

Abstract
Unpredictable and uncontrollable protein adsorption on nanoparticles remains a considerable challenge to achieving effective application of nanotechnologies within biological environments. Nevertheless, engineered nanoparticles offer unprecedented functionality and control in probing and altering biological systems. In this review, we highlight recent advances in harnessing the “protein corona” formed on nanoparticles as a handle to tune functional properties of the protein-nanoparticle complex. Towards this end, we first review nanoparticle properties that influence protein adsorption and design strategies to facilitate selective corona formation, with the corresponding characterization techniques. We next focus on literature detailing corona-mediated functionalities, including stealth to avoid recognition and sequestration while in circulation, targeting of predetermined in vivo locations, and controlled activation once localized to the intended biological compartment. We conclude with a discussion of biocompatibility outcomes for these protein-nanoparticle complexes applied in vivo. While formation of the nanoparticle-corona complex may impede our control over its use for the projected nanobiotechnology application, it concurrently presents an opportunity to create improved protein-nanoparticle architectures by exploiting natural or guiding selective protein adsorption to the nanoparticle surface.
Graphical Abstract
A review of recent literature on rational design, development, and biological applications of engineered nanoparticles with an adsorbed protein corona.
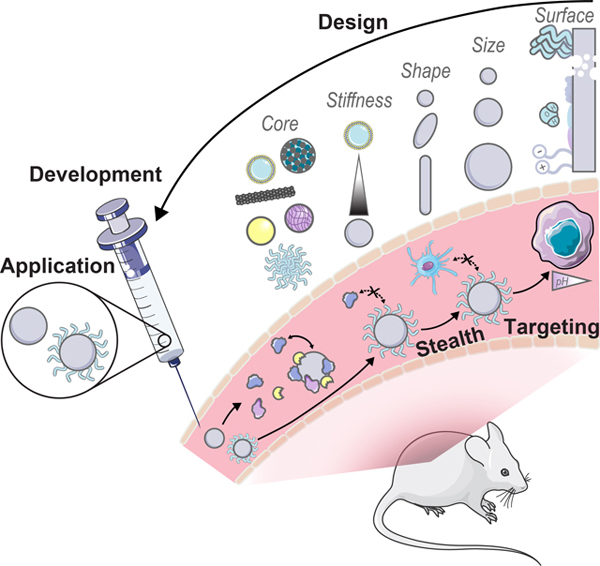
Introduction
When engineered nanoparticles are introduced into a biological medium, proteins swiftly adsorb to and coat the nanoparticle surfaces. This phenomenon is at present well-established, duly termed formation of the nanoparticle’s “protein corona” to provoke imagery of the corona surrounding the sun during a solar eclipse, with tendrils of light (or rather, adsorbed proteins) reaching outwards. As our repertoire of engineered nanoparticles becomes ever-more diverse, these nanoparticles are continually applied for broader functions across vastly differing biological environments. A comprehensive understanding of the protein corona remains one of the greatest challenges in successfully developing and implementing nanobiotechnologies. Moreover, by delving into the fundamental interactions governing protein corona formation, we realize the opportunities to be had in taking advantage of this phenomenon.
In this review, we begin by discussing the unpredictable protein corona formed upon exposure of nanoparticles to biological environments, then expand into how recent work has employed this information towards a priori design of corona-mediated functionalities. We highlight certain corona design examples, alongside the relevant development and characterization techniques. Corona design discussion is centered on applications towards corona-mediated nanoparticle stealth, targeting, and activation, with a corresponding discussion of nanoparticle construct biocompatibility to follow.
1. Corona-based Nanoparticle Design
Nanoparticles have emerged as an ideal platform upon which to develop biological sensing, imaging, and delivery tools.1–3 However, pristine nanoparticles undergo significant transformations once injected into biological environments: biomolecules, most notably proteins, rapidly coat the nanoparticle surface in the energetically favorable process of corona formation.4,5 The abruptness of protein adsorption on foreign nanosurfaces causes proteins to interact in unusual modes, contrary to the normal protein-protein interactions governed by precise genetic control, and often produces undesirable outcomes such as protein denaturation.6,7 Further, corona formation unpredictably changes the nanoparticle identity and fate, as the adsorbed proteins mask original surface characteristics and endow new biochemical properties to the nanoparticle.8–10 As a result, how the nanoparticle-corona complex interacts with biological machinery is impacted and in vivo circulation, bioaccumulation, and biocompatibility outcomes are drastically modified.11,12 Consequently, protein corona formation can lead to reduced or abolished nanoparticle efficacy and contradiction of expected in vitro results, whereby the nanoparticle is no longer able to carry out its designated function.13,14 On the contrary, the protein corona can be taken advantage of, where in situ protein adsorption may facilitate stealth targeting and delivery, with improved functionality or therapeutic effect to follow.15,16 Thus, the surface-adsorbed corona may be exploited by avoiding nonselective, deleterious protein adsorption in favor of selective, advantageous protein-surface assembly. Regardless, the protein corona display17ed on the nanoparticle surface must be appreciated as one of the principle design parameters to ensure successful applications of nanobiotechnologies in vivo.
Nanoparticle physicochemical properties and the surrounding bioenvironment are inherent variables affecting protein corona formation. Many studies classify the protein corona around specific nanoparticles in specific biological systems of interest, although findings often lack generality or are contradictory as to which nanoparticle or protein properties drive protein corona composition, dynamics, and subsequent biological outcomes. Additionally, the protein corona is dependent upon a convolution of parameters carrying varying weights, and often these parameters are difficult to decouple without meticulous experimental design. Therefore, while we summarize recent findings and generic design rules as depicted in Fig. 1, we note that these generalities may not always hold depending on the intricacies of the nanoparticle-biosystem under consideration.
Figure 1. Factors governing protein corona formation include intrinsic nanoparticle characteristics and extrinsic biological characteristics.

Intrinsic nanoparticle properties (top) can be employed as design handles during rational nanoparticle-corona design and extrinsic biofactors (bottom) must be carefully considered to ensure the complex will function properly within the intended biological environment. Some images in this figure are adapted with permission from Servier Medical Art by Servier (http://smart.servier.com), licensed under a Creative Commons Attribution 3.0 Unported License.
1.1. Nanoparticle Characteristics
Nanoparticle surface properties are most significant in governing protein corona formation, as compared to the nanoparticle core.8,18,19 These nanoparticle surface features include: (i) electrostatic charge, (ii) hydrophobicity, and (iii) surface structure. These attributes are functions of nanoparticle surface chemistry and ligand functionalization.
(i). Electrostatic charge.
Nanoparticle charge affects protein corona composition and packing density.20–24 Many studies conclude that proteins possessing opposite charges from the nanoparticles are enriched in the corona. As most proteins are negatively charged at physiological pH, it is often found that cationic nanoparticles adsorb the highest number of proteins.10,20–22,25 However, there are examples in which proteins are able to overcome electrostatically adverse conditions and adsorb by nonelectrostatic driving forces.26–28 It is important to consider that even if nanoparticles are synthesized with appropriate surface chemistries, these nanosurfaces may only retain these engineered features transiently within biosystems.8 Nanoparticle charge thus impacts surface packing, where higher magnitude charge leads to more proteins in the corona.18 Other studies conclude that more cationic surfaces increase conformational changes of adsorbed proteins.28 These results are contradictory, in that post-adsorptive protein structural changes generally take place under lower surface packing densities, where proteins have more accessible area to spread out and denature on the surface.6,29 It is unlikely that such conformational changes would be able to occur in a highly crowded corona environment. Beyond the individual nanoparticle, surface charge also directly impacts colloidal stability because neutral surfaces (or surfaces neutralized by protein and ion adsorption) tend to aggregate in the absence of intervening electrostatic repulsions.30 As will be discussed below, the surrounding solution ionic strength determines the importance of electrostatic interactions, as these interactions are screened and play less of a role in high-salt systems. Accordingly, nanoparticles must be designed in such a manner that they are not only colloidally stable as a homogeneous solution, but retain colloidal stability in the presence of proteins in the surrounding bulk and surface-adsorbed state.20
Manipulating nanoparticle charge offers a useful means to tune nanoparticle interactions with biological cells, with regards to cell internalization and toxicity:31 positively charged nanoparticles have enhanced cell internalization due to interactions with the negatively charged cell membrane,2,32–35 especially enhanced for the case of cancer cells.21 Yet, if positively charged nanoparticles bind too many proteins, this leads to colloidal instability, aggregation, and downstream toxicity.20,36
(ii). Hydrophobicity.
Nanoparticle surface chemistry also dictates hydrophobicity, where hydrophobic nanoparticles exhibit increased protein adsorption capacity,9,18 more stable protein adsorption,9,23,30 and cause more protein conformational changes.9,30 Nanoparticles with hydrophobic surfaces are more likely to produce deleterious effects on protein structures, as protein unfolding is driven by the favorable interactions of the protein hydrophobic core with the nanoparticle surface.9 Frequently, such unfolding is irreversible, leading to larger scale aggregates, nonfunctional nanoparticles, and immune activation/clearance.25,30 Again, these conclusions of high packing density and high conformational changes are at odds with each other, for the same reasons as specified for electrostatic charge.
(iii). Surface structure.
Features of nanoparticle surface topography that impact protein corona formation include surface roughness, porosity, and sterics. A rough or porous surface creates more available surface area for proteins to coat and so allows proteins to minimize lateral repulsive forces in the adsorbed state. Functionalization with polymers, targeting ligands, or other moieties that extend outward into solution also affects nanoparticle surface topography. The grafting density and conformation of such attached ligands impacts accessibility of proteins to the nanoparticle surface.37,38 Based on the principles of both hydrophobicity and surface structure, corona mitigation techniques often involve surface-grafting of hydrophilic polymers such as polyethylene glycol (PEG) to abrogate protein adsorption and sterically stabilize the nanoparticle.8,39,40 Higher antiadhesive polymer coverage is associated with alleviated protein corona formation, while lower coverage or linear conformations are less effective in deterring protein binding.38,41,42 Combining these concepts of surface roughness and sterically stabilizing polymers, Piloni et al. demonstrated that a patchy polymer-grafted nanoparticle reduced protein adsorption in comparison to a smooth polymer-grafted surface by six-fold (protein content per nanoparticle).11 PEG strategies and efficacies will be discussed further in Section 3.1.1.
Beyond surface characteristics, other nanoparticle factors that impact corona formation include: (iv) size, (v) curvature or shape, (vi) stiffness, and, to a much lesser extent, (vii) core material composition.
(iv). Size.
Size is found to quantitatively, though not qualitatively, impact corona formation: larger nanoparticles present more high surface free energy, net exposed area and permit higher protein loading per nanoparticle,22,43 although the corona constituents are often no different than those on smaller nanoparticles of identical material properties.30 Moreover, smaller nanoparticles possess a higher surface area to volume ratio, hence smaller nanoparticles adsorb more proteins on an area normalized basis. Optimizing the metric of protein loading on a nanoparticle number or area basis depends on the desired application, as will be discussed in subsequent sections. Further, increasing nanoparticle size decreases surface curvature (though dependent on the geometry), therefore there exists a threshold above which larger particles do not necessarily adsorb more proteins.44
Another important consideration is the effect of nanoparticle size on targeting and localization efficiencies: biological barrier crossing efficiency and mechanism are both size-dependent, where successful crossing scales inversely with size.34,45 Here, it is critical to consider the hydrodynamic, in situ nanoparticle-corona complex size; an adsorbed protein corona may add up to hundreds of nanometers to in vitro particle size.46 Biological barriers range from vasculature walls to cell membranes, with typical cutoff dimensions including <6 nm for renal clearance24 vs. >300 nm for liver or spleen filtration,47 ~20–200 nm for tumor penetration and retention,3,47,48 <50–100 nm for blood-brain barrier (BBB) crossing and extracellular matrix (ECM) navigation,49,50 and 10–100 nm for cell internalization.34 On the cellular level, Shadmani et al. applied a mathematical model based on diffusion of membrane-mobile receptors to examine how protein corona formation impacts internalization of gold nanoparticles by receptor-mediated endocytosis.45 From this model, optimal values for nanoparticle-corona radius (40 nm bare diameter increasing to 60 nm upon corona formation) and targeting ligand density (~1500 μm−2 on a 100 nm gold nanoparticle) are described to minimize endocytosis time through a balance of membrane tension energy and ligand-receptor interaction density, demonstrating how in silico models are useful towards nanoparticle design.
(v). Curvature/Shape.
Reiterating the earlier discussion of lateral interactions, now considering nanoparticle shape, a higher curvature surface minimizes adverse lateral protein-protein interactions. Thus, a more curved nanoparticle surface would be expected to adsorb more proteins if unfavorable protein-protein interactions are preeminent (e.g. lateral, repulsive electrostatic interactions). However, if favorable nanoparticle-protein interactions dominate (e.g. attractive dispersion forces), a flatter surface would be advantageous to facilitate more adsorption. This latter case is manifested as higher protein adsorption (per unit surface area) on higher aspect ratio nanoparticles, such as nanorods relative to nanospheres.51 Other studies find that curvature impacts adsorbed protein orientation to result in lower packing.52 Therefore, no generalizable rules can be deduced with regards to the effect of nanoparticle shape on protein adsorption. Once applied in vivo, shape also dictates how nanoparticles behave within convective flow, such as how they interact with bounding walls, and internalization efficiencies and mechanisms, since membrane bending energy is dependent on nanoparticle form factor.34
(vi). Stiffness.
As with shape, nanoparticle stiffness has been shown to affect modes of cell internalization and bioaccumulation: less stiff nanoparticles generally exhibit lower cell internalization across many cell types, and correspondingly longer circulation times due to the more difficult uptake and clearance by macrophages.33,53,54 Stiffness here refers to the nanomaterial’s ability to resist deformation under applied force, related to the material’s Young’s Modulus and geometry. Yet, the impact of nanoparticle stiffness on protein corona formation remains relatively under-studied. While proteins are considered soft matter and may be expected to increase the inherent nanoparticle softness, this may not be the dominant factor, as higher protein adsorption often leads to the opposite downstream outcomes as those reported for less stiff nanoparticles. The effect of nanoparticle stiffness on protein adsorption remains an open question.
(vii). Core composition.
Finally, while the core material composition does influence corona formation to some extent,22 the core is mostly shielded from direct exposure with the biofluid and thus plays a minor role in determining protein corona formation. However, use of exogenous nanoparticle core materials can lead to immune activation and toxicity during attempted clearance.24
1.2. Biological Environment Factors
In addition to the influence of innate nanoparticle variables on protein corona formation, the bioenvironment of the intended application must be taken into account. Environmental parameters include: (i) biomolecular components, (ii) solution conditions, and (iii) surrounding dynamics.
(i). Biomolecular components.
Native biomolecule concentration and composition within a biological environment influences the consequent protein corona formed on nanoparticles. Higher protein concentration in the surrounding fluid frequently leads to more protein adsorption on nanoparticles, as suggested by ideal-solution thermodynamics, and witnessed experimentally.7,55 Nonetheless, relative corona protein concentration does not necessarily correlate with native circulating protein concentration due to preferential protein partitioning into surface vs. bulk solution phases.26,56 More complex mechanisms often govern protein corona formation, giving rise to surprising magnitudes of protein enrichment or depletion on nanoparticles relative to the native biofluid. A frequent example of this phenomenon is the Vroman effect, where highly abundant proteins initially adsorbed to nanoparticles competitively exchange with and are eventually replaced by lower abundance, higher surface-affinity proteins.7,28,57 Cooperative adsorption is another mechanism leading to corona composition unanticipated from circulating concentrations, where initially bound corona proteins provide a scaffold promoting successive protein adsorption.57,58 Regarding native biomolecule composition, the observed corona in the presence of proteases may be a convolution of protease degradation of and exchange with the existing corona.59 These higher order mechanisms offer an explanation as to why corona constituents and kinetics resulting from single protein adsorption experiments are often not representative of whole biofluid experiments.26 These findings also stress the importance of testing nanoparticles within physiologically relevant biological fluids. A prominent example is the proliferous use of blood serum (absent of blood coagulation proteins) instead of blood plasma (which contains blood coagulation proteins) to test nanoparticles designed for intravenous administration, where coronas formed from plasma proteins have been shown to be different from those of serum and more strongly adhered.22,48,57,60 An additional consideration in terms of biomolecule composition arises in that nanoparticles may be subject to harsh conditions such as enzymatic degradation in the gastrointestinal tract61 and cancer cells,59 or immobilized, tenacious biomolecules in the mucus layer61 and brain extracellular matrix.62 Presence of these biomolecules introduce physical obstacles to penetration and routes to irreversible corona formation with subsequent toxicity. It is of further consequence that disease states alter endogenous protein concentrations and compositions, which leads to deleterious effects if the same such protein is pre-conjugated on nanoparticles for targeting purposes.63
(ii). Solution conditions.
Corona formation is a function of surrounding conditions, such as temperature, ionic strength, and pH. Increasing temperature increases the weighting of the entropic term within the net Gibbs free energy change of adsorption (ΔG = ΔH – TΔS) This results in proteins that are entropically favorable to adsorb (+ΔS) becoming more favorable at higher temperature (−ΔS), and vice versa.26,43 Within this analysis, it is key to note that each term is the net system, therefore a function of the protein, nanoparticle, and solution initial and final states during binding. For solution ionic strength, electrostatic forces scale inversely with the square-root of salt ionic strength in solution. These electrostatic forces originate from interactions between electric double layers surrounding the charged colloidal nanoparticles and proteins. This underscores how nanoparticle surface charge is inherently coupled with solution ionic strength, and the two parameters must be co-designed appropriately. In high ionic strength conditions (high salt concentration), nanoparticles and proteins do not “see” each other in solution until they are in closer proximity. Closer approach between entities bearing the same electrostatic charge results in more protein adsorption. In addition, once adsorbed, charge shielding ensures less unfavorable lateral repulsions between adsorbed proteins (again, most bearing the same negative charge at physiological pH). Both phenomena imply more protein adsorption with more salt present. However, when ionic strength becomes too elevated, charge screening leads to undesirable protein-nanoparticle complex aggregation.30,48 Accordingly, ionic strength and even ionic composition are important considerations in protein adsorption and potential downstream toxicity due to aggregation, such as high free calcium ion concentrations in the brain microenvironment leading to nanoparticle aggregation.50 pH is another relevant solution condition in that it governs the protonation state of surface chemistries on the nanoparticle, again influencing aggregation tendency,50 and impacts adsorbed protein extent and stability. As the solution pH approaches the protein isoelectric point, proteins become less stable in solution and tend to self-aggregate or adsorb to available nanoparticle surfaces.64 In terms of corona stability, the pH range that the nanoparticle will encounter is crucial to consider as a design parameter, as biological compartments cover a significant pH range at the organ and cell levels, and can differ as a function of disease state, such as the acidic pH of tumors.7,65 Many protein or peptide drugs externally loaded on nanoparticles may not survive severe conditions,61 or their expected release profile can be negatively impacted.49
(iii). Dynamics.
Finally, temporal dynamics and hydrodynamics should be considered for protein corona formation. Protein adsorption occurs within seconds of contact with biofluids,57 and may either display a dynamic nature, with fast and reversible protein association/dissociation events on the nanoparticle surface, or enter an irreversibly aggregated state.6,30,66 Proteins adsorbed directly to the nanoparticle surface are termed the “hard”, inner corona, characterized by a longer (if not indefinite) residence time in the corona phase and often more prominent conformational changes.7,28,29,57 Proteins interacting predominantly with other adsorbed proteins, instead of directly with the nanoparticle surface, constitute the “soft”, outer corona, and frequently maintain their native conformation as they undergo continuous exchange with proteins in the surrounding media.7,46 Protein corona composition is impacted by the contact time and history of nanoparticles in biofluids: the former, reiterating the likes of the Vroman effect, and the latter, in that nanoparticles evolve to carry a “fingerprint” of adsorbed proteins as they progress from one biological compartment to the next.10,67
Nanoparticles must endure flow conditions during transit, navigating channels or regions of characteristic tortuosity, permeability, and hydrodynamics. A classic example of nanoparticles maneuvering through a highly tortuous path is within the porous extracellular matrix (ECM) of the brain, relevant for neurosensors or brain-targeted therapeutics.49,62 The ECM is a mesh-like structural and biochemical scaffold for brain cells, with channels of widths ~40–200 nm, that acts as an adhesive and steric barrier for nanoparticles attempting to pass.49,50 Work within the Nance lab has pioneered brain-penetrating nanoparticles, with design principles to ensure that the nanoparticles exhibit minimized electrostatic, hydrophobic, or hydrogen bonding interactions with the ECM.62 Towards nanoparticle permeability, crossing of biological barriers is of paramount importance. Again considering the brain, the blood-brain barrier (BBB) functions as a selective barrier to protectively isolate the brain from an influx of potentially harmful entities within blood circulation, as will be discussed in Section 4.3. It must also be taken into consideration that the brain’s tortuous ECM and selective BBB vary with pathology and developmental age.13,62 Finally, regarding hydrodynamics, dynamic flow can result in a more rapidly formed and compositionally diverse corona.57,68 From a design perspective, shear stresses imposed on nanoparticle-loaded cargoes must be considered a priori: the required stability of the corona-nanoparticle complex will depend on whether the dominant transport mechanism will be passive diffusion or active convection.57
In sum, a host of intrinsic nanoparticle-based and extrinsic bioenvironmental factors affect protein corona formation (Fig. 1). These relevant factors should be considered beforehand to aid appropriate experimental design and implementation towards rational protein-nanoparticle complex construction. There are frequent discrepancies between in vitro and in vivo corona characterizations that arise from negligence of these factors, such as flow dynamics present in circulation that are absent for in vitro tests.10,17,69 Validation in vivo is preferable to gauge nanoparticle functionality or efficacy, and accordingly, we focus mainly on in vivo studies for the remainder of this review.
2. Corona-based Nanoparticle Development and Characterization
With these design considerations in mind, we now discuss how recent work has leveraged these interactions to develop engineered protein-nanoparticle constructs. Next, we detail characterization methods, both conventional and new, to properly assess protein-nanoparticle complex formation and function.
2.1. Nanoparticle-Corona Complexation Considerations and Techniques
Development of functional protein-nanoparticle constructs requires special attention to the packing, conformation, and orientation of proteins on the nanoparticle surface. As detailed earlier, less tightly packed proteins may experience damaging post-adsorptive transitions including spreading, reorientation, and denaturation.6,29,70 This control over interfacial protein conformation is critical, in that denatured proteins are generally not functional and increase the risk of triggering nanoparticle aggregation or immune system recognition and clearance.7,9 In turn, packing densities and patterns of biomolecules on nanoparticles can significantly affect targeting abilities23 and clearance pathways.58 Protein orientation on the surface also directly impacts protein function, in that particular protein domains must be outwardly displayed in solution, such as enzymatic active sites and targeting moieties for receptor or molecular recognition.52,64,70
Protein adsorption on nanoparticle surfaces is accomplished by either noncovalent or covalent means. Within the subset of noncovalent corona formation techniques, we describe both post factum and ab initio routes of protein corona formation. With post factum corona formation, the in vivo corona formed on nanoparticles is characterized, and this knowledge is applied to the advantage of the construct towards specific means.31 For example, spontaneous apolipoprotein adsorption onto peptide-functionalized liposomes assists in brain targeting of drug-loaded liposomes.15 The ab initio protein corona is achieved by initially pre-coating nanoparticles with the desired protein, resulting in noncovalent attachment of the protein on the nanoparticle. Examples of passive incubation to pre-coat nanoparticles with functional protein coronas include: albumin for extended circulation or biobarrier crossing;61,71 antibodies for targeted cell uptake;71 apolipoprotein E for extended circulation or brain targeting;9,72 and transferrin for cancer cell targeting.73 Proteins may also be actively adsorbed, i.e. requiring energy input, such as high-intensity sonication to disperse hydrophobic carbon nanotubes with various plasma protein coatings.74,75 Another aim of passive ab initio corona formation is to passivate exposed nanoparticle surface for biocompatibility,76 or retain the folded protein conformation or orientation of the functional corona components.6
Noncovalent methodologies are simple and rapid, yet inherently less stable than a covalent attachment and thus prone to exchange with other proteins in solution.77 When Zhang et al. probed pre-coated protein stability on spherical nucleic acids, they discovered that ~45% of the initial corona is removed upon exposure to blood serum.71 Additionally, passive incubation techniques generally lack control over the resulting spatial distribution and functional orientation of proteins on nanoparticle surfaces.3,17 It is difficult to control monolayer vs. multilayer protein corona formation on nanoparticles, where monolayers may be desired, but multilayers often result from a combination of the hard and soft coronas.7 To overcome unpredictable corona organization, Mout et al. present a rational design strategy taking advantage of directed electrostatic assembly to form hierarchical protein-nanoparticle superstructures via coengineering recombinant proteins with ligand-tagged nanoparticles.78 Noncovalent assembly is also ideal in some cases for preserving the intrinsic nanoparticle properties.75,79 A clever bridge between retaining nanoparticle properties and enabling controlled protein attachment has been done by Mann et al., where DNA is noncovalently adsorbed on the surface of single-walled carbon nanotubes, then nanobodies are covalently attached to the DNA.79 This strategy preserves the intrinsic, near-infrared fluorescence of the underlying nanotube by avoiding protein-nanotube covalent attachment chemistries, and simultaneously confers more controlled protein orientation and packing that in turn enables successful nanobody targeting.
Covalent conjugation of proteins to nanoparticles offers another attachment route. While covalent functionalization is more stable and controlled than noncovalent adsorption, the former requires introducing new covalent bonds on both the nanoparticle surface and the protein. Examples of covalent corona attachment methods include maleimide-thiol chemistry,33,40,49 photochemical cross-linking,24 N-hydroxysuccinimide (NHS) active ester reactions,21,64 and thiol-ene click chemistry.3 These chemistries can be applied sequentially, such as gold nanoparticle-thiol surface chemistry followed by NHS ester chemistry with a linker to tether the protein.63 New conjugation protocols such as those from Lee et al. offer promising, facile chemistries for more direct gold nanoparticle-PEG linker-peptide attachments.33 New covalent chemistries have also enabled protein attachment to carbon nanotubes, with re-aromatization of the graphitic sidewalls to retain the desired near-infrared fluorescence for nanosensor readout.1
2.2. Nanoparticle-Corona Characterization Methods
With the formation of these protein-nanoparticle complexes, their physical, biological, and dynamic properties must be characterized, alongside testing in suitable ex vivo or in vivo systems (Fig. 2). Many requisite bioanalytical methods are well-established for this purpose and can be directly applied or adapted to study corona formation and outcomes.80 We also highlight novel methodologies being developed towards this.
Figure 2. Modes of characterizing protein-nanoparticle complex formation and performance.

(a) Physical properties include complex size and colloidal morphology (preferably characterized in solution over on surface), surface charge, corona adsorption, and corona composition and amount. (b) Biological properties include surface-adsorbed protein conformation and function. (c) Dynamics include kinetics (preferably characterized in solution over on surface), thermodynamics, and interactions. (d) in vivo function can be assessed in model organisms, organs or organoids, cells, and cellular barriers. Some images in this figure are adapted with permission from Servier Medical Art by Servier (http://smart.servier.com), licensed under a Creative Commons Attribution 3.0 Unported License.
Techniques commonly applied to assess in-solution physical properties of protein corona formation include: zeta potential to assess surface charge;22,44,49 dynamic light scattering (DLS)22,44,49 or fluorescence correlation spectroscopy (FCS)30,81 for hydrodynamic size; absorbance spectroscopy for colloidal morphology and concentration;8,22,76 fluorescence quenching to track adsorption,56 with Stern-Volmer analysis of the mechanism;52,63 and fluorescence microscopy to confirm adsorption via colocalization.14,61,64 Although zeta potential is not rigorously equivalent to the electric surface potential nor the Stern potential and there are implicit geometry assumptions in the calculation, zeta potential still provides a proxy for colloidal charge and stability, where often the zeta potential tends to zero in the presence of destabilizing protein adsorbates. Small-angle x-ray scattering (SAXS) has recently been applied to acquire in-solution colloidal morphology of nanoparticle-corona systems, including protein-nanoparticle complexation to verify binding and higher order aggregate formation to examine potential routes of in vivo toxicity.10,26,78 Diffusion nuclear magnetic resonance (NMR) has also been employed to infer protein adsorption on nanoparticles via increasing hydrodynamic radius, offering the advantage of in situ characterization in turbid bioenvironments by virtue of not being an optics-based measurement.82 Regarding the aforementioned techniques used to measure protein-nanoparticle size (DLS, FCS, etc.), the readout must be carefully interpreted. Large increases in hydrodynamic size may indicate aggregation of the nanoparticles in the presence of proteins via polymer bridging or other noncovalent interactions, rather than formation of protein multilayers on individual nanoparticles.30,68,81 Surface techniques are also applied to assess dried-state physical properties of protein corona formation, including: electron microscopy (EM),49,70,74 atomic force microscopy (AFM),9,75 and energy dispersive X-ray spectroscopy (EDS mapping)83 for size and morphology. However, these methods all require drying samples on a substrate for observation, which results in conclusions not representative of the solubilized system. Recent work has also implemented cryogenic transmission electron microscopy (cryo-TEM) to enable visualization of protein-nanoparticle morphology in a closer-to-native state.17,84
Beyond the whole-complex attributes, the composition of the protein corona is of paramount importance to take advantage of post factum corona formation. To study corona constituents, the protein-nanoparticle complexes are first isolated from non-binding entities, typically accomplished by some variation of a pull-down assay.14 After corona proteins are unbound from the nanoparticle, characterization methods to identify the protein constituents include gel electrophoresis (GE)61,63,71 and proteomic mass spectrometry (MS).8,29,46 Separation techniques to isolate the soft, more loosely bound corona from the hard corona are currently in development, such as asymmetric field-flow fractionation by Weber et al.85 The Sutherland lab has also developed an in situ click-chemistry reaction to separately characterize the soft and hard coronas formed on model nanoparticles.29 To gauge whether corona loading or mitigation strategies are successful, net protein adsorption can be measured by protein assays such as the bicinchoninic assay (BCA) for protein loading,8,49,72 gel electrophoresis again, immunoblotting (e.g. Western blots),59 and enzyme-linked immunosorbent assays (ELISAs).59 The accuracy of colorimetric protein assays such as BCA in the presence of nanoparticles must be critically assessed prior to experiments, as nanoparticles often interfere by adsorbing the reporter molecule or absorbing the output light used to quantify protein concentration. Moreover, the specific chemistry of the assay will determine whether proteins in solution, in the adsorbed state, or both are being measured.
Techniques applied to assess biological function in the corona include: circular dichroism (CD)52,69 and solution NMR86,87 spectroscopy for bound protein structure and conformation; Förster resonance energy transfer (FRET) for measurement of protein-protein interactions61 and conformational changes;7 nano differential scanning fluorimetry (nanoDSF) for protein stability and conformational changes;64 and immunoblotting to evaluate accessibility and function in the corona.63,83,88 To study corona structural organization and functionality at the nanoparticle surface, Herda et al. developed a method to characterize adsorbed protein orientation by exploiting antibody-conjugated gold nanoparticles to map available epitopes.70 When they applied this method towards transferrin proteins covalently conjugated to PEGylated silicon dioxide nanoparticles, they found that only ~4% of corona proteins adopt the correct orientation to facilitate receptor binding, highlighting the need for more homogenous and controlled protein grafting methodologies. Recently, the Chan lab developed a modified-ELISA workflow to similarly probe protein corona organization and binding functionality when adsorbed from blood serum onto gold nanoparticles, establishing that merely a third of the adsorbed proteins remain functional for binding to their target proteins.89 Imaging advances have led to the development of various techniques to assess protein interactions on surfaces, including single molecule high resolution imaging with photobleaching (SHRImP) by Warning et al. to measure protein conformational changes on a surface.6
Methods to study dynamics of protein corona formation include: isothermal titration calorimetry (ITC) to extract thermodynamic binding energies and equilibrium parameters,38,43,90 as reviewed extensively elsewhere,91 and surface plasmon resonance (SPR),16,29,46 biolayer interferometry (BLI),92 and quartz crystal microbalance with dissipation monitoring (QCM-d)52 to determine binding kinetics. Recently, Kari et al. designed a custom biosensor system for in situ determination of protein corona structure and composition by coupling SPR and proteomic MS, enabling differentiation of the hard and soft corona formed on liposomes under physiologically relevant conditions.46 Super-resolution microscopy presents a single-molecule technique with requisite sensitivities to monitor individual protein-nanoparticle binding events, avoiding ensemble-averaged methods of studying corona formation.93 However, it is important to note that application of surface techniques such as SPR and microscopy to study nanoparticles again requires surface immobilization of the nanoparticles. Surface immobilization introduces topographical constraints that affect kinetics and transport, giving rise to sampling artifacts and changing the in-solution nanoparticle properties. Ideally, protein-nanoparticle complexes are studied in solution with physiologically relevant parameters that are known to affect corona formation (including ionic strength, temperature, pH, etc.). Accordingly, in-solution kinetic corona methods have been developed, including fluorescence assays to monitor protein fall-off71 and exchange56 on solubilized nanoparticle surfaces. To expand upon the use of surface charge changes as a proxy for protein corona formation, Zhao and colleagues measured in-solution protein binding dynamics onto nanoparticles using pulsed streaming potential, resulting in knowledge of adsorption rates and equilibria under varying buffer conditions.94 Further, Weiss et al. have developed a microfluidic system to simulate a flow environment, with control over fluid flow and shear applied to nanoparticles and proteins.57 This microfluidic system has elucidated the more complex corona formed in dynamic rather than static conditions.
While prior studies provide insight into bio-corona formation, numerous techniques and model fits are ill-applied and present conclusions not representative of the system. The protein corona is often treated as existing at thermodynamic equilibrium, despite a body of literature providing evidence otherwise.95–97 One frequent manifestation of this equilibrium assumption is the erroneous application of the Langmuir adsorption isotherm to mechanistically describe proteins adsorbing to nanoparticles, despite the fact that many of the model conditions are not satisfied.98 A key point here is that Langmuir-like binding profile does not necessitate that the binding mechanism is indeed a Langmuir isotherm: this profile shape for protein-surface adsorption processes often emerges as the result of adsorption-induced protein spreading/denaturation, reorientation, and aggregation as a function of bulk protein concentration,7 in contrast to originating from the dynamic equilibrium adsorption process required for Langmuirian adsorption.98 Therefore, while the Langmuir isotherm does provide a simple functional form that may fit data, it should only be applied towards extraction of relative binding affinity measures rather than true thermodynamic parameters or underlying adsorption mechanisms. For instance, ITC is often a method applied with intent to assess protein-nanoparticle binding events. Instead, ITC often measures a convolution of protein binding to individual nanoparticles, to aggregated nanoparticles, and nanoparticle aggregation.26 Aggregation is a kinetically controlled, non-equilibrium process that violates the central assumption of ITC that each titration step is equilibrated, observed as visible aggregation and baseline drifting during the run. Accordingly, the reported free energies and equilibria values must be taken with the perspective that these are whole-system energy changes, often with higher-order processes occurring simultaneously. The suitability of such models and experimental methodologies to describe certain nanoparticle-protein corona formation processes should be carefully considered prior to application.
Finally, molecular dynamics (MD) simulations provide insight into fundamental interactions driving adsorption to surfaces and protein structural changes upon adsorption, as reviewed extensively elsewhere.99–101 Atomistic MD models convey a detailed picture of protein-nanoparticle interactions, including the individual amino acids responsible for association.102 Alternatively, coarse-grained MD models trade such detail for access to longer time and length scales, increasing approximately an order of magnitude from the millisecond and nanometer scales in atomistic models.99 Although coarse-grained models are inherently lower resolution, such as lacking physicochemical details of the nanoparticle surface,103 these models can facilitate the study of protein-protein interactions and adsorption onto smaller nanoparticles, with explicit curvature effects. Both scenarios are unfeasible in atomistic models, which instead highlight interfacial phenomena in dilute protein settings. Some particular MD studies of interest involving protein-nanoparticle systems include: atomistic MD simulations of amyloidogenic peptides on gold nanoparticles (modelled as a gold surface)102 and plasma proteins on model nanomaterials;9,28 hybrid MD simulations, with an atomistic nanoparticle description and a coarse-grained, solvent-explicit protein description;23 and multiscale MD simulations, adopting coarse-grained or meso-scale models for single vs. simultaneous protein adsorption on small gold nanoparticles, respectively.104 MD simulations extend our understanding of dynamic protein-nanoparticle interactions, yet require further refinement and validation against experimental results prior to use as purely predictive tools, due to the underlying complexity of nanoparticles interacting with proteins.99
Towards applied nanoparticle-corona technologies, in vivo studies provide compelling evidence for sustained engineered nanoparticle function or therapeutic efficacy within complex bioenvironments. Animal models such as mice and rats provide the means to study not only function, but also systems-level clearance profiles, bioaccumulation, and toxicity.42,61,83 Ex vivo organ slices enable measurement of nanoparticle diffusion by particle tracking studies, providing insight into unfavorable adhesive interactions with the surrounding biological matrix.40 At the cellular level, fluorescence (often confocal) microscopy,40,61,63 immunofluorescence,49,83 and flow cytometry40,41,63 provide information on cellular uptake, spatial localization, cell morphology, and cytotoxicity. As an intermediate between achieving in vitro experimental control and assessing in vivo translatability, transwells offer a useful cellular model for biological barriers such as the blood-brain barrier67 and three-dimensional organoids or tumor spheroids offer a scaled-down organ model for assessing efficacy and toxicity.20,105
3. Development of Stealth Nanoparticles
After creation and in vitro characterization of nanoparticle-corona complexes, several challenges still lie between administration and successful use of nanoparticles in vivo. Many such obstacles stem from the recognition of these synthetic nanomaterials by the body. Nanoparticles often trigger an immune response, resulting in immune cell recruitment, antibody and chemokine release, and activation of the mononuclear phagocytic system (MPS) (Fig. 3a). Briefly, the MPS entails the recognition, engulfment, and subsequent clearance of nanoparticles from blood circulation through the action of phagocytic cells, such as Kupffer cells in the liver, dendritic cells in major organs, microglia in the nervous system, and alveolar macrophages in the air spaces of the lungs.106 Consequently, nanoparticles are often found to accumulate in the liver and spleen. Numerous studies have shown that the protein corona plays a critical role in modulating the MPS.3,77,107 Specifically, proteins termed opsonins promote phagocytosis, and include complement proteins and immunoglobulins (IgG, IgA, and IgM). Conversely, dysopsonins are proteins that aid evasion of phagocytosis, and include albumin and apolipoproteins. The protein corona can thus be tuned to mediate challenges that the nanoparticle faces from injection to localization.
Figure 3. Nanoparticle stealth, strategies and outcomes.

(a) Administration of nanoparticles into the body leads to formation of the protein corona that can trigger an immune response or clearance of nanoparticles. Rational design of the protein corona can promote the enrichment of favorable, dysopsonin proteins or mitigate the adsorption of unfavorable, opsonin proteins to promote nanoparticle stealth. (b) Strategies that utilize polymer, protein, or biomimetic coatings have been developed to design the protein corona for better nanoparticle stealth. Protein images (PDB ID 1E7I, 1LE2, and 1AV1)111–113 are reproduced with permission from the RCSB PDB (rcsb.org). Some images in this figure are adapted with permission from Servier Medical Art by Servier (http://smart.servier.com), licensed under a Creative Commons Attribution 3.0 Unported License.
To prevent activation of the immune response and nanoparticle recognition by the body, several strategies can be implemented to provide nanoparticles with stealth properties. In literature, “stealth” is often used to indicate resistance to biofouling, referring to the low nonspecific adsorption of proteins on nanoparticles. Although less adsorption of certain proteins such as opsonins correlates with better biological compatibility, more factors are involved in nanoparticle stealth for biological applications.77 We therefore refer to stealth herein as the ability to evade recognition by the body. In the discussion to follow, we highlight studies that report longer nanoparticle retention time in vivo and lower titer of biomolecules that indicate immune response. Studies have demonstrated this phenomenon through the design of the nanoparticle corona using polymer, protein, or biomimetic coatings (Fig. 3b).
3.1. Polymer Coatings for Stealth
Attachment of polymers to nanoparticle surfaces provides a facile approach to modify hydrophilicity, size, and other nanomaterial properties, as detailed in Section 1.1, that may modify protein corona formation in comparison to the bare nanoparticle and confer stealth in vivo.
3.1.1. Polyethylene Glycol Coatings for Stealth.
Polyethylene glycol (PEG) is one of the most studied polymer coatings for use as a stealth agent on nanoparticles, and we point readers to previous reviews with more in-depth discussion on the efforts of PEG use in biological settings.108,109 PEG is water soluble and capable of extending the half-lives of nanoparticle carriers in circulation,40,110 presumably due to the water solvation effect whereby it is less energetically favorable for proteins to exchange with water adsorbed to the highly hydrophilic PEG chains. One important consideration is the PEG grafting density on the nanoparticle surface, which controls surface roughness and PEG orientation, and subsequently impacts protein corona formation. PEG in a dense, brush conformation better repels protein adsorption than a less dense, mushroom conformation.38,41 Recent work implemented a two-layer PEG system, where the first layer is a dense polymer brush to prevent protein adsorption, followed by a second layer that approaches the mushroom-to-brush transition to reduce liver uptake.38 This study also highlights that certain aspects of the PEG-driven stealth mechanism are still under investigation. It was originally thought that PEG enables nanoparticle stealth by repressing protein adsorption that in turn triggers MPS clearance. However, recent work shows that PEGylated nanoparticle surfaces can exhibit substantial adsorption of proteins, and it is the repressed adsorption of specific opsonin proteins and enhanced adsorption of dysopsonin proteins that enables stealth (referred to as the PEG “harvesting” effect).46,114 In the case of polystyrene nanocarriers, a PEGylated surface enriched selective binding of a dysopsonin protein clusterin, which results in shielding of the nanoparticles from macrophage uptake.114 Thus, PEGylation could serve to recruit selective proteins to the nanoparticle surface towards desired applications, such as avoiding macrophage internalization as shown here. 46,114
Recent studies are moving away from the use of PEG as a stealth agent, as fundamental challenges of using PEG-nanoparticle conjugates come to light. The ubiquitous use of PEG in nanomedicine has led to the formation of anti-PEG antibodies in the body and rapid clearance of PEGylated nanoparticles from the body, termed the “accelerated blood clearance” phenomenon.115,116 Furthermore, use of PEG does not necessarily suppress unfavorable protein adsorption onto all nanoparticles,27,117 such as nanosomes with PEG linkers shown to irreversibly aggregate after protein corona formation in whole serum.66 Due to these findings, researchers are investigating other polymer coatings for nanoparticle stealth.109
3.1.2. Zwitterionic Polymer Coatings for Stealth.
Zwitterionic polymers, containing both positive and negative charges, are promising for stealth nanoparticle applications because they behave similarly to PEG in preventing protein corona formation in vitro.18,65,77 It is known that surface charge affects in vivo nanoparticle fate: cationic polymer coatings promote cellular adhesion and uptake, yet exhibit higher clearance as compared to their anionic and zwitterionic counterparts.35 Zwitterionic polymer coatings, such as sulfobetaines,2,36 phosphorylcholine,77,118 and peptides119,120 have been increasingly investigated and have shown efficacy in vivo. A zwitterionic peptide coating of alternating negatively charged glutamic acid and positively charged lysine on gold nanoparticles showed prolonged circulation in vivo in tumor-bearing nude mice.120 Compared to PEG-coated gold nanoparticles, these zwitterionic peptide-coated nanoparticles were inert to the immune system and did not elicit elevated levels of immune proteins, such as IgM and IgG. Similarly, a gold nanocage system functionalized with acylsulfonamide-based pH responsive zwitterionic ligands showed four-fold longer circulation lifetime and tumor accumulation in BALB/c mice bearing 4T1 murine breast tumors than a neutrally charged polyvinylpyrrolidone-functionalized gold nanocage.65
3.1.3. Carbohydrate Coatings for Stealth.
Researchers are increasingly turning towards biologically derived polymers, such as carbohydrate coatings, to prolong nanoparticle circulation in vivo. Hydroxyethyl starch (HES)-linked nanoparticles have created drug nanocarriers with prolonged in vivo circulation half-life of several hydrophobic chemotherapy drugs.47,121 HES-conjugated polydopamine nanoparticles were shown to have similar circulation half-life and drug-loading capability as PEGylated polydopamine mannosylated PPE-nanocarriers were shown to avoid protein adsorption and better target dendritic cells for immunotherapy.123 This noncovalent PPE adsorption and sugar passivation is generalizable to other nanocarrier systems, and as different sugar-coated systems are shown to have varying responses in the body, there is a need for further investigation on how carbohydrate polymers interact with the protein corona to modulate stealth.19
3.2. Protein Coatings for Stealth
Another solution towards constructing stealth nanoparticles is to engineer the protein corona itself to avoid triggering the immune system and MPS detection.10 As all nanoparticles are expected to develop coronas in vivo and the existence of these coronas often promotes immune cell association,77 directed adsorption of dysopsonins and/or reduced adsorption of opsonins on the nanoparticle surface can be employed to reduce clearance of nanoparticles.90
Nanoparticle surface properties may be altered to direct adsorption of desired proteins or repel unwanted proteins. In a study of peptide-embedded liposomes, it was shown that the adsorption of IgM correlates with rapid clearance through MPS and accumulation in the lymph nodes.116 By modifying the length of the peptide displayed on the liposome, adsorption of IgM decreased, leading to longer nanoparticle half-life in circulation. To encourage dysopsonin adsorption, nanogels were created using molecular imprinting, a method that templated nanogels to bear a binding site for native dysopsonin protein: albumin.16 Upon injection into a tumor xenograft model, it was shown that the molecularly imprinted nanogels (MIP-NGs) had a higher half-life in blood (6.8 hours), compared to the non-imprinted nanogels (3 hours). Furthermore, these MIP-NGs were observed to circulate in the liver without aggregation or capture for over 10 hours, demonstrating that nanoparticle surface modifications can be utilized to adsorb necessary stealth proteins.
Protein corona shields can be made through the design of the nanoparticle surface a priori. Oh et al. created a protein corona shield for mesoporous silica nanoparticles using a recombinant fusion protein of glutathione-S-transferase genetically combined with Her2-binding affibody.3 This outer corona shield led to the reduction of protein corona formation and subsequent higher retention in plasma. In another study, pre-incubation of charged polystyrene nanoparticles in IgG-depleted plasma formed a corona enriched in vitronectin and fibrinogen on negatively charged polystyrene nanoparticles or enriched in clusterin and hemopexin on positively charged polystyrene nanoparticles.123 These nanoparticle-corona complexes showed reduced uptake by RAW264.7 macrophages and remained stable when reintroduced into whole plasma. Preincubation with the dysopsonin apolipoprotein E on graphene, gold nanoparticles, and iron oxide nanoparticles showed markedly improved blood circulation and better biocompatibility than opsonin IgE-coated nanoparticles.9 Using the growing database of corona proteins, it is increasingly possible to tailor nanoparticle surfaces for avoidance of premature clearance.
3.3. Biomimetic Coronas for Stealth
Similar to plasma-derived protein coatings for stealth applications, other biomimetic solutions to maintain nanoparticle biocompatibility include employing cell membrane proteins to shield nanoparticles from recognition. To keep nanoparticles in circulation and curtail recognition from immune cells, blood cells are a template for nanoparticle stealth. Corbo et al. have utilized white blood cell (leukocyte) proteins, such as macrophage receptors, to decorate liposomes and produce a new class of nanoparticles called leukosomes.17 They showed that leukosomes have lower accumulation in MPS organs and have reduced uptake by macrophages. Similarly, Meng et al. extract red blood cell membranes to coat immunomagnetic micro and nanoparticles.14 This coating prevents the formation of a protein corona in whole blood for better retention.
The use of biomimetic coronas could also accelerate the development of personalized nanomedicine with low immunogenicity. Personalized protein nanoparticles can be made from patient-derived proteins extracted from a variety of human sources such as serum, tears, saliva, or breast milk.24 The proteins are initially cast on a metal nanoparticle core, then extracted to create a biodegradable nanoparticle made up of only proteins. These protein nanoparticles were used in vivo without any inflammation or immune cell recruitment. Biomimetic solutions in creating stealth nanoparticles leverage specific cell-type proteins or personalized proteins to evade detection.
4. Targeting and Activation of Functional Nanoparticles to Biological Systems
After bypassing recognition and clearance from circulation, nanoparticles must overcome additional barriers towards successful localization and function. As such, targeted delivery of nanoparticles remains a major challenge in the clinical adoption of nanomedicine, and recent literature reveals that efficacious nanoparticles can manipulate protein corona engineering towards this purpose. In this section, we discuss the targeting strategies of nanoparticles and subsequent activation of nanoparticles once they arrive at a biological target of interest.
4.1. Challenges and Considerations in Nanoparticle Targeting
Nanoparticle surfaces can be engineered for targeting through the addition of different synthetic and biological ligands, such as small molecules, peptides, and antibodies. However, nanoparticle targeting elements incorporated on bare nanoparticle surfaces and validated in vitro may show different functionality in vivo, where the formation of the protein corona upon administration could inhibit the accessibility of these targeting ligands.13 Some studies show that cellular uptake of nanoparticles is controlled by the outermost protein corona as opposed to the surface ligands meant to target receptor-expressing cells.11,124 A notable exception of this phenomenon was seen in poly(beta-aminoester) polymer nanoparticles with variable terminal targeting peptides.124 The nanoparticles were coated with retinol, a hepatic targeting moiety, and the protein corona formed dictated organ biodistribution, yet cellular uptake was determined by the terminal peptides independent of the corona. For many other cases, however, the in vivo protein corona attenuates the targeting properties of nanoparticles. Serum proteins were shown to decrease association of transferrin-labeled liposomes with glioblastoma cancer cells, although transferrin-labeled liposomes still exhibited better association, tumor uptake, and tumor growth inhibition than unlabeled liposomes.73 There is currently a dearth of literature on the mechanism of these targeting moieties post factum, and work is moving towards understanding the strategies for ab initio nanoparticle design. Certain properties such as size, conformation, and mobility of targeting ligand have come to light as important design parameters for targeting applications.
Size of targeting ligands could play a substantial role in targeting potency. An example is transferrin, an 80-kilodalton glycoprotein used in many targeting studies for its well documented ability to promote clathrin-mediated endocytosis, and subsequent intracellular trafficking through recycling pathways.125 Transferrin, peptide LT7 (CHAIYPRH), and DT7 (the d-amino acid analogue of LT7) are all targeting ligands for transferrin receptors that are overexpressed in several cancer types.69,125 Investigation of targeting and uptake of polystyrene nanoparticles functionalized with these ligands revealed that the transferrin-passivated nanoparticles out-performed the peptide-passivated ones.69 Analysis of the protein corona formed around each of these nanoparticles revealed differences in composition, and underscored a size and conformation effect on ligand targeting.
As introduced in Section 2.1, the conformation of targeting ligands on nanoparticle surfaces can affect the ability of the nanoparticle to carry out its intended function.23,52 Fibronectin, a protein that binds cell-adhesion receptors called integrins and extracellular matrix components, can undergo pronounced conformational changes when adsorbed onto bare gold nanoparticle surfaces compared to when adsorbed to the nanoparticle surface through protein-protein interactions, leading to loss of function in the former.7,126 The function of targeting modalities on nanoparticles must be preserved in the surface-adsorbed state and during in vivo application. Finally, beyond simply optimizing ligand avidity towards the intended target, Figueroa et al. highlights how increasing mobility of ligands tethered to nanoparticle surfaces drives more elevated cellular uptake.127
4.2. Protein Corona Strategies in Nanoparticle Targeting
Several nanoparticle targeting schemes are validated with applications in vivo. Cancer therapies often require targeted approaches because treatments, such as chemotherapy, are cytotoxic to both cancerous and non-diseased cells and have a limited dosing range. Hence, most literature examples of nanoparticle targeting are devoted to designing nanoparticles for cancer therapy. These design principles could be extended to other diseases and tissue types. This section highlights some targeting modalities that can be attached to a variety of nanoparticles through different conjugation chemistries.
Targeting strategies that activate transport pathways or bind overexpressed biomarkers are promising for in vivo applications because they increase uptake of the nanoparticles by the target cell. To activate transport pathways, nanoparticle surfaces can be functionalized with ligands that bind to requisite receptors or proteins on the target cell (Fig. 4a). The use of polypeptides is frequently employed to deliver nanomedicine to tumor cells, such as the arginine-glycine-aspartic acid (RGD) peptide motif that binds to integrin transmembrane proteins.110,128,129 Other ligands include synthetically malleable polymers that are readily incorporated through bioconjugation chemistry or layer-by-layer synthesis; a prominent example is hyaluronic acid that binds to CD44 receptors overexpressed in many cancers.105 Additionally, the use of hyaluronic acid is shown to reduce the immunogenicity through the selective adsorption of anti-inflammatory proteins to the formed protein corona.130
Figure 4. Corona-mediated targeting and activation.

(a) Nanoparticles can be targeted to cell receptors through the surface decoration of ligands such as polymers, peptides, and proteins. (b) Delivery of nanoparticles through biological barriers is difficult due to environmental factors such as pH gradients and physical forces that destabilizes the outer protein corona. Nanoparticles can pass through barriers by permeation or the targeting of nanoparticles to cells for transcytosis. (c) Strategies for the activation of nanoparticles include the use of pH, light, enzymes, and redox reactions. Protein images (PDB ID 5UE3 and 3AI8)131,132 are reproduced with permission from the RCSB PDB (rcsb.org). Some images in this figure are adapted with permission from Servier Medical Art by Servier (http://smart.servier.com), licensed under a Creative Commons Attribution 3.0 Unported License.
Dual stealth and targeting surface functionalization prevent the adsorption of plasma proteins and thus retains nanoparticle targeting capability. Koide and colleagues designed nanosomes, consisting of core metals covered by an anti-adhesive mixed self-assembled monolayer, capable of preventing protein adsorption while outwardly displaying n-acetylglucosamine (GlcNAc).66 This outer layer triggered uptake through the cancer-specific GlcNAc salvage pathway, and resulted in body circulation, accumulation in the tumor, and reduced tumor size. Similarly, mesoporous silica nanoparticles with a protein shield of glutathione-S-transferase fused with Her2-binding affibodies were shown to adsorb few corona proteins and resulted in increased uptake and growth inhibition of breast cancer in vivo in SK-BR3 xenograft mice.3 Corona proteins adsorbed during nanoparticle transport can also be used for targeting, and as mentioned in Section 3, redirecting accumulation from the liver and spleen leads to better accumulation of nanoparticles in other organs. An interesting case of nanoparticles for cancer therapy is blood-triggered generation of platinum nanoparticles as anti-cancer agents. Platinum, originating from the chemotherapy drug cisplatin, is triggered by albumin to assemble in vivo to function as an anti-cancer agent.83 This native corona of albumin then promotes targeting tumors with better efficacy in leukemia xenograft mice than commercial albumin-platinum conjugates.
4.3. Protein Corona Strategies in Nanoparticle Passage Across Biological Barriers
Targeting also aids efficient delivery of nanoparticles through biological barriers such as the blood-brain barrier (BBB), mucous membranes, and epithelial barriers (Fig. 4b). These barriers impose certain limitations on the physical characteristics of the nanoparticles, as introduced in Section 1.1. For example, the BBB excludes passage on the basis of size and surface properties including charge and hydrophilicity.13,133 Nanoparticles have been shown to pass the BBB via transcytosis-mediated routes, mediated by immune cells134,135 or the presence of apolipoproteins,15,72 transferrin,49 or other proteins within the adsorbed corona (as reviewed extensively elsewhere133,136,137). Importantly, traversing biological barriers can lead to alterations in the nanoparticle corona. A study of the nanoparticle protein corona was conducted in an in vitro cellular transwell model of the BBB, demonstrating evolution of the protein corona as well as a stabilizing effect after BBB crossing.67 It is shown for gold nanoparticles that only 9 of the 20 most abundant proteins in the corona are retained after passage through this BBB model, where serum albumin and α−2-macroglobulin remain abundant, with enrichment of complement C9. For efficient design of targeting elements to cross biological barriers, it is important to ensure stable attachment such that targeting functionality can be maintained across different environments.
Strategies exist at the intersection of nanoparticle surface design and biological environment considerations in designing nanoparticles towards targeted biological barrier crossing. For oral delivery, nanocarriers must withstand acidic pH, enzymatic degradation, and differing surface charge requirements during passage through the mucous membrane and intestinal epithelium. Passage across the negatively charged mucus barrier is best achieved with neutral, hydrophobic molecules, while passage across the intestinal epithelium is optimal with cationic, hydrophobic molecules. With these system constraints in mind, Wang et al. aimed to overcome this issue of ineffective oral administration and uptake of insulin through rational corona design.61 Ultimately, pre-coating albumin on cationic liposomes enabled increased penetration across both mucosal and epithelial barriers: the protein coating is enzymatically hydrolyzed as the liposomes cross the mucus layer, resulting in exposure of the underlying positively charged liposome that subsequently improves transepithelial transport. In vivo experiments show that the uptake amounts and transepithelial permeability of these liposomes carrying insulin were 3.24- and 7.91-fold higher, respectively, than that of free insulin. Continuing this same idea, Zeng et al. suggest that the presence of other proteins such as protease inhibitors in the nanoparticle corona, in addition to pre-loaded albumin, serves the role of protecting albumin from hydrolysis prior to reaching the intended destination (in this case, tumors).83
4.4. Activation of Nanoparticle Functions upon Localization
As discussed in previous sections, nanoparticle systems can be designed to increase their bioavailability, circulation time, and ability to target and localize to desired areas such as specific organs or tumors. However, surface functionalizations that prove beneficial for these purposes can be detrimental once these nanoparticles arrive at their target site.138 It is thus desirable to alter nanoparticle composition in a controlled manner through various cleavable bonds and mechanisms upon nanoparticle localization. Several environmental triggers have been used for this purpose in recent years, with pH, light, enzymes, and redox environments being the most common (Fig. 4c). Other triggers including temperature139 and electrostimulation140 have been demonstrated, but are less common due to the inherent difficulty of applying these external stimuli to in vivo systems in a controlled manner to avoid unintended side effects.
4.4.1. pH-Responsive Nanoparticles.
The use of pH as a trigger stems from the range of distinct pH values that occur within the body: blood has a pH of 7.4,65 tumor environments range from pH 6.5–7,128 the gastrointestinal tract fluctuates from 5.7–7.4,61 and lysosomes have a pH ~5.141 These characteristic pHs have been exploited to design activatable nanoparticles in several examples over recent years. As introduced in Section 3.1.1, PEGylation of nanoparticles often confers low-biofouling properties to nanoparticles desirable for circulation, however, the “PEG dilemma” arises in that the same properties that help biotransport also render the constructs less susceptible to cell internalization once localized.138 Towards this problem, Lim et al. employed mesoporous silica nanoparticles conjugated to the chemotherapy drug doxorubicin via a pH-sensitive hydrazine linker.142 These nanoparticles were encapsulated with a polyaspartamide-PEG-biotin coating to inhibit burst drug release, increase hydrophilicity, and increase cell penetration, respectively. Once these nanoparticles were endocytosed by MCF-7 breast cancer cells, the acidic lysosome environment promoted cleavage of the hydrazine linker and released doxorubicin, resulting in decreased cell viability than free doxorubicin. Likewise, Wang et al. designed polymeric nanoparticles to shed their protective PEG coating, needed for stable transit, upon exposure to the acidic tumoral microenvironment, exposing a targeting iRGD peptide to facilitate tumor penetration and cellular uptake of the doxorubicin prodrug.128
In addition to dePEGylation, other modes of pH activation have been leveraged in recent years. Li et al. developed a peptide-assembling nanoparticle system loaded with immune checkpoint inhibitors to both effectively target breast cancer cells and release the cargo once internalized.143 This result was achieved by designing a peptide polymer with cholesterol, a histidine domain for endosomal escape, and a targeting peptide sequence. Once the drug-loaded nanoparticle was endocytosed, the drop in pH led to protonation of the histidine domain, which facilitated endosomal escape and resulted in successful in vivo drug release. Naidu et al. showed different release kinetics of ion channel antagonists from transferrin-functionalized polymeric nanoparticles in various pH environments, finding faster drug release at lower pH.49 This result suggests that pH-responsive systems can be beneficial for treatment of neurotrauma by maintaining drug cargo within the nanoparticles until they enter the acidic (pH ~5) endosomal environment of damaged central nervous system cells. Overall, these nanoparticle-drug systems benefit from pH activation by controlling drug release to occur at the predetermined location.
4.4.2. Light-Activated Nanoparticles.
Light-activation has also been implemented to enhance the efficacy of nanoparticle systems because external light triggers offer greater spatiotemporal control of activation compared to other methods.144 Zhou et al. demonstrated the utility of near-infrared-(NIR)-triggered dePEGylation of polymeric nanoparticles to both decrease the nanoparticle size, aiding tumor penetration, and expose RGD peptides, for enhanced tumor uptake.110 Kong et al. analogously used UV light-triggered dePEGylation of liposomal nanoparticles functionalized with cancer-targeting peptide E.39 Their findings showed that the PEGylated nanoparticles remained freely circulating within the zebrafish xenograft cancer cell model until triggered dePEGylation caused accumulation and uptake by cancer cells due to the targeting peptide. Further, Feng and co-workers used NIR irradiation to produce reactive oxygen species (ROS) that cleaved a thioketal bond between a cancer prodrug and a PEG moiety adsorbed onto a self-assembled nanoparticle composed of a photosensitizer and an immunoinhibitory compound.145
Taking advantage of a protein corona stealth effect rather than that of PEG, Yeo et al. evaluated the use of gold nanorods coated with mouse serum proteins and the photosensitizer molecule Chlorin e6 to accumulate in and subsequently eliminate tumors in mice.76 The serum protein corona effectively shielded the nanoparticles from immune system clearance and increased their bioavailability. Once accumulated at the tumor site, visible-light laser irradiation induced the production of ROS by Chlorin e6 which, when combined with the temperature increase of the nanorods themselves, led to complete tumor regression within 19 days and no significant regrowth after 31 days. Also utilizing the nanoparticle-adsorbed protein corona, Fukuda et al. demonstrated the potential of single-walled carbon nanotubes suspended in apolipoprotein A-I to produce ROS under NIR illumination.74 These ROS led to both lower HeLa cancer cell viability and disintegration of the neurotoxic peptide aggregate amyloid beta, which is implicated in neurodegenerative diseases such as Alzheimer’s. As light can be easily controlled both spatially and temporally, these light-activated systems show promise for effective in vivo applications.
4.4.3. Enzyme-Activated Nanoparticles.
Enzymatic activation has been demonstrated to be an effective method to trigger nanoparticle function, as nanoparticles encounter various intracellular and extracellular enzymes including proteases, phospholipases, and glycosidases. Rodriguez-Quijada et al. observed enzymatic degradation of the protein corona formed on doxorubicin-loaded gold nanoparticles by matrix metalloproteinases, in turn affecting the doxorubicin release rate into pancreatic cancer cells.35 Various corona proteins were degraded at different rates, leading to varying levels of cytotoxicity depending on the identity of the in vitro pre-formed corona proteins. Matrix metalloproteinases were also used by Gao et al. to dePEGylate their prodrug nanoparticles once accumulated at the target tumor site.146 Another enzyme of interest is cathepsin B, used frequently in the realm of antibody-drug conjugates. Cathepsin B has the potential to augment nanoparticle efficacy through its abundance in lysosomes and consistent activity. Han et al. used a dual enzyme strategy to increase the efficacy of their drug-loaded quantum dots. First, they used the aforementioned matrix metalloproteinases to dePEGylate their quantum dots and expose a cyclic RGD targeting peptide, simultaneously increasing cellular targeting and uptake. Once within the lysosome, native cathepsin B cleaved the cancer drug gemcitabine from the quantum dot surface, thereby increasing drug release into the cell and thus nanoparticle efficacy.129 There remains further work to be done in this area to utilize the array of endogenous enzymes found within biological systems of interest for enzyme-activation of nanoparticles.
4.4.4. Redox-Responsive Nanoparticles.
Redox chemistry offers another trigger to activate nanoparticle systems, as the nanoparticle travels between oxidative and reductive environments found in the extracellular and intracellular spaces, respectively. For example, the second component of the cleavable system used by Feng et al. takes advantage of redox chemistry with a photosensitizer and an immunoinhibitory compound linked by a reducible disulfide bond. Once within the cellular environment, the abundant antioxidant, glutathione, reduces the disulfide bond and causes release of the nanoparticle components, proving effective for tumor ablation during in vivo mouse studies.145 Nanoparticle dePEGylation has also been accomplished by a reducible disulfide attachment that is cleaved to release drug cargo once internalized.147,148 Similarly, Yu et al. demonstrated the utility of ceria nanoparticles encapsulated by a poly(lactic-co-glycolic acid) (PLGA)-PEG coating linked by a thioketal bond and loaded with the kidney injury drug, atorvastatin.149 The PEG coating was removed by thioketal bond cleavage once nanoparticles accumulated at the kidney injury site where ROS production was elevated, resulting in the release of atorvastatin for treatment. Interestingly, the ceria nanoparticles were also functionalized with triphenylphosphine to target the injured cell’s mitochondria and scavenge the ROS produced by the injured mitochondria. The use of redox-responsive activation in nanoparticle systems gives rise to multifunctional modalities that would be more efficacious when used synergistically.
5. Biocompatibility
The presence of foreign objects such as bacteria, viruses, and nanoparticles within the body can induce a response by the immune system. Depending on the nanoparticle’s properties, this immune response can both render the nanoparticles ineffective and also lead to inflammation and systemic complications.150 Although many advancements have been made in preventing this response as detailed in Section 3, it is still important to understand the scope of possible interactions between nanoparticles and the immune system to ensure nanoparticle efficacy and inform rational design of the protein corona (Fig. 5).
Figure 5. Biocompatibility considerations for nanoparticle administration.

Proteins known as opsonins often adsorb to nanoparticles in vivo and elicit an immune response. This response involves the innate immune system (complement and mononuclear phagocyte systems) and/or adaptive immune system (lymphocytes and antibodies). Protein corona design can be employed to guide immunostimulation (increase in cytokine and leukocyte levels), whereby the immune system is activated intentionally towards eliminating harmful cells or combatting immunodeficiency. Conversely, the nanoparticle-corona construct can be manipulated towards immunosuppression (decrease in cytokine and leukocyte levels), and nanoparticle clearance or an inflammatory response are avoided. Some images in this figure are adapted with permission from Servier Medical Art by Servier (http://smart.servier.com), licensed under a Creative Commons Attribution 3.0 Unported License.
The human immune system is composed of two branches: the innate immune system and the adaptive immune system. The innate immune system is composed of the complement system and the mononuclear phagocyte system (MPS) working in tandem to identify and eliminate pathogens. This process is activated almost immediately after an infection is detected. The adaptive immune system uses lymphocytes known as T cells and B cells in combination with antibodies to eliminate foreign and native infected cells. In contrast to the innate immune system, the adaptive response requires about one week post-infection to fully develop. Due to this discrepancy in time scale, the innate immune response is the body’s first line of defense against perceived foreign invaders and is canonically observed in assaying nanoparticle biocompatibility. Improper nanoparticle design that neglects the innate immune system could lead to untimely clearance, unintended immune response, and complications including systemic toxicity.151 Conversely, over-use of nanoparticle coatings such as PEG can generate anti-PEG antibodies by the adaptive immune system as discussed in Section 3.1.1. It is thus of great importance to consider the potential mounted immune responses, particularly the innate response, when designing nanoparticle systems for use in vivo.
5.1. Immune System Activation and Suppression
The involvement of nanoparticles with the innate immune system can be categorized into immune activation and suppression. Activation is better studied than suppression due to it being easier to elicit with nanoparticles, although both have been demonstrated in recent years.8,152 Immune activation can be further divided into unintended activation via the complement system and intended activation through elevated cytokine levels and leukocyte activation. Immunosuppression aims to mitigate the immune response by depleting inflammatory cytokine levels and limiting leukocyte migration, which can result in longer nanoparticle circulation time from lower MPS clearance and reduced inflammation in hypersensitive systems.153,154
5.1.1. Complement Activation.
It is generally accepted that nanoparticles possessing different physicochemical properties will activate different innate immune response pathways.27,150 As part of the innate immune response, the complement system is composed of a series of soluble proteins produced by hepatocytes in the liver that amplify or “complement” the function of antibodies in the adaptive immune system. Complement proteins often contribute to the formation of the in vivo protein corona when a nanoparticle enters the body, which places the complement system at the forefront of the immune response to nanoparticles.25 The complement system can be activated by three different pathways: classical, lectin, and alternative. The classical pathway is activated by the binding of antibodies to antigens present on the surface of pathogens. The lectin pathway is initiated by the binding of mannan-binding lectin with mannose and fucose residues found in the cell wall of bacteria. The alternative pathway is activated by the binding of complement protein C3 to the pathogen. All three pathways converge at the point where this C3 protein is cleaved into anaphylatoxins C3a and C3b, which leads the complement cascade to ultimately recruit phagocytes and lymphocytes to the site of infection. Nanoparticles typically activate the classical and alternative pathways, as their surfaces provide ample area for antibodies and complement proteins to bind and trigger the respective cascades.155,156
Nanoparticle surface properties are key to eliciting immune activation. For instance, Coty et al. demonstrated that dextran-coated poly(isobutylcyanoacrylate) nanoparticles activate different complement pathways depending on the architecture of the dextran coating itself.150 They concluded that the density and length of the dextran coating modulated the ability of different complement proteins such as C3 and mannose-binding lectin to bind to the nanoparticle surface, in turn affecting which pathway was activated. Fülöp et al. explored the complement activation effect of various coating materials on superparamagnetic iron oxide nanoparticles (SPIONs), similarly determining that dextran coating leads to complement activation, in this case by the alternative pathway.151 They incubated SPIONs noncovalently adsorbed to starch, carboxymethyldextran, chitosan, phosphatidylcholine, citric acid, and dextran coatings with human serum samples in vitro and measured the levels of the complement pathway-specific marker SC5b-9. Phosphatidylcholine and chitosan showed no reaction, starch and carboxymethyldextran showed minor effects, and dextran caused massive complement activation. Escamilla-Rivera et al. conducted a similar study, comparing complement activation of iron oxide nanoparticles with bare surfaces, with a polyvinylpyrrolidone (PVP) coating, and with a PEG coating.27 Interestingly, the PEG coating resulted in doubled complement protein adsorption levels in vitro and higher levels of inflammatory cytokines in vivo, while the bare and PVP-coated nanoparticles showed no significant increase in either case. Quach and Kah studied the effect of gold nanoparticle size, shape, and polyelectrolyte ligand on complement activation.25 By detecting the endpoint complement marker SC5b-9 concentration, they determined that polyethyleneimine ligand induced the most complement activation and that there is a negative correlation between nanoparticle surface hydrophilicity and complement activation. Based on the literature, there is little predictability with which nanoparticle materials or coatings will elicit an immune response, necessitating that each be tested individually.12 Due to the importance of the complement pathway in understanding the biocompatibility of nanoparticle systems, more studies are required to fully understand the mechanisms and dependencies of this cascade within the context of nanoparticle activation.
5.1.2. Immunostimulation.
Rather than avoiding immune activation, nanoparticles can also be applied to stimulate an immune response. Immunostimulation is beneficial in the context of harnessing the immune system to clear infected cells, including cancer cells, and combating immunodeficiency.152 Since the protein corona influences the interactions between nanoparticles and cells of the immune system, it plays a crucial role in mediating this stimulatory response. For example, Dai et al. explored the effect of different in vitro protein coronas formed on poly(methylacrylic acid) (PMA) nanoparticles on cytokine production in THP-1 monocytes.157 In particular, they found that serum-incubated PMA nanoparticles showed increased levels of inflammatory cytokines including interleukin-8 and interleukin-1ß, demonstrating the importance of the protein corona in immunostimulation responses to nanoparticle introduction.
Several nanoparticle systems in recent years have been designed to purposefully leverage the protein corona to activate the immune system. Mo et al. exploited the serum protein corona formed on black phosphorus nanosheets to polarize M0 macrophages into M1 macrophages, stimulating the immune system to eliminate cancer cells.152 Similarly, Kouser et al. investigated the inflammatory response of functionalized carbon nanotubes with adsorbed human properdin, a protein that upregulates the alternative complement pathway.88 The authors adsorbed either the full properdin protein or only the binding domain, thrombospondin type I repeat 4 and 5 (TSR4+5), to cellulose-coated and oxidized carbon nanotubes. Upon in vitro incubation in blood serum, TSR4+5-coated nanotubes inhibited complement activation due to the lack of available surface area for native properdin to adsorb, while properdin-nanotubes maintained complement activation. Furthermore, pre-adsorbed properdin enhanced the uptake of carbon nanotubes by THP-1 macrophages, stimulating the production of pro-inflammatory cytokines, while pre-adsorbed TSR4+5 was ineffective in producing this immunostimulatory response. Taken together, these various studies demonstrate the potential design of nanoparticles to either stimulate immune cell activation directly or rationally engage immunostimulation through the complement system.
Employing nanoparticles that produce ROS can induce an oxidative stress response in cells as another form of immunostimulation. These ROS include hydrogen peroxide, superoxide anion radicals, singlet oxygen, and free hydroxyl radicals, which lead to a variety of oxidative stress responses, including inflammation, apoptosis, DNA damage, and lipid peroxidation.158 While nanoparticles themselves can produce an oxidative stress response, Jayaram et al. demonstrated that the in vitro protein corona also plays a role in the oxidative stress experienced by cells.159 In this study, titanium dioxide nanoparticles were found to produce ROS that caused oxidation of corona proteins, including complement C3, serum albumin, and plasminogen. The oxidized corona proteins subsequently caused downregulation of peroxiredoxin expression, enzymes responsible for clearing peroxide species, thus resulting in an oxidative stress response to the cell. They also concluded that an increase in nanoparticle surface defects exacerbates the oxidative stress effect. Due to the potential of the protein corona to produce an oxidative stress response in cells, it is important to further study nanoparticles in this light in addition to other immunological effects.
5.1.3. Immunosuppression.
Nanoparticles can also function to limit activation of the immune system by suppressing cytokine levels to lower MPS activity and reduce inflammatory effects.160 Cai et al. connected the nanoparticle protein corona to a decrease in cytokine production by macrophages, showing a decrease in level of proinflammatory cytokine, interleukin-6 (IL-6), produced due to the presence of the protein corona.8 Similarly, Dai et al. discovered that their PMA nanoparticles had immunosuppressive effects in THP-1 monocytes depending on the source of their in vitro protein corona.157 They found that PMA nanoparticles incubated with HeLa cell-conditioned media reduced the production of IL-6, IL-1b, IL-8, and TNF-a cytokines. In addition to reducing cytokine production, nanoparticles can also suppress an overactive immune system by targeting and eliminating specific immune cell populations. In certain autoimmune diseases, B cells can become overactive, leading to the destruction of healthy cells. Luk et al. demonstrated the possibility of targeting these hypersensitive B cells by coating polymeric nanoparticles with red blood cell membranes containing B cell receptor-targeted antigens.154 By purposefully designing the protein corona of this nanoparticle system, the authors were able to successfully target and visualize autoimmune B cells, opening the door to the development of targeted immunosuppressive treatments. Overall, these findings demonstrate the ability of the protein corona to induce an immunosuppressive response in vivo.
5.2. Cytotoxicity Assays
The assessment of these various immune activation and suppression functionalities, and more broadly the extent to which a nanoparticle is deemed biocompatible, relies on cytotoxicity assays. These assay outputs depend not only on the nanoparticle properties and interactions, but also on the specific assay used in the study. Common techniques for toxicity assessment include nanoparticle incubation with representative cell systems, such as HeLa cancer cells and THP-1 monocytes to assess cell viability,40 endpoint cytokine level measurements to determine inflammation,25,76 and live mice and rat models to compare in vitro analysis with in vivo efficacy.27,76,160 Although these techniques can be useful tools for characterization, literature increasingly suggests that the adaptation of standardized in vitro toxicity assays to assessing nanoparticle outcomes must be done with care. For example, common cell viability assays include the MTT,142,149,159 LDH,161,162 Trypan Blue,160 and CCK-8 assays.8,74 However, nanoparticles have been shown to interfere with such assays by either adsorbing the reagent or readout molecules, or in the case of colorimetric assays, absorbing or contributing to the output signal being quantified.161–163 This leads to false cytotoxicity or efficacy predictions because the nanoparticle presence alone vastly modulates the assay output, often confirmed by seeing negligible in vitro toxicity yet drastic changes in cell morphology. As such, it is imperative to include the necessary controls for these assays and run multiple, orthogonal assays to avoid reporting misleading results.
Conclusions
Nanoparticles offer a promising platform towards studying and manipulating biological systems. Yet, formation of the protein corona on nanoparticle surfaces upon introduction into biological environments remains a considerable barrier between in vitro design and in vivo application. Beyond characterizing the protein corona formed on varying nanomaterials, recent developments have sought to elegantly exploit and rationally design the protein corona to achieve improved nanomaterial functionality. In this review, we have described intrinsic nanoparticle and extrinsic biological factors that govern unpredictable protein corona formation, such that these insights may be used towards either mitigation of unfavorable adsorption or enhancement of desired adsorption. Many such factors are co-dependent, therefore work remains in separating out nuances among these variables. To extend experimental data into broader design rules, recent work has involved ensemble machine learning approaches to develop predictive models of protein corona fingerprints formed on nanoparticles based on protein, nanoparticle, and solution characteristics.164 Such models could better inform corona-based design. Moreover, future work should move towards characterization under biologically relevant conditions, such as under flow for intravenous applications, in biofluids most representative of the intended application. Based on these design principles, work has implicated noncovalent adsorption and covalent coupling strategies. Characterization techniques have been adapted from tangential fields and developed anew. Recent advances continue to give insight into minutia of the protein corona, such as the hard vs. soft corona constituents and kinetics. However, particular care must be taken in characterizing individual particle vs. aggregate size, and future characterization should be done primarily in solution rather than in dried or immobilized surface settings. Additionally, models of protein-nanoparticle association should be applied with caution regarding assumptions and limitations, especially if mechanistic or quantitative conclusions are to be made from the data.
Corona-mediated nanoparticle functionalities include stealth, targeting, and activation. Stealth continues to be mechanistically explored, as more studies demonstrate the difficulty in fully eradicating protein corona formation. Instead, achieving stealth seems to rely on adsorption of specific proteins to mask the foreign nanoparticle presence. While common strategies rely on hydrophilic, zwitterionic, or carbohydrate shells, new work recognizes the promise of protein or biomimetic coatings towards attaining stealth. It is increasingly recognized that controlling, instead of eliminating, protein adsorption will further benefit stealth aims in nanomedicine. After evading recognition during circulation, targeting enables specific localization. Nanoparticle targeting stands to benefit from the remarkable molecular specificity of protein interactions by taking advantage of endogenous protein interactions with their target ligands. Finally, activatable properties have been applied to induce or guide specific nanoparticle function within the targeted area. Activation of nanoparticle function typically relies on biological or externally applied triggers, though future work is required to use such cleavable strategies synergistically with the protein corona. Combining such concepts, recent work has demonstrated modular nanoparticle constructs that are capable of tumor targeting (with cleavable reporters to indicate success) and gene delivery.165 Other exciting work has moved to multiplexed testing of the targeting and function of many nanoparticle chemistries simultaneously via DNA-barcoding.166 Finally, biocompatibility of such nanoparticle-corona constructs has inspired uses in both immune stimulation (both intended and unintended) and immune suppression, where the assays to assess biocompatibility stand to be refined. In sum, design of the protein corona on nanoparticles presents a functional handle to tune construct properties and attain improved outcomes towards in vivo stealth, targeting, and function.
Acknowledgements
M.P.L. acknowledges support of Burroughs Wellcome Fund Career Award at the Scientific Interface (CASI), the Simons Foundation, NIH NIDA CEBRA award # R21DA044010, Stanley Fahn PDF Junior Faculty Grant with Award #PF-JFA-1760, Beckman Foundation Young Investigator Award, DARPA Young Faculty Award, FFAR New Innovator Award, an IGI award, support from CITRIS and the Banatao Institute, and a USDA award. M.P.L. is a Chan Zuckerberg Biohub Investigator and an Innovative Genomics Institute Investigator. R.L.P. and F.L. acknowledge the support of NSF Graduate Research Fellowships (NSF DGE 1752814), and L.C. acknowledges the support of National Defense Science and Engineering Graduate (NDSEG) Fellowship. We would like to acknowledge the use of medical clipart from Servier Medical Art by Servier (http://smart.servier.com).
Footnotes
Conflicts of interest
There are no conflicts to declare.
References
- 1.Chio L, Pinals RL, Murali A, Goh NS and Landry MP, Advanced Functional Materials, 2020, 30, 1910556. [Google Scholar]
- 2.Gupta A, Das R, Yesilbag Tonga G, Mizuhara T and Rotello VM, ACS Nano, 2018, 12, 89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oh JY, Kim HS, Palanikumar L, Go EM, Jana B, Park SA, Kim HY, Kim K, Seo JK, Kwak SK, Kim C, Kang S and Ryu J-H, Nature Communications, 2018, 9, 4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hadjidemetriou M and Kostarelos K, Nature Nanotechnology, 2017, 12, 288–290. [DOI] [PubMed] [Google Scholar]
- 5.Ke PC, Lin S, Parak WJ, Davis TP and Caruso F, ACS Nano, , DOI: 10.1021/acsnano.7b08008. [DOI] [PubMed] [Google Scholar]
- 6.Warning LA, Zhang Q, Baiyasi R, Landes CF and Link S, J. Phys. Chem. Lett, , DOI: 10.1021/acs.jpclett.9b03446. [DOI] [PubMed] [Google Scholar]
- 7.Raoufi M, Hajipour MJ, Kamali Shahri SM, Schoen I, Linn U and Mahmoudi M, Nanoscale, 2018, 10, 1228–1233. [DOI] [PubMed] [Google Scholar]
- 8.Cai R, Ren J, Ji Y, Wang Y, Liu Y, Chen Z, Farhadi Sabet Z, Wu X, Lynch I and Chen C, ACS Appl. Mater. Interfaces, 2020, 12, 1997–2008. [DOI] [PubMed] [Google Scholar]
- 9.Lu X, Xu P, Ding H-M, Yu Y-S, Huo D and Ma Y-Q, Nat Commun, 2019, 10, 4520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giulimondi F, Digiacomo L, Pozzi D, Palchetti S, Vulpis E, Capriotti AL, Chiozzi RZ, Laganà A, Amenitsch H, Masuelli L, Mahmoudi M, Screpanti I, Zingoni A and Caracciolo G, Nat Commun, 2019, 10, 3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Piloni A, Wong CK, Chen F, Lord M, Walther A and Stenzel MH, Nanoscale, 2019, 11, 23259–23267. [DOI] [PubMed] [Google Scholar]
- 12.Ding T and Sun J, Pharm Res, 2020, 37, 10. [Google Scholar]
- 13.Naidu PSR, Gavriel N, Gray CGG, Bartlett CA, Toomey LM, Kretzmann JA, Patalwala D, McGonigle T, Denham E, Hee C, Ho D, Taylor NL, Norret M, Smith NM, Dunlop SA, Iyer KS and Fitzgerald M, ACS Appl. Mater. Interfaces, 2019, 11, 22085–22095. [DOI] [PubMed] [Google Scholar]
- 14.Meng Q-F, Cheng Y-X, Huang Q, Zan M, Xie W, Sun Y, Li R, Wei X, Guo S-S, Zhao X-Z, Rao L and Liu W, ACS Appl. Mater. Interfaces, 2019, 11, 28732–28739. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Z, Guan J, Jiang Z, Yang Y, Liu J, Hua W, Mao Y, Li C, Lu W, Qian J and Zhan C, Nat Commun, 2019, 10, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takeuchi T, Kitayama Y, Sasao R, Yamada T, Toh K, Matsumoto Y and Kataoka K, Angew. Chem. Int. Ed, 2017, 56, 7088–7092. [DOI] [PubMed] [Google Scholar]
- 17.Corbo C, Molinaro R, Taraballi F, Toledano Furman NE, Hartman KA, Sherman MB, De Rosa E, Kirui DK, Salvatore F and Tasciotti E, ACS Nano, 2017, 11, 3262–3273. [DOI] [PubMed] [Google Scholar]
- 18.Perng W, Palui G, Wang W and Mattoussi H, Bioconjugate Chem, 2019, 30, 2469–2480. [DOI] [PubMed] [Google Scholar]
- 19.Stepien G, Moros M, Pérez-Hernández M, Monge M, Gutiérrez L, Fratila RM, de las Heras M, Menao Guillén S, Puente Lanzarote JJ, Solans C, Pardo J and de la Fuente JM, ACS Appl. Mater. Interfaces, 2018, 10, 4548–4560. [DOI] [PubMed] [Google Scholar]
- 20.Zhu Y, Meng T, Tan Y, Yang X, Liu Y, Liu X, Yu F, Wen L, Dai S, Yuan H and Hu F, Mol. Pharmaceutics, 2018, 15, 5374–5386. [DOI] [PubMed] [Google Scholar]
- 21.Zhao J, Wu S, Qin J, Shi D and Wang Y, ACS Appl. Mater. Interfaces, 2018, 10, 41986–41998. [DOI] [PubMed] [Google Scholar]
- 22.Lai W, Wang Q, Li L, Hu Z, Chen J and Fang Q, Colloids and Surfaces B: Biointerfaces, 2017, 152, 317–325. [DOI] [PubMed] [Google Scholar]
- 23.Simonelli F, Rossi G and Monticelli L, J. Phys. Chem. B, 2019, 123, 1764–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lazarovits J, Chen YY, Song F, Ngo W, Tavares AJ, Zhang Y-N, Audet J, Tang B, Lin Q, Tleugabulova MC, Wilhelm S, Krieger JR, Mallevaey T and Chan WCW, Nano Lett, 2019, 19, 116–123. [DOI] [PubMed] [Google Scholar]
- 25.Quach QH and Kah JC, RSC Adv, 2018, 8, 6616–6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pinals RL, Yang D, Rosenberg DJ, Chaudhary T, Crothers AR, Iavarone AT, Hammel M and Landry MP, bioRxiv, 2020, 2020.01.13.905356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Escamilla-Rivera V, Solorio-Rodriguez A, Uribe-Ramirez M, Lozano O, Lucas S, Chagolla-López A, Winkler R and De Vizcaya-Ruiz A, IJN, 2019, Volume 14, 2055–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee H, Small, 2020, 16, 1906598. [Google Scholar]
- 29.Mohammad-Beigi H, Hayashi Y, Zeuthen CM, Eskandari H, Scavenius C, Juul-Madsen K, Vorup-Jensen T, Enghild JJ and Sutherland DS, bioRxiv, 2020, 2020.02.05.924480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang H, Ma R, Nienhaus K and Nienhaus GU, Small, 2019, 15, 1900974. [DOI] [PubMed] [Google Scholar]
- 31.Chen D, Parayath N, Ganesh S, Wang W and Amiji M, Nanoscale, 2019, 11, 18806–18824. [DOI] [PubMed] [Google Scholar]
- 32.Guo P, Liu D, Subramanyam K, Wang B, Yang J, Huang J, Auguste DT and Moses MA, Nat Commun, 2018, 9, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee JC, Donahue ND, Mao AS, Karim A, Komarneni M, Thomas EE, Francek ER, Yang W and Wilhelm S, ACS Appl. Nano Mater, , DOI: 10.1021/acsanm.9b02541. [DOI] [Google Scholar]
- 34.Francia V, Montizaan D and Salvati A, Beilstein J. Nanotechnol, 2020, 11, 338–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gafur A, Kristi N, Maruf A, Wang G and Ye Z, Biomater. Sci, 2019, 7, 3581–3593. [DOI] [PubMed] [Google Scholar]
- 36.Affonso de Oliveira JF, Scheffer FR, Landis RF, Teixeira Neto É, Rotello VM and Cardoso MB, ACS Appl. Mater. Interfaces, 2018, 10, 41917–41923. [DOI] [PubMed] [Google Scholar]
- 37.Champanhac C, Simon J, Landfester K and Mailänder V, Biomacromolecules, 2019, 20, 3724–3732. [DOI] [PubMed] [Google Scholar]
- 38.Zhou H, Fan Z, Li PY, Deng J, Arhontoulis DC, Li CY, Bowne WB and Cheng H, ACS Nano, 2018, 12, 10130–10141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kong L, Chen Q, Campbell F, Snaar-Jagalska E and Kros A, Adv. Healthcare Mater, 2020, 1901489. [DOI] [PubMed] [Google Scholar]
- 40.Dancy JG, Wadajkar AS, Connolly NP, Galisteo R, Ames HM, Peng S, Tran NL, Goloubeva OG, Woodworth GF, Winkles JA and Kim AJ, Science Advances, 2020, 6, eaax3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li X, Lu C, Xia W, Quan G, Huang Y, Bai X, Yu F, Xu Q, Qin W, Liu D and Pan X, AAPS PharmSciTech, 2020, 21, 78. [DOI] [PubMed] [Google Scholar]
- 42.Bertrand N, Grenier P, Mahmoudi M, Lima EM, Appel EA, Dormont F, Lim J-M, Karnik R, Langer R and Farokhzad OC, Nat Commun, 2017, 8, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gal N, Schroffenegger M and Reimhult E, J. Phys. Chem. B, 2018, 122, 5820–5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lima T, Bernfur K, Vilanova M and Cedervall T, Sci Rep, 2020, 10, 1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shadmani P, Mehrafrooz B, Montazeri A and Naghdabadi R, J. Phys.: Condens. Matter, 2020, 32, 115101. [DOI] [PubMed] [Google Scholar]
- 46.Kari OK, Ndika J, Parkkila P, Louna A, Lajunen T, Puustinen A, Viitala T, Alenius H and Urtti A, Nanoscale, 2020, 12, 1728–1741. [DOI] [PubMed] [Google Scholar]
- 47.Yu C, Zhou Q, Xiao F, Li Y, Hu H, Wan Y, Li Z and Yang X, ACS Appl. Mater. Interfaces, 2017, 9, 10481–10493. [DOI] [PubMed] [Google Scholar]
- 48.Ho YT, Azman N. ‘Ain, Loh FWY, Ong GKT, Engudar G, Kriz SA and Kah JCY, Bioconjugate Chem, 2018, 29, 3923–3934. [DOI] [PubMed] [Google Scholar]
- 49.Naidu PSR, Denham E, Bartlett CA, McGonigle T, Taylor NL, Norret M, Smith Nicole. M., Dunlop SA, Iyer KS and Fitzgerald M, RSC Adv, 2020, 10, 2856–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Curtis C, Toghani D, Wong B and Nance E, Colloids and Surfaces B: Biointerfaces, 2018, 170, 673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Visalakshan RM, MacGregor MN, Sasidharan S, Ghazaryan A, Mierczynska-Vasilev AM, Morsbach S, Mailänder V, Landfester K, Hayball JD and Vasilev K, ACS Appl. Mater. Interfaces, 2019, 11, 27615–27623. [DOI] [PubMed] [Google Scholar]
- 52.Wang X, Wang M, Lei R, Zhu SF, Zhao Y and Chen C, ACS Nano, 2017, 11, 4606–4616. [DOI] [PubMed] [Google Scholar]
- 53.Palomba R, Palange AL, Rizzuti IF, Ferreira M, Cervadoro A, Barbato MG, Canale C and Decuzzi P, ACS Nano, 2018, 12, 1433–1444. [DOI] [PubMed] [Google Scholar]
- 54.Anselmo AC and Mitragotri S, Advanced Drug Delivery Reviews, 2017, 108, 51–67. [DOI] [PubMed] [Google Scholar]
- 55.Francia V, Yang K, Deville S, Reker-Smit C, Nelissen I and Salvati A, ACS Nano, 2019, 13, 11107–11121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pinals RL, Yang D, Lui A, Cao W and Landry MP, J. Am. Chem. Soc, 2020, 142, 1254–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weiss ACG, Kempe K, Förster S and Caruso F, Biomacromolecules, 2018, 19, 2580–2594. [DOI] [PubMed] [Google Scholar]
- 58.Chen F, Wang G, Griffin JI, Brenneman B, Banda NK, Holers VM, Backos DS, Wu L, Moghimi SM and Simberg D, Nature Nanotechnology, 2017, 12, 387–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rodriguez-Quijada C, de Puig H, Sánchez-Purrà M, Yelleswarapu C, Evans JJ, Celli JP and Hamad-Schifferli K, ACS Appl. Mater. Interfaces, 2019, 11, 14588–14596. [DOI] [PubMed] [Google Scholar]
- 60.Mahmoudi M, Trends in Biotechnology, 2018, 36, 755–769. [DOI] [PubMed] [Google Scholar]
- 61.Wang A, Yang T, Fan W, Yang Y, Zhu Q, Guo S, Zhu C, Yuan Y, Zhang T and Gan Y, Adv. Healthcare Mater, 2019, 8, 1801123. [DOI] [PubMed] [Google Scholar]
- 62.Nance E, in Biomedical Nanotechnology, Humana Press, New York, NY, 2017, pp. 91–104. [Google Scholar]
- 63.Reza Sepand M, Ghavami M, Zanganeh S, Stacks S, Ghasemi F, Montazeri H, Corbo C, Derakhshankhah H, Nasser Ostad S, Hossein Ghahremani M and Mahmoudi M, Nanoscale, , DOI: 10.1039/C9NR08784B. [DOI] [PubMed] [Google Scholar]
- 64.Tonigold M, Simon J, Estupiñán D, Kokkinopoulou M, Reinholz J, Kintzel U, Kaltbeitzel A, Renz P, Domogalla MP, Steinbrink K, Lieberwirth I, Crespy D, Landfester K and Mailänder V, Nature Nanotechnology, 2018, 13, 862–869. [DOI] [PubMed] [Google Scholar]
- 65.Piao J-G, Gao F, Li Y, Yu L, Liu D, Tan Z-B, Xiong Y, Yang L and You Y-Z, Nano Res, 2018, 11, 3193–3204. [Google Scholar]
- 66.Koide R and Nishimura S-I, Angew. Chem. Int. Ed, 2019, 58, 14513–14518. [DOI] [PubMed] [Google Scholar]
- 67.Cox A, Andreozzi P, Dal Magro R, Fiordaliso F, Corbelli A, Talamini L, Chinello C, Raimondo F, Magni F, Tringali M, Krol S, Jacob Silva P, Stellacci F, Masserini M and Re F, ACS Nano, 2018, 12, 7292–7300. [DOI] [PubMed] [Google Scholar]
- 68.Palchetti S, Pozzi D, Capriotti AL, Barbera GL, Chiozzi RZ, Digiacomo L, Peruzzi G, Caracciolo G and Laganà A, Colloids and Surfaces B: Biointerfaces, 2017, 153, 263–271. [DOI] [PubMed] [Google Scholar]
- 69.Zhang H, Wu T, Yu W, Ruan S, He Q and Gao H, ACS Appl. Mater. Interfaces, 2018, 10, 9094–9103. [DOI] [PubMed] [Google Scholar]
- 70.Herda LM, Hristov DR, Lo Giudice MC, Polo E and Dawson KA, J. Am. Chem. Soc, 2017, 139, 111–114. [DOI] [PubMed] [Google Scholar]
- 71.Zhang W, Meckes B and Mirkin CA, ACS Cent. Sci, 2019, 5, 1983–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dal Magro R, Albertini B, Beretta S, Rigolio R, Donzelli E, Chiorazzi A, Ricci M, Blasi P and Sancini G, Nanomedicine: Nanotechnology, Biology and Medicine, 2018, 14, 429–438. [DOI] [PubMed] [Google Scholar]
- 73.Jhaveri A, Deshpande P, Pattni B and Torchilin V, Journal of Controlled Release, 2018, 277, 89–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fukuda R, Umeyama T, Tsujimoto M, Ishidate F, Tanaka T, Kataura H, Imahori H and Murakami T, Carbon, 2020, 161, 718–725. [Google Scholar]
- 75.Di Giosia M, Valle F, Cantelli A, Bottoni A, Zerbetto F, Fasoli E and Calvaresi M, Carbon, 2019, 147, 70–82. [Google Scholar]
- 76.Yeo ELL, Thong PSP, Soo KC and Kah JCY, Nanoscale, 2018, 10, 2461–2472. [DOI] [PubMed] [Google Scholar]
- 77.Weiss ACG, Kelly HG, Faria M, Besford QA, Wheatley AK, Ang C-S, Crampin EJ, Caruso F and Kent SJ, ACS Nano, 2019, 13, 4980–4991. [DOI] [PubMed] [Google Scholar]
- 78.Mout R, Yesilbag Tonga G, Wang L-S, Ray M, Roy T and Rotello VM, ACS Nano, 2017, 11, 3456–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mann FA, Lv Z, Grosshans J, Opazo F and Kruss S, Angewandte Chemie International Edition, , DOI: 10.1002/anie.201904167. [DOI] [Google Scholar]
- 80.Wang C, Wang Z and Dong L, Trends in Biotechnology, , DOI: 10.1016/j.tibtech.2018.02.016. [DOI] [Google Scholar]
- 81.Silvestri A, Di Silvio D, Llarena I, Murray RA, Marelli M, Lay L, Polito L and Moya SE, Nanoscale, 2017, 9, 14730–14739. [DOI] [PubMed] [Google Scholar]
- 82.Carril M, Padro D, del Pino P, Carrillo-Carrion C, Gallego M and Parak WJ, Nat Commun, 2017, 8, 1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zeng X, Sun J, Li S, Shi J, Gao H, Leong WS, Wu Y, Li M, Liu C, Li P, Kong J, Wu Y-Z, Nie G, Fu Y and Zhang G, Nat Commun, 2020, 11, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kokkinopoulou M, Simon J, Landfester K, Mailänder V and Lieberwirth I, Nanoscale, 2017, 9, 8858–8870. [DOI] [PubMed] [Google Scholar]
- 85.Weber C, Simon J, Mailänder V, Morsbach S and Landfester K, Acta Biomaterialia, 2018, 76, 217–224. [DOI] [PubMed] [Google Scholar]
- 86.Perera YR, Hill RA and Fitzkee NC, Israel Journal of Chemistry, 2019, 59, 962–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ceccon A, Schmidt T, Tugarinov V, Kotler SA, Schwieters CD and Clore GM, J. Am. Chem. Soc, 2018, 140, 6199–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kouser L, Paudyal B, Kaur A, Stenbeck G, Jones LA, Abozaid SM, Stover CM, Flahaut E, Sim RB and Kishore U, Front. Immunol, 2018, 9, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang Y, Wu JLY, Lazarovits J and Chan WCW, J. Am. Chem. Soc, 2020, 142, 8827–8836. [DOI] [PubMed] [Google Scholar]
- 90.Simon J, Müller LK, Kokkinopoulou M, Lieberwirth I, Morsbach S, Landfester K and Mailänder V, Nanoscale, 2018, 10, 10731–10739. [DOI] [PubMed] [Google Scholar]
- 91.Prozeller D, Morsbach S and Landfester K, Nanoscale, 2019, 11, 19265–19273. [DOI] [PubMed] [Google Scholar]
- 92.Park J, Park JE, Hedrick VE, Wood KV, Bonham C, Lee W and Yeo Y, Small, 2018, 14, 1703670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Feiner-Gracia N, Beck M, Pujals S, Tosi S, Mandal T, Buske C, Linden M and Albertazzi L, Small, 2017, 13, 1701631. [DOI] [PubMed] [Google Scholar]
- 94.Zhao L, Zhao L, Li H, Sun P, Wu J, Li K, Hu S, Wang X and Pu Q, Anal. Chem, 2019, 91, 15670–15677. [DOI] [PubMed] [Google Scholar]
- 95.Horbett TA, Journal of Biomedical Materials Research Part A, , DOI: 10.1002/jbm.a.36460. [DOI] [Google Scholar]
- 96.Milani S, Baldelli Bombelli F, Pitek AS, Dawson KA and Rädler J, ACS Nano, 2012, 6, 2532–2541. [DOI] [PubMed] [Google Scholar]
- 97.Monopoli MP, Åberg C, Salvati A and Dawson KA, Nature Nanotechnology, 2012, 7, 779–786. [DOI] [PubMed] [Google Scholar]
- 98.Latour RA, J. Biomed. Mater. Res, 2015, 103, 949–958. [DOI] [PubMed] [Google Scholar]
- 99.Casalini T, Limongelli V, Schmutz M, Som C, Jordan O, Wick P, Borchard G and Perale G, Front. Bioeng. Biotechnol, , DOI: 10.3389/fbioe.2019.00268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhdanov VP, Current Opinion in Colloid & Interface Science, 2019, 41, 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Malaspina DC, Pérez-Fuentes L, Drummond C, Bastos-González D and Faraudo J, Current Opinion in Colloid & Interface Science, 2019, 41, 40–49. [Google Scholar]
- 102.Brancolini G, Bellucci L, Maschio MC, Di Felice R and Corni S, Current Opinion in Colloid & Interface Science, 2019, 41, 86–94. [Google Scholar]
- 103.Brancolini G, Lopez H, Corni S and Tozzini V, International Journal of Molecular Sciences, 2019, 20, 3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tavanti F, Pedone A and Menziani MC, International Journal of Molecular Sciences, 2019, 20, 3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Correa S, Boehnke N, Barberio AE, Deiss-Yehiely E, Shi A, Oberlton B, Smith SG, Zervantonakis I, Dreaden EC and Hammond PT, ACS Nano, 2020, 14, 2224–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hume DA, Irvine KM and Pridans C, Trends in Immunology, 2019, 40, 98–112. [DOI] [PubMed] [Google Scholar]
- 107.Cai R and Chen C, Adv. Mater, 2019, 31, 1805740. [DOI] [PubMed] [Google Scholar]
- 108.Huckaby JT and Lai SK, Advanced Drug Delivery Reviews, 2018, 124, 125–139. [DOI] [PubMed] [Google Scholar]
- 109.Sanchez-Cano C and Carril M, IJMS, 2020, 21, 1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhou M, Huang H, Wang D, Lu H, Chen J, Chai Z, Yao SQ and Hu Y, Nano Lett, 2019, 19, 3671–3675. [DOI] [PubMed] [Google Scholar]
- 111.Bhattacharya AA, Grüne T and Curry S, Journal of Molecular Biology, 2000, 303, 721–732. [DOI] [PubMed] [Google Scholar]
- 112.Wilson C, Mau T, Weisgraber KH, Wardell MR, Mahley RW and Agard DA, Structure, 1994, 2, 713–718. [DOI] [PubMed] [Google Scholar]
- 113.Borhani DW, Rogers DP, Engler JA and Brouillette CG, Proceedings of the National Academy of Sciences, 1997, 94, 12291–12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Müller J, Bauer KN, Prozeller D, Simon J, Mailänder V, Wurm FR, Winzen S and Landfester K, Biomaterials, 2017, 115, 1–8. [DOI] [PubMed] [Google Scholar]
- 115.Zhang P, Sun F, Liu S and Jiang S, Journal of Controlled Release, 2016, 244, 184–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Guan J, Shen Q, Zhang Z, Jiang Z, Yang Y, Lou M, Qian J, Lu W and Zhan C, Nat Commun, 2018, 9, 2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Papi M, Caputo D, Palmieri V, Coppola R, Palchetti S, Bugli F, Martini C, Digiacomo L, Pozzi D and Caracciolo G, Nanoscale, 2017, 9, 10327–10334. [DOI] [PubMed] [Google Scholar]
- 118.Li S, Cai Y, Cao J, Cai M, Chen Y and Luo X, Polym. Chem, 2017, 8, 2472–2483. [Google Scholar]
- 119.Zhang C, Lu J, Hou Y, Xiong W, Sheng K and Lu H, ACS Appl. Mater. Interfaces, 2018, 10, 17463–17470. [DOI] [PubMed] [Google Scholar]
- 120.Zhao J, Qin Z, Wu J, Li L, Jin Q and Ji J, Biomater. Sci, 2018, 6, 200–206. [DOI] [PubMed] [Google Scholar]
- 121.Zhao K, Li D, Xu W, Ding J, Jiang W, Li M, Wang C and Chen X, Biomaterials, 2017, 116, 82–94. [DOI] [PubMed] [Google Scholar]
- 122.Wu H, Hu H, Wan J, Li Y, Wu Y, Tang Y, Xiao C, Xu H, Yang X and Li Z, Chemical Engineering Journal, 2018, 349, 129–145. [Google Scholar]
- 123.Simon J, Bauer KN, Langhanki J, Opatz T, Mailänder V, Landfester K and Wurm FR, Adv. Sci, 2019, 6, 1901199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fornaguera C, Guerra-Rebollo M, Lázaro MÁ, Cascante A, Rubio N, Blanco J and Borrós S, Adv. Healthcare Mater, 2019, 8, 1900849. [DOI] [PubMed] [Google Scholar]
- 125.Shen Y, Li X, Dong D, Zhang B, Xue Y and Shang P, Am J Cancer Res, 2018, 8, 916–931. [PMC free article] [PubMed] [Google Scholar]
- 126.Han Z and Lu Z-R, J. Mater. Chem. B, 2017, 5, 639–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Figueroa SM, Fleischmann D, Beck S and Goepferich A, Macromolecular Bioscience, 2020, 20, 1900427. [DOI] [PubMed] [Google Scholar]
- 128.Wang T, Wang D, Liu J, Feng B, Zhou F, Zhang H, Zhou L, Yin Q, Zhang Z, Cao Z, Yu H and Li Y, Nano Lett, 2017, 17, 5429–5436. [DOI] [PubMed] [Google Scholar]
- 129.Han H, Valdepérez D, Jin Q, Yang B, Li Z, Wu Y, Pelaz B, Parak WJ and Ji J, ACS Nano, 2017, 11, 1281–1291. [DOI] [PubMed] [Google Scholar]
- 130.Almalik A, Benabdelkamel H, Masood A, Alanazi IO, Alradwan I, Majrashi MA, Alfadda AA, Alghamdi WM, Alrabiah H, Tirelli N and Alhasan AH, Sci Rep, 2017, 7, 10542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Scannevin RH, Alexander R, Haarlander TM, Burke SL, Singer M, Huo C, Zhang Y-M, Maguire D, Spurlino J, Deckman I, Carroll KI, Lewandowski F, Devine E, Dzordzorme K, Tounge B, Milligan C, Bayoumy S, Williams R, Schalk-Hihi C, Leonard K, Jackson P, Todd M, Kuo LC and Rhodes KJ, J. Biol. Chem, 2017, 292, 17963–17974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mirković B, Renko M, Turk S, Sosič I, Jevnikar Z, Obermajer N, Turk D, Gobec S and Kos J, ChemMedChem, 2011, 6, 1351–1356. [DOI] [PubMed] [Google Scholar]
- 133.Agrahari V, Burnouf P-A, Burnouf T and Agrahari V, Advanced Drug Delivery Reviews, 2019, 148, 146–180. [DOI] [PubMed] [Google Scholar]
- 134.Wu M, Zhang H, Tie C, Yan C, Deng Z, Wan Q, Liu X, Yan F and Zheng H, Nature Communications, 2018, 9, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Xue J, Zhao Z, Zhang L, Xue L, Shen S, Wen Y, Wei Z, Wang L, Kong L, Sun H, Ping Q, Mo R and Zhang C, Nature Nanotechnology, 2017, 12, 692–700. [DOI] [PubMed] [Google Scholar]
- 136.Tang L, Wang Y and Li J, Chemical Society Reviews, 2015, 44, 6954–6980. [DOI] [PubMed] [Google Scholar]
- 137.Nabi B, Rehman S, Khan S, Baboota S and Ali J, Brain Research Bulletin, 2018, 142, 384–393. [DOI] [PubMed] [Google Scholar]
- 138.Zhu Y, Chen C, Cao Z, Shen S, Li L, Li D, Wang J and Yang X, Theranostics, 2019, 9, 8312–8320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kwon Y, Choi Y, Jang J, Yoon S and Choi J, Pharmaceutics, 2020, 12, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Puiggalí-Jou A, del Valle LJ and Alemán C, ACS Biomater. Sci. Eng, , DOI: 10.1021/acsbiomaterials.9b01794. [DOI] [PubMed] [Google Scholar]
- 141.Yu J, Chu X and Hou Y, Chem. Commun, 2014, 50, 11614–11630. [DOI] [PubMed] [Google Scholar]
- 142.Lim C, Cho E-B and Kim D, Korean J. Chem. Eng, 2019, 36, 166–172. [Google Scholar]
- 143.Li G, Gao Y, Gong C, Han Z, Qiang L, Tai Z, Tian J and Gao S, ACS Appl. Mater. Interfaces, 2019, 11, 39513–39524. [DOI] [PubMed] [Google Scholar]
- 144.Jin G, He R, Liu Q, Lin M, Dong Y, Li K, Tang BZ, Liu B and Xu F, Theranostics, 2019, 9, 246–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Feng B, Hou B, Xu Z, Saeed M, Yu H and Li Y, Adv. Mater, 2019, 31, 1902960. [DOI] [PubMed] [Google Scholar]
- 146.Gao A, Chen B, Gao J, Zhou F, Saeed M, Hou B, Li Y and Yu H, Nano Lett, 2020, 20, 353–362. [DOI] [PubMed] [Google Scholar]
- 147.Li J, Xu R, Lu X, He J and Jin S, IJN, 2017, Volume 12, 8043–8056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Yang Z, Guo Q, Cai Y, Zhu X, Zhu C, Li Y and Li B, Journal of Orthopaedic Translation, 2020, 21, 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yu H, Jin F, Liu D, Shu G, Wang X, Qi J, Sun M, Yang P, Jiang S, Ying X and Du Y, Theranostics, 2020, 10, 2342–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Coty J-B, Eleamen Oliveira E and Vauthier C, International Journal of Pharmaceutics, 2017, 532, 769–778. [DOI] [PubMed] [Google Scholar]
- 151.Fülöp T, Nemes R, Mészáros T, Urbanics R, Kok RJ, Jackman JA, Cho N-J, Storm G and Szebeni J, Journal of Controlled Release, 2018, 270, 268–274. [DOI] [PubMed] [Google Scholar]
- 152.Mo J, Xu Y, Wang X, Wei W and Zhao J, Nanoscale, 2020, 12, 1742–1748. [DOI] [PubMed] [Google Scholar]
- 153.Gabizon A, Tzemach D, Mak L, Bronstein M and Horowitz AT, Journal of Drug Targeting, 2002, 10, 539–548. [DOI] [PubMed] [Google Scholar]
- 154.Luk BT, Jiang Y, Copp JA, Hu C-MJ, Krishnan N, Gao W, Li S, Fang RH and Zhang L, Mol. Pharmaceutics, 2018, 15, 3723–3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Pondman KM, Sobik M, Nayak A, Tsolaki AG, Jäkel A, Flahaut E, Hampel S, ten Haken B, Sim RB and Kishore U, Nanomedicine: Nanotechnology, Biology and Medicine, 2014, 10, 1287–1299. [DOI] [PubMed] [Google Scholar]
- 156.Wang G, Chen F, Banda NK, Holers VM, Wu L, Moghimi SM and Simberg D, Front. Immunol, , DOI: 10.3389/fimmu.2016.00418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Dai Q, Guo J, Yan Y, Ang C-S, Bertleff-Zieschang N and Caruso F, Biomacromolecules, 2017, 18, 431–439. [DOI] [PubMed] [Google Scholar]
- 158.Ray PD, Huang B-W and Tsuji Y, Cellular Signalling, 2012, 24, 981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Jayaram DT, Runa S, Kemp ML and Payne CK, Nanoscale, 2017, 9, 7595–7601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.de Araújo RF, de Araújo AA, Pessoa JB, Freire Neto FP, da Silva GR, Leitão Oliveira ALCS, de Carvalho TG, Silva HFO, Eugênio M, Sant’Anna C and Gasparotto LHS, Pharmacological Reports, 2017, 69, 119–129. [DOI] [PubMed] [Google Scholar]
- 161.Gioria S, Caputo F, Urbán P, Maguire CM, Bremer-Hoffmann S, Prina-Mello A, Calzolai L and Mehn D, Nanomedicine, 2018, 13, 539–554. [DOI] [PubMed] [Google Scholar]
- 162.Riaz Ahmed KB, Nagy AM, Brown RP, Zhang Q, Malghan SG and Goering PL, Toxicology in Vitro, 2017, 38, 179–192. [DOI] [PubMed] [Google Scholar]
- 163.Wörle-Knirsch JM, Pulskamp K and Krug HF, Nano Lett, 2006, 6, 1261–1268. [DOI] [PubMed] [Google Scholar]
- 164.Findlay MR, Freitas DN, Mobed-Miremadi M and Wheeler KE, Environmental Science: Nano, 2018, 5, 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Boehnke N, Correa S, Hao L, Wang W, Straehla JP, Bhatia SN and Hammond PT, Angewandte Chemie International Edition, 2020, 59, 2776–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Dahlman JE, Kauffman KJ, Xing Y, Shaw TE, Mir FF, Dlott CC, Langer R, Anderson DG and Wang ET, PNAS, 2017, 114, 2060–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]