains (Fig. 1A), and is highly related to BS69 (aka ZMYND11), which has recently been identified as a specific reader of histone H3.3K36me3 (13, 14). Unlike BS69, RACK7 does not bind H3.3K36me3 due to the replacement of two amino acids in the binding pocket that are critical for H3.3K36me3 recognition (25). A recent study suggests that RACK7 reads the dual histone mark H3K4me1 and H3K14ac (12). While searching for histone modifications/mutations that RACK7 recognizes, we unexpectedly found that RACK7, but not BS69, preferentially bound the histone H3.3G34R peptide in vitro (Fig. 1B and fig. S1A). To confirm this finding, we assembled oligonucleosomes carrying either wild-type or mutant histone H3. Consistently, full-length RACK7 purified from Sf9 insect cells preferentially bound H3.3G34R oligonucleosomes, but not those carrying K27M, K36M, or unmodified H3.3 or H3.1 (Fig. 1C). The binding specificity of RACK7 for H3.3G34R was further supported by in vitro pull-down assays using peptides carrying substitutions of G34 to other amino acids including V, D, H, S, K, L, and W. Only G34R and G34K peptides showed binding with RACK7 (fig. S1B). Further binding experiments showed that RACK7 did not discriminate between H3.1 and H3.3 as evidenced by its binding to both H3.1G34R and H3.3G34R (fig. S1C).
Fig. 1. RACK7 recognizes H3.3G34R mutation.

(A) Schematic representation of the domain structure of human RACK7. Numbers indicate amino acid positions at the boundaries of various domains. (B) In vitro peptide pull-down assays using various H3.3 peptides and HA-tagged full-length RACK7 purified from insect cells. (C) Nucleosome-binding assays using different designer nucleosomes assembled in vitro and HA-tagged full-length RACK7 purified from insect cells. (D and E) In vitro peptide pull-down assays using wild-type H3.3 or H3.3G34R peptide and various RACK7 internal deletion mutants purified from insect cells (D) and isolated domains of RACK7 fused with GST (E). (F) MST analysis of the interaction between GST-PHDRACK7 with H3.3G34R (left) and H3.3 wild-type (right) peptides. The Kd value is indicated. Data are represented as means ± SD from three biological replicates. (G) In vitro peptide pull-down assays using H3.3G34R peptide with wild-type or D104A mutated PHDRACK7 fused with GST purified from Escherichia coli. (H and I) Peptide pull-down assays using long peptides (amino acids 1 to 44) as indicated with HA-tagged RACK7 full length purified from insect cells (H) and GST-tagged PHDRACK7 purified from E. coli (I). All experiments were repeated three times.
To identify the domain of RACK7 that mediates H3.3G34R recognition, we purified RACK7 deletion mutant proteins lacking each of the three potential reader domains. As shown in Fig. 1D, removal of the PHD domain abrogated binding to H3.3G34R peptides in vitro, suggesting that the PHD domain is necessary for RACK7 to bind H3.3G34R. Deletion of Bromo domain also reduced the binding to H3.3G34R peptides, suggesting that this domain may be necessary for optimal binding of RACK7 to H3.3G34R. We next purified the three reader domains individually and performed in vitro binding assays. As shown in Fig. 1E, the PHD domain alone (PHDRACK7) bound H3.3G34R, while the Bromo and PWWP domains showed barely detectable interactions with the H3.3G34R peptide. These results suggest that the PHD domain is necessary and sufficient to mediate H3.3G34R recognition by RACK7. Using microscale thermophoresis (MST) (26, 27), we found that PHDRACK7 bound to H3.3G34R peptide with a dissociation constant (Kd) of approximately 6 μM but showed no detectable binding to wild-type H3.3 peptides under the same assay conditions (Fig. 1F). To further define the binding property of PHDRACK7, we mutated and analyzed each and every amino acid in the PHD domain and found a mutation (D104A) that appears to disrupt the binding of RACK7 to the H3.3G34R peptide in vitro (Fig. 1G). In contrast to the 6 μM Kd of the wild-type PHDRACK7 binding to the H3.3G34R peptide, the MST assay failed to fit a binding curve for PHDRACK7 (D104A), suggesting that PHDRACK7 (D104A) has very little, if at all, binding activity for H3.3G34R peptide (fig. S1D). It should be noted that the same mutation, D104A, reduces the binding activity of RACK7 to the H3 N terminus by approximately fourfold (12). Together, our results suggest that D104A has a more pronounced impact on PHDRACK7 binding to H3.3G34R than to the N-terminal tail of H3.3.
As discussed above, RACK7 has previously been reported to bind histone to the N-terminal tail of histone H3 (H3K4me0 and H3K4me1) (12). Our in vitro binding assays further showed that RACK7 appeared to bind H3.3G34R (amino acids 22 to 44) more strongly than H3K4me0 (H3), H3K4me1, and H3K4me2 (amino acids 1 to 21) (fig. S1E, compare lanes 1, 2, and 3 with lane 9, pull-down ratio is shown at the bottom). Consistently, with a longer histone H3.3 peptide (amino acids 1 to 44), the G34R mutation also increased the binding ability of both full-length RACK7 (Fig. 1H and fig. S1F) and PHDRACK7 (Fig. 1I and fig. S1F), further supporting that RACK7 preferentially binds H3.3G34R. Introducing H3K14ac into a histone peptide containing the G34R mutation (H3.3K14acG34R; 1 to 44) had no influence on the binding to G34R (fig. S1G). In addition, the mono-, di-, and trimethylation of H3.3K36 also do not interfere with the interaction of G34R with RACK7 in vitro (fig. S1H). Collectively, our biochemical data support the notion that PHDRACK7 specifically recognizes H3.3G34R, primarily through binding the key residue G34R in the mutant histone H3 tail.
RACK7 binds H3.3G34R chromatin in cells
We next wished to determine whether RACK7 also preferentially binds H3.3G34R-decorated chromatin in cells. To do this, we used three human pediatric GBM (pGBM) cell lines (3). SJ-HGGx6c and SJ-HGGx42c are heterozygous for the G34R mutation in H3F3A (referred to as R6, R42), which encodes H3.3, while SJ-HGGx39c has wild-type H3F3A (referred to as WT39). Immunofluorescence staining showed that RACK7 is localized in the nucleus in all three tumor cell lines, indicating that the H3.3G34R mutation did not alter RACK7 subcellular localization (fig. S2A). We carried out RACK7 chromatin immunoprecipitation sequencing (ChIP-seq) and identified 9844, 8843, and 452 RACK7 peaks in R6, R42, and WT39, respectively. By removing the peaks shared with the H3.3 wild-type cell line, WT39, we identified 9632 and 8660 unique RACK7 peaks in the two H3.3G34R cell lines, R6 and R42, respectively (fig. S2B). We randomly selected six RACK7 peaks and confirmed their enrichment in both R42 and R6 cells as compared with the WT39 cells by ChIP–quantitative polymerase chain reaction (qPCR) (Fig. 2A). Snapshots of RACK7 enrichment peaks in all three pGBM lines are shown in Fig. 2B. A snapshot of RACK7 ChIP-seq biological repeats in all three pGBM lines is shown in fig. S2C. These results suggest that RACK7 has more extensive chromatin binding in H3.3G34R pGBM cells when compared with the H3.3 wild-type pGBM cells.
Fig. 2. RACK7 binds H3.3G34R chromatin in cells.
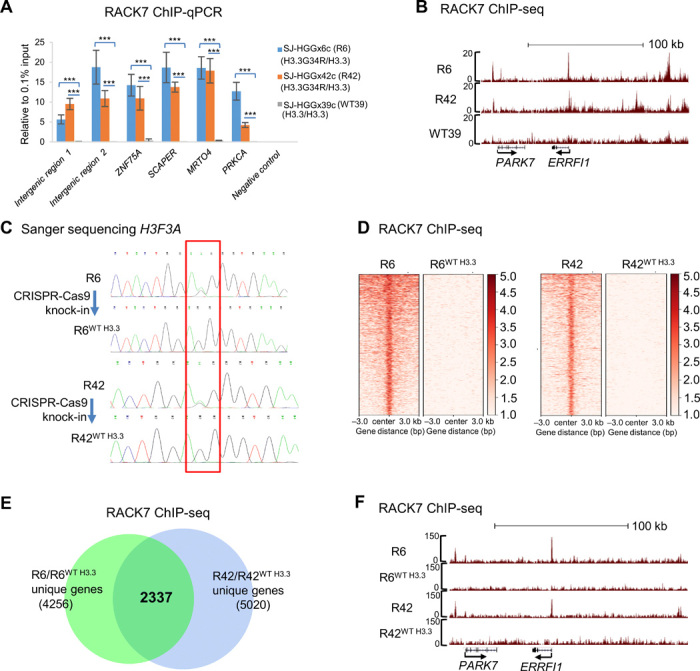
(A) ChIP-qPCR validation of selected RACK7-bound peaks in pGBM R6, R42, and WT39 cells. Data are represented as means ± SD from three biological replicates, ***P < 0.001, two-tailed Student’s t-test. (B) Genome browser snapshot of selected RACK7 ChIP-seq signals in pGBM R42, R6, and WT39 cells. (C) Sanger sequencing result shows the sequence of H3F3A in R6 and R6WT H3.3 (top) cells and R42 and R42WT H3.3 (bottom) cells. (D) Heatmap analysis of RACK7 ChIP-seq signals in R6 and R6WT H3.3 (left) and R42 and R42WT H3.3 (right) cells. bp, base pair. (E) Venn diagram analysis of RACK7 ChIP-seq unique genes between two comparisons, R6 with R6WT H3.3 and R42 with R42WT H3.3, respectively. (F) Genome browser snapshot of selected RACK7 ChIP-seq signals in R6, R6WT H3.3, R42, and R42WT H3.3 cells.
RACK7 was previously shown to bind active enhancers in the breast cancer cell line ZR-75-30, and RACK7 knockout (KO) decreases the level of H3K4me1 codecorated by RACK7, which is a histone mark associated with enhancers (15). Our ChIP-seq analysis in these three pGBM cells found no notable differences of H3K4me1 at genomic locations cobound by RACK7 between the two H3.3G34R-containing (R6 and R42) and the wild-type H3.3WT (WT39) pGBM cells (fig. S2D). In addition, while 73.2% of RACK7 peaks overlap with both H3K4me1 and H3K27ac in ZR-75-30 cells, we found only 14.1 and 29.3% coenrichment of the RACK7 peaks with H3K4me1 in R6 and R42, respectively. Furthermore, RACK7 does not appear to associate with histone demethylases (data not shown), as was shown in ZR-75-30 cells. Together, these findings suggest that molecular mechanisms by which RACK7 functions may be cell type dependent.
To determine whether the increased RACK7 enrichment in R6 and R42 cells is due to the histone mutation, H3.3G34R, we corrected H3.3G34R in R6 and R42 cells to wild-type H3.3 by CRISPR-Cas9–mediated knock-in (generating R6WT H3.3 and R42WT H3.3) (Fig. 2C). The correction of H3.3G34R to H3.3 wild type abrogated RACK7 enrichment (Fig. 2D), indicating that H3.3G34R is necessary for the formation of the unique RACK7 peaks in both R6 and R42 cell lines. After mapping these unique peaks to the nearest gene, we identified 2337 genes with RACK7 bound nearby that are common to R6 and R42 cells but are absent in the corresponding corrected lines, R6WT H3.3 and R42WT H3.3 (Fig. 2E). As a control, we found comparable RACK7 protein levels in these cell lines (R6, R6WT H3.3, R42, and R42WT H3.3) (fig. S2E), and their nuclear localizations are unaltered by the genetic manipulation of the histone mutation (fig. S2F). We again confirmed the genome-wide ChIP-seq data by ChIP-qPCR analysis of RACK7 at the same six genomic regions (fig. S2G). A snapshot of the genomic regions is shown in Fig. 2F. Together, these findings suggest that H3.3G34R promotes RACK7 binding to H3.3G34R-decorated chromatin regions, consistent with our in vitro biochemical data that RACK7 preferentially binds H3.3G34R histones.
RACK7 represses gene transcription through its binding to H3.3G34R
To explore the function of the histone mutation H3.3G34R, we performed genome-wide transcriptomic analysis of R6, R42, R6WT H3.3, and R42WT H3.3 by RNA sequencing (RNA-seq). Compared with H3.3G34R, we found 1882 up-regulated and 1510 down-regulated genes that are in common in both corrected, knock-in cells (R6WT H3.3 and R42WT H3.3) (Fig. 3A). Among the 1882 up-regulated genes, we found RACK7 occupancy of 253 genes in the two H3.3G34R pGBM lines, but not in H3.3-corrected, knock-in cells (fig. S3A). These genes were enriched in vesicle (P = 7.77 × 10−8) and membrane-bounded vesicle (P = 2.95 × 10−8) Gene Ontology (GO) pathways (Fig. 3B). In contrast, the 154 down-regulated genes decorated by RACK7 in two H3.3G34R lines (fig. S3B) are associated with less significant GO terms (Fig. 3C). These results suggest that loss of RACK7 chromatin occupancy gives rise to transcriptional up-regulation of vesicle pathway–related genes in R6WT H3.3 and R42WT H3.3 cells, which is consistent with previous reports that RACK7 acts as a transcriptional repressor via its chromatin binding (12, 15).
Fig. 3. RACK7 represses gene transcription through its binding to H3.3G34R mutation.
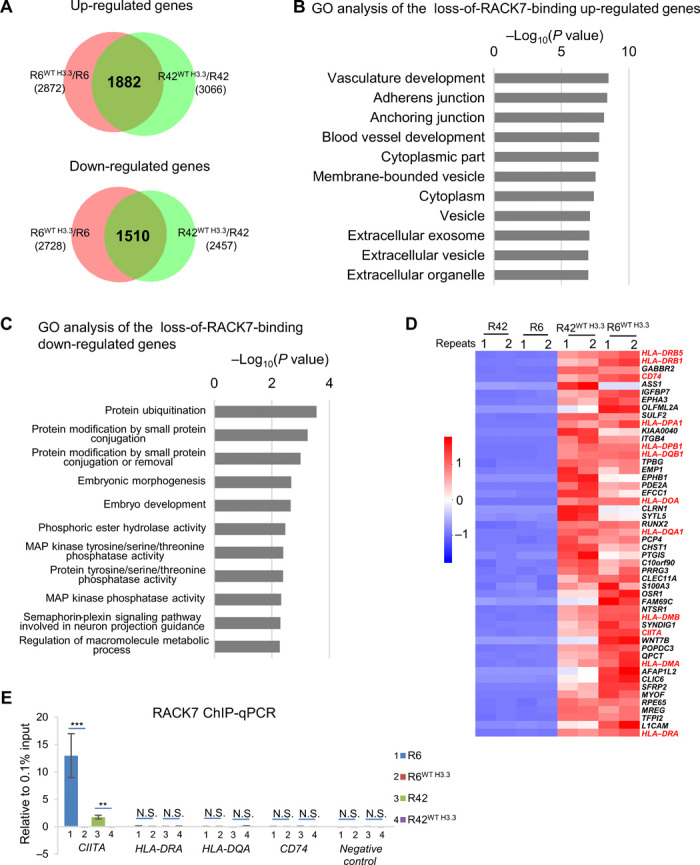
(A) Venn diagram analysis of the up-regulated genes [1.5-fold (P < 0.05)] (top) and down-regulated genes [1.5-fold (P < 0.05)] (bottom) in R6WT H3.3 and R42WT H3.3 cells, relative to R6 and R42, respectively. (B and C) Gene Ontology (GO) analysis of up-regulated genes [1.5-fold (P < 0.05)] (B) and down-regulated genes [1.5-fold (P < 0.05)] (C) that have lost RACK7 binding in R6WT H3.3 and R42WT H3.3 cells. GO terms are ordered by P values from biological proces, cellular component, and molecular function analysis. MAP, mitogen-activated protein. (D) Heatmap analysis of the top 50 up-regulated genes based on the RNA-seq data in R6WT H3.3 and R42WT H3.3 cell lines compared with those of their parental cells (R6 and R42), respectively. Sample order is based on the sum of log2(fold change). Scale indicated Z score, defined as Δ (FPKM − mean FPKM)/SD. Two biological repeats for each cell lines were used. (E) ChIP-qPCR validation of RACK7-bound CIITA peaks, HLA-DRA, HLA-DQA, and CD74 in R6 (line 1), R6WT H3.3 (line 2), R42 (line 3), and R42WT H3.3 (line 4) cells. Data are represented as means ± SD from three biological replicates, **P < 0.01 and ***P < 0.001, two-tailed Student’s t test. N.S., not significant.
To further analyze the RNA-seq data implicating the vesicle pathways in the H3.3 knock-in cells, we plotted heatmap of the top 50 up-regulated genes from the 1882 genes up-regulated in common in both H3.3 knock-in cell lines. Unexpectedly, we also found up-regulation of MHC class II genes, their chaperones, CD74, and their master regulator, CIITA (Fig. 3D). In contrast, the transcription level of MHC class I genes appeared unaffected (fig. S3C). We found no evidence that RACK7 binds to MHC class II genes, or their chaperones and CD74 in H3.3G34R cells (Fig. 3E and fig. S3D), suggesting that they are not directly regulated by RACK7. All these genes are known to be controlled by CIITA (16). We found that CIITA gene is decorated by RACK7 in H3.3G34R cells (R6 and R42) (Fig. 3E and fig. S3E). This region was reported to be involved in regulating CIITA transcription (28). Our results suggest that RACK7 indirectly suppresses MHC class II gene expression by directly repressing transcription of the master regulator, CIITA. Because MHC class II molecules were known to be delivered from the ER to the cell surface through vesicles (22), our results further suggest that RACK7 and H3.3G34R work together to suppress transcription of genes involved in the MHC class II complex and their delivery pathways.
RACK7 KO has similar effects on gene expression as does H3.3 knock-in, which corrects the G34R mutation
If the biological effects of H3.3G34R are mediated primarily by the recruitment of RACK7, then we would predict that the impact of RACK7 removal will be akin to that of correcting the histone G34R genetic mutation. We addressed this hypothesis by knocking out RACK7 in R6 and R42 (generating R6RACK7 KO and R42RACK7 KO), respectively (fig. S4A), and next performed the genome-wide RNA-seq analysis. As a result, abrogating RACK7 (by KO) had a significant correlation with correcting the G34R mutation (wild-type H3 knock-in) when each transcriptome was compared with its parental line (R6 and R42) (Fig. 4A). Knocking out RACK7 in R6 and R42 results in 2484 and 2523 up-regulated genes when compared with each parental cell line, respectively (fig. S4B). Among them, 1293 genes were up-regulated in both R6RACK7 KO and R42RACK7 KO cells (fig. S4B). When compared with the 1882 co–up-regulated genes in H3.3 knock-in cells (Fig. 3A), we identified 765 genes that were co–up-regulated in all R6RACK7 KO, R42RACK7 KO, R6WT H3.3, and R42WT H3.3 cells (Fig. 4B). These 765 co–up-regulated genes were enriched in GO terms of vesicle (P = 9.27 × 10−11), membrane-bounded vesicle (P = 1.09 × 10−11), and MHC class II protein complex (P = 1.13 × 10−8) pathways (Fig. 4C). Similarly, MHC class II genes, their chaperones, CD74, and CIITA show high transcriptional changes in the RACK7 KO cells (fig. S4C) as well, while in contrast, MHC class I genes were unaffected (fig. S4D). Collectively, our results suggest that knocking out RACK7 and knocking in H3.3 to correct the G34R mutation both activate the same groups of genes involved in MHC class II protein complex and vesicle pathways.
Fig. 4. RACK7 KO has similar effects on gene expression as does H3.3 knock-in, which corrects the G34R mutation.

(A) Scatter plot of log2(fold change) gene expression in R6WT H3.3 and R6RACK7 KO cells (left) and R42WT H3.3 and R42RACK7 KO cells (right), relative to R6 and R42, respectively. Pearson’s product-moment correlation was analyzed by R. (B and C) Venn diagram (B) and GO (C) analysis of co–up-regulated genes [1.5-fold(P < 0.05)] in wild-type H3.3 knock-in and RACK7 KO cells relative to their parental cells. (D) RT-qPCR analysis of mRNA levels of genes in indicated cells. ACTB, actin beta. (E) Western blot analysis of HLA-DRA and HLA-DRB5 in indicated cells. (F) R6, R6WT H3.3, and R6RACK7 KO, which, stably expressing YFP (yellow), were stained for CD74 (red) and 4′,6-diamidino-2-phenylindole (DAPI) (blue) and imaged by confocal microscopy. (G) Flow cytometric analysis of the cell surface expression of HLA-DR in indicated cells. (H) RT-qPCR analysis of select genes in R6, R6RACK7 KO, and R6RACK7 KO rescued with a RACK7 transgene (left), and in R42, R42RACK7 KO, and R42RACK7 KO rescued with a RACK7 transgene (right) cells. Data in (D) and (H) are represented as means ± SD from three biological replicates, **P < 0.01 and ***P < 0.001, two-tailed Student’s t test.
We next confirmed up-regulation of MHC class II genes, their chaperone genes, CD74, and CIITA by reverse transcription qPCR (RT-qPCR) not only in R6RACK7 KO, R42RACK7 KO but also in R6WT H3.3, R42WT H3.3 cells (Fig. 4D). Up-regulation of two representative MHC class II genes was also confirmed by Western blotting (Fig. 4E). Confocal microscopy showed the appearance of CD74 in the perinuclear region vesicles but not cell surface in R6RACK7 KO and R6WT H3.3 cells (Fig. 4F), which is consistent with previous reports that CD74 works as a chaperone at the beginning step of the MHC class II molecule delivery process (17). Up-regulation of MHC class II genes appears to translate to more MHC class II proteins on the cell surface in either RACK7 KO or H3.3 knock-in cells (Fig. 4G), which is likely to be assisted by the increased expression of vesicle transport genes in these cells. To further confirm the role of RACK7 in H3.3G34R cells, we reintroduced wild type (fig. S4E) and the binding-defective mutant, RACK7D104A (fig. S4F), into R6RACK7 KO and R42RACK7 KO cells, respectively. Reintroducing wild-type RACK7, but not the binding-defective RACK7D104A, suppressed the up-regulation of CIITA and MHC class II genes in both R6RACK7 KO and R42RACK7 KO cells (Fig. 4H and fig. S4G). ChIP-qPCR of the tagged H3.3G34R showed that CIITA was decorated by H3.3G34R (fig. S4H) in the H3.3G34R cells, consistent with our model that RACK7 regulates gene expression in these pGBM cells by binding histone H3.3G34R. Furthermore, our ChIP-qPCR analysis of chromatin purified from RACK7 KO cells rescued by either wild-type or RACK7D104A shows that wild-type RACK7 associates better with the CIITA gene locus than the binding-defective mutant RACK7D104A (fig. S4I). Collectively, these findings demonstrate that RACK7 occupies the CIITA locus through binding H3.3G34R. As a control, knocking out RACK7 in WT39 cells (fig. S4J) did not appear to up-regulate MHC class II genes (under real-time PCR detectability). Collectively, these findings suggest that H3.3G34R and RACK7 collaborate to suppress the entire molecular program that ensures the expression and presence of MHC class II gene products on the cell surface.
RACK7 indirectly regulates MHC class II genes via controlling its master regulator CIITA
Because RACK7 only decorates the CIITA gene but not the MHC class II genes in H3.3G34R cells (R6 and R42) (Fig. 3E and fig. S3, D and E), we next investigated whether up-regulation of MHC class II genes observed in RACK7 KO and H3.3 knock-in cells was dependent on the CIITA gene. We used CRISPR-Cas9 to KO CIITA in R6RACK7 KO and R6WT H3.3 cells and found decreased levels of HLA-DR on the cell surface in R6RACK7 KO + CIITA KO and R6WT H3.3 + CIITA KO when compared with the R6RACK7 KO and R6WT H3.3 cells (Fig. 5A). This result suggests that CIITA is necessary for the up-regulation of MHC class II genes and its associated genes in RACK7 KO and H3.3 knock-in cells.
Fig. 5. RACK7 directly regulates CIITA and genes involved in vesicular transportation through its chromatin binding.

(A) Flow cytometric analysis of HLA-DR antibody–stained R6, R6WT H3.3, R6RACK7 KO, R6CIITA KO, R6WT H3.3 + CIITA KO, and R6RACK7 KO + CIITA KO cells. All cells contain YFP transgene (detailed in Materials and Methods). YFP-positive cells were used to analyze the cell surface expression of HLA-DR. (B) RT-qPCR of gene expressions in R6, R6WT H3.3, and R6RACK7 KO cells. Data are represented as means ± SD from three biological replicates, ***P < 0.001, two-tailed Student’s t test. (C) Flow cytometric analysis of HLA-DR antibody–stained R6QKI KO, R6WT H3.3 + QKI KO, and R6RACK7 KO + QKI KO cells, compared with their parental cells in (A). All cells contain YFP transgene (detailed in Materials and Methods). YFP-positive cells were used to analyze the cell surface expression of HLA-DR. (D) Flow cytometric analysis of the HLA-DR antibody–stained R6 (top), R6WT H3.3 (middle), and R6RACK7 KO (bottom) lenti-CRISPR KO GFAP, VIM, and OCIAD2, respectively. All cells contain YFP transgene (detailed in Materials and Methods). YFP-positive cells were used to analyze the cell surface expression of HLA-DR.
RACK7 directly regulates genes involved in vesicular transportation of MHC class II molecules to the cell surface
To determine whether the RACK7 target genes up-regulated in both H3.3 knock-in and RACK7 KO cells participated in the regulation of the transportation of MHC class II molecules, we chose four genes, QKI, GFAP, VIM, and OCIAD2, which are likely direct targets of RACK7 (fig. S5A) for further investigation. Their transcriptional up-regulation in R6RACK7 KO and R6WT H3.3 was validated by RT-qPCR (Fig. 5B). Among these genes, QKI (quaking) was an RNA binding protein and was recently identified as a regulator of endolysosomes, as its deletion down-regulates endolysosomes and helps glioma stem cells to maintain their stemness (29). GFAP (glial fibrillary acidic protein) and VIM (vimentin) are intermediate filament proteins, and astrocytes deficient in GFAP and VIM have less vesicle mobility and nondirectional movement, thus reduced MHC class II molecules delivery (30). OCIAD2 (OCIA domain containing 2) is a cancer-related protein that localizes to early endosomes (31). We first knocked out QKI in R6, R6RACK7 KO, and R6WT H3.3 cells, respectively. Knocking out QKI decreased the HLA-DR molecules in the cell surface of these cells (R6RACK7 KO and R6WT H3.3 cells) (Fig. 5C) when compared with R6, R6RACK7 KO, and R6WT H3.3 cells (Fig. 5A, top). Likewise, knocking out the additional RACK7 target genes, GFAP, VIM, and OCIAD2, also led to a reduction in the MHC class II molecules on the cell surface in R6RACK7 KO and R6WT H3.3 cells (Fig. 5D). Together, these results suggest that RACK7 directly suppresses the transcription of CIITA and genes involved in vesicle pathways to potentially inhibit the MHC class II immune response.
RACK7 KO or H3.3 knock-in up-regulates cell differentiation genes and inhibits cell migration and invasion
In addition to MHC class II molecules, we also found up-regulation of genes (Fig. 4B) associated with cell differentiation in R6RACK7 KO, R42RACK7 KO, R6WT H3.3, and R42WT H3.3 cells (P = 1.83 × 10−4; 158 genes). Thirty-three of these genes (158) appear to be direct targets of RACK7, as they were decorated by RACK7 on chromatin in both H3.3G34R cells (R6 and R42) but not in the H3.3 knock-in cells, in which the G34R mutation was corrected to wild type (Fig. 6A and fig. S6A). We validated transcriptional up-regulation of some of these genes by RT-qPCR (Fig. 6B). One of these RACK7 target genes, GFAP, which is expressed in astrocytes (32), was further confirmed by Western blotting (fig. S6B) and immunofluorescence (Fig. 6C). The up-regulation of GFAP was rescued by wild-type RACK7 but not by the binding-defective mutant, RACK7D104A, in the RACK7 KO cells (Fig. 6D). RACK7 chromatin binding in the RACK7 KO cells was restored by reintroducing wild-type RACK7 but not the D104A mutant (fig. S4I). Collectively, our results demonstrate that knocking out RACK7 or knocking in H3.3 in H3.3G34R pGBM enhances expression of differentiation genes such as GFAP and enhances MHC class II immune response. Reduced GFAP expression is associated with tumor progression in a mouse glioma model (32). However, these human GBM cell lines failed to generate tumors in vivo (both intracranial and subcutaneous injections) within a period of 2 to 3 months. Although longer-term observations may be necessary to determine whether these cell lines form tumor in vivo, we find that correction of the G34R mutation or knocking out RACK7 both reduced the migration and invasion capabilities of these cells (Fig. 6E). The migration and invasion phenotypes were rescued by wild-type RACK7 but not by the binding-defective mutant, RACK7D104A (Fig. 6E), suggesting that binding of RACK7 to H3.3G34R may be important for increased migration and invasion of these pGBM cells.
Fig. 6. RACK7 KO or H3.3 knock-in up-regulates cell differentiation genes and inhibits cell migration and invasion.

(A) Heatmap analysis of RACK7-decorated genes involved in cell differentiation (GO: 0030154) and up-regulated in R42WT H3.3, R42RACK7 KO, R6WT H3.3, and R6RACK7 KO cell lines. Scale indicated Z score, defined as Δ (FPKM − mean FPKM)/SD. Two biological repeats for each cell lines were used. (B) RT-qPCR of gene expression in R6, R6WT H3.3, and R6RACK7 KO and R42, R42WT H3.3, and R42RACK7 KO cells. (C) Immunofluorescence of GFAP in R6, R6WT H3.3, and R6RACK7 KO (lines 1 and 2) and R42, R42WT H3.3, and R42RACK7 KO (lines 3 and 4) cells. (D) RT-qPCR of the GFAP mRNA expression level in R42RACK7 KO and R42RACK7 KO cells rescued with wild-type or D104A mutated RACK7 transgene. (E) In vitro migration (top) and invasion (bottom) assays examining the migration and invasion abilities of the R42, R42WT H3.3, R42RACK7 KO, and R42RACK7 KO rescued with wild-type or D104A-mutated RACK7 transgene cells. Data in (B), (D), and (E) are represented as means ± SD from three biological replicates, *P < 0.05, **P < 0.01, and ***P < 0.001, two-tailed Student’s t test.
DISCUSSION
We identified RACK7 as a reader for H3.3G34R, and this conclusion is supported by multiple biochemical and ChIP-seq data. Furthermore, we demonstrate that the preferential association of RACK7 with the H3.3G34R mutant histone in pGBM cell lines helps to suppress gene expression programs, including the entire program important for the expression and delivery of MHC class II proteins to the cancer cell surface.
We have provided multiple lines of evidence that RACK7 preferentially binds H3.3G34R in vitro. We also described several independent experiments that support binding of RACK7 to histone H3.3G34R–decorated chromatin regions in vivo. Specifically, we demonstrate that RACK7 ChIP-seq peaks are significantly reduced when H3.3G34R is corrected to wild-type H3.3 (Fig. 2D). Consistently, there are more RACK7 ChIP-seq peaks in the H3.3G34R-containing pGBM (R6 and R42) than in H3.3 wild-type pGBM cells (WT39). To circumvent the lack of a validated, ChIP-grade H3.3G34R antibody, we introduced H3.3G34R–hemagglutinin epitope YPYDVPDYA (HA) transgene into R6 and R42 cells and performed ChIP-qPCR using an HA antibody. Our results showed that RACK7-bound regions around CIITA, GFAP, and other four genes were also decorated by H3.3G34R (fig. S4H), consistent with our hypothesis that RACK7 binds H3.3G34R in vivo.
To determine whether the ability of RACK7 to regulate gene expression is dependent on its binding to H3.3G34R-decorated chromatin in vivo, we attempted to identify a RACK7 mutant that fails to bind H3.3G34R. However, this effort was complicated by the fact that RACK7 was also known to bind the N-terminal tail of histone H3 (12). We were unable to find a mutation that strictly affects binding to G34R but not the N-terminal tail of histone H3. Nevertheless, we succeeded in finding a mutation (D104A in the PHD domain of RACK7), which appears to have a more profound effect on binding to G34R than to the N-terminal tail of H3. Specifically, the D104A mutation reduces binding to G34R from a Kd of 6 μM to essentially no binding as opposed to about only a fourfold reduction in binding to the N-terminal H3. The D104A mutation failed to restore suppression of RACK7 target genes such as CIITA (fig. S4G) (which we show to be decorated by H3.3G34R histone in the H3.3G34R-containing pGBM cells) and also displayed a reduced chromatin enrichment on these genes (fig. S4I). RACK7D104A also failed to rescue the cell migration and invasion phenotypes (Fig. 6E). We therefore favor the hypothesis that the effect of RACK7 in vivo is mediated primarily by binding to G34R, although we cannot completely exclude possible contributions of RACK7 binding to the N-terminal tail of histone H3.
The transportation of MHC class II molecules by vesicles has been well demonstrated previously (22). During this process, MHC class II molecules are delivered from the ER through the Golgi to endosome compartments. Early endosomes (21), the late endosome/lysosome (17), and the multivesicular body (17) are all involved in this process. Besides these vesicles, MHC class II molecules were detected in exosomes as well (22). In our results, a cohort of up-regulated genes, upon RACK7 KO or H3.3 knock-in, are clustered in the vesicle GO pathway (P = 9.27 × 10−11, 186 genes), specifically on ER-to-Golgi transport vesicle membrane (P = 6.53 × 10−5, 10 genes), trans-Golgi network (P = 7.96 × 10−5, 19 genes), endosome (P = 6.33 × 10−5, 48 genes), lysosome (P = 4.92 × 10−8, 44 genes), and extracellular exosome pathway (P = 1.39 × 10−8, 142 genes). Our results therefore suggest that the H3.3G34R mutation in conjunction with RACK7 regulate genes that participate in multiple steps of the biogenesis and delivery of MHC class II molecules.
The role of the MHC II immune response in antitumor immunity is becoming increasingly appreciated (33). In diffuse large B cell lymphoma (DLBCL), the MHC class II gene expression signature is correlated with a good outcome (34), while HLA-DM expression predicts improved survival in patients with breast carcinoma (35). MHC II–positive melanomas have improved response rates and clinical benefit to PD-1 (programmed cell death 1)/PD-L1 (programmed cell death ligand 1) inhibition (36). Agents that are used to treat cancer, such as the histone deacetylase (HDAC) and DNA methyltransferase inhibitors, increase the expression of MHC class II (summarized in table S1) (37–40), suggesting that an elevated MHC class II immune response may have a role in therapeutic responses. Thus, inhibiting the function of RACK7 in H3.3G34R-containing pGBM may help to increase the effect of immunotherapy or other therapy approaches. Last, we found that loss of RACK7 chromatin binding at its target genes in RACK7 KO or H3.3 knock-in cells switches the cells to a more differentiated state. The expression of the differentiation-associated genes in the RACK7 KO and H3.3 knock-in cells may therefore lower their capacity to generate brain tumor.
What is the clinical significance of our finding that H3.3G34R/RACK7 mediates repression of the MHC class II genes in H3.3G34R-containing pGBM? To begin to address this issue, we compared expression of the master regulator of MHC class II genes, CIITA, in pediatric high-grade gliomas (HGGs) with H3.3G34R/V, H3K27M, or wild-type H3 across all brain locations (n = 210) using data from Mackay et al. (41). H3.3G34R/V tumors showed lower expression compared with H3K27M (P = 0.0006, t test). However, these tumors arise in different brain regions, and H3.3G34R/V tumors are typically found in older adolescent through young adulthood ages. In a more refined cohort (n = 99) trying to match location of H3.3G34R/V HGGs, the trend to lower expression in H3.3G34R/V tumors (n = 19), compared with cortical pediatric HGGs with wild-type H3 (n = 80), did not reach statistical significance (P = 0.19, t test). Matching the tumor locations is our best estimate of an appropriate control comparison. However, the distinct DNA methylation signatures of H3.3G34R/V tumors likely reflect differences in tumor cell origin, at least in part. Other HGGs may have different basal expression of MHC due to cell of origin and may also have alternate mutations that influence MHC class II–related expression patterns. These data highlight the power of our experimental approach using isogenic cells that have identical genetic background except for the status of H3.3 (one carries H3.3G34R, and the other is wild-type H3.3). This controlled comparison removes the noise contributed by different cells of origin and heterogeneity of other mutations and allowed us to conclude that H3.3G34R plays a role in regulating these important genes. Future experiments are necessary to investigate the in vivo significance of our finding.
RACK7 was reported to suppress tumorigenesis, in part by repressing S100A family oncogenes (15). It also has an antimetastasis function by inhibiting multiple metastasis–associated genes (12). However, we did not find global transcriptional up-regulation of these genes upon RACK7 KO or H3.3 knock-in except for S100A3 and epidermal growth factor receptor (EGFR), which were decorated by RACK7 in H3.3G34R cells. S100A3 and S100A5 expression differed markedly in the solid tumor tissues in relation to the astrocytic tumor types and grades (42). EGFR is frequently mutated and/or overexpressed in different types of human cancers (41), but not in pediatric H3.3G34R GBM (41). Although up-regulation of S100A3 and EGFR by inhibiting RACK7 is implicated in increasing invasive ability in previous reports (12, 15), their roles in H3.3G34R-associated pGBM remain to be determined in the future.
In addition to G34R, H3.3G34V mutation is also associated with human pGBMs (2). However, unlike H3.3G34R, H3.3G34V does not bind recombinant RACK7 directly (Fig. 2B). We recently identified an H3.3G34V-binding protein, which also interacts with RACK7 in the human H3.3G34V cells [KNS42 (43)], and histone H3.3G34V peptides pulled down RACK7 from lysate of an H3.3G34V cell line (data not shown), suggesting that RACK7 may bind H3.3G34V indirectly in H3.3G34V pGBM cells. Much work remains to be done to understand the mechanism of action of the H3G34V mutation, but our preliminary results suggest that both mutations may recruit RACK7, although the underlying biochemical mechanisms may differ.
Together, our findings identify RACK7 as a reader of H3.3G34R and demonstrate that the H3.3G34R mutation functions by recruiting RACK7 to suppress the expression of MHC class II genes (indirectly) and transport genes (directly), resulting in decreased expression of MHC class II molecules at the cell surface. Association of RACK7 with H3.3G34R also suppresses expression of genes associated with differentiation functions. Abrogation of RACK7 enhances expression of MHC class II genes and (possibly enhanced MHC class II–mediated immune response) differentiation genes (possibly increased differentiation) in these tumor cells. Although the cell origin for H3.3G34R-associated pGBM is still unclear, the fact that normal neural stem cells, astrocytes, and microglia cells (18–20) could express MHC class II genes supports our hypothesis that suppression of the MHC class II gene expression and delivery in the yet-to-be-identified cells due to the H3.3G34R mutation may contribute to tumorigenesis. Collectively, our findings not only provide molecular insights into H3.3G34R function but also suggest that targeting RACK7 may be an effective way to treat G34R-carrying pGBM.
MATERIALS AND METHODS
Protein expression and purification
The DNA fragments corresponding to various truncations of human RACK7 were subcloned into the modified pGEX-6P1 or pGEX-4T-1 vector, and the plasmids were transformed into Escherichia coli strain Rosetta (DE3). The transformants were grown at 37°C to OD600 (optical density at 600 nm) of 0.8, and protein expression was induced by adding 0.2 mM isopropyl-β-d-thiogalactopyranoside (IPTG). After further culturing at 16°C for 16 hours, cells were harvested and lysed with the buffer containing 20 mM tris-HCl (pH 8.0), 150 mM NaCl, 1 mM EDTA, 0.2% Triton X-100, 1 mM phenylmethylsulfonyl fluoride (PMSF), and 1 mM dithiothreitol (DTT). The supernatant was incubated with Glutathione Sepharose (GE) beads for 2 hours, and glutathione S-transferase (GST)–tagged proteins were eluted with the elution buffer containing 50 mM tris-HCl (pH 8.0) and 20 mM glutathione.
The HA-tagged full-length and deleted human RACK7 were subcloned into the pFastBac HT-A vector, and viruses were generated according to the Bac-to-Bac Baculovirus Expression System (Invitrogen). The Sf9 cells infected by virus were cultured at 28°C for 48 hours and then harvested and lysed with the buffer containing 20 mM tris-HCl (pH 7.4), 300 mM NaCl, 0.2% Triton X-100, 10% glycerol, 0.5 mM EDTA, 1 mM PMSF, and 1 mM DTT. The HA infinity beads were added into the supernatant and incubated at 4°C for 2 hours, and HA-tagged protein was eluted with the elution buffer containing 200 mM tris-HCl (pH 7.9), 150 mM KCl, 0.2 mM EDTA, 5 mM MgCl2, 0.1% NP-40, and HA-tag peptide (0.2 mg/ml; amino acid: YPYDVPDYA). The HA-tag peptide was synthesized by Beijing Scilight Biotechnology LLC.
In vitro binding assays
For peptide pull-down assays, recombinant, full-length HA-tagged RACK7 and mutants were purified from insect cell Sf9, while PHD, Bromo, and PWWP domains of RACK7 were purified from E. coli. Two micrograms of full-length RACK7 or mutant proteins was incubated with 2 μl (concentration, 0.1 mM) of various biotinylated histone peptides in the binding buffer [20 mM tris-HCl (pH 7.3), 150 mM NaCl, 0.1% NP-40] at 4°C for 4 hours. The protein-peptide complexes were immobilized to streptavidin beads (Millipore) at 4°C for 1 hour. The bound proteins were washed with binding buffer and separated on 10% SDS–polyacrylamide gel electrophoresis (PAGE) followed by Coomassie blue staining. Modified histone peptides were synthesized by Beijing Scilight Biotechnology LLC.
MST analysis
The determination of the binding capacity of PHDRACK7 and PHDRACK7 (D104A) to wild-type or G34R-mutated histone peptides (amino acids 22 to 44) was performed by MST (NanoTemper, Monolith NT.115) according to the manufacturer’s instruction with 20% light-emitting diode (LED) and 40% MST power (27). The purified recombinant GST-PHDRACK7 and PHDRACK7 (D104A) protein were labeled with NHS-647 (NanoTemper Technologies) and applied at a final concentration of 50 nM. The final concentrations of peptides ranged from 15.259 nM to 500 μM. Both proteins and peptides were diluted in 20 mM tris-HCl (pH 8.0), 100 mM NaCl, 0.5% NP-40. Samples were filled into standard, treated capillaries (NanoTemper Technologies). The MST data were analyzed by thermophoresis with temperature jump.
Recombinant oligonucleosomes
The open reading frames of histone H2A, H2B, H4, wild-type or mutant histone H3.3 were subcloned into the pET28a vector, and the plasmids were transformed into E. coli strain Rosetta (DE3). The transformants were grown at 37°C to OD600 of 0.8, and protein expression was induced by adding 0.2 mM IPTG. After further culturing at 37°C for 2 hours, cells were harvested and lysed with lysis buffer containing 50 mM tris-HCl (pH 7.5), 100 mM NaCl, 1 mM EDTA, and 1 mM PMSF. The bacterial lysates were then sonicated at 200-W “power,” 30 s “on” and 30 s “off” for 20 “cycles.” The pellets were washed three times with lysis buffer plus 1% Triton X-100 and three times with lysis buffer; resuspended in unfolding buffer containing 7 M guanidinium-HCl, 20 mM tris-HCl (pH 7.5), and 10 mM DTT; and mixed gently for 1 hour at room temperature. The purified histones were quantitated by 15% SDS-PAGE followed by Coomassie blue staining.
Histone octamers were obtained by mixing the four unfolded recombinant histones isolated as described above in equimolar amounts with approximately 4 mg of total protein in about 1 ml of volume. Then, the mixtures were dialyzed at 4°C against 2 liters of refolding buffer [10 mM tris-HCl (pH 7.5), 2 M NaCl, 1 mM EDTA, and 5 mM β-mercaptoethanol] with at least three buffer changes. Either the second or third dialysis step was performed overnight. The samples were centrifuged to remove any precipitates, and proteins were concentrated to 250 μl with a Millipore microconcentrator (10-kDa cutoff; Millipore, UFV4BGC00). Then, the samples were loaded onto a HiLoad Superdex 200 HR 10/30 column equilibrated with refolding buffer. Recombinant core histone octamers were eluted with a relative molecular mass of 100 kDa and were analyzed by 15% SDS-PAGE followed by Coomassie blue staining.
Recombinant oligonucleosomes were obtained by mixing the recombinant core histone octamers and plasmid containing tandem 147–base pair DNA fragments in a mass ratio of 1:1.2 at room temperature for 15 min. Then, the mixtures were dialyzed at 4°C against 1 liter of TEN buffers containing 10 mM tris-HCl (pH 7.5), 1 mM EDTA, and 5 mM β-mercaptoethanol and different concentrations of NaCl—1.4, 1.2, 1.0, 0.8, and 0.6 M. The mixtures were dialyzed at 4°C against 1 liter of TE buffer containing 10 mM tris-HCl (pH 7.5) and 1 mM EDTA overnight.
Nucleosome-binding assays
For nucleosome-binding assays, 2 μg of full-length recombinant HA-tagged RACK7, expressed and purified from insect cell Sf9, was immobilized on HA beads first, which were then incubated with 5 μg of nucleosomes in binding buffer [20 mM tris-HCl (pH 7.9), 150 mM NaCl, 0.05% NP-40, 1 mM MgCl2, and 5% glycerol] at 4°C for 6 hours. After three washes with binding buffer, the bound proteins were separated on 15% SDS-PAGE followed by Coomassie blue staining.
Cell culture
All three of these lines (SJ-HGGx6c, SJ-HGGx42c, and SJ-HGGx39c) were established from pGBMs from the cerebral cortex. The cells were maintained under a neural stem cell condition. Briefly, the cells were cultured in Neurobasal:KnockOut DMEM/F-12 (1:2) with 0.5% B27 (minus vitamin A, Thermo Fisher Scientific), 0.5% StemPro Neural Supplement (Thermo Fisher Scientific), 0.5% N-2 (Thermo Fisher Scientific), recombinant human epidermal growth factor (EGF) and FGF-b (20 ng/ml; PeproTech), recombinant human PDGF-AA and PDGF-BB (10 ng/ml; PeproTech), 0.5 mM sodium pyruvate (Thermo Fisher Scientific), 0.5% nonessential amino acid (Thermo Fisher Scientific), 1% GlutaMAX (Thermo Fisher Scientific), and 0.0002% heparin sodium salt (STEMCELL). The culture surface was coated with 1% Geltrex LDEV (lactose dehydrogenase elevating virus) free, growth factor reduced, human embryonic stem cell (hESC) qualified (Thermo Fisher Scientific), which was modified from previous research (44, 45). The SJ-HGGx6c and SJ-HGGx42c were transduced with a lentiviral vector (vCL20SF2-luc2a-YFP) expressing luciferase and yellow fluorescent protein (YFP) (46).
Establishment of knock-in and KO cell lines
RACK7 and QKI KO cells were generated by using the CRISPR-Cas9 system as described (15). Briefly, guide RNAs were cloned into vector PX335 (hSpCas9 + guide RNA; Addgene, 42335), and the single clones were established by dilution cloning. The KO efficiencies were confirmed by Western blotting. Guide RNA sequences used were as follows: RACK7, 5′-GTGATGTGTCCTGCGGCGAG-3′; QKI, 5′-GGATCTTCAACCACCTCGAG-3′.
CIITA, GFAP, and VIM KO cells were generated by using the lentivirus CRISPR-Cas9 system as described (47). Briefly, guide RNAs were cloned into lentiCRISPRv2 (Addgene, 49535), which were transfected cells together with the package plasmid pVSVG and psPAX2. These cells were then selected by puromycin for 10 days for other assays. Guide RNA sequences used were as follows: CIITA, 5′-GTGACAGGTAGGACCCAGCA-3′; GFAP, 5′-GCTCGATGTAGCTGGCAAAG-3′; VIM, 5′-GGTAGTCACGTAGCTCCGGC-3′.
H3.3WT knock-in cells were generated by using the CRISPR-Cas9 system as described (48). Briefly, guide RNA and donor plasmids were cloned into the vector, pX335 (hSpCas9 + guide RNA; Addgene, 42335) and pMD19-T (TaKaRa, 6013), and single clones were isolated by dilution cloning. The knock-in efficiencies were confirmed by PCR of the DNA fragment, followed by Sanger sequencing. Guide RNA sequence used was 5′-TCTTCACCCTTCCAGTAGA-3′
Immunoblotting analysis
Immunoblotting analysis was done according to standard procedures, and images were acquired using Bio-Rad ChemiDoc Touch Imaging System. Antibodies used for immunoblotting analysis are listed in table S2.
Immunofluorescence assay
A total of 2 × 105 cells in six-well plates with coverslips were used in the immunofluorescence assays. The cells were fixed with 3% paraformaldehyde solution at room temperature for 30 min and washed with phosphate-buffered saline (PBS) solution three times. Cells were permeabilized with 0.2% Triton X-100 in PBS for 5 min at room temperature. After three washes with PBS, cells were blocked with block buffer (1% bovine serum albumin in PBS) at room temperature for 30 min. The cells were then incubated with the first antibody at 4°C overnight and second antibody at room temperature for 1 hour. Cells were washed three times with PBST buffer (0.1% Tween 20 in PBS), and images were acquired using fluorescence confocal (Leica) microscopy or Olympus fluorescence microscopy. The antibodies used are listed in table S2.
ChIP and ChIP-seq
ChIP analyses of RACK7 were carried out using antibodies listed in table S2. Briefly, cells were cross-linked with 1% formaldehyde for 10 min at room temperature and stopped with 125 mM glycine. Cells were lysed with ChIP lysis buffer [50 mM Hepes (pH 7.5), 500 mM NaCl, 1 mM EDTA, 1% Triton, 0.1% Na-deoxycholate, 0.05% SDS, 1 mM PMSF, and 1 mM DTT]. Whole-cell lysate was sonicated with a Bioruptor sonicator (Diagenode). Chromatin samples were incubated with 2 μg of RACK7 antibody overnight at 4°C. The protein-DNA complexes were immobilized onto protein A/G beads (Invitrogen, 10 μl per reaction). The bound fractions were washed three times with the lysis buffer and three times with radioimmunoprecipitation assay buffer (50 mM Hepes, 300 mM LiCl, 1 mM EDTA, 0.5% NP-40, 0.7% Na-deoxycholate), and once with 50 mM NaCl in tris-EDTA. Elution and reverse cross-linking were carried out in the elution buffer [50 mM tris-HCl (pH 8.0), 10 mM EDTA, and 1% SDS] at 65°C for 6 hours. After RNase A and Proteinase K digestion, DNA was purified using the PCR Extraction Kit from Qiagen. Each ChIP-seq was repeated three times. Enrichment at specific loci was confirmed by ChIP-qPCR. The primers used in ChIP-qPCR are listed in table S3. The sequencing libraries for ChIP-seq were prepared from 2 to 5 ng of DNA using the KAPA HyperPrep Kits (KAPA, KK8504) according to the manufacturer’s instructions. Completed ChIP-seq libraries were qualified by Bioanalyzer 2100 (Agilent) and sequenced at the Base Pair Biotechnology Co. Ltd. The software used to collect and analyze the ChIP-seq data was Bowtie2.2.5. The ChIP-seq profiles were normalized to 1 million total tag numbers, and peaks were called by MACS2.1.1. Duplicated reads of ChIP-seq datasets were removed using the SAMtools.
mRNA capture and sequencing
The mRNA capture and sequencing were performed by the Base Pair Biotechnology Co. Ltd. For bioinformatics analysis, the mRNA sequencing reads were mapped to human reference genome (Ensembl, release 91) using the software Hisat2. Differentially expressed genes are calculated by the ballgown program (version 2.12.0) and DESeq program (version 1.32.0) with cutoffs as follows: FPKM (fragments per kilobase of transcript per million mapped reads) ≥0.5 (removal of genes with FPKM <0.5 in all samples), P < 0.05, and fold change ≥1.5. Two biological repeats for each cell line were used.
Heatmap analysis
The heatmap analysis was performed by R, and column was normalized by Z score, defined as Δ (FPKM − mean FPKM)/SD. The sample order of the top 50 up-regulated genes is based on the sum of log2(fold change). Two biological repeats for each cell line were used.
Correlation analysis
Pearson’s product-moment correlation of log2(fold change) RNA expression between KO and knock-in cells relative to their parental cells was analyzed by R command “cor.test.”
Quantitative PCR
Total RNA was extracted using the Simply P total RNA extraction kit (Bioflux), and then complementary DNAs were synthesized by PrimeScript RT Reagent Kit with gDNA Eraser (TaKaRa). The PCR mixture was prepared on a 384-well plate using a FastStart Universal SYBR Green Master Mix according to the manufacturer’s instructions (Roche). They were run in duplicate on a LightCycler 480 II Real-Time PCR System (Roche). Expression level of each mRNA was normalized to ACTB. Primers for each mRNA were tested with melting curve and listed in table S4.
Flow cytometry
Cells were washed with PBS and blocked with Human BD FC Block (BD Biosciences) in staining buffer (BD Biosciences) for 10 min on ice. Cells were then incubated with APC Mouse Anti-Human HLA-DR antibody (BD Biosciences) for 30 min on ice protected from light. The cells were washed and resuspended in PBS for flow cytometry. The detailed antibody’s information is listed in table S2.
FACSCalibur (BD) and CellQuest software were used to get 15,000 cells for each sample. Two fluorescence excitation wavelengths were used for each sample: one is to detect APC HLA-DR, and another is to detect YFP. SJ-HGGx6c (R6) contained a luciferase-YFP transgene. Other cells including R6WT H3.3, R6RACK7 KO, R6CIITA KO, R6WT H3.3 + CIITA KO, R6RACK7 KO + CIITA KO, R6QKI KO, R6WT H3.3 + QKI KO, R6RACK7 KO + QKI KO, R6GFAP KO, R6WT H3.3 + GFAP KO, R6RACK7 KO + GFAP KO, R6VIM KO, R6WT H3.3 + VIM KO and R6RACK7 KO + VIM KO, R6OCIAD2 KO, R6WT H3.3 + OCIAD2 KO, and R6RACK7 KO + OCIAD2 KO were all generated from SJ-HGGx6c by CRISPR-Cas9. All of them contained luciferase-YFP transgenes. YFP-positive cells were used to analyze the cell surface expression of HLA-DR. Data were analyzed by FlowJo software. The flow cytometry was performed at the Key Laboratory of Medical Molecular Virology, Ministry of Education and Public Health, School of Basic Medical Sciences, Fudan University.
Migration and invasion assays
A total of 8 × 104 or 2 × 104 cells were used in the migration and invasion assays, respectively. Cells were suspended in Neurobasal Medium containing EGF (200 μg/ml), b-FGF (200 μg/ml), PDGF-AA (100 μg/ml), PDGF-BB (100 μg/ml), and 0.0002% heparin sodium salt in PBS and then were plated in the upper chamber with Matrigel noncoated and coated membranes (24-well insert; pore size, 8 mm; BD Biosciences) for Transwell migration and invasion assays, respectively. The bottom chamber contained full medium. After 24 (migration assay) or 48 (invasion assay) hours, the bottom of the chamber insert was fixed and stained with crystal violet and counted microscopically. Each membrane was calculated three microscope fields, and the means were used for representation. All assays were performed in at least biological triplicates.
Statistical analyses
All the P values of real-time qPCR, migration, and invasion assays in the main figures and supplementary figures were calculated by two-tailed Student’s t test, and three biological repeats were used. The P values of the correlation analysis were Pearson’s product-moment correlation analyzed by R.
DNA oligonucleotides
DNA oligonucleotides used in this study were synthesized by Sangon Biotech (Shanghai) Co. Ltd. and are listed in tables S3 and S4.
Data availability
All data generated are included in the figures, supplementary figures, and supplementary files. All sequencing datasets have been deposited in the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus under accession numbers GSE138060 and GSE138077.
Supplementary Material
Acknowledgments
We thank L. Kasper, X. Zhu, and J. Zhang of St. Jude Children’s Research Hospital for help with pGBM cell culture. We thank J. Partridge from St. Jude Children’s Research Hospital for experimental help. We thank Y. Wang from School of Basic Medical Sciences, Fudan University for help with flow cytometry. Funding: This work was supported by grants to R.G. from the Ministry of Science and Technology of China (2016YFA0500700 and 2018YFA0108700) and the National Natural Science Foundation of China Program (31571318 and 31771450); to S.J.B., in part, from American Lebanese and Syrian Associated Charities (ALSAC); to Y.X. from the Ministry of Science and Technology of China (2016YFA0500700) and the National Natural Science Foundation of China (31830107 and 31821002); and to Z.L. from the National Natural Science Foundation of China (31630002 and 31470725) and the Shanghai Municipal Commission of Health and Family Planning (20144Y0103). Y.S. is an American Cancer Society Research professor and a recipient of the National Cancer Institute Outstanding Investigator Award (R35 CA210104). Author contributions: R.G. and Y.S. conceived and designed the project. S.J.B. provided critical discussions and advice for experimental design. R.G. and F.J. carried out in vitro binding, ChIP-seq, CRISPR, FACS, and MST assays. G.Y., W.X., F.J., and R.G. carried out the bioinformatics analysis. Z.L. carried out part of the biochemical assays supervised by Y.X. T.L., Y.Y., J.N.A., J.C., and Y.M. provided technical assistance and discussion. C.J. analyzed CIITA expression in pediatric HGGs. S.J.B. and C.H. established the three human patient pGBM cell lines. H.S., F.L., and Y.G.S. provided discussion of RACK7 function and experimental help. R.G., Y.S., and S.J.B. cowrote the manuscript with input from all authors. Competing interests: Y.S. is a cofounder/equity holder of Constellation Pharmaceuticals Inc. and Athelas Therapeutics Inc., an equity holder of Imago Biosciences, and a consultant for Active Motif. The other authors declare that they have no competing interests. Data and materials availability: ChIP-seq and RNA-seq data are deposited in NCBI (accession numbers GSE138060 and GSE138077). All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. Requests for patient-derived cell lines should be submitted to S. Baker at St. Jude Children’s Research Hospital and will be provided pending scientific review and a completed material transfer agreement.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/29/eaba2113/DC1
REFERENCES AND NOTES
- 1.Behjati S., Tarpey P. S., Presneau N., Scheipl S., Pillay N., Van Loo P., Wedge D. C., Cooke S. L., Gundem G., Davies H., Nik-Zainal S., Martin S., Laren S. M., Goodie V., Robinson B., Butler A., Teague J. W., Halai D., Khatri B., Myklebost O., Baumhoer D., Jundt G., Hamoudi R., Tirabosco R., Amary M. F., Futreal P. A., Stratton M. R., Campbell P. J., Flanagan A. M., Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat. Genet. 45, 1479–1482 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwartzentruber J., Korshunov A., Liu X.-Y., Jones D. T. W., Pfaff E., Jacob K., Sturm D., Fontebasso A. M., Quang D.-A. K., Tönjes M., Hovestadt V., Albrecht S., Kool M., Nantel A., Konermann C., Lindroth A., Jäger N., Rausch T., Ryzhova M., Korbel J. O., Hielscher T., Hauser P., Garami M., Klekner A., Bognar L., Ebinger M., Schuhmann M. U., Scheurlen W., Pekrun A., Frühwald M. C., Roggendorf W., Kramm C., Dürken M., Atkinson J., Lepage P., Montpetit A., Zakrzewska M., Zakrzewski K., Liberski P. P., Dong Z., Siegel P., Kulozik A. E., Zapatka M., Guha A., Malkin D., Felsberg J., Reifenberger G., von Deimling A., Ichimura K., Collins V. P., Witt H., Milde T., Witt O., Zhang C., Castelo-Branco P., Lichter P., Faury D., Tabori U., Plass C., Majewski J., Pfister S. M., Jabado N., Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231 (2012). [DOI] [PubMed] [Google Scholar]
- 3.St. Jude Children's Research Hospital–Washington University Pediatric Cancer Genome Project , Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 44, 251–253 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nacev B. A., Feng L., Bagert J. D., Lemiesz A. E., Gao J. J., Soshnev A. A., Kundra R., Schultz N., Muir T. W., Allis C. D., The expanding landscape of 'oncohistone' mutations in human cancers. Nature 567, 473–478 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lewis P. W., Müller M. M., Koletsky M. S., Cordero F., Lin S., Banaszynski L. A., Garcia B. A., Muir T. W., Becher O. J., Allis C. D., Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stafford J. M., Lee C.-H., Voigt P., Descostes N., Saldaña-Meyer R., Yu J.-R., Leroy G., Oksuz O., Chapman J. R., Suarez F., Modrek A. S., Bayin N. S., Placantonakis D. G., Karajannis M. A., Snuderl M., Ueberheide B., Reinberg D., Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Sci. Adv. 4, eaau5935 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Larson J. D., Kasper L. H., Paugh B. S., Jin H., Wu G., Kwon C.-H., Fan Y., Shaw T. I., Silveira A. B., Qu C., Xu R., Zhu X., Zhang J., Russell H. R., Peters J. L., Finkelstein D., Xu B., Lin T., Tinkle C. L., Patay Z., Onar-Thomas A., Pounds S. B., McKinnon P. J., Ellison D. W., Zhang J., Baker S. J., Histone H3.3 K27M accelerates spontaneous brainstem glioma and drives restricted changes in bivalent gene expression. Cancer Cell 35, 140–155.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pathania M., De Jay N., Maestro N., Harutyunyan A. S., Nitarska J., Pahlavan P., Henderson S., Mikael L. G., Richard-Londt A., Zhang Y., Costa J. R., Hébert S., Khazaei S., Ibrahim N. S., Herrero J., Riccio A., Albrecht S., Ketteler R., Brandner S., Kleinman C. L., Jabado N., Salomoni P., H3.3K27M cooperates with Trp53 loss and pdgfra gain in mouse embryonic neural progenitor cells to induce invasive high-grade gliomas. Cancer Cell 32, 684–700.e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Funato K., Major T., Lewis P. W., Allis C. D., Tabar V., Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science 346, 1529–1533 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu C., Jain S. U., Hoelper D., Bechet D., Molden R. C., Ran L., Murphy D., Venneti S., Hameed M., Pawel B. R., Wunder J. S., Dickson B. C., Lundgren S. M., Jani K. S., De Jay N., Papillon-Cavanagh S., Andrulis I. L., Sawyer S. L., Grynspan D., Turcotte R. E., Nadaf J., Fahiminiyah S., Muir T. W., Majewski J., Thompson C. B., Chi P., Garcia B. A., Allis C. D., Jabado N., Lewis P. W., Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 352, 844–849 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fang D., Gan H., Lee J.-H., Han J., Wang Z., Riester S. M., Jin L., Chen J., Zhou H., Wang J., Zhang H., Yang N., Bradley E. W., Ho T. H., Rubin B. P., Bridge J. A., Thibodeau S. N., Ordog T., Chen Y., van Wijnen A. J., Oliveira A. M., Xu R.-M., Westendorf J. J., Zhang Z., The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science 352, 1344–1348 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li N., Li Y., Lv J., Zheng X., Wen H., Shen H., Zhu G., Chen T.-Y., Dhar S. S., Kan P.-Y., Wang Z., Shiekhattar R., Shi X., Lan F., Chen K., Li W., Li H., Lee M. G., ZMYND8 reads the dual histone mark H3K4me1-H3K14ac to antagonize the expression of metastasis-linked genes. Mol. Cell 63, 470–484 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo R., Zheng L., Park J. W., Lv R., Chen H., Jiao F., Xu W., Mu S., Wen H., Qiu J., Wang Z., Yang P., Wu F., Hui J., Fu X., Shi X., Shi Y. G., Xing Y., Lan F., Shi Y., BS69/ZMYND11 reads and connects histone H3.3 lysine 36 trimethylation-decorated chromatin to regulated Pre-mRNA processing. Mol. Cell 56, 298–310 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wen H., Li Y., Xi Y., Jiang S., Stratton S., Peng D., Tanaka K., Ren Y., Xia Z., Wu J., Li B., Barton M. C., Li W., Li H., Shi X., ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature 508, 263–268 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen H., Xu W., Guo R., Rong B., Gu L., Wang Z., He C., Zheng L., Hu X., Hu Z., Shao Z.-M., Yang P., Wu F., Shi Y. G., Shi Y., Lan F., Suppression of enhancer overactivation by a RACK7-histone demethylase complex. Cell 165, 331–342 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ting J. P.-Y., Trowsdale J., Genetic control of MHC class II expression. Cell 109, S21–S33 (2002). [DOI] [PubMed] [Google Scholar]
- 17.Wubbolts R., Fernandez-Borja M., Oomen L., Verwoerd D., Janssen H., Calafat J., Tulp A., Dusseljee S., Neefjes J., Direct vesicular transport of MHC class II molecules from lysosomal structures to the cell surface. J. Cell Biol. 135, 611–622 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vagaska B., New S. E. P., Alvarez-Gonzalez C., D’Acquisto F., Gomez S. G., Bulstrode N. W., Madrigal A., Ferretti P., MHC-class-II are expressed in a subpopulation of human neural stem cells in vitro in an IFNγ-independent fashion and during development. Sci Rep 6, 24251 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gosselin D., Skola D., Coufal N. G., Holtman I. R., Schlachetzki J. C. M., Sajti E., Jaeger B. N., O'Connor C., Fitzpatrick C., Pasillas M. P., Pena M., Adair A., Gonda D. D., Levy M. L., Ransohoff R. M., Gage F. H., Glass C. K., An environment-dependent transcriptional network specifies human microglia identity. Science 356, eaal3222 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dong Y., Benveniste E. N., Immune function of astrocytes. Glia 36, 180–190 (2001). [DOI] [PubMed] [Google Scholar]
- 21.Furuta K., Walseng E., Roche P. A., Internalizing MHC class II-peptide complexes are ubiquitinated in early endosomes and targeted for lysosomal degradation. Proc. Natl. Acad. Sci. U.S.A. 110, 20188–20193 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.ten Broeke T., Wubbolts R., Stoorvogel W., MHC class II antigen presentation by dendritic cells regulated through endosomal sorting. Cold Spring Harb Perspect. Biol. 5, a016873 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Denzin L. K., Sant'Angelo D. B., Hammond C., Surman M. J., Cresswell P., Negative regulation by HLA-DO of MHC class II-restricted antigen processing. Science 278, 106–109 (1997). [DOI] [PubMed] [Google Scholar]
- 24.Denzin L. K., Cresswell P., HLA-DM induces CLIP dissociation from MHC class II αβ dimers and facilitates peptide loading. Cell 82, 155–165 (1995). [DOI] [PubMed] [Google Scholar]
- 25.Adhikary S., Sanyal S., Basu M., Sengupta I., Sen S., Srivastava D. K., Roy S., Das C., Selective recognition of H3.1K36 dimethylation/H4K16 acetylation facilitates the regulation of all-trans-retinoic acid (ATRA)-responsive genes by putative chromatin reader ZMYND8. J. Biol. Chem. 291, 2664–2681 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seidel S. A. I., Dijkman P. M., Lea W. A., van den Bogaart G., Jerabek-Willemsen M., Lazic A., Joseph J. S., Srinivasan P., Baaske P., Simeonov A., Katritch I., Melo F. A., Ladbury J. E., Schreiber G., Watts A., Braun D., Duhr S., Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods 59, 301–315 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wienken C. J., Baaske P., Rothbauer U., Braun D., Duhr S., Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Commun. 1, 100 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Lohsen S., Majumder P., Scharer C. D., Barwick B. G., Austin J. W., Zinzow-Kramer W. M., Boss J. M., Common distal elements orchestrate CIITA isoform-specific expression in multiple cell types. Genes Immun. 15, 543–555 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shingu T., Ho A. L., Yuan L., Zhou X., Dai C., Zheng S., Wang Q., Zhong Y., Chang Q., Horner J. W., Liebelt B. D., Yao Y., Hu B., Chen Y., Fuller G. N., Verhaak R. G. W., Heimberger A. B., Hu J., Qki deficiency maintains stemness of glioma stem cells in suboptimal environment by downregulating endolysosomal degradation. Nat. Genet. 49, 75–86 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vardjan N., Gabrijel M., Potokar M., Svajger U., Kreft M., Jeras M., de Pablo Y., Faiz M., Pekny M., Zorec R., IFN-γ-induced increase in the mobility of MHC class II compartments in astrocytes depends on intermediate filaments. J. Neuroinflammation 9, 144 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sinha S., Bheemsetty V. A., Inamdar M. S., A double helical motif in OCIAD2 is essential for its localization, interactions and STAT3 activation. Sci. Rep. 8, 7362 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Friedmann-Morvinski D., Bushong E. A., Ke E., Soda Y., Marumoto T., Singer O., Ellisman M. H., Verma I. M., Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science 338, 1080–1084 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Axelrod M. L., Cook R. S., Johnson D. B., Balko J. M., Biological consequences of MHC-II expression by tumor cells in cancer. Clin. Cancer Res. 25, 2392–2402 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosenwald A., Wright G., Chan W. C., Connors J. M., Campo E., Fisher R. I., Gascoyne R. D., Muller-Hermelink H. K., Smeland E. B., Giltnane J. M., Hurt E. M., Zhao H., Averett L., Yang L., Wilson W. H., Jaffe E. S., Simon R., Klausner R. D., Powell J., Duffey P. L., Longo D. L., Greiner T. C., Weisenburger D. D., Sanger W. G., Dave B. J., Lynch J. C., Vose J., Armitage J. O., Montserrat E., López-Guillermo A., Grogan T. M., Miller T. P., Blanc M. L., Ott G., Kvaloy S., Delabie J., Holte H., Krajci P., Stokke T., Staudt L. M.; Lymphoma/Leukemia Molecular Profiling Project , The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N. Engl. J. Med. 346, 1937–1947 (2002). [DOI] [PubMed] [Google Scholar]
- 35.Oldford S. A., Robb J. D., Codner D., Gadag V., Watson P. H., Drover S., Tumor cell expression of HLA-DM associates with a Th1 profile and predicts improved survival in breast carcinoma patients. Int. Immunol. 18, 1591–1602 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Johnson D. B., Estrada M. V., Salgado R., Sanchez V., Doxie D. B., Opalenik S. R., Vilgelm A. E., Feld E., Johnson A. S., Greenplate A. R., Sanders M. E., Lovly C. M., Frederick D. T., Kelley M. C., Richmond A., Irish J. M., Shyr Y., Sullivan R. J., Puzanov I., Sosman J. A., Balko J. M., Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat. Commun. 7, 10582 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Charette M., Marabelle A., Houot R., Turning tumour cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur. J. Cancer 68, 134–147 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Turner T. B., Meza-Perez S., Londoño A., Katre A., Peabody J. E., Smith H. J., Forero A., Norian L. A., Michael Straughn J. Jr., Buchsbaum D. J., Randall T. D., Arend R. C., Epigenetic modifiers upregulate MHC II and impede ovarian cancer tumor growth. Oncotarget 8, 44159–44170 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith H. J., McCaw T. R., Londono A. I., Katre A. A., Meza-Perez S., Yang E. S., Forero A., Buchsbaum D. J., Randall T. D., Michael Straughn J. Jr., Norian L. A., Arend R. C., The antitumor effects of entinostat in ovarian cancer require adaptive immunity. Cancer 124, 4657–4666 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tarafdar A., Hopcroft L. E. M., Gallipoli P., Pellicano F., Cassels J., Hair A., Korfi K., Jørgensen H. G., Vetrie D., Holyoake T. L., Michie A. M., CML cells actively evade host immune surveillance through cytokine-mediated downregulation of MHC-II expression. Blood 129, 199–208 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mackay A., Burford A., Carvalho D., Izquierdo E., Fazal-Salom J., Taylor K. R., Bjerke L., Clarke M., Vinci M., Nandhabalan M., Temelso S., Popov S., Molinari V., Raman P., Waanders A. J., Han H. J., Gupta S., Marshall L., Zacharoulis S., Vaidya S., Mandeville H. C., Bridges L. R., Martin A. J., Al-Sarraj S., Chandler C., Ng H.-K., Li X., Mu K., Trabelsi S., H’mida-Ben Brahim D., Kisljakov A. N., Konovalov D. M., Moore A. S., Carcaboso A. M., Sunol M., de Torres C., Cruz O., Mora J., Shats L. I., Stavale J. N., Bidinotto L. T., Reis R. M., Entz-Werle N., Farrell M., Cryan J., Crimmins D., Caird J., Pears J., Monje M., Debily M.-A., Castel D., Grill J., Hawkins C., Nikbakht H., Jabado N., Baker S. J., Pfister S. M., Jones D. T. W., Fouladi M., von Bueren A. O., Baudis M., Resnick A., Jones C., Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell 32, 520–537.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Camby I., Lefranc F., Titeca G., Neuci S., Fastrez M., Dedecken L., Schäfer B. W., Brotchi J., Heizmann C. W., Pochet R., Salmon I., Kiss R., Decaestecker C., Differential expression of S100 calcium-binding proteins characterizes distinct clinical entities in both WHO grade II and III astrocytic tumours. Neuropathol. Appl. Neurobiol. 26, 76–90 (2000). [DOI] [PubMed] [Google Scholar]
- 43.Bjerke L., Mackay A., Nandhabalan M., Burford A., Jury A., Popov S., Bax D. A., Carvalho D., Taylor K. R., Vinci M., Bajrami I., McGonnell I. M., Lord C. J., Reis R. M., Hargrave D., Ashworth A., Workman P., Jones C., Histone H3.3. mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discov. 3, 512–519 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gangemi R. M. R., Griffero F., Marubbi D., Perera M., Capra M. C., Malatesta P., Ravetti G. L., Zona G. L., Daga A., Corte G., SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells 27, 40–48 (2009). [DOI] [PubMed] [Google Scholar]
- 45.Pollard S. M., Yoshikawa K., Clarke I. D., Danovi D., Stricker S., Russell R., Bayani J., Head R., Lee M., Bernstein M., Squire J. A., Smith A., Dirks P., Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 4, 568–580 (2009). [DOI] [PubMed] [Google Scholar]
- 46.Alexander T. B., Gu Z., Iacobucci I., Dickerson K., Choi J. K., Xu B., Payne-Turner D., Yoshihara H., Loh M. L., Horan J., Buldini B., Basso G., Elitzur S., de Haas V., Zwaan C. M., Yeoh A., Reinhardt D., Tomizawa D., Kiyokawa N., Lammens T., De Moerloose B., Catchpoole D., Hori H., Moorman A., Moore A. S., Hrusak O., Meshinchi S., Orgel E., Devidas M., Borowitz M., Wood B., Heerema N. A., Carrol A., Yang Y.-L., Smith M. A., Davidsen T. M., Hermida L. C., Gesuwan P., Marra M. A., Ma Y., Mungall A. J., Moore R. A., Jones S. J. M., Valentine M., Janke L. J., Rubnitz J. E., Pui C.-H., Ding L., Liu Y., Zhang J., Nichols K. E., Downing J. R., Cao X., Shi L., Pounds S., Newman S., Pei D., Auvil J. M. G., Gerhard D. S., Hunger S. P., Inaba H., Mullighan C. G., The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature 562, 373–379 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanjana N. E., Shalem O., Zhang F., Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Platt R. J., Chen S., Zhou Y., Yim M. J., Swiech L., Kempton H. R., Dahlman J. E., Parnas O., Eisenhaure T. M., Jovanovic M., Graham D. B., Jhunjhunwala S., Heidenreich M., Xavier R. J., Langer R., Anderson D. G., Hacohen N., Regev A., Feng G., Sharp P. A., Zhang F., CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/29/eaba2113/DC1
Data Availability Statement
All data generated are included in the figures, supplementary figures, and supplementary files. All sequencing datasets have been deposited in the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus under accession numbers GSE138060 and GSE138077.