

Abstract
Background
Dyskeratosis congenita (DKC) and idiopathic aplastic anemia (AA) are bone marrow failure syndromes that share characteristics of premature aging with severe telomere attrition. Aging is also reflected by DNA methylation changes, which can be utilized to predict donor age. There is evidence that such epigenetic age predictions are accelerated in premature aging syndromes, but it is yet unclear how this is related to telomere length. DNA methylation analysis may support diagnosis of DKC and AA, which still remains a challenge for these rare diseases.
Results
In this study, we analyzed blood samples of 70 AA and 18 DKC patients to demonstrate that their epigenetic age predictions are overall increased, albeit not directly correlated with telomere length. Aberrant DNA methylation was observed in the gene PRDM8 in DKC and AA as well as in other diseases with premature aging phenotype, such as Down syndrome and Hutchinson-Gilford-Progeria syndrome. Aberrant DNA methylation patterns were particularly found within subsets of cell populations in DKC and AA samples as measured with barcoded bisulfite amplicon sequencing (BBA-seq). To gain insight into the functional relevance of PRDM8, we used CRISPR/Cas9 technology to generate induced pluripotent stem cells (iPSCs) with heterozygous and homozygous knockout. Loss of PRDM8 impaired hematopoietic and neuronal differentiation of iPSCs, even in the heterozygous knockout clone, but it did not impact on epigenetic age.
Conclusion
Taken together, our results demonstrate that epigenetic aging is accelerated in DKC and AA, independent from telomere attrition. Furthermore, aberrant DNA methylation in PRDM8 provides another biomarker for bone marrow failure syndromes and modulation of this gene in cellular subsets may be related to the hematopoietic and neuronal phenotypes observed in premature aging syndromes.
Graphical abstract
Keywords: PRDM8, Epigenetic clock, DNA methylation, Telomere, Aging, Dyskeratosis congenita, Aplastic anemia, iPSC, Hematopoietic differentiation, Neuronal differentiation
Background
Premature aging syndromes, such as Down syndrome, Werner syndrome, or dyskeratosis congenita (DKC), are often associated with impaired neurologic development or hematological abnormalities [1–3]. Down syndrome is caused by trisomy 21, Werner syndrome results from mutations in the Werner syndrome ATP-dependent helicase (WRN), and DKC is a paradigmatic disease for studying the consequences of critical short telomeres [1, 2]. Premature telomere shortening in DKC is caused by mutations in genes of the telomerase complex such as telomerase reverse transcriptase (TERT) or telomerase RNA component (TERC), resulting in impaired telomere maintenance [4]. Clinically, DKC patients show a triad of skin hyperpigmentation, nail dystrophy, and oral leukoplakia [1, 5]. Severe forms of DKC are characterized by a neurological impairment such as disturbed cerebellar development, whereas cryptic forms develop slowly without manifestation of characteristic symptoms before adulthood and may develop bone marrow failure, which can be observed in up to 90% of all DKC patients by the age of 30 [1, 6, 7]. The bone marrow is then hypocellular and indistinguishable from patients with acquired aplastic anemia (AA), a disease characterized by a T cell-mediated autoimmune destruction of the hematopoietic stem cell compartment [8, 9]. Diagnosis of DKC is often based on premature telomere shortening in peripheral blood leukocytes compared to healthy individuals, followed by genetic analysis of the relevant telomere maintenance-associated genes [10]. Of note, in acquired AA, telomere length in the granulocyte compartment can also show substantial attrition and was shown to reflect the degree of autoimmune-mediated damage to the hematopoietic stem cell compartment [11]. This may hamper correct diagnosis of DKC [12], which is of utmost importance since DKC, in contrast to acquired AA, is not responding to immunosuppressive therapy and requires optimized conditioning protocols in case of allogeneic stem cell transplantation [13].
Another hallmark of aging, besides telomere shortening, is epigenetic changes. Over the life time, the DNA methylation pattern changes continuously at specific CG dinucleotides (CpG sites) in the genome. Analysis of such age-associated DNA methylation changes can be used as a biomarker for aging, often referred to as “epigenetic clock” [14]. Various different epigenetic signatures have been described, which were often based on DNA methylation profiles with Illumina BeadChip technology [15, 16]. For more cost-effective high throughput analysis, we derived a targeted epigenetic age-predictor based on three age-associated CpG sites, which was specifically trained for blood [17]. In a preliminary analysis of 5 DKC and 15 AA patients, we observed that this epigenetic clock might be accelerated in bone marrow failure syndromes [17]. However, while DNA methylation profiles of three DKC patients in a further study did not reveal unequivocal acceleration of epigenetic age, we did observe a significant hypermethylation within the promoter region of the short transcript of the PR Domain Zinc Finger Protein 8 (PRDM8) [18]. PRDM8 belongs to the PRDM family characterized by a conserved N-terminal PR domain related to the catalytic SET domains, which represent a large group of histone methyltransferases [19–22] that seem to be generally involved in regulation of stem cell function, developmental processes, and malignant transformation [23, 24].
In this study, we demonstrate that epigenetic aging is overall moderately accelerated in DKC and AA, and this is independent from telomere attrition. Furthermore, DKC and AA revealed aberrant DNA methylation within the gene PRDM8, which was also observed for other premature aging syndromes. To gain further insight into the biological function of this gene in induced pluripotent stem cells (iPSCs), we created PRDM8 knockout clones, resulting in impaired hematopoietic and neuronal differentiation.
Results
Telomere attrition and accelerated epigenetic aging in bone marrow failure syndromes
Telomere length was measured in granulocytes of 65 AA and 17 DKC patients. All DKC patients and most AA patients revealed significant telomere attrition (Fig. 1a). In comparison to the age-adjusted linear model of the 105 healthy donors [17], the predicted age based on telomere length in granulocytes revealed a significant offset (delta telomere age) in AA (mean absolute error [MAE] = 35.03 years; P = 0.0003), which were on average predicted 19.05 years older than their chronological age, and DKC (MAE = 84.14 years; P < 0.0001; Fig 1b), which were also on average predicted 84.14 years older than their chronological age. Similar results were observed when telomere length was analyzed in lymphocytes (Additional file 1: Fig. S1a, b). This is in line with previous observations on 11 independent DKC and 27 independent AA samples [18].
Fig. 1.

Telomere age and epigenetic age are increased in dyskeratosis congenita and aplastic anemia. a Telomere lengths of 105 healthy donors [17], 65 aplastic anemia (AA), and 17 dyskeratosis congenita (DKC) patients were measured in granulocytes and correlated to chronological age. DKC and AA patients show a reduced telomere length compared to healthy controls. b Offset of predicted telomere age (delta age) was higher for DKC and AA patients than for healthy controls [17]. t test: *** P < 0.001, **** P < 0.0001. c Epigenetic age predictions of 70 AA and 18 DKC samples revealed much lower correlation to chronological age than 243 healthy controls [17, 25]. d The difference between predicted epigenetic age and chronological age (delta age) was higher for DKC and AA than for healthy controls, as described for other samples before [17]. t test: ** P < 0.01, **** P < 0.0001. e, f Telomere age and epigenetic age (e), as well as delta telomere age and delta epigenetic age (f), do not correlate in 62 AA and 13 DKC samples
We then analyzed epigenetic age in these samples using our previously described epigenetic aging signature based on DNA methylation at three CpGs within the genes phosphodiesterase 4C (PDE4C; CpG next to cg17861230, which is not presented by the Illumina BeadChip), integrin alpha 2b (ITGA2B; cg25809905), and aspartoacylase (ASPA; cg02228185) [17]. For blood samples of 243 healthy controls [17, 25], we observed a good correlation with chronological age (MAE = 5.45 years; R2 = 0.83), whereas there was a significant offset in epigenetic age predictions for the 70 AA (MAE = 10.50 years; R2 = 0.65; P < 0.0001) and 18 DKC patients (MAE = 17.38 years; R2 = 0.15; P < 0.0001). On average, AA and DKC samples were predicted 6.06 years, and 16.36 years older than their chronological age (Fig. 1c, d), which is in line with previous observations [18]. We then analyzed if telomere attrition was correlated with accelerated epigenetic aging, but this was not the case (Fig. 1e, f and Additional file 1: Fig. S1c, d). Thus, telomere attrition and epigenetic aging seem to reflect two independent processes of premature cellular aging in these bone marrow failure syndromes.
Aberrant DNA methylation patterns within the gene PRDM8 in AA and DKC
The gene of PR Domain Zinc Finger Protein 8 (PRDM8) was previously shown to be hypermethylated in DKC and AA patients [18]. In continuation of this work, we analyzed if DNA methylation at this gene is a suitable biomarker for these bone marrow failure syndromes. In fact, two independent DNA methylation assays for two CpGs (cg19409579 and cg27242132) demonstrated that the majority of AA and DKC samples revealed significantly higher DNA methylation as compared to blood samples of healthy controls (Fig. 2a and Additional file 1: Fig. S2a). Hypermethylation at PRDM8 did not correlate with accelerated epigenetic age or telomere attrition, indicating that these processes are not directly associated with each other (Fig. 2b, c and Additional file 1: Fig. S2b, c). Furthermore, there was no correlation of DNA methylation in the PRDM8 assays with blood counts (Additional file 1: Fig. S3a, b).
Fig. 2.
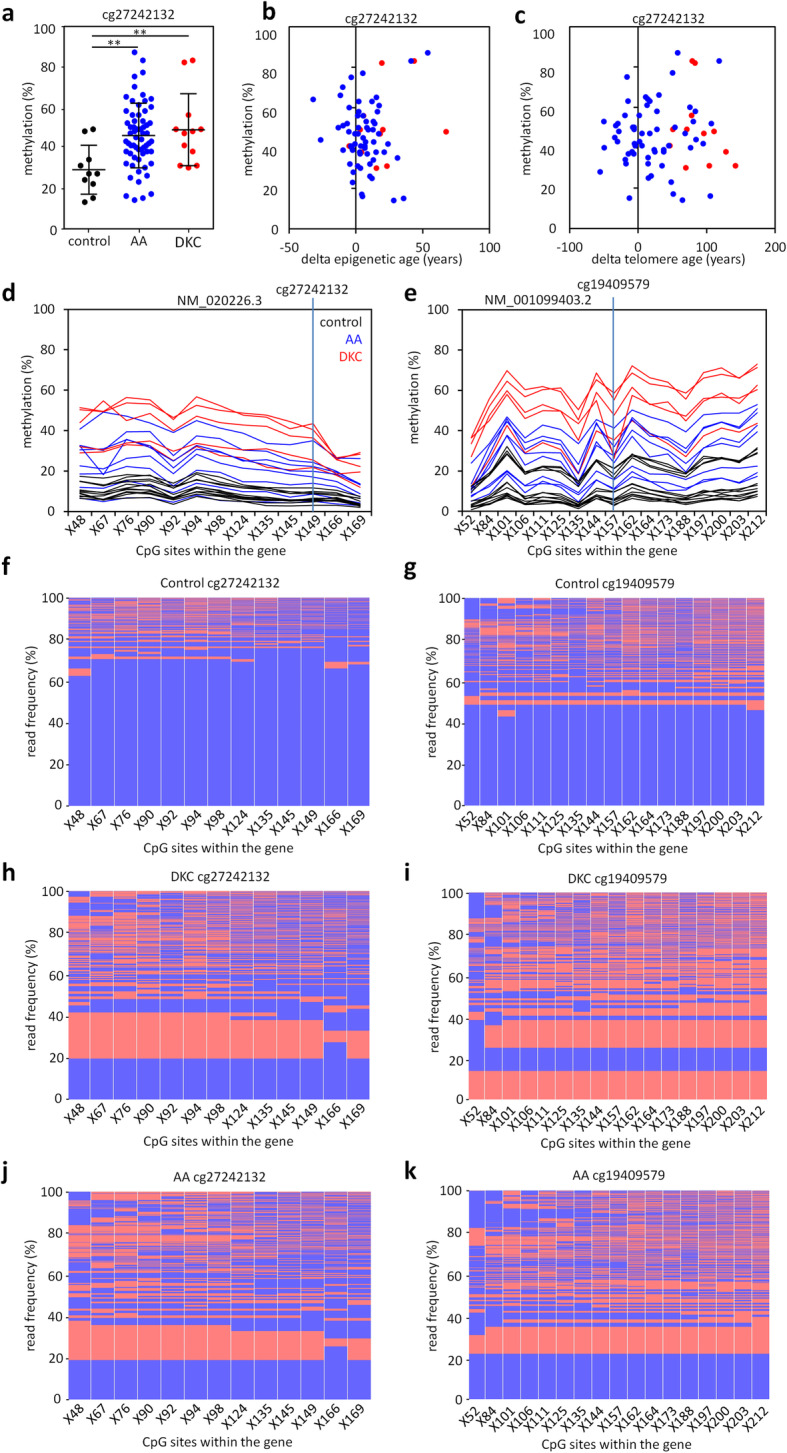
Hypermethylation in PRDM8 in dyskeratosis congenita and aplastic anemia. a DNA methylation was measured by MassARRAY at the CpG site cg27242132 in blood samples of 62 new aplastic anemia (AA) and 12 new dyskeratosis congenita (DKC) patients, as compared to 10 previously described healthy controls [18]. t test: ** P < 0.01. b, c DNA methylation in cg27242132 does not correlate to epigenetic age (b) or telomere age (c). d, e Barcoded bisulfite amplicon sequencing (BBA-seq) was used to determined DNA methylation around the two relevant CpGs (cg27242132 and cg19409579) in independent samples: 12 controls, 8 AA, and 5 DKC validated higher methylation in DKC and AA compared to controls across the amplicons with 13 (assay 1) and 17 (assay 2) neighboring CpGs. f–k The frequencies of DNA methylation patterns within individual BBA-seq reads are exemplarily depicted for both PRDM8 amplicons in a healthy donor (f, g), a DKC patient (h, i) and a AA patient (j, k)
The region in PRDM8 is rich in CpG sites and therefore these measurements were performed with MassARRAY technology to facilitate analysis of longer amplicons than allowed for pyrosequencing. However, both methods only provide average DNA methylation levels across all cells within a sample. Therefore, we utilized barcoded bisulfite amplicon sequencing (BBA-seq) to obtain individual reads by deep sequencing that reflect the heterogeneity of DNA methylation patterns within the cells of a given sample. We exemplarily analyzed 12 healthy, 8 AA, and 5 DKC samples that were not included in the above analysis with BBA-seq. For both PRDM8 regions (around cg27242132 and cg19409579), the results clearly validated aberrant DNA methylation within 13 (assay 1) and 17 (assay 2) neighboring CpG sites (Fig. 2d, e). We then analyzed the DNA methylation patterns within individual BBA-seq reads of single DNA strands. In healthy samples, we observed that the CpGs within these regions were predominately non-methylated, whereas about 25 to 60% of the reads provided stochastic patterns of DNA methylation at neighboring CpGs (Fig. 2f, g). The fraction of methylated reads was higher in AA and DKC samples, and many of them comprised reads that were consistently methylated across all CpGs (Fig. 2 h–k). There was no clear difference in the DNA methylation patterns of AA and DKC—while the DNA methylation level was overall higher in these samples, they also comprised entirely non-methylated strands, reads with stochastic DNA methylation at neighboring CpGs, and often reads that are consistently methylated.
Aberrant DNA methylation in PRDM8 is not reflected on average gene expression levels
Since DKC is one of various prominent premature aging syndromes, we subsequently analyzed if PRDM8 is also aberrantly methylated in other premature aging syndromes. To this end, we utilized DNA methylation profiles of Werner syndrome (GSE42865), Hutchinson-Gilford-Progeria syndrome (HGPS, GSE42865), Down syndrome (GSE52588), and profiles of DKC patients (GSE75310). Aberrant hypermethylation of the two relevant CpG sites cg19409579 and cg27242132 was particularly observed in DKC, while there was also some offset in other premature aging diseases (Fig. 3a and Additional file 1: Fig. S4a). Furthermore, aberrant hypomethylation within the promoter region of the longer of the two PRDM8 transcripts (NM_020226.3) was observed in all premature aging syndromes, albeit this was less prominent in DKC and Werner syndrome (Additional file 1: Fig. S4b). These results indicate that aberrant epigenetic patterns in PRDM8 are frequently observed in premature aging syndromes.
Fig. 3.

DNA methylation in PRDM8 in premature aging syndromes and differential gene expression. a To evaluate aberrant DNA methylation in PRDM8 (at cg27242132) in other premature aging syndromes, we used available DNA methylation profiles of 103 healthy controls (GSE36054, GSE32148, GSE49064), 4 DKC samples (GSE75310), 29 Down syndrome samples (GSE52588), 4 Werner syndrome samples (GSE42865), and 3 Hutchinson-Gilford-Progeria syndrome (HGPS) samples (GSE42865). DNA methylation levels (β values) revealed hypermethylation in all premature aging syndromes. t test: * P < 0.05, ** P < 0.01, *** P < 0.001. b–d Expression of all PRDM8 transcripts (b), the long PRDM8 transcript (NM020026.3; c), and the short transcript (NM001099403.2; d) was analyzed by qRT-PCR in 10 controls, 27 AA, and 14 DKC patients. t test: * P < 0.05; ns, not significant
We have subsequently analyzed if PRDM8 gene expression is also aberrantly regulated in these syndromes. Our previous analysis with quantitative RT-PCR revealed downregulation of PRDM8 in five DKC samples [18]. We have now performed qRT-PCR for a new set of more patient samples (10 healthy controls, 27 AA patients, and 14 DKC patients) and did not observe a significant difference in gene expression levels of either the long (NM_020226.3) or the short transcript of PRDM8 (NM_001099403.2; Fig. 3b–d). Gene expression levels for all transcripts were slightly higher in AA than in controls, but expression was generally rather low in the blood samples. In addition, we have analyzed public gene expression profiles of Down syndrome (blood; GSE35665, n = 15 [26]), Werner syndrome (fibroblasts; GSE48761, n = 10 [27]), and HGPS (fibroblasts; GSE69391, n = 6 [28] and GSE3860, n = 3 [29]), but none of these datasets revealed significant gene expression changes in PRDM8 (data not shown). This finding might be attributed to the fact that PRDM8 was also hardly expressed in those datasets. Furthermore, as suggested by the BBA-seq data, aberrant DNA methylation patterns are not observed in all DNA molecules of a sample and hence a potential epigenetic effect might be masked by other cell populations without differential DNA methylation. Either way, PRDM8 expression might be relevant during cellular differentiation or in specific cellular subsets.
PRDM8 knockout impairs hematopoietic differentiation
To gain insight into the functional relevance of PRDM8, we generated clonal induced pluripotent stem cell (iPSC) lines with homozygous and heterozygous gene knockout of PRDM8. Deletion of the start codon with CRISPR/Cas9 nickase resulted in loss of PRDM8 (Fig. 4a). Sequencing indicated that deletion of the intron/exon boundary led to a reading frame shift and formation of a premature stop codon, thus generating a complete knockout of the PRDM8 protein. The PRDM8+/− and PRDM8−/− iPSCs maintained expression of the pluripotency markers OCT4 and TRA-1-60, they could be culture expanded over many passages (up to 60 passages), and they revealed a positive Epi-Pluri-Score [30], indicating that PRDM8 knockout clones remained pluripotent (Additional file 1: Fig. S5a, b). Furthermore, upon differentiation in embryoid bodies (EBs) for 2 weeks, qRT-PCR analysis validated upregulation of marker genes for ectodermal, mesodermal, and endodermal lineages, albeit upregulation of the neuronal markers nestin (NES) and paired box 6 (PAX6) was reduced for the PRDM8 knockouts (Additional file 1: Fig. S5c). Expression of PRDM8 was hardly detectable in the knockout cells by qRT-PCR upon 14 days of differentiation in the embryoid body assay (Fig. 4b).
Fig. 4.

Impaired hematopoietic differentiation of PRDM8−/− iPSCs. a Scheme of two main transcripts of PRDM8 (NM_020226.3 and NM_001099403.2) and sites of genomic editing. Two pairs of guide RNAs (gRNAs) were designed targeting the intron/exon boundary at the start codon of both transcripts. b Genome editing was confirmed by gene expression analysis after 14 days of embryoid body assay (normalized to GAPDH and PRDM8 expression in the undifferentiated control cells). c Phase contrast pictures of EBs after 16 days of hematopoietic differentiation. The control EB produces hematopoietic progenitor cells, whereas this is not the case for the PRDM8+/− clone and the PRDM8−/− clone, which consistently revealed enhanced growth. Scale bar, 500 μM. d Knockout of PRDM8 resulted in a significantly lower number of hematopoietic progenitor cells. t test: * P < 0.05; ns, not significant. e Cytospins supported impaired hematopoiesis after PRDM8 knockout. Scale bar, 500 μM. f The colony forming unit (CFU) potential is lost in PRDM8 knockout clones. g Flow cytometry substantiates hematopoietic differentiation of the control iPSCs (read line: autofluorescence; blue line: with antibodies)
We have then differentiated three iPSC lines, including the isogenic iPSC line from which we generated the knockout clones, as well as the PRDM8+/− and PRDM8−/− iPSCs toward the hematopoietic lineage using an EB-based differentiation protocol [31]. After 16 days, we observed production of hematopoietic cells from the EBs in control iPSCs. In contrast, hematopoietic differentiation was hardly observed with the PRDM8+/− and particularly the PRDM8−/− iPSCs and their EBs acquired a much larger size than EBs of control iPSCs (Fig. 4c, d). The few iPSC-derived hematopoietic cells of the knockout clones did not reveal typical hematopoietic morphology in cytospins (Fig. 4e). Furthermore, colony formation potential was lost upon PRDM8 knockout (Fig. 4f and Additional file 1: Fig. S6). PRDM8+/− and PRDM8−/− iPSCs did not produce enough progenitor cells for flow cytometry analysis, but for control iPSC expression of hematopoietic surface markers was confirmed (Fig. 4g). Thus, control iPSCs revealed clear hematopoietic differentiation, whereas differentiation was abrogated upon PRDM8 knockout.
PRDM8 knockout impairs neuronal differentiation
We then differentiated iPSCs toward the neuronal lineage because PRDM8 was reported to be involved in neuronal development in mice [32, 33]. After 2 weeks of neuronal differentiation, microscopic analyses revealed typical ganglion-like structures with bridging neurites in the control cells. In contrast, these neuronal agglomerates were hardly observed in the derivatives of PRDM8+/− and PRDM8−/− iPSCs (Fig. 5a). Furthermore, after 10 days of neuronal differentiation, qRT-PCR analysis of the neuronal marker genes tachykinin precursor 1 (TAC1), voltage-gated sodium channel subunit alpha (SCN9A), nestin (NES), neurofilament heavy polypeptide (NEFH), and SRY-box transcription factor 1 (SOX1) revealed clear upregulation in the isogenic iPSC control, whereas this was not observed in the PRDM8+/− and PRDM8−/− iPSCs (Fig. 5b). We then compared global gene expression profiles of the syngenic control and PRDM8−/− iPSCs upon neuronal differentiation: in the PRDM8−/− cells, 1280 genes were more than 4-fold less expressed, whereas 1769 genes were at least 4-fold higher expressed than in controls (Fig. 5c; Additional file 2: Table S1). These genes were particularly associated with gene ontology categories for cell adhesion and neuronal development (Additional file 1: Fig. S7a). Overall, these results clearly support the notion that loss of PRDM8 impairs neuronal differentiation of iPSCs.
Fig. 5.

PRDM8 knock out leads to impaired neuronal differentiation. a Representative phase contrast pictures of neuronal differentiations on day 13. Control cells reveal typical aggregates of immature sensory neurons, which are absent in the PRDM8 knockout cells. Scale bar, 200 μM. b Quantitative RT-PCR analysis after 10 days of neuronal differentiation shows upregulation of neuronal markers in control cells, but not in PRDM8 knockouts (color code depicts mean fold change versus GAPDH). c Gene expression changes in RNA-Seq after 27 days of peripheral neuron differentiation (RPM, reads per million; genes with a log2 fold change above 2 or below − 2 are depicted). d CpG sites that are either 20% hypermethylated (red, 1511 CpGs) or hypomethylated (blue, 1738 CpGs) in the PRDM8−/− clone compared to the isogenic control. e Epigenetic age of control and knockout iPSCs after neuronal differentiation was close to 0 year.
PRDM8 knockout does not impact on epigenetic age
Subsequently, we analyzed DNA methylation profiles of a syngenic control clone and the PRDM8−/− iPSC clone upon 10 days of neuronal differentiation with the Illumina Infinium MethylationEPIC BeadChip. Filtering for CpGs with more than 20% change in DNA methylation levels between control and knockout cells showed that 1738 CpGs were hypomethylated and 1511 CpGs were hypermethylated in PRDM8−/− versus control (Fig. 5d; Additional file 3: Table S2). We then analyzed if loss of PRDM8 significantly accelerated the epigenetic clock in iPSC-derived neuronal cells. Here, we used the epigenetic clock by Horvath [34] because it was trained on multiple different cell types, while our 3 CpG age predictor utilized for targeted analysis of DKC and AA samples was specifically trained for blood. There was no difference in epigenetic age predictions upon 10 days of neuronal differentiation between three control lines, PRDM8+/−, and PRDM8−/− clones (Fig. 5e). In fact, all iPSC-derived cell preparation was estimated close to 0 year, indicating that loss of PRDM8 does not clearly accelerate the epigenetic clock in our iPSC model system.
Discussion
Diagnosis of bone marrow failure syndromes remains a challenge. In this study, we further validated that leukocytes of DKC and AA patients reveal telomere attrition and accelerated epigenetic aging. Moderate acceleration of epigenetic aging has also been described for Down syndrome [34], HGPS [35], or Werner syndrome [36]. Notably, telomere attrition and epigenetic aging seem to be independent, which is in line with previous studies [37–40]. It should be noted that epigenetic age can be influenced by various external parameters, such as gender, alcohol consumption, or body mass index [17]. However, the prominent increase in epigenetic age in AA and DKC patients argues for a direct effect by premature aging. Therefore, analysis of epigenetic age, in addition to telomere length analysis, can provide added value to identify DKC and AA patients because not all patients revealed shortened telomeres or disease-associated mutations [41].
Furthermore, we analyzed DNA methylation within PRDM8 in 62 independent AA and 13 independent DKC samples with MassARRAY and could confirm that this region frequently reveals aberrant hypermethylation. This was further validated with BBA-seq in 8 additional AA and 5 DKC samples. Thus, we provide a new deep-sequencing-based assay that may be better applicable by other scientists and that facilitates longer amplicons. Furthermore, the individual reads provide insight into the DNA methylation pattern: aberrant gain of DNA methylation seems to be increased in cellular subsets, but the neighboring CpGs do not always appear to be coherently modified, as might be expected by binding of DNA methyltransferases (e.g., DNMT3A or DNMT3B) to a specific DNA strand. On the other hand, there was no evidence that DNA methylation in PRDM8 was associated with blood counts. In future studies, it will be interesting to better understand how the aberrant and heterogeneous DNA methylation within PRDM8 is evoked and controlled.
Notably, genome wide DNA methylation profiles of other premature aging syndromes provided similar hypermethylation within PRDM8 and hypomethylation in the promoter of the long transcript of PRDM8. In fact, one of the top 25 differentially methylated regions (DMRs) reported in Werner syndrome is located within PRDM8 [42]. It is generally anticipated that aberrant DNA methylation is accompanied with changes in gene expression. However, our qRT-PCR experiments of AA and DKC did not reveal significant downregulation of either the short or the long transcript of PRDM8. Furthermore, we did not observe significant changes in gene expression of PRDM8 in datasets of premature aging syndromes. This might be partly attributed to the fact that PRDM8 is hardly expressed in blood and fibroblasts. Moreover, as reflected by the BBA-seq results, the aberrant hypermethylation in PRDM8 was not consistent across all DNA molecules of a sample and this might occlude differential gene expression in specific subpopulations. A transient expression of PRDM8 might still be relevant for regulation of developmental processes or premature aging phenotypes, even if the gene is hardly expressed in the adult tissues. However, this functional link cannot be directly proven by our analysis.
So far, little is known about the biological function of PRDM8—particularly in humans. It has been suggested that different isoforms of PRDM proteins (with or without the PR domain) play opposing roles in malignancies: The longer transcript was suggested to function as tumor suppressor, whereas the shorter variant acts as an oncogene [43]. In the murine model system, it was reported that Prdm8 is expressed in brain tissues [33, 44]. Prdm8 seems to function as a repressor for Cadherin-11 to ensure proper neural circuit formation [32]. Inoue et al. described that Prdm8 knockout mice reveal growth retardation and abnormal generation of upper-layer neocortical neurons [33]. When analyzing human iPSCs with PRDM8 knockout, we observed impaired neural differentiation potential—even for the heterozygous knockout iPSCs. This may indicate that aberrant regulation of PRDM8 may contribute to impaired neurological development, which is characteristic for many premature aging syndromes [45].
In addition, iPSCs with PRDM8 knockout revealed impaired hematopoietic differentiation—another key feature of premature aging syndromes. Other PRDM family members are also involved in hematopoiesis and leukemic transformation. For example, PRDM1 seems to be involved in differentiation, maintenance and function of different myeloid cell types [46], B cells [47], T cells [48], and NK cells [49]. Loss of Prdm3 results in a decreased hematopoietic stem cell pool [50, 51]. PRDM8 and PRDM16 might have partly redundant function. Both of them seem to be involved in methylation of lysine 9 of histone 3 (H3K9) [52, 53], a histone mark that is tightly associated with the DNA methylation pattern. Prdm16 was shown to be critical for establishing and maintaining hematopoietic stem cells and it is aberrantly expressed in AML [54–56]. Furthermore, Prdm16 is also an important factor for neuronal stem cells [55] and, like Prdm8, plays a role in neocortical development [57]. Notably, in our previous work, we demonstrated that not only PRDM8, but also PRDM16, is aberrantly methylated in DKC [18], and thus both of them might be relevant for the pathophysiology of the disease.
Conclusions
Diagnosis of AA and DKC patients remains a challenge. Our results support the notion that both premature aging syndromes frequently reveal telomere attrition, accelerated epigenetic aging, and aberrant DNA methylation in PRDM8. There is little correlation between these biomarkers. Therefore, analysis of epigenetic age or aberrant DNA methylation in PRDM8 might be advantageous in patients without significant telomere attrition or specific mutations to identify these bone marrow failure syndromes. In the future, it will be important to also consider samples with other differential diagnoses to better define the specificity and sensitivity of our assays. BBA-seq analysis for PRDM8 and epigenetic aging [25] can provide insight into the heterogeneity of aberrant DNA methylation within a sample and it will be interesting to better understand how these processes are regulated. While the functional relevance of aberrant DNA methylation in PRDM8 needs to be further explored, our results indicate that the gene plays an important role for hematopoietic and neuronal differentiation. Thus, it might contribute to the phenotype of premature aging syndromes, albeit the functional link remains to be demonstrated.
Methods
Sample collection and next generation sequencing (NGS)
Blood samples were obtained from the Registry for Telomeropathies and Aplastic Syndromes of RWTH Aachen University and participating hospitals. The study was approved by the local ethic committee and all samples were taken after written consent (EK206/09). All DKC patients revealed lymphocyte telomere length below 1% percentile and diagnosis was complemented by clinical and genotypic characteristics [12, 58]. All AA and DKC samples were screened by NGS using a self-customized gene panel containing the entire coding sequence of genes that are known to play a relevant role in DKC (CTC1, DKC1, NHP2, NOP10, RTEL1, TERC, TERT, TCAB1, USB1, and exon 6 of TINF2, which is a known hot spot region) [12]. Library preparation and sequencing were performed with the TruSeq Custom Amplicon Kit and the MiSeq Reagent Kit v2 (all from Illumina) using a MiSeq Illumina platform. Sequencing was performed with 250 bp paired end and data was first analyzed with the Illumina RTA software. Afterwards, the SeqNEXT software (version 4.3.0, JSI medical systems GmbH, Ettenheim, Germany) was used for alignment and variant calling. A mean coverage of 100 × was reached and since we looked for germline variants, a cutoff of mutant allele frequency of > 30% or ≥ 10 absolute mutant reads was chosen. Further information about the patients is provided in Additional file 4: Table S3.
Analysis of telomere length and telomere age prediction
Telomere length of granulocytes and lymphocytes was analyzed in 105 samples from healthy donors as described before [17], 70 independent patients with AA, and 18 independent patients with DKC. In 5 AA patients and 1 DKC patient, telomere length measurement of the granulocytes was not possible due to insufficient cell number. Flow-FISH for telomere length was performed as described in detail before [12, 59]. In brief, samples were mixed with a FITC-labeled or Alexa488-labled telomere-specific (CCCTAA)3-peptide nucleic acid FISH probe (Panagene, Daejeon, South Korea) for DNA hybridization followed by DNA counterstaining with LDS 751 (Sigma Aldrich, St. Louis, MO, USA). An FC 500 flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA) was used for data acquisition. Bovine thymocytes were used as internal controls to calculate telomere length in kilobases and samples were measured in triplicates. The cow thymocytes as well as granulocytes and lymphocytes from human samples were identified based on forward scatter and LDS 751 binding to double-stranded DNA. Telomere age was estimated by linear regression on the age-adjusted samples from healthy donors. Mean average error [MAE] was calculated as follows:
with x = delta age. Mean delta age () was calculated as follows:
Epigenetic age prediction by pyrosequencing of three CpGs
Pyrosequencing of the three age-associated CpGs was measured in 243 blood samples of healthy controls and 105 [17] and 80 [25] of these were also mentioned in previous work. Furthermore, we analyzed 70 independent AA and 18 independent DKC patients. Pyrosequencing was described in detail before [17] and performed at Cygenia GmbH (Aachen, Germany). In short, DNA was isolated from patient samples with the QIAamp DNA Mini and Blood Mini kit (Qiagen, Hilden, Germany) and bisulfite converted with the EZ DNA Methylation Kit (Zymo Research, Freiburg, Germany). Converted DNA was PCR amplified with the PyroMark PCR kit 800 (Qiagen) and 20 μL PCR product was immobilized to 60 μL Streptavidin Sepharose HP beads (GE Healthcare, Chicago, IL, USA), annealed to 0.8 μL 20 μM sequencing primers for 2 min at 80 °C (see all primer sequences in Additional file 1: Table S4). Samples were measured with the PyroMark Gold Q96 Reagents on a PyroMark Q96 ID and analyzed with the Pyro-Q-CpG 1.0.9 software (all from Qiagen). Epigenetic age was calculated as follows:
Predicted age (in years) = 38.0 − 26.4 × β value (cg02228185) − 23.7 × β value (cg25809905) + 164.7 × β value (CpG upstream of cg17861230)
MassARRAY analysis of DNA methylation in PRDM8
Measurements of all patient data on PRDM8 and its two CpG sites cg19409579 and cg27242132 have been performed with MassARRAY analysis instead of pyrosequencing. This is due to the fact that the PRDM8 region has many CpG sites, and pyrosequencing primers overlapping with CpGs were temperature sensitive and did not provide reliable measurements. With MassARRAY, longer amplicons can be measured thus making primer design outside of CpG-rich regions and analysis of these PRDM8 regions possible. MassARRAY analyses were performed with a MassARRAY Analyzer 4 System (Agena Bioscience, Hamburg, Germany) as previously described [18] at Varionostic GmbH (Ulm, Germany).
Barcoded bisulfite amplicon sequencing of DNA methylation in PRDM8
Two BBA-seq assays were designed around the CpG sites cg27242132 (assay 1) and cg19409579 (assay 2; Additional file 1: Fig. S8) and amplified with the PyroMark PCR kit (Qiagen). The forward and reverse primers contain handle sequences for the subsequent barcoding step (Additional file 1: Tables S5). PCR conditions were 95 °C for 15 min; 40 cycles of 94 °C for 30 s, 53 °C for 30 s, 72 °C for 30 s; and then final elongation at 72 °C for 10 min. The two amplicons of each donor were pooled at equal concentrations, quantified with Qubit (Invitrogen, Carlsbad, CA, USA), and cleaned up with paramagnetic beads from Agencourt AMPure PCR Purification system (Beckman Coulter, Brea, CA, USA). Four microliters of PCR products were subsequently added to 21 μL PyroMark Master Mix (Qiagen) containing 0.4 μM of barcoded primers (adapted from NEXTflexTM 16S V1-V3 Amplicon Seq Kit, Bioo Scientific, Austin, USA) for a second PCR (95 °C for 15 min; 16 cycles of 95 °C for 30 s, 60 °C for 30 s, 72 °C for 30 s; final elongation at 72 °C for 10 min). PCR products were again quantified with the Qubit, combined in equimolar ratios, and cleaned by Select-a-Size DNA Clean & Concentrator Kit (Zymo Research). A 12-pM DNA library was diluted with 15% PhiX spike-in control and eventually subjected to 250 bp paired-end sequencing on a MiSeq lane using the Miseq reagent V2 Nano kit (both from Illumina, San Diego, CA, USA). Obtained FastQ files from the MiSeq were aligned to the reference genome hg19 using the Bismark tool [60] and DNA methylation values and patterns were extracted with the Bismark methylation extractor. The average number of reads across all samples was 4088 for assay 1 and 3242 for assay 2. For heatmaps, the frequencies of DNA methylation patterns in individual reads were calculated by the number of reads containing the pattern divided by the total number of reads of the target region per sample. The most abundant reads of similar patterns within neighboring CpGs were grouped together and visualization was performed with Python’s package seaborn [61].
qRT-PCR measurement of patient samples
Total RNA was isolated from 10 healthy controls, 27 AA, and 14 DKC patients with the miRNeasy Kit (Qiagen). One hundred nanograms of RNA was then converted into cDNA with the High Capacity cDNA Reverse Transcription Kit (Thermo Scientific, Waltham, MA, USA). cDNA was analyzed in qPCR with specific primers for either the long transcript of PRDM8 (NM020226.3), the short transcript of PRDM8 (NM 001099403.2), or all PRDM8 transcripts (Primers see in Additional file 1: Table S6) using the Power SYBR Green PCR Master Mix (Thermo Scientific) and the StepOnePlus Real-Time PCR System (Applied Biosystems, Waltham, MA, USA). Data was normalized to GAPDH.
Generation, cultivation, and genome editing of human iPSCs
Mesenchymal stromal cells were reprogrammed into iPSCs with episomal plasmids and characterized with Epi-Pluri-Score as described before [30, 62]. iPSCs were cultured on tissue culture plastic (TCP) coated with Vitronectin XF (0.5 mg/cm2; Stemcell Technologies, Vancouver, Canada) in StemMACS iPS-Brew XF (Miltenyi Biotec, Bergisch Gladbach, Germany). For genome editing, we used a CRISPR/Cas9n double nicking approach [63]. In brief, two pairs of guide RNA (gRNA) were designed targeting the intron/exon boundary at the start codon of the PRDM8 gene (Fig. 3a, Additional file 1: Table S6). Deletion of the intron/exon boundary leads to a reading frame shift and premature stop codon, thus generating a complete knockout of the PRDM8 protein. gRNA oligonucleotides were cloned individually into a variant of vector pX335 (Addgene #42335, Addgene, Watertown, MA, USA) carrying a Puromycin-GFP selection cassette. gRNA plasmids were transfected into iPSCs using the NEON transfection system (1500 V, 20 ms pulse width, 1 pulse, Thermo Fisher Scientific, Waltham, MA, USA). Transfected cells were selected by puromycin treatment (0.4 μg/mL) for 24 h. Ten days later, individual colonies were picked and screened for deletions in the PRDM8 target region by PCR (Primers see Additional file 1: Table S7). To further validate pluripotency of iPSCs, we determined DNA methylation at three pluripotency-associated CpGs by pyrosequencing to estimate the Epi-Pluri-Score, as described in detail before [30].
Embryoid body assay
To test for three-lineage potential of iPSC clones, cells were spontaneously differentiated via the EB assay. iPSCs were incubated with 1 mg/mL collagenase IV (Thermo Fisher Scientific) for 5–15 min and rinsed off with KO-DMEM (Thermo Fisher Scientific). Resulting cell clusters sedimented by gravity at 37 °C for 10 min and were resuspended in EB culture medium containing 20% FCS, 100 μM non-essential amino acids, 2 mM L-glutamine, and 0.2% ß-mercaptoethanol (all Thermo Fisher Scientific) and then transferred to ultra-low attachment plates (Corning, NY, USA) to form EBs in a 3D culture. On day 5, EBs were transferred to plates coated with 0.1% gelatin (Stemcell Technologies) for 2D culture. Cells were cultured for additional 9 days. Gene expression of marker genes was analyzed by quantitative RT-PCR (depicted as mean fold change versus GAPDH). Primer sequences are listed in Additional file 1: Tab. S8.
Hematopoietic differentiation
Hematopoietic differentiation of human iPSCs was induced with a Spin-EB protocol [31]. iPSCs were cultured on Matrigel Matrix (Corning) and detached as single cells with Accutase (Stemcell Technologies). Three thousand single cells per well were seeded in a U-bottom 96-well plate (Greiner Bio-One, Kremsmünster, Austria) in serum free medium containing 50% IMDM, 50% Ham’s F12, 0.5% BSA, 1% chemically defined lipid concentrate, 2 mM GlutaMAX (all Thermo Fisher Scientific), 400 μM 1-thioglycerol, 50 μg/mL L-ascorbic acid, and 6 μg/mL holo-transferrin (all Sigma Aldrich) supplemented with 10 ng/mL FGF-2 (Peprotech, Hamburg, Germany), 10 ng/mL BMP-4 (Miltenyi Biotec), and 10 μM Y-27632 (Abcam, Cambridge, Great Britain). The plates were centrifuged at 350g for 5 min and cells formed EBs on the next day. From day 2 to 16, the EBs were cultured in serum free medium supplemented with 10 ng/mL FGF-2, 10 ng/mL BMP-4, 50 ng/mL SCF (Peprotech), 10 ng/mL VEGF-A (Peprotech), and 10 U/mL penicillin/streptomycin (Thermo Fisher Scientific). Hematopoietic progenitors were harvested after 16 days of differentiation, separated with a 40 μM cell strainer, centrifuged at 200g for 5 min, and then used for colony forming unit assay, cytospin, and immunophenotypic analysis.
Colony forming unit assay
After 16 days of differentiation, 5000 hematopoietic progenitor cells were seeded in 500 μL of methylcellulose-based medium (HSC-CFU lite with EPO; Miltenyi Biotec) in 24-well plates. Colonies were quantified after 12 days.
Cytospin
Cellular morphology of hematopoietic progenitors was analyzed by cytospins with 1% neutral benzidine (Sigma Aldrich) and Diff Quik staining (Medion Grifols Diagnostics, Düdingen, Switzerland). Cytospins were analyzed with a Leica DMRX microscope (Leica Microsystems, Wetzlar, Germany).
Immunophenotypic analysis
Flow cytometric analysis of hematopoietic surface markers was performed with a FACS Canto II (BD Biosciences) with the following antibodies: CD3-APC (clone HIT3a), CD31-PE (clone WM59), CD34-APC (clone 581), CD43-FITC (clone 1G10), CD66b-PE (clone G10F5) (all BD Biosciences, Franklin Lakes, USA), CD11c-PE-Cy7 (clone 3.9), HLA-DR-FITC (clone LN3), CD235a-PE (clone HIR2/GA-R2), cKIT-PE-Cy7 (clone 104D2), (all Thermo Fisher Scientific), CD45-APC-Vio770 (clone 5B1), CD33-APC (clone AC104.3E3) (all Miltenyi Biotec), and CD41-PE-Cy7 (clone HIP8; Biolegend, San Diego, CA, USA). Data was analyzed using the FlowJo software (Tree Star, Ashland, OR, USA).
Neuronal differentiation of iPSCs
Differentiation of iPSCs toward peripheral neurons was performed as previously described [64, 65]. iPSCs were seeded as single cells at a density of 105 cells/cm2 in presence of 10 μM Y-27632. At 80–90% confluency, neural conversion was induced by dual-SMAD inhibition: for the first 5 days, LDN-193189 (100 nM or 1 μM, Sigma Aldrich) and SB431542 (10 μM, Miltenyi Biotec) were added to the basal culture medium consisting of knockout DMEM/F-12 containing 15% knockout serum replacement, 1 mM L-glutamine, 100 μM non-essential amino acids, 100 μM β-mercaptoethanol, 100 U/mL penicillin, and 100 μg/mL streptomycin (all Thermo Fisher Scientific). To accelerate neural crest specification, three small molecules (3 μM CHIR99021, 10 μM DAPT, and 10 μM SU5402, all Tocris, Bristol, UK) were added between days 2 to 10. After 4 days, the medium was supplemented in increasing percentages with DMEM/F-12, containing 10 ml/L N2, 20 ml/L B27 minus vitamin A supplements and 100 U/mL penicillin, 100 μg/mL streptomycin (all Thermo Fisher Scientific). N2/B27 medium was added to the basal medium at 25% between days 4 and 5, 50% between days 6 and 7, and 75% between days 8 and 10. On day 10, cells were dissociated using Accutase and seeded on glass coverslips coated with 50 μg/mL Poly-L-Ornithine (Sigma Aldrich) and 5 μg/mL Laminin (Thermo Fisher Scientific) and further cultured for up to 3 weeks with N2/B27 medium supplemented with 20 ng/mL NGF (R&D Systems, Minneapolis, MN, USA), BDNF, GDNF (all PeproTech), and 200 ng/mL L-ascorbic acid (Sigma Aldrich). Medium was changed every 3–4 days.
DNA methylation and gene expression analyses
DNA methylation analysis of samples after 10 days of neural differentiation was performed with the Infinium MethylationEPIC BeadChip (Illumina). Therefore, DNA was isolated with the NucleoSpin Tissue kit (Macherey-Nagel, Düren, Germany) and bisulfite conversion as well as hybridization was performed at Life and Brain GmbH (Bonn, Germany). Data was preprocessed with the Bioconductor Illumina Minfi package for R [66–72] and normalized with quantile normalization. Data of DNA methylation profiles has been deposited at Gene Expression Omnibus (GEO) under the reference ID GSE141106. For differential DNA methylation analysis, CpGs in X and Y chromosomes as well as SNPs were excluded and we then filtered for beta-values with a difference between PRDM8−/− and WT of <− 0.2 or > 0.2.
RNA-Seq of samples after 27 days of neural differentiation was performed at Life and Brain GmbH (Bonn, Germany) on a HiSeq 2500 sequencer (Illumina). Adapter trimming and rRNA removal in raw fastq files was performed using TrimGalore (Babraham Bioinformatics, Cambridge, UK) and SortMeRNA [73], respectively. Reads were aligned to the human genome build GRCh38 with STAR aligner [74]. Read counts were retrieved by HTSeq-count and further analyzed with the R package DESeq2. RNA-Seq data has been deposited at Gene Expression Omnibus (GEO) under the reference ID GSE141107. Gene ontology enrichment analyses of differentially expressed genes were performed using the PANTHER software [75]. Categories with more than 1000 genes were excluded and similar categories are only listed once.
Additional datasets of premature aging syndromes
We utilized 450 k Illumina Bead Chip datasets of DKC (GSE75310 [18]), Down syndrome (GSE52588 [34, 76, 77]), Werner syndrome (GSE42865 [78]), Hutchinson-Gilford-Progeria (GSE42865), and healthy controls (GSE36054 [79], GSE32148 [80], GSE49064 [81]) to analyze DNA methylation patterns within the PRDM8 gene. Gene expression analyses for PRDM8 were performed with the following available datasets with corresponding controls: Down syndrome (GSE35665 [26]), Werner syndrome (GSE48761 [27]), and HGPS (GSE69391 [28], GSE3860 [29]).
Statistical analysis
Statistical significance of delta telomere age, delta epigenetic age, methylation values of specific CpG sites, and absolute cell counts were estimated by unpaired t tests. CFU colonies and absolute cell counts of iPS-derived hematopoietic progenitor cells are presented as means ± standard deviation.
Supplementary information
Additional file 1: Combined PDF of Supplemental Figures S1 – S6 and Supplemental Tables S3 – S4. Fig. S1: Telomere age of lymphocytes in dyskeratosis congenita and aplastic anemia. Fig. S2: The CpG site cg19409579 in PRDM8 is hypermethylated in AA and DKC. Fig. S3: Aberrant DNA methylation in PRDM8 is not correlated with blood counts. Fig. S4: Aberrant DNA methylation patterns of PRDM8 in premature aging diseases. Fig. S5: Characterization of PRDM8 knockout induced pluripotent stem cells. Fig. S6: Colony forming unit assay of iPSC-derived hematopoietic progenitor cells. Fig. S7: Gene ontology analysis after neuronal differentiation. Fig. S8: Genomic regions for the BBA-seq assays within PRDM8. Tab. S4: Primers used for pyrosequencing of the epigenetic aging signature. Tab S5: Primers for bisulfite barcoded amplicon sequencing of PRDM8. Tab. S6: Quantitative RT-PCR primers used for PRDM8 gene expression analysis. Tab. S7: Guide RNAs used for creating CRISPR knockouts of PRDM8 in iPSCs. Tab. S8: Quantitative RT-PCR primers used for analysis of embryoid body assays.
Additional file 2: Tab. S1: Differentially expressed genes in PRDM8-/- versus control. Genes that are either four-fold downregulated (1280 CpG sites) or upregulated (1769 CpG sites; log2 fold change <-2 or > 2) in PRDM8-/- compared to control iPSCs with ENSEMBL ID, reads per million (RPM) of the knockout and control cells, and log2 fold change of knockout and control.
Additional file 3: Tab. S2: Differentially methylated CpG sites in PRDM8-/- versus control. CpG sites that are either hypermethylated (1511 CpG sites) or hypomethylated (1, 738 CpG sites; delta β-value >0.2 or < -0.2) in PRDM8-/- compared to control iPSCs with β-values of the knockout and control cells, and the difference in methylation between knockout and control.
Additional file 4:. Tab. S3: Patient data of all DKC, AA samples and healthy controls. List of all patient samples and healthy controls with their according gender, chronological age, diagnosis, mutated genes, and blood counts.
Acknowledgements
Not applicable
Abbreviations
- AA
Aplastic anemia
- BBA-seq
Barcoded bisulfite amplicon sequencing
- CpG site
CG dinucleotide
- DKC
Dyskeratosis congenita
- EB
Embryoid body
- gRNA
Guide RNA
- HGPS
Hutchinson-Gilford-Progeria syndrome
- iPSC
Induced pluripotent stem cell
- MAE
Mean absolute error
- qRT-PCR
Quantitative reverse transcription PCR
- TCP
Tissue culture plastic
Authors’ contributions
ME and OC performed experiments. JF and CCK performed data processing and analysis of methylation and gene expression data. VT and MN performed and analyzed BBA-seq experiments. CR and AL performed neuronal differentiation of iPSCs. TB, MV, and MK provided and characterized patient samples. MZ contributed to study design. FB set up patient cohort and together with WW designed the study. OC, ME, and WW wrote the first draft of the manuscript and all authors approved the final manuscript.
Funding
This work was supported by the German Research Foundation (DFG; WA 1706/8-1, WA 1706/12-1, BR 1782/5-1, LA 2740/3–1, and 363055819/GRK2415), the Interdisciplinary Center for Clinical Research (IZKF) within the faculty of Medicine at the RWTH Aachen University (O3-3), Deutsche Krebshilfe (TRACK-AML), the Federal Ministry of Education and Research (VIP+ project “Epi-Blood-Count”), and by the Flow Cytometry Facility, a core facility of the IZKF within the Faculty of Medicine at RWTH Aachen University.
Availability of data and materials
Data of DNA methylation profiles has been deposited at Gene Expression Omnibus (GEO) under the reference ID GSE141106. RNA-Seq data has been deposited at GEO under the reference ID GSE141107.
Ethics approval and consent to participate
Blood samples were obtained from the Registry for Telomeropathies and Aplastic Syndromes of RWTH Aachen University and participating hospitals. The study was approved by the local ethic committee and all samples were taken after written consent (EK206/09).
Consent for publication
Not applicable
Competing interests
WW and JF are involved in Cygenia GmbH (www.cygenia.com) that may provide service for epigenetic signatures applied in this study. WW, FB, and THB are inventors of a patent application on PRDM8 as a biomarker (DE 10 2015 121 969 A1 2017.06.22). Apart from this, the authors have nothing to disclose.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Olivia Cypris, Monika Eipel, Fabian Beier and Wolfgang Wagner contributed equally to this work.
Contributor Information
Olivia Cypris, Email: ocypris@ukaachen.de.
Monika Eipel, Email: monika-eipel@hotmail.de.
Julia Franzen, Email: jufranzen@ukaachen.de.
Corinna Rösseler, Email: croesseler@ukaachen.de.
Vithurithra Tharmapalan, Email: vtharmapalan@ukaachen.de.
Chao-Chung Kuo, Email: chao-chung.kuo@rwth-aachen.de.
Margherita Vieri, Email: mvieri@ukaachen.de.
Miloš Nikolić, Email: mnikolic@ukaachen.de.
Martin Kirschner, Email: mkirschner@ukaachen.de.
Tim H. Brümmendorf, Email: tbruemmendorf@ukaachen.de
Martin Zenke, Email: martin.zenke@rwth-aachen.de.
Angelika Lampert, Email: alampert@ukaachen.de.
Fabian Beier, Email: fbeier@ukaachen.de.
Wolfgang Wagner, Email: wwagner@ukaachen.de.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s13148-020-00914-5.
References
- 1.Dokal I. Dyskeratosis congenita. Hematology Am Soc Hematol Educ Program. 2011;2011:480–486. doi: 10.1182/asheducation-2011.1.480. [DOI] [PubMed] [Google Scholar]
- 2.Oshima J, Sidorova JM, Monnat RJ., Jr Werner syndrome: clinical features, pathogenesis and potential therapeutic interventions. Ageing Res Rev. 2017;33:105–114. doi: 10.1016/j.arr.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roizen NJ, Patterson D. Down’s syndrome. Lancet. 2003;361(9365):1281–1289. doi: 10.1016/S0140-6736(03)12987-X. [DOI] [PubMed] [Google Scholar]
- 4.Calado RT, Young NS. Telomere maintenance and human bone marrow failure. Blood. 2008;111(9):4446–4455. doi: 10.1182/blood-2007-08-019729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alter BP, Giri N, Savage SA, Rosenberg PS. Telomere length in inherited bone marrow failure syndromes. Haematologica. 2015;100(1):49–54. doi: 10.3324/haematol.2014.114389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walne AJ, Dokal I. Advances in the understanding of dyskeratosis congenita. Br J Haematol. 2009;145(2):164–172. doi: 10.1111/j.1365-2141.2009.07598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamaguchi H, Sakaguchi H, Yoshida K, Yabe M, Yabe H, Okuno Y, et al. Clinical and genetic features of dyskeratosis congenita, cryptic dyskeratosis congenita, and Hoyeraal-Hreidarsson syndrome in Japan. Int J Hematol. 2015;102(5):544–552. doi: 10.1007/s12185-015-1861-6. [DOI] [PubMed] [Google Scholar]
- 8.Townsley DM, Dumitriu B, Young NS. Bone marrow failure and the telomeropathies. Blood. 2014;124(18):2775–2783. doi: 10.1182/blood-2014-05-526285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Young NS. Aplastic anemia. N Engl J Med. 2018;379(17):1643–1656. doi: 10.1056/NEJMra1413485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fernandez Garcia MS, Teruya-Feldstein J. The diagnosis and treatment of dyskeratosis congenita: a review. J Blood Med. 2014;5:157–167. doi: 10.2147/JBM.S47437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brummendorf TH, Maciejewski JP, Mak J, Young NS, Lansdorp PM. Telomere length in leukocyte subpopulations of patients with aplastic anemia. Blood. 2001;97(4):895–900. doi: 10.1182/blood.v97.4.895. [DOI] [PubMed] [Google Scholar]
- 12.Ferreira MSV, Kirschner M, Halfmeyer I, Estrada N, Xicoy B, Isfort S, et al. Comparison of flow-FISH and MM-qPCR telomere length assessment techniques for the screening of telomeropathies. Ann N Y Acad Sci. 2020;1466(1):93–103. doi: 10.1111/nyas.14248. [DOI] [PubMed] [Google Scholar]
- 13.Savage SA, Alter BP. Dyskeratosis congenita. Hematol Oncol Clin North Am. 2009;23(2):215–231. doi: 10.1016/j.hoc.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Field AE, Robertson NA, Wang T, Havas A, Ideker T, Adams PD. DNA methylation clocks in aging: categories, causes, and consequences. Mol Cell. 2018;71(6):882–895. doi: 10.1016/j.molcel.2018.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49(2):359–367. doi: 10.1016/j.molcel.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10):R115. doi: 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weidner CI, Lin Q, Koch CM, Eisele L, Beier F, Ziegler P, et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014;15(2):R24. doi: 10.1186/gb-2014-15-2-r24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weidner CI, Lin Q, Birkhofer C, Gerstenmaier U, Kaifie A, Kirschner M, et al. DNA methylation in PRDM8 is indicative for dyskeratosis congenita. Oncotarget. 2016;7(10):10765–10772. doi: 10.18632/oncotarget.7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang S, Shao G, Liu L. The PR domain of the Rb-binding zinc finger protein RIZ1 is a protein binding interface and is related to the SET domain functioning in chromatin-mediated gene expression. J Biol Chem. 1998;273(26):15933–15939. doi: 10.1074/jbc.273.26.15933. [DOI] [PubMed] [Google Scholar]
- 20.Schneider R, Bannister AJ, Kouzarides T. Unsafe SETs: histone lysine methyltransferases and cancer. Trends Biochem Sci. 2002;27(8):396–402. doi: 10.1016/s0968-0004(02)02141-2. [DOI] [PubMed] [Google Scholar]
- 21.Sun XJ, Xu PF, Zhou T, Hu M, Fu CT, Zhang Y, et al. Genome-wide survey and developmental expression mapping of zebrafish SET domain-containing genes. PLoS One. 2008;3(1):e1499. doi: 10.1371/journal.pone.0001499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu H, Min J, Lunin VV, Antoshenko T, Dombrovski L, Zeng H, et al. Structural biology of human H3K9 methyltransferases. PLoS One. 2010;5(1):e8570. doi: 10.1371/journal.pone.0008570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morishita K. Leukemogenesis of the EVI1/MEL1 gene family. Int J Hematol. 2007;85(4):279–286. doi: 10.1532/IJH97.06174. [DOI] [PubMed] [Google Scholar]
- 24.Fog CK, Galli GG, Lund AH. PRDM proteins: important players in differentiation and disease. Bioessays. 2012;34(1):50–60. doi: 10.1002/bies.201100107. [DOI] [PubMed] [Google Scholar]
- 25.Han Y, Franzen J, Stiehl T, Gobs M, Kuo CC, Nikolic M, et al. New targeted approaches for epigenetic age predictions. BMC Biol. 2020;18(1):71. doi: 10.1186/s12915-020-00807-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li C, Jin L, Bai Y, Chen Q, Fu L, Yang M, et al. Genome-wide expression analysis in Down syndrome: insight into immunodeficiency. PLoS One. 2012;7(11):e49130. doi: 10.1371/journal.pone.0049130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheung HH, Liu X, Canterel-Thouennon L, Li L, Edmonson C, Rennert OM. Telomerase protects werner syndrome lineage-specific stem cells from premature aging. Stem Cell Reports. 2014;2(4):534–546. doi: 10.1016/j.stemcr.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kubben N, Zhang W, Wang L, Voss TC, Yang J, Qu J, et al. Repression of the antioxidant NRF2 pathway in premature aging. Cell. 2016;165(6):1361–1374. doi: 10.1016/j.cell.2016.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Csoka AB, English SB, Simkevich CP, Ginzinger DG, Butte AJ, Schatten GP, et al. Genome-scale expression profiling of Hutchinson-Gilford progeria syndrome reveals widespread transcriptional misregulation leading to mesodermal/mesenchymal defects and accelerated atherosclerosis. Aging Cell. 2004;3(4):235–243. doi: 10.1111/j.1474-9728.2004.00105.x. [DOI] [PubMed] [Google Scholar]
- 30.Lenz M, Goetzke R, Schenk A, Schubert C, Veeck J, Hemeda H, et al. Epigenetic biomarker to support classification into pluripotent and non-pluripotent cells. Sci Rep. 2015;5:8973. doi: 10.1038/srep08973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y, Wang Y, Gao Y, Forbes JA, Qayyum R, Becker L, et al. Efficient generation of megakaryocytes from human induced pluripotent stem cells using food and drug administration-approved pharmacological reagents. Stem Cells Transl Med. 2015;4(4):309–319. doi: 10.5966/sctm.2014-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ross SE, McCord AE, Jung C, Atan D, Mok SI, Hemberg M, et al. Bhlhb5 and Prdm8 form a repressor complex involved in neuronal circuit assembly. Neuron. 2012;73(2):292–303. doi: 10.1016/j.neuron.2011.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Inoue M, Iwai R, Yamanishi E, Yamagata K, Komabayashi-Suzuki M, Honda A, et al. Deletion of Prdm8 impairs development of upper-layer neocortical neurons. Genes Cells. 2015;20(9):758–770. doi: 10.1111/gtc.12274. [DOI] [PubMed] [Google Scholar]
- 34.Horvath S, Garagnani P, Bacalini MG, Pirazzini C, Salvioli S, Gentilini D, et al. Accelerated epigenetic aging in Down syndrome. Aging Cell. 2015;14(3):491–495. doi: 10.1111/acel.12325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horvath S, Oshima J, Martin GM, Lu AT, Quach A, Cohen H, et al. Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging. 2018;10(7):1758–1775. doi: 10.18632/aging.101508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maierhofer A, Flunkert J, Oshima J, Martin GM, Haaf T, Horvath S. Accelerated epigenetic aging in Werner syndrome. Aging. 2017;9(4):1143–1152. doi: 10.18632/aging.101217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Banszerus VL, Vetter VM, Salewsky B, Konig M, Demuth I. Exploring the relationship of relative telomere length and the epigenetic clock in the LipidCardio cohort. Int J Mol Sci. 2019;20(12). [DOI] [PMC free article] [PubMed]
- 38.Belsky DW, Moffitt TE, Cohen AA, Corcoran DL, Levine ME, Prinz JA, et al. Eleven telomere, epigenetic clock, and biomarker-composite quantifications of biological aging: do they measure the same thing? Am J Epidemiol. 2018;187(6):1220–1230. doi: 10.1093/aje/kwx346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Breitling LP, Saum KU, Perna L, Schottker B, Holleczek B, Brenner H. Frailty is associated with the epigenetic clock but not with telomere length in a German cohort. Clin Epigenetics. 2016;8:21. doi: 10.1186/s13148-016-0186-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marioni RE, Harris SE, Shah S, McRae AF, von Zglinicki T, Martin-Ruiz C, et al. The epigenetic clock and telomere length are independently associated with chronological age and mortality. Int J Epidemiol. 2016. [DOI] [PMC free article] [PubMed]
- 41.Dokal I, Vulliamy T, Mason P, Bessler M. Clinical utility gene card for: dyskeratosis congenita - update 2015. Eur J Hum Genet. 2015;23(4). [DOI] [PMC free article] [PubMed]
- 42.Maierhofer A, Flunkert J, Oshima J, Martin GM, Poot M, Nanda I, et al. Epigenetic signatures of Werner syndrome occur early in life and are distinct from normal epigenetic aging processes. Aging cell. 2019:e12995. [DOI] [PMC free article] [PubMed]
- 43.Mzoughi S, Tan YX, Low D, Guccione E. The role of PRDMs in cancer: one family, two sides. Curr Opin Genet Dev. 2016;36:83–91. doi: 10.1016/j.gde.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 44.Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, et al. Towards a knowledge-based Human Protein Atlas. Nat Biotechnol. 2010;28(12):1248–1250. doi: 10.1038/nbt1210-1248. [DOI] [PubMed] [Google Scholar]
- 45.Kubben N, Misteli T. Shared molecular and cellular mechanisms of premature ageing and ageing-associated diseases. Nat Rev Mol Cell Biol. 2017;18(10):595–609. doi: 10.1038/nrm.2017.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chang DH, Angelin-Duclos C, Calame K. BLIMP-1: trigger for differentiation of myeloid lineage. Nat Immunol. 2000;1(2):169–176. doi: 10.1038/77861. [DOI] [PubMed] [Google Scholar]
- 47.Shapiro-Shelef M, Lin KI, McHeyzer-Williams LJ, Liao J, McHeyzer-Williams MG, Calame K. Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity. 2003;19(4):607–620. doi: 10.1016/s1074-7613(03)00267-x. [DOI] [PubMed] [Google Scholar]
- 48.Rutishauser RL, Martins GA, Kalachikov S, Chandele A, Parish IA, Meffre E, et al. Transcriptional repressor Blimp-1 promotes CD8(+) T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity. 2009;31(2):296–308. doi: 10.1016/j.immuni.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kallies A, Carotta S, Huntington ND, Bernard NJ, Tarlinton DM, Smyth MJ, et al. A role for Blimp1 in the transcriptional network controlling natural killer cell maturation. Blood. 2011;117(6):1869–1879. doi: 10.1182/blood-2010-08-303123. [DOI] [PubMed] [Google Scholar]
- 50.Zhang Y, Stehling-Sun S, Lezon-Geyda K, Juneja SC, Coillard L, Chatterjee G, et al. PR-domain-containing Mds1-Evi1 is critical for long-term hematopoietic stem cell function. Blood. 2011;118(14):3853–3861. doi: 10.1182/blood-2011-02-334680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goyama S, Yamamoto G, Shimabe M, Sato T, Ichikawa M, Ogawa S, et al. Evi-1 is a critical regulator for hematopoietic stem cells and transformed leukemic cells. Cell Stem Cell. 2008;3(2):207–220. doi: 10.1016/j.stem.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 52.Pinheiro I, Margueron R, Shukeir N, Eisold M, Fritzsch C, Richter FM, et al. Prdm3 and Prdm16 are H3K9me1 methyltransferases required for mammalian heterochromatin integrity. Cell. 2012;150(5):948–960. doi: 10.1016/j.cell.2012.06.048. [DOI] [PubMed] [Google Scholar]
- 53.Eom GH, Kim K, Kim SM, Kee HJ, Kim JY, Jin HM, et al. Histone methyltransferase PRDM8 regulates mouse testis steroidogenesis. Biochem Biophys Res Commun. 2009;388(1):131–136. doi: 10.1016/j.bbrc.2009.07.134. [DOI] [PubMed] [Google Scholar]
- 54.Corrigan DJ, Luchsinger LL, Justino de Almeida M, Williams LJ, Strikoudis A, Snoeck HW. PRDM16 isoforms differentially regulate normal and leukemic hematopoiesis and inflammatory gene signature. J Clin Invest. 2018;128(8):3250–3264. doi: 10.1172/JCI99862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chuikov S, Levi BP, Smith ML, Morrison SJ. Prdm16 promotes stem cell maintenance in multiple tissues, partly by regulating oxidative stress. Nat Cell Biol. 2010;12(10):999–1006. doi: 10.1038/ncb2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aguilo F, Avagyan S, Labar A, Sevilla A, Lee DF, Kumar P, et al. Prdm16 is a physiologic regulator of hematopoietic stem cells. Blood. 2011;117(19):5057–5066. doi: 10.1182/blood-2010-08-300145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Inoue M, Iwai R, Tabata H, Konno D, Komabayashi-Suzuki M, Watanabe C, et al. Prdm16 is crucial for progression of the multipolar phase during neural differentiation of the developing neocortex. Development. 2017;144(3):385–399. doi: 10.1242/dev.136382. [DOI] [PubMed] [Google Scholar]
- 58.Kirschner M, Maurer A, Wlodarski MW, Ventura Ferreira MS, Bouillon AS, Halfmeyer I, et al. Recurrent somatic mutations are rare in patients with cryptic dyskeratosis congenita. Leukemia. 2018;32(8):1762–1767. doi: 10.1038/s41375-018-0125-x. [DOI] [PubMed] [Google Scholar]
- 59.Bouillon AS, Ventura Ferreira MS, Awad SA, Richter J, Hochhaus A, Kunzmann V, et al. Telomere shortening correlates with leukemic stem cell burden at diagnosis of chronic myeloid leukemia. Blood advances. 2018;2(13):1572–1579. doi: 10.1182/bloodadvances.2018017772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. bioinformatics. 2011;27(11):1571-1572. [DOI] [PMC free article] [PubMed]
- 61.Waskom M, Botvinnik O, drewokane, Hobson P, Halchenko Y, Lukauskas S, et al. Seaborn: v0.7.0 (January 2016): Zenodo; 2016. Available from: 10.5281/zenodo.45133.
- 62.Willmann CA, Hemeda H, Pieper LA, Lenz M, Qin J, Joussen S, et al. To clone or not to clone? Induced pluripotent stem cells can be generated in bulk culture. PLoS One. 2013;8(5):e65324. doi: 10.1371/journal.pone.0065324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154(6):1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chambers SM, Qi Y, Mica Y, Lee G, Zhang XJ, Niu L, et al. Combined small-molecule inhibition accelerates developmental timing and converts human pluripotent stem cells into nociceptors. Nat Biotechnol. 2012;30(7):715–720. doi: 10.1038/nbt.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Meents JE, Bressan E, Sontag S, Foerster A, Hautvast P, Rosseler C, et al. The role of Nav1.7 in human nociceptors: insights from human induced pluripotent stem cell-derived sensory neurons of erythromelalgia patients. Pain. 2019;160(6):1327–1341. doi: 10.1097/j.pain.0000000000001511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30(10):1363–1369. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maksimovic J, Gordon L, Oshlack A. SWAN: Subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol. 2012;13(6):R44. doi: 10.1186/gb-2012-13-6-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fortin JP, Labbe A, Lemire M, Zanke BW, Hudson TJ, Fertig EJ, et al. Functional normalization of 450 k methylation array data improves replication in large cancer studies. Genome Biol. 2014;15(12):503. doi: 10.1186/s13059-014-0503-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fortin JP, Hansen KD. Reconstructing A/B compartments as revealed by Hi-C using long-range correlations in epigenetic data. Genome Biol. 2015;16:180. doi: 10.1186/s13059-015-0741-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fortin JP, Triche TJ, Jr, Hansen KD. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics. 2017;33(4):558–560. doi: 10.1093/bioinformatics/btw691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Triche TJ, Jr, Weisenberger DJ, Van Den Berg D, Laird PW, Siegmund KD. Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res. 2013;41(7):e90. doi: 10.1093/nar/gkt090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Andrews SV, Ladd-Acosta C, Feinberg AP, Hansen KD, Fallin MD. “Gap hunting” to characterize clustered probe signals in Illumina methylation array data. Epigenetics Chromatin. 2016;9:56. doi: 10.1186/s13072-016-0107-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kopylova E, Noe L, Touzet H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. 2012;28(24):3211–3217. doi: 10.1093/bioinformatics/bts611. [DOI] [PubMed] [Google Scholar]
- 74.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mi H, Muruganujan A, Ebert D, Huang X, Thomas PD. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019;47(D1):D419–DD26. doi: 10.1093/nar/gky1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bacalini MG, Boattini A, Gentilini D, Giampieri E, Pirazzini C, Giuliani C, et al. A meta-analysis on age-associated changes in blood DNA methylation: results from an original analysis pipeline for Infinium 450 k data. Aging. 2015;7(2):97–109. doi: 10.18632/aging.100718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bacalini MG, Gentilini D, Boattini A, Giampieri E, Pirazzini C, Giuliani C, et al. Identification of a DNA methylation signature in blood cells from persons with Down syndrome. Aging. 2015;7(2):82–96. doi: 10.18632/aging.100715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Heyn H, Moran S, Esteller M. Aberrant DNA methylation profiles in the premature aging disorders Hutchinson-Gilford Progeria and Werner syndrome. Epigenetics. 2013;8(1):28–33. doi: 10.4161/epi.23366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Alisch RS, Barwick BG, Chopra P, Myrick LK, Satten GA, Conneely KN, et al. Age-associated DNA methylation in pediatric populations. Genome Res. 2012;22(4):623–632. doi: 10.1101/gr.125187.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Harris RA, Nagy-Szakal D, Pedersen N, Opekun A, Bronsky J, Munkholm P, et al. Genome-wide peripheral blood leukocyte DNA methylation microarrays identified a single association with inflammatory bowel diseases. Inflamm Bowel Dis. 2012;18(12):2334–2341. doi: 10.1002/ibd.22956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Steegenga WT, Boekschoten MV, Lute C, Hooiveld GJ, de Groot PJ, Morris TJ, et al. Genome-wide age-related changes in DNA methylation and gene expression in human PBMCs. Age. 2014;36(3):9648. doi: 10.1007/s11357-014-9648-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Combined PDF of Supplemental Figures S1 – S6 and Supplemental Tables S3 – S4. Fig. S1: Telomere age of lymphocytes in dyskeratosis congenita and aplastic anemia. Fig. S2: The CpG site cg19409579 in PRDM8 is hypermethylated in AA and DKC. Fig. S3: Aberrant DNA methylation in PRDM8 is not correlated with blood counts. Fig. S4: Aberrant DNA methylation patterns of PRDM8 in premature aging diseases. Fig. S5: Characterization of PRDM8 knockout induced pluripotent stem cells. Fig. S6: Colony forming unit assay of iPSC-derived hematopoietic progenitor cells. Fig. S7: Gene ontology analysis after neuronal differentiation. Fig. S8: Genomic regions for the BBA-seq assays within PRDM8. Tab. S4: Primers used for pyrosequencing of the epigenetic aging signature. Tab S5: Primers for bisulfite barcoded amplicon sequencing of PRDM8. Tab. S6: Quantitative RT-PCR primers used for PRDM8 gene expression analysis. Tab. S7: Guide RNAs used for creating CRISPR knockouts of PRDM8 in iPSCs. Tab. S8: Quantitative RT-PCR primers used for analysis of embryoid body assays.
Additional file 2: Tab. S1: Differentially expressed genes in PRDM8-/- versus control. Genes that are either four-fold downregulated (1280 CpG sites) or upregulated (1769 CpG sites; log2 fold change <-2 or > 2) in PRDM8-/- compared to control iPSCs with ENSEMBL ID, reads per million (RPM) of the knockout and control cells, and log2 fold change of knockout and control.
Additional file 3: Tab. S2: Differentially methylated CpG sites in PRDM8-/- versus control. CpG sites that are either hypermethylated (1511 CpG sites) or hypomethylated (1, 738 CpG sites; delta β-value >0.2 or < -0.2) in PRDM8-/- compared to control iPSCs with β-values of the knockout and control cells, and the difference in methylation between knockout and control.
Additional file 4:. Tab. S3: Patient data of all DKC, AA samples and healthy controls. List of all patient samples and healthy controls with their according gender, chronological age, diagnosis, mutated genes, and blood counts.
Data Availability Statement
Data of DNA methylation profiles has been deposited at Gene Expression Omnibus (GEO) under the reference ID GSE141106. RNA-Seq data has been deposited at GEO under the reference ID GSE141107.