

SpyCas9 and its engineered variants can recognize NYGG PAMs, affecting their use for genome editing and off-target predictions.
Abstract
CRISPR technologies have overwhelmingly relied on the Streptococcus pyogenes Cas9 (SpyCas9), with its consensus NGG and less preferred NAG and NGA protospacer-adjacent motifs (PAMs). Here, we report that SpyCas9 also recognizes sequences within an N(A/C/T)GG motif. These sequences were identified on the basis of preferential enrichment in a growth-based screen in Escherichia coli. DNA binding, cleavage, and editing assays in bacteria and human cells validated recognition, with activities paralleling those for NAG(A/C/T) PAMs and dependent on the first two PAM positions. Molecular-dynamics simulations and plasmid-clearance assays with mismatch-intolerant variants supported induced-fit recognition of an extended PAM by SpyCas9 rather than recognition of NGG with a bulged R-loop. Last, the editing location for SpyCas9-derived base editors could be shifted by one nucleotide by selecting between (C/T)GG and adjacent N(C/T)GG PAMs. SpyCas9 and its enhanced variants thus recognize a larger repertoire of PAMs, with implications for precise editing, off-target predictions, and CRISPR-based immunity.
INTRODUCTION
Despite an overabundance of available CRISPR nucleases derived from CRISPR-Cas systems (1), the type II-A Cas9 nuclease from Streptococcus pyogenes (SpyCas9) dominates the landscape of CRISPR technologies. Part of the reason is historical, as SpyCas9 was one of the original single-effector CRISPR-associated (Cas) proteins to be characterized and demonstrated for programmable DNA cleavage (2). In addition, SpyCas9 exhibits robust nuclease activities in various prokaryotic and eukaryotic organisms. Last, the consensus “NGG” protospacer-adjacent motif (PAM) represents one of the simplest PAMs recognized by natural CRISPR nucleases (3). Regardless of the motivation, SpyCas9 remains the overwhelming favorite for CRISPR-based applications, including genome editing, gene regulation, base editing, imaging, and gene drives (4).
While NGG represents the consensus PAM for SpyCas9, NAG and NGA have also been validated as PAMs, albeit with reduced recognition compared to NGG (5, 6). Rather than representing a unique feature of SpyCas9, most Cas nucleases are known to recognize less preferred PAMs that extend beyond the reported consensus sequence. For instance, the Cas9 from the CRISPR1 locus in Streptococcus thermophilus (Sth1Cas9) has been assigned the consensus PAM 5′-NNAGAAW (W = A/T) but was shown to recognize NNGGAAA and NNAGSAT (S = C/G) as part of gene repression in bacteria (7, 8). Separately, the type V-A Cas12a nuclease from Acidaminococcus sp. has been assigned the consensus TTTV (V = A/C/G) but was shown to accommodate G at the −4 position and C at the −3 position as part of indel formation in human cells (9, 10). Recognizing a larger set of PAMs generally expands the targeting space of Cas nucleases, but it also increases the number of potential off-target sites (11). Therefore, determining the full set of recognized PAM sequences, particularly for widely used nucleases such as SpyCas9, remains an important endeavor that affects the broad application space in which CRISPR technologies are applied.
Here, we identified and validated a wider set of PAMs recognized by SpyCas9. The motifs were identified using an enrichment-based screen dubbed PAM-SEARCH that links a PAM to carbon catabolism in Escherichia coli. When applied to SpyCas9, the screen unexpectedly enriched for noncanonical PAM sequences within an NHGG (H = A/C/T) motif, with the canonical NGG motif exhibiting lower but substantial enrichment. An NNGG motif was mentioned in passing based on an E. coli clearance–based PAM screen (5, 12), but the motif was never subsequently validated or considered. Preferential enrichment of these sequences in our growth-based screen therefore spurred further investigation. Using a collection of assays in bacteria and in human cells, we confirmed that Cas9 could recognize the motif in different contexts, with targeting activity often paralleling that for the established NAG PAM and dependent on the identity of the first 2 nucleotides (nts) in the motif. We also explored the underlying molecular mechanism, where molecular-dynamics (MD) simulations and plasmid clearance by Cas9 variants with reduced mismatch tolerance both supported induced-fit recognition of an extended PAM rather than recognition of a canonical PAM with an imperfect R-loop. Last, we showed practical utility for the motif by separately demonstrating that these motifs are present in a prior unbiased off-target analysis and that the location of base editing could be shifted by merely designing a guide using a (C/T)GG PAM or an N(C/T)GG PAM. Thus, our findings extend the set of PAMs recognized by this widely used CRISPR nuclease as well as its enhanced variants warranting the inclusion of NHGG PAMs in off-target prediction algorithms and facilitating shifts to the window of base editing.
RESULTS
PAM-SEARCH: A positive, growth-based screen for PAMs recognized by CRISPR nucleases
We had previously developed and implemented PAM-SCANR (PAM SCreen Achieved by NOT-gated Repression), a positive fluorescence-based screen in E. coli to comprehensively determine CRISPR nuclease PAMs (8). Within the screen, a catalytically dead nuclease blocked transcription of the lacI repressor only in the presence of a functional PAM, while expressed LacI blocked transcription of the downstream gfp. The target sequence overlaps with the lacI promoter to maximize separation between the PAM library and the promoter sequence while still achieving robust repression. Although PAM-SCANR was successfully used to identify the PAM landscape for CRISPR nucleases from type I, II, and V CRISPR-Cas systems, we sought to develop a simplified version of the screen that linked recognized PAM sequences to growth rather than fluorescence-activated cell sorting (FACS).
After determining that replacing gfp with the β-lactamase gene bla did not confer the expected growth dependence on ampicillin (fig. S1), we instead used the xylAB genes responsible for the first two enzymatic steps of d-xylose catabolism (Fig. 1A). We then assessed circuit functionality in an E. coli strain lacking the endogenous xylAB genes and constitutively expressing the high-affinity transporter encoded by xylFGH (13). This configuration resulted in robust growth with d-xylose as the sole carbon source, but only when xylAB expression was induced with isopropyl-β-d-thiogalactopryanoside (IPTG) (fig. S2A). Therefore, triggering xylAB expression through CRISPR-based repression of LacI would be expected to link a recognized PAM to growth on d-xylose. We call the resulting screen PAM-SEARCH (PAM Sequences Enriched by Associating Repression with CarboHydrate consumption).
Fig. 1. A growth-based PAM screen yielded alternative motifs for the S. pyogenes Cas9.

(A) Genetic circuit for linking PAM sequences to growth on d-xylose in E. coli. We call the resulting screen PAM-SEARCH. gRNA, guide RNA. (B) Schematic of the growth-based screen. NGS, next-generation sequencing. (C) Output of the screen performed with SpyCas9. The data are displayed as a nucleotide depletion plot, PAM wheel (8), and motif plot. Each dot represents a distinct 5-nt sequence within the library. See data file S1 for an interactive version of the PAM wheel based on the Krona plot. Results from a biological replicate of the screen can be found in fig. S3 and data file S2.
We selected the well-characterized SpyCas9 (hereafter referred to as Cas9) for this growth-based screen because the nuclease’s PAM preferences have been well established (5, 12, 14, 15) and this Cas9 was not previously subjected to the PAM-SCANR screen (8). We used the same target site from PAM-SCANR given the performance of the prior screen. Supporting the compatibility of the catalytically dead nuclease with the screen, we found that cells containing dCas9 and the xylAB growth circuit with a canonical NGG PAM (AGGTG) and a targeting single-guide RNA (sgRNA) exhibited robust growth on d-xylose with and without IPTG induction (fig. S2B). By contrast, cells with a non-PAM (TTTTT) or different nontargeting sgRNAs exhibited robust growth only in the presence of IPTG (fig. S2B).
PAM-SEARCH revealed putative, noncanonical PAM sequences for SpyCas9
We next subjected Cas9 to the growth-based PAM screen. As part of the screen, we randomized the 5 base pairs (bp) flanking the sgRNA target site in the genetic circuit to capture the standard 3-nt PAM and any additional sequence variations (Fig. 1A). We then conducted the screen by culturing cells encoding the PAM library, dCas9, and the targeting sgRNA in d-xylose medium with and without IPTG induction (Fig. 1B). The resulting extracted PAM libraries were subjected to next-generation sequencing to quantify the enrichment of each potential PAM sequence relative to the IPTG control, which confers growth regardless of PAM sequence.
The screens revealed a variety of enriched sequences that captured established 3-nt PAMs while also revealing a distinct 4-nt PAM (Fig. 1C, fig. S3A, and data files S1 and S2). For the known PAMs recognized by Cas9, NGG was highly enriched as expected for the established consensus PAM, while NAG was variably enriched and NGA was not enriched, in line with their weaker recognition by Cas9. Beyond these motifs, the most enriched sequences fell within a motif represented by NHGG (H = A/C/T), with some sequences exhibiting enrichment scores even greater than those for NGGH sequences. The first 2 nt of the motif correlated with the degree of enrichment, with consistently higher enrichment scores if A/C/T is at the first position of NAGG and generally higher enrichment scores if A/C is at the first position of NYGG (Y = C/T). These motifs were unexpected given that the PAM for Cas9 is always reported as 3 nt, and a 4-nt motif representing NNGG was at most noted but never validated as part of two PAM screens both based on plasmid clearance in E. coli (5, 12).
Beyond observing the NHGG motif, we explored why the associated sequences were more highly enriched than canonical NGG sequences. We first ruled out an intermediate level of xylAB expression for optimal growth, as increasing IPTG concentrations only served to increase the final turbidity of a culture harboring the xylAB growth circuit and grown on d-xylose (fig. S2A). We then evaluated whether IPTG induced similar levels of growth for different PAM sequences, which could skew the enrichment scores (fig. S3D). We found that NYGG sequences yielded reduced turbidities in the presence of IPTG compared to that in the absence of IPTG, resulting in ratios that were significantly higher than a baseline NGG sequence (P = 0.00014 and 0.037 for CTGGA and CAGGT, respectively, compared to AGGTG using a two-tailed t test assuming equal variance, n = 3 to 4). These results therefore suggest that the use of IPTG to normalize the library played a role in the higher enrichment scores for NHGG sequences.
Identified motifs validated as suboptimal PAMs in E. coli and human cells
As NHGG sequences were enriched over the consensus NGG PAM, we asked to what extent Cas9 could recognize different NHGG sequences outside the context of our PAM screen. We focused on three such NHGG sequences (TAGGG, CCGGG, and GTGGC) compared to the consensus NGG PAM (AGGTG) and the less recognized NAG PAM (AAGTG and GAGCG) (Fig. 2, A to D). GAGCG was specifically selected as one of the few NAGH sequences that were modestly enriched in the screen. We first evaluated growth with the xylAB growth circuit with cloned PAM sequences. All three NHGG sequences resulted in turbidity ratios that were at least equal to and statistically indistinguishable from that of the NGG sample (P = 0.054 to 0.70 compared to AGGTG using a two-tailed t test assuming equal variance, n = 3) (Fig. 2A), in line with the PAM screen.
Fig. 2. Validation of individual PAM sequences in E. coli and U2OS cells.

Assessing individual PAM sequences in E. coli (A) using the growth-based genetic circuit and measuring turbidity of the culture, (B) using the growth-based circuit with gfp replacing xylAB and measuring GFP fluorescence, (C) through direct transcriptional repression of gfp under the control of the lacZ promoter by measuring GFP fluorescence, and (D) through plasmid clearance by targeting the same construct from (B) with catalytically active SpyCas9. A nontargeting Sth1 sgRNA served as the nontargeting control. Assessing individual PAM sequences by targeting (E) gfp in U2OS-GFP cells and (F) genomic sites in HEK293T cells. Indels were measured with TIDE (17), CRISPResso analysis (18), and flow cytometry analysis. The NYGG data are the same in both (E) panels. Indel formation calculated by TIDE is in reference to a nontargeting control sample. Values are based on independent experiments starting from separate E. coli colonies or separate cell culture wells. Bars represent the mean of each set of triplicate or quadruplicate measurements. Statistical significance was calculated in comparison to the AGGTG sequence for (A) to (D) or the nontargeting sgRNA control in (E) using a two-tailed t test assuming equal variance with cutoffs of 0.05 (*) or 0.01 (**).
We then conducted a series of experiments that progressively moved away from the PAM screen and toward traditional uses of CRISPR technologies. First, we replaced xylAB with gfp in the growth circuit, decoupling PAM recognition from cellular growth. Fluorescence measurements with the resulting constructs yielded strong gfp induction for the NHGG and NAGH sequences enriched by the screen in comparison to a nontargeting sgRNA that was slightly but significantly lower than that of the NGG sequence (1.5- to 2.1-fold, P = 3.3 × 10−6 to 1.7 × 10−4, two-tailed t test assuming equal variance, n = 3) (Fig. 2B). The values for the enriched sequences were modestly lower than those for NGG but significantly higher than those for the poorly enriched NAGH sequence (224- to 341-fold, P = 6.6 × 10−7 to 3.7 × 10−5, two-tailed t test assuming equal variance, n = 3), suggesting that linking PAM functionality to d-xylose catabolism at least partially contributed to the enrichment of NHGG sequences over the canonical NGG motif in our screen. Next, we evaluated recognition of NHGG PAM sequences outside the context of the derepression circuit through two approaches: directly repressing gfp expression again using dCas9 through a different target site (Fig. 2C) and evaluating plasmid clearance with active Cas9 using the original target site (Fig. 2D). In both experiments, the NGG sequences yielded the highest average clearance activity, with only one NHGG sequence (TAGGG) that exhibited statistically indistinguishable activity by plasmid clearance (P = 0.0999, two-tailed t test assuming equal variance, n = 3), while the remaining NHGG and NAGH sequences performed similarly. The trends were similar whether relying on the catalytically dead or active Cas9, arguing against differential recognition of the NHGG sequences for DNA binding versus cleavage. As these last experiments in E. coli represent the largest deviation from the PAM screen and better approximate typical uses of Cas9, we conclude that NHGG sequences can be recognized as nonoptimal PAMs similarly to NAG in E. coli. The enrichment of some NHGG sequences over NGG within the PAM screen instead appeared to be an artifact of the screen tied to d-xylose catabolism and targeting within the derepression construct.
Beyond the validation experiments in E. coli, we asked to what extent Cas9 could recognize NYGG sequences in human cells as part of indel formation, where NYGG does not overlap with the traditional PAMs (NGG, NAG, and NGA) for this nuclease. We used a previously reported assay in which U2OS.EGFP cells expressing a destabilized version of enhanced green fluorescent protein (EGFP) (16) are transiently transfected with a plasmid encoding Cas9 and an sgRNA designed to target the gfp open reading frame. Editing activity was then assessed by flow cytometry analysis and sequencing of the target site 72 hours after transfection (Fig. 2E and fig. S4). Flow cytometry analysis revealed that all but one of the tested targets with a flanking NYGG motif (CCGGG as the exception) yielded significantly more GFP-negative cells than the nontargeting control (P = 0.17 for CCGGG, P = 2.1 × 10−5 to 0.0054 for the other five compared to the nontargeting sgRNA sample using a two-tailed t test assuming equal variance, n = 4) (Fig. 2E). The efficiency of GFP disruption varied across the tested targets, with one (GTGGC1) being statistically indistinguishable from the NGGH target (P = 0.11, two-tailed t test assuming equal variance, n = 4) and all but two (CCGGG and CTGGG) being statistically indistinguishable from the NAGH target (P = 0.067 to 0.98, two-tailed t test assuming equal variance, n = 4). Similar trends were observed when assessing indel frequencies determined through Tracking Indels by DEcomposition (TIDE) analysis (17) and amplicon sequencing. The measured indel frequencies were consistently lower for TIDE than for next-generation sequencing, which we attribute to TIDE’s limited ability to detect large indels as well as potential noise in the Sanger sequencing chromatograph used to establish the baseline for editing (17). We also tested target sites shifted by 1 bp from each other: one site with a YGG PAM and the shifted site with an NYGG PAM (Fig. 2E, right). In two of three sites tested, the NYGG site yielded significantly more GFP-negative cells than the nontargeting sgRNA control (P = 1.5 × 10−4 to 5.4 × 10−3, two-tailed t test assuming equal variance, n = 4) but at values 1.4- to 3.4-fold lower than the corresponding YGG sites. We observed similar trends when quantifying indel formation as before. As part of the deep sequencing, we evaluated whether the types of indels within the next-generation sequencing results diverged between NGG and NYGG PAMs using the online algorithm CRISPResso (18), although we did not notice any consistent differences (fig. S4D). Last, we targeted NYGG and corresponding YGG sites from EMX1, FANCF, and human embryonic kidney (HEK) site 3 (within the long noncoding RNA locus LINC01509) in HEK293T cells (Fig. 2F). Targeting the NYGG sites resulted in measurable indel formation from 2.1 to 35.2% at all of the tested sites, with each measurement above the threshold of significance for TIDE analyses. Therefore, Cas9 can target genomic sites in mammalian cells using an NYGG PAM, albeit with efficiencies lower than those for an NGG PAM.
The N(A/C/T)GG motif is present in previously published datasets
While Cas9 is normally thought to recognize a 3-nt PAM, numerous studies have applied unbiased, high-throughput screens with Cas9 that output recognized PAM sequences extending to four or more nucleotides (5, 12, 15). These datasets include not only PAM screens but also spacer acquisition assays and identification of off-target cleavage sites across a genome (19, 20). We thus investigated to what extent the NAGG and NYGG motifs were present in these published datasets.
We first evaluated previous high-throughput PAM screens performed with Cas9 that used PAM libraries with at least five randomized base pairs flanking the target (fig. S5, A to E) (5, 12, 15). Two studies applied screens based on depletion through plasmid clearance in E. coli and had noted but not validated an NNGG motif within the datasets (5, 12). Breaking the resulting depletion scores into different motifs confirmed modest to strong depletion of sequences associated with NAGG and NYGG, yet it also revealed strong influences of the first nucleotide of each motif. Another study applied an in vitro screen based on sequencing cleaved DNA (15). This study did not note either motif, although our own analysis of the dataset revealed that NYGG is partially enriched at the higher applied concentration of the ribonucleoprotein (RNP) complex, corresponding to weaker recognition (fig. S5E).
We next evaluated a high-throughput screen for spacers acquired by the type II-A CRISPR-Cas system from S. pyogenes (19). The screen revealed NGG as the overwhelmingly preferred PAM adjacent to newly acquired spacers (Fig. 3A, top). However, other motifs appeared, albeit at much lower frequencies of acquisition. Within these motifs, NAGG and NYGG were not only present but also appeared more frequently than either NAGH or NGAN—particularly when accounting for their frequency in random DNA (Fig. 3A, bottom). The acquisition dataset therefore provided a distinct verification of the NAGG and NYGG motifs and could serve as a unique means to identify PAMs for type II CRISPR-Cas systems.
Fig. 3. Analysis of previously published, high-throughput datasets for Cas9.
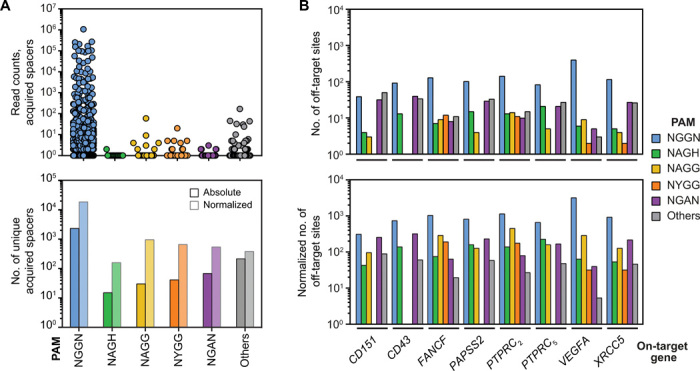
(A) Analysis of a previous high-throughput spacer acquisition assay in Staphylococcus aureus RN4220 (19). Acquired spacer sequences were mapped to the phage genome to determine the frequency of sequences within different motifs (19). Each dot represents the number of unique spacers that were acquired and mapped to a protospacer adjacent to the displayed PAM motif. Bars indicate the total number of unique, acquired spacers adjacent to the indicated motif. The normalized numbers represent the total number normalized to the frequency of the motif in random DNA. (B) Analysis of a previous genome-wide off-target screen (20). The assays were previously conducted in vitro with isolated human genomic DNA and different concentrations of the Cas9:sgRNA RNP complex. The PAMs flanking each identified off-target cleavage site were sorted into different motifs and grouped on the basis of the indicated on-target site. The values represent the total number of distinct off-target sites for each sgRNA that adhere to a given motif that were identified as part of the screen. The normalized numbers represent the total number normalized to the frequency of the motif in random DNA.
Last, we turned to unbiased off-target analyses used to identify genomic sites bearing homology to the guide sequence that can undergo cleavage and editing. While the exact methodology used for these screens has varied, all involve either inserting sequence tags into cleaved sites in vivo or cleaving genomic DNA in vitro. Within the screens performed using Cas9, the vast majority only reported 3 nt for the identified PAMs. Fortunately, one recent study that relied on biochemical cleavage of genomic DNA (SITE-Seq) (20) offers an open repository of annotated and analyzed sequencing data, which allowed us to evaluate the extent to which the screen identified off-target sites flanked by NYGG or NAGG motifs across eight different target sites in the human genome with different concentrations of Cas9. Of the eight targets previously evaluated with SITE-Seq, four included off-target sites flanked by the NYGG motif. (Fig. 3B, top). Furthermore, for each of these targets, the number of off-target sites flanked by NYGG was similar to those flanked by NAGH when normalizing for the frequency of each motif in random DNA (Fig. 3B, bottom). Therefore, NYGG can be associated with off-target sites detected as part of unbiased screens. In further support, initial manual curation of off-targets flanked in two other screens (GUIDE-Seq and CIRCLE-Seq) (21, 22) revealed numerous sites flanked by NYGG motifs. Separately, off-target sites with the NAGG motif were present for all but one of the SITE-Seq targets. For five of these eight targets, the number of off-target sites was notably higher for NAGG than for NAGH after normalizing for the frequency of each motif in random DNA, suggesting some preference for NAGG over NAGH by Cas9 for imperfect targets (Fig. 3B). In total, we conclude that the NHGG motif is present in different high-throughput screens, further supporting the capacity of Cas9 to recognize these sequences as PAMs in ranging contexts.
MD simulations suggest induced-fit recognition of N(C/T)GG-flanked targets over recognition of an NGG PAM with a bulged R-loop
Given the evidence that Cas9 can recognize and cleave targets flanked by NHGG sequences, we asked how Cas9 is recognizing these sequences at the molecular level. To begin answering this question, we used MD simulations to predict molecular interactions between Cas9 and an NGG, NAG, or NYGG PAM and to determine whether these interactions could lead to stable R-loop formation and target cleavage. As a starting point, we selected the crystal structure of Cas9 complexed with an R-loop of a TGGC-flanked target captured in the precleavage state [Protein Data Bank (PDB) code: 5F9R]. To simulate NAG and NYGG target recognition, both strands of TGGC were mutated to TAGC or TCGG, respectively (see Materials and Methods for details). For each simulation, we calculated the binding free energies and identified interactions between the PAM-interacting (PI) domain and the PAMs. The simulations predicted that bi-arginine residues in the PI domain (R1333 and R1335) form four and three hydrogen bonds, respectively, with the last 2 nt of the NGG PAM (ΔG = −15.6 ± 1.0 kcal/mol; table S1) and NAG PAM (ΔG = −8.5 ± 1.8 kcal/mol; table S1) as well as a salt-bridge interaction between R1335 and E1219 (Fig. 4A). For NYGG, one of the bi-arginine residues (R1333) switched between fully engaging the first G in the motif and partially engaging the same nucleotide along with E1219 (Fig. 4A). This switching resulted in an intermediate binding affinity (ΔG = −13.3 ± 2.7 kcal/mol; table S1) between that for NGG and NAG, implying that Cas9 can energetically favor interactions with NYGG at least over NAG.
Fig. 4. Induced-fit recognition of an NYGG PAM by Cas9 revealed by MD simulations.
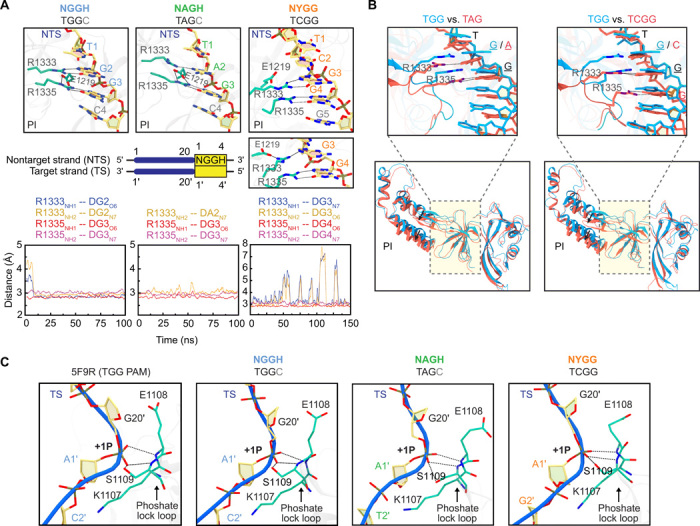
(A) Zoomed-in view of the TGG, TAG, and TCGG PAMs recognized by the Cas9 PI domain. The protein residues and nontarget DNA strand nucleotides are represented as a stick model and colored by cyan and yellow, respectively. The dashed lines denote hydrogen bonds and salt bridges. Two panels are shown for NYGG, where the simulations predicted R1333 switches between fully engaging the first G in the PAM and partially engaging this same nucleotide and E1219. Below each panel is the temporal evolution of the hydrogen-bonding distances between the bi-arginine and the PAM calculated from one representative simulation trajectory. (B) Structural superposition of the conformations for TAG and TCGG (cyan) onto TGG (red). The cleft of the PI domain accommodating the PAM and its flanking sequences is highlighted by a dashed rectangle and magnified in the above panel with the PAM. For clarity, dashed lines are only used to denote hydrogen bonds associated with recognition of NAG and NYGG. (C) Location of phosphate lock loop maintained across simulations. The corresponding interactions in the crystal structure (PDB code: 5F9R) used for the simulations are shown for comparison.
We next superimposed the conformations with the three PAMs and determined the main structural differences in the PI domain of Cas9 (Fig. 4B). For all conformations, the phosphate lock loop was engaged with the phosphodiester group at the +1 PAM position on the target strand (Fig. 4C), indicating that recognition of all three PAMs would lead to R-loop formation and target cleavage (23). Comparing NAG to NGG, the C-terminal cleft of the PI domain underwent a prominent lateral shift, while the bi-arginine and PAM nucleotides were shifted downward by a root mean square displacement (RMSD) of ~2.8 Å. Furthermore, the side chain of R1335 adopted an extended pose for NAG rather than a bent pose for both NGG and NYGG. Comparing NYGG to NGG, the C-terminal cleft underwent an even more pronounced shift, while the bi-arginine slid downward by an RMSD of ~3.8 Å to interact with the G’s in the NYGG PAM. The downward-shifted arginines for both NAG and NYGG resembled the induced fit exhibited by Cas9 variants recognizing alternative PAMs (24, 25).
While our simulations support recognition of an extended NYGG PAM through conformational changes in Cas9, an alternative hypothesis is that the R-loop formed between the guide and target DNA bulges to mimic an NGG-flanked target. Accordingly, Cas9 is known to accommodate bulged or mismatched targets to varying degrees depending on the location and type of bulge (26). One scenario is that the N of NYGG bulges out to condense the distance between a perfect R-loop and a standard NGG PAM. However, this scenario is less likely given that the most stable simulated structure for the NYGG PAM positioned the phosphate lock loop at the N rather than the Y (Fig. 4C).
Another scenario is that a bulge forms at the PAM-proximal end of the target strand, thereby coupling formation of an imperfect R-loop with recognition of a YGG PAM (fig. S6A). The most stable version of this R-loop would involve base pairing between the N of NYGG and the 20th nucleotide of the guide, forming a 1-nt bulge at the PAM-proximal nucleotide of the target strand. In practice, this could occur when the 20th nucleotide of the guide and the N of NYGG are the same base—equivalent to the PAM-proximal base, and the next downstream base on the target strand are the same. We explored this scenario in the context of the MD simulations by inserting a G nucleotide between the 19th position (C) and 20th position (G) in the target strand of the NGG structure and then repeating the simulations (see Materials and Methods for details) (fig. S6B). Of four independent simulations, three yielded conformations in which the bi-arginine residues completely lost hydrogen bond contacts with NGG or the phosphate lock loop was disengaged (fig. S6B), either of which would prevent R-loop formation and DNA cleavage. The fourth simulation yielded an energetically favorable interaction between the bi-arginine residues and NGG (table S1) as well as engagement of the phosphate lock loop. However, the bulged section of the target strand was in a highly kinked orientation that was distinct from all other simulations that we conducted and would be expected to be structurally unstable (fig. S6B). These results argue against recognition of NYGG-flanked targets through a bulged R-loop coupled to recognition of a canonical NGG PAM.
Targeting of N(C/T)GG-flanked targets by engineered variants of SpyCas9 supports induced-fit recognition of an extended PAM
Our simulation results favored recognition of NYGG-flanked targets through a conformational change in Cas9 rather than formation of bulges in the DNA target. One ramification is that high-specificity variants of Cas9 known to reject imperfect targets (27–29) would be expected to recognize NYGG-flanked targets similarly to the wild-type (WT) nuclease. To evaluate this experimentally, we selected three recently reported variants of Cas9 engineered to exhibit reduced off-targeting [eSpyCas9(1.1), SpyCas9HF1, and eSpyCas9(1.1)HF1] (27–29). We also assessed the D1135E variant reported to exhibit reduced recognition of non-NGG PAMs compared to the WT Cas9 (Fig. 5A) (12).
Fig. 5. Recognition of the NYGG motif by engineered SpyCas9 variants.
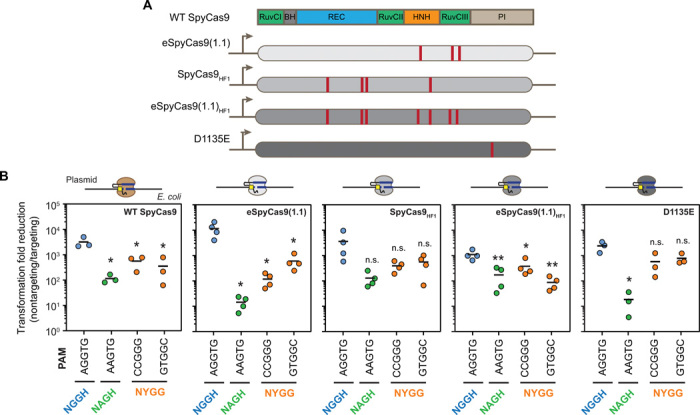
(A) Mutations present in the four tested variants eSpyCas9(1.1), SpyCas9HF1, eSpyCas9(1.1)HF1, and D1135E. (B) Assessing individual PAM sequences in E. coli through plasmid clearance. See Fig. 2D for details. For SpyCas9HF1 and eSpyCas9(1.1)HF1, transformed cells were grown in liquid culture without selecting for the transformed plasmid between recovery from electroporation and plating on solid agar. See fig. S6 (C and D) for plasmid clearance with SpyCas9HF1 and eSpyCas9(1.1)HF1 in the absence of outgrowth without selection. The data presented for WT Cas9 were collected separately from those in Fig. 2D. Values represent measurements from independent experiments starting from separate E. coli colonies. Bars represent the mean of each set of triplicate or quadruplicate measurements. Statistical significance was calculated in comparison to the AGGTG sequence for each variant using a two-tailed t test assuming equal variance with a maximal value of 0.05 (*) or 0.01 (**).
We assessed PAM recognition with these variants by performing the plasmid clearance assay in E. coli, as it provided a simple readout of cleavage activity by Cas9 and allowed us to directly compare the recognition of NYGG relative to NGG and NAG using the same target sequence (Fig. 5B). We specifically tested two sequences that adhered to the NYGG motif (CCGGG and GTGGC) downstream of an identical target site. The 20th nucleotide of the target sequence is a C, which allowed us to directly compare sequences that theoretically could (CCGGG) or could not (GTGGC) generate a bulged R-loop. We initially found that SpyCas9HF1 and eSpyCas9(1.1)HF1 exhibited weak plasmid clearance activity (fig. S6, C and D), potentially reflecting how the HNH domain of the HF1 variants slowly transitions between inactive and active states (27). We therefore altered the sensitivity of the plasmid clearance assay similar to prior work by culturing the transformed cells in liquid media without selecting for the transformed plasmid before plating (30). Performing the standard clearance assay with eSpyCas9(1.1) and the D1135E variant and the modified clearance assay with SpyCas9HF1 and eSpyCas9(1.1)HF1 revealed that the two NYGG motifs led to clearance of the target plasmid by all three engineered variants at efficiencies between that of NGG and NAG (Fig. 5B). Separately, the D1135E variant demonstrated reduced plasmid clearance only for NAG, suggesting that this variant recognizes NYGG differently from NAG. The results provide further evidence against recognition of NYGG-flanked targets by Cas9 through formation of a bulged R-loop.
N(C/T)GG PAM sequences generally preserve the DNA cleavage site and allow for a shifted base editing window
We next asked how the introduction of an additional nucleotide between the target of the guide sequence and the NGG PAM affects the location of DNA cleavage. To directly identify the cleavage site, we digested linear double-stranded DNA (dsDNA) with a preformed Cas9:sgRNA RNP complex in vitro and applied run-off Sanger sequencing to determine the cut site of an NGG and NYGG target (Fig. 6A). We found that the target strands of the NGG and NYGG targets were both nicked principally between the 17th and 18th nucleotides. Nicking of the nontarget strand was more variable, consistent with previous work targeting short, linear DNA substrates in vitro (2). The nontarget strand of the NGG target was nicked principally between the 17th and 18th nucleotides, resulting in blunt cleavage of the dsDNA. In contrast, the nontarget strand of the NYGG target exhibited a broader spectrum of nicking locations and was principally nicked between the 13th and 14th nucleotides. Therefore, the cleavage site is principally dictated by the location of the guide rather than the two G’s in the PAM, although an NYGG could lead to more variable nicking of the nontarget strand.
Fig. 6. Assessing the cleavage site and base editing location for NYGG-flanked targets.

(A) Mapping the in vitro DNA cleavage site. Digested fragments were subjected to run-off Sanger sequencing. Chromatographs were inverted to match the indicated strand’s sequence. Horizontal bars indicate A-tailing from sequencing. (B) Expected impact of using NGG versus the adjacent NYGG on base editing. (C) Determining the base editing location in E. coli. BE3 (31) was coexpressed with an sgRNA targeting the poly-C sequence flanked by different PAMs on a multicopy plasmid. C-to-T conversion was quantified with the Sequalizer algorithm (34) in comparison to a nontargeting control. Error bars represent the SD from three independent experiments starting from separate E. coli colonies. (D) Evaluating base editing efficiency when targeting the genome of HEK293T cells. C-to-T conversion was quantified with EditR on each of the three biological replicates. See Materials and Methods for how C-to-T edits (%) were calculated. Boxes outlined in black represent (C), while white spaces represent A/G/T in the target sequence. Gray boxes indicate values below the statistically significant editing threshold. Raw editing values and the EditR output can be found in fig. S7B.
The relationship between the cleavage site and two G’s in each PAM led us to ask how these different PAMs affect the location of base editing. Base editors modify one or a few nucleotide bases within a defined window often using a mutated Cas9 that only nicks the target strand (D10A) and fused to a single-stranded DNA–modifying domain (31). The editors act on a defined stretch of the nontarget strand displaced as part of R-loop formation, where the length of the editable sequence can vary between a single, specific base pair to approximately 6 bp depending on the selected nuclease, modifying domain, and target sequence (32). Because the nicking site in the target strand was determined by the guide location for both the NGG and NYGG target (Fig. 6A), we hypothesized that shifting the guide sequence by 1 bp with a YGG or adjacent NYGG PAM would, in turn, shift the location of editing by the same distance (Fig. 6B). If so, this simple strategy would expand the range of targetable sequences for the many existing Cas9-derived base editors (31, 33).
To test our hypothesis, we used the base editor BE3, which converts a cytidine to thymidine on the nontarget strand primarily within a 4-bp window (31). We inducibly expressed BE3 and an sgRNA targeting a C-rich sequence with different flanking PAM sequences on a plasmid vector. Paired YGG and NYGG PAMs were used to simulate shifting the distance between the two G’s in the PAM and the target without changing the target sequence. We then assessed the frequency of C-to-T transitions at different time points after inducing expression of the base editor using the Sequalizer algorithm (34) (Fig. 6C and fig. S7A). In line with our hypothesis, we observed primary editing over a ~4-bp stretch that was different distances from the two G’s in the PAM sequence: 13 bp for the YGG PAM and 14 bp for the NYGG PAM. Selecting the NYGG PAM versus a YGG PAM thus offers a simple strategy to shift the base editing window with SpyCas9.
As an extension, we evaluated the extent to which BE3 could perform base editing at NYGG target sites in the genome of HEK293T cells. Following transient transfection of plasmids encoding BE3 and different sgRNAs, we detected measurable C-to-T edits within the standard base editing window for three of the five NYGG sites. The frequencies of editing at these sites varied between 0.2 and 26.3% above the threshold of significance determined by EditR (35), while nearly 40% C-to-T editing above the threshold of significance was detected for the NGG site (Fig. 6D and fig. S7B). We thus conclude that the noncanonical NYGG PAM can be used for base editing at endogenous sites in human cells.
DISCUSSION
In this work, we developed and applied a positive, growth-based PAM screen (PAM-SEARCH) to characterize the PAM landscape recognized by CRISPR nucleases. PAM-SEARCH relies on DNA binding rather than cleavage to identify recognized PAM sequences and therefore offers a distinct means of determining PAM preferences compared to other existing techniques (5, 12, 15). PAM-SCANR, the predecessor of PAM-SEARCH, also offered a positive output through DNA binding. However, PAM-SEARCH offered a number of unique benefits, including a simplified workflow that relies on growth rather than cell sorting and roughly 10-fold higher enrichment scores that could be further boosted with an extended period of selective growth. PAM-SEARCH should thus be a useful addition to the growing toolbox of high-throughput techniques for PAM determination.
One unexpected outcome from applying PAM-SEARCH to Cas9 was the preferential enrichment of sequences that fell within an NHGG motif. Our follow-up experiments in E. coli suggested that the enrichment was due, in part, to reduced growth for NHGG PAMs in the presence of IPTG, our control condition for the screen, and the effect was linked to targeting the lacI promoter within the inverted-repression circuit. Further work will be required to interrogate the exact mechanism, which we speculate could be due to specific PAM sequences influencing the promoter activity of lacI or recognition of an endogenous transcriptional regulator. Future work could also evaluate the extent to which PAM-SEARCH preferentially enriches suboptimal PAMs, particularly for PAMs lacking G’s.
When interrogating how Cas9 could recognize NYGG-flanked targets, we considered two models: induced-fit recognition of an extended PAM or recognition of a canonical NGG PAM coupled to a bulged R-loop. We collected multiple lines of evidence arguing for the induced-fit model. The MD simulations predicted stable recognition of the two shifted G’s and engagement of the phosphate lock loop by Cas9 at the N position of NYGG, whereas simulated structures with a bulged target strand within the NGG conformation lacked one of the two features necessary for R-loop formation or was predicted to be structurally unstable. We also found that engineered Cas9 variants that reject mismatches within the R-loop recognized NYGG sequences. Last, the “bulged R-loop” model would imply that base pairing between the 20th nucleotide in the guide and the 1st nucleotide of the NYGG PAM on the target strand would be critical for recognition. However, our collective experimental data in E. coli and human cells contained ample instances (14 of 23) in which an NYGG target incapable of forming this base pair led to moderate or high targeting activity (table S1). The fraction of instances in which an NYGG target was capable of forming this base pair was higher (10 of 14) though, suggesting that the bulged R-loop model cannot be entirely ruled out and may contribute to target recognition. Future work could resolve the crystal structure of Cas9 bound to an NYGG-adjacent target to fully determine how Cas9 recognizes these targets.
The potential ability of Cas9 to recognize sequences effectively representing a shifted consensus PAM begs the question whether other CRISPR nucleases exhibit this same ability. One prior study of the Cas9 from S. thermophilus LMG18311 reported similar flexibility by accommodating an extra N between the target and its canonical NNGTAAA PAM (36), where a high-throughput PAM screen could help distinguish between a shifted PAM and an unshifted but less preferred PAM. Separately, biochemical characterization of the Cas9 from Neisseria meningitidis showed no flexibility between the target and the canonical NNNNGATT PAM (37). Combining these studies with our work would suggest that some but not all Cas9s can accommodate an extra nucleotide between the target and canonical PAM. More direct experiments will be required to assess the prevalence of this phenomenon across Cas9s and other CRISPR nucleases and whether it applies to nucleases commonly used for genome editing and other applications (4). The standard visualization schemes for PAM screens (e.g., sequence logo, PAM wheel) can also mask shifted PAMs, although this could be remedied by supplementing the visualization scheme with motif plots (e.g., Fig. 1C and fig. S3A) that include shifted core sequences or motifs.
Through this work, we identified two technological ramifications for SpyCas9 recognizing NYGG PAMs: off-target predictions and base editors. A range of tools for predicting off-target sites in silico have proliferated in the past few years, where virtually all the tools predefine the Cas9 PAMs as NGG or NAG (38). Our results would suggest that NYGG PAMs can yield targeting levels similar to that of NAG PAMs and thus should be integrated into these algorithms. Separately, we showed through plasmid editing in E. coli that selecting NYGG versus YGG as the PAM can shift the stretch of base pairs targeted for base editing with Cas9 by 1 bp. The shift emerged from a 1-bp difference in the guide targets (relative to the two G’s in the PAM), where the guide target still dictates the stretch of single-stranded DNA that undergoes base editing. A 1-bp shift may seem small, but the shift could prove particularly critical when using editors with narrowed windows (33) and aiming for just one or two specific base pairs. In total, the expanded set of recognized motifs has both positive and negative consequences for the future implementation of Cas9 and its many engineered variants.
MATERIALS AND METHODS
Strains, plasmids, gene fragments, and oligonucleotides
Table S1 contains all strains, plasmids, gene fragments [Integrated Data Technologies (IDT) gBlocks], and oligonucleotides used in this work. The xylose knockout strain was generated by amplifying cat cassette from the previously used ∆xylAB Pcon-xylFGH strain and recombining it into BW25113 ∆CRISPR ∆lacI-lacZ using λ-red–mediated recombineering in NM500. Successful recombinants were screened by colony polymerase chain reaction (PCR) and sequenced. The cat resistance in each recombined mutant was excised by flippase recognition target (FRT)–mediated recombination, resulting in BW25113 ΔCRISPR1 ΔlacI-lacZ ∆xylAB Pcon-xylFGH. This strain was used for screening and validation for all E. coli experimental datasets.
Plasmid design and construction
The growth-based circuit (CB618) used as part of PAM-SEARCH was created by amplifying xylA through xylB from E. coli K-12 MG1655, amplifying the original PAM-SCANR backbone to replace the gfp gene with xylAB using Gibson cloning. Silent point mutations were introduced to remove two Aat II cut sites in xylA and xylB with Q5 mutagenesis. To remain consistent with previous work, the AGGT and AAGT PAMs were inserted into the PAM-SEARCH_xylAB growth circuit (CB621 and CB620, respectively) downstream of the target sequence. The 5-nt PAM library (CB622) was inserted into the target plasmid backbone (CB618) with Q5 mutagenesis (New England Biolabs, catalog no. E0554S). Additional PAMs were inserted into the PAM-SEARCH_xylAB growth circuit (CB620, CB621, CB625, CB627 to CB629, CBS639, and CBS1365 to CBS1368), the PAM-SCANR gfp-based construct (CB630 to CB632 and CB634), and the direct repression gfp circuit (CB635, CB637 to CB639, CB641, and CB642) with site-directed mutagenesis. The guide targeting the direct repression circuit (CB643) was inserted as annealed oligos into the sgRNA scaffold (CB454). The plasmids expressing the SpyCas9 sgRNA (CB455) and Sth1Cas9 sgRNA (CB450) were created in our prior work (8) and used here. The guide sgRNA (CB643) for the direct repression experiments was added by inserting annealed oligos into the sgRNA scaffold construct that also encodes for dCas9. The nontargeting Sth1Cas9 sgRNA (CBS157) construct was created by cleaving the gene fragment gbDC09, containing the St1Cas9 sgRNA, and inserting it into the backbone of CB624. The nontargeting SpyCas9 sgRNA (CBS778) was generated by inserting the nontarget spacer and sgRNA sequence into the CBS157 backbone via Gibson assembly.
The WT SpyCas9 gene (Addgene no. 42876) was used for plasmid clearance experiments and also as a backbone plasmid (CB339) for the high-fidelity variants. The high-fidelity SpyCas9 genes (Addgene nos. 89961, 101215, and 101217) were inserted into the backbone SpyCas9 plasmid (CB339) with Gibson assembly to produce the high-fidelity SpyCas9 plasmids (CBS154 to CBS156). The D1135E variant (CBS455) was generated via Q5 mutagenesis of CB339. Gibson assembly was used to insert a Bbs I gfp dropout in the spacer region of the plasmid (CBS1203) containing the sgRNA and human codon-optimized SpyCas9 for mammalian expression (Addgene no. 42230). This cloning vector was then digested with Bbs I, and spacers were inserted by phosphorylating and annealing oligos with complementary overhangs to produce the plasmids used to target regions through EGFP in U2OS (CBS138 to CBS145 and CBS147 to CBS153) and Emx1, FANCF, and HEK site 3 in HEK293T (CBS881 to CBS889, CBS891, CBS948, CBS1074 to CBS1082, CBS1084, and CBS1085). The BE3 plasmid and puromycin/EGFP plasmids were obtained from Addgene (nos. 73021 and 45561, respectively). The Cas9 expression cassette was removed from CBS1203 via Q5 mutagenesis to create the cloning vector for spacer insertion into the sgRNA handle for mammalian expression for the BE experiments (CBS483). This cloning vector was then digested with Bbs I, and spacers were inserted by phosphorylating and annealing oligos with complementary overhangs to produce the plasmids used to target regions in Emx1, FANCF, and HEK site 3 in HEK293T (CBS541, CBS551, CBS564, CBS565, CBS1031, and CBS1135).
To generate the E. coli base editor expression plasmid, the BE3 base editor gene (31) was constructed by Gibson assembly using gbSPC054, gbSPC055, and CB453. The operator sequences recognized by LacI and AraC were introduced by Gibson assembly from pBAD33 and SPC120. The promoter sequence for the base editor was introduced by Gibson assembly using gb220. The ribosome-binding site (RBS) of the base editor, the araC promoter, and the H840A mutation to change dCas9 into nCas9 was introduced by Q5 site-directed mutagenesis.
E. coli growth conditions
All E. coli strains used for data generation were derivatives of E. coli BW25113. They were cultured at 37°C and 220 RPM in LB medium [NaCl (10 g/liter), yeast extract (5 g/liter), and tryptone (10 g/liter)] in 14-ml round-bottom polypropylene or glass tubes in volumes of 5 ml or less. Larger volumes were grown in Erlenmeyer glass flasks with baffles. M9 minimal medium (1× M9 base salts, 2 mM MgSO4, and 0.1 mM CaCl2) was used as the base for all growth assays, with concentrations of casAA and carbon source specific to each experiment. Phosphate-buffered saline (PBS; 1×) was used for washing cells for the xylose growth experiments. M9 glycerol medium consists of 1× M9 base salts, 2 mM MgSO4, 0.1 mM CaCl2, 0.4% glycerol, and 0.2% casamino acids. M9 xylose medium consists of 1× M9 base salts, 2 mM MgSO4, 0.1 mM CaCl2, 0.2% d-xylose, and 0.02% casamino acids. Plasmids were maintained with ampicillin, chloramphenicol, and/or kanamycin in concentrations of 100, 34, and 50 μg/ml, respectively, on both solid and liquid medium.
d-xylose growth assay
Single colonies were inoculated overnight in LB medium with appropriate chloramphenicol and kanamycin antibiotics. The following day, 1.5 ml of cells was spun down and washed three times with 1 ml of 1× PBS. After washing, cells were resuspended in 1 ml of M9 base media. The ABS600 (absorbance at 600 nm) of the culture was measured, and these cells were back-diluted to an ABS600 of ~0.001 into an overnight M9 glycerol medium with kanamycin and chloramphenicol. The following day, these cells were again washed three times in 1× PBS. The cells were resuspended in M9 base medium to measure the ABS600. The cultures were then back-diluted to an ABS600 of ~0.001 in 200 μl of M9 xylose medium, kanamycin, chloramphenicol, and ± 1 mM IPTG for overnight culturing and ABS600 measurements in a plate reader. The final ABS600 of these cultures were then used.
To enrich for recognized PAMs within the nucleotide library, the same culture conditions were used. Instead of single colonies, the nucleotide library was freshly transformed into the host strain that contained the dCas9 and Spy sgRNA plasmid (CB624) and recovered in SOC (super optimal broth with catabolite repression) medium for 1 hour at 37°C. After recovery, 250 μl of the recovering cells was back-diluted into a 25-ml overnight LB culture with kanamycin and chloramphenicol to ensure that the entire library diversity was preserved. The following morning, the cells were washed three times in 1× PBS and then resuspended in M9 base medium. The ABS600 was measured and the culture was then back-diluted to an ABS600 ~ 0.001 into 25 ml of overnight M9 glycerol medium with kanamycin and chloramphenicol. After 15.5 hours of incubation, the culture was split equally into two flasks, and 1 mM IPTG was added to one culture. The cultures were then returned to the shaking incubator for 30 more minutes of growth at 37°C. The cultures were then washed three times in 1× PBS and resuspended in M9 base medium. The ABS600 of the cultures was measured and the cultures were back-diluted to an ABS600 ~ 0.001 in 25 ml of overnight M9 xylose medium with kanamycin and chloramphenicol. IPTG (1 mM) was added to the culture that had been previously induced to maintain growth of all the cells in xylose. After 16 hours, the plasmids were then midi-prepped to extract the plasmids from the surviving cells.
Next-generation sequencing for PAM-SEARCH
DNA from the PAM screen was prepared for deep sequencing by first amplifying a 135-bp region from the reporter plasmid isolated from IPTG-induced and uninduced cultures grown in M9 xylose medium. These oligos that contained the necessary adapters for analysis by Illumina MiniSeq are prDC739 and prDC740. Once amplified, the DNA fragments were purified with AMPure XP magnetic beads (Beckman Coulter). A 0.9× bead purification was used followed by two 80% ethanol washes. After washing, the beads were dried and the DNA was eluted in twice the original volume with nuclease-free water. These amplified DNA fragments were then indexed, and phase-shifting staggers were inserted downstream of the Illumina sequencing primer site using an 8-cycle PCR (prDC731 to prDC738). Nextera indices 501, 502, 503, 504, 701, 702, 703, and 704 were used in this work, and staggers were added to increase the diversity. Once amplified, the PCR products were purified with AMPure XP magnetic beads similarly to the first PCR, but instead eluting in the original PCR volume. These samples were quantified on a NanoDrop 2000c spectrophotometer and by agarose gel electrophoresis. The samples were then quantified on a QuBit, analyzed with the Bioanalyzer HS DNA chip, and pooled. Samples were run on the MiniSeq Mid Output Kit with 300 cycles (150-bp paired end) with 20% PhiX spike-in on an Illumina MiniSeq.
PAM wheel representation
A detailed procedure for generating Krona plots (39) can be found in the Detailed Protocol document of the previous manuscript (8). The PAMs adjacent to a perfectly matching target site were extracted, and the number of reads per sequence was counted. The enrichment score was then calculated between the control and test sample. PAM enrichment counts were inserted into the Krona Microsoft Excel template document, PAMs were separated into individual nucleotides in every column of the Excel sheet, and the corresponding enrichment was used to generate the plot. The log2(fold enrichment) was calculated for each PAM sequence and plotted by nucleotide position in the PAM to generate the nucleotide plots. The same log2(fold enrichment) values were used to plot PAM sequences according to motifs to generate the motif plots.
Flow cytometry analysis of bacterial cells
For GFP activation, single colonies were inoculated and cultured overnight in base M9 media. Transcriptional repression was performed in LB medium. The next morning, the ABS600 was measured on a NanoDrop 2000c spectrophotometer. Cells were diluted to an ABS600 of 0.01 and grown for ~4.5 hours, reaching an ABS600 of ~0.2. Cultures were then diluted 1:50 in 1× PBS and analyzed on an Accuri C6 Flow Cytometer (Becton Dickinson) equipped with a CFlow plate sampler, a 488-nm laser, and a 530 ± 15-nm band-pass filter. Forward scatter (cutoff of 18,500) and side scatter (cutoff of 600) were used to cut out noncellular events, and a gate was set for E. coli cells based on previous work. Fluorescence of gated events was recorded using FL1-H, with a minimum number of 30,000 collected events for data analysis.
Plasmid clearance assays
The SpyCas9 (Figs. 2D and 5B), eSpyCas9(1.1) (Fig. 5B), SpyCas9HF1 (fig. S6C), eSpyCas9(1.1)HF1 (fig. S6D), and SpyCas9D1135E (Fig. 5B) plasmids were used to assess plasmid clearance on the PAM-SCANR (CB456, CB457, CB631, and CB634) plasmids containing various PAMs from prior work (8). In short, strains containing both a Cas9 plasmid (CB339 and CBS154 to CBS156) and the reporter plasmids were electroporated with 50 ng of targeting sgRNA plasmid (CB455) or nontargeting Sth1 sgRNA plasmid (CB450). Following transformation, cells were recovered in SOC medium for 1 hour and plated on LB agar containing ampicillin, kanamycin, and chloramphenicol. The fold reduction in the transformation efficiency was calculated as the ratio of the number of transformants for the nontargeting plasmid divided by that of the targeting plasmid.
The SpyCas9HF1 and eSpyCas9(1.1)HF1 variants exhibited limited clearance under the conditions described above (fig. S6, C and D), so an additional nonselective outgrowth was included to enhance the stringency of the assay (Fig. 5B) (30) as performed previously. As part of the modified clearance assay, 20 μl of the recovered SOC media was back-diluted into 2 ml of LB media with chloramphenicol and kanamycin. Cultures were grown overnight and plated on LB agar containing all three antibiotics (ampicillin, chloramphenicol, and kanamycin) the next day. The fold reduction in the transformation efficiency was calculated as the ratio of the number of transformants for the nontargeting plasmid divided by that of the targeting plasmid.
Mammalian cell culture
The U2OS.EGFP cell line was maintained in Dulbecco’s modified Eagle’s medium (DMEM) high glucose, 10% fetal bovine serum, and 1% penicillin and streptomycin media and incubated at 37°C with 5% CO2 using standard tissue culture techniques. The HEK293T cell line was maintained in high-glucose DMEM, 10% fetal calf serum, and 1% penicillin and streptomycin media and incubated at 37°C with 5% CO2 using standard tissue culture techniques.
Indel formation experiments in mammalian cell culture
Plasmids for transfection were isolated from bacterials stocks using either the Qiagen EndoFree Plasmid Maxi-Prep Kit (catalog no. 12362) or ZymoPURE II Midiprep Kit (catalog no. D4200). U20S.EGFP cells were transfected using the GenJet transfection reagent for U2OS cells version II (SignaGen Laboratories, catalog no. SL100489-OS) and following the manufacturer’s protocol with slight deviations. The cells were seeded at 200,000 cells per well in a 12-well dish 1 day before transfection. The preparation of the DNA encapsulated liposomes was carried out according to the manufacturer’s protocol (SignaGen Laboratories) in serum-free DMEM. Twenty hours after transfection, the media was removed, cells were washed with 1× PBS, and fresh media was added. Seventy-two hours after transfection, the cells were washed with PBS and then trypsinized. Once detached from the surface, the solution was neutralized with complete media. The cells were then pelleted by centrifugation at 200g for 5 min. The supernatant was then removed and the cells were resuspended in 1× PBS for flow cytometry and/or genomic DNA extraction. The genomic DNA was extracted using the GeneJET Genomic DNA Extraction Kit (Thermo Fisher Scientific, catalog no. K0721) following the manufacturer’s instructions. The genomic DNA was then amplified and sent for Sanger or deep sequencing.
HEK293T cells were seeded at 200,000 cells per well in a 12-well dish 1 day before transfection. A total of 1488 nanograms of plasmid encoding Cas9 and the sgRNA was mixed with 116 ng of the pGFP/puro and transfected into cells with Lipofectamine 2000 (catalog no. 11668) following the manufacturer’s protocol. The media was changed to fresh complete media supplemented with puromycin (6 μg/ml; Life Technologies, catalog no. A1113803) 1 day after transfection. Seventy-two hours after transfection, the adhered cells were washed with PBS and trypsinized. Once detached from the surface, the cells were neutralized with complete media and pelleted by centrifugation at 800g for 3 min. The supernatant was removed and the cells were resuspended in 1× PBS for genomic DNA extraction using the PureLink Genomic DNA Mini Kit (Life Technologies, catalog no. K182002). The genomic DNA was amplified, sent for Sanger sequencing, and analyzed with TIDE (https://tide.deskgen.com/) using suggested parameters.
Flow cytometry analyses of U2OS cells
U2OS cells were passed through a 100-μm strainer before analysis on the flow cytometer. Forward scatter (cutoff at 500,000) and side scatter (cutoff at 50,000) were used to cut out noncellular events, and a gate was set for the fluorescence (FL1-H) of the cells based on untransfected controls. Fluorescence of gated events was recorded using FL1-H, with a minimum number of 10,000 collected events for analysis.
Quantification of indel formation by next-generation sequencing
U2OS genomic DNA was prepared for deep sequencing by first amplifying short regions of the chromosomally encoded egfp. The oligos contained the necessary adapters for analysis by the Illumina MiSeq 250PE Nanokit are prDC174 to prDC179. Once amplified, the DNA fragments were purified with AMPure XP magnetic beads (Beckman Coulter). A 1.8× bead purification was used followed by two 80% ethanol washes. After washing, the DNA was eluted in 20 μl of sterile, nuclease-free water. These amplified DNA fragments were then indexed using an 8-cycle PCR amplification. Nextera indices 501, 502, 503, 504, 707, 708, and 709 were used in this work and are encoded within prDC185, prDC186, prDC180, prDC181, prDC187, prDC188, and prDC189, respectively. Once amplified, the PCR products were purified with AMPure XP magnetic beads similar to the first cleanup. These samples were quantified on a NanoDrop 2000c spectrophotometer and by a Qubit Fluorometer. DNA then diluted to 15 nM and samples with separate indices were pooled to a total volume of 20 μl. Samples were run on the 250-bp paired-end read Illumina MiSeq Nano kit. The sequencing results for each sample were then analyzed with CRISPResso using the standard parameters to evaluate indel formation (https://github.com/pinellolab/crispresso).
Analysis of previously published datasets
The raw data from previous datasets were obtained through the Supplementary Materials. For Fig. 3A (top), the PAM sequences were sorted by the motifs NGGN, NAGH, NAGG, NYGG, NGAN, and Others, and the read counts of unique, acquired spacers were then plotted by each motif. The number of unique, acquired spacers was plotted by the same motif for Fig. 3A (bottom) along with the number of acquired spacers normalized to the frequency each motif would be found in a random DNA sequence. For Fig. 3B (top), the PAM sequences were sorted by the motifs NGGN, NAGH, NAGG, NYGG, NGAN, and Others, and the number of off-target sites for each motif was plotted and grouped by the on-target gene. For Fig. 3B (bottom), the number of off-target sites per motif was normalized to the frequency each motif would be found in a random DNA sequence. For fig. S5, the PAM sequences were sorted by the motifs NGGH, NGGG, NAGH, NAGG, NYGH, NYGG, NGAN, and Others. For fig. S5 (A to C), the log2(fold change) for each motif was plotted. The read counts for each sequence were plotted by each motif. For fig. S7 (D and E), the read counts for each motif were plotted.
System setup and MD simulations
The initial coordinates for simulating the TGG PAM system were taken from the crystal structure of SpyCas9/R-loop complex solvated at 3.4 Å (PDB code: 5F9R) (40). This structure contains an intact nontarget DNA strand bearing a TGG PAM sequence (numbered 21 to 23 in 5F9R). The missing heavy atoms and the hydrogen atoms were added by the leap program within AmberTools16 (41), and two Mg2+ ions were docked at the RuvC active center of SpyCas9. The above system was then immersed in a cubic water box with a minimal thickness of 13.5 Å from each edge. Additional Mg2+ ions were added to generate a 5 mM concentration as applied in standard reaction buffer (2). The ionic strength of NaCl was set to 150 mM to mimic the physiological setting.
All simulations were performed by the CUDA-accelerated version of AMBER16 pmemd engine (pmemd.cuda), using the amber force fields ff14SBonlysc, ff99bsc0_chiOL3, and ff99bsc0_OL15 for describing the protein, RNA, and DNA solutes, respectively (41). The Joung-Cheatham parameter sets (42) were chosen for modeling the monovalent metal ions and the recent multisite ion model with a modified Lennard-Jones potential (43) for the Mg2+ ions in combination with the TIP4PEw water model. The nonbonded interactions were truncated at 10 Å, and the long-range electrostatics were calculated through the particle mesh Ewald summation method (44), with a grid spacing of 1 Å. The bonds involving hydrogens were constrained via the SHAKE algorithm (45). The time step of integration was set to 1 fs during the equilibration stage and 2 fs in the production run. The temperature was maintained at 310.15 K through the Langevin thermostat, and the pressure was controlled at 1.013 bar using the Monte Carlo barostat. The structural snapshots were stored at 50-ps intervals.
The TGG PAM–containing system was subjected to a sequence of steps to reach stability following our previous protocol (46), involving thorough energy minimization and slow heating to 310.15 K followed by 50-ns equilibration. The equilibrated system was used as the starting point for the TAG and TCGG PAM systems as well as the bulge system with a canonical TGG PAM (see below). The TAG system was obtained by mutating the first G in the PAM on the nontarget strand to A (G2 to A2) and the complementary C in the PAM on the target strand to T (C2′ to T2′). The TCGG system was obtained by mutating 2 nt in the PAM on the nontarget strand (G2 to C2 and C4 to G4) and mutating the corresponding base substitution in the target strand to meet base pair complementarity. The TAG and TCGG systems underwent a further equilibration lasting 50 and 100 ns, respectively. During the first half of equilibration, the hydrogen-bonding distances between the bi-arginine and the recognized nucleotides (i.e., AG in TAG PAM and GG in TCGG PAM) were restrained around 3.0 Å with an increasing force constant from 0 to 10 kcal/mol. The force constant was gradually diminished to 0 kcal/mol in the second equilibration stage. Two independent simulations under the NPT ensemble were carried out for each PAM system. The production run was extended to 100 ns for the TGG and TAG PAM systems and 150 ns for the TCGG PAM system. The last 50 ns of the simulation trajectory was extracted for analysis.
The bulge system was constructed by inserting an extra G between the C and G at the end of the target DNA strand (C19′ and G20′) without altering the TGG PAM. Four simulation replicas were performed in the NPT ensemble, each reaching 2000 ns. The last 500 ns of each simulation trajectory was taken for analysis.
Binding energy calculations by molecular mechanics–generalized Born surface area
The binding energy between the bi-arginine (i.e., R1333 and R1335) and the recognized nucleotides in the PAM was estimated through the end-point molecular mechanics–generalized Born surface area (MM-GBSA) approach (47). This method is computationally more efficient than the alternative MM-PBSA (molecular mechanics–Poisson-Boltzmann surface area), yet it has been proven to gain comparable or better accuracy in ranking ligand-binding affinities (48). The MM-GBSA calculations were enabled by the MMPBSA.py program in AmberTools16 (47). The pairwise GB model of Hawkins, Cramer, and Truhlar (GBHCT) was used, adopting the parameters developed by Tsui and Case (49). Other parameters were set following our previous studies (46). For the bulge-free systems, the last 50 ns of the replicated trajectories were aggregated for calculations. For the bulge-containing system, the individual energy values from the four independent simulations were reported, given the divergent results among the replicates. The entropic contribution was not considered here because of high computational demand and potential convergence problem. However, omission of this term does not qualitatively affect the overall trend.
In vitro cleavage assay
The targeting sgRNA was produced using NEB’s HiScribe T7 High Yield RNA Synthesis kit (catalog no. E2040S) following the manufacturer’s protocol for transcription of short transcripts from an amplified gBlock DNA template (gbDC10). The RNA was purified with the RNA Clean & Concentrator kit (Zymo Research, catalog no. R1015) following deoxyribonuclease treatment. SpyCas9 protein was purchased from New England Biolabs (catalog no. M0386T) and diluted with NEBuffer 3.1 according to the manufacturer’s protocol. The target plasmids (pCB621 and pCB625) were amplified using prDC211 and prDC212. The amplicons were visualized and gel-extracted from a 2% agarose gel. The sgRNA was incuated at 95°C for 3 min and then allowed to cool to room temperature. To form RNP complexes, SpyCas9 was then incubated with sgRNA at a 1:1 molar ratio at room temperature for 10 min. Digestion reactions were then carried out with a molar ratio of RNP:DNA of 50:1 and incubated at 37°C for 45 min. Proteinase K was then added to the reaction and incubated at room temperature for 10 min. DNA fragments were then purified using the Zymo DNA Clean & Concentrator kit (catalog no. D4013) and submitted for Sanger sequencing. The sequenced fragments were then aligned to the reference plasmids to determine the cut site.
Base editing in E. coli
Base editing experiments were carried out in MG1655 ΔaraBAD and Pcon-araF (strain CB330) transformed with the sgRNA plasmid (CBS252). The cells were then cotransformed with the base editor plasmid (CBS253) and target plasmids (CBS254 to CBS259) and selected on LB plates with chloramphenicol, kanamycin, and ampicillin. Three colonies were picked, resuspended in 750 μl of LB media with chloramphenicol, kanamycin, and ampicillin, and grown overnight. Fifty microliters of overnight cultures was back-diluted into 750 μl of LB with chloramphenicol, kanamycin, ampicillin, 1 mM IPTG, and 0.2% arabinose to induce the base editor. Colony PCRs were initiated directly from liquid cultures after 2, 4, 5, 12, and 16 hours using prSPC120 and prSPC121. The PCR products were then sequenced with Sanger sequencing. The Sequalizer algorithm was then used to normalize the sequencing peaks for quantification of editing (34).
Base editing in HEK293T
HEK293T cells were seeded at 200,000 cells per well in a 12-well dish 1 day before transfection. Plasmid encoding BE3 (1116 ng) was mixed with 116 ng of the pGFP/puro and 371 ng of the sgRNA plasmid and transiently transfected into cells with Lipofectamine 2000 (catalog no. 11668) following the manufacturer’s protocol. The media was changed to fresh complete media supplemented with puromycin (6 μg/ml; Life Technologies, catalog no. A1113803) 1 day after transfection. Seventy-two hours after transfection, the adhered cells were washed with 1× PBS and trypsinized. Once detached from the surface, the cells were neutralized with complete media and pelleted by centrifugation at 800g for 3 min. The supernatant was removed and the cells were resuspended in 1× PBS. The cells were then passed through a 70-μm filter and kept on ice. Sorting gates were determined using cells transfected without the GFP plasmid using a live/dead stain. The top 10% GFP signal was used as a cutoff, and cells above this threshold were sorted. Genomic DNA was then extracted from the sorted population using the PureLink Genomic DNA Mini Kit. The genomic DNA was amplified, sent for Sanger sequencing, and analyzed with EditR (35). The critical cutoff value for each nucleotide was calculated by EditR with a P value set to 0.01. The percentage of C-to-T edits was calculated by subtracting the critical cutoff value from the reported percent total peak height for T (or A when sequencing the opposite strand). The resulting C-to-T edit (%) was then averaged across three biological replicates for each sample and reported in Fig. 6D.
Statistical analyses
Statistical significance was calculated for samples in Figs. 2 and 5B and figs. S2A, S3D, and S6 (C and D), and the ensuing P values can be found in table S1. All comparisons were made using a two-tailed t test assuming equal variance using a requirement value of 0.05 and 0.01 as the cutoff for statistical significance. The P values below 0.05 are indicated with (*), below 0.01 are indicated with (**), and above 0.05 are indicated with (n.s.) in each of the appropriate figures. For Fig. 2, (A to D), comparisons were made to the NGG-positive control sample (AGGTG). For Fig. 2B, the samples were additionally compared to the AAGTG sample, which is only reported in the text. For the lower panel in Fig. 2E, comparisons were made to the nontargeting sgRNA control. Further comparisons were made for Fig. 2E, comparing the NHGGs to the NAGH (AAGT) sequence to determine the statistically indistinguishable sequences (P > 0.05) and comparing the NHGGs to the NGG (TGGTG) sample to determine the statistically indistinguishable sequences (P > 0.05). These comparisons are only reported in the text. For Fig. 5B and figs. S3D (top) and S6 (C and D), comparisons were made to the NGG-positive control sample (AGGTG). For Fig. 6D, the frequency of C-to-T edits was calculated using the EditR algorithm, which calculated the critical value for each nucleotide and assigned the cutoff value (P < 0.01). The final C-to-T edits (%) were calculated by subtracting the total editing by the assigned cutoff value for each nucleotide.
Supplementary Material
Acknowledgments
We thank A. Keung (NCSU) and R. Smyth (HIRI) for use of laboratory space and equipment as part of the mammalian-cell experiments. We thank T. Krammer for helping with FACS. The SpyCas9 plasmid was a gift from L. Marraffini (Addgene no. 42876). The SpyCas9HF1 plasmid was a gift from M. Lynch (Addgene no. 89961). The eSpyCas9(1.1) and eSpyCas9(1.1)HF1 plasmids were gifts from J. Doudna and K. Joung (Addgene nos. 101215 and 101217, respectively). The human codon-optimized SpyCas9 and sgRNA expression plasmid was a gift from F. Zhang (Addgene plasmid: 42230). The Puromcyin/EGFP expression plasmid was a gift from M. McVoy (Addgene plasmid: 45561). The BE3 expression construct was a gift from D. Liu (Addgene plasmid: 73021). Computational resources were provided by the UNT office of High-Performance Computing. Funding: This work was supported through funding from the National Science Foundation (MCB-1452902 to C.L.B.), the NIH (1R35GM119561 to C.L.B., 9R15HL147265 to J.L.), Agilent Technologies (gift no. 3926 to C.L.B.), and the Camille Dreyfus Teacher-Scholar Award (T7-17-038 to C.L.B.). Author contributions: R.T.L. and C.L.B. devised PAM-SEARCH. R.T.L., D.C., and C.L.B. developed the experiments. D.C., R.T.L., R.A.S., S.P.C., and B.A.M. performed the experiments. D.C. generated the PAM wheel and nucleotide enrichment plot. Z.Z. and J.L. developed the MD simulations while Z.Z. performed the simulations. D.C. and C.L.B. wrote the manuscript, with input from all other authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The next-generation sequencing data for the PAM assay conducted on SpyCas9 as well as the data for the indel formation in U2OS cells will be available through NCBI SRA (GSE125959). All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/29/eabb4054/DC1
REFERENCES AND NOTES
- 1.Makarova K. S., Wolf Y. I., Iranzo J., Shmakov S. A., Alkhnbashi O. S., Brouns S. J. J., Charpentier E., Cheng D., Haft D. H., Horvath P., Moineau S., Mojica F. J. M., Scott D., Shah S. A., Siksnys V., Terns M. P., Venclovas Č., White M. F., Yakunin A. F., Yan W., Zhang F., Garrett R. A., Backofen R., van der Oost J., Barrangou R., Koonin E. V., Evolutionary classification of CRISPR-Cas systems: A burst of class 2 and derived variants. Nat. Rev. Microbiol. 18, 67–83 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J. A., Charpentier E., A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leenay R. T., Beisel C. L., Deciphering, communicating, and engineering the CRISPR PAM. J. Mol. Biol. 429, 177–191 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lau C.-H., Applications of CRISPR-Cas in bioengineering, biotechnology, and translational research. CRISPR J. 1, 379–404 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Jiang W., Bikard D., Cox D., Zhang F., Marraffini L. A., RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 31, 233–239 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Y., Ge X., Yang F., Zhang L., Zheng J., Tan X., Jin Z.-B., Qu J., Gu F., Comparison of non-canonical PAMs for CRISPR/Cas9-mediated DNA cleavage in human cells. Sci. Rep. 4, 5405 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rock J. M., Hopkins F. F., Chavez A., Diallo M., Chase M. R., Gerrick E. R., Pritchard J. R., Church G. M., Rubin E. J., Sassetti C. M., Schnappinger D., Fortune S. M., Programmable transcriptional repression in mycobacteria using an orthogonal CRISPR interference platform. Nat. Microbiol. 2, 16274 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leenay R. T., Maksimchuk K. R., Slotkowski R. A., Agrawal R. N., Gomaa A. A., Briner A. E., Barrangou R., Beisel C. L., Identifying and visualizing functional PAM diversity across CRISPR-Cas systems. Mol. Cell 62, 137–147 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zetsche B., Gootenberg J. S., Abudayyeh O. O., Slaymaker I. M., Makarova K. S., Essletzbichler P., Volz S. E., Joung J., van der Oost J., Regev A., Koonin E. V., Zhang F., Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–771 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacobsen T., Liao C., Beisel C. L., The Acidaminococcus sp. Cas12a nuclease recognizes GTTV and GCTV as non-canonical PAMs. FEMS Microbiol. Lett. 366, fnz085 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nishimasu H., Shi X., Ishiguro S., Gao L., Hirano S., Okazaki S., Noda T., Abudayyeh O. O., Gootenberg J. S., Mori H., Oura S., Holmes B., Tanaka M., Seki M., Hirano H., Aburatani H., Ishitani R., Ikawa M., Yachie N., Zhang F., Nureki O., Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science 361, 1259–1262 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kleinstiver B. P., Prew M. S., Tsai S. Q., Topkar V. V., Nguyen N. T., Zheng Z., Gonzales A. P. W., Li Z., Peterson R. T., Yeh J.-R. J., Aryee M. J., Joung J. K., Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 523, 481–485 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Afroz T., Biliouris K., Boykin K. E., Kaznessis Y., Beisel C. L., Trade-offs in engineering sugar utilization pathways for titratable control. ACS Synth. Biol. 4, 141–149 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsu P. D., Scott D. A., Weinstein J. A., Ran F. A., Konermann S., Agarwala V., Li Y., Fine E. J., Wu X., Shalem O., Cradick T. J., Marraffini L. A., Bao G., Zhang F., DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karvelis T., Gasiunas G., Young J., Bigelyte G., Silanskas A., Cigan M., Siksnys V., Rapid characterization of CRISPR-Cas9 protospacer adjacent motif sequence elements. Genome Biol. 16, 253 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Händel E.-M., Gellhaus K., Khan K., Bednarski C., Cornu T. I., Müller-Lerch F., Kotin R. M., Heilbronn R., Cathomen T., Versatile and efficient genome editing in human cells by combining zinc-finger nucleases with adeno-associated viral vectors. Hum. Gene Ther. 23, 321–329 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brinkman E. K., Chen T., Amendola M., van Steensel B., Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, e168 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pinello L., Canver M. C., Hoban M. D., Orkin S. H., Kohn D. B., Bauer D. E., Yuan G.-C., Analyzing CRISPR genome-editing experiments with CRISPResso. Nat. Biotechnol. 34, 695–697 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heler R., Samai P., Modell J. W., Weiner C., Goldberg G. W., Bikard D., Marraffini L. A., Cas9 specifies functional viral targets during CRISPR-Cas adaptation. Nature 519, 199–202 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cameron P., Fuller C. K., Donohoue P. D., Jones B. N., Thompson M. S., Carter M. M., Gradia S., Vidal B., Garner E., Slorach E. M., Lau E., Banh L. M., Lied A. M., Edwards L. S., Settle A. H., Capurso D., Llaca V., Deschamps S., Cigan M., Young J. K., May A. P., Mapping the genomic landscape of CRISPR-Cas9 cleavage. Nat. Methods 14, 600–606 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Tsai S. Q., Zheng Z., Nguyen N. T., Liebers M., Topkar V. V., Thapar V., Wyvekens N., Khayter C., Iafrate A. J., Le L. P., Aryee M. J., Joung J. K., GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 33, 187–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsai S. Q., Nguyen N. T., Malagon-Lopez J., Topkar V. V., Aryee M. J., Joung J. K., CIRCLE-seq: A highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat. Methods 14, 607–614 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anders C., Niewoehner O., Duerst A., Jinek M., Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 513, 569–573 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirano S., Nishimasu H., Ishitani R., Nureki O., Structural basis for the altered PAM specificities of engineered CRISPR-Cas9. Mol. Cell 61, 886–894 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Anders C., Bargsten K., Jinek M., Structural plasticity of PAM recognition by engineered variants of the RNA-guided endonuclease Cas9. Mol. Cell 61, 895–902 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin Y., Cradick T. J., Brown M. T., Deshmukh H., Ranjan P., Sarode N., Wile B. M., Vertino P. M., Stewart F. J., Bao G., CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 42, 7473–7485 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen J. S., Dagdas Y. S., Kleinstiver B. P., Welch M. M., Sousa A. A., Harrington L. B., Sternberg S. H., Joung J. K., Yildiz A., Doudna J. A., Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 550, 407–410 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Slaymaker I. M., Gao L., Zetsche B., Scott D. A., Yan W. X., Zhang F., Rationally engineered Cas9 nucleases with improved specificity. Science 351, 84–88 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kleinstiver B. P., Pattanayak V., Prew M. S., Tsai S. Q., Nguyen N. T., Zheng Z., Joung J. K., High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529, 490–495 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xue C., Seetharam A. S., Musharova O., Severinov K., Brouns S. J. J., Severin A. J., Sashital D. G., CRISPR interference and priming varies with individual spacer sequences. Nucleic Acids Res. 43, 10831–10847 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Komor A. C., Kim Y. B., Packer M. S., Zuris J. A., Liu D. R., Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eid A., Alshareef S., Mahfouz M. M., CRISPR base editors: Genome editing without double-stranded breaks. Biochem. J. 475, 1955–1964 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim Y. B., Komor A. C., Levy J. M., Packer M. S., Zhao K. T., Liu D. R., Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 35, 371–376 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Farzadfard F., Gharaei N., Higashikuni Y., Jung G., Cao J., Lu T. K., Single-nucleotide-resolution computing and memory in living cells. Mol. Cell 75, 769–780.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kluesner M. G., Nedveck D. A., Lahr W. S., Garbe J. R., Abrahante J. E., Webber B. R., Moriarity B. S., EditR: A method to quantify base editing from sanger sequencing. CRISPR J. 1, 239–250 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen H., Choi J., Bailey S., Cut site selection by the two nuclease domains of the Cas9 RNA-guided endonuclease. J. Biol. Chem. 289, 13284–13294 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y., Rajan R., Seifert H. S., Mondragón A., Sontheimer E. J., DNase H activity of Neisseria meningitidis Cas9. Mol. Cell 60, 242–255 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsu P. D., Lander E. S., Zhang F., Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–1278 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ondov B. D., Bergman N. H., Phillippy A. M., Interactive metagenomic visualization in a Web browser. BMC Bioinformatics. 12, 385 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang F., Taylor D. W., Chen J. S., Kornfeld J. E., Zhou K., Thompson A. J., Nogales E., Doudna J. A., Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science 351, 867–871 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salomon-Ferrer R., Case D. A., Walker R. C., An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. 3, 198–210 (2013). [Google Scholar]
- 42.Joung I. S., Cheatham T. E. III, Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B 112, 9020–9041 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liao Q., Pabis A., Strodel B., Kamerlin S. C. L., Extending the nonbonded cationic dummy model to account for ion-induced dipole interactions. J. Phys. Chem. Lett. 8, 5408–5414 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Darden T., York D., Pedersen L., Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993). [Google Scholar]
- 45.Miyamoto S., Kollman P. A., Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 13, 952–962 (1992). [Google Scholar]
- 46.Zuo Z., Zolekar A., Babu K., Lin V. J. T., Hayatshahi H. S., Rajan R., Wang Y.-C., Liu J., Structural and functional insights into the bona fide catalytic state of Streptococcus pyogenes Cas9 HNH nuclease domain. eLife 8, e46500 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miller B. R. 3rd, McGee T. D. Jr., Swails J. M., Homeyer N., Gohlke H., Roitberg A. E., MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 8, 3314–3321 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Li C. H., Zuo Z. C., Su J. G., Xu X. J., Wang C. X., The interactions and recognition of cyclic peptide mimetics of Tat with HIV-1 TAR RNA: A molecular dynamics simulation study. J. Biomol. Struct. Dyn. 31, 276–287 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Tsui V., Case D. A., Theory and applications of the generalized born solvation model in macromolecular simulations. Biopolymers 56, 275–291 (2000). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/29/eabb4054/DC1