

Scaden enables robust cell type deconvolution of complex samples across data types, using a deep learning–based model.
Abstract
We present Scaden, a deep neural network for cell deconvolution that uses gene expression information to infer the cellular composition of tissues. Scaden is trained on single-cell RNA sequencing (RNA-seq) data to engineer discriminative features that confer robustness to bias and noise, making complex data preprocessing and feature selection unnecessary. We demonstrate that Scaden outperforms existing deconvolution algorithms in both precision and robustness. A single trained network reliably deconvolves bulk RNA-seq and microarray, human and mouse tissue expression data and leverages the combined information of multiple datasets. Because of this stability and flexibility, we surmise that deep learning will become an algorithmic mainstay for cell deconvolution of various data types. Scaden’s software package and web application are easy to use on new as well as diverse existing expression datasets available in public resources, deepening the molecular and cellular understanding of developmental and disease processes.
INTRODUCTION
The analysis of tissue-specific gene expression using next-generation sequencing [RNA sequencing (RNA-seq)] is a centerpiece of the molecular characterization of biological and medical processes (1). A well-known limitation of tissue-based RNA-seq is that it typically measures average gene expression across many molecularly diverse cell types that can have distinct cellular states (2). A change in gene expression between two conditions can therefore be attributed to a change in the cellular composition of the tissue or a change in gene expression in a specific cell population, or a mixture of the two. To deconvolve the cell type composition from a change in gene expression is especially important in systems with cellular proliferation (e.g., cancer) or cellular death (e.g., neuronal loss in neurodegenerative diseases) due to systematic cell population differences between experimental groups (3).
To account for this problem, several computational cell deconvolution methods have been proposed during the last years (4, 5). These algorithms use gene expression profiles (GEPs) of cell type–specifically expressed genes to estimate cellular fractions using linear regression to detect, interpret, and possibly correct for systematic differences in cellular abundance between samples (4). While the best-performing linear regression algorithms for deconvolution seem to be variations of support vector regression (6–10), the selection of an optimal GEP is a field of active research (10, 11). It has been recently shown that the design of the GEP is the most important factor in most deconvolution methods, as results from different algorithms strongly correlate given the same GEP (11).
In theory, an optimal GEP should contain a set of genes that are predominantly expressed within each cell population of a complex sample (12). They should be stably expressed across experimental conditions, for example, across health and disease, and resilient to experimental noise and bias. However, bias is typically inherent to biomedical data and is imparted, for instance, by intersubject variability, variations across species, different data acquisition methods, different experimenters, or different data types. The negative impact of bias on deconvolution performance can be partly improved by using large, heterogeneous GEP matrices (11). It is therefore expected that recent advancement in cell deconvolution relied almost exclusively on sophisticated algorithms to normalize the data and engineer optimal GEPs (10).
While GEP-based approaches lay the foundational basis of modern cell deconvolution algorithms, we hypothesize that deep neural networks (DNNs) could create optimal features for cell deconvolution, without relying on the complex generation of GEPs. DNNs such as multilayer perceptrons are universal function approximators that achieve state-of-the-art performance on classification and regression tasks. Whereas this feature is of little importance for strictly linear input data, it makes DNNs superior to linear regression algorithms as soon as data deviate from ideal linearity. This means, for instance, that as soon as data are noisy or biased and classical linear regression algorithms may falter, the hidden layer nodes of the DNN learn to represent higher-order latent representations of cell types that do not depend on input noise and bias. We theorize, therefore, that by using gene expression information as network input, hidden layer nodes of the DNN would represent higher-order latent representations of cell types that are robust to input noise and technical bias.
An obvious limitation of DNNs is the requirement for large training data to avoid overfitting of the machine learning model. While ground-truth information on tissue RNA-seq cell composition is scarce, one can use single-cell RNA-seq (scRNA-seq) data to obtain large numbers of in silico tissue datasets of predefined cell composition (7–9, 13–15). We do this by subsampling and subsequently merging cells from scRNA-seq datasets, this approach being limited only by the availability of tissue-specific scRNA-seq data. It is to be noted that scRNA-seq data suffer from biases, such as dropout, to which RNA-seq data are not subject (16). While this complicates the use of scRNA-seq data for GEP design (8), we surmise that latent network nodes could represent features that are robust to these biases.
On the basis of these assumptions, we developed a single cell–assisted deconvolutional DNN (Scaden) that uses simulated bulk RNA-seq samples for training and predicts cell type proportions for input expression samples of cell mixtures. Scaden is available as downloadable software package and web application (https://scaden.ims.bio). Scaden is trained on publicly available scRNA-seq and RNA-seq data, does not rely on specific GEP matrices, and automatically infers informative features. Last, we show that Scaden deconvolves expression data into cell types with higher precision and robustness than existing methods that rely on GEP matrices.
RESULTS
Scaden overview, model selection, and training
In this part, we focus on the design and optimization of Scaden by training, validation, and testing on in silico data. Note that the generation of in silico data is a strictly linear mathematical operation. Our aim in this context, to corroborate Scaden’s basic functionality, is to show that Scaden’s performance compares with (but not necessarily exceeds) that of state-of-the-art algorithms.
The basic architecture of Scaden is a DNN that takes gene counts of RNA-seq data as input and outputs predicted cell fractions (Fig. 1). To optimize the performance of the DNN, it is trained on data that contain both the gene expression and the real cell type fraction information (Fig. 1B). The network then adjusts its weights to minimize the error between the predicted cell fractions and the real cell fractions (Fig. 1C). We restricted feature selection to the removal of “uninformative” genes that have either zero expression or an expression variance below 0.1, leaving ~10,000 genes for training. In our hands, this feature selection step decreases training time and memory usage.
Fig. 1. Overview of training data generation and cell type deconvolution with Scaden.
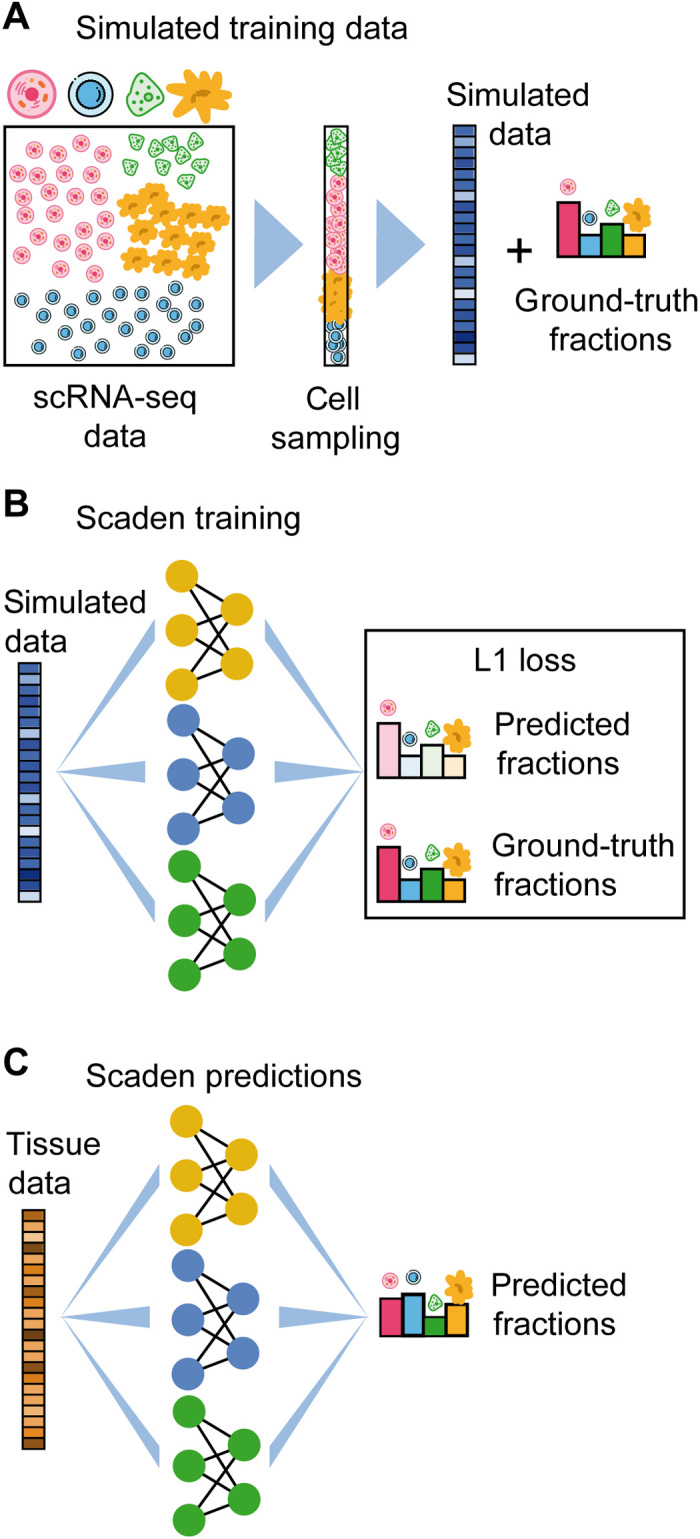
(A) Artificial bulk samples are generated by subsampling random cells from an scRNA-seq dataset and merging their expression profiles. (B) Model training and parameter optimization on simulated tissue RNA-seq data by comparing cell fraction predictions to ground-truth cell composition. (C) Cell deconvolution of real tissue RNA-seq data using Scaden.
For the model selection and training, we made use of the large numbers of artificial bulk RNA-seq datasets with defined composition that can be generated in silico from published scRNA-seq and RNA-seq datasets (simulated tissues; Fig. 1A and tables S1 and S2). The only constraint is that the scRNA-seq and RNA-seq data must come from the same tissue as the bulk data subject to deconvolution.
To find the optimal DNN architecture for cell deconvolution, we generated bulk peripheral blood mononuclear cell (PBMC) RNA-seq data from four publicly available scRNA-seq datasets (tables S1 and S3). We performed leave-one-dataset-out cross-validation, training Scaden on mixtures of synthetic datasets from three scRNA-seq datasets and evaluating the performance on simulated tissue from a fourth scRNA-seq dataset.
We used the root mean square error (RMSE), Pearson’s correlation coefficient (r), the slope and intercept of the regression fitted for ground-truth and predicted cell fractions, and Lin’s concordance correlation coefficient (CCC) (17) to assess algorithmic performance. The CCC is a measure sensitive not only to scatter but also to deviations from linearity (slope and intercept). Within the main text, we report on CCC and RMSE values only; other metrics can be found in the Supplementary Materials.
The final Scaden model is an ensemble of the three best-performing models (table S4), and the final cell type composition estimates are the averaged predictions of all three ensemble models (Fig. 1 and fig. S1). Using an ensemble of models increased the deconvolution performance as compared to single best models (table S6). Details of the model and hyperparameters are given in table S5. We also evaluated the effect of the size of the training dataset on Scaden deconvolution performance, repeating leave-one-dataset-out cross-validation on PBMC data with training dataset sizes from 150 up to 15,000 samples (fig. S2). The increase in CCC value starts to level off from about 1500 simulated samples for this dataset but continues to increase slowly with sample size. We specifically addressed the question to what degree the DNN, trained on simulated samples, tends to overfit, failing to generalize to real bulk RNA-seq data. To understand after how many steps a model trained on in silico data overfits on real RNA-seq data, we trained Scaden on simulated data from an ascites scRNA-seq dataset (table S1; 6000 samples) and evaluated the loss function on a corresponding annotated RNA-seq dataset (18) (table S2; three samples) as a function of the number of steps (fig. S3). All models converged after approximately 5000 steps and slightly overfit when trained for longer. On the basis of this result, we opted for an early-stop approach after 5000 steps for evaluation on real bulk RNA-seq data.
We then compared Scaden to four state-of-the-art GEP-based cell deconvolution algorithms, CIBERSORT (CS) (6), CIBERSORTx (CSx) (7), Multi-subject Single Cell deconvolution (MuSiC) (8), and Cell Population Mapping (CPM) (9). While CS relies on hand-curated GEP matrices, CSx, MuSiC, and CPM can generate GEPs using scRNA-seq data as input.
To get an initial estimate of Scaden’s deconvolution fidelity, we trained the model on 24,000 simulated PBMC RNA-seq samples from three datasets and tested its performance in comparison to CS, CSx, MuSiC, and CPM on a fourth dataset of 500 samples each (e.g., training on data6k, data8k, and donorA and evaluation on donorC). We used corresponding scRNA-seq datasets for the construction of GEPs as input for CSx and MuSiC, and CPM. For CS, we used the PBMC-optimized LM22 GEP matrix (6), which was developed by the CS authors for the deconvolution of human PBMC data.
For two of four test datasets (donorA and donorC), Scaden obtained the highest CCC and lowest RMSE, followed by CSx, MuSiC, CS, and CPM (fig. S4 and table S7). CSx and MuSiC obtained the highest CCC values for the data8k and data6k datasets, respectively. Scaden obtained the highest average CCC and lowest RMSE (0.88 and 0.08, respectively), followed by MuSiC (0.85 and 0.10), CSx (0.83 and 0.11), CS (0.63 and 0.15), and CPM (0 and 0.20; fig. S4). As expected, all algorithms that use scRNA-seq data as reference performed well, with the notable exception of CPM. We want to mention that CPM focuses on the reconstruction of continuous spectra of cellular states, while it incorporates cell deconvolution as an additional feature. We therefore report CPM’s deconvolution performance in the Supplementary Materials from here on. On average, Scaden also obtained the highest correlation and the best intercept and slope values on simulated PBMC data (table S7). A closer inspection on a per–cell type basis (Fig. 2A) revealed that Scaden yields consistently higher CCC values and lower RMSEs when compared to the other algorithms.
Fig. 2. Performance comparison of deconvolution algorithms on simulated tissue data.
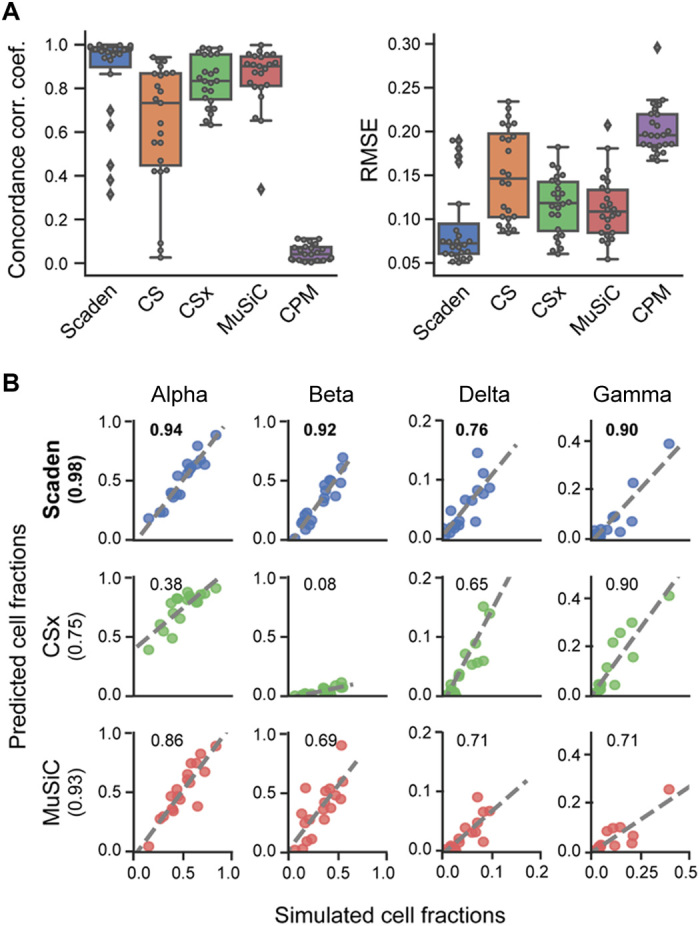
(A) Boxplots of the cell type prediction CCC and RMSE for four simulated PBMC datasets. Tables S14 and S16 contain information on the five (six for CS) cell types used. (B) Scatterplots for four pancreas cell types of ground-truth (x axis) and predicted values (y axis) for Scaden, CSx, and MuSiC on artificial pancreas data (20). Numbers inside the plotting area and in parenthesis signify CCC values.
A specific feature of the MuSiC algorithm is that it preferentially weighs genes according to low intersubject and intracell cluster variability for its GEP, which increases deconvolution robustness when high-expression heterogeneity is observed between human participants, for example (8). To understand whether Scaden can use multisubject information to increase its deconvolution performance, we trained Scaden, CSx, and MuSiC on scRNA-seq pancreas data from several participants (19) and assessed the performance on a separate simulated pancreas RNA-seq dataset (20). To allow for direct comparison, we chose the same pancreas training and test datasets that were used in the original MuSiC publication (table S1). To enable Scaden to leverage the heterogeneity of multisubject data, training data were generated separately for every participant in the dataset (see Methods). CSx cannot profit from multisubject data but performed well on the artificial PBMC datasets and was therefore included in the comparison. The best average performance (across cell types) is achieved by Scaden (CCC = 0.98), closely followed by MuSiC (CCC = 0.93), while CSx does not perform as well (CCC = 0.75; Fig. 2B and table S8). On a per–cell type basis, Scaden’s predictions are clearly superior to the other two algorithms for all cell types. This provides strong evidence that Scaden, by separating training data generation for each participant, can learn intersubject heterogeneity and outperform specialized multisubject algorithms such as MuSiC on the cell type deconvolution task.
In addition, we wanted to test how the best-performing deconvolution algorithms Scaden, MuSiC, and CSx behave when unknown cell content is part of the mixture. To test this, all cells falling into the “Unknown” category were removed from the training or reference PBMC datasets but added to the simulated mixture samples at fixed percentages (5, 10, 20, and 30%; see Methods). Scaden obtains the highest CCC for all tested percentages of unknown cell content (fig. S5 and table S9). The general deconvolution performance declines linearly with increasing percentage of unknown content for all tested algorithms, indicating that Scaden, MuSiC, and CSx have a similar robustness against unknown mixture content.
We next compared the runtime and memory footprint of Scaden and MuSiC on an Intel Xeon six-core central processing unit (CPU) to the runtime of the CSx web application. Scaden is the only algorithm that requires the generation of in silico training data, which takes 13 min for 2000 samples with a peak memory usage of 8 GB. Similar values were obtained for the human brain data. Next, we used the PBMC data to benchmark the runtime and memory consumption of the deconvolution task. For Scaden, model training took ~11 min and cell fraction prediction ~8 s for 500 samples, using less than 1-GB memory. We used the web application of CSx with batch correction to deconvolve the 500 PBMC samples in 35 min. MuSiC took only 2 min and 15 s to deconvolve all 500 samples, with the memory usage peaking at 4.5 GB. As Scaden can take advantage of a graphics processing unit (GPU), we additionally compared training duration on an AMD Ryzen 5 2600 CPU and GeForce RTX 2600 GPU on the same machine. Training on the CPU took 9 min and 39 s, while it took only 3 min and 2 s on the GPU, corresponding to a roughly three times shorter runtime for Scaden if a GPU is available.
Robust deconvolution of bulk expression data
The true use case of cell deconvolution algorithms is the cell fraction estimation of tissue RNA-seq data. In particular for noisy and biased bulk RNA-seq data, we hypothesize that Scaden’s latent feature representations might help it to more robustly predict cell fractions as compared to GEP-based algorithms.
We therefore assessed the performance of Scaden, CS, CSx, and MuSiC to deconvolve two publicly available human PBMC bulk RNA-seq datasets, for which curated GEP matrices and RNA-seq data with associated ground-truth cell type compositions from flow cytometry are available (see the “Data availability” section). We will refer to these datasets that consists of 12 samples each as PBMC1 (21) and PBMC2 (10) (table S2). Both datasets have similar cell type compositions across samples, with CD4 and CD8 T cells making up the biggest fractions. Deconvolution for all methods was performed as described in the previous section, with the difference that data from all four PBMC scRNA-seq datasets were now deployed for Scaden training. Results are given in Fig. 3 (A to C) and tables S10 and S11.
Fig. 3. Comparison of deconvolution algorithms on PBMC tissue RNA-seq data.
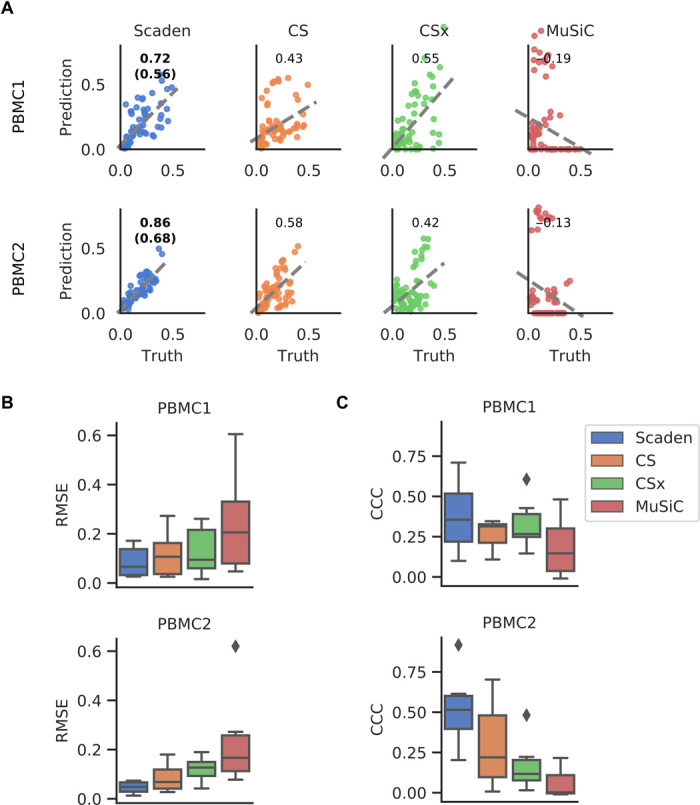
(A) Per–cell type scatterplots of ground-truth (x axis) and predicted values (y axis) for Scaden, CS, CSx, and MuSiC on real PBMC1 and PBMC2 cell fractions. Numbers inside the plotting area signify CCC values. For Scaden, the CCC using only scRNA-seq training data is shown in parenthesis, and the CCC using mixed scRNA-seq and RNA-seq training data is shown without parentheses. (B) Boxplots of RMSE values for real PBMC1 and PBMC2 data. (C) CCC values for real PBMC1 and PBMC2 data.
On the PBMC1 dataset and using all cell types, Scaden obtained the highest CCC and lowest RMSE (0.56 and 0.13), while CSx (0.55 and 0.16) and CS (0.43 and 0.15) performed well yet notably worse than Scaden (Fig. 3A and tables S10 and S11). CPM (0 and 0.18) and MuSiC (−0.19 and 0.32) both failed to deconvolve the cell fractions of the PBMC1 data. Scaden also obtained the best CCC and RMSE (0.68 and 0.08) on the PBMC2 dataset, while CS (0.58 and 0.10) and CSx (0.42 and 0.13) obtained good deconvolution results. Similar to the PBMC1 data deconvolution results, CPM (−0.16 and 0.11) and MuSiC (−0.13 and 0.30) did not perform well on the PBMC2 deconvolution task. In addition to CCC and RMSE metrics, Scaden achieves the best correlation, intercept, and slope on both PBMC datasets (tables S10 and S11).
In particular, Scaden outperforms classical algorithms on a per–cell type basis (Fig. 3, B and C). These results show weaker correlations and a strong dependence on the cell type. A closer examination of the metrics in table S11 and fig. S6 shows that the largest variations are found in the slope and intercept.
We further evaluated how good the Scaden ensemble performs compared to the best single DNN model (M512, 512 nodes input layer). While the M512 model shows good deconvolution performance on the PBMC1 (CCC, 0.57) and PBMC2 (CCC, 0.68) datasets, the ensemble model achieves the best average cross-validation performance (table S6). We therefore opted to use the ensemble method to reduce interdataset performance variation observed with M512 and other single models.
An additional algorithmic feature of Scaden is that it seamlessly integrates increasing amounts of training data, which can be of different types, such as a combination of simulated tissue and real tissue data with cell fraction information. In theory, even limited real tissue training data could make Scaden robust to data type bias and consequently improve Scaden’s deconvolution performance on real tissue data. We therefore trained Scaden on a mix of simulated PBMC and real PBMC2 (12 samples) data and evaluated its performance on real PBMC1 data (Fig. 3, A and B, fig. S6, and tables S10 and S11). While the training contained very little (~2%) real data, Scaden’s CCC increased from 0.56 to 0.72, and the RMSE decreased from 0.13 to 0.10. We observed similar performance increases when Scaden was trained on simulated PBMC and real PBMC1 data and evaluated on real PBMC2 data (Fig. 3, A and B, fig. S6, and tables S10 and S11). Next, we wanted to investigate how a Scaden model trained on only few real samples compares to the models trained on simulated or simulated and real data. While a Scaden model trained on only bulk PBMC1 samples (n = 12) deconvolves PBMC2 data with a CCC of 0.62, it does not reach the CCC of models trained on simulated data (CCC of 0.68) or on simulated and bulk data (CCC of 0.86). We would also not advise training models on so few training samples, as these models are usually overfit.
This further validates that Scaden reliably deconvolves tissue RNA-seq data into the constituent cell fractions and that very accurate deconvolution results can be obtained if reference and target datasets are from the same experiment.
We next wanted to test how the algorithm performs on postmortem human brain tissue of a subsample from the Religious Orders Study and Memory and Aging Project (ROSMAP) study (22), for which ground-truth cell composition information was recently measured by immunohistochemistry (41 samples with all cell types given) (23). The data provided by this study consist of bulk RNA-seq data from the dorsolateral prefrontal cortex and pose a special challenge due to the complexity of its cell type composition, which is further complicated by the fact that the data originate from brains of healthy individuals as well as patients with Alzheimer’s disease (AD) at various stages of neuronal loss. As reference datasets, we used the scRNA-seq dataset provided by Darmanis et al. (24) from the anterior temporal lobe of living patients and the Lake dataset that isolates nuclei of neurons from two (visual and frontal) cortical regions from a postmortem brain and subjects them to RNA-seq (25). From these, we generated 2000 training samples (Darmanis) and 4000 samples (two regions from the Lake dataset).
Figure 4A shows the deconvolution results for all three algorithms with the Darmanis (scRNA-seq) reference dataset. Scaden achieves the highest CCC value (0.92) followed by MuSiC (0.87) and CSx (0.81; table S12). Compared to Scaden, MuSiC and CSx overestimate neural percentages, leading to higher RMSE values of 0.09 and 0.12, respectively (Scaden, 0.06). Notably, all methods showed a lower CCC on the per–cell type level (Fig. 3B), demonstrating that some per–cell type correlations are poor, either in slope, intercept, variance, or a combination of them. This emphasizes the need for a cell type–specific inspection of results and highlights that, depending on the dataset, cell type–specific deconvolution results can be far from perfect.
Fig. 4. Deconvolution performance comparison on brain tissue RNA-seq data.
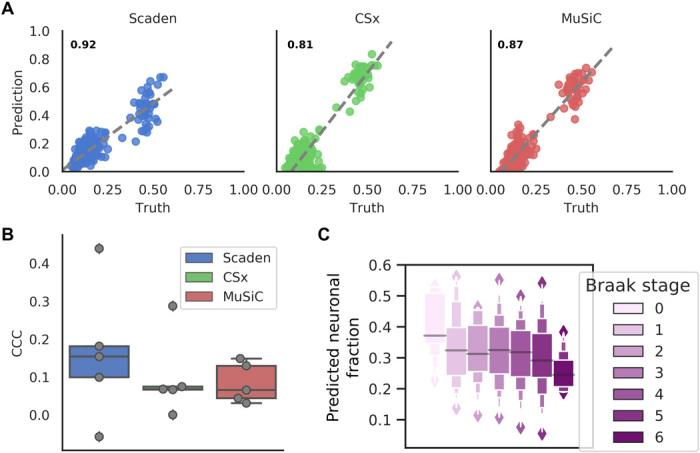
(A) Prediction of human brain cell fractions of the ROSMAP dataset using the Darmanis dataset as a reference: scatterplots of ground-truth (x axis) and predicted values (y axis) for Scaden, CSx, and MuSiC of data. CCC values are shown as inserts. (B) Per–cell type CCC values for ROSMAP using the Darmanis data as a reference. (C) Neuronal content determined by Scaden trained on mouse brain data and evaluated on the Braak stage of the ROSMAP study.
In addition to comparing the predictive power of Scaden, CSx, and MuSiC on human brain tissue with different reference datasets, we also tested how the choice of reference datasets affected Scaden’s deconvolution results. Notably, all methods substantially drop in performance when the Lake single-nucleus RNA-seq dataset is used as a reference as we had presumed (fig. S7A). We want to emphasize that Scaden, in contrast to CSx and MuSiC, has the possibility to simultaneously use both datasets as reference, whereas for CSx and MuSiC, the user has to choose one of the two, unaware of which will give the correct results.
We found that the performance of Scaden was almost unaffected when the Lake dataset was added to the Darmanis training samples (CCC = 0.90, RMSE = 0.06; fig. S7A and table S12). These results show that cell deconvolution with Scaden is robust to training data bias (Darmanis single-cell versus Lake single-nucleus data). An added benefit of Scaden is that it allows for the inclusion and mixing of different scRNA-seq experiments in the training dataset, further increasing its robustness (fig. S7A). Last, when calculating the CCC values on a per-sample basis, Scaden achieves the best scores for most samples (fig. S7B).
In a next step, we wanted to assess whether Scaden’s deconvolution performance was robust across species by trying to predict the cell fractions of the ROSMAP study (22) with a Scaden model trained on in silico data from five mouse brain scRNA-seq datasets (table S1). Intriguingly, Scaden was able to achieve a CCC value of 0.83 and an RMSE of 0.079, showing that Scaden can reliably deconvolve RNA-seq data across related species.
The ROSMAP study also contains information on the Braak stages (26) corresponding to 390 human postmortem prefrontal cortex samples, which correlate with the severity and progression stage of AD and the degree of neuronal loss. We used the Scaden model trained on artificial data generated from five mouse brain scRNA-seq datasets to predict neuronal cell fractions of this larger human dataset. Overall, Scaden’s cell fraction predictions capture the increased neuronal loss with increasing Braak stage (Fig. 4C). The largest drop in neural percentage is observed at stage 5, when the neurodegeneration typically reaches the prefrontal cortex of the brain.
Given the robustness with which Scaden predicts tissue RNA-seq cell fractions using scRNA-seq training data, even across species, we next wanted to investigate whether an scRNA-seq–trained Scaden model can also deconvolve other data types. To this end, we measured the deconvolution performance on a bulk PBMC microarray dataset (20 samples) (6) of a Scaden model trained on scRNA-seq and RNA-seq PBMC data (see above). We compared Scaden to CS using the microarray-derived LM22 matrix. CS achieved a slightly higher CCC and slightly lower total RMSE (0.72 and 0.11) than Scaden (0.71 and 0.13), while Scaden obtained the highest average CCC (0.50) compared to CS (0.39; fig. S8 and table S13). Notably, in this scenario, Scaden was trained entirely on simulated scRNA-seq and RNA-seq data, while CS’s LM22 GEP was optimized on PBMC microarray data.
Overall, we provide strong evidence that Scaden robustly deconvolves tissue data across tissues, species, and even data types.
DISCUSSION
Scaden is a novel deep learning–based cell deconvolution algorithm that, in many instances, compares favorably in both prediction robustness and accuracy to existing deconvolution algorithms that rely on GEP design and linear regression. We believe that Scaden’s performance relies to a large degree on the inherent feature engineering of the DNN. The network does not only select features (genes) for regression but also creates new features that are optimal for the regression task in the nodes of the hidden layers. These hidden features are nonlinear combinations of the input features (gene expression), which makes it notoriously difficult to explain how a DNN works (27). It is important to highlight that this feature creation is fundamentally different from all other existing cell deconvolution algorithms, which rely on heuristics that select a defined subset of genes as features for linear regression.
Another advantage of this inherent feature engineering is that Scaden can be trained to be robust to input noise and bias (e.g., batch effects). Noise and bias are all prevalent in experimental data, because of different sample quality, sample processing, experimenters, and instrumentation, for example. If the network is trained on different datasets of the same tissue, however, then it learns to create hidden features that are robust to noise and bias, such as batch effects. This robustness is pivotal in real-world cell deconvolution use cases, where the bulk RNA data for deconvolution and the training data (and therefore the network and GEP) contain different noise and biases. In this study, we tested Scaden with training data from scRNA-seq datasets generated with a variety of different protocols and could not identify a specific protocol that is not suitable. While especially recent cell deconvolution algorithms include batch correction heuristics before GEP construction, Scaden optimizes its hidden features automatically when trained on data from various batches. Potential protocol-specific biases can therefore be alleviated when employing training data from multiple protocols.
The robustness to noise and bias, which might be due to hidden feature generation, is especially evident in Scaden’s ability to deconvolve across data types. A network trained on in silico bulk RNA-seq data can seamlessly deconvolve microarray data of the same tissue. This is quite noteworthy, as microarray data are known to have a reduced dynamic range and several hybridization-based biases compared to RNA-seq data. In other words, Scaden can deconvolve bulk data of types that it has never been trained on, even in the face of strong data type bias. This raises the possibility that Scaden trained on scRNA-seq data might reliably deconvolve other bulk omics data as well, such as proteomic and metabolomic data. This assumption is strengthened by the fact that Scaden, trained on scRNA-seq data, attains state-of-the-art performance on the deconvolution of bulk RNA-seq data, two data types with very distinct biases (16).
As highlighted in the introduction, a drawback for many DNNs is the large amount of training data required to obtain robust performance. Here, we used scRNA-seq data to create in silico bulk RNA-seq data of predefined type (target tissue) with known composition, across datasets. This immediately highlights Scaden’s biggest limitation, the dependency on scRNA-seq data of the target tissue. In this study, we have shown that Scaden, trained solely on simulated data from scRNA-seq datasets, can outperform GEP-based deconvolution algorithms. We did observe, however, that the addition of labeled RNA-seq samples to the training data did substantially improve deconvolution performance in the case of PBMC data. We therefore believe that efforts to increase the similarity between simulated training data and the target bulk RNA-seq data could increase Scaden’s performance further. Mixtures of in silico bulk RNA-seq data and publically available RNA-seq data, of purified cell types, for example, could further increase the deconvolution performance of Scaden. Furthermore, domain adaptation methods can be used to improve performance of models that are trained on data (here, scRNA-seq data) that are similar to the target data (here, RNA-seq data) (28). In future versions, Scaden’s simple multilayer perceptron architecture could leverage domain adaptation to further stabilize and improve its cell deconvolution performance.
Scaden uses an ensemble approach by averaging the predictions of three different models to increase performance and improve generalization. Increasing the number of models per ensemble would allow for the estimation of the prediction uncertainty. While not implemented in this study, this could be an interesting extension to Scaden’s ensemble architecture.
Recent cell deconvolution algorithms have used cell fraction estimates to infer cell type–specific gene expression from bulk RNA-seq data. It is straightforward to use Scaden’s cell fraction estimates to infer per-group (3) and per-sample (7) cell type–specific gene expression using simple regression or non-negative matrix factorization, respectively. We would like to add a note of caution, however, as the error of cell fraction estimates, which can be quite large, is propagated into the gene expression calculations and will affect any downstream statistical analysis.
While Scaden achieves good performance on the samples and tissues used in this study, it is important to keep in mind that cell type similarity, sample heterogeneity, and complexity, as well as experimental noise and bias, can severely limit deconvolution accuracy. Furthermore, Scaden is currently not attempting to model cell size differences in its algorithm, which might be useful to consider for the interpretation of prediction results.
In summary, the deconvolution performance, robustness to noise and bias, and the flexibility to learn from large numbers of in silico datasets, across data types (scRNA-seq and RNA-seq mixtures) and potentially even tissues, make us believe that DNN-based architectures will become an algorithmic mainstay of cell type deconvolution.
METHODS
Datasets and preprocessing
scRNA-seq datasets
The following human PBMC scRNA-seq datasets were downloaded from the 10X Genomics data download page: 6k PBMCs from a Healthy Donor, 8k PBMCs from a Healthy Donor, Frozen PBMCs (Donor A), and Frozen PBMCs (Donor C) (29). Throughout this paper, these datasets are referred to with the handles data6k, data8k, donorA, and donorC, respectively. It was not intended to incorporate as many datasets as possible. Instead, these four datasets were chosen with the goal to dispose of a set of samples with consistent cell types and gene expression. This limited our choice to datasets that displayed clearly identifiable cell types for the majority of cells. The Ascites scRNA-seq dataset was downloaded from https://figshare.com as provided by Schelker et al. (18). Pancreas and mouse brain datasets were downloaded from the scRNA-seq dataset collection of the Hemberg laboratory (https://hemberg-lab.github.io/scRNA.seq.datasets/). The human brain datasets from Darmanis et al. (24) and Lake et al. (25) where downloaded from Gene Expression Omnibus (GEO) with accession numbers GSE67835 and GSE97930, respectively. A table listing all datasets including references to the original publications can be found in table S1.
scRNA-seq preprocessing and analysis
All datasets were processed using the Python package Scanpy (v. 1.2.2) (30) following the Scanpy’s reimplementation of the popular Seurat’s clustering workflow. First, the corresponding cell-gene matrices were filtered for cells with less than 500 detected genes and genes expressed in less than five cells. The resulting count matrix for each dataset was filtered for outliers with high or low numbers of counts. Gene expression was normalized to library size using the Scanpy function “normalize_per_cell.” The normalized matrix of all filtered cells and genes was saved for the subsequent data generation step.
The following processing and analysis steps had the sole purpose of assigning cell type labels to every cell. All cells were clustered using the louvain clustering implementation of the Scanpy package. The louvain clustering resolution was chosen for each dataset, using the lowest possible resolution value (low-resolution values lead to less clusters) for which the calculated clusters appropriately separated the cell types. The top 1000 highly variable genes were used for clustering, which were calculated using Scanpy’s “filter_genes_dispersion” function with parameters min_mean = 0.0125, max_mean = 3, and min_disp = 0.5. Principal components analysis was used for dimensionality reduction.
To identify cell types, marker genes were investigated for all cell types in question. For PBMC datasets, useful marker genes were adopted from public resources such as the Seurat tutorial for 2700 PBMCs (31). Briefly, interleukin-7 receptor (IL7R) was taken as marker for CD4 T cells, LYZ for monocytes, MS4A1 for B cells, GNLY for natural killer cells, FCER1A for dendritic cells, and CD8A and CCL5 as markers for CD8 T cells. For all other scRNA-seq datasets, marker genes and expected cell types were inferred from the original publication of the dataset. For instance, to annotate cell types of the mouse brain dataset from Zeisel et al. (32), we used the same marker genes as Zeisel and colleagues. We did not use the same cell type labels from the original publications because a main objective was to assure that cell type labeling is consistent between all datasets of a certain tissue.
Cell type annotation was performed manually across all the clusters for each dataset, such that all cells belonging to the same cluster were labeled with the same cell type. The cell type identity of each cluster was chosen by crossing the cluster’s highly differentially expressed genes with the curated cell type’s marker genes. Clusters that could not be clearly identified with a cell type were grouped into the “Unknown” category.
Tissue datasets for benchmarking
To assess the deconvolution performance on real tissue expression data, we used datasets for which the corresponding cell fractions were measured and published. The first dataset is the PBMC1 dataset, which was obtained from Zimmermann et al. (21). The second dataset, PBMC2, was downloaded from GEO with accession code GSE107011 (10). This dataset contains both RNA-seq profiles of immune cells (S4 cohort) and from bulk individuals (S13 cohort). As we were interested in the bulk profiles, we only used 12 samples from the S13 cohort from these data. Flow cytometry fractions were collected from the Monaco et al. publication (10).
In addition to the above mentioned two PBMC datasets, we used Ascites RNA-seq data. This dataset was provided by the authors, and cell type fractions for this dataset were taken from the supplementary materials of the publication (18).
For the evaluation on pancreas data, artificial bulk RNA-seq samples created from the scRNA-seq dataset of Xin et al. (20) were used. This dataset was downloaded from the resources of the MuSiC publication (8). The artificial bulk RNA-seq samples used for evaluation were then created using the “bulk_construct” function of the MuSiC tool.
To assess how Scaden and the GEP algorithms deal with the presence of unknown cell types, we generated PBMC bulk RNA samples from the four scRNA-seq datasets (6000 each). The undefined amount of unknown cells that was generated by this approach was removed to be replaced by defined amounts of 5, 10, 20, and 30% of unknown cells, respectively. Cell fractions of all four samples were predicted with Scaden trained on the other three.
Performance on these samples was then assessed to test robustness against unseen cell types in the bulk mixture. Scaden was trained on samples from all datasets but the test dataset, while CSx and MuSiC used data8k as a reference.
The microarray dataset GSE65133 was downloaded from GEO, and cell type fractions were taken from the original CS publication (6).
Last, we wanted to get insights into neurodegenerative cell fraction changes in the brain. While it is known that neurodegenerative diseases like AD are accompanied by a gradual loss of brain neurons, stage-specific cell type shifts are still hard to come by. Here, we use the ROSMAP study cortical RNA-seq dataset along with the corresponding clinical metadata, to infer cell type composition over six clinically relevant stages of neurodegeneration (22). Furthermore, to assess deconvolution accuracy on postmortem human brain tissue, we used 41 samples from the ROSMAP, for which cell composition information from immunohistochemistry (23) was recently released and for which fractions for all cell types were reported. The ROSMAP RNA-seq data were downloaded from www.synapse.org/. The cell composition values were provided by the authors of the study (23).
RNA-seq preprocessing and analysis
For the RNA-seq datasets analyzed in this study, we did not apply any additional processing steps but used the obtained count or expression tables directly as downloaded for all datasets except the ROSMAP dataset. For the latter, we generated count tables from raw FastQ files using Salmon (33) and the GRCh38 reference genome. FastQ files from the ROSMAP study were downloaded from Synapse (www.synapse.org).
Simulation of bulk RNA-seq samples from scRNA-seq data
Scaden’s DNN requires large amounts of training RNA-seq samples with known cell fractions. This explains why the generation of artificial bulk RNA-seq data is one of the key elements of the Scaden workflow.
To generate the training data, preprocessed scRNA-seq datasets were used (see the “Datasets and preprocessing” section), comprising the gene expression matrix and the cell type labels. Artificial RNA-seq samples were simulated by subsampling cells from individual scRNA-seq datasets; cells from different datasets were not merged into samples to preserve within-subject relationships. Datasets generated from multiple participants were split according to participant, and each subsampling was constrained to cells from one participant to capture the cross-subject heterogeneity and keep subject-specific gene dependencies.
The exact subsampling procedure is described in the following. First, for every simulated sample, random fractions were created for all different cell types within each scRNA-seq dataset using the random module of the Python package NumPy. Briefly, a random number was chosen from a uniform distribution between 0 and 1 using the NumPy function “random.rand()” for each cell type, and then this number was divided by the sum of all random numbers created to ensure the constraint of all fractions adding up to 1
where rc is the random number created for cell type c and Call is the set of all cell types. Here, fc is the calculated random fraction for cell type c. Then, each fraction was multiplied with the total number of cells selected for each sample, yielding the number of cells to choose for a specific cell type
where Nc is the number of cells to select for the cell type c, and Ntotal is the total number of cells contributing to one simulated RNA-seq sample (500, in this study). Next, Nc cells were randomly sampled from the scRNA-seq gene expression matrix for each cell type c. Afterward, the randomly selected single-cell expression profiles for every cell type are then aggregated by summing their expression values, to yield the artificial bulk expression profile for this sample.
Using the above-described approach, cell compositions that are strongly biased toward a certain cell type or are missing specific cell types are rare among the generated training samples. To account for this and to simulate cell compositions with a heavy bias to and the absence of certain cell types, a variation of the subsampling procedure was used to generate samples with sparse compositions, which we refer to as sparse samples. Before generating the random fractions for all cell types, a random number of cell types was selected to be absent from the sample, with the requirement of at least one cell type constituting the sample. After these leave-out cell types were chosen, random fractions were created and samples generated as described above. The average cell type proportions of the training dataset generated as described above are equal for all cell types. This allows for unbiased deconvolution as the true cell composition of a given tissue is not known beforehand. Using different sampling distributions (e.g., Gaussian and Uniform) or excluding sparse samples did not change Scaden’s deconvolution performance notably on the simulated PBMC datasets. This shows that Scaden is relatively robust to training data generated by different sampling procedures.
Using this procedure, we generated 32,000 samples for the human PBMC training dataset, 14,000 samples for the human pancreas training dataset, 6000 samples for human brain, and 30,000 samples for the mouse brain training dataset (table S3).
Artificial bulk RNA-seq datasets were stored in “h5ad” format using the Anndata package (30), which allows to store the samples together with their corresponding cell type ratios while also keeping information about the scRNA-seq dataset of origin for each sample. This allowed to access samples from specific datasets, which is useful for cross-validation.
Scaden overview
The following section contains an overview of the input data preprocessing, the Scaden model, model selection, and how Scaden predictions are generated.
Input data preprocessing
The data preprocessing step is aimed to make the input data more suitable for machine learning algorithms. To achieve this, an optimal preprocessing procedure should transform any input data from the simulated samples or from the bulk RNA-seq to the same feature scale. Before any scaling procedure can be applied, it must be ensured that both the training data and the bulk RNA-seq data subject to prediction share the same features. Therefore, before scaling, both datasets are limited to contain features (genes) that are available in both datasets. In addition, uninformative genes that have either zero expression or an expression variance below 0.1 were removed, leaving ~10,000 genes for model training and inference. The two-step processing procedure used for Scaden is described in the following:
First, to account for heteroscedasticity, a feature inherent to RNA-seq data, the data were transformed into logarithmic space by adding a pseudocount of 1 and then taking the Logarithm (base 2).
Second, every sample was scaled to the range [0,1] using the MinMaxScaler() class from the Sklearn preprocessing module. Per-sample scaling, unlike per-feature scaling that is more common in machine learning, assures that intergene relative expression patterns in every sample are preserved. This is important, as our hypothesis was that a neural network could learn the deconvolution from these intergene expression patterns
where xscaled, i is the log2 expression value of gene x in sample i, Xi is the vector of log2 expression values for all genes of sample i, min(Xi) is the minimum gene expression of vector Xi, and max(Xi) is the maximum gene expression of vector Xi.
Note that all training datasets are stored as expression values and are only processed as described above. In the deployment use case, the simulated training data should contain the same features as in the bulk RNA-seq sample that shall be deconvolved.
Model selection
The goal of model selection was to find an architecture and hyperparameters that robustly deconvolve simulated tissue RNA-seq data and, more importantly, real bulk RNA-seq data. Because of the very limited availability of bulk RNA-seq datasets with known cell fractions, model selection was mainly optimized on the simulated PBMC datasets. To capture interexperimental variation, we used leave-one-dataset-out cross-validation for model optimization: A model was trained on simulated data from all but one dataset, and performance was tested on simulated samples from the left-out dataset. This allows to simulate batch effects between datasets and helps to test the generalizability of the model. In the process of model selection and (hyper-) parameter optimization, performed on PBMC and Ascites datasets, we found three models with different architectures and dropout rates but comparable performance. To address overfitting in individual models, we decided to use a combination of models, expecting this to serve as another means of regularization. We did not test multiple combinations but rather used an informed choice with varying layer sizes and dropout regularization, with the goal to increase model diversity. We observed that the average of an ensemble of models generalized better to the test sets than individual models. Model training and prediction is done separately for each model, with the prediction averaging step combining all model predictions (fig. S1 and tables S4 and S6). We provide a list of all tested parameters in the Supplementary Materials (table S5).
Final Scaden model
The Scaden model learns cell type deconvolution through supervised training on datasets of simulated bulk RNA-seq samples simulated with scRNA-seq data. To account for model biases and to improve performance, Scaden consists of an ensemble of three DNNs with varying architectures and degrees of dropout regularization. All models of the ensemble use four layers of varying sizes between 32 and 1024 nodes, with dropout regularization implemented in two of the three ensemble models. The exact layer sizes and dropout rates are listed in table S4. The rectified linear unit is used as activation function in every internal layer. We used a Softmax function to predict cell fractions, as we did not see any improvements in using a linear output function with consecutive non-negativity correction and sum-to-one scaling. Python (v. 3.6.6) and the TensorFlow library (v. 1.10.0) were used for implementation of Scaden. A complete list of all software used for the implementation of Scaden is provided in table S15.
Training and prediction
After the preprocessing of the data, a Scaden ensemble can be trained on simulated tissue RNA-seq data or mixtures of simulated and real tissue RNA-seq data. Parameters are optimized using Adam with a learning rate of 0.0001 and a batch size of 128. We used an L1 loss as optimization objective
where yi is the vector of ground-truth fractions of sample i and is the vector of predicted fractions of sample i. Each of the three ensemble models is trained independently for 5000 steps. This “early stopping” serves to avoid domain overfitting on the simulated tissue data, which would decrease the model performance on the real tissue RNA-seq data. We observed that training for more steps lead to an average performance decrease on real tissue RNA-seq data. To perform deconvolution with Scaden, a bulk RNA-seq sample is fed into a trained Scaden ensemble, and three independent predictions for the cell type fractions of this sample are generated by the trained DNNs. These three predictions are then averaged per cell type to yield the final cell type composition for the input bulk RNA-seq sample
where is the final predicted fraction for cell type c and is the predicted fraction for cell type c of model i.
Scaden requirements
Currently, a disadvantage of the Scaden algorithm is the necessity to train a new model for deconvolution if no perfect overlap in the feature space exists. This constraint limits the usefulness of pretrained models. Once trained, however, the prediction runtime scales linearly with sample numbers and is usually in the order of seconds, making Scaden a useful tool if deconvolution is to be performed on very large datasets. While the requirements are dataset dependent, the Scaden demo was profiled to require a peak of 3.2 GB of random-access memory (RAM) during the DNN training process, so a computer with 8 GB of RAM should be able to run it smoothly. In our tests with an Intel(R) Xeon(R) CPU E5-1630 workstation, the demo could run in 22 min, spending most of the CPU time in the DNN training process. The most prominent and obvious issue of Scaden is the difference between simulated scRNA-seq data used for training and the bulk RNA-seq data subject to inference. While Scaden is able to transfer the learned deconvolution between the two data types and achieves state-of-the-art performance, we hypothesize that efforts to improve this translatability could improve Scaden’s prediction accuracy even further. Algorithmic improvements are therefore likely to address this issue and are planned for future releases.
Algorithm comparison
We used several performance measures to compare Scaden to four existing cell deconvolution algorithms, CS with LM22 GEP, CSx, MuSiC, and CPM. To compare the performance of the five deconvolution algorithms, we measured the RMSE, Lin’s CCC, Pearson product moment correlation coefficient r, and R2 values, comparing real and predicted cell fractions estimates. In addition, to identify systematic prediction errors and biases, slope and intercept for the regression lines were calculated. These metrics are defined as follows
where y are the ground-truth fractions, are the prediction fractions, σx is the SD of x, is the covariance of y and , and are the mean of the predicted and ground-truth fractions, respectively.
All metrics were calculated for all data points of a dataset and separately for all data points of a specific cell type. For the latter approach, we then averaged the resulting values to recover single values. While the metrics calculated on all data points might be sufficient, we deem that the cell type–specific deconvolution might, in many instances, be of even greater interest. It is noteworthy in this context that cell type–specific deconvolution performance can be quite weak, depending on the dataset. This is true for all tested deconvolution algorithms, while Scaden achieves best performance.
CIBERSORT
CS is a cell convolution algorithm based on specialized GEPs and support vector regression. Cell composition estimations were obtained using the CS web application (https://cibersort.stanford.edu/). For all deconvolutions with CS, we used the LM22 GEP, which was generated by the CS authors from 22 leukocyte subsets profiled on the HGU133A microarray platform.
Because the LM22 GEP matrix contains cell types at a finer granularity than what was used for this study, predicted fractions of subcell types were added together. For cell grouping, we used the mapping of subcell types to broader types given by figure 6 from Monaco et al. (10). We provide a table with the exact mappings used here in the Supplementary Materials (table S13). The deconvolution was performed using 500 permutations with quantile normalization disabled for all datasets but GSE65133 (Microarray), as is recommended for RNA-seq data. We used default settings for all other CS parameters.
CIBERSORTx
CSx is a recent variant of CS that can generate GEP matrices from scRNA-seq data and use these for deconvolution. For additional deconvolution robustness, it applies batch normalization to the data. All signature matrices were created by uploading the labeled scRNA-seq expression matrices and using the default options. Quantile normalization was disabled. For deconvolution on simulated data, no batch normalization was used. For all bulk RNA-seq datasets, the S-Mode batch normalization was chosen. All PBMC datasets were deconvolved using a GEP matrix generated from the data6k dataset (for simulated samples from data6k, a donorA GEP matrix was chosen).
MuSiC
MuSiC is a deconvolution algorithm that uses multisubject scRNA-seq datasets as GEP matrices in an attempt to include heterogeneity in the matrices to improve generalization. While MuSiC tries to address similar issues of previous deconvolution algorithms by using scRNA-seq data, the approach is very different. For deconvolution, MuSiC applies a sophisticated GEP-based deconvolution algorithm that uses weighted non-negative least-squares regression with an iterative estimation procedure that imposes more weight on informative genes and less weight on noninformative genes.
The MuSiC R package contains functionality to generate the necessary GEP matrix given an scRNA-seq dataset and cell type labels. To generate MuSiC deconvolution predictions on PBMC datasets, we used the data8k scRNA-seq dataset as reference data for MuSiC and follow the tutorial provided by the authors to perform the deconvolution. For deconvolution of artificial samples generated from the data8k dataset, we provided MuSiC with the data6k dataset as a reference instead.
MuSiC was developed with a focus on multisubject scRNA-seq datasets, in which the algorithm tries to take advantage from the added heterogeneity that these datasets contain, by calculating a measure of cross-subject consistency for marker genes. To assess how Scaden performs on multisubject datasets compared to MuSiC, we evaluated both methods on artificial bulk RNA-seq samples from human pancreas. We used the bulk_construct function from MuSiC to combine the cells from all 18 participants contained in the scRNA-seq dataset from Xin et al. (20) to generate artificial bulk samples for evaluation. Next, as a multisubject reference dataset, we used the pancreas scRNA-seq dataset from Segerstolpe et al. (19), which contains single-cell expression data from 10 different participants, 4 of which with type 2 diabetes. For Scaden, the Segerstolpe scRNA-seq dataset was split by participants, and training datasets were generated for each participant, yielding in total 10,000 samples. For MuSiC, a processed version of this dataset was downloaded from the resources provided by the MuSiC authors (8) and used as an input reference dataset for the MuSiC deconvolution. Deconvolution was then performed according to the MuSiC tutorial, and performance was compared according to the above-defined metrics.
Cell Population Mapping
CPM is a deconvolution algorithm that uses single-cell expression profiles to identify a so-called “cell population map” from bulk RNA-seq data (9). In CPM, the cell population map is defined as composition of cells over a cell-state space, where a cell state is defined as a current phenotype of a single cell. Contrary to other deconvolution methods, CPM tries to estimate the abundance of all cell states and types for a given bulk mixture, instead of only deconvolving the cell types. As input, CPM requires an scRNA-seq dataset and a low-dimensional embedding of all cells in this dataset, which represents the cell-state map. As CPM estimates abundances of both cell states and types, it can be used for cell type deconvolution by summing up all estimated fractions for all cell states of a given cell type, a method that is implemented in the scBio R package, which contains the CPM method. To perform deconvolution with CPM, we used the data6k PBMC scRNA-seq dataset as an input reference for all PBMC samples. For samples simulated from the data6k dataset, we used the data8k dataset as a reference. According to the CPM paper, a dimension reduction method can be used to obtain the cell-state space. We therefore used Uniform Manifold Approximation and Projection (UMAP), a dimension reduction method widely used for scRNA-seq data, to generate the cell-state space mapping for the input scRNA-seq data. Deconvolution was then performed using the CPM function of the scBio package with an scRNA-seq dataset and accompanying UMAP embedding as input.
Code and software availability
The source code for Scaden is available at https://github.com/KevinMenden/scaden. Documentation is published at https://scaden.readthedocs.io. Code to generate the figures along with the training datasets used in this study is published at figshare: https://figshare.com/projects/Scaden/62834. The Scaden web application can be accessed at https://scaden.ims.bio.
Supplementary Material
Acknowledgments
We would like to thank the people of the Genome Biology of Neurodegenerative Diseases group and Institute of Medical Systems Biology for helpful discussions and suggestions. Funding: This study was supported, in part, by RiMod-FTD, an EU Joint Programme–Neurodegenerative Disease Research (JPND), and the NOMIS Foundation to K.M., A.D., and P.H. and BMBF Integrative Data Semantics for Neurodegenerative research (IDSN), ERA-Net E-Rare MAXOMOD, CRC 1286/Z2, CRU 296 P8, and CRU 306 P-C to M.M., S.O., D.S.M., K.K., and S.B. Author contributions: K.M. and S.B. initiated the project. K.M., P.H., and S.B. designed the study, deep learning models, and analysis. K.M., M.M., and S.O. built the deep learning models. K.M., M.M., K.K., and A.D. analyzed the data. D.S.M. and S.O. built the Scaden web application. K.M., K.K., and S.B. wrote the manuscript. M.M., A.D., and P.H. contributed to the manuscript writing. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. Only publicly available datasets were used during this study. The scRNA-seq PBMC datasets donorA, donorC, data6k, and data8k were all downloaded from 10X Genomics (https://support.10xgenomics.com/single-cell-gene-expression/datasets), where they are listed as “Frozen PBMCs (Donor A),” “Frozen PBMCs (Donor C),” “6k PBMCs from a Healthy Donor,” and “8k PBMCs from a Healthy Donor,” respectively. The pancreas scRNA-seq dataset from Segerstolpe et al. (19) was downloaded from ArrayExpress with accession code E-MTAB-5061. The scRNA-seq datasets from Baron et al. (34) (pancreas), Tasic et al. (35), Zeisel et al. (32), Romanov et al. (36), Campbell et al. (37), and Chen et al. (38) (all mouse brain) were all downloaded from https://hemberg-lab.github.io/scRNA.seq.datasets/. The ascites scRNA-seq dataset was downloaded from https://figshare.com/s/711d3fb2bd3288c8483a. The bulk RNA-seq dataset PBMC1 is accessible from ImmPort with accession code SDY67. The PBMC2 dataset was downloaded from GEO with accession code GSE107011. The ROSMAP human brain RNA-seq dataset was downloaded from Synapse (ID: syn3219045). The bulk RNA-seq data from ascites was provided by Schelker et al. (18). The pancreas scRNA-seq dataset from Xin et al. (20) was accessed from the MuSiC tutorial site (https://xuranw.github.io/MuSiC/articles/pages/data.html).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/30/eaba2619/DC1
REFERENCES AND NOTES
- 1.Hrdlickova R., Toloue M., Tian B., RNA-Seq methods for transcriptome analysis. Wiley Interdiscip. Rev. RNA 8, e1364 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Egeblad M., Nakasone E. S., Werb Z., Tumors as organs: Complex tissues that interface with the entire organism. Dev. Cell 18, 884–901 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuhn A., Thu D., Waldvogel H. J., Faull R. L. M., Luthi-Carter R., Population-specific expression analysis (PSEA) reveals molecular changes in diseased brain. Nat. Methods 8, 945–947 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Avila Cobos F., Vandesompele J., Mestdagh P., De Preter K., Computational deconvolution of transcriptomics data from mixed cell populations. Bioinformatics 34, 1969–1979 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Mohammadi S., Zuckerman N., Goldsmith A., Grama A., A critical survey of deconvolution methods for separating cell types in complex tissues. Proc. IEEE 105, 340–366 (2017). [Google Scholar]
- 6.Newman A. M., Liu C. L., Green M. R., Gentles A. J., Feng W., Xu Y., Hoang C. D., Diehn M., Alizadeh A. A., Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Newman A. M., Steen C. B., Liu C. L., Gentles A. J., Chaudhuri A. A., Scherer F., Khodadoust M. S., Esfahani M. S., Luca B. A., Steiner D., Diehn M., Alizadeh A. A., Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 37, 773–782 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang X., Park J., Susztak K., Zhang N. R., Li M., Bulk tissue cell type deconvolution with multi-subject single-cell expression reference. Nat. Commun. 10, 380 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frishberg A., Peshes-Yaloz N., Cohn O., Rosentul D., Steuerman Y., Valadarsky L., Yankovitz G., Mandelboim M., Iraqi F. A., Amit I., Mayo L., Bacharach E., Gat-Viks I., Cell composition analysis of bulk genomics using single-cell data. Nat. Methods 16, 327–332 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Monaco G., Lee B., Xu W., Mustafah S., Hwang Y. Y., Carré C., Burdin N., Visan L., Ceccarelli M., Poidinger M., Zippelius A., Pedro de Magalhães J., Larbi A., RNA-Seq signatures normalized by mRNA abundance allow absolute deconvolution of human immune cell types. Cell Rep. 26, 1627–1640.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vallania F., Tam A., Lofgren S., Schaffert S., Azad T. D., Bongen E., Haynes W., Alsup M., Alonso M., Davis M., Engleman E., Khatri P., Leveraging heterogeneity across multiple datasets increases cell-mixture deconvolution accuracy and reduces biological and technical biases. Nat. Commun. 9, 4735 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Venet D., Pecasse F., Maenhaut C., Bersini H., Separation of samples into their constituents using gene expression data. Bioinformatics 17, S279–S287 (2001). [DOI] [PubMed] [Google Scholar]
- 13.Shapiro E., Biezuner T., Linnarsson S., Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat. Rev. Genet. 14, 618–630 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Tabula Muris Consortium , Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelley K. W., Nakao-Inoue H., Molofsky A. V., Oldham M. C., Variation among intact tissue samples reveals the core transcriptional features of human CNS cell classes. Nat. Neurosci. 21, 1171–1184 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hicks S. C., Townes F. W., Teng M., Irizarry R. A., Missing data and technical variability in single-cell RNA-sequencing experiments. Biostatistics 19, 562–578 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin L. I., A concordance correlation coefficient to evaluate reproducibility. Biometrics 45, 255–268 (1989). [PubMed] [Google Scholar]
- 18.Schelker M., Feau S., Du J., Ranu N., Klipp E., MacBeath G., Schoeberl B., Raue A., Estimation of immune cell content in tumour tissue using single-cell RNA-seq data. Nat. Commun. 8, 2032 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Segerstolpe Å., Palasantza A., Eliasson P., Andersson E. M., Andréasson A. C., Sun X., Picelli S., Sabirsh A., Clausen M., Bjursell M. K., Smith D. M., Kasper M., Ämmälä C., Sandberg R., Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab. 24, 593–607 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xin Y., Kim J., Okamoto H., Ni M., Wei Y., Adler C., Murphy A. J., Yancopoulos G. D., Lin C., Gromada J., RNA sequencing of single human islet cells reveals type 2 diabetes genes. Cell Metab. 24, 608–615 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Zimmermann M. T., Oberg A. L., Grill D. E., Ovsyannikova I. G., Haralambieva I. H., Kennedy R. B., Poland G. A., System-wide associations between DNA-methylation, gene expression, and humoral immune response to influenza vaccination. PLOS ONE 11, e0152034 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bennett D. A., Buchman A. S., Boyle P. A., Barnes L. L., Wilson R. S., Schneider J. A., Religious orders study and rush memory and aging project. J. Alzheimers Dis. 64, S161–S189 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patrick E., Taga M., Ergun A., Ng B., Casazza W., Cimpean M., Yung C., Schneider J. A., Bennett D. A., Gaiteri C., De Jager P. L., Bradshaw E. M., Mostafavi S., Deconvolving the contributions of cell-type heterogeneity on cortical gene expression. bioRxiv 2019, 566307 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Darmanis S., Sloan S. A., Zhang Y., Enge M., Caneda C., Shuer L. M., Gephart M. G. H., Barres B. A., Quake S. R., A survey of human brain transcriptome diversity at the single cell level. Proc. Natl. Acad. Sci. U.S.A. 112, 7285–7290 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lake B. B., Ai R., Kaeser G. E., Salathia N. S., Yung Y. C., Liu R., Wildberg A., Gao D., Fung H. L., Chen S., Vijayaraghavan R., Wong J., Chen A., Sheng X., Kaper F., Shen R., Ronaghi M., Fan J. B., Wang W., Chun J., Zhang K., Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 352, 1586–1590 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Braak H., Braak E., Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259 (1991). [DOI] [PubMed] [Google Scholar]
- 27.J. Yosinski, J. Clune, A. Nguyen, T. Fuchs, H. Lipson, Understanding Neural Networks Through Deep Visualization (2015); http://arxiv.org/abs/1506.06579.
- 28.Athiwaratkun B., Finzi M., Izmailov P., Wilson A. G., Improving consistency-based semi-supervised learning with weight averaging. Jmlr 17, 1–35 (2018). [Google Scholar]
- 29.M. Zhang, K. T. Ma, J. H. Lim, Q. Zhao, J. Feng, Deep future gaze: Gaze anticipation on egocentric videos using adversarial networks, in 2017 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Honolulu, HI, USA, 21 to 26 July 2017. [Google Scholar]
- 30.Wolf F. A., Angerer P., Theis F. J., SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Satija R., Farrell J. A., Gennert D., Schier A. F., Regev A., Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33, 495–502 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zeisel A., Hochgerner H., Lönnerberg P., Johnsson A., Memic F., van der Zwan J., Häring M., Braun E., Borm L. E., La Manno G., Codeluppi S., Furlan A., Lee K., Skene N., Harris K. D., Hjerling-Leffler J., Arenas E., Ernfors P., Marklund U., Linnarsson S., Molecular architecture of the mouse nervous system. Cell 174, 999–1014.e22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Love M. I., Soneson C., Patro R., Swimming downstream: Statistical analysis of differential transcript usage following Salmon quantification. F1000Res. 7, 952 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baron M., Veres A., Wolock S. L., Faust A. L., Gaujoux R., Vetere A., Ryu J. H., Wagner B. K., Shen-Orr S. S., Klein A. M., Melton D. A., Yanai I., A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cell Syst. 3, 346–360.e4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tasic B., Menon V., Nguyen T. N., Kim T. K., Jarsky T., Yao Z., Levi B., Gray L. T., Sorensen S. A., Dolbeare T., Bertagnolli D., Goldy J., Shapovalova N., Parry S., Lee C., Smith K., Bernard A., Madisen L., Sunkin S. M., Hawrylycz M., Koch C., Zeng H., Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat. Neurosci. 19, 335–346 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Romanov R. A., Zeisel A., Bakker J., Girach F., Hellysaz A., Tomer R., Alpár A., Mulder J., Clotman F., Keimpema E., Hsueh B., Crow A. K., Martens H., Schwindling C., Calvigioni D., Bains J. S., Máté Z., Szabó G., Yanagawa Y., Zhang M.-D., Rendeiro A., Farlik M., Uhlén M., Wulff P., Bock C., Broberger C., Deisseroth K., Hökfelt T., Linnarsson S., Horvath T. L., Harkany T., Molecular interrogation of hypothalamic organization reveals distinct dopamine neuronal subtypes. Nat. Neurosci. 20, 176–188 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Campbell J. N., Macosko E. Z., Fenselau H., Pers T. H., Lyubetskaya A., Tenen D., Goldman M., Verstegen A. M. J., Resch J. M., McCarroll S. A., Rosen E. D., Lowell B. B., Tsai L. T., A molecular census of arcuate hypothalamus and median eminence cell types. Nat. Neurosci. 20, 484–496 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen R., Wu X., Jiang L., Zhang Y., Single-cell RNA-seq reveals hypothalamic cell diversity. Cell Rep. 18, 3227–3241 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/30/eaba2619/DC1