

Summary
γ-Secretases are a family of intramembrane cleaving proteases involved in various signaling pathways and diseases, including Alzheimer’s disease (AD). Cells co-express differing γ-secretase complexes, including two homologous presenilins (PSEN). We examined the significance of this heterogeneity and identified a unique motif in PSEN2 that directs this γ-secretase to late endosomes/lysosomes via a phosphorylation-dependent interaction with the AP-1 adaptor complex. Accordingly, PSEN2 selectively cleaves late endosomal/lysosomal localized substrates and generates the prominent pool of intracellular Aβ that contains longer Aβ; familial AD (FAD)-associated mutations in PSEN2 increased the levels of longer Aβ further. Moreover, a subset of FAD mutants in PSEN1, normally more broadly distributed in the cell, phenocopies PSEN2 and shifts its localization to late endosomes/lysosomes. Thus, localization of γ-secretases determines substrate specificity while FAD-causing mutations strongly enhance accumulation of aggregation-prone A 42 in intracellular acidic compartments. The findings reveal potentially important roles for specific intracellular, localized reactions contributing to AD-pathogenesis.
Introduction
γ-Secretases constitute a family of biologically and pathologically relevant intramembrane cleaving proteases (i-Clips) (Jurisch-Yaksi et al., 2013; Wolfe, 2009). They consist of four subunits and are structurally very different from other i-Clips such as the rhomboids (Freeman, 2014) and signal peptide peptidases (SPPs & SPPLs) (Voss et al., 2013), which are monomeric or dimeric proteins. The catalytic activity of the complex is provided by the PSEN1 or PSEN2 subunit isoforms, while three additional subunits, APH1A or B/C, nicastrin (NCT), and PEN-2 are needed to build a functional enzyme (De Strooper and Annaert, 2010; Edbauer et al., 2003; Takasugi et al., 2003). Thus, and disregarding alternative splicing of PSEN and APH1 subunits, at least four distinct γ-secretase complexes can coexist in the same cell (De Strooper and Annaert, 2010). To date, it is unclear why cells express so many different γ-secretases, since overexpression studies suggest that the different complexes can cleave the same substrates, and mutations in both PSEN genes can cause Alzheimer’s disease (AD). On the other hand, murine knockouts of different subunits show major phenotypical differences suggesting biologically diverse functions (Jurisch-Yaksi et al., 2013; Serneels et al., 2005). For instance, PSEN1 or APH1A knockout mice show severe embryonic phenotypes due to deficient Notch signaling, while PSEN2 or APH1B/C knockout mice display a normal life span (Serneels et al., 2005). Biochemical evidence also suggests that the four different γ-secretase complexes process the Amyloid Precursor Protein (APP) differently, generating shorter or longer Aβ peptides depending on the subunit composition (Acx et al., 2014).
Other i-Clips make use of the compartmentalization of the cell to control cleavage of their various substrates (Freeman, 2014; Golde et al., 2009; Krawitz et al., 2005). However, it is remarkable how little is known about the subcellular compartmentalization of γ-secretase complexes (De Strooper and Annaert, 2010). Here, we used a variety of approaches to establish that two major γ-secretase classes, comprising PSEN1 and PSEN2 as alternative subunits, have very different subcellular locations. We identify a motif in PSEN2 that restricts its location to late endosomes/lysosomes (LE/LYS) and interacts specifically and in a phosphorylation-dependent manner with the trans-Golgi network (TGN)/endosomal adaptor complex protein (AP) complex AP-1 (Bonifacino, 2014). This more restricted localization of PSEN2 is conserved in a wide range of cell lines, in primary neurons, and in brain. PSEN1, instead, is more broadly distributed in the cell, including the plasma membrane. This distinct distribution explains the differential preference for substrates of PSEN1-versus PSEN2-complexes. In addition, we find that PSEN2 produces a distinct intracellular pool of Aβ that has been inferred to be pathological relevant, but whose origin was never precisely determined (Friedrich et al., 2010; Gouras et al., 2010; Pensalfini et al., 2014). Moreover, familial AD (FAD) mutations in PSEN2 strongly increase intracellular aggregation-prone Aβ42 production, and some FAD-PSEN1 mutations phenocopy FAD-PSEN2 with respect to localization, substrate specificity and intracellular Aβ42/40 ratio.
Results
PSEN2/γ-secretase is restricted to late endosomes and lysosomes.
To analyze the intracellular distribution of PSEN1 and PSEN2, we fractionated a postnuclear supernatant of wild-type (WT) mouse embryonic fibroblasts (MEFs) by discontinuous sucrose/D2O gradient centrifugation. PSEN1 immunoreactivity broadly distributed in all fractions whereas PSEN2 displayed a strong enrichment in fractions containing the LE/LYS markers Rab7 and Cathepsin D. The different fractions all contained active γ-secretase, as demonstrated by de novo Aβ production using an in vitro γ-secretase assay (Figures 1A and 1A’). Magnetic isolation of LE/LYS using superparamagnetic iron oxide nanoparticles (SPIONs) confirmed that PSEN2, but not PSEN1, co-enriched with Rab7 and LAMP1 (Figure 1B). Finally, cell surface biotinylation of WT MEFs revealed a significant level of PSEN1 (9% ± 2.8, n=4) at the cell surface whereas PSEN2 was barely detected (1% ± 0.8, n=3) (Figures 1C and1C’). Similar results were obtained with several independent cell lines, underscoring a broad conservation of the distinct distribution of PSEN1 and PSEN2 (Figures 1D and 1E).
Figure 1: PSEN1 and PSEN2 have distinct subcellular distributions.
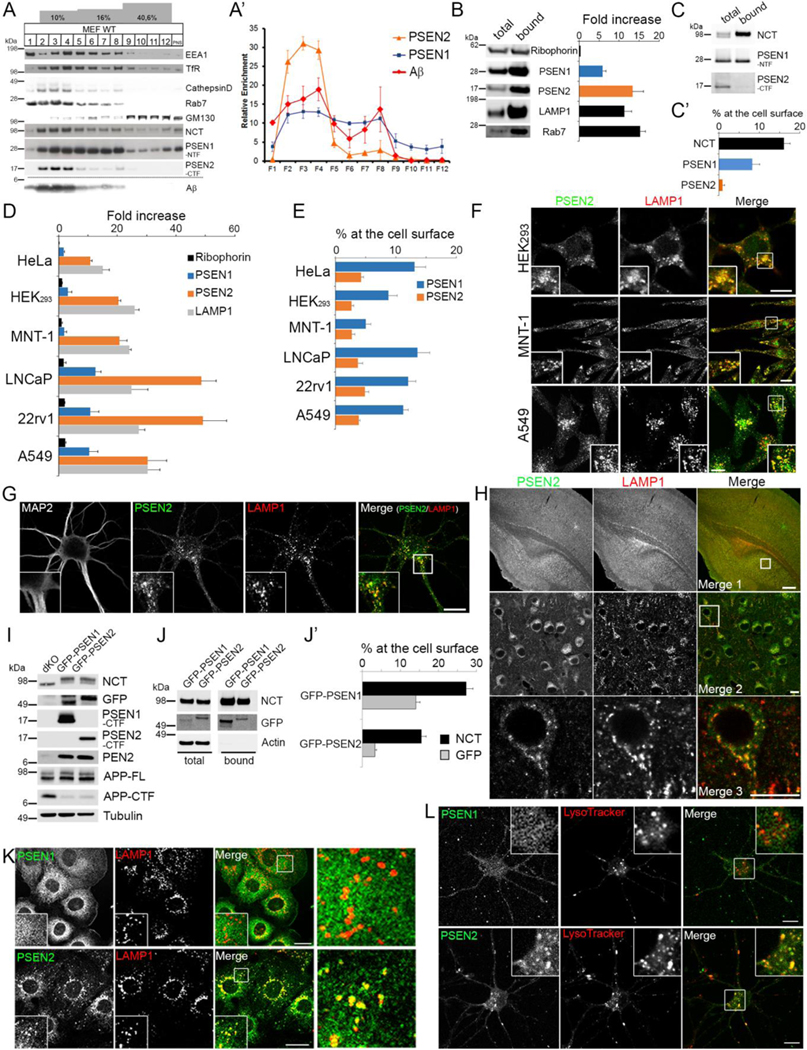
(A) Discontinuous sucrose/D2O flotation gradient of WT MEF postnuclear supernatant analyzed by Western blot using compartment specific antibodies. De novo Aβ production measured using a cell free assay.
(A’) Quantification of the enrichments from (A) for PSEN1 (blue), PSEN2 (orange) and γ-secretase activity (Aβ, red, mean ± SEM, n=2 for PSENs, n=4 for Aβ).
(B-C) Quantitative Western blot of LE/LYS isolated using SPIONs (B) and cell surface biotinylated PSENs and nicastrin (NCT). (B-C’) (mean ± SEM, n=3).
(D-E) as in (B-C) for the indicated cell lines (mean ±SEM, n=2–4).
(F) Double immunostaining of endogenous PSEN2 and LAMP1 confirming LE/LYS enrichment for PSEN2 in indicated cell lines. Bar= 10 μm.
(G-H) Endogenous PSEN2 co-localizes with LAMP1 in murine primary hippocampal neurons (DIV 7, G) and in brain cerebral cortex area (P10, H). Inset in G: zoomed area. Bar= 10 μm. (H) Merge 2 (subicular area) and 3: respective zoomed area of the square in merge 1 and 2. Merge 1: Bar= 200 μm, merge 2, 3: Bar= 10 μm.
(I) Western blot analysis of PSEN dKO MEFs and dKO MEFs stably rescued with GFP-PSEN1 or GFP-PSEN2 demonstrates restored γ-secretase assembly and activity.
(J-J’) Quantitative Western blot of cell surface biotinylated GFP-PSEN1 and -PSEN2 shows more GFP-PSEN1 at the cell surface (mean ± SEM, n=4).
(K) Co-localization of GFP-PSEN1 (top) and GFP-PSEN2 (bottom) with endogenous LAMP1. Insets and right panel: zoomed areas. Bar=10 μm.
(L) Primary hippocampal neurons (DIV7) expressing GFP-PSEN1 (green, top), or GFP-PSEN2 (green, bottom) labeled with Lysotracker red (50 nM). Inset: GFP-PSEN2 co-localizes with Lysotracker. Bar=10 μm.
See also Figure S1.
We further confirmed the restricted localization of PSEN2 using confocal microscopy. The higher expression of PSEN2 in HEK293, MNT-1 and A549 cells (Figure S1A) allowed us to demonstrate the enrichment of endogenous PSEN2 in LAMP1 positive LE/LYS (Figure 1F). Also in murine primary hippocampal neurons (Figure 1G) and in brain tissue (Figure 1H), endogenous PSEN2 strongly co-localized with LAMP1, particularly in somatodendritic compartments. Because of the unavailability of antibodies that detect low levels of endogenous PSEN1 and in order to generate a cell model to study transport regulation, we engineered N-terminally fluorescent protein (GFP or TagRFP)-tagged PSEN1 and PSEN2 constructs and retrovirally expressed them in PSEN1 and PSEN2 single and double knockout (dKO) MEFs. We selected cell lines that expressed physiological (i.e., stoichiometric to other γ-secretase subunits) levels of PSENs, as demonstrated by endoproteolysis of GFP-PSEN, mature N-glycosylation of NCT, PEN-2 stabilization and processing of APP-CTF, the direct substrate of γ-secretase (Figure 1I). Of note, NCT was more abundant (1.7 fold) at the cell surface in PSEN1-versus PSEN2-rescued dKO MEFs (Figures 1J and 1J’), confirming the presence of PSEN1 complexes at the cell surface. GFP-PSEN2 was as expected mainly restricted to LAMP1-positive LE/LYS compartments (Figure 1K). Similar patterns were observed when PSEN1 and PSEN2 were rescued in single knockout MEFs. GFP-PSEN1 co-localized with the ER-marker BIP and with transferrin receptor at the cell surface (Figure S1B), and GFP-PSEN2 with LAMP1 (Figure S1C). We also co-expressed both PSENs in dKO MEFs with identical results, implying that the overall localization of one PSEN is not influenced by the other (Figures S1B to S1D). Moreover, analysis of isolated plasma membrane sheets of GFP-PSEN1- and TagRFP-PSEN2-rescued dKO MEF lines confirmed the higher level of PSEN1 and the sparse presence of PSEN2 at the cell surface (Figure S1E). GFP-PSEN1 was as well more abundant in cell surface biotinylated fractions while GFP-PSEN2 co-purified with LAMP1 in LE/LYS isolates (Figures 1J, 1J’ and S1F). Finally, when expressed in primary hippocampal neurons, GFP-PSEN2, but not GFP-PSEN1, localized to LysoTracker-positive compartments (Figure 1L), confirming that the tagged-PSENs behave as their endogenous counterparts also in neurons.
The N-terminus of PSEN2 contains a unique acidic-dileucine sorting motif
We hypothesized that the restricted localization of PSEN2 complexes might be the consequence of a specific sorting motif. Transmembrane domains (TMDs) are highly conserved between PSEN1 and PSEN2 while the cytosolic N-terminus and loop between TMD6 and 7 are highly divergent. We swapped these domains between the PSENs (Figure 2A) and stably expressed the hybrid proteins in dKO MEFs. We found that all hybrids rescued γ-secretase maturation and activity (Figure 2B). However, whereas the cytosolic loop swap (hybrids 2 and 3) did not alter PSEN1 localization, the exchange of its N-terminus with the one of PSEN2 (hybrid 4) was sufficient to direct PSEN1 to LE/LYS, as shown by co-localization with LAMP1 (Figures 2C and C’). Conversely, replacing the N-terminus of PSEN2 by that of PSEN1 (hybrid 5) resulted in a WT PSEN1-like distribution. Thus, crucial sorting information is located within amino acids 1–76 of PSEN2.
Figure 2: Cytosolic N-terminus of PSEN2 is responsible for its localization to LE/LYS.
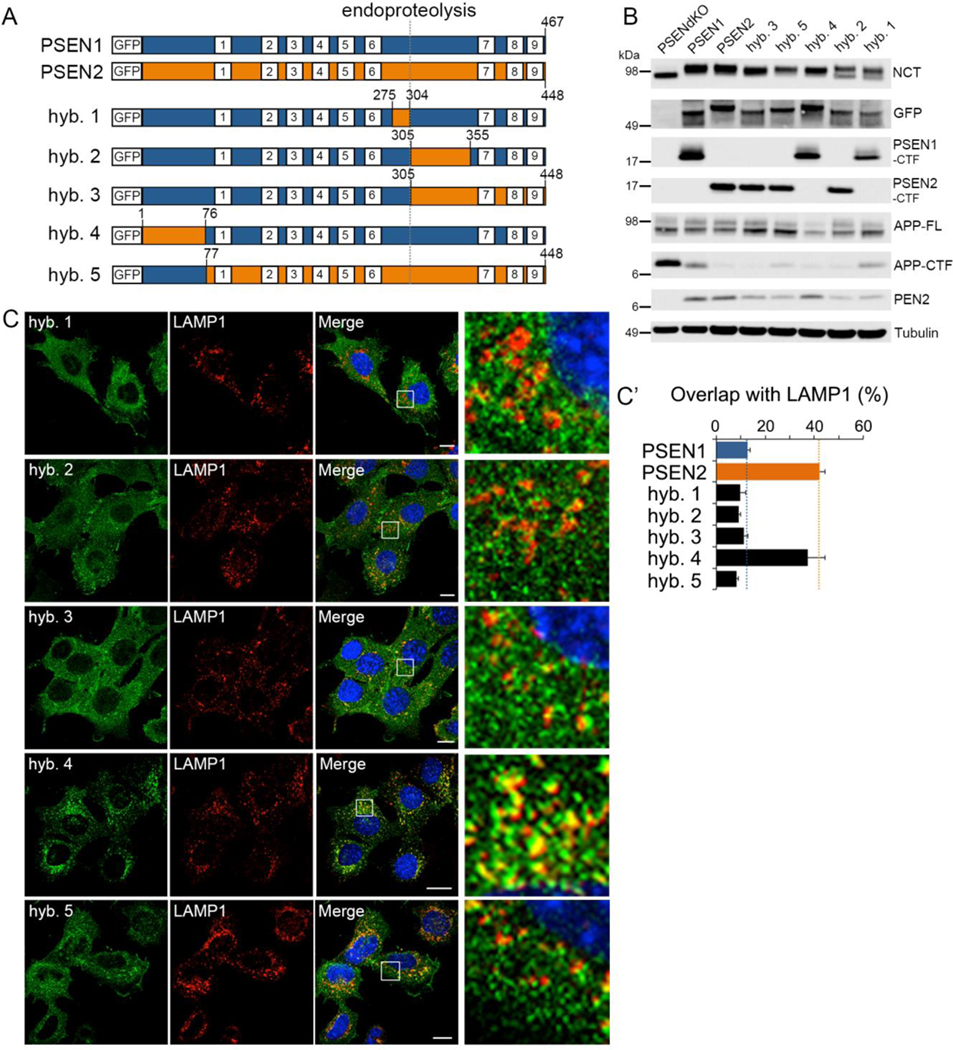
(A) Scheme of GFP-PSEN1 (blue) and GFP-PSEN2 (orange) WT and hybrids (1 to 5, see supplemental info), with nine transmembrane domains (white squares) and endoproteolytic cleavage site (dotted line).
(B) Western blot of dKO MEFs rescued with GFP-PSEN hybrids. All hybrids rescue γ-secretase assembly and activity.
(C) Co-localization of GFP-PSEN hybrids with endogenous LAMP1. Right: zoomed area. Bar=10 μm.
(C’) Quantification of (C) using Manders’ coefficient (mean ± SEM, n=3, ≥30 cells per experiment).
Inspection of the PSEN2 N-terminal segment revealed a highly conserved E16RTSLM21 sequence (Figure 3A), which fits the acidic-dileucine motif [D/E]xxxL[L/I/M] present in other proteins that are transported to LE/LYS (Traub and Bonifacino, 2013). This motif is known to bind to the heterotetrameric AP complexes AP-1, AP-2 and AP-3, which sort cargo at different stages of the endomembrane system. To determine whether the PSEN2 motif binds to these complexes, we first used a yeast 3-hybrid (Y3H) assay (Mattera et al., 2011) (Figure 3B, 3C). We found that the PSEN2 N-terminus interacted with the AP-1 γ-σ1, but not the AP-2α-σ2 and AP-3 δ-σ3, hemicomplexes (Figure 3C). This interaction was lost when essential amino acids in the PSEN2 motif (Glu16, Leu20-Met21 or combinations of them) were mutated to Ala (Figure 3C, 3D). Interestingly, interaction with the AP-2 hemicomplex was detected only when Met21 was substituted by Leu (Figure S2A), indicating that the C-terminal –LM configuration of the motif is responsible for its selective binding to AP-1.
Figure 3: Adaptor complex AP-1 binds the N-terminal conserved E16RTSLM21 motif in PSEN2.
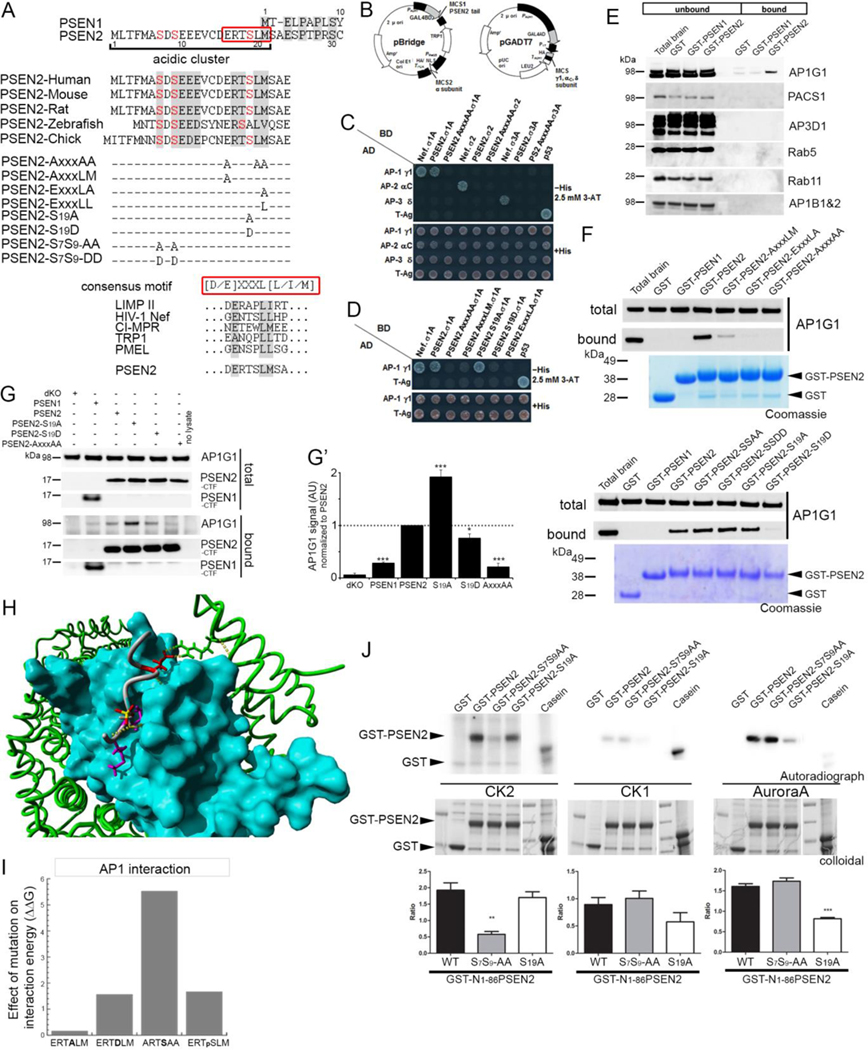
(A) Alignment of human PSEN1 and PSEN2 N-termini (top). PSEN2 N-terminus includes an acidic cluster with a conserved ERTSLM sequence (red box and lower panel) resembling the consensus [D/E]XXXL[L/I/M] and three conserved phosphorylatable serines (red). Scheme shows point mutations used in this study.
(B) Two vectors used in Y3H.
(C) Y3H shows that PSEN2 tail interacts with AP-1 γ1-σ1A but not with AP-2 αC-σ2 or AP-3 δ-σ3A. This interaction is impaired by combined replacement of Glu16, Leu20 and Met21 for Ala (PSEN2-AxxxAA). Note selective interaction of the control Nef dileucine signal with AP-1 γ1-σ1A, AP-2 αC-σ2 and AP-3 δ-σ3A and not with mismatched combinations of AP subunits.
(D) PSEN2 tail-AP-1 γ1-σ1A interaction is impaired by individual Ala substitution in the PSEN2 motif. While the S19A substitution is inconsequential, the phosphorylation-mimicking S19D substitution abolishes interaction.
(E) Western blot analysis of pull-downs of rat brain extracts with recombinant GST, GST-N1–81PSEN1 and GST-N1–86PSEN2.
(F) Same as in (E) but for single or triple Ala substitutions in the ERTSLM motif (top) and Ser to Ala/Asp mutations in N-terminus (bottom) of GST-N1–86PSEN2. Coomassie staining shows equal loading of GST-fusion proteins.
(G-G’) Co-immunoprecipitation of CHAPSO-extracts from dKO MEFs (control) and rescued cells using anti-GFP antibody. Western blot (G, quantified in G’) with anti-AP-1-γ1 shows increased binding to GFP-PSEN2-S19A compared to PSEN2 WT (mean ± SEM n=3, *** p<0.001, *p<0.05).
(H) Surface representation of the σ1 subunit (blue) and γ1subunit (green) of AP-1 hemicomplex with the VCDERTSLMS peptide from PSEN2 (gray) in the binding groove of the σ1 subunit. Glu (red) and Leu/Met (purple) interact with the γ1 and σ1 subunit, respectively. The phospho group on Ser19 which disfavors interaction, is marked in yellow/red.
(I) Theoretical effect of mutations on the interaction energy between the ‘CDERTSLMS’ motif and the AP-1 hemicomplex calculated using FoldX forcefield. All mutations, except S19A, strongly destabilize interaction (high (>1) positive ΔΔG (kcal.mol-1)).
(J) In vitro kinase assays using purified GST-PSEN2 WT and mutants as substrates incubated with CKII, CKI or Aurora A. Top: autoradiograph of phosphorylated GST fusion proteins. Note very low labeling of fusion proteins by CK1 compared to control (Casein). Middle: colloidal staining shows equal loading. Bottom: quantification of 32P incorporation in GST-fusion proteins as a ratio to colloidal gold staining (mean ± SEM n=3, *** p<0.001). See also Figures S2.
We next performed pull-down experiments from rat brain extracts using GST fused to the PSEN N-termini and again observed selective co-isolation of endogenous AP-1 only with GST-N1–86PSEN2, as detected by immunoblotting for the γ subunit (AP1G1) (Figure 3E). This interaction was not seen when critical residues in the PSEN2 motif were mutated to Ala (Figures 3F top). Finally, co-immunoprecipitation from extracts of cell lines expressing higher endogenous PSEN2 levels (Figure S1A), also confirmed interaction with AP-1 (Figure S2B).
Phosphorylation of the E16RTSLM21 motif regulates PSEN2 interaction with AP-1
Intriguingly Ser19 in the E16RTSLM21 sorting motif (Figure 3A) has been shown to undergo phosphorylation in vivo (Walter et al., 1996). We mutated this residue, and also two other conserved proximal serines (Ser7 and Ser9) in GST-N1–86PSEN2 to a non-phophorylatable Ala or a phosphomimetic Asp. Mutation of Ser19 to Asp (S19D) but not Ala (S19A) abrogated AP-1 interaction in Y3H (Figure 3D) and GST pull-down assays (Figure 3F lower panel), suggesting that the interaction of PSEN2 with AP-1 is regulated by Ser19 phosphorylation. In contrast, the other mutations did not affect interaction, again confirming the specificity of the effects of the Ser19 mutation. We then tested the physiological importance of these interactions in intact dKO cells stably expressing GFP-PSEN2 WT or the AxxxAA, S19A, and S19D mutants. All constructs were expressed at physiological levels as shown by the restoration of γ-secretase assembly and activity (Figure S2C). In line with Y3H and GST pull-down experiments, co-immunoprecipitation analysis showed that AP-1 binds to PSEN2 WT and PSEN2-S19A, but not to PSEN2-AxxxAA and only weakly to PSEN2-S19D (Figures 3G–G’). Note that under these conditions, significantly more endogenous AP-1 bound to PSEN2-S19A, indicating a stronger interaction.
The canonical [D/E]xxxL[L/I/M] motif binds to AP-1 through interaction of the C-terminal L[L/I/M] residues with σ1 (AP1S1) and through the N-terminal D/E residues with γ (AP1G1), as shown by structural and biochemical analysis (Jia et al., 2014; Kelly et al., 2008; Mattera et al., 2011). Based on the available crystal structure of AP-1 (Jia et al., 2014) we calculated the interaction energies (ΔΔG values in kcal.mol−1) for the different mutant versus WT PSEN2 motifs using FoldX forcefield (Figure 3I). We obtained high positive ΔΔG-values when substituting Leu20-Met21 and Glu16 residues to Ala, consistent with their critical importance for the binding of the PSEN2 motif to the groove of the 1 and 1 subunits, respectively (Figure 3H). In this analysis, the S19D mutation or the addition of a phosphate to Ser19 (ERTpSLM) also disfavored the interaction by more than 1.5 kcal.mol−1 (Figure 3I) as a result of non-optimal hydrogen-bonding and electrostatic interactions with the surrounding residues in the σ1 subunit. Several mutations at this position are predicted to be detrimental for the interaction in particular -and next to Asp-Glu, which is as well phosphomimetic, and the structurally related Asn (D, E and N in Figure S2D).
Although casein kinase 1 (CK1) was previously suggested to phosphorylate Ser19 (Walter et al., 1996), this was not confirmed by a kinase-substrate predicting algorithm (scansite3.mit.edu). Instead, Aurora A appeared to be a more likely candidate to phosphorylate Ser19 while Ser9 was predicted to be a CK2 site. To explore this prediction we performed in vitro kinase assays using recombinant kinases and either GST or GST-N1–86PSEN2 as substrates (Figure 3J). These data conclusively demonstrated that Ser19 is preferentially phosphorylated by Aurora A and also confirmed the phosphorylation of Ser7/Ser9 by CK2. The direct phosphorylation of Ser19 by Aurora A was furthermore confirmed by MS/MS analysis of in vitro phosphorylated trypsin digested GST-N1–86PSEN2 (Figure S2E).Taken together, these data strongly support the phosphorylation-regulated interaction of PSEN2 with AP-1.
The E16RTSLM21 motif regulates the targeting of PSEN2 to LE/LYS
We next assessed the effect of different E16RTSLM21 mutations on the localization of GFP-PSEN2 to LE/LYS. The phosphomimetic GFP-PSEN2-S19D mutant co-localized less well with LAMP1 than did WT PSEN-2 (Figure 4A and 4A’). The sorting mutant GFP-PSEN2-AxxxAA displayed an even lower co-localization with LAMP1 and exhibited a broad distribution resembling that of GFP-PSEN1 (Figure 1K). In accordance, using cell surface biotinylation, GFP-PSEN2-AxxxAA was, like GFP-PSEN1 (Figure 1J’), more abundantly localized at the plasma membrane (Figures 4B and B’). Interestingly, in primary hippocampal neurons, AP-1 regulates sorting of receptors, such as the transferrin receptor, to the somatodendritic domain (Farias et al., 2012). Using specific axonal and dendritic markers, we measured the polarity index of PSEN2 in neurons. This clearly showed that GFP-PSEN2 is exclusively present in the somatodendritic compartment, while GFP-PSEN1 localizes to both axons and dendrites (Figures 4C and 4C’). Interestingly, GFP-PSEN2-AxxxAA exhibited a non-polarized distribution, indeed suggesting that AP-1 sorts PSEN2 to somatodendritic LE/LYS in neurons (Figure 4C’). Altogether these results further validate the role of AP-1 in the sorting and localization of PSEN2.
Figure 4: E16RTSLM21 motif in PSEN2 is sufficient for its LE/LYS localization.
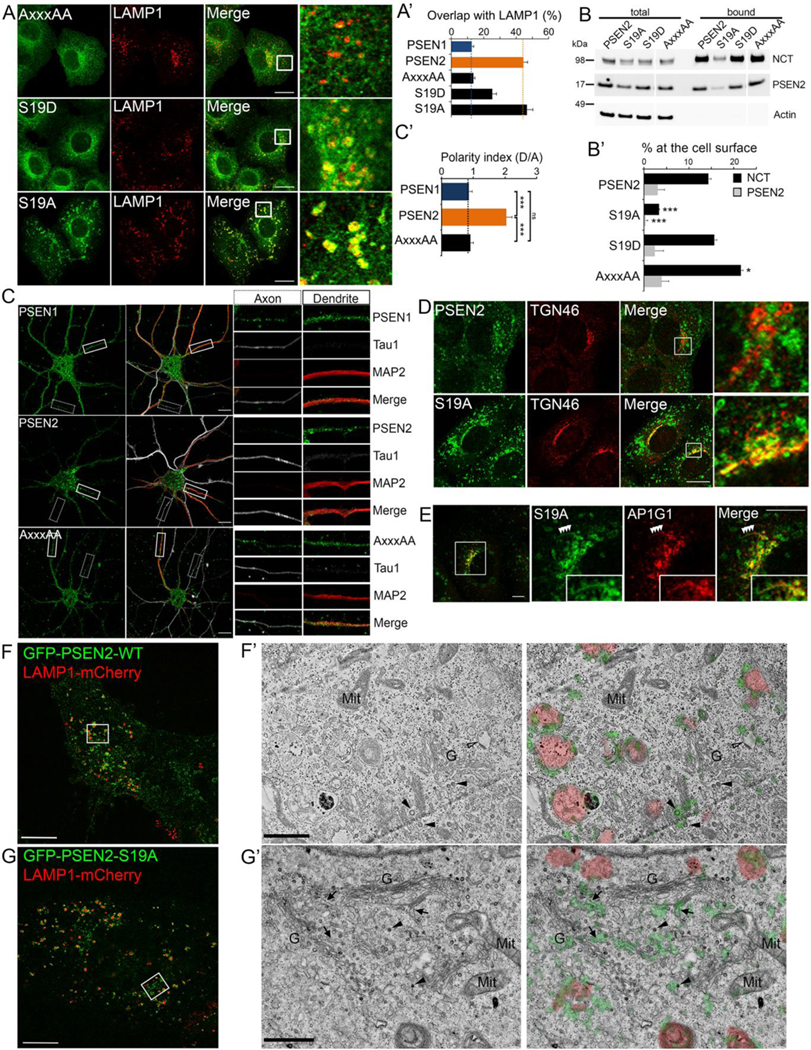
(A) Double immunostaining shows strong overlap of GFP-PSEN2-S19A but not -AxxxAA with LAMP1. Right: zoomed area. Bar=10 μm. (A’) Quantification using Mander’s coefficient (mean ± SEM, n=3, ≥30 cells per experiment).
(B-B’) Quantitative Western blot of cell surface biotinylated NCT and PSEN2-CTF for GFP-PSEN2 WT and mutant rescued cells shows low amount of S19A at the cell surface (mean ± SEM, n=4, *p<0.05, ***p<0.001).
(C) Triple fluorescent labeling of primary hippocampal neurons (DIV7) shows PSEN1 localization in axons (tau1) and dendrites (MAP2) while PSEN2 (but not PSEN2-AxxxAA) is solely present in dendrites. Bar=10μm. (C’) Polarity index (axonal/dendritic signal) calculated for GFP-PSEN1 (n=28), GFP-PSEN2 (n=31) and GFP-PSEN-AxxxAA (n=22) (mean ± SEM, ***p<0.001).
(D) Partial co-localization of GFP-PSEN2-S19A with TGN46. Right: Zoomed area. Bar=10 μm.
(E) GFP-PSEN2-S19A co-localizes with endogenous AP-1 (AP1G1) at the TGN and on emanating tubular structures (inset). Bar= 5 μm.
SIM image of GFP-PSEN2-WT (F) and -S19A cells (G) co-transfected with LAMP1-mCherry: area indicated by a square is analyzed by CLEM (F’ and G’). G: Golgi; Mit: mitochondria; arrowheads: clathrin coated vesicles; empty arrowhead: endosome with flat clathrin-coated domain; arrows: tubular structures. (F, G) Bar= 10 μm (F,G) and 1 μm (F’, G’).
Intriguingly, the non-phosphorylatable GFP-PSEN2-S19A, accumulated not only in LE/LYS but also prominently in the TGN, as shown by the co-localization with TGN46 (Figure 4D) and AP-1 (Figure 4E). Occasionally, co-localization could be observed over long tubules emanating from the TGN (Figure 4E, inset). To further support the TGN localization of GFP-PSEN2-S19A, we co-expressed LAMP1-mCherry and correlated 2-color structured illumination microscopy imaging with electron microscopy (SIM-CLEM). GFP-PSEN2 WT is mainly localized to the limiting membrane of LAMP1-positive organelles that could be identified at the ultrastructural level as LE/MVBs and lysosomes (Figure 4F and 4F’). In contrast, GFP-PSEN2-S19A localized additionally to a juxtanuclear area (Figure 4G) that at the EM level associated with tubular structures and vesicles of the TGN, as evidenced by the presence of clathrin coats and their close vicinity to Golgi stacks (<500nm,(Peden et al., 2004)) (Figure 4G’; and Movie S1 and S2 using 2-color SIM-CLEM with LAMP1-mCherry and MitoTracker, respectively).
The above results suggest that AP-1 plays a role in sorting PSEN2 from the TGN to LE/LYS, although this role may not necessarily involve direct delivery but rather routing via endosomal compartments (Braulke and Bonifacino, 2009; Saftig and Klumperman, 2009). To test the involvement of early endosomes (EE) in this routing, we overexpressed Rab5Q79L, a GTP-locked mutant blocking transport from EE. GFP-PSEN2, but not GFP-PSEN1, readily accumulated in enlarged endosomes under these conditions (Figure S3A). Moreover, overexpression of Rab11, which causes cargo to accumulate in recycling endosomes, resulted in trapping of GFP-PSEN1, but not GFP-PSEN2, in this compartment (Figure S3B). Under the same conditions, GFP-PSEN2 AxxxAA, but not the non-phosphorylatable S19A mutant, co-localized with overexpressed Rab11 (Figure S3B). Overall, these data indicate that the interaction with AP-1 directs PSEN2 via EE to LE/LYS while PSEN1 traffics through the Rab11 recycling compartment likely en route to the cell surface.
The subcellular localization of PSENs affects substrate specificity
We wondered whether the different subcellular localization of PSEN1- and PSEN2-γ-secretases could provide a basis for substrate specificity. Among candidate substrates that localize to LE/LYS, two proteins involved in the biogenesis of melanosomes (a type of lysosome-related organelle), tyrosinase-related protein (TRP1) and premelanosome protein (PMEL) (Wang et al., 2006; Watt et al., 2013). Indeed, confocal microscopy showed localization of endogenous PSEN2 to LAMP1-positive LE/LYS in MNT-1, a human melanocyte cell line (Raposo and Marks, 2007) (57% overlap, Figure 1F), while exogenous PMEL co-localized with GFP-PSEN2 in rescued dKO MEFs (Figure S4A), conclusively demonstrating that substrate and enzyme are present within the same organelles.
We next used siRNA to silence PSEN1 or PSEN2 expression in MNT-1 cells. Knockdown of PSENs did not affect the overall maturation of PMEL or TRP1 (Figure S4B). However, the γ-secretase cleavage products PMEL-CTF and TRP1-CTF accumulated dramatically in PSEN2 knockdown MNT-1 cells while only a moderate effect was seen in PSEN1 knockdown cells (Figure 5A and 5A’). Similar results were obtained with single PSEN1-or PSEN2-knockout MNT-1 cells generated by CRISPR/Cas9 genome editing (Figure 5C). Of note, the knockout cell lines showed accumulation of APP-CTF demonstrating that APP can be processed by either one of the PSENs (Figure S4C). The physiological relevance of PSEN2-catalyzed cleavage was evident from further EM analysis showing that the maturation of melanosomes was impaired in PSEN2 knockdown and knockout cells (Figures 5B, 5D, 5D’), along with a sharp decrease of stage III and IV mature melanosomes and accumulation of stage II immature melanosomes (Watt et al., 2013). Finally, melanosomes in retinal pigment epithelium (RPE) of Psen2−/− mice appeared significantly rounder, less numerous, and surrounded by an enlarged limiting membrane (Figure S4D). These data indicated that PSEN2-selective substrate processing is required for proper melanosome maturation both in culture cells and in mice.
Figure 5: Subcellular localization of γ-secretase regulates substrate accessibility.
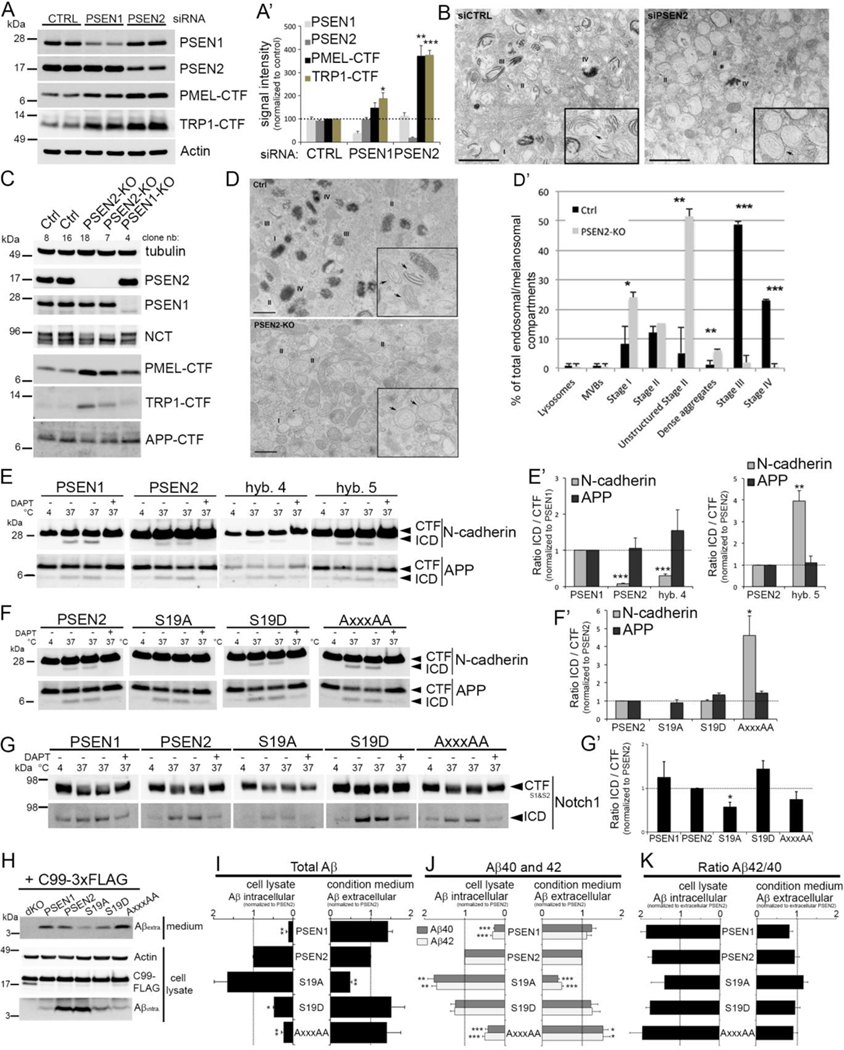
(A-A’) Quantitative Western blot of siRNA-treated MNT-1 cells (mean ± SEM, n=3).
(B) Transmission EM of WT and PSEN2-siRNA treated MNT-1 cells. I to IV indicate different stages of melanosome maturation. PSEN2 knockdown results in more immature stages. Arrows in insets specify stage II melanosomes containing normal and very thin fibrils in WT (Ctrl), and PSEN2 knockdown cells, respectively. Bar=1μm.
(C) Western blot analysis of WT MNT-1 (Ctrl) and single PSEN2 (two clones) and PSEN1 (one clone) KO cells generated using CRISPR/Cas9.
(D) Transmission EM of WT (Ctrl) and PSEN2-KO (clone 18) MNT-1 cells. Insets: round shaped unstructured immature melanosomes (arrows) accumulate in PSEN2-KO cells at the expense of mature stage II, III and IV melanosomes. Bar=500nm.
(D’) Quantification of (D) as percentage of each compartment relative to total compartments. (E-G) Western blot demonstrating ICD production from endogenous N-cadherin-, APP- and Notch1-CTFs from membrane fractions of dKO MEFs rescued with GFP-PSEN1 or -PSEN2 as indicated. (E’-G’) Quantification of (E-G) (mean ± SEM, n=4–5).
(H-I) Western blot of intracellular and secreted A from total lysate (bottom) and conditioned media (top) (quantified in I).
(J-K) ELISA of intracellular and extracellular A 40 and 42, including Aβ42/40 (mean ± SEM, n=7). For all graphs: *p<0.05, *p<0.01, ***p<0.001. See also Figure S4.
We next analyzed the distribution of more ubiquitously expressed substrates such as APP, N-cadherin and Notch. APP and Notch are distributed in intracellular compartments, and are also present at the cell surface (Perez et al., 1999; Sannerud and Annaert, 2009; Tagami et al., 2008), while N-cadherin mainly resides at the cell surface (De Strooper and Annaert, 2010). We isolated crude membrane fractions in the absence of detergent, reasoning that under these conditions the different γ-secretases would only cleave substrates that are in the same membrane compartment in vitro. We analyzed membranes isolated from cells expressing only GFP-PSEN1 or -PSEN2, or from the different cell lines expressing sorting mutants. Cell surface expressed N-cadherin-CTF was less efficiently cleaved by PSEN2/γ-secretase than by PSEN1/γ-secretase (Figure 5E and 5E’). Swapping of the sorting motif between PSEN1 and PSEN2 (hybrids 4 & 5, respectively, Figure 2A) largely reversed this preference. Expression of the other sorting mutants confirmed this observation. GFP-PSEN2-S19A, which is restricted to TGN & LE/LYS failed to process N-cadherin while GFP-PSEN2-AxxxAA showed increased N-cadherin-cleavage (Figures 5F and 5F’), reminiscent of WT PSEN1. APP-CTF was processed to the APP-intracellular domain fragment (AICD) to similar extents by both γ-secretase complexes, as expected. Also, the PSEN2 and PSEN1 sorting mutants did not affect significantly this cleavage, in agreement with the broad and rather uniform distribution of APP in various cellular membranes (Figures 5E to 5F’). PSEN1 and PSEN2 localization did not affect endogenous Notch ICD (NICD) production from its precursor substrate NEXT (following ectodomain shedding of mature Notch1 by ADAM family of metalloproteases), neither in conditions of naïve or delta ligand-induced Notch signaling (Figure 5G–G’, S4E–E‘). Thus, regulated intramembrane proteolysis of Notch1 is not confined to the cell surface. Instead, and following ectodomain shedding, NEXT is either cleaved at the cell surface or is internalized and sorted to LE/MVBs for further processing to NICD as demonstrated (Maes et al., 2014; Tagami et al., 2008). As opposed to APP, however, expression of PSEN2-S19A, which is even more depleted from the cell surface and restricted to LE/LYS and TGN, decreased NICD production, while the S19D mutation displayed more processing (Figure 5G’ and S4E‘). These observations suggest that Notch1 processing is affected by subtle changes in PSEN2 localization. This might be of relevance to cancer-related Notch mutations and/or the observed high variability in PSEN2 expression levels in different cancer cell lines (Figure S1A).
When we analyzed Aβ production in intact cells transduced with APP-CTF (APP-C99–3xFLAG), the direct substrate of γ-secretase, we confirmed that PSEN2-expressing cells secrete less Aβ40 and Aβ42 compared to PSEN1-expressing cells (Figure 5H to 5J) (Bentahir et al., 2006). Unexpectedly, this was accompanied by a significant increase in intracellular Aβ (Figure 5H–I: total Aβ; Figure 5 J: Aβ40 and Aβ42). In addition, for both PSEN1- and PSEN2-expressing cells, the intracellular ratios of long to short Aβ tended to be higher than those of the secreted peptides (Figure 5K), indicating that even in cells expressing WT PSENs relatively more aggregation-prone Aβ42 remains intracellularly or in the LE/LYS. Increases in intracellular Aβ and decreases in secreted Aβ peptides were even more pronounced with the PSEN2-S19A but not the S19D mutant, indicating that PSEN2-generated Aβ in LE/LYS does not get secreted efficiently. In agreement, the AxxxAA mutant, which fails to be delivered to LE/LYS, produced low intracellular and higher extracellular amounts of Aβ peptides reminiscent of PSEN1 processing (Figure 5J–J’). Similar results for total Aβ were obtained in cells transduced with the APPswedish mutant (Figure S4F). Thus, our data demonstrate that the differential localization of PSEN1- and PSEN2γ-secretases results in differential processing of N-cadherin and Notch1, and also has important implications for the processing of APP towards intracellular Aβ.
FAD mutations in PSEN1 and PSEN2 generate high intracellular Aβ 42/40 ratios
The >180 familial AD (FAD) mutations reported for PSEN1 are in great contrast to the limited number of FAD-PSEN2 mutations. So far, only 29 have been identified, 13 of which are pathogenic ((Canevelli et al., 2014); http://www.molgen.vib-ua.be/ADMutations, (Cruts et al., 2012)). No clear reason for this discrepancy currently exists. Our findings break new ground for studying the effect of FAD mutations on localization and production of intracellular Aβ peptides. We introduced four FAD mutations, T122P, N141I, M239V, or M239I, into GFP-PSEN2, and expressed these constructs stably in PSEN dKO MEFs (Figure S5A). FAD mutations did not affect the subcellular localization of PSEN2, as these constructs displayed identical co-localizations with LAMP1 (Figure 6A–B) but differently affected substrate processing. Both the PSEN2 T122P and N141I mutations strongly decreased N-cadherin- and Notch-CTF processing but had no effect on AICD production (Figure 6C). The opposite effects were seen for the related FAD M239V and M239I, which mainly affected AICD production while sparing other substrates. All FAD mutations significantly decreased extracellular Aβ which is caused by a major drop in Aβ40 (Figure 6D–E). While this is compensated by increased Aβ42, what was unanticipated however is that this compensation is much more pronounced in the intracellular pool resulting in a dramatic rise in Aβ42/40 ratios, with FAD-N141I being the most extreme (Aβ42/40>50 fold compared to WT PSEN2) (Figure 6F). Hence, the FAD mutations in PSEN2 suggest that the effects on carboxypeptidase-like processing of γ-secretase are more strongly enhanced in the LE/LYS compartments, indicating that the localization modulates this important pathogenic aspect of PSEN function.
Figure 6: Most intracellular Aβ is generated by PSEN2 γ-secretase.
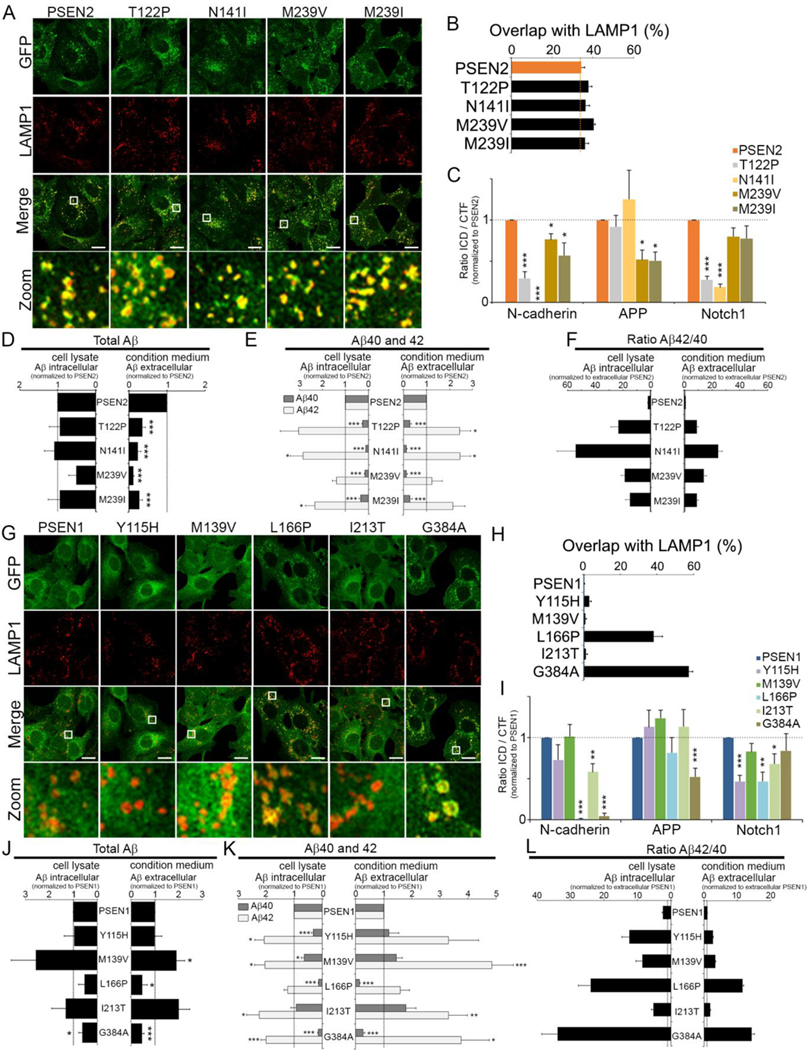
(A-B) FAD mutations do not affect co-localization of PSEN2 with LAMP1 in LE/LYS. Bottom: zoomed area. Bar=10 μm. (B) Overlap quantified using Mander’s coefficient (mean ± SEM, n=3, ≥30 cells per experiment).
(C) Quantification of ICD production from indicated substrates starting from membrane fractions of PSEN2 WT or PSEN2-FAD rescued cell lines (mean ± SEM; n=5).
(D) Quantitative Western blot of total secreted and intracellular Aβ from PSEN2-WT and -FAD rescued cells (mean ± SEM; n=6).
(E) as in (D) but using ELISA to quantify secreted and intracellular Aβ40 and −42 as well as Aβ42/40 ratios (F) (mean ± SEM; n=6).
(G-H) as in (A-B) but for GFP-PSEN1 WT and FAD mutants.
(I) as in (C): Note that FAD PSEN1-L166P and G384A are unable to process N-cadherin CTF. (J-L) as in (D-F). PSEN1 mutations most strongly increase intracellular 42/40 ratio with highest ratio for L166P and G384A.
(C, D, E, I, J, K) *p<0.05; **p<0.01; ***p<0.001. See also Figures S5.
We next wondered whether FAD-PSEN1 mutations might affect localization and intracellular Aβ generation too. We selected five common mutations and generated stably rescued dKO MEFs (Figure S5B). To our surprise, two out of five mutations (L166P and G384A) caused re-localization of GFP-PSEN1 to LAMP1-positive organelles indistinguishable from PSEN2 (Figures 6G and 6H). Similar to PSEN2, they failed to process N-cadherin-CTF to ICD while affecting other substrates to a lesser extent. Interestingly, several FAD mutations decreased NICD production while not affecting AICD or N-cadherin ICD (Figure 6I), underscoring that individual mutations differently affect major substrates which could contribute to pathology.
Again, the loss of Aβ40 concomitant with increases in Aβ42 was strongly observed in the mutations that affected the localization of PSEN1 and this most noticeably in the intracellular pool, resulting here also in the highest Aβ42/40 ratios (Figures 6K and 6L). In conclusion, we demonstrated that some FAD-PSEN1 mutations additionally act strongly on the localization of γ-secretase which apparently significantly promotes the generation of intracellular long Aβ42. We also obtained fibroblasts from one FAD-PSEN2 N141I patient. Remarkably, quantitative effects of total Aβ were less pronounced, in line with the fact that three normal psen alleles are present (Fig. S5C-D). However high intracellular Aβ42 was observed, suggesting that the pathogenic allele acts as expected. In these fibroblasts we find thus a selective high intracellular Aβ42, which is not observed in control fibroblasts and independent FAD-PSEN1 patient fibroblast lines, and correlates with the more restricted LE/LYS localization of PSEN2. Importantly, the effect of such mutation is unlikely to become clear from studies limited to extracellular Aβ as the main effects occur in the intracellular compartment.
Discussion
We demonstrate here that PSEN2/γ-secretase is mainly restricted to a specific subcellular compartment, i.e. LE/LYS, in contrast to PSEN1/γ-secretase, which is broadly distributed in the cell including plasma membrane. This differential subcellular compartmentalization of the enzymes provides a mechanism for the elusive substrate specificity of the different γ-secretases and contradicts the dogma that γ-secretases indiscriminately cleave many substrates. The more restricted compartmentalization of PSEN2, in contrast to PSEN1, contributes to the intracellular pool of Aβ peptide previously associated with the AD disease process (Bayer and Wirths, 2011; Gouras et al., 2010; Pensalfini et al., 2014). The fact that all FAD-PSEN2 mutations studied here most dramatically increase intracellular Aβ42/40 ratios, and the discovery of FAD-PSEN1 mutations that phenocopy FAD-PSEN2 mutations, put our findings also in the context of the understanding of the pathophysiology of AD, stressing the importance of the intracellular generation of aggregation-prone Aβ42.
Our work focused mainly on the cellular pathways followed by the PSEN2 complex, which is targeted to LE/LYS via the selective interaction with the AP-1 adaptor complex (Figure 7). PSEN1-complexes that lack such AP-1 interaction motif follow different trafficking routes, frequenting Rab11-positive recycling endosomes, and are more abundant at the cell surface. The molecular basis for this different cell biological behavior between PSEN1 and PSEN2 is provided by a sorting signal E16RTSLM21 in the PSEN2 N-terminus that had escaped attention until now. Such motif binds the γ1-σ1 (AP1G1-AP1S1) hemicomplex of the AP-1 adaptor complex (Mattera et al., 2011) and mediates transport of PSEN2-complexes from the TGN likely via early endosomal compartments to their final residency in LE/LYS (Figures 3, 4 and 7). Mutating essential amino acids in this motif abrogates binding to AP-1, as shown by Y3H assays, pull-down experiments, and in silico structural analysis. This same interaction also explains the somatodendritic polarity of PSEN2 in LE/LYS compartments of primary hippocampal neurons (Figures 1G and 4C) (Bonifacino, 2014; Farias et al., 2012). Failure of PSEN2 to bind to AP-1 as evidenced by mutations in the E16RTSLM21 motif results in a non-polarized distribution of PSEN2 in dendrites and axons (Figure 4C).
Figure 7: Distinct endosomal transport itineraries of PSEN1 and PSEN2.
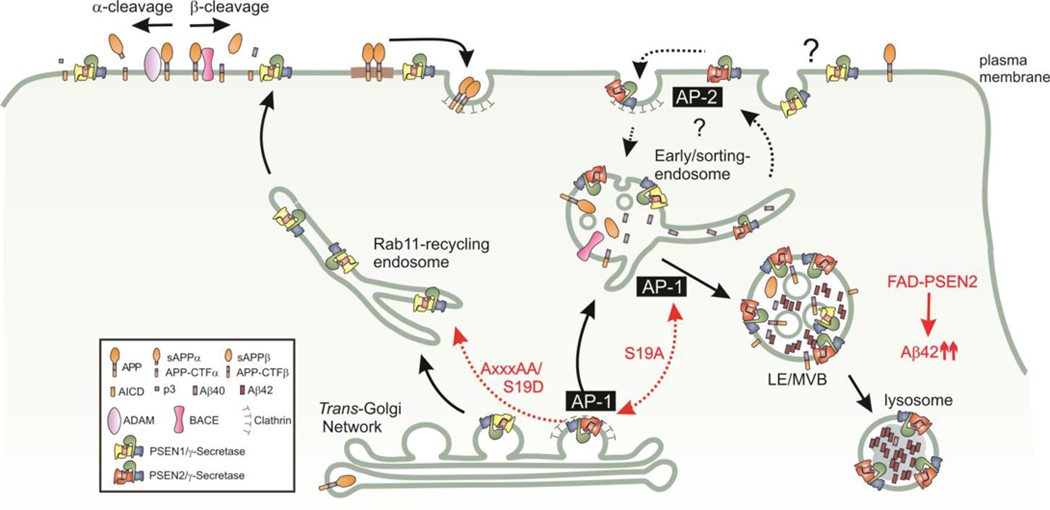
PSEN1 (yellow) complexes are sorted from the TGN to the cell surface likely via the Rab11-positive recycling compartments. In contrast, interaction with AP-1 via a conserved ERTSLM-motif allows PSEN2 (red) complexes to be sorted to LE/LYS, probable via early/sorting endosomes. Phosphorylation of Ser19 affects PSEN2 sorting. Mutation to Ala confers a more strict localization in LE/LYS and TGN. Substituting Ser19 to Asp (phosphomimetic) decreases binding and results in a more randomized distribution. Changing critical residues in the motif (AxxxAA) fully prevents AP-1 binding, resulting in “default” sorting of PSEN2/γ-secretase along the PSEN1/γ-secretase transport route (red dashed arrow). LE/LYS Localization of PSEN2/γ-secretase accounts for a major pool of intracellular Aβ, wherein relative Aβ42 levels become strongly increased by FAD mutations.
Two features of this motif emerge from the current study. First, the C-terminal hydrophobic Met determines specificity for AP-1 interaction. Y3H assays demonstrated that mutation of this residue to Leu confers binding to AP-2 but not AP-3, suggesting that yet other structural elements exist that prevent it from interacting with AP-3 (Figure S2A). Second, the interaction with AP-1 is abrogated by phosphomimetic mutations of Ser19 in the E16RTSLM21 motif. We found that Ser19 behaves as an Aurora A kinase target. Although Aurora A is predominantly known for its role in cell cycle regulation and mitosis (Nikonova et al., 2013), this kinase has recently been linked to other functions such as neurite outgrowth (Takitoh et al., 2012) and mitochondria-associated ER membrane control (Kashatus et al., 2011). Moreover, calcium release from the ER, which is affected in AD pathogenesis, might activate Aurora A (Plotnikova et al., 2010).
The Ser19 to Ala mutation provides the other extreme situation. This mutant shows enhanced binding to AP-1 in co-IP experiments and displays a significant TGN localization different from the localization of wild-type PSEN2 (Figures 3G’, 3G’, 4D). This suggests that PSEN2 is dynamically sorted between TGN and LE/LYS and that AP-1 mediates bi-directional transport (Bonifacino, 2014). The effect of the Ser19 to Ala mutation might then be explained by the enhanced interaction with AP-1, which keeps the PSEN2 mutant associated longer with exit carriers from the TGN and LE/LYS. The apparent correlation of GFP-PSEN2 S19A fluorescence with clathrin-coated tubules and vesicles in the TGN as shown by SIM-CLEM at the ultrastructural level (Figure 4G–G’) supports this interpretation.
The different mutants and the transplantation experiments of the E16RTSLM21 motif to PSEN1 indicate that this signal is necessary and sufficient to traffic PSEN2 to LE/LYS. Apparently the other subunits, and in particular the variable APH-1 subunit, are not required in this regulation. Other lines of evidence suggest that the APH1 subunits are responsible for the biochemical differences and/or conformations of the respective γ-secretases with APH-1B favoring the generation of longer Aβ peptides (Acx et al., 2014; Serneels et al., 2009).
An interesting finding of our study is that localization of the PSEN2/γ-secretase plays a major and unanticipated role in the building up of an intracellular Aβ pool. Previous work has indicated that an intraneuronal Aβ progressively accumulates in AD mice models and in human AD brain (Gouras et al., 2010). Since this feature precedes the appearance of tangles and amyloid plaques, accumulation of intracellular Aβ peptide might be a very early event in the pathological cascade leading to AD (Takahashi et al., 2002). We show here that PSEN2 γ-secretase generates the main source of this intracellular pool which is instigated by its more restricted localization in LE/LYS. Interestingly intracellular accumulation of Aβ is shown to impair proper LE/MVB sorting (Almeida et al., 2006). Moreover, high local concentrations of Aβ in these organelles, together with an acidic pH has profound effects on the conformation, aggregation and probably toxicity of Aβ (Esbjorner et al., 2014; Hu et al., 2009) which may be further accelerated by relative increases in more toxic Aβ42 (Kuperstein et al., 2010). PSEN FAD mutations essentially affect the processivity of γ-secretase, resulting in release of longer Aβ (Chavez-Gutierrez et al., 2012); however our work here suggests that some mutations in PSEN1 can also shift the subcellular localization to LE/LYS which is directly correlated with increased intracellular Aβ42 at the expense of Aβ40 much alike mutations in PSEN2. This adds an important and completely unexpected cellular dimension that potentially could impact our understanding of the pathological effects of FAD-PSEN and in particular FAD-PSEN2 mutations. Given the strikingly congruent effects of all PSEN2 mutations studied here, we speculate that intracellular Aβ toxicity may lead to LE/LYS dysfunction causing a profound disturbance of proteostasis in the affected cells. It is too early to speculate that this mechanisms could ultimately also play in other forms of AD. At least several FAD-PSEN1 mutations caused re-localization to LE/LYS thereby mimicking the FAD-PSEN2 effects, also with respect to substrate processing. It is possible that in other mutations the effects on production of Aβ dominate the picture, but even there it is possible that long Aβ gets endocytosed and accumulates in the intracellular compartment.
Furthermore, it should be realized that almost all FAD-PSEN1 mutations studied so far result in enzymes that are relatively less efficient in processing than their wild-type counterparts (Chavez-Gutierrez et al., 2012). This may cause spillover of APP-CTF to the PSEN2 complex, resulting in the production of more Aβ in acidic compartments, where Aβ is more prone to aggregation (Esbjorner et al., 2014; Hu et al., 2009). Secondly, it should be noted that early disturbances in endo/lysosomal compartments have been documented as an early characteristic of AD brain ((Cataldo et al., 2000; Ginsberg et al., 2010), reviewed in (Peric and Annaert, 2015)) and GWAS studies have also evidenced the crucial role of the endolysosomal sorting system in sporadic AD (Karch and Goate, 2015). Our work may indeed provide an important clue towards the understanding of a crucial pathogenic circle in AD: abnormal processing of APP-CTF (as occurs in FAD) in endo/lysosomes might affect their function; reversibly subtle alterations in trafficking of APP or in endo/lysosomal biology might deviate Aβ generation to these compartments, increasing the pool of intracellular Aβ and causing AD in a pernicious feed-forward cycle.
The physiologically most important conclusion from the current work is that sub-compartmentalization of the different γ-secretases and their substrates provides specificity as well as spatial and temporal control of their proteolytic activities. Instead of being specific “proteasomes of the membrane” (Kopan and Ilagan, 2004), γ-secretases appear to function as intriguingly spatially regulated enzymes with precise functions in different physiological pathways. Additional work in proper cellular and mouse models is needed to further support the potential importance for AD pathogenesis and which may provide a rationale for the development of PSEN2/γ-secretase specific inhibitors that will act at the heart of intracellular Aβ generation.
Experimental procedures
Additional procedures and details are in Supplemental Experimental Procedures.
SIM-CLEM
GFP-PSEN2 rescued cells grown on glass bottom dishes co-expressing LAMP1-mCherry or labeled with MitoTracker were fixed and Z-sections generated by SIM followed by EM processing.
Molecular Modeling
The structure of the AP1 core (PDBID: 4P6Z) was used as template for modelling binding with the PSEN2 peptide motif VCDERTSLMS using FoldX force field (Schymkowitz et al., 2005). Effects of mutations on interaction energies was analyzed using BuildModel and Analyze Complex command. Molecular graphics were generated with YASARA (Krieger et al., 2002).
PSEN1 and PSEN2 knockout MNT-1 cells
CRISPR Design Tool (http://www.genome-engineering.org/crispr/) was used to select the genomic sequence target in hPSEN1 (5’-ggatggactgcgtggctcat-3’) and hPSEN2 (5’-gaccgctatgtctgtagtgg3’). Oligo pairs encoding 20 nucleotides guide sequences were annealed and ligated into the plasmid pX330 (Addgene). MNT-1 cells were transfected using JetPRIME and KO clones selected by serial dilution.
Isolation of LE/LYS
Cells were incubated (15 min, 37°C) with DMSA-coated SPIONs (0.2 mg/ml), washed with PBS, and incubated in fresh medium (4 h, 37°C). Harvested cells were centrifuged (180g, 10 min) and pellets resuspended in homogenization buffer (HB; 250 mM sucrose, 5 mM Tris and 1 mM EGTA pH 7.4 supplemented with PI). After cell cracking a postnuclear supernatant was loaded on a LS column placed in a strong magnetic field (0.5 T, SuperMACSII, Miltenyi). Nonmagnetic material was removed and after detachment from the magnet, the bound LE/LYS were eluted, centrifuged (126,000g, 1 h) and analyzed.
Aβ
Cells were processed 24 h after transfection with C99-3xFLAG (or APPsw). Conditioned media were cleared by centrifugation and cells lysed in lysis buffer (50 mM Tris-HCL, pH 7.4; 150 mM NaCl; 1% Triton X-100; 0.1% SDS; 0.5% sodium deoxycholate). Total Aβ was detected by Western blot (6E10 antibody). Aβ40 and −42 were quantified by ELISA (JRF/cAb40/28 antibody for Aβ1–40, JRF/cAb42/26 for Aβ1–42, and detection antibody JRF/AbN/25).
Supplementary Material
Acknowledgements
Financial support was from VIB (WA, BDS, FR, JS), KU Leuven (C16/15/073 and IDO/12/020 to WA), federal government (IAP P7/16: WA, BDS, FR, JS), Hercules (AKUL/09/037, 11/30, 13/39 to WA), SAO (S#14017 to WA) and NICHD Intramural Program (ZIA HD001607 to JSB). AKT and GDB held PhD fellowships of VIB and IWT. We thank K. Ivanova for initial FP-PSEN rescue. PE was a Marie-Curie postdoc and RW is a FWO fellow. AG, RH and MV are supported by EU-ERC (208259); LR and GvN by “Fondation ARC” (n°DOC20130606986 and # PJA20131200286). We thank PICT-IBiSA (Institut Curie, Paris) and France-BioImaging (ANR-10-INSB-04).
Footnotes
Supplemental information includes Supplemental Experimental procedures, five figures, and two movies.
References
- Acx H, Chavez-Gutierrez L, Serneels L, Lismont S, Benurwar M, Elad N, and De Strooper B (2014). Signature amyloid beta profiles are produced by different gamma-secretase complexes. The Journal of biological chemistry 289, 4346–4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida CG, Takahashi RH, and Gouras GK (2006). Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. The Journal of neuroscience : the official journal of the Society for Neuroscience 26, 4277–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer TA, and Wirths O (2011). Intraneuronal Abeta as a trigger for neuron loss: can this be translated into human pathology? Biochemical Society transactions 39, 857–861. [DOI] [PubMed] [Google Scholar]
- Bentahir M, Nyabi O, Verhamme J, Tolia A, Horre K, Wiltfang J, Esselmann H, and De Strooper B (2006). Presenilin clinical mutations can affect gamma-secretase activity by different mechanisms. Journal of neurochemistry 96, 732–742. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS (2014). Adaptor proteins involved in polarized sorting. The Journal of cell biology 204, 7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braulke T, and Bonifacino JS (2009). Sorting of lysosomal proteins. Biochimica et biophysica acta 1793, 605–614. [DOI] [PubMed] [Google Scholar]
- Canevelli M, Piscopo P, Talarico G, Vanacore N, Blasimme A, Crestini A, Tosto G, Troili F, Lenzi GL, Confaloni A, et al. (2014). Familial Alzheimer’s disease sustained by presenilin 2 mutations: systematic review of literature and genotype-phenotype correlation. Neuroscience and biobehavioral reviews 42, 170–179. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, and Nixon RA (2000). Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. The American journal of pathology 157, 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez-Gutierrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H, et al. (2012). The mechanism of gamma-Secretase dysfunction in familial Alzheimer disease. The EMBO journal 31, 2261–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruts M, Theuns J, and Van Broeckhoven C (2012). Locus-specific mutation databases for neurodegenerative brain diseases. Human mutation 33, 1340–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, and Annaert W (2010). Novel research horizons for presenilins and gamma-secretases in cell biology and disease. Annual review of cell and developmental biology 26, 235–260. [DOI] [PubMed] [Google Scholar]
- Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, and Haass C (2003).Reconstitution of gamma-secretase activity. Nature cell biology 5, 486–488. [DOI] [PubMed] [Google Scholar]
- Esbjorner EK, Chan F, Rees E, Erdelyi M, Luheshi LM, Bertoncini CW, Kaminski CF, Dobson CM, and Kaminski Schierle GS (2014). Direct observations of amyloid beta self-assembly in live cells provide insights into differences in the kinetics of Abeta(1–40) and Abeta(1–42) aggregation. Chemistry & biology 21, 732–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farias GG, Cuitino L, Guo X, Ren X, Jarnik M, Mattera R, and Bonifacino JS (2012). Signal-mediated, AP-1/clathrin-dependent sorting of transmembrane receptors to the somatodendritic domain of hippocampal neurons. Neuron 75, 810–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman M (2014). The Rhomboid-Like Superfamily: Molecular Mechanisms and Biological Roles. Annual review of cell and developmental biology. [DOI] [PubMed] [Google Scholar]
- Friedrich RP, Tepper K, Ronicke R, Soom M, Westermann M, Reymann K, Kaether C, and Fandrich M (2010). Mechanism of amyloid plaque formation suggests an intracellular basis of Abeta pathogenicity. Proceedings of the National Academy of Sciences of the United States of America 107, 1942–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD, Alldred MJ, Counts SE, Cataldo AM, Neve RL, Jiang Y, Wuu J, Chao MV, Mufson EJ, Nixon RA, et al. (2010). Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer’s disease progression. Biological psychiatry 68, 885–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golde TE, Wolfe MS, and Greenbaum DC (2009). Signal peptide peptidases: a family of intramembrane-cleaving proteases that cleave type 2 transmembrane proteins. Seminars in cell & developmental biology 20, 225–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouras GK, Tampellini D, Takahashi RH, and Capetillo-Zarate E (2010). Intraneuronal beta-amyloid accumulation and synapse pathology in Alzheimer’s disease. Acta neuropathologica 119, 523–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Crick SL, Bu G, Frieden C, Pappu RV, and Lee JM (2009). Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proceedings of the National Academy of Sciences of the United States of America 106, 20324–20329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia X, Weber E, Tokarev A, Lewinski M, Rizk M, Suarez M, Guatelli J, and Xiong Y (2014). Structural basis of HIV-1 Vpu-mediated BST2 antagonism via hijacking of the clathrin adaptor protein complex 1. eLife 3, e02362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurisch-Yaksi N, Sannerud R, and Annaert W (2013). A fast growing spectrum of biological functions of gamma-secretase in development and disease. Biochimica et biophysica acta 1828, 2815–2827. [DOI] [PubMed] [Google Scholar]
- Karch CM, and Goate AM (2015). Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biological psychiatry 77, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashatus DF, Lim KH, Brady DC, Pershing NL, Cox AD, and Counter CM (2011). RALA and RALBP1 regulate mitochondrial fission at mitosis. Nature cell biology 13, 1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly BT, McCoy AJ, Spate K, Miller SE, Evans PR, Honing S, and Owen DJ (2008). A structural explanation for the binding of endocytic dileucine motifs by the AP2 complex. Nature 456, 976–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopan R, and Ilagan MX (2004). Gamma-secretase: proteasome of the membrane? Nature reviews Molecular cell biology 5, 499–504. [DOI] [PubMed] [Google Scholar]
- Krawitz P, Haffner C, Fluhrer R, Steiner H, Schmid B, and Haass C (2005). Differential localization and identification of a critical aspartate suggest non-redundant proteolytic functions of the presenilin homologues SPPL2b and SPPL3. The Journal of biological chemistry 280, 39515–39523. [DOI] [PubMed] [Google Scholar]
- Krieger E, Koraimann G, and Vriend G (2002). Increasing the precision of comparative models with YASARA NOVA--a self-parameterizing force field. Proteins 47, 393–402. [DOI] [PubMed] [Google Scholar]
- Kuperstein I, Broersen K, Benilova I, Rozenski J, Jonckheere W, Debulpaep M, Vandersteen A, Segers-Nolten I, Van Der Werf K, Subramaniam V, et al. (2010). Neurotoxicity of Alzheimer’s disease Abeta peptides is induced by small changes in the Abeta42 to Abeta40 ratio. The EMBO journal 29, 3408–3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes H, Kuchnio A, Peric A, Moens S, Nys K, De Bock K, Quaegebeur A, Schoors S, Georgiadou M, Wouters J, et al. (2014). Tumor vessel normalization by chloroquine independent of autophagy. Cancer cell 26, 190–206. [DOI] [PubMed] [Google Scholar]
- Mattera R, Boehm M, Chaudhuri R, Prabhu Y, and Bonifacino JS (2011). Conservation and diversification of dileucine signal recognition by adaptor protein (AP) complex variants. The Journal of biological chemistry 286, 2022–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikonova AS, Astsaturov I, Serebriiskii IG, Dunbrack RL Jr., and Golemis EA (2013). Aurora A kinase (AURKA) in normal and pathological cell division. Cellular and molecular life sciences : CMLS 70, 661–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peden AA, Oorschot V, Hesser BA, Austin CD, Scheller RH, and Klumperman J(2004). Localization of the AP-3 adaptor complex defines a novel endosomal exit site for lysosomal membrane proteins. The Journal of cell biology 164, 1065–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pensalfini A, Albay R 3rd, Rasool S, Wu JW, Hatami A, Arai H, Margol L, Milton S, Poon WW, Corrada MM, et al. (2014). Intracellular amyloid and the neuronal origin of Alzheimer neuritic plaques. Neurobiology of disease 71, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez RG, Soriano S, Hayes JD, Ostaszewski B, Xia W, Selkoe DJ, Chen X, Stokin GB, and Koo EH (1999). Mutagenesis identifies new signals for beta-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including Abeta42. The Journal of biological chemistry 274, 18851–18856. [DOI] [PubMed] [Google Scholar]
- Peric A, and Annaert W (2015). Early etiology of Alzheimer’s disease: tipping the balance toward autophagy or endosomal dysfunction? Acta neuropathologica 129, 363–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotnikova OV, Pugacheva EN, Dunbrack RL, and Golemis EA (2010). Rapid calcium-dependent activation of Aurora-A kinase. Nature communications 1, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo G, and Marks MS (2007). Melanosomes--dark organelles enlighten endosomal membrane transport. Nature reviews Molecular cell biology 8, 786–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saftig P, and Klumperman J (2009). Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nature reviews Molecular cell biology 10, 623–635. [DOI] [PubMed] [Google Scholar]
- Sannerud R, and Annaert W (2009). Trafficking, a key player in regulated intramembrane proteolysis. Seminars in cell & developmental biology 20, 183–190. [DOI] [PubMed] [Google Scholar]
- Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, and Serrano L (2005). The FoldX web server: an online force field. Nucleic acids research 33, W382–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serneels L, Dejaegere T, Craessaerts K, Horre K, Jorissen E, Tousseyn T, Hebert S, Coolen M, Martens G, Zwijsen A, et al. (2005). Differential contribution of the three Aph1 genes to gamma-secretase activity in vivo. Proceedings of the National Academy of Sciences of the United States of America 102, 1719–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serneels L, Van Biervliet J, Craessaerts K, Dejaegere T, Horre K, Van Houtvin T, Esselmann H, Paul S, Schafer MK, Berezovska O, et al. (2009). gamma-Secretase heterogeneity in the Aph1 subunit: relevance for Alzheimer’s disease. Science 324, 639–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagami S, Okochi M, Yanagida K, Ikuta A, Fukumori A, Matsumoto N, Ishizuka-Katsura Y, Nakayama T, Itoh N, Jiang J, et al. (2008). Regulation of Notch signaling by dynamic changes in the precision of S3 cleavage of Notch-1. Molecular and cellular biology 28, 165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi RH, Nam EE, Edgar M, and Gouras GK (2002). Alzheimer beta-amyloid peptides: normal and abnormal localization. Histology and histopathology 17, 239–246. [DOI] [PubMed] [Google Scholar]
- Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, Thinakaran G, and Iwatsubo T (2003). The role of presenilin cofactors in the gamma-secretase complex. Nature 422, 438–441. [DOI] [PubMed] [Google Scholar]
- Takitoh T, Kumamoto K, Wang CC, Sato M, Toba S, Wynshaw-Boris A, and Hirotsune S (2012). Activation of Aurora-A is essential for neuronal migration via modulation of microtubule organization. The Journal of neuroscience : the official journal of the Society for Neuroscience 32, 11050–11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub LM, and Bonifacino JS (2013). Cargo recognition in clathrin-mediated endocytosis. Cold Spring Harbor perspectives in biology 5, a016790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss M, Schroder B, and Fluhrer R (2013). Mechanism, specificity, and physiology of signal peptide peptidase (SPP) and SPP-like proteases. Biochimica et biophysica acta 1828, 2828–2839. [DOI] [PubMed] [Google Scholar]
- Walter J, Capell A, Grunberg J, Pesold B, Schindzielorz A, Prior R, Podlisny MB, Fraser P, Hyslop PS, Selkoe DJ, et al. (1996). The Alzheimer’s disease-associated presenilins are differentially phosphorylated proteins located predominantly within the endoplasmic reticulum. Mol Med 2, 673–691. [PMC free article] [PubMed] [Google Scholar]
- Wang R, Tang P, Wang P, Boissy RE, and Zheng H (2006). Regulation of tyrosinase trafficking and processing by presenilins: partial loss of function by familial Alzheimer’s disease mutation. Proceedings of the National Academy of Sciences of the United States of America 103, 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt B, van Niel G, Raposo G, and Marks MS (2013). PMEL: a pigment cell-specific model for functional amyloid formation. Pigment cell & melanoma research 26, 300–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe MS (2009). Intramembrane-cleaving proteases. The Journal of biological chemistry 284, 13969–13973. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.