

Abstract
Approaches for the creation of soft materials, particularly hydrogels, with hierarchical structure are of interest in a variety of applications owing to their unique properties. In the context of tissue mimics, hydrogels with multiscale structures more accurately capture the complexities of tissues within the body (e.g., fibrous collagen-rich microenvironments). However, cytocompatible fabrication of such materials with hierarchical structures and independent control of mechanical and biochemical properties remains challenging and is needed for probing and directing cell-microenvironment interactions for three-dimensional (3D) cell encapsulation and culture applications. To address this, we have designed innovative multifunctional assembling peptides: these unique peptides contain a core block that mimics the structure of collagen for achieving relevant melting temperatures; ‘sticky’ ends to promote assembly of long fibrils; and a biocompatible reactive handle that is orthogonal to assembly to allow the formation of desired multiscale structures and their subsequent rapid, light-triggered integration within covalently crosslinked synthetic hydrogels. Nano- to micro-fibrils were observed to form in physiologically-relevant aqueous solutions, where both underlying peptide chemical structure and assembly conditions were observed to impact the resulting fibril sizes. These assembled structures were ‘locked’ into place and integrated as linkers within cell-degradable, bioactive hydrogels formed with photoinitiated thiol-ene ‘click’ chemistry. Hydrogel compositions were identified for achieving robust mechanical properties like those of soft tissues while also retaining higher ordered structures after photopolymerization. The utility of these innovative materials for 3D cell culture was demonstrated with human mesenchymal stem cells, where cell morphologies reminiscent of responses to assembled native collagen were observed now with a fully synthetic material. Using a bottom-up approach, a new materials platform has been established that combines the advantageous properties of covalent and assembling chemistries for the creation of synthetic hydrogels with controllable nanostructure, mechanical properties, and biochemical content.
Introduction
Synthetic hydrogels, often covalently crosslinked water-swollen polymer networks, have been designed for mimicking a range of soft tissues through relevant mechanical properties (Young’s modulus E ~ 0.1–100 kPa) and facile incorporation of a variety of biochemical cues, particularly for controlled 3D cell culture and hypothesis testing.1–4 However, these synthetic hydrogels with relatively homogenous structures down to the nanoscale often lack the fibrillar structure of native tissues, such as the lung interstitium or skin, where fibrillar collagen I is highly abundant.5–7 Collagens, amongst other proteins that reside in these tissues, exhibit multiscale assembly, dictated by their underlying chemical structure, to produce hierarchically structured tissues with wide-ranging properties that allow them to serve as effective junctions between different cell and tissue types.5, 6 This multiscale structure often is not matched by soft synthetic hydrogels, despite being shown to influence both cell-matrix and cell-cell interactions.8–10 For probing and directing such cell-microenvironment interactions, innovative methods are needed for creating robust hierarchically structured hydrogels that mimic collagenous tissues with independent control of structure, mechanical properties, and biochemical cues.11
Inspired by native tissues, both top-down and bottom-up approaches have been established for creating fibrous materials with controlled nano- and micro-structure to influence cell behavior. Physically assembled hydrogels formed with harvested collagen offer a 3D platform for cell culture that provides relevant multiscale structures and biochemical content; however, the mechanical properties of these hydrogels (E ~ 0.1–1 kPa) can be variable between batches and often are an order of magnitude lower than those of native collagenous tissues.12, 13 To address this, collagen has been encapsulated within covalently crosslinked synthetic hydrogels to increase the Young’s modulus closer to what is observed in native tissues while still retaining the fibrillar structure of assembled collagen;14 while useful, structure and cell binding sites are inherently coupled, limiting hypothesis testing, and batch-to-batch variability and potential immunogenic effects of harvested collagen can be confounding.15 To create well-defined soft fibrous materials, pulling or electrospinning methods have been established for forming soft biopolymer microfibers upon which cells are seeded,16–18 and recently these have been incorporated within covalently crosslinked hydrogels for 3D culture.19 Moving to the nanoscale, peptide amphiphiles and other polypeptides have been used to create nanofibers or α-helical structures for 2D cell culture.20, 21 Approaches for the self-assembly of short peptides, with both native and non-native amino acids (e.g., Fmoc-FF-OH, FVD, AYFIL, F-α,β-dehydrophenylalanine-F), into hydrogels have been established for the encapsulation and 3D culture of cells; however, mechanical properties and structure are coupled.22–25 Despite these advances, a great need remains for hydrogel systems that contain hierarchical structure reminiscent of fibrous, collagen-rich tissues and allow independent control of mechanical properties and biochemical content for elucidating key regulators of cell function in multidimensional microenvironments.4
Toward addressing this, a range of assembling peptides, called collagen mimetic peptides (CMPs) or collagen like peptides, have been designed to mimic the structure of collagen through variations of the repeat X-Y-G, with the most common repeat in human collagen being P-O-G (proline-hydroxyproline-glycine).26 Modulating the number of P-O-G units ((POG)n) controls hydrogen bonding between the backbones of these CMPs and thereby ‘melting temperature’ (Tm) of resulting triple helices, for example, 37 °C and 65 °C for (POG)7 and (POG)10.26, 27 Synthetic or recombinantly expressed (POG)-based peptides have been appended to multi-arm poly(ethylene glycol) (PEG) to form physical hydrogels28 or with monofunctional PEG acrylate for integration within covalently crosslinked hydrogels.29, 30 Yet, the strength of the interactions between these traditional (POG)n repeats often is insufficient for the controlled formation of long fibrils to mimic collagen on the micro-scale. To combat this, modifications to this base sequence have been demonstrated for increasing interactions between peptide strands and triple helices for achieving longer fibrils. Incorporating non-native amino acids increases the stability of the resulting assembled structures: these include integration of aza-glycines for increased hydrogen bond propensity,31 moieties for end-to-end interactions between triple helices (e.g., charged amino acids,32, 33 aromatic stacking groups34 for ‘sticky’ ends), and linkers for their stabilization (e.g., cysteine knots, metal ligands).35, 36 These seminal approaches allow the formation of stable fibrils and, in some cases, weak physically-crosslinked hydrogels (E ~ 0.01–0.1 kPa); however, the mechanical properties of such materials is an order of magnitude lower than that of collagenous tissues.12, 13 Further, the reactive handles integrated to date are either not orthogonal to peptide assembly (e.g., cysteines) or lack biocompatibility (e.g., metal ligands), limiting the opportunity to control structure independent of mechanical properties in 3D cell cultures.
In this work, we have designed innovative multifunctional assembling peptides with a core block that mimics the structure of collagen for achieving physiologically-relevant melting temperatures, ‘sticky’ ends to promote the formation of long fibrils, and a reactive handle that is orthogonal to assembly to allow facile integration of assembled structures within covalently crosslinked hydrogels. For the reactive handle, an accessible lysine protected with an allyloxycarbonyl group (K(alloc)) was used. This reactive functional group has previously been used by our group and others to integrate linear non-assembling peptides within synthetic hydrogels;37, 38 here, we exploit its orthogonality to assembly to allow the formation of long fibrils reminiscent of collagen followed by their covalent stabilization. Specifically, two different designs of multifunctional collagen mimetic peptides (mfCMPs) were assembled in aqueous solutions over days. Upon achieving the desired nano- to micro-structure in solution, the assembled fibrils were ‘locked’ into place by photoinitiated thiol–ene ‘click’ chemistry, using tetra-thiol PEG and bis- and mono-alkene linear bioactive peptides, to form hydrogels with multiscale structures under physiologically relevant conditions. These building blocks and processing steps permitted facile tuning of resulting properties relevant for mimicking collagen-rich loose connective tissues (e.g., healthy to diseased lung, E ~ 1–10 kPa).3, 37, 39 Overall, this strategy enabled the creation of robust fully-synthetic hydrogels with controlled multiscale structure, mechanical properties, and biochemical content for 3D cell culture, where cell morphologies are consistent with those in harvested collagen systems are observed. The approaches and building blocks established here could prove useful for the fabrication of a variety of hierarchically structured materials of relevance for a range of applications, including controlled cell culture, regenerative medicine, and therapeutic delivery.
Results and Discussion
Approach for creating hierarchically structured hydrogels using multifunctional assembling peptides
In this work, we investigated an approach for the creation of hierarchically structured hydrogels comprised of assembled peptides stabilized and integrated within covalently crosslinked polymer networks for 3D cell culture (Figure 1a). To create assembling peptides that could be integrated within a well-defined synthetic hydrogel framework and impart a fibrous structure reminiscent of collagen-rich tissues, two mfCMP peptide designs were investigated. Specifically, mfCMP-1a and mfCMP-2a were constructed by making multiple substitutions and perturbations to the base sequence of (POG)n to impart different melting temperatures and achieve desired assembled structures, informed by seminal works in CMP design.32, 34 For mfCMP-1a, aspartic acid (D) and lysine (K) residues were substituted for P and O on the C- and N-terminal ends of a peptide, respectively, to encourage end-to-end assembly through ionic interactions. Hartgerink and coworkers have previously shown the formation of long fibrils in solution with non-reactive (PKG)4(POG)4(DOG)4.32 Importantly, we improved the hydrogen bonding potential of this sequence relative to similar previously reported ‘sticky’ ended sequences by lengthening the (POG)n block (from n=4 to n=6) for enhanced stability and, thereby, increased melting temperature of the triple helix (Figure 1b). For mfCMP-2a, aromatic residues were incorporated on the termini to promote aromatic stacking, specifically a phenylalanine (F) residue and a pentafluorophenylalanine residue (F(F5)) (Figure 1c).34 In both sequences, we added a K(alloc) residue to provide a reactive handle in the peptide backbone that was orthogonal to assembly and allowed triggered covalent modification under cytocompatible conditions at any time of interest, ultimately enabling incorporation within hydrogels with robust and tunable mechanical properties and biochemical content as shown below. The alloc group was placed in the middle of these sequences, between hydrogen bonding and end-to-end interaction ‘blocks’ for mfCMP-1a and between two hydrogen bonding blocks for mfCMP-2a, toward limiting any potential interference with triple helix and fibrillar assembly of the peptide strands, respectively.
Fig. 1:
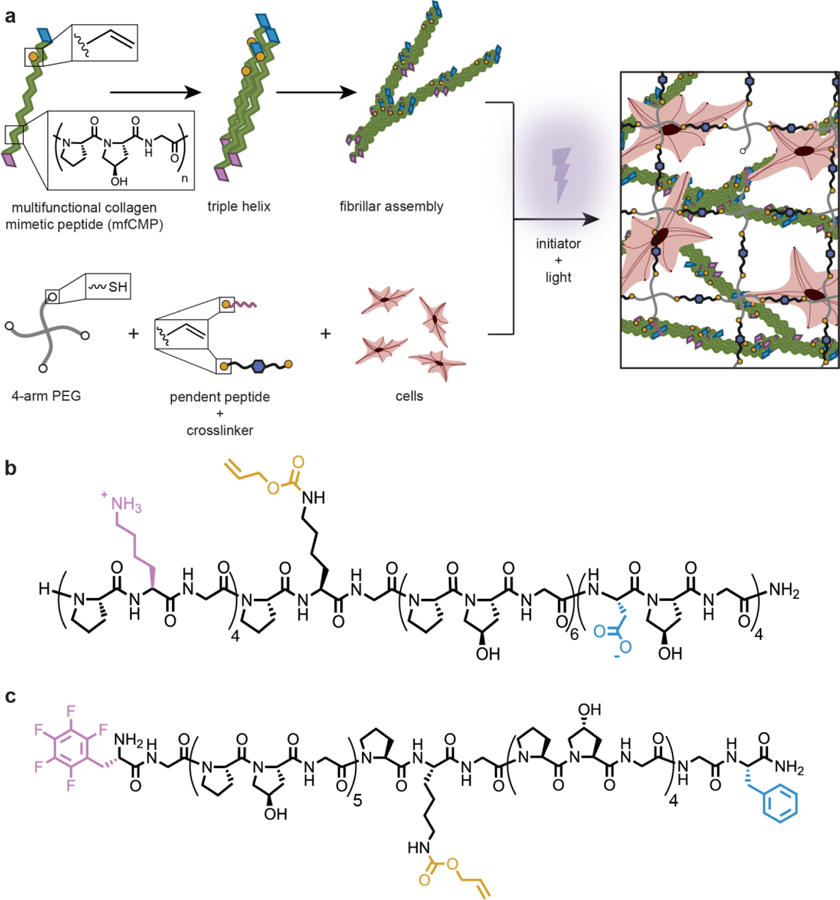
Approach to creating robust hierarchically-structured hydrogels. a) Multifunctional CMPs (mfCMPs) were designed for assembly and subsequent integration within covalently crosslinked hydrogels formed by light-triggered thiol–ene click chemistry, with a 4-arm PEG-SH macromer, cell-degradable peptide linker, photoinitiator, and integrin-binding pendant peptide. This approach allows for the encapsulation and 3D culture of cells within a robust, synthetic hydrogel with multiscale structures. Two mfCMP designs were established: b) mfCMP-1a (‘sticky’ ends lysine (pink) and aspartic acid (blue); reactive handle alloc-protected lysine (yellow)) and c) mfCMP-2a (aromatic stacking ends pentafluorophenylalanine (pink) and phenylalanine (blue)).
Both mfCMP-1a and mfCMP-2a were synthesized by solid phase peptide synthesis (SPPS) using Fmoc chemistry. Notably, while an automated synthesizer with some refinement of standard protocols could be used effectively to make mfCMP-1a, low purity and yields were observed for mfCMP-2a, potentially owing to its larger number of (POG) repeat units. To address this, we adapted a protocol for synthesis of a tri-amino acid fragment Fmoc-GPO(tBu)-OH40 and built mfCMP-2a by fragment condensation on resin. This approach provides opportunities for facile scaling of peptide synthesis through gram-scale synthesis of the Fmoc-GPO(tBu)-OH starting material. The resulting peptides were purified by high performance liquid chromatography (HPLC) and identified by mass spectrometry (Figures S1 and S2): (PKG)4PK(alloc)G(POG)6(DOG)4 (mfCMP-1a) and F(F5)G(POG)5PK(alloc)G(POG)4GF (mfCMP-2a).
mfCMPs assemble into stable triple helices and larger fibrillar structures
To assess the first level of hierarchical structure within these materials, the assembly of peptide strands into triple helices was examined in solution using circular dichroism (CD). The samples were heated to 85 °C for 15 minutes to ‘melt’ any assembled structures and then allowed to cool to room temperature and assemble for 48 hours prior to analysis or subsequent processing. Wavelengths scans with CD provided clear observation of the characteristic peak of a polyproline type II helix at 225 nm, indicating triple helix formation (Figure S3), and subsequent temperature scans at 225 nm established the melting temperature, Tm, of the triple helices (Figures 2b and 2c). The Tm is defined as the point where 50% of the triple helices are disassembled and is attributed to the inflection point of the melting curve. Importantly, a Tm at or above physiological temperature was observed for both mfCMP designs: mfCMP-1a at approximately 37 °C and mfCMP-2a at approximately 62 °C, where mfCMP-2a exhibits a higher Tm largely owing to the 3 additional (POG) triplets it contains.
Fig. 2:
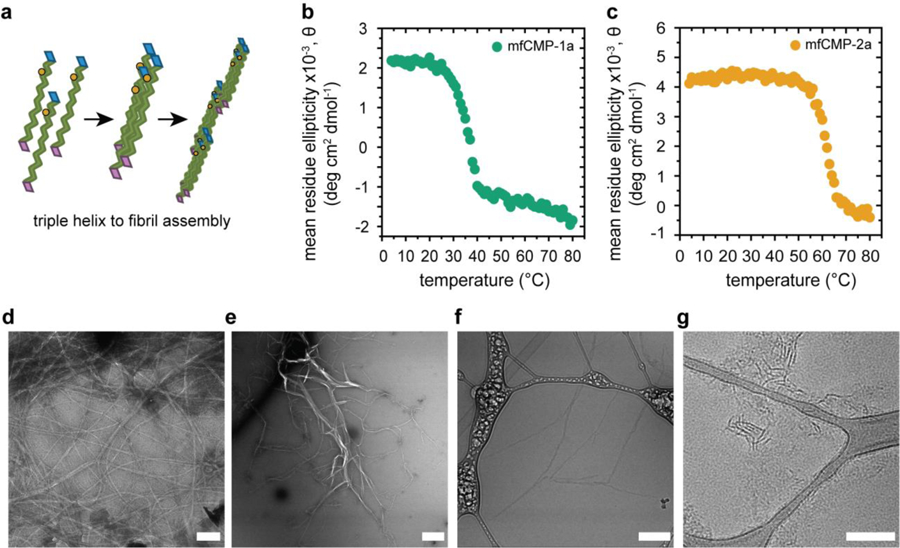
Assembly of mfCMP into triple helices and fibrils in solution. a) Schematic: three peptide strands assemble into triple helices as a result of hydrogen bonding, and these subsequently assemble into fibrils, facilitated by end-to-end interactions imparted by the charged or aromatic amino acid substitutions. CD of b) mfCMP-1a and c) mfCMP-2a measuring the ellipticity of the sample at a wavelength of 225 nm over a range of temperatures to determine the melting temperature of each peptide. TEM of d) mfCMP-1a and e) mfCMP-2a after assembly at 1 mM concentration and negatively stained with 2% uranyl acetate. Scale bars = 500 nm. Cryo-TEM of f) mfCMP-1a and g) mfCMP-2a assembled at higher concentrations and diluted to 1 mM immediately before preparing samples, mirroring conditions used for hydrogel formation. Scale bars = 200 nm.
To examine higher-order assembly of each peptide into fibrils, we utilized transmission electron microscopy (TEM) and cryogenic TEM (cryo-TEM). First, TEM was performed on cast films of assembled peptides for assessing the types and sizes of assembled structures (assembled at 1 mM, uranyl acetate counterstain). mfCMP-1a formed fibrils that were long and rope-like and appeared interconnected throughout the grid (Figure 2d), with an average width of 30 nm. mfCMP-2a formed fibrils with a larger distribution of widths and lengths, with large and small width populations on average of 65 and 10 nm. Additionally, a D-periodic banding structure was observed when this peptide was assembled at lower concentrations (0.3 mM, ~ 1 mg/mL, of mfCMP-2a), reminiscent of the banded structure observed in native collagen (Figure S4).33 These average widths for mfCMP-1a and mfCMP-2a are consistent with reports of similar CMPs and, more importantly, native collagen fibrils that range in diameter from ~10 nm to 500 nm.8, 32, 41
Assembled morphologies in the hydrated state and without the use of any counterstain were examined with cryo-TEM, enabling a more precise assessment of fibril sizes and the assembled nanostructures incorporated within the covalently crosslinked hydrogels. Cryo-TEM samples were prepared using peptides that had been heated and assembled at higher concentrations (5 mM mfCMP-1a and 20 mM mfCMP-2a, selected based on respective solubilities) and subsequently were diluted to 1 mM directly before imaging, which is the same procedure used for incorporation of each mfCMP into the hydrogels. In these fully hydrated samples, mfCMP-1a fibrils measured at approximately half of the width observed in the cast films (~14 nm), and mfCMP-2a fibrils were more reflective of the smaller width population that was observed in the cast films (~10 nm), where the larger widths observed in cast films were likely an artifact of drying. mfCMP-1a fibrils were ~400 nm long between junction points with an interconnected network spanning the field of view, consistent with our prior observations, whereas mfCMP-2a fibril lengths were consistently shorter in length at ~100 nm long. This length for mfCMP-2a fibrils differs from a prior report for other aromatic stacking CMP sequences;34 a notable difference between these systems is the processing conditions. Specifically, prior reports performed peptide assembly with temperature cycling (4 °C, room temperature, and 37 °C) to yield fibrils that were microns long versus ours that were heated and then held at room temperature to facilitate direct comparison to mfCMP-1a. Overall, these data and related observations supported the formation of robust assembled nanostructures reminiscent of collagen and the use of not only peptide chemical structure, but also assembly and processing conditions in solution to influence the resulting fibrillar structures.
Covalent incorporation of mfCMPs did not impact final hydrogel mechanical properties
With sequences and assembly conditions identified for achieving a range of biologically relevant melting temperatures and hierarchical structures, we set out to determine concentrations of assembled mfCMPs that could be integrated within synthetic hydrogels while still preserving desired mechanical properties in the range of different soft tissues. Here, mfCMPs were assembled at higher concentrations and diluted, as described in cryo-TEM, now diluting into hydrogel precursor solutions immediately prior to light exposure for triggering photopolymerization. Using in situ photorheometry, a hydrogel precursor solution formulation without mfCMP first was identified for achieving a modulus in the range of loose connective tissues (E ~ 1–10 kPa), where E ~ 3*G’ ~ 10 kPa for these hydrogels based on rubber elasticity.42 The specific formulation identified with a modulus in this range both before and after equilibrium swelling was 6 wt% 4-arm PEG-SH, di-alloc cell-degradable linker peptide (1:1 alloc:thiol), and 2.2 mM photoinitiator LAP in phosphate buffered saline (PBS) irradiated with 10 mW cm−2 at 365 nm. Assembled mfCMPs subsequently were diluted into this precursor solution formulation, replacing a portion of the non-assembling di-alloc peptide linker with up to 2.5 mM mfCMP (the upper limit allowed with an assembled 5 mM mfCMP-1a stock solution) (Figure 3a). Hydrogel precursor solutions with and without assembled mfCMPs were photopolymerized on a rheometer, and the resulting mechanical properties were measured via oscillatory rheometry.
Fig. 3:
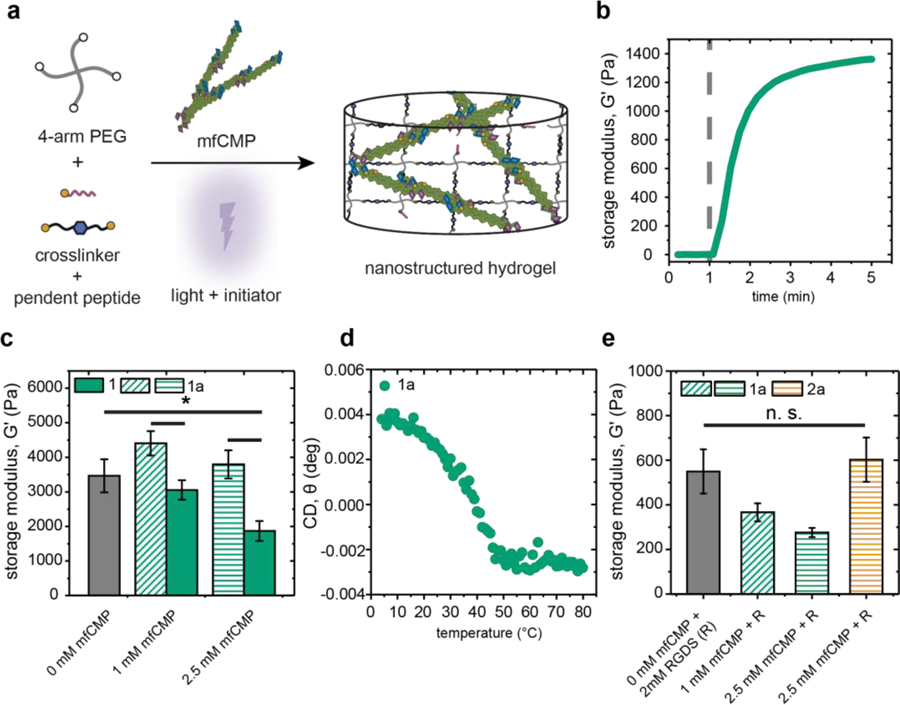
Mechanical properties of nanostructured hydrogels containing mfCMPs. a) Schematic of approach for incorporation of assembled mfCMPs within hydrogels using K(alloc) handle that is orthogonal to assembly: mfCMPs were assembled in solution for 48 h, diluted into a hydrogel precursor solution, and immediately irradiated in situ to form robust, nanostructured hydrogels. b) In situ oscillatory rheometry was used to monitor the rapid formation of these hydrogels (dashed gray line indicates when irradiation commenced; here, hydrogel formed with 2.5 mM mfCMP-1a as representative example), and their resulting mechanical properties compared. Hydrogels formed with c) increasing amounts of mfCMPs with or without an alloc group (mfCMP-1a or mfCMP-1, respectively). d) CD on an in situ hydrogel containing 2.5 mM mfCMP-1a to determine the melting temperature of assembled mfCMPs within the covalently crosslinked network. e) Moduli of hydrogels formed with mfCMPs and integrin binding peptide RGDS equilibrated in DPBS at 37 °C, which were the formulations used in 3D cell culture studies. The data shown illustrate the mean with error bars showing the standard error (n ≥ 3; * p < 0.05).
These hydrogels formed rapidly with a time to complete gelation of approximately 2 minutes (Figure 3b), which was estimated following established protocols,37 representing a relevant time scale for encapsulation of live cells. Next, increasing amounts of mfCMPs were incorporated into the hydrogel to determine if there was a maximum amount that could be added without compromising the final modulus of the material. Notably, the average storage moduli for hydrogels with 1 and 2.5 mM mfCMP-1a were statistically the same as the 0 mM mfCMP control (Figure 3c). To further assess if the assembled mfCMP-1a fibrils were serving as covalent crosslinks within hydrogels, an additional control was included: mfCMP-1a was replaced with an analogous assembled peptide without the alloc reactive handle (mfCMP-1, (PKG)4(POG)6(DOG)4) to present a similar nanofibrillar structure but without the ability to form covalent crosslinks (Figure S5). We observed that, with increasing amounts of mfCMP-1, a statistical difference in moduli emerged for the resulting photopolymerized hydrogels, with a G’ decrease of ~50%. Hydrogels with 2.5 mM mfCMP-2a consistently formed, with a small yet statistical difference in the modulus relative to control hydrogels (0 mM mfCMP) (Figure S6). These data supported the hypothesis that inclusion of the alloc reactive handle was allowing the mfCMPs to serve as crosslinks within the photopolymerized network, enabling the formation of hydrogels with integrated assembled mfCMP nanostructures while maintaining control over mechanical properties. Further, the observation of decreasing modulus with higher concentrations of mfCMP-1 suggested that assembled fibrils missing the alloc reactive handle disrupted the crosslinking of the covalent hydrogel.
Retention of the assembled structure upon formation of the covalent hydrogel was examined using CD. Specifically, a peak associated with the assembled triple helix within a hydrogel formed in situ within a CD cuvette was identified and tracked as a function of temperature (Figure S7). With this approach, a melting curve for assembled mfCMP-1a within the hydrogel was observed similar to that in solution, with the same Tm ~37 °C (Figure 3d). These data confirm the formation of nanostructured hydrogels with this innovative approach.
To determine the moduli of hydrogels used in 3D cell culture, mfCMP-1a and mfCMP-2a hydrogels were formed in molds off the rheometer and equilibrium swollen in DPBS overnight at 37 °C to mimic cell encapsulation and culture conditions. Hydrogels with different concentrations of mfCMPs were synthesized with an alloc-functionalized, integrin-binding peptide RGDS (2 mM) to allow cell adhesion to the hydrogel network,1, 43 and the final swollen moduli measured using oscillatory rheometry. No statistical differences were observed between the mfCMP hydrogels and the no mfCMP control. Importantly, the Young’s moduli of the resulting equilibrium swollen hydrogels were E ~ 1–1.5 kPa, which is similar to our inspiration collagen-rich, loose connective tissues such as the lung interstitium (E ~ 1.5 kPa reported on average for healthy lung).39
Presence of mfCMPs did not impact cell viability and promoted cell elongation in 3D culture within nanostructured hydrogels
Finally, we sought to establish the utility of these materials for human mesenchymal stem cell (hMSC) encapsulation and 3D culture, and if the incorporation of mfCMPs had any effect on hMSC viability and morphology. To investigate this, hMSCs were encapsulated as a single cell suspension within hydrogels that contained no mfCMP (0 mM), a low concentration of mfCMP (1 mM), or a high concentration of mfCMP (2.5 mM), using a low cell density to promote primarily cell-matrix interactions initially (3.75x106 cells/mL). Encapsulated cells were cultured within these hydrogels for up to 10 days, and differences in cell viability and morphology were examined between hydrogel compositions using quantitative imaging, where the effects of mfCMP-1a and mfCMP-2a were assessed independently.
Cells exhibited good viability in all conditions (greater than ~ 85%), where by day 3 no statistical differences in viability were observed between the three hydrogel compositions (Figure 4). The effects of these multiscale mfCMP-1a hydrogels on hMSCs were further examined through the quantification of cell morphology with 3D shape factor measurements after 10 days of culture. Confocal z-stacks of fixed and stained cells were used to create a 3D representation of the cells in Volocity® image analysis software, and the shape factor of each object determined, where “object” was defined as a single cell or group of cells in contact. Based on observations of subpopulations within distributions of these data, cell morphology results were divided into three categories: elongated (0 ≤ Ψ ≤ 0.25), spread (0.25 < Ψ ≤ 0.5), and rounded (0.5 < Ψ ≤ 1), allowing comparisons between hydrogel conditions (Figure S6). Interestingly, with the higher mfCMP-1a concentration, we observed a significant shift in cell morphology from simply spread to highly elongated for over 25% of the population as compared to less than 5% in 0 and 1 mM compositions (Figure 4d). In addition to this increase in elongated cells and cell clusters, we also observed the appearance of ‘holes’ in hMSC clusters at high mfCMP-1a concentrations (indicated by arrowheads in Fig. 4 a,b), where the cells appear to be encircling objects. This ‘hole’ formation within cell clusters is not a typical behavior of hMSCs in cell-degradable hydrogels formed with non-assembling peptides and PEG, where most typically a rounded to spread cell morphology is observed as evidenced in the 0 mM control in our study.3 In this context, we speculate that the cells are wrapping around the multiscale structures within the 2.5 mM mfCMP-1a synthetic matrix. Subsequently, cell response to mfCMP-2a hydrogels was investigated, where hMSCs were encapsulated into hydrogels containing 1 or 2.5 mM of pre-assembled mfCMP-2a. We observed that mfCMP-2a hydrogels supported cell viability (Figure S7a, S7c) and spreading (Figure S7b, S7d) yet without the highly elongated and unique cell morphologies observed with mfCMP-1a hydrogels.
Fig. 4:
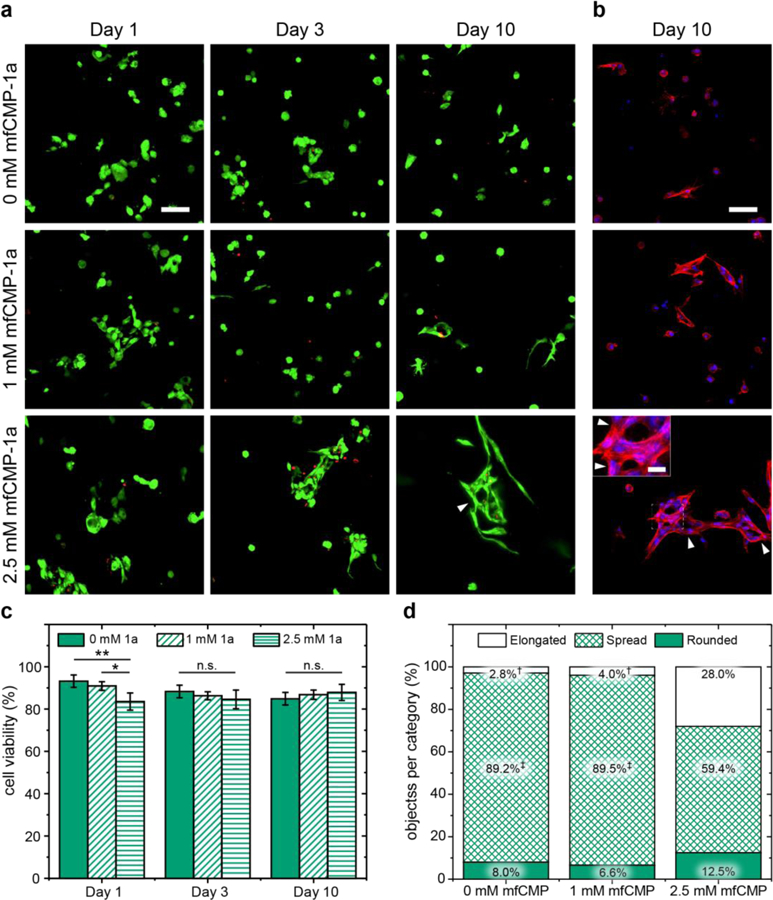
Effects of nanostructured mfCMP-1a hydrogels on hMSC viability and morphology in 3D cell culture. Representative maximum intensity projections from confocal z-stacks (200 μm thick) of hMSCs encapsulated within hydrogels without mfCMP-1a (0 mM), at a low concentration of mfCMP-1a (1 mM), or at a high concentration (2.5 mM) of mfCMP-1a for a) Live/Dead cytotoxicity assay (live cells, green; dead cell nuclei, red) and b) fixed stained samples at day 10 (F-actin cytoskeleton, red; cell nuclei, blue). Scale bars: large images = 100 μm, inset image = 25 μm. Arrowheads note unique ‘holes’ that were consistently observed within cell clusters when cultured in 2.5 mM mfCMP-1a hydrogels. From these images, c) cell viability was quantified (error bars represent the standard error around the mean; n = 3 hydrogels, > 250 cells counted per condition; *p < 0.05, **p < 0.01). d) Cell morphology was analyzed through quantification of 3D shape factor (Ψ) of individual or clusters of cells at day 10 with a focus on analysis of cell elongation († indicates a significant difference in the percent of elongated objects relative to the experimental condition 2.5 mM mfCMP-1a, p < 0.05; ‡ indicates a significant difference in the percentage of spread objects relative to the experimental condition 2.5 mM mfCMP-1a, p < 0.05); full set of quantitative results and statistics are available in Tables S1–S4).
Our observations of cell responses to the different compositions of hydrogels with and without mfCMPs have interesting implications when combined with our observations of the assembled mfCMP multiscale structures and hydrogel properties. Different fibril lengths were achieved based on the chemical structure of the mfCMP: mfCMP-1a with charged ‘sticky’ ends assembled into long fibrils (~400 nm between fibril junctions), whereas mfCMP-2a with aromatic ends assembled into shorter fibrils (~100 nm); both exhibited similar fibril widths (~10 nm). In this context, we can deduce that increased concentrations of longer fibrils are most conducive to cell elongation within these well-defined hierarchically structured hydrogel-based synthetic matrices. These observations of increased cell elongation with increased fibril length are consistent with reports for hMSCs and other human cell types seeded on collagen hydrogels14, 30 or electrospun fibers,17, 19 respectively, now within a fully synthetic hydrogel system. Uniquely, the bottom-up control of fibrillar structure that these innovative mfCMP materials afford along with independent tuning of matrix modulus or integrin binding can be exploited in future investigations, building upon studies performed within harvested collagen hydrogels and going beyond them without their inherent limitations. Further, mfCMPs with a variety of different amino acid substitutions or other shorter peptide sequences that are designed to form a range of nano- and micro-structures could be integrated within this framework to enable long-term 3D cultures for probing different cell responses. Additionally, combining the innovative approaches established here with methods to promote alignment of assembled structures (e.g., convective assembly or tensile loading)44, 45 could enable future studies for promoting specific morphologies, orientations, and functions or fates of hMSCS or other cell types within such soft synthetic ECMs.
In conclusion, these studies have not only established an approach for the design of multifunctional assembling peptides and their integration within covalently crosslinked synthetic hydrogels, but also demonstrated the utility of these soft materials with multiscale structure for controlled 3D cell culture. These mfCMPs integrated a non-native alloc-protected lysine residue as a reactive handle orthogonal to assembly for triggered crosslinking using thiol–ene click chemistry along with a large hydrogen bonding block and ‘sticky’ end groups for the formation of long fibrils with physiologically-relevant melting temperatures and sizes consistent with that of native collagen. By inclusion of the alloc reactive group, a significant amount of assembled mfCMPs could be covalently incorporated during formation of the hydrogels while still preserving the robust mechanical properties. This afforded a handle for the independent control of nanostructure and modulus within synthetic hydrogels. These materials allowed the encapsulation and 3D culture of primary human cells, where highly elongated morphologies were observed over time reminiscent of cell responses to collagen now with a fully synthetic approach. This work provides an innovative strategy for the integration of assembled peptides within synthetic hydrogels and a new biomimetic platform for studying cell-matrix interactions. The robust nature of this platform offers future opportunities for both fundamental studies of structure-property-function relationships and their translation into a variety of applications, from regenerative medicine to nanostructured membranes.
Methods
General materials.
All general organic reagents were purchased from commercial sources, Thermo Fisher Scientific (Waltham, MA) and Millipore Sigma (St. Louis, MO), and used as received unless otherwise stated. Amino acids and 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) were purchased from ChemPep (Wellington, FL) unless stated otherwise. Human mesenchymal stem cells (hMSCs) were purchased from Lonza (Basel, Switzerland). General cell culture materials, including Dulbecco’s modified eagle medium (DMEM), Penicillin-Streptomycin (5,000 U/mL), Fungizone, Fetal Bovine Serum (FBS, 16000044), Trypsin-EDTA (0.5%), and Dulbecco’s phosphate-buffered saline (DPBS), were purchased from Thermo Fisher Scientific (Waltham, MA), and recombinant human basic fibroblast growth factor (bFGF) was obtained from PeproTech (Rocky Hill, NJ). Reagents for assessing cell viability and morphology, including the LIVE/DEAD™ Viability/Cytotoxicity Kit for mammalian cells, methanol-free formaldehyde, Triton™ X-100, and 4’,6-diamidino-2-phenylindole dihydrochloride (DAPI), were purchased from Thermo Fisher Scientific (Waltham, MA). Bovine serum albumin (BSA), TWEEN® 20, and Phalloidin-Tetramethylrhodamine B isothiocyanate (Phalloidin-TRITC) were obtained from MilliporeSigma (Burlington, MA). ImageJ from the National Institutes of Health (Rockville, MD), Volocity® from PerkinElmer (Waltham, MA), and MATLAB from MathWorks (Natick, MA) were used for image analysis and computational processing.
Synthesis of multifunctional collagen mimetic peptides 1a and 1 (mfCMP-1a & mfCMP-1).
Both mfCMP-1a ((PKG)4PK(alloc)G(POG)6(DOG)4) and a control sequence, mfCMP-1 ((PKG)4(POG)6(DOG)4), were synthesized by standard solid phase peptide synthesis (SPPS) using Fmoc chemistry on TentaGel® Resin (Peptides International, Louisville, KY; 0.19 meq/g) with a Tribute peptide synthesizer (Gyros Protein Technologies, Tucson, AZ), where K(alloc) is an allyloxycarbonyl protected lysine residue, O is hydroxyproline, and all other amino acids are standard abbreviations. Amino acids and HBTU were loaded into cartridges, and all amino acids were double coupled on resin. Fmoc deprotections were carried out in 20% piperidine in N,N-dimethylformamide (DMF) for 30 minutes. Amino acid couplings were done using 0.4 M 4-methylmorpholine (4-MMP) in DMF for 1 hour. The Fmoc of the final amino acid was also deprotected after the peptide was synthesized. The peptide was then cleaved from the resin in a solution of trifluoroacetic acid (TFA), triisopropylsilane (TIPS), and water at 95%, 2.5%, and 2.5% v/v, respectively, for 2 hours. After cleavage the peptides were precipitated in ethyl ether at 10x excess in 50 mL conical tubes and refrigerated overnight. They were then centrifuged at 4400 RPM at 4 °C for 10 minutes and washed 3x with ethyl ether, resuspending and centrifuging after each wash, and allowed to dry at room temperature overnight. Peptides were then desiccated for at least 1 hour under vacuum prior to purification by reverse phase high performance liquid chromatography using a Jasco co-2060 Plus column heater set to 65 °C (HPLC) (XBridge C18 OBD 5 μm column, linear gradient from 12 to 22% acetonitrile over 9 minutes) (Waters Corporation, Milford, MA). The collected fraction was frozen then lyophilized and the identity determined using electrospray ionization (ESI+) mass spectrometry (Acquity UPLC H-class/SQD2) (Waters Corporation, Milford, MA). After lyophilization, the peptides were dissolved in DI water at a dilute concentration and dialyzed against DI water for 24 hours to remove any remaining associated ions. The peptide solution was then re-lyophilized prior to sample preparation.
Synthesis of Fmoc-GPO(tBu)-OH fragment.
The small molecule fragment Fmoc-GPO(tBu)-OH was synthesized through a series of reactions using N-Hydroxysuccinimide (HOSu) chemistry adapted from previously published work.40 The details of each reaction step are described below.
Reaction 1:
Briefly, Fmoc-G-OH (12.5 g, 0.042 mol) and HOSu (4.84 g, 0.042 mol) were dissolved in a mixture of ethyl acetate (100 mL) and DMF (40 mL) and stirred on ice for 10 minutes in a round bottom flask. A similar molar equivalent of dicyclohexylcarbodiimide (DCC) (8.66 g, 0.042 mol) was dissolved in ethyl acetate (60 mL) in a scintillation vial and added dropwise. The reaction mixture was stirred at room temperature overnight before it was filtered to remove the dicyclohexylurea by-product (DCU). The filtrate was concentrated by rotary evaporation and then redissolved in a minimal amount of ethyl acetate and precipitated into hexanes (10x excess). The Fmoc-G-OSu was collected by filtration after precipitation and dried under vacuum overnight. 1HNMR was used to confirm this product and all subsequent products (Bruker AVIII 600) (Bruker Daltonics, Billerica, MA).
Reaction 2:
The Fmoc-G-OSu (10 g, 0.025 mol) was dissolved in DMF (100 mL) and added to a round bottom flask. H-Pro-OH (2.92 g, 0.025 mol) was dissolved in DMF (10 mL) in a scintillation vial then added to the Fmoc-G-OSu solution. Diisopropylethylamine (DIPEA) (2.18 mL, 0.0125 mol) was then added to the reaction, while stirring. The reaction mixture was stirred at room temperature overnight and then diluted with ethyl acetate (50 mL) and washed with 5 wt% sodium bicarbonate solution (NaHCO3) (50 mL). The aqueous layer was then acidified to approximately pH 1 with 1 M HCl, assessed using pH paper, and extracted 4x with ethyl acetate (100 mL each time). The organic layers were combined, washed 2x with water (100 mL each time) to remove any non-organic compounds, and dried over magnesium sulfate (MgSO4). The filtrate was concentrated through rotary evaporation and dissolved in a minimal amount of ethyl acetate before precipitation into hexanes (10x excess). The product was collected via filtration. The Fmoc-GP-OH was then dried overnight under vacuum, and 1HNMR was used to confirm the product.
Reaction 3:
Fmoc-GP-OH (7.14 g, 0.018 mol) was dissolved in ethyl acetate (100 mL) and HOSu (2.07 g, 0.018 mol) was added to the Fmoc-GP-OH solution with additional ethyl acetate (12 mL) and stirred on ice for 10 minutes in a round bottom flask. DCC (3.71 g, 0.018 mol) was dissolved in ethyl acetate (56 mL) in beaker and added dropwise. The reaction mixture was stirred at room temperature overnight before it was filtered to remove the DCU by-product. The filtrate was concentrated by rotary evaporation and then re-dissolved in a minimal amount of ethyl acetate and precipitated into hexanes (10x excess). The Fmoc-GP-OSu was collected by filtration after precipitation and dried under vacuum overnight. 1HNMR was used to confirm the product.
Reaction 4:
The Fmoc-GP-OSu (3.49 g, 0.007 mol) and H-Hyp(tBu)-OH (1.46 g, 0.008 mole) were added to a round bottom flask and dissolved in DMF (100 mL), DIPEA (0.62 mL, 0.004 mol) was then added to the reaction, while stirring. The reaction mixture was stirred at room temperature overnight and then diluted with ethyl acetate (50 mL) and washed with 5 wt% sodium bicarbonate solution (NaHCO3) (50 mL). The aqueous layer was then acidified to approximately pH 1 with 1 M HCl, assessed using pH paper, and extracted 4x with ethyl acetate (300 mL each time). The organic layers were combined, washed 2x with water (300 mL each time) to remove any non-organic compounds, and dried over magnesium sulfate (MgSO4). The filtrate was concentrated through rotary evaporation, and the final product was purified by flash chromatography over silica gel using pure hexanes to remove by-products before switching to ethyl acetate to elute the product (CombiFlash Rf, Teledyne Isco). The final Fmoc-GPO(tBu)-OH was collected, dried under vacuum, and confirmed by 1HNMR.
Synthesis of mfCMP-2a by fragment condensation using Fmoc-GPO(tBu)-OH.
The mfCMP-2a peptide (F(F5)G(POG)5PK(alloc)G(POG)4GF) was synthesized by SPPS manually, employing a fragment condensation approach, where F(F5) represents pentafluorophenylalanine. TentaGel® Resin (0.2 mmol) (Peptides International; 0.19 meq/g) was placed in a 25 mL glass peptide synthesis vial with a built-in glass frit (ChemGlass) and placed on a wrist action shaker (Lab-Line, Melrose Park, IL). Approximately 10 mL of dichloromethane (DCM) was added to the reaction vessel to swell for 10 minutes while stirring. After swelling the resin was drained, 10–15 mL of fresh DMF was added 5x and stirred for 1–2 minutes, draining after each wash to ensure complete DCM removal. Deprotections were performed with 20% piperidine in DMF 3x for 30 minutes, where each time used piperidine was drained and fresh piperidine solution was added. A 2 μL aliquot of the drained solution was diluted into 1 mL DMF in a 1 cm pathlength cuvette and measured using UV-VIS spectroscopy. The peak at 301 nm, which indicates the presence of the Fmoc group, was measured. Coupling was considered complete if absorbance was ~ 0.1 or less; if needed, additional deprotection steps were performed until that level was reached. Amino acids or fragments (Fmoc-GPO(tBu)-OH) were added sequentially to manually synthesize mfCMP-2a. Specifically, for amino acid and fragment couplings, 2 molar equivalents were dissolved in just enough DMF to cover the resin (~2 mL), and 2 molar equivalents of DIPEA was then added to the solution and the vessel shaken to mix. The coupling solution was then added to the reaction vessel and stirred for 30 minutes, and a Kaiser test for primary amines and a chloranil test for secondary amines were used to monitor the coupling progress. Each amino acid or fragment was double coupled, and additional couplings were performed if Kaiser or chloranil tests showed a positive result for the presence of amines. When synthesis was complete, the peptide was then cleaved from the resin in a solution and precipitated in ethyl ether as described previously for mfCMP-1a. The peptide product was desiccated for at least 1 hour under vacuum prior to purification by reverse phase HPLC (XBridge C18 OBD 5 μm column, linear gradient from 5 to 45% acetonitrile at 0.5 %/min, column preheated to 65 °C) (Waters Corporation, Milford, MA). The collected fraction was frozen and lyophilized, and its identity determined using ESI+ mass spectrometry (Acquity UPLC H-class/SQD2) (Waters Corporation, Milford, MA).
Synthesis of crosslinker and integrin binding peptides.
The cell-degradable linker peptide sequence KK(alloc)GGPQG↓IWGQGK(alloc)K, a tryptophan variant of a wild type sequence derived from collagen, was chosen as the peptide linker for these hydrogels because it responds to a variety of matrix metalloproteinases secreted by a range of cell types including hMSCs.37 An integrin binding sequence derived from vitronectin and fibronectin, K(alloc)GWGRGDS, that binds a variety of integrins was incorporated within the matrix to promote cell adhesion.46 Both sequences were made on an automated Liberty Blue microwave peptide synthesizer (CEM Corporation, Matthews, NC). MBHA rink-amide resin (0.25 mmol equivalents) (NovaBiochem, 0.52 meq/g) was used added to the reaction vessel on the Liberty Blue and swollen in DMF for 10 minutes. Deprotections were performed with 20% piperidine in DMF for 8 minutes at 75 °C, and coupling reactions were performed using 5 molar equivalents of amino acid (0.2 M in DMF), N,N’-diisopropylcarbodiimide (DIC) (0.5 M in DMF), and ethyl(hydroxyimino) cyanoacetate (OxymaPure) (1 M in DMF) for 8 minutes at 75 °C. The peptide was then cleaved from the resin and precipitated in ethyl ether as described previously for mfCMP-1a. Peptides were desiccated for at least 1 hour under vacuum prior to purification by reverse phase HPLC (XBridge C18 OBD 5 μm column, linear gradient from 25 to 35% acetonitrile for linker peptide and 20 to 30% for RGDS peptide) (Waters Corporation, Milford, MA). The collected fraction was frozen then lyophilized and its identity determined using ESI+ mass spectrometry (Acquity UPLC H-class/SQD2) (Waters Corporation, Milford, MA).
Once fractions were confirmed through ESI+ mass spectrometry, stock solutions of linker peptide and integrin binding peptides were prepared in DPBS. Concentrations were confirmed using UV-VIS spectrometry, converting absorbance at 280 nm from the tryptophan in both peptides to concentration through Beer’s Law.
PEG-SH macromer synthesis.
Four arm poly(ethylene glycol) tetrathiol (PEG-SH) was synthesized starting from four arm PEG-OH (JenKem Technology, USA) following a modified version of a previously published protocol37. Briefly, four arm PEG-OH (20,000 g/mol; 10 g) was functionalized with allyl ether end groups; the allyl ether end groups subsequently were reacted with thioacetic acid using radically-mediated thiol–ene chemistry to form PEG-thioacetate; and the resulting thioacetate groups were cleaved using concentrated sodium hydroxide (NaOH) to produced PEG-SH. 1HNMR was used to confirm successful functionalization in each step, including the PEG-SH product (Bruker AVIII 600) (Bruker Daltonics, Billerica, MA). Prior to use, PEG-SH was dissolved in water (~30 mL per 1g of PEG) and treated with tris(2-carboxyethyl)phosphine (TCEP) (350 mg per 1 g of PEG-SH) on a shaker overnight to break any disulfide bonds that may be present. After TCEP treatment, the PEG-SH was dialyzed (MWCO 1 kDa, Spectrum Laboratories) for 24 hours against deionized water (pH 4) to remove TCEP and preserve free thiol groups. After dialysis, PEG-SH was collected in a 50 mL conical, frozen, and lyophilized to yield a white powder. Stock solutions of 80 mM thiol groups were made and final concentration was confirmed by Ellman’s assay.
LAP photoinitiator synthesis.
The photoinitiator lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) was synthesized following a published protocol.37 Briefly, dimethyl phenylphosphinite was added to an equivalent amount of 2,4,6-trimethylbenzoyl chloride dropwise and allowed to stir overnight. Four times molar excess of lithium bromide in 2-butanone was then added to the reaction, heated to 50 °C, and stirred for ~10 minutes. The white precipitate was filtered and rinsed 3x with fresh 2-butanone, dried under vacuum, and analyzed by 1HNMR (Bruker AVIII 600) (Bruker Daltonics, Billerica, MA).
General procedure for peptide assembly.
Solutions for peptide assembly were prepared in 1x DPBS (pH 7.4) or deionized water (pH 7) for mfCMP-1a and mfCMP-2a, respectively. The peptide solutions were then heated to 85 °C for 15 minutes and allowed to slowly cool to room temperature and assemble for 48 hours prior to experiments.
Circular dichroism (CD).
Triple helix formation along with melting temperatures of mfCMPs were measured using a CD spectropolarimeter (J-810 JASCO Corporation, Easton, MD). Solutions were prepared in DPBS or deionized water for mfCMP-1a and mfCMP-2a, respectively, with 0.1 mM to 0.3 mM of peptide, and respective buffer or water without dissolved peptide was used for background correction. The solutions were measured in a quartz cuvette with a 1 mm path length. Measurements at different wavelengths were taken from 250 nm to 195 nm at desired temperatures with a scan rate of 50 nm/minute and averaged over 3 scans. Data points were collected every nanometer and converted to mean residue ellipticity [θ] (deg cm2 dmol−1) using the formula:
where θ is the measured ellipticity in millidegrees, L is the pathlength in mm, c is the molar concentration of the solution, and N is the number of residues in the peptide. A similar protocol was employed to look at the values of the peak at 225 nm, which indicates a polyproline type II helix, between the temperatures of 4 °C and 80 °C to determine the ‘melting temperature’ (Tm) of the peptide, which is the temperature at which approximately 50% of the peptide strands are assembled and disassembled. Similar data conversion to [θ] was performed as stated above, and data points were taken every degree with a heating rate of 10 °C per hour. To determine the melting temperature the data was fit to a Boltzmann curve, commonly used to determine thermal unfolding temperatures of proteins in solution. The first derivative of the Boltzmann fit was then taken to determine the melting temperature of the peptides in solution, which occurs at the inflection point.
Similar protocols were performed on in situ polymerized hydrogels containing mfCMP-1a. Briefly, hydrogel precursor solutions with assembled mfCMP-1a were prepared (see Hydrogel formation section), loaded into a 0.1 mm path length demountable cuvette, and polymerized using 10 mW cm−2 365 nm light for 4 minutes. The cuvette was then loaded into the machine, and similar wavelength and temperature scans as described above were performed, monitoring the peak value associated with the triple helix at 231 nm. Note, in the case of the temperature scan, the characteristic peak for the triple helix was observed to shift from 225 nm in free solution to 231 nm upon integration within the hydrogel; this wavelength shift is consistent with other reports of wavelength shifts in CD spectra when proteins with higher ordered structures are confined within a polymer network.47
Transmission electron microscopy (TEM).
Peptide solutions were assembled at a concentration of 1 mM for 48 hours prior to sample preparation as described above. Carbon-coated copper grids (200 mesh; Electron Microscopy Sciences, Hatfield, PA) were treated using a glow discharge plasma cleaner (PDC-32G, Harrica Plasma Inc., Ithica, NY) for 1 minute. An aliquot of assembled peptide solution (4 µL) was then pipetted on to the grid and, after 1 minute, excess solution was blotted with filter paper on all sides of the grid to create an even thin film across the grid. The resulting cast film of the sample was air dried for 10 minutes. Once dry, the sample was stained with uranyl acetate, which provides contrast during imaging with electron microscopy. Uranyl acetate in deionized water (4 µL of 2%) was dropped onto the grid and blotted away immediately with filter paper. The stained cast films were allowed to air dry for 1 hour prior to imaging using a Talos TEM (FEI Company, Hillsboro, OR). In the resulting images fibril widths were measured using FIJI.48
Cryogenic transmission electron microscopy (cryo-TEM).
Peptide solutions were assembled at concentrations of 5 mM and 20 mM for mfCMP-1a and mfCMP-2a, respectively, based on their solubility. mfCMP-1a rapidly formed large insoluble higher ordered structures with increased peptide concentration; however, no insoluble structures were observed in the case of mfCMP-2a at higher concentrations. After assembly, peptides were diluted to a concentration of 1 mM prior to sample preparation, mirroring the procedure used for preparation of mfCMP hydrogels. Lacey carbon film grids (200 mesh; Electron Microscopy Sciences, Hatfield, PA) were treated using a glow discharge plasma cleaner (PDC-32G, Harrica Plasma Inc., Ithica, NY) for 1 minute. The grids were loaded onto a Vitrobot, and 4 µL of assembled peptide solution was pipetted onto the grid, blotted twice for 2 seconds, and plunged into liquid ethane. The grids were transferred manually into holders immersed in liquid nitrogen and stored there until imaging. Imaging was performed at 200 kV on a Talos TEM with a Falcon 3 camera (FEI Company, Hillsboro, OR) using a Gatan 626 cryo holder at −176 °C. In the resulting images fibril widths and lengths were measured with FIJI.
Hydrogel formation.
Monomer, peptides, and photoinitiator were prepared in DPBS prior to polymerization. Solutions of 6 wt% PEG-SH, linker peptide, mfCMPs, and RGDS were prepared in stoichiometric ratios of thiol groups to alloc groups (12 mM SH:12 mM alloc). The RGDS was incorporated at a concentration of 2 mM, a concentration previously observed to effectively promote hMSC adhesion on and within these types of PEG-peptide hydrogels, and the remaining 10 mM of alloc groups were distributed between the linker peptide and the mfCMPs depending on the desired concentration of mfCMP to be incorporated. For example, if the desired mfCMP concentration was 2.5 mM, then the balance of non-assembly linker peptide was 7.5 mM. Each solution also contained 2.2 mM of LAP photoinitiator, and hydrogels were formed through irradiation of the complete precursor solution using cytocompatible doses of long wavelength UV light (10 mW cm−2 at 365 nm; Exfo Omnicure Series 2000 light source with 365 nm bandpass filter, Excelitas Technologies Corp., Waltham, MA) for 4 minutes to ensure complete gelation, even though a plateau in the storage modulus was observed after ~2 minutes.
Rheology on in situ hydrogels.
Hydrogels were formed on a rheometer with UV-visible light accessory (TA AR-G2, TA Instruments, New Castle, DE) that was attached to a filtered mercury lamp using a liquid-filled light guide (Exfo Omnicure Series 2000 light source with 365 nm bandpass filter, Excelitas Technologies Corp., Waltham, MA). Hydrogel precursor solution (10 µL) was pipetted on to the bottom quartz plate and the top plate (8 mm flat plate) lowered to fill the geometry. Measurements were collected at room temperature for 1 minute before the light was turned on, and the gels were polymerized for 4 minutes (10 mW cm−2 at 365 nm). The storage (G’) and loss (G’’) modulus were measured at 1% strain and a frequency of 6 rad/s, which was determined to be in the linear viscoelastic regime for these hydrogels. Approximate gelation time was defined to be when the percent change in modulus between 2 data points was less than 10%.37, 49
Rheology on equilibrium swollen hydrogels.
Hydrogel precursor solutions (22 µL) with 0 mM mfCMP, 1 mM mfCMP-1a, 2.5 mM mfCMP-1a, or 2.5 mM mfCMP-2a were prepared and pipetted into molds (1 mL syringes with the tips removed). Each hydrogel composition also contained 2 mM alloc-functionalized RGDS to remain consistent with hydrogel formulations that would be used in cell culture. The syringe molds were then placed under the collimated light source and irradiated (10 mW cm−2 at 365 nm) (Exfo Omnicure Series 2000 with 365 nm bandpass filter, Excelitas Technologies Corp., Waltham, MA). The hydrogels were polymerized for 4 minutes, the time determined by time sweep measurements to be sufficient for complete gelation of all formulations. After polymerization, the hydrogels were removed from the molds and placed in a 48-well non-tissue culture treated plate (CELLTREAT, Pepperell, MA). Buffer (500 µL of 1x DPBS) was added to each well, and the hydrogels were placed in an incubator at 37 °C and allowed to swell overnight. The diameter of each hydrogel was measured, and subsequently measurements of G’ and G’’ were performed as described for in situ measurements, replacing the quartz plate used for irradiation with a Peltier plate at 37 °C for temperature control (1% strain, frequency 6 rad/s).
Culture and encapsulation of human mesenchymal stem cells.
Bone marrow-derived human mesenchymal stem cells (hMSCs) were expanded under sterile conditions at 37 °C and 5% CO2 on tissue culture treated polystyrene flasks (182 cm2; CELLTREAT, Pepperell, MA). From thaw, hMSCs were cultured in growth media of low glucose DMEM supplemented with 50 U/mL penicillin, 50 μg/mL streptomycin, 0.2% v/v Fungizone, 10% v/v fetal bovine serum (FBS), and 1 ng/mL basic fibroblast growth factor (bFGF). Growth media was replaced every 3 days, and cells were passaged at approximately 85% confluency. Twenty-four hours prior to cell encapsulation, growth media was replaced with bFGF-free media (DMEM supplemented with 50 U/mL penicillin, 50 μg/mL streptomycin, 0.2% v/v Fungizone, 10% v/v FBS). hMSCs (passage 7) were encapsulated as a single cell suspension at a density of 3.75 x 106 cells/mL within 20 μL of gel precursor solution; this low cell density was selected to promote primarily cell-matrix interactions at the start of the experiment while supporting cell viability. For encapsulation, cells were incubated with 1x Trypsin for 8 minutes to detach from the tissue culture treated flasks, diluted into bFGF-free media during removal from plates, centrifuged for 5 minutes at 1000 RPM, and counted using a hemocytometer. Cells were resuspended in 1x DPBS (supplemented with 50 U/mL penicillin, 50 μg/mL streptomycin, and 0.2% v/v Fungizone) and added to the gel precursor solution to obtain a final solution of 6% w/w PEG-SH, 2 mM alloc-functionalized RGDS peptide, and 10 mM total linker peptides with a balance between the non-assembling linker and assembled mfCMPs as noted earlier. Precursor solution with suspended cells (20 μL) was pipetted into syringe molds and irradiated as before for hydrogel formation (10 mW cm−2 at 365 nm light for 4 minutes). The resulting cell-gel constructs were then placed into non-tissue culture treated 48 well plates (CELLTREAT, Pepperell, MA), washed with bFGF-free media for 30 minutes at 37 °C and then replaced with fresh bFGF-free media. bFGF-free media was then replaced every 2 days for the remainder of the experiment.
Viability of hMSCs in mfCMP hydrogels.
Viability of hMSCs encapsulated within hydrogels containing 0, 1, or 2.5 mM mfCMP-1a was assessed after 1, 3, or 10 days of culture using a LIVE/DEAD™ Viability/Cytotoxicity Kit for mammalian cells, following the protocol provided by the manufacturer. Early time points were used to determine the impact of the encapsulation on the cells, whereas late time points indicated cell response to the materials. Briefly, hydrogels were washed 2x in 500 μL DPBS for 5 minutes (37 °C, 5% CO2), followed by a 25 minute incubation in 350 μL DPBS containing 2 μM Calcein AM and 4 μM Ethidium homodimer-1 (37 °C, 5% CO2), and finally washed 2x in 500 μL DPBS for 5 minutes (37 °C, 5% CO2). Hydrogels were then transferred to a chambered coverglass slide (Nunc™ Lab-Tek™ II Chambered Coverglass, 8 well; ThermoFisher Scientific, Waltham, MA), covered with fresh DPBS, and imaged using a confocal microscope (Zeiss LSM 800; Zeiss, Oberkochen, Germany). Three gels were imaged for each condition, and 3 z-stack images were taken for each gel. Orthogonal projections were made of each 200 μm thick z-stack, and ImageJ was used to count the number of live (Calcein AM, green) and dead (Ethidium homodimer-1, red) cells in each projected image.
Cytoskeletal staining and imaging of hMSCs in mfCMP hydrogels.
Cell morphology of encapsulated hMSCs was assessed though confocal imaging of F-actin labeled with a TRITC fluorophore after sample fixation after 10 days of culture to visualize the cell body and perform a 3D analysis of cell shape. Hydrogels were rinsed 2x in 500 μL DPBS for 5 minutes, followed by cell fixation in 300 μL 4% PFA for 15 minutes. Gels were then rinsed 3x in 500 μL DPBS for 5 minutes. Cells were blocked in 400 μL 5% w/v BSA overnight at 4 °C and washed 4x with 400 μL TWEEN® 20 wash solution (0.2% v/v TWEEN® 20 and 1.5% w/v BSA in DPBS) for 5 minutes. Cells were permeabilized with 400 μL 0.2% v/v Triton™ X-100 in DPBS for 30 minutes, followed by 3x wash in 400 μL of the previously described TWEEN® 20 wash solution (5 minutes). Gels were incubated overnight at 4 °C with 300 μL Phalloidin-TRITC (1:250 dilution) in DPBS with 1.5% w/v BSA. Gels were then washed 2x in 400 μL TWEEN® 20 wash solution for 30 minutes, then 2x for 15 minutes, followed by a 1 hour incubation with DAPI (700 nM) in DPBS at room temperature. Gels were washed 3x in 400 μL TWEEN® 20 wash solution for 5 minutes prior to transfer to a chambered coverglass slide. Gels were covered with fresh DPBS, and imaged using a confocal microscope. Three gels were imaged for each condition, and three 200 μm-thick z-stack images were taken for each gel.
Cell morphology analysis using Velocity software.
Cell morphology of stained cells (phalloidin-TRITC, staining F-actin) was quantitatively analyzed with Volocity® 3D image analysis software. Specifically, z-stack images were imported into Volocity®, and cell bodies were detected by searching for objects in the red channel (TRITC-labeled F-actin). Here, an object is defined as a single cell or a group of cells in direct contact. Filters were used to improve the accuracy of object volume measurements (filter to close the objects, 4 iterations; filter to fill holes in objects). Noise was removed from the objects (fine filter) in order to improve the precision of object surface area measurements. Finally, objects with volumes below 1000 μm3 were considered to be debris and were excluded in measurements, and the shape factor of each object was recorded, where “object” was defined as either a single cell or cluster of cells in contact. Shape factor is a measurement of object sphericity, lies between 0 and 1, and is defined by the equation:
where Ψ represents shape factor, V is the object volume, and A is the measured object surface area. The resulting shape factors (where 0 is a more irregularly shaped object and 1 is a perfect sphere) were split into three categories: Rounded, Spread, and Elongated. MATLAB was used to determine the average percent of objects belonging to each category, as well as the standard errors. Results for each hydrogel composition are shown as the percent of elongated, spread, or rounded objects (cells or cell clusters) within the sampled population (n = 3 hydrogels, > 50 objects or 100 cells counted per condition).
Statistical analysis.
All values reported are averages (mean) ± standard error for each condition for three independent samples unless otherwise stated. Statistical significance was determined by a two-sided Student’s t-test after the evaluation of the variances of each population.
Supplementary Material
Acknowledgements
We acknowledge financial support from The National Science Foundation (NSF) DMR BMAT (1253906), Pew Charitable Trusts (26178), and the NSF SBE2 IGERT Program. This publication also was made possible by the Delaware COBRE programs supported by grants from the National Institute of General Medical Sciences (NIGMS) from the National Institutes of Health (NIH) (P20GM104316, 5 P30 GM110758–02). The authors would like to acknowledge the Keck Center for Advanced Microscopy and Microanalysis, the University of Delaware NMR and Mass Spectrometry Core facilities, and the Millicent Sullivan group for their use of equipment. We would specifically like to acknowledge Mark LaRue for assistance with peptide synthesis and Orlando Walker Jr. for his early contributions to the fibril width analysis.
Footnotes
Additional information
Supplementary information that accompanies this paper includes HPLC and mass spectrometry analysis for all peptides; 1HNMR analysis for each peptide fragment, each step in the PEG macromer synthesis, and the photoinitiator synthesis; CD analysis for mfCMP-1, mfCMP-1a, and mfCMP-2a; additional TEM images for mfCMP-2a as well as for mfCMP-1; quantitative image analysis details including shape factor binning histograms, characterization of hMSCs within hydrogels containing mfCMP-2a, cell shape factor analysis, and statistical analysis for cell viability and shape factor.
References
- 1.Hilderbrand AM, Ovadia EM, Rehmann MS, Kharkar PM, Guo C and Kloxin AM, Current Opinion in Solid State and Materials Science, 2016, 20, 212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown TE and Anseth KS, Chemical Society Reviews, 2017, 46, 6532–6552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murphy WL, McDevitt TC and Engler AJ, Nature Materials, 2014, 13, 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rose JC and De Laporte L, Advanced Healthcare Materials, 2018, 7, 1701067. [DOI] [PubMed] [Google Scholar]
- 5.Abraham LC, Zuena E, Perez-Ramirez B and Kaplan DL, Journal of Biomedical Materials Research Part B: Applied Biomaterials, 2008, 87B, 264–285. [DOI] [PubMed] [Google Scholar]
- 6.Ricard-Blum S, Cold Spring Harbor Perspectives in Biology, 2011, 3. [DOI] [PMC free article] [PubMed]
- 7.Pailler-Mattei C, Bec S and Zahouani H, Medical Engineering & Physics, 2008, 30, 599–606. [DOI] [PubMed] [Google Scholar]
- 8.Shoulders MD and Raines RT, Annual Review of Biochemistry, 2009, 78, 929–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wade RJ, Bassin EJ, Rodell CB and Burdick JA, Nature Communications, 2015, 6, 6639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cai L and Heilshorn SC, Acta Biomaterialia, 2014, 10, 1751–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Madl CM and Heilshorn SC, Annual Review of Biomedical Engineering, 2018, 20, 21–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Oosten ASG, Vahabi M, Licup AJ, Sharma A, Galie PA, MacKintosh FC and Janmey PA, Scientific Reports, 2016, 6, 19270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Motte S and Kaufman LJ, Biopolymers, 2013, 99, 35–46. [DOI] [PubMed] [Google Scholar]
- 14.Munoz-Pinto DJ, Jimenez-Vergara AC, Gharat TP and Hahn MS, Biomaterials, 2015, 40, 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lynn AK, Yannas IV and Bonfield W, Journal of Biomedical Materials Research Part B: Applied Biomaterials, 2004, 71B, 343–354. [DOI] [PubMed] [Google Scholar]
- 16.Liu S, Zhang H, Remy RA, Deng F, Mackay ME, Fox JM and Jia X, Advanced Materials, 2015, 27, 2783–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wade RJ, Bassin EJ, Gramlich WM and Burdick JA, Advanced Materials, 2015, 27, 1356–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baker BM, Trappmann B, Wang WY, Sakar MS, Kim IL, Shenoy VB, Burdick JA and Chen CS, Nature Materials, 2015, 14, 1262–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matera DL, Wang WY, Smith MR, Shikanov A and Baker BM, ACS Bio Sci Eng, 2019, 5, 2965–2975. [DOI] [PubMed] [Google Scholar]
- 20.Sur S, Tantakitti F, Matson JB and Stupp SI, Biomaterials Science, 2015, 3, 520–532. [DOI] [PubMed] [Google Scholar]
- 21.Ahrens CC, Welch ME, Griffith LG and Hammond PT, Biomacromolecules, 2015, 16, 3774–3783. [DOI] [PubMed] [Google Scholar]
- 22.Sahoo JK, Nazareth C, VandenBerg M and Webber M, Biomaterials Science, 2017, DOI: 10.1039/C7BM00304H. [DOI] [PubMed] [Google Scholar]
- 23.Jayawarna V, Ali M, Jowitt TA, Miller AF, Saiani A, Gough JE and Ulijn RV, Advanced Materials, 2006, 18, 611–614. [Google Scholar]
- 24.Tang JD, Mura C and Lampe KJ, Journal of the American Chemical Society, 2019, 141, 4886–4899. [DOI] [PubMed] [Google Scholar]
- 25.Yadav N, Chauhan MK and Chauhan VS, Biomaterials Science, 2020, DOI: 10.1039/C9BM01304K. [DOI] [PubMed] [Google Scholar]
- 26.Kar K, Amin P, Bryan MA, Persikov AV, Mohs A, Wang Y-H and Brodsky B, The Journal of Biological Chemistry, 2006, 281, 33283–33290. [DOI] [PubMed] [Google Scholar]
- 27.Kersteen E. a. and Raines RT, Biopolymers, 2001, 59, 24–28. [DOI] [PubMed] [Google Scholar]
- 28.Stahl PJ, Romano NH, Wirtz D and Yu SM, Biomacromolecules, 2010, 11, 2336–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cosgriff-Hernandez E, Hahn MS, Russell B, Wilems T, Munoz-Pinto D, Browning MB, Rivera J and Höök M, Acta Biomaterialia, 2010, 6, 3969–3977. [DOI] [PubMed] [Google Scholar]
- 30.Parmar PA, Chow LW, St-Pierre J-P, Horejs C-M, Peng YY, Werkmeister JA, Ramshaw JAM and Stevens MM, Biomaterials, 2015, 54, 213–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kasznel AJ, Zhang Y, Hai Y and Chenoweth DM, Journal of the American Chemical Society, 2017, 139, 9427–9430. [DOI] [PubMed] [Google Scholar]
- 32.O’Leary LER, Fallas J. a., Bakota EL, Kang MK and Hartgerink JD, Nature Chemistry, 2011, 3, 821–828. [DOI] [PubMed] [Google Scholar]
- 33.Rele S, Song Y, Apkarian RP, Qu Z, Conticello VP and Chaikof EL, Journal of the American Chemical Society, 2007, 129, 14780–14787. [DOI] [PubMed] [Google Scholar]
- 34.Gottlieb D, Morin SA, Jin S and Raines RT, Journal of Materials Chemistry, 2008, 18, 3865–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krishna OD and Kiick KL, Biomacromolecules, 2009, 10, 2626–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pires MM, Przybyla DE and Chmielewski J, Angewandte Chemie International Edition, 2009, 48, 7813–7817. [DOI] [PubMed] [Google Scholar]
- 37.Sawicki LA and Kloxin AM, Biomaterials Science, 2014, 2, 1612–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deforest CA, Sims EA and Anseth KS, Chemistry of Materials, 2010, 22, 4783–4790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Booth AJ, Hadley R, Cornett AM, Dreffs AA, Matthes SA, Tsui JL, Weiss K, Horowitz JC, Fiore VF, Barker TH, Moore BB, Martinez FJ, Niklason LE and White ES, American Journal of Respiratory and Critical Care Medicine, 2012, 186, 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dai N, Wang XJ and Etzkorn FA, Journal of the American Chemical Society, 2008, 130, 5396–5397. [DOI] [PubMed] [Google Scholar]
- 41.Kyle S, Aggeli A, Ingham E and McPherson MJ, Trends in Biotechnology, 2009, 27, 423–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anseth KS, Bowman CN and Brannon-Peppas L, Biomaterials, 1996, 17, 1647–1657. [DOI] [PubMed] [Google Scholar]
- 43.DeForest CA and Anseth KS, Annual Review of Chemical and Biomolecular Engineering, 2012, 3, 421–444. [DOI] [PubMed] [Google Scholar]
- 44.Susilo Monica E, Paten Jeffrey A, Sander Edward A, Nguyen Thao D and Ruberti Jeffrey W, Interface Focus, 2016, 6, 20150088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kureshi A, Cheema U, Alekseeva T, Cambrey A and Brown R, Journal of The Royal Society Interface, 2010, 7, 707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tibbitt MW and Anseth KS, Biotechnology and Bioengineering, 2009, 103, 655–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liang Y, Coffin MV, Manceva SD, Chichester JA, Jones RM and Kiick KL, Journal of Biomedical Materials Research Part A, 2016, 104, 113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P and Cardona A, Nature Methods, 2012, 9, 676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ovadia EM, Colby DW and Kloxin AM, Biomaterials Science, 2018, 6, 1358–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.