

Tryptamine-derived lipidoid-doped lipid nanoparticle formulations facilitate cargos crossing the blood-brain barrier.
Abstract
Safe and efficient delivery of blood-brain barrier (BBB)–impermeable cargos into the brain through intravenous injection remains a challenge. Here, we developed a previously unknown class of neurotransmitter–derived lipidoids (NT-lipidoids) as simple and effective carriers for enhanced brain delivery of several BBB-impermeable cargos. Doping the NT-lipidoids into BBB-impermeable lipid nanoparticles (LNPs) gave the LNPs the ability to cross the BBB. Using this brain delivery platform, we successfully delivered amphotericin B (AmB), antisense oligonucleotides (ASOs) against tau, and genome-editing fusion protein (−27)GFP-Cre recombinase into the mouse brain via systemic intravenous administration. We demonstrated that the NT-lipidoid formulation not only facilitates cargo crossing of the BBB, but also delivery of the cargo into neuronal cells for functional gene silencing or gene recombination. This class of brain delivery lipid formulations holds great potential in the treatment of central nervous system diseases or as a tool to study the brain function.
INTRODUCTION
The treatment of central nervous system (CNS) diseases, such as neurodegenerative disorders, brain tumors, brain infections, and stroke, is severely constrained by the blood-brain barrier (BBB) because it prevents the transfer of most of small-molecule drugs and macromolecules (e.g., peptides, gene drugs, and protein drugs) into the brain (1–6). To date, extensive efforts have been undertaken to enhance brain delivery efficiency, including direct CNS administration, disruption of the BBB, and carrier vehicle–mediated delivery (1, 6, 7). However, direct CNS administration is invasive, which may cause infection and tissue damage, and is also limited by diffusion distance and rapid efflux of drugs out of the CNS within hours (8–10). Disruption of the BBB, using techniques such as osmotic disruption, biochemical disruption, and ultrasound-mediated disruption, is effective to introduce drugs into the brain; however, these transient BBB openings also allows for the leakage of plasma proteins into the brain, leading to neurotoxicity, vascular pathology, and chronic neuropathologic changes in the brain (1, 7, 11). Therefore, approaches for safe and efficient delivery of BBB-impermeable cargos, particularly for gene and nucleic acid therapy, into CNS through intravenous injection remain to be desired.
The carrier vehicle–mediated brain drug delivery is considered a promising and versatile brain delivery system. For decades, various carrier vehicles, such as viral vectors, exosomes, molecular Trojan horses, and sundry nanoparticle formulations, have been developed to enhance brain delivery (12–15). Viral vectors are effective for gene delivery to brain but have limitations such as production cost and safety concerns (7, 16). Exosomes have been used to deliver small molecules, proteins, and nucleic acids to the brain due to their nonimmunogenic nature; however, there still exist many challenges in the isolation methods, cargo loading procedure, in vivo toxicity, and pharmacokinetics (7, 13). The molecular Trojan horse approach, relying on the receptor-specific monoclonal antibodies or peptides to ferry the genetically fused cargo into the brain, is promising in delivery of biologics across the BBB. However, the manufacturing process needs to be tailored specifically for each for different biologic cargo, and the stability, safety, and immunogenicity are challenges to clinical development (17). Crossing the BBB with various nanoparticles, such as liposomes, cationic polymers, inorganic nanoparticles, and nanocapsules, has shown promise in delivery of various cargos into the CNS, but complicated modifications are always needed to ensure that the particles produced are BBB-permeable (15, 18, 19). Here, we demonstrated a simple and effective approach for delivering cargos into the brain using neurotransmitter (NT)–derived synthetic lipids. This approach is very robust, successfully delivering three classes of cargos (i.e., small molecule, nucleic acid, and protein cargos) all using the same, simple nanoparticle design.
NTs are endogenous chemicals that enable neurotransmission. Notably, some NTs have been demonstrated to cross the BBB (20). For example, dimethyltryptamine and other tryptamine derivatives have been shown to cross the BBB by active transport across the endothelial plasma membrane (21). We hypothesized that the synthetic lipids derived from these NT derivatives will retain their capability for crossing BBB and may be useful as drug carriers for brain delivery.
In this study, we synthesized a series of lipidized NT derivatives, termed “NT-lipidoids,” through Michael addition reaction between the primary amine in the NTs and alkyl acrylate. We found that the tryptamine-derived lipidoids (NT1-lipidoids) could carry fluorescent dye to cross the BBB efficiently without any additional targeting ligands. The NT1-lipidoid can be doped into otherwise BBB-impermeable lipid formulations, and the resulting lipid nanoparticles (NT1-LNPs) also gained the ability to cross the BBB. Using NT1-lipidoid as a new BBB-penetrating moiety, we successfully delivered both small-molecule drug [amphotericin B (AmB)] and biomacromolecules, including antisense oligonucleotides (Tau-ASOs) and gene editing protein [green fluorescent protein (GFP)–Cre], into mouse brain through intravenous injection (Fig. 1A). Delivery of both Tau-ASOs and GFP-Cre resulted in functional intracellular delivery, as indicated by a reduction in both tau mRNA and protein levels in the case of ASO delivery, and the induction of red fluorescence in neurons using a floxed tdTomato mouse line (Ai14) in the case of GFP-Cre delivery. This is the first observation of effective genome recombination in neuronal cells of the Ai14 tdTomato mouse brain through intravenous injection of Cre protein delivery vehicles. The successful delivery of Tau-ASO and GFP-Cre protein, which both require LNPs to facilitate delivery across the cell membrane, proves that the LNP formulation and cargo could remain complexed and functional even after crossing the BBB. In summary, we developed a new, simple, and effective brain delivery system, which may find wide applications in treating CNS diseases or provide tools to study the brain function.
Fig. 1. Design of NT-lipidoids for effective brain delivery.

(A) Schematic illustration of formulating NT-lipidoid–doped LNPs for cargo delivery into the brain. (B) Synthesis route, lipid nomenclature, and chemical structure of NTs used for lipidoid synthesis. (C) Representative ex vivo fluorescence images of the dissected brain 1 hour after one-time intravenous injection of DiR-labeled NT-LNPs (1 mg/kg). DiR was doped into the NT-LNPs with a 10% weight ratio. The mice were perfused with saline before dissection.
RESULTS
NT-lipidoid synthesis and BBB permeability study of the NT-LNPs
We selected the NTs tryptamine, phenethylamine, and phenylethanolamine as the structural basis of our synthetic lipidoids. We synthesized the NT-lipidoids through Michael addition between the primary amine of the NTs and acrylate-containing hydrophobic tails in glass vials at 70°C for 48 hours (Fig. 1B) using an approach similar to our previously published combinatorial lipid library synthesis strategy (22). The result is a combinatorial library of NT-lipidoids, each containing one particular NT as the head group and one particular bioreducible hydrophobic structure as the tail group. The NT-lipidoids were named “NTn-O[x]B” (n = 1, 2, and 3), where NT1 is tryptamine, NT2 is phenethylamine, and NT3 is phenylethanolamine, and O[x]B represents the bioreducible hydrophobic tail where [x] indicates the number of carbon atoms in the hydrophobic tail of the acrylate shown in Fig. 1B. For example, NT1-O12B indicates a lipidoid containing a tryptamine head group and a hydrophobic tail group containing 12 carbon atoms. All the NT-lipidoids were purified using flash chromatography and characterized by electrospray ionization mass spectrometry (ESI-MS) (fig. S1). The resulting NT-lipidoids are amphiphilic and thus should be capable of self-assembly into either micelles or liposomes when prepared in aqueous solution. Dynamic light scattering (DLS) and transmission electron microscopy (TEM) of the NT-lipidoids indicated that these structures indeed self-assembled into spherical liposome-like structures (fig. S2).
We studied whether these NT-lipidoids can cross the BBB upon systemic intravenous delivery, using a fluorescent dye [DiR (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide)] as a model cargo. Hydrophobic small molecules such as DiR can partition into the hydrophobic region of micelles and liposomes and have often been used to track the biodistribution of these structures. To formulate the DiR-loaded NT-lipidoids, we mixed the NT-lipidoid and DiR in ethanol in a 10:1 (w/w) ratio, added the mixture dropwise to sodium acetate buffer (25 mM, pH 5.2), and then removed the ethanol through dialysis. The DiR-loaded NT-lipidoid nanoparticle solution was injected into mouse via tail vein injection. After 1 hour, the animals were sacrificed and thoroughly perfused with saline to wash away any free LNPs or DiR remaining in the blood vessels. The skull was removed, and the brain was imaged using an IVIS imaging device (PerkinElmer) at the excitation wavelength of 750 nm.
As shown in Fig. 1C, strong DiR fluorescence signal is observed in the mouse brain treated with DiR/NT1-lipidoid nanoparticles, compared to the mouse brain treated with DiR/NT2-lipidoids and DiR/NT3-lipidoids where the fluorescent signal was very weak. This result showed that NT1-derived lipidoids exclusively had the capability to cross the BBB. We also observed that the length of the aliphatic tail chain notably influenced the observed fluorescent intensity, in which NT1-lipidoids containing a shorter aliphatic chain length result in a greater fluorescence intensity (fig. S3). There is no notable difference of the physical properties, such as hydrodynamic sizes, polydispersity index (PDI), zeta potential, and morphologies between these NT1-derived lipidoids (fig. S2).
We hypothesized that doping NT1-lipidoids such as NT1-O12B into other BBB-impermeable lipid formulations led to the resulting lipid formulation crossing the BBB. We tested the ability of our previously published synthetic lipids, 76-O16B, EC16-80, and 113-O16B, to deliver DiR to the brain (22–24). We found that none of these lipids were effective in delivering the DiR into the mouse brain by themselves; however, after doping these lipids with the NT1-O12B, strong DiR signals could be observed in the mouse brain (fig. S4).
These results demonstrated that the NT1-lipidoids, but not NT2 or NT3, can effectively facilitate crossing of the BBB. The chemical structure of NT1 is based on the NT dimethyltryptamine, which has been reported to cross the BBB by active transport across the endothelial plasma membrane (21). We further investigated whether the chemistry of the linker that connects the NT-derived head group with the lipidoid tail group affects the NT-lipidoid derivatives for brain delivery. As shown in fig. S5, we synthesized a series of NT derivatives with different linkers. We found that the change in the chemical structure of the NT1-lipidoid affected the DiR signal detected in the brain, indicating that the successful delivery of DiR into the brain is highly dependent on the specific chemical structure. No strong DiR signals were observed from the mouse brain treated with NT2- and NT3-derived lipidoids with any linkers. These results support our hypothesis that the specific structure of the NT-lipidoid is driving the ability to cross the BBB, although the specific mechanism for NT-lipidoid–mediated brain delivery is not known yet.
Delivery of small-molecule AmB into the mouse brain
As shown above, we identified NT1-derived lipidoids that are able to deliver a hydrophobic dye (DiR) into the brain, either when used alone or when doped into other LNPs. We next aimed to test whether these NT1-derived lipidoids can deliver therapeutically relevant hydrophobic drug molecules into the brain. We chose AmB as the model drug in this study. AmB is a classic polyene antifungal drug and is the gold standard for the treatment of severe systemic fungal infections. However, it cannot be used clinically for the treatment of brain fungal infections due to its BBB impermeability (25, 26). The ability to successfully deliver AmB into the brain upon systemic administration would expand its range of therapeutic applications. Recently, we formulated AmB in synthetic lipidoid nanoparticles and conducted a thorough pharmacokinetics (PK) and biodistribution study of the AmB formulations using traditional synthetic LNPs, but in that study, none of our LNPs were capable of permeating the BBB to deliver AmB into the mouse brain (27).
We first encapsulated AmB in pure NT1-lipidoids (namely NT1-O12B, NT1-O14B, NT1-O16B, and NT1-O18B) using a procedure similar to the DiR encapsulation. The AmB-loaded NT1-lipidoid nanoparticles were injected into mice via tail vein at a dose of AmB (5 mg/kg) per mouse. After 24 hours, animals were sacrificed, and the brains were harvested, perfused with saline, and homogenized. The AmB concentration in the brain tissue was quantified using high-performance liquid chromatography (HPLC) according to the method that previously reported (27). As shown in fig. S6, the AmB concentration in the brain tissue of all the four groups was around 150 ng/g (AmB/tissue). Notably, in our previous report, AmB was undetectable in the brain after systemic delivery with traditional synthetic lipidoids, indicating that the NT1-lipidoid formulations enhanced the AmB delivery into the mouse brain. However, the AmB formulated in NT-lipidoid formulation showed opaque solution (fig. S7A), indicating the large size of the particles in the solution. DLS results (fig. S7B) showed that the nanoparticles are in range of 750 to 800 nm in diameter. We hypothesized that making NT1-lipidoid nanoparticle smaller may help improve the brain delivery efficiency. In the previous report, we found that the quaternized lipidoids provided stable AmB formulation with smaller particle size, comparing with the nonquaternized lipids (27). Thus, we hypothesized that doping a quaternized lipidoid with NT1-lipidoid may result in a smaller nanoparticle size while maintaining or improving the ability to penetrate the BBB.
Here, we synthesized a new phenylboronic acid quaternized lipidoid, PBA-Q76-O16B (Fig. 2A), for AmB encapsulation. We chose NT1-O12B as the dopant for enhanced brain delivery because it showed the highest DiR fluorescence intensity (Fig. 1C) among all NT-lipidoids. The AmB was formulated in the mixture of NT1-O12B and PBA-Q76-O16B, with the two lipidoids mixed at different weight ratios (7:3, 5:5, 3:7, 1:9, and pure PBA-Q76-O16B). As shown in Fig. 2B, the AmB encapsulates gradually became homogeneous transparent yellow solution with the increasing percentage of PBA-Q76-O16B lipidoid in the formulations. The hydrodynamic sizes also decreased from 800 to 100 nm (Fig. 2C). Using DiR as a cargo, we observed that lipidoids containing NT1-O12B and PBA-Q76-O16B at a 3:7 (w/w) ratio provided the strongest fluorescent signal in mouse brain when compared with all other lipid ratios (Fig. 2D). The fluorescent signal intensity at a 3:7 ratio was 4.5-fold higher than that of brain treated with DiR formulated in pure NT1-O12B (fig. S8). We then studied the AmB delivery using the mixed lipids and determined the AmB concentration in the mouse brain tissue 24 hours after intravenous injection of AmB (5 mg/kg) per mouse. As shown in Fig. 2E, the amount of AmB detected in the brain increased as the doping ratio of PBA-Q76-O16B increased from 0% (i.e., pure NT1-O12B) to 70% (i.e., 3:7 ratio) and reached a highest concentration of around 300 ng/g, which was about twofold higher than pure NT1-O12B. When the doping ratio increased further to 90% (i.e., 1:9), the AmB concentration was slightly lower but was still higher than of that treated with AmB formulated in pure NT1-O12B. Thus, the results for AmB delivery closely matched the results for DiR delivery (Fig. 2, D and E). Without doping with NT1-lipidoid, the AmB was nearly undetectable in the brain after intravenous injection of pure PBA-Q76-O16B/AmB. These results showed the key role of NT1-lipidoids in facilitating the brain delivery and the importance of finding the optimal doping ratio.
Fig. 2. NT-LNPs delivering AmB into the mouse brain.
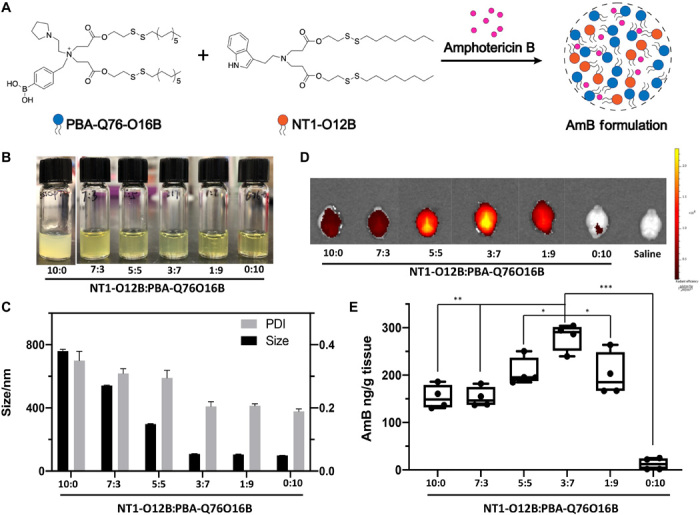
(A) Chemical structure of PBA-Q76-O16B, NT1-O12B, and schematic illustration of the doped NT1-lipidoid AmB formulation. (B) Photographs of AmB formulations in NT1-O12B doped with different amounts of PBA-Q76O16B (weight ratio is used). The pure NT1-O12B/AmB encapsulates appeared as an opaque suspension, while the appearance of the encapsulates changed from translucent solutions to homogeneous transparent yellow solutions as the doping ratio of PBA-Q76-O16B lipidoid increased. Photo credit: Feihe Ma, Tufts University. (C) Hydrodynamic diameters and PDIs of different NT-LNP/AmB formulations determined by DLS measurements. (D) Representative fluorescence images of the dissected mouse brain 1 hour after one-time intravenous injection of DiR-loaded NT1-O12B/PBA-Q76-O16B LNPs (1 mg/kg). The weight ratio of DiR in LNPs is 10%. (E) AmB concentration in brain tissues 24 hours after intravenous injection of AmB (5 mg/kg) in various NT1-O12B/PBA-Q76-O16B LNP formulations measured using HPLC (n = 4 per group). The mice were perfused with saline before dissected. One-way analysis of variance (ANOVA), Sidak post hoc analysis, *P < 0.05, **P < 0.001, or ***P < 0.0001. Graphical data are represented as box and whisker plots with individual points overlaid, where error bars represent maximum and minimum values and the boxed line represents the median.
Delivery of nucleic acid Tau-ASOs into the mouse brain for gene knockdown
ASO-mediated therapies showed great promise in treating many diseases. However, the fact that ASOs do not readily cross the BBB remains one of the substantial obstacles for their clinical application in treating CNS disorders (28, 29). For functional ASO delivery to the brain through intravenous injection, there are two physical barriers to overcome, namely the BBB and the target cell membrane. The ASO cargo has to first cross the BBB to reach the brain tissue and then must be delivered efficiently into the target cells. In a recent study, we identified lipidoid 306-O12B-3 that can efficiently deliver ASO both in vitro and in vivo (30). However, this lipid formulation mainly delivered the ASO into the liver and could not cross the BBB through intravenous injection. Here, we hypothesized that a mixed lipidoid formulation containing both NT1-lipidoid and 306-O12B-3 would be able to deliver ASO into the brain and facilitate gene silence through intravenous injection (Fig. 3A).
Fig. 3. NT-LNP facilitates the delivery of Tau-ASO into the mouse brain and for gene knockdown in both mRNA and protein levels.

(A) Chemical structure of 306-O12B-3, NT1-O14B, and schematic illustration of the doped NT-lipidoid Tau-ASO formulation for brain delivery. (B) GFP silencing efficiency of HEK-GFP cells treated with or without ASO/NT-LNP complexes. The NT1-O14B LNPs alone showed no silencing efficacy, while doping NT1-lipidoid into 306-O12B-3 LNPs led to successful gene silencing in vitro. *P < 0.01 versus all other samples in the same group. (C) Tau-ASOs formulated with NT1-O14B doped with different ratios of 306-O12B-3, saline, or scrambled Tau-ASO-LNPs were intravenously injected into C57BL/6J mice (n = 6 per group) via the tail vein, and the brain was analyzed for total tau mRNA levels. Graphical data are represented as box and whisker plots with individual points overlaid, where error bars represent maximum and minimum values and the boxed line represents the median, *P < 0.05 or **P < 0.001. (D) Total tau protein levels of the NT1-O14B/306-O12B-3 = 3:7 group, comparing to that of saline or scrambled Tau-ASO, **P < 0.001. One-way ANOVA, Sidak post hoc analysis.
We first evaluated the efficiency of the mixed lipidoid formulations for ASO delivery in vitro by delivering ASO targeting GFP mRNA into human embryonic kidney (HEK) cells stably expressing GFP (Fig. 3B), using the same approach we reported in our recent study (30). Here, we chose the NT1-O14B lipidoid; although the shorter-tail NT1-O12B was most effective for small-molecule delivery, our previous experience with biomacromolecules (22) indicated that the slightly longer tail length would be more appropriate here. The NT1-O14B alone showed no GFP silencing effect (10:0 ratio in Fig. 3B), indicating that this lipidoid alone is not effective for delivering ASO intracellularly. However, GFP silencing was observed when the ASO was delivered using LNPs containing a mixture of NT1-O14B and 306-O12B-3. When the doping ratio of 306-O12B-3 was greater than 50% (i.e., 5:5 weight ratio or more in favor of 306-O12B-3), we observed the GFP silencing in GFP-HEK cells, silencing efficiency increasing as the 306-O12B-3 doping ratio increased. Scrambled ASO delivered by Lipofetamine 2000 showed no GFP silencing, indicating that the GFP silencing was truly ASO sequence–specific.
We next explored whether the mixed lipidoid formulation (NT1-O14B and 306-O12B-3) could deliver ASO into the brain and mediate the gene knockdown in vivo. We chose tau as a therapeutic target and designed ASO targeting tau mRNA, because ASO-mediated tau reduction has shown promising results in the treatment of Alzheimer’s disease after the local injection of the Tau-ASO using an intracerebroventricular (ICV) pump (31, 32).
The sequence of Tau-ASO was chosen according to the published literature (31) and was synthesized by Integrated DNA Technologies (IDT). Tau-ASO was supplied containing chemical modification with phosphorothioate groups between each nucleic acid and 2′-O-methoxyethyl in the five nucleotides on the 5′ and 3′ termini of ribose to improve efficacy. To formulate the ASO for intravenous injection, we mixed the ASO with the formulated LNP solution (detailed formulation procedure was described in Materials and Methods) at a weight ratio of 1:15 (ASO to total lipids). Each mouse received five injections of ASO (1 mg/kg), with each injection spaced 3 days apart. The mice were sacrificed 4 days after the last injection and perfused, and the brain tissues were harvested and homogenized to extract the total RNA. The total tau mRNA levels were analyzed by quantitative polymerase chain reaction (qPCR). As shown in Fig. 3C, when ASO was delivered using either pure NT1-O14B or pure 306-O12B-3, no tau mRNA reduction in the brain tissues was detected. For the mixed lipidoid formulations, only NT1-O14B and 306-O12-3 with a w/w ratio of 5:5 and 3:7 displayed tau mRNA reduction in the brain. These two formulations resulted in ~25 and ~50% mRNA reduction, respectively. No tau mRNA silencing was observed in the mixed lipidoid formulations in other ratios (i.e., 7:3 and 1:9). We hypothesize that this is due to the necessity of precisely balancing the need to cross both the BBB and the cell membrane. For example, in the case of the 7:3 ratio, it may be the case that the particles can effectively cross the BBB due to the high NT1-O14B content; however, the low 306-O12B-3 content resulted in the particles being unable to cross the cell membrane. By contrast, in the case of the 1:9 ratio, the NT1-O14B content may be too low for the particles to even cross the BBB. At the intermediate weight ratios, we see successful tau knockdown, indicating that the ASO can be delivered across both the BBB and the cell membrane.
To confirm that the ASO delivery resulted in functional knockdown of tau, we also checked the tau protein level of the ASO-treated mice using enzyme-linked immunosorbent assay (ELISA) (Fig. 3D). Comparing with the untreated group, mice treated with Tau-ASO formulated in NT1-O14B/306-O12B-3 (3:7, w/w) showed substantially reduced total tau protein level. Furthermore, we delivered scrambled Tau-ASO with the best-performing ratios (NT1-O14B/306-O12B-3 at 3:7, w/w) using the exact same method as that of functional ASO. As shown here, neither tau mRNA silencing effect nor tau protein reduction was detected, demonstrating that the tau knockdown is specifically due to sequence-specific ASO silencing. Together, these results showed that doping the BBB-impermeable lipidoid formulation with NT1-lipidoid imparted these particles with the ability to penetrate the BBB and enabled the successful intracellular delivery of ASO for gene knockdown in mouse brain upon intravenous injection.
Delivery of GFP-Cre fusion protein for gene recombination in the Ai14 mouse brain
Delivery of genome-editing proteins for genome modification in the brain has therapeutic potential for the treatment of CNS disorders. However, current protein delivery strategies are mainly through local injection to the brain, which typically requires invasive drilling through the skull to access the tissue and thus restricts the clinical applications. We aim to evaluate whether NT1-lipidoids can assist the protein delivery into the brain cells through systemic (intravenous) administration. If successful, this would markedly improve the clinical feasibility of these genome-editing approaches. GFP-fused Cre recombinase was chosen as a model protein for the study using the Ai14 model mouse line (Fig. 4A). The Ai14 mouse line contains a flox-stop-flox tdTomato construct. The successful intracellular delivery of Cre protein into the cells of Ai14 mouse leads to the gene recombination and turns on the tdTomato expression, which can be directly visualized as red fluorescence signal without additional staining. Here, we used (−27)GFP-Cre protein as we previously reported for LNP delivery (22). We chose to use NT1-O14B LNPs doped with PBA-Q76-O16B, as these nanoparticles could successfully deliver (−27)GFP-Cre. The weight ratio of NT1-O14B and PBA-Q76-O16B was fixed at 3:7, based on the results we observed from the AmB and ASO delivery. Lipid formulations were prepared using the approaches described for the formulation for ASO delivery. Briefly, we mixed the (−27)GFP-Cre protein with LNPs at a weight ratio of 1:4 and incubated the solution at room temperature for 15 min before intravenous injection. Mice were injected four times with a dose of 50 μg of protein per injection. Five days after the last injection, the mice were sacrificed, and brain tissues were collected, fixed, and dehydrated. Then, the tissues were cryo-sectioned into 15-μm slices and counterstained with 4′,6-diamidino-2-phenylindole (DAPI) for fluorescence imaging. As shown in Fig. 4B, strong tdTomato signals were observed in multiple regions of the brain, including cerebral cortex, hippocampus, and cerebellum. The red fluorescence signal from cells in hippocampus is weaker than that in cerebral cortex and cerebellum, which could be due to the delivery efficiency to hippocampus is lower than to cortex and cerebellum. In contrast, no tdTomato expression in the brain was observed for the mice injected with LNP formulations using either pure NT1-O14B (10:0) or pure PBA-Q76-O16B (0:10). These results again demonstrated the importance of engineering the LNPs to balance the need to cross both the BBB and the cell membrane to achieve the successful gene recombination in neuronal cells upon systemic administration. The successful gene recombination was observed in multiple regions in the mouse brain, which showed the advantage of systemic delivery over local injection to reach a wide area in the brain.
Fig. 4. In vivo delivery of Cre recombinase to the Ai14 mouse brain to induce gene recombination.
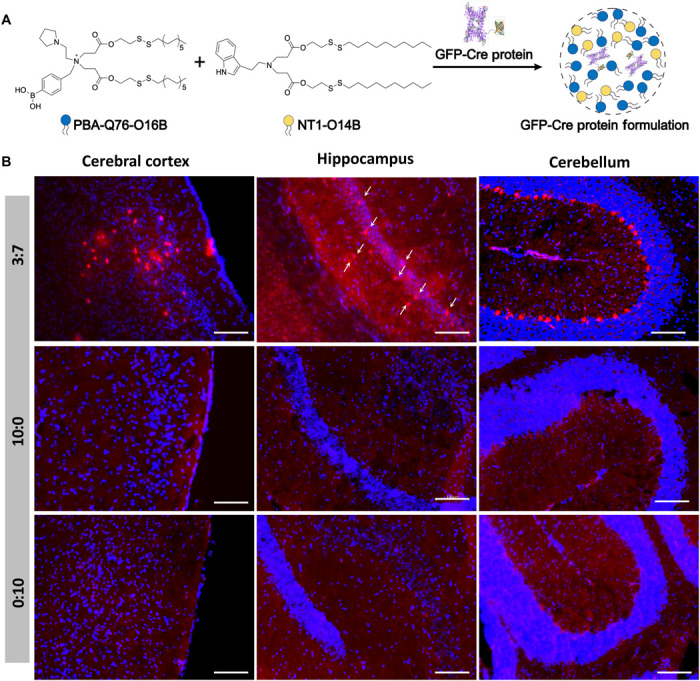
(A) Schematic illustration of mixed LNP formulation using NT1-O14B and PBA-Q76-O16B for GFP-Cre protein delivery into the brain. (B) Fluorescence image of the brain slices of Ai14 mice treated with (−27)GFP-Cre in different LNP formulations. Ai14 mouse was intravenously injected with (−27)GFP-Cre complexed with NT1-O14B:PBA-Q76-O16B = 3:7, 10:0, or 0:10 LNPs. After 3 weeks, the group of NT1-O14B:PBA-Q76-O16B = 3:7 showed tdTomato expression indicative of Cre-mediated recombination in cerebral cortex, hippocampus, and cerebellum. Red, tdTomato. The white arrows in the hippocampus 3:7 subpanel indicate tdTomato+ cells. Blue, DAPI. Scale bars, 100 μm.
DISCUSSION
The BBB is a major obstacle for delivery of therapeutics into the brain to treat the CNS diseases. Although extensive efforts have been undertaken to enhance brain delivery efficiency, each method has both advantages and disadvantages; thus, the development of safe and efficient delivery of BBB-impermeable cargos into the brain through intravenous injection remains a big challenge. Among a number of approaches, crossing the BBB with various nanoparticles has shown promise in delivery of various cargos into the CNS, but complicated modifications are always needed to ensure that the particles produced are BBB-permeable (15, 18, 19). The desired brain delivery platforms should be simple, efficient, and able to deliver different types of BBB-impermeable cargos, such as small molecular drugs and biologics.
We designed and synthesized a new class of NT-lipidoids. As a starting point and proof of concept, we selected three NTs, namely tryptamine (NT1), phenethylamine (NT2), and phenylethanolamine (NT3), and synthesized NT-lipidoids from these precursors. These precursors were selected, in part, due to the compelling evidence in the literature that tryptamine and phenethylamine can effectively cross the BBB via active transport (33, 34). We found that the NT1-lipidoid can serve as a new BBB-penetrating moiety. Doping the NT1-lipidoids into BBB-impermeable LNPs resulted in LNPs that can cross BBB effectively. While both tryptamine (NT1) and phenethylamine (NT2) can cross the BBB (33, 34), only NT1-lipidoids could effectively facilitate crossing of the BBB. The results indicated that even if the functional heads themselves can cross BBB, their derived lipidoids may not (Fig. 1C). Notably, the physical properties of the NT1 and NT2 lipidoids such as size, zeta potential, and PDI were very similar (fig. S2), indicating that difference in delivery capability is likely a result of chemical rather than physical properties. The NT1-lipidoids may cross the BBB through the receptor-mediated transcytosis; however, further studies are necessary to understand the underlying mechanism, including identifying the receptors.
Using this brain delivery platform, we successfully delivered several different types of BBB-impermeable cargos, including small-molecular drug (AmB), nucleic acid (Tau-ASOs), and genome-editing protein [(−27)GFP-Cre] into the mouse brain through intravenous injection. Doping NT1-O12B to the BBB-impermeable lipidoid PBA-Q76-O16B resulted in an AmB formulation that could cross BBB. Using this approach, the AmB concentration in the brain tissue reached as high as 300 ng/g (AmB/tissue) with a delivery efficiency of about 0.135% of injected dose after 24 hours of intravenous injection with AmB (5 mg/kg). Notably, this concentration of AmB is higher than that which has previously been reported (26). Although successful, the delivery efficiency to brain needs to be improved as the concentration in brain remains low compared with the concentration of AmB in other organs, such as liver and spleen (fig. S9D).
Doping NT1-lipidoids to otherwise BBB-impermeable lipidoids also resulted in the functional delivery of both ASO and Cre protein into the brain. Unlike AmB delivery, the functional delivery of either ASO or Cre protein into the brain needs to cross two barriers, one is the BBB and another is the cell membrane barrier. It is notable that the delivery of these biomacromolecules was functional. For example, ASOs must be present inside a cell expressing the target mRNA to functionally knock down the expression of that target, and the GFP-Cre protein needs to get into cell nucleus for successful gene recombination.
Our results showed that these NT1-lipidoid–doped LNPs not only could facilitate the delivery of the cargo across the BBB but also could facilitate efficient intracellular delivery to brain cells for both gene knockdown and gene recombination (Figs. 3 and 4). Notably, in the previous study, ~80% reduction of tau mRNA and ~70% reduction of tau protein were achieved by continuously dosing 28 days, and the dose was 25-μg Tau-ASO per day through local injection by ICV pump (31). In comparison, ~50% reduction of tau mRNA and ~30% reduction of tau protein were achieved by only five times intravenous injection of 20-μg Tau-ASO/LNP complex in this study. The tail vein intravenous method developed here appears to be both more efficient and less invasive than the implanted ICV pump and thus shows more promise for future clinical applications. Furthermore, this is the first report of effective genome editing in neuronal cells of the Ai14 mouse brain through intravenous injection of Cre protein delivery vehicles, and the genome editing was observed in various different regions of the mouse brain, including cerebral cortex, hippocampus, and cerebellum.
It is particularly notable that, while each delivery application used a different BBB-impermeable LNP carrier, we simply doped with the same NT1-lipidoid for each application. This demonstrates a unique simplicity and flexibility of this system. We would theorize that simply adding NT1-lipidoid to any LNP system may provide that system with a similar BBB permeability, provided appropriate optimization. This may prove to be a very powerful and versatile platform for therapeutic drug delivery to the brain. In summary, we developed a new, simple, universal, and effective brain delivery system, which may find wide applications in treating CNS diseases or providing a tool to study the brain function.
MATERIALS AND METHODS
General
All chemicals used for lipid synthesis were purchased from Sigma-Aldrich and directly used as received. All ASOs and DNA fragments were purchased from IDT. The ASOs were used as provided from IDT, and when noted, we used the ASO product that is provided by the company to contain chemical modifications to improve stability. HeLa-DsRed and GFP-HEK cells were maintained in Dulbecco’s modified Eagle’s medium (Sigma-Aldrich) complemented with 10% fetal bovine serum (Sigma-Aldrich) and 1% penicillin-streptomycin (Gibco). The fluorescence intensity for GFP-HEK cells was analyzed by flow cytometer (BD FACSCalibur, BD Biosciences, CA). The (−27)GFP-Cre (no. 89253, Addgene) protein was expressed and extracted from BL21 Escherichia coli and further purified by Ni-NTA (nitrilotriacetic acid) column (Qiagen). Nanoparticle size and zeta potential were recorded on a ZetaPALS particle size analyzer. TEM images were captured by a FEI Tecnai Spirit transmission electron microscope. All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committees of the Tufts University.
Lipid synthesis
All head amines used for lipid synthesis are commercially available from Sigma-Aldrich. All the cationic lipidoids (NT1-O12B~O18B, NT2-O12B~O18B, NT3-O12B~O18B, NT1-EC16, NT1-C18, NT1-1E, NT2-EC16, NT2-1E, NT3-EC16, NT3-1E, 306-O12B-3, and PBA-Q76-O16B) were synthesized according to our previous reports (22, 24, 35). Briefly, to synthesize lipid, head amine and acrylate were mixed and heated at 1:2.4 molar ratio in Teflon-lined glass screw-top vials for 48 hours. The crude product was purified using flash chromatography on silica gel. The lipid structure was confirmed using ESI-MS. The NT1-Neu was synthesized according to the reported procedure (36). Briefly, into a dry flask, palmitic-acid-5-hydroxy-stearic-acid and dichloromethane (DCM) were added, and then dropwise addition of (COCl)2 that was dissolved in DCM. After reacting for 2 hours, the excess (COCl)2 was removed by rotary evaporation, and then into the dry flask, tryptamine dissolved in DCM was added and reacted for 12 hours. Then, the solution was concentrated and was purified using flash chromatography on silica gel.
Biodistribution of DiR-labeled NT-LNPs in mouse brain
We dissolved the NT-lipidoids and DiR, at a weight ratio of 10:1, together in 100% ethanol. One hundred microliters of solution was then added dropwise to 300 μl of sodium acetate buffer (25 mM, pH 5.2) and vortexed briefly. Last, we removed the ethanol in the formulation by dialysis against diH2O (deionized water) [molecular weight cutoff (MWCO), 35 kDa; Thermo Fisher Scientific] for 12 hours. Then, the DiR-labeled LNPs were intravenously injected into BALB/C mice (female, 6 weeks age). After 1 hour, mice were anesthetized and perfused with saline. Afterward, mouse brains were collected. The fluorescent signal distribution was visualized using the SpectrumCT Biophotonic Imager (PerkinElmer, Boston, MA).
Preparation of AmB/NT-lipidoid nanoparticle formulations
The AmB encapsulates were prepared according to our previous report (27). Briefly, 1 mg of each lipidoid (solid) was mixed with 1 mg of AmB in 300 μl of dimethyl sulfoxide (DMSO). The mixtures were sonicated for 30 min and then vortexed for 10 min until completely dissolved. The solution was added dropwise to a glass bottle containing 600 μl of sodium acetate buffer (pH 5.0) and vortexed briefly. The solutions were further dialyzed against distilled water to remove DMSO and nonencapsulated AmB using dialysis tubing (MWCO, 35 kDa) overnight.
Characterization of AmB/NT-lipidoid nanoparticle formulations
The particle sizes and PDI of all encapsulates were measured using DLS. Zeta potential was recorded on a ZetaPALS particle size analyzer (fig. S7C). The DLCs (drug loading content) of AmB were calculated according to our previous report (fig. S7D) (27). TEM images were captured by a FEI Tecnai Spirit transmission electron microscope (fig. S7E).
Tissue biodistributions of AmB
The tissue biodistributions of AmB were measured using HPLC analysis with a modular liquid chromatograph system (Agilent) according to our previous report (27). The calibration curve of AmB concentration measured using HPLC was shown in fig. S9 (A and B). The mAU (milli-Absorbance Unit) time graphs of AmB concentrations in brain tissues 24 hours after intravenous treatment with NT1-O12B/PBA-Q76O16-LNP (ratio, 3:7)/AmB complex at a single dose of 5 mg of AmB per kilogram were shown in fig. S9C. The AmB concentrations in heart, liver, spleen, lung, and kidney were shown in fig. S9D.
In vitro and in vivo delivery of ASO/LNP complexes
For the in vitro study, we first dissolved the NT-lipidoid and the bioreducible lipid, at weight ratios of 10:0, 7:3, 5:5, 3:7, or 0:10, together in 100% ethanol. One hundred microliters of solution was then added dropwise to 300 μl of sodium acetate buffer (25 mM, pH 5.2) and vortexed briefly. Last, we removed the ethanol in the formulation by dialysis against diH2O (MWCO, 35 kDa) for 12 hours. For the intracellular delivery, cells stably expressing GFP, termed GFP-HEK cells, were seeded in a 48-well plate at a density of 20,000 cells per well 24 hours before the LNP delivery. We mixed the ASO targeting the GFP mRNA and the lipid sample at a 1:15 (w/w ratio) in Opti-MEM medium and incubated the solution at room temperature for 15 min. The ASO/LNP complexes were then added to the cells (the final ASO concentrations were 16.7 nM in each well), and the treated cells were then cultured for 16 hours before flow cytometric analysis. The GFP silencing efficiency of each treatment group was calculated on the basis of the mean fluorescence intensity (MFI) of the cells as measured by flow cytometry. In short, first, the MFI of ASO-treated cells was calculated as a percentage of the MFI of untreated cells (i.e., MFItreated/MFIuntreated × 100%). Then, the GFP silencing efficiency was defined as the inverse of this MFI percentage (i.e., [1 – MFItreated/MFIuntreated] × 100%). The GFP-ASO sequence was as follows: 5′-TTGCCGGTGGTGCAGATAAA-3′. The scrambled GFP-ASO sequence was as follows: 5′-GGAGTACACTATATCGGTGG-3′ (30). For the in vivo study, in addition to the NT-lipids and bioreducible lipids, PEGylated lipid {1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG2000)} was also included in the LNP formulation to improve the circulation time in body. The DSPE-PEG2000 was dissolved in ethanol and mixed with synthetic lipids at a weight ratio of 1:24 [DSPE-PEG2000:(NT1-O14B + 306-O12B-3)]. The LNPs were then formulated with ASO using the same procedure as described for the in vitro ASO delivery study. For in vivo studies, ASO targeting tau mRNA were purchased containing chemically modified phosphorothioate groups in the nucleic acid backbone and 2′-O-methoxyethyl–modified nucleotides in the first and last five nucleotides of the ASO sequence (IDT). The murine Tau-ASO sequence was as follows: 5′-ATCACTGATTTTGAAGTCCC-3′. The scrambled Tau-ASO sequence was as follows: 5′-CCTTCCCTGAAGGTTCCTCC-3′ (31). We mixed the ASO with the formulated LNP solution at a weight ratio of 1:15 (ASO to total lipids) and incubated at room temperature for 15 min before intravenous injection to mice. All ASO-treated mice were C57BL/6J nontransgenic mice. The mice received either saline or ASO in lipid formulations via tail vein injection at 1 mg ASO/kg body weight on days 0, 4, 8, 12, and 16. On day 20, mice were sacrificed, and brain tissue was collected. Total RNA was extracted from the brain tissue using TRIzol. Reverse transcription was executed by ImProm-II reverse transcriptase (Promega) and 250-ng oligo(dT)12-18 primers (Thermo Fisher Scientific) at 42°C for 60 min with 1 μg of total RNA. The resulting complementary DNA (cDNA) library was analyzed by qPCR for 40 cycles and 58° to 60°C annealing temperature on a CFX96 Touch Real-Time PCR Detection System (Bio-Rad). Tau mRNA levels were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA and presented relative to saline controls. Primer/probe sequences were as follows: total murine Tau: 5′-GAACCACCAAAATCCGGAGA-3′ (forward) and 5′-CTCTTACTAGCTGATGGTGAC-3′ (reverse); probe: 5′-/56-FAM/CCAA-GAAGGTGGCAGTGGTCC/3IABkFQ/-3′; murine GAPDH: 5′-TGCCCCCATGTTTGTGATG-3′ (forward) and 5′-TGTGGTCATGAGCCCTTCC-3′ (reverse); probe: 5′-/56-FAM/AATGCATCCTGCACCACCAACTGCTT/3IABkFQ/-3′ (Invitrogen) (32).The total tau protein levels in brain were examined with the Tau (Total) Mouse ELISA Kit (Invitrogen), and the total protein concentration was tested using the Pierce Bicinchoninic Acid Protein Assay Kit (Thermo Fisher Scientific).
In vivo delivery of GFP-Cre protein to Ai14 mouse via intravenous administration
We prepared the lipid formulations similarly to as described in the ASO delivery section. For this experiment, we fixed the weight ratio of NT1-O14B and PBA-Q76-O16B at 3:7. We mixed the (−27)GFP-Cre protein with LNPs at a weight ratio of 1:4 and incubated the solution at room temperature for 15 min. All GFP-Cre–treated mice were Ai14 mice (the Jackson laboratory). Three mice in each group were intravenously injected with saline or (−27)GFP-Cre/LNP complexes on days 0, 5, 10, and 15, with 50 μg of protein for each injection. The brain tissues were collected on day 20. We fixed the brain tissues in 4% paraformaldehyde overnight at 4°C and dehydrated the fixed tissue in 30% sucrose. Then, the tissues were cryo-sectioned into 15-μm slices and counterstained with DAPI for fluorescence imaging. The red fluorescence from the tdTomato expression was imaged directly, without any additional antibody staining or amplification.
Characterization of ASO/NT1-lipidoid and GFP-Cre/NT1-lipidoid nanoparticle formulations
The particle size, PDI, zeta potential, and TEM images of blank and ASO-loaded NT1-O14B/306-O12B-3 (ratio, 3:7) nanoparticles or the blank and (−27)GFP-Cre–loaded NT1-O14B/PBA-Q76O16B (ratio, 3:7) nanoparticles were shown in fig. S10.
Statistical analysis
Statistical analysis was performed using one-way analysis of variance (ANOVA), followed by the Tukey-Kramer multiple comparisons test for more than two groups. Student’s t test was used for comparing two groups using Prism (v.8, GraphPad Software, La Jolla, CA). Values of P < 0.05 were considered as significance.
Supplementary Material
Acknowledgments
Funding: We acknowledge the support from the NIH grants R01 EB027170-01 and UG3 TR002636-01. Author contributions: Q.X. conceived the original idea of using NTs as functional heads to construct lipidoids, supervised the project, and revised the manuscript. F.M. designed the experiments and wrote the manuscript. F.M. and L.Y. conducted the experiments, analyzed the data, and revised the manuscript. Z.S. helped in the HPLC experiment and X.R. helped in the synthesis. J.C. conducted the TEM imaging. Z.G. helped revise the manuscript. Competing interests: Q.X. is an inventor on a patent application related to this work filed by the Tufts University (no. 63/019,530, filed 4 May 2020). Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/30/eabb4429/DC1
REFERENCES AND NOTES
- 1.Pardridge W. M., Blood–brain barrier delivery. Drug Discov. Today 12, 54–61 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Barchet T. M., Amiji M. M., Challenges and opportunities in CNS delivery of therapeutics for neurodegenerative diseases. Expert Opin. Drug Deliv. 6, 211–225 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Collins P. Y., Patel V., Joestl S. S., March D., Insel T. R., Daar A. S., Bordin I. A., Costello E. J., Durkin M., Fairburn C., Glass R. I., Hall W., Huang Y., Hyman S. E., Jamison K., Kaaya S., Kapur S., Kleinman A., Ogunniyi A., Otero-Ojeda A., Poo M.-M., Ravindranath V., Sahakian B. J., Saxena S., Singer P. A., Stein D. J., Anderson W., Dhansay M. A., Ewart W., Phillips A., Shurin S., Walport M., Grand challenges in global mental health. Nature 475, 27–30 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Obermeier B., Daneman R., Ransohoff R. M., Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 19, 1584–1596 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khorkova O., Wahlestedt C., Oligonucleotide therapies for disorders of the nervous system. Nat. Biotechnol. 35, 249–263 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patel M. M., Patel B. M., Crossing the blood–brain barrier: Recent advances in drug delivery to the brain. CNS Drugs 31, 109–133 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Dong X., Current strategies for brain drug delivery. Theranostics 8, 1481–1493 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fung L. K., Shin M., Tyler B., Brem H., Saltzman W. M., Chemotherapeutic drugs released from polymers: Distribution of 1,3-bis(2-chloroethyl)-l-nitrosourea in the rat brain. Pharm. Res. 13, 671–682 (1996). [DOI] [PubMed] [Google Scholar]
- 9.Rubenstein J. L., Fridlyand J., Abrey L., Shen A., Karch J., Issa E. W. S., Damon L., Prados M., McDermott M., O’Brien J., Haqq C., Shuman M., Phase I study of intraventricular administration of rituximab in patients with recurrent CNS and intraocular lymphoma. J. Clin. Oncol. 25, 1350–1356 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Lu C.-T., Zhao Y.-Z., Wong H. L., Cai J., Peng L., Tian X.-Q., Current approaches to enhance CNS delivery of drugs across the brain barriers. Int. J. Nanomedicine 9, 2241–2257 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Villringer K., Cuesta B. E. S., Ostwaldt A.-C., Grittner U., Brunecker P., Khalil A. A., Schindler K., Eisenblätter O., Audebert H., Fiebach J. B., DCE-MRI blood–brain barrier assessment in acute ischemic stroke. Neurology 88, 433–440 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Fu H., McCarty D. M., Crossing the blood–brain-barrier with viral vectors. Curr. Opin. Virol. 21, 87–92 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Ha D., Yang N., Nadithe V., Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: Current perspectives and future challenges. Acta Pharm. Sin. B 6, 287–296 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pardridge W. M., Drug and gene targeting to the brain with molecular Trojan horses. Nat. Rev. Drug Discov. 1, 131–139 (2002). [DOI] [PubMed] [Google Scholar]
- 15.Zhou Y., Peng Z., Seven E. S., Leblanc R. M., Crossing the blood-brain barrier with nanoparticles. J. Control. Release 270, 290–303 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Mingozzi F., High K. A., Immune responses to AAV vectors: Overcoming barriers to successful gene therapy. Blood 122, 23–36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pardridge W. M., Delivery of biologics across the blood–brain barrier with molecular Trojan horse technology. BioDrugs 31, 503–519 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Blanco E., Shen H., Ferrari M., Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 33, 941–951 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wen J., Wu D., Qin M., Liu C., Wang L., Xu D., Vinters H. V., Liu Y., Kranz E., Guan X., Sun G., Sun X., Lee Y. J., Martinez-Maza O., Widney D., Lu Y., Chen I. S. Y., Kamata M., Sustained delivery and molecular targeting of a therapeutic monoclonal antibody to metastases in the central nervous system of mice. Nat. Biomed. Eng. 3, 706–716 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Snowman A. M., Snyder S. H., Cetirizine: Actions on neurotransmitter receptors. J. Allergy Clin. Immunol. 86, 1025–1028 (1990). [DOI] [PubMed] [Google Scholar]
- 21.Carbonaro T. M., Gatch M. B., Neuropharmacology of N,N-dimethyltryptamine. Brain Res. Bull. 126, 74–88 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang M., Zuris J. A., Meng F., Rees H., Sun S., Deng P., Han Y., Gao X., Pouli D., Wu Q., Georgakoudi I., Liu D. R., Xu Q., Efficient delivery of genome-editing proteins using bioreducible lipid nanoparticles. Proc. Natl. Acad. Sci. U.S.A. 113, 2868–2873 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang J., Chen X., Glass Z., Gao F., Mao L., Wang M., Xu Q., Integrating combinatorial lipid nanoparticle and chemically modified protein for intracellular delivery and genome editing. Acc. Chem. Res. 52, 665–675 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang M., Alberti K., Sun S., Arellano C. L., Xu Q., Combinatorially designed lipid-like nanoparticles for intracellular delivery of cytotoxic protein for cancer therapy. Angew. Chem. Int. Ed. 53, 2893–2898 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Ostrosky-Zeichner L., Marr K. A., Rex J. H., Cohen S. H., Amphotericin B: Time for a new “gold standard”. Clin. Infect. Dis. 37, 415–425 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Xu N., Gu J., Zhu Y., Wen H., Ren Q., Chen J., Efficacy of intravenous amphotericin B-polybutylcyanoacrylate nanoparticles against cryptococcal meningitis in mice. Int. J. Nanomedicine 6, 905–913 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu F., Yang L., Li Y., Junier A., Ma F., Chen J., Han H., Glass Z., Zhao X., Kumamoto C. A., Sang H., Xu Q., In vitro and in vivo study of Amphotericin B formulation with quaternized bioreducible lipidoids. ACS Biomater Sci. Eng. 6, 1064–1073 (2020). [DOI] [PubMed] [Google Scholar]
- 28.Rinaldi C., Wood M. J., Antisense oligonucleotides: The next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 14, 9–21 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Talbot K., Wood M. J., Wrangling RNA: Antisense oligonucleotides for neurological disorders. Sci. Transl. Med. 11, eaay2069 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Yang L., Ma F., Liu F., Chen J., Zhao X., Xu Q., Efficient delivery of antisense oligonucleotides using bioreducible lipid nanoparticles in vitro and in vivo. Mol. Ther. Nucleic Acids 19, 1357–1367 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeVos S. L., Goncharoff D. K., Chen G., Kebodeaux C. S., Yamada K., Stewart F. R., Schuler D. R., Maloney S. E., Wozniak D. F., Rigo F., Bennett C. F., Cirrito J. R., Holtzman D. M., Miller T. M., Antisense reduction of tau in adult mice protects against seizures. J. Neurosci. 33, 12887–12897 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeVos S. L., Miller R. L., Schoch K. M., Holmes B. B., Kebodeaux C. S., Wegener A. J., Chen G., Shen T., Tran H., Nichols B., Zanardi T. A., Kordasiewicz H. B., Swayze E. E., Bennett C. F., Diamond M. I., Miller T. M., Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci. Transl. Med. 9, eaag0481 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paley E. L., Tryptamine-induced tryptophanyl-tRNAtrp deficiency in neurodifferentiation and neurodegeneration interplay: Progenitor activation with neurite growth terminated in Alzheimer’s disease neuronal vesicularization and fragmentation. J. Alzheimer’s Dis. 26, 263–298 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Mosnaim A. D., Callaghan O. H., Hudzik T., Wolf M. E., Rat brain-uptake index for phenylethylamine and various monomethylated derivatives. Neurochem. Res. 38, 842–846 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Tang Q., Liu J., Jiang Y., Zhang M., Mao L., Wang M., Cell-selective messenger RNA delivery and CRISPR/Cas9 genome editing by modulating the interface of phenylboronic acid-derived lipid nanoparticles and cellular surface sialic acid. ACS Appl. Mater. Interfaces 11, 46585–46590 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Ma F.-H., An Y., Wang J., Song Y., Liu Y., Shi L., Synthetic nanochaperones facilitate refolding of denatured proteins. ACS Nano 11, 10549–10557 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/30/eabb4429/DC1