

Abstract
Lung cancer is a heterogeneous disease in which patient-specific treatments are desirable and the development of targeted therapies has been effective. Though mutations in KRAS are frequent in lung adenocarcinoma, there are currently no targeted agents against KRAS. Using a mouse lung adenocarcinoma cell line with a KRAS mutation (CMT167) we previously showed that PPARγ activation in lung cancer cells inhibits cell growth in vitro yet promotes tumor progression when activated in myeloid cells of the tumor microenvironment. Here we report PPARγ activation in myeloid cells promotes the production of TGF-β1, which in turn acts on CMT167 cancer cells to increase migration and induce an epithelial-mesenchymal transition (EMT). Targeting TGF-β1 signaling in CMT167 cells prevented their growth and metastasis in vivo. Similarly, another mouse lung adenocarcinoma cell line with a KRAS mutation, LLC, induced TGF-β1 in myeloid cells through PPARγ activation. However, LLC cells are more mesenchymal and did not undergo EMT in response to TGF-β1; nor did LLC require TGFβ1 signaling for metastasis in vivo. Converting CMT167 cells to a mesenchymal phenotype through overexpression of ZEB1 made them unresponsive to TGFβ1 receptor inhibition. The ability of TGF-β1 to induce EMT in lung tumors may represent a critical process in cancer progression. We propose that TGFβ receptor inhibition could provide an additional treatment option for KRAS mutant epithelial lung tumors.
Keywords: lung cancer, macrophages, TGF-β, PPARγ, EMT
INTRODUCTION
During the past several years, new therapeutic approaches for the treatment of lung cancer have been developed using targeted therapies against oncogenic drivers [1]. Oncogenic driving mutations in lung adenocarcinoma include KRAS, EGFR, and ALK fusions [2]. Specific inhibitors such as erlotinib and gefitinib targeting drivers like EGFR have resulted in good initial responses, although tumors develop resistance mechanisms that ultimately result in relapse and progression of the disease [3]. However, many lung adenocarcinomas lack distinguishable oncogenic drivers, or have mutations in KRAS, for which targeted therapies remain elusive. Therefore, development of additional therapeutic options, specifically for KRAS driven lung tumors is greatly needed.
Developing new therapeutic strategies will require a better understanding of the interactions between cancer cells and the surrounding tumor microenvironment (TME) [4–6]. The TME is a complex niche including cancer associated-fibroblasts, inflammatory cells, adaptive immune cells, vascular cells, and extracellular matrix. Defining the critical cell types and mechanisms whereby cells of the TME regulate cancer progression remains a challenge. In lung cancer, progress in studying the TME has been limited by the lack of mouse models allowing dissection of the role of cancer cells vis-à-vis cells of the TME. To investigate the role of the TME in lung cancer progression, our laboratory has employed an immunocompetent orthotopic mouse model where murine cancer cells derived from lung adenocarcinomas in C57BL/6 mice are directly injected into the left lung of syngeneic mice [7–12]. These cancer cells form primary tumors, which over several weeks progress to form secondary pulmonary tumors and metastasize to other organs such as the liver and brain. This model allows us to selectively manipulate pathways in either the cancer cells, using shRNA approaches, or in the TME, using the appropriate targeted knockout host.
We previously focused on the role of peroxisome proliferator-activated receptor γ (PPARγ) in KRAS mutant lung cancer [8]. PPARγ is a member of the nuclear receptor superfamily, which is a group of ligand-activated transcription factors that has been implicated in a variety of human diseases, including cancer [13, 14]. Studies from our laboratory and others have demonstrated that activation of PPARγ in human non-small cell lung cancer (NSCLC) cell lines inhibits transformed growth and promotes differentiation to a less invasive phenotype [8, 15–18]. Therefore we hypothesized that PPARγ activation would exert protective effects. However, we previously demonstrated that systemic PPARγ activation increases cancer progression due to activation of PPARγ in myeloid cells [8]. Therefore, while PPARγ activation directly in cancer cells appears to be protective, PPARγ activity in the TME promoted tumor metastasis. This demonstrated the importance of the TME in tumor progression and metastasis.
The goal of the present study was to define how PPARγ activation in myeloid cells affects cancer cell progression. We report that lung tumors activate PPARγ in myeloid cells to induce production of transforming growth factor-β1 (TGF-β1), which then acts on cancer cells to mediate progression. The effects of TGF-β1 on tumor progression are dependent on the differentiation state of the cancer cells. Epithelial cancer cells require TGF-β1 to undergo epithelial-mesenchymal transition (EMT), which is critical for progression, whereas mesenchymal cells do not, despite also promoting production of TGF-β1 by myeloid cells. This suggests that targeting TGF-β1 signaling may represent a therapeutic strategy for a subset of KRAS dependent epithelial lung tumors.
MATERIALS AND METHODS
Cells:
CMT167 cells [19] originally provided by Dr. Alvin Malkinson (University of Colorado), were stably transfected with firefly luciferase as previously described [12]. Luciferase expressing Lewis Lung Carcinoma (LLC-luc) cells were purchased from Caliper Life Sciences (LL/2-luc-M38). Both cell lines were maintained in DMEM with 4.5 g/L glucose containing 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 500 μg/ml G418.
CMT luciferase expressing cells (referred to as CMT-luc) were used to make the TGF-β receptor 2 (TGFβRII) knockdowns using short hairpin RNA (shRNA) specific to TGFβRII (TRCN0000294529, TRCN0000294530, TRCN0000294531, TRCN0000294600, TRCN0000294602). All shRNA constructs were purchased from the University of Colorado Functional Genomics Facility. Lentivirus produced in HEK 293FT cells was transferred to CMT-luc or LLC-luc cells with polybrene (Sigma). Cells expressing shRNA were selected using 2 μg/mL puromycin (Sigma). TGFβRII (fwd: GGGATTGCCATAGCTGTCAT, rev: TGATGGCACAATTGTCACTG) mRNA levels were measured by qRT-PCR and normalized to β–actin mRNA levels (fwd: TGATGGCACAATTGTCACTG, rev: CCAGTTGGTAACAATGCCATG). TGFβRII protein levels were determined by western blot using anti-TGFβRII antibody (Cell Signaling). TGFβRII response to 5 ng/mL recombinant mouse TGF-β1 (R&D Systems) or vehicle control was measured by western blot using anti-pSMAD2 (Cell Signaling #3108S) and total SMAD2/3 (Cell Signaling #8828S) antibodies.
CMT-luc cells were stably transfected with ZEB1 overexpression vector, ORF expression clone for ZEB1 (GeneCopoeia, EX-Mm05622-Lv-105) or an empty vector control (GeneCopoeia, EX-NEG-Lv105). Lentivirus produced in HEK293FT cells was transferred to CMT cells with polybrene (Sigma). Cells overexpressing ZEB1 were selected using 2 μg/ml puromycin (Sigma). Overexpression efficiency was confirmed by qRT-PCR Zeb1 (fwd: CCCGTGCGTTGAGATTTGAT rev: CTTCCCATTTAAAGGCTGGTCTAC). Individual clones were screened for expression by western blot using anti- ZEB1 antibodies (Novus Biologicals #NBP-105987). Protein levels for E-cadherin (BD Biosciences #610182) and Vimentin (Santa Cruz #sc-73259) were measured by western blot. mRNA levels were measured using qPCR for Zeb1 (fwd: CCCGTGCGTTGAGATTTGAT rev: CTTCCCATTTAAAGGCTGGTCTAC), E-cadherin (fwd:GTCCTGGGCAGAGTGAGATT rev: TGGAGCTTTAGATGCCGCTT), and Vimentin (fwd: CGGAAAGTGGAATCCTTGCA rev: CACATCGATCTGGACATGCTGT).
Mouse model:
Mice were maintained in the vivarium of the University of Colorado Anschutz Medical Campus. All procedures were performed under protocols approved by the Institutional Animal Care and Use Committee at the University of Colorado Anschutz Medical Campus. PPARγ myeloid cell knockout (PPARγ-MKO) mice were generated by crossing PPARγflox/flox mice with LysMCre transgenic mice. PPARγflox/flox mice lacking LysMCre expression were used as controls (PPARγ-WT). In experiments using the TGFβRI inhibitor SB431542 (Selleck Chemicals), mice were treated with 10 mg/kg SB431542 by intraperitoneal (IP) injection at the time of cancer cell injection. Treatment was continued daily, five days a week until the time of euthanasia.
Cancer cells were prepared in Hanks Buffered Salt Solution (HBSS) containing 1.35 mg/mL Matrigel (Corning) and injected directly into the left lung as described previously [8, 12, 20]. Directly prior to euthanasia, mice were injected IP with 300 mg/kg D-luciferin (PerkinElmer). Following euthanasia, lungs were perfused and inflated with PBS/heparin (80 U/mL, Sigma). The lungs, liver, and brain were removed for direct ex vivo bioluminescent imaging using an IVIS 50 imaging system (Caliper). Metastatic incidence was identified by positive bioluminescence in each organ. Primary tumor size was measured by digital caliper. The lungs were fixed in 10% formalin for immunohistochemistry and hematoxylin and eosin (H&E) staining. Secondary lung metastases were counted by clusters of cancer cells identified in H&E stained lung sections.
Flow cytometry:
For flow cytometric analysis, tumor bearing left lungs from four mice were pooled and digested to form single cell suspensions as previously described [20]. Cells were blocked with anti-CD16/32 prior to staining with marker specific antibodies. The specific antibodies used were: CD11b-FITC (clone: M1/70), SiglecF-PE (clone:E50–2440), Ly6G-PECy7(clone 1A8), CD64-Alexa647(clone:X54–5/7.1), CD11c-APC-Cy7(clone:HL3) (BD Biosciences). Flow cytometry was performed using the Gallios flow cytometer (Beckman Coulter) by the University of Colorado Cancer Center Flow Cytometry Core. The data represent the average of four separate flow analyses.
TGF-β1 immunohistochemistry:
Whole left lungs preserved in 10% formalin were paraffin embedded and processed into tissue sections (5 μm) by the Pathology Core in the Pulmonary Division at the University of Colorado Anschutz Medical Campus. Tissue sections were stained with rabbit polyclonal TGF-β1 antibody (Santa Cruz #sc-146). TGF-β1 positive cells were identified by brown staining and counted as a percent of the total cells per field. Three tumors and three surrounding lung fields were counted per mouse. A total of eight PPARγ-WT and five PPARγ-MKO mice were analyzed for CMT tumors and five for LLC tumors.
Isolation and treatment of bone marrow-derived macrophages:
Bone marrow cells isolated from the femur and tibia of PPARγ-MKO or PPARγ-WT mice were matured in RPMI with 20% FBS, 1% Pen-Strep and 50 ng/mL recombinant mouse M-CSF for seven days, which results in a population that is >95% macrophages[12]. Following differentiation, bone marrow-derived macrophages (BMDM) were treated with the PPARγ agonist pioglitazone (10 μM), the PPARγ antagonist T0070907 (T007: 10 μM, Cayman), a combination of pioglitazone and T007, or vehicle control (DMSO) for 48 hours. In experiments where tumor homogenate was used to stimulate BMDM, whole left lungs were collected from tumor-bearing or naïve mice and snap frozen in liquid nitrogen. Tissues were homogenized in phosphate-buffered saline (PBS) using an overhead stirrer (Wheaton). A final concentration of 0.5 mg/mL protein each from tumor or control homogenate was added to BMDM for 48 hours. BMDM were then washed three times with HBSS and placed into fresh RPMI for 24 hours prior to collecting the culture media. TGF-β1 released into the media was measured by ELISA (R&D Systems). TGF-β1 levels were normalized to total BMDM RNA or protein concentration.
Treatment of Macrophages with CMT-Conditioned Media:
Conditioned media was collected from CMT cells after 48 hours. Media incubated for 48 hours in the absence of cells was used as a control. RAW 264.7 cells were treated with CMT-conditioned media (30%) and supplemented with pioglitazone (10μM) or DMSO control for 48 hours. Cells were washed and placed into fresh media and TGF-β1 released into the media was measured by ELISA (R&D Systems) at 24 hours. TGF-β1 levels were normalized to total RAW 264.7 protein concentration.
Measurement of EMT, cell migration and proliferation:
To measure changes in EMT, parental, control shRNA, or TGFβRII knockdown CMT-luc or LLC-lu cells were treated with 5 ng/mL recombinant mouse TGF-β1 (R&D Systems) or vehicle control. For experiments with the TGFβRI inhibitor, 1 μM SB431542 was added simultaneously. Protein and RNA were collected after 72 hours. Vimentin, Zeb1, and E-cadherin were measured by qRT-PCR and normalized to the geometric mean of β–actin, GAPDH, and ubiquitin C. In CMT-luc cells, E-cadherin was measured by Western blot using an anti-E-cadherin antibody (BD Biosciences #610182). Migration was measured using cells pre-treated with 5 ng/mL TGF-β1 or vehicle for 48 hours. Cells were suspended into 0.1% serum media with vehicle or 5 ng/mL TGF-β1 to match the original treatment and placed in the top of an 8 μm transwell chamber. The bottom chamber contained media with 10% FBS with vehicle or TGF-β1. Migration toward the serum gradient was measured 24 hours later by DAPI staining. The number of cells that migrated was counted in five 10X fields per transwell. For measurement of cell proliferation, cells were treated with 5 ng/mL TGF-β1 or vehicle for 72 hours prior to plating in equal numbers. After an overnight incubation, a zero time point was collected. Cells were harvested after 72 hours. Cell number was quantified by measuring luciferase activity normalized to the zero time point.
Statistical Analysis:
All data are presented as mean ± SEM. Multi-group analyses were conducted using non-parametric 1-way ANOVA (Kruskal-Wallis) followed by Dunn’s post hoc tests for multiple comparisons. Unpaired Student’s t-test was used to compare differences between two variables. For metastasis incidence, Fisher’s exact test was used to compare variables to control conditions. P values less than 0.05 were considered statistically significant.
RESULTS
PPARγ activation induces TGF-β1 expression in myeloid cells
To examine how the lung tumor microenvironment contributes to tumor growth we have used an orthotopic immunocompetent model of lung cancer. This model has the advantage of allowing us to study the interaction of the immune system with lung tumors in the correct microenvironment, which is imperative to accurately recapitulating these interactions in vivo [8, 11, 12, 20]. For these studies we have employed two C57BL/6 cell lines, CMT167 [7, 9–12, 21] and Lewis lung carcinoma (LLC) cells, both of which express oncogenic KRAS and are derived from C57BL/6 mice [9, 22]. These cell lines have been engineered to express luciferase, which allows for efficient tracking of these cells in mice. CMT-luc and LLC-luc have different states of differentiation. CMT-luc cells appear more epithelial in vitro, growing in tightly bound clusters, while LLC-luc cells appear more mesenchymal, growing more as individual cells with spindly protrusions (Supplemental Figure S1A). Human lung adenocarcinoma cell lines have been classified as epithelial or mesenchymal based on their E-cadherin to vimentin ratio, with the log2 transformed value of greater than zero corresponding to epithelial cells and less than zero to mesenchymal cells [23]. As a quantitative measure of their state of differentiation, the levels of E-cadherin and vimentin in each cell line were measured by qRT-PCR. CMT-luc cells have high levels of E-cadherin with low levels of vimentin, resulting in a log2 transformed E-cadherin/Vimentin ratio of 1.44. LLC-luc cells have virtually undetectable levels of E-cadherin and high levels of vimentin resulting in a ratio of −4.85 (Supplemental Figure S1B and S1C).
We previously showed that loss of PPARγ in mouse myeloid cells decreases metastasis of CMT-luc cells [8]. To define the mechanisms whereby activation of PPARγ in myeloid cells regulates CMT-luc progression, we employed mice with a targeted deletion of PPARγ in myeloid cells (PPARγ-MKO) or wild type controls (PPARγ-WT) and sought to determine if deletion of PPARγ altered myeloid cell recruitment to the primary tumor. PPARγ-WT or PPARγ-MKO mice were injected with CMT-luc cells as described previously [9]. Myeloid cell populations within the tumors were quantified by flow cytometry. Consistent with our previous studies [10], the percentage of neutrophils and recruited macrophages were increased within tumor-bearing lungs compared to normal lung tissue. However, loss of expression of PPARγ did not affect myeloid cell recruitment, as there was no difference in the percentage of neutrophils (CD11b+Ly6G+), alveolar macrophages (CD11c+SigF+) or recruited macrophages (CD11b+CD64+SigF-) when comparing tumor-bearing lungs from PPARγ wild type (PPARγ-WT) or PPARγ myeloid cell knockout (PPARγ-MKO) mice (Supplemental Figure S2A and S2B).
Since there was no difference in myeloid cell recruitment in the PPARγ-WT or PPARγ-MKO mice, we examined factors released by myeloid cells that could influence tumor progression. PPARγ activation induces an “M2” phenotype in macrophages [8, 24], and the “M2” phenotype is associated with TGF-β1 production in tumor settings [25]. Additionally, TGF-β1 increases tumor cell metastasis by promoting EMT and a more invasive phenotype [26, 27]. TGF-β1 expression, as determined by immunohistochemistry, was detected in CMT-luc tumors and in the surrounding lung tissue of PPARγ-WT mice. These levels were markedly decreased in both compartments in PPARγ-MKO mice (Figure 1A, 1B). To determine if myeloid PPARγ activation directly increases TGF-β1 production by myeloid cells, bone marrow-derived macrophages (BMDM) from PPARγ-WT or PPARγ-MKO mice were treated with the PPARγ agonist pioglitazone (Pio). In PPARγ-WT BMDM, pioglitazone significantly increased TGF-β1 release. This was not observed in PPARγ-MKO BMDM, or when treating PPARγ-WT BMDM in combination with the PPARγ antagonist, T0070907 (T007) (Figure 1C). Together these data demonstrate that PPARγ activation in myeloid cells induces TGF-β1 production in vitro and is contributing to TGF-β1 production within the TME in vivo.
Figure 1: TGF-β1 is produced by myeloid cells in response to PPARγ activation.
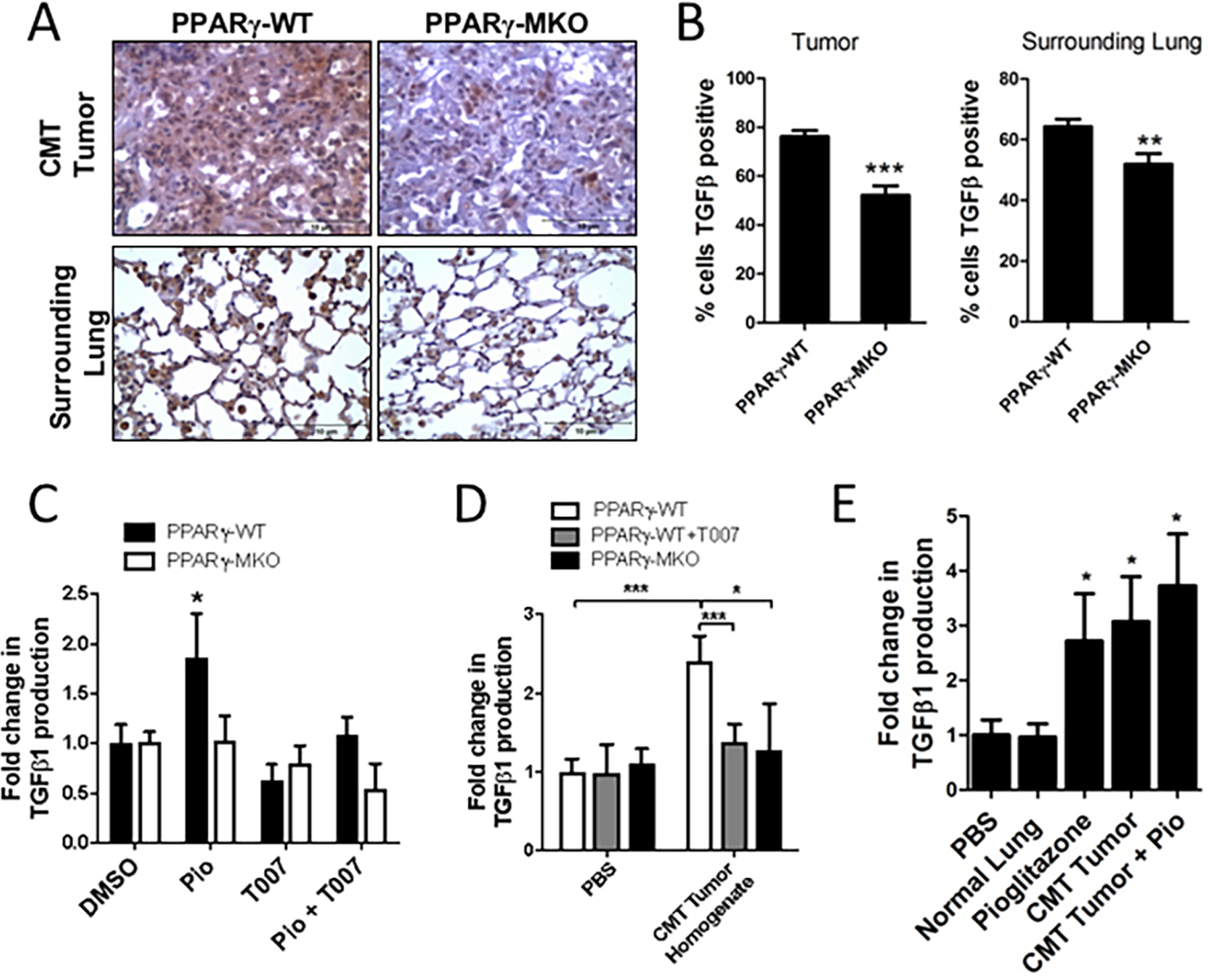
(A) PPARγ-WT or PPARγ-MKO mice were orthotopically injected with CMT-luc cells. Tumor bearing lungs were collected 4 weeks later. IHC for TGF-β1 was performed on tissue sections and representative images at 40X magnification are shown. (B) Quantification of TGF-β1 positive cells within tumor and surrounding lung tissue in IHC sections, reported as a percent of the total cells. (C) TGF-β1 released from bone marrow derived macrophages (BMDM) from PPARγ-WT or PPARγ-MKO mice treated with pioglitazone (Pio), PPARγ inhibitor (T007), a combination of both, or vehicle control (DMSO) and normalized to WT DMSO control. (D) TGF-β1 released from PPARγ-WT or PPARγ-MKO BMDM treated with media supplemented with CMT-luc tumor homogenate and normalized to PPARγ-WT PBS control. (E) TGF-β1 released from PPARγ-WT BMDM treated with media supplemented with homogenate from normal lung or lung with CMT-luc tumor (CMT), pioglitazone (Pio) or a combination of tumor homogenate and pioglitazone and normalized to WT vehicle control (*p<0.05, **p<0.01, ***p<0.001).
Since TGF-β1 expression within the tumor and surrounding tissue in vivo is dependent on PPARγ expression in myeloid cells, even in the absence of a pharmacological activator such as pioglitazone (Figure 1A), we hypothesized that CMT-luc tumors produce endogenous activators of PPARγ that act on myeloid cells to increase TGF-β1 production. We therefore examined TGF-β1 production in BMDM treated with CMT-luc tumor homogenate. CMT-luc tumor homogenate increased TGF-β1 production in PPARγ-WT BMDM compared to the PBS control; this increase was blocked by the PPARγ inhibitor T007 and was not detected in PPARγ-MKO BMDM (Figure 1D). Importantly, tissue homogenate from normal lungs did not increase TGF-β1 production by BMDM (Figure 1E). Additionally, the combination of CMT-luc tumor homogenate and pioglitazone did not result in additional increases in the levels of TGF-β1 produced by PPARγ-WT BMDM, indicating maximal release of TGF-β1 is achieved using either agent (Figure 1E). We also investigated the effect of CMT-conditioned media on TGF-β1 production by the mouse macrophage cell line, RAW 264.7. Stimulation of RAW 264.7 cells with pioglitazone increased TGF-β1 production. Conditioned media from CMT cells modestly induced TGF-β1 production by RAW macrophages and this appeared to be additive with pioglizatone stimulation (Supplemental Figure S2C). These data indicate that factors produced by CMT-luc tumors activate PPARγ in myeloid cells, leading to elevated production of TGF-β1.
Responsiveness to TGF-β1 is necessary for progression of CMT-luc tumors
To assess the effect of increased myeloid cell production of TGF-β1 on cancer cells, TGFβRII was silenced in CMT-luc cells to block TGF-β1 signaling. Two lentiviral shRNAs targeting TGFβRII (531 and 602) achieved greater than 50% knockdown by mRNA and protein levels when compared to non-transduced parental cells or control non-targeting shRNA (Figure 2A, 2B), and were used for further studies. TGFβRII knockdown shRNA 602 slowed down the kinetics of p-SMAD2/3 induction in response to TGF-β1 compared to the parental control cells or the control shRNA (Supplemental Figure S3A). To investigate the lack of TGF-β1 signaling on tumor cell progression, we injected CMT-luc control vector and CMT-luc cells with the TGFβRII knockdown shRNA 602 into the left lung of WT mice. CMT-luc cells with TGFβRII knockdown formed smaller primary tumors when compared to control CMT-luc cells (Figure 2C). In fact, the ability of TGFβRII knockdown cells to establish detectable tumors after injection was lower than that of control cells (TGFβRII knockdown: 8/19: 42% vs control: 16/16:100%). Mice injected with TGFβRII knockdown CMT-luc cells also had significantly lower incidence of metastases to the other lobes of the lung and trends towards lower metastasis to the liver (Figure 2D).
Figure 2: Inhibition of TGFβRII signaling reduces primary tumor size and metastasis of CMT-luc cells.
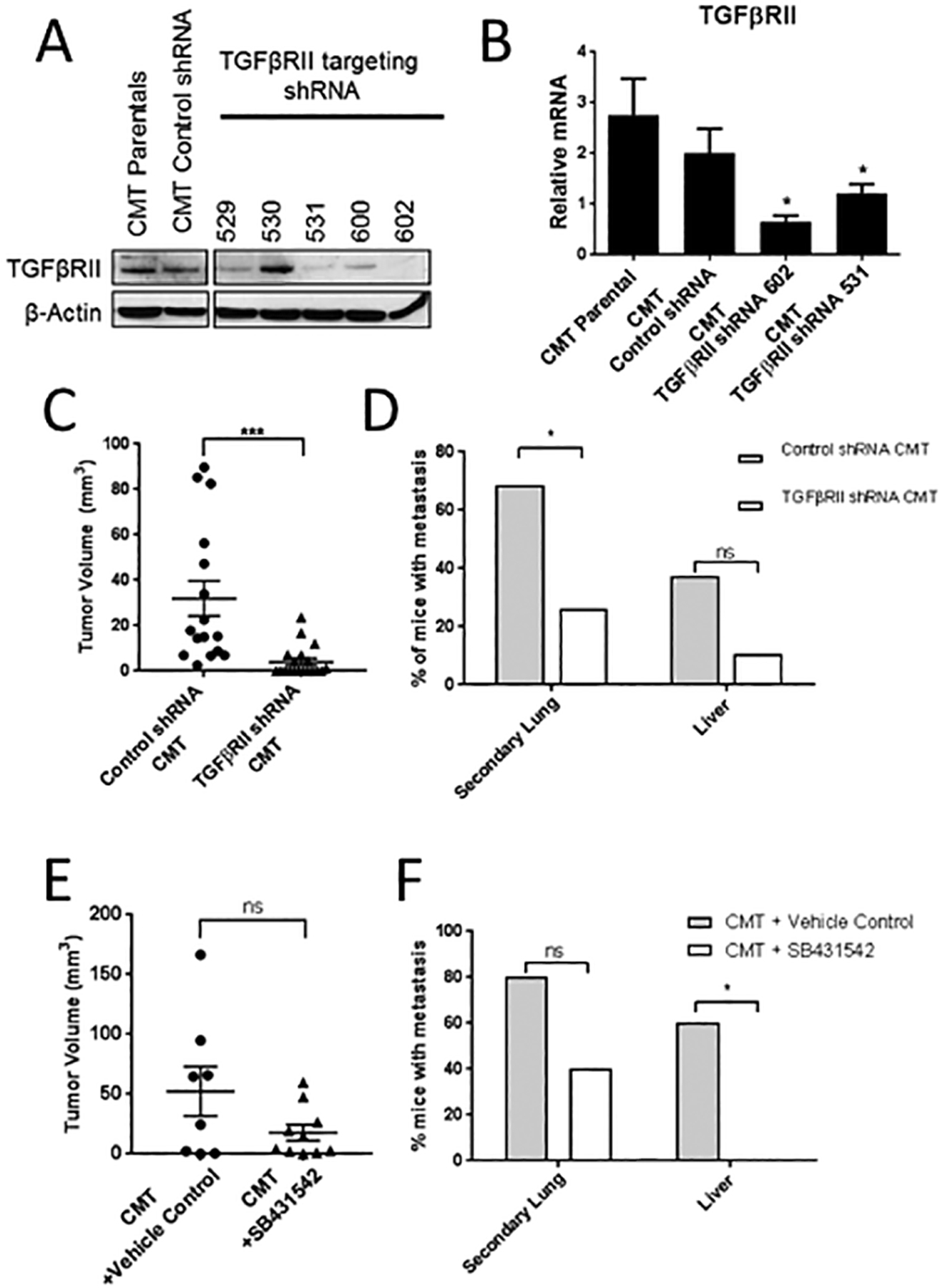
(A) Western blot for TGFβRII in untransduced parental CMT-luc cells or CMT-luc cells transduced with control or TGFβRII targeting shRNA. (B) qRT-PCR of TGFβRII expression in parental CMT-luc and CMT-luc cells transduced with control shRNA or TGFβRII targeting shRNA (602 or 531). (C,D) WT mice were orthotopically injected with CMT-luc cells expressing control shRNA or the TGFβRII targeting shRNA 602. Mice were harvested to measure metastasis 5 weeks post injection. There were 16 mice injected with the control shRNA CMT-luc cells and 19 mice injected with the TGFβRII targeting shRNA CMT-luc cells. The data for all mice injected is reported. (E,F) WT mice were orthotopically injected with CMT-luc cells and treated with the TGFβRI inhibitor SB431542 (10 mg/kg) or vehicle control (1:1 DMSO:PBS) starting at the time of injection and continuing 5 days a week for 5 weeks. (D,F) Primary tumor size was measured by caliper and reported as the tumor volume in mm3. (E,F) Incidence of metastasis reported as the percent of mice with metastasis to the other lobes of the lung (secondary lung) or liver, as measured by bioluminescence (*p<0.05).
To confirm these results, we tested the effects of the TGFβRI inhibitor SB431542 on CMT-luc tumor progression in vivo. TGFβRI dimerizes with TGFβRII following ligand binding to initiate downstream signaling from the receptor. Therefore, inhibiting TGFβRI should have similar effects to the knockdown of TGFβRII. Mice were injected with CMT-luc cells and treated with SB431542 or vehicle control. Primary tumor incidence did not vary between control and treated groups (9/10:90% for each). Importantly, mice treated with SB431542 trended toward smaller primary tumors, similar to tumors from TGFβRII knockdown CMT-luc cells (Figure 2E). In addition, mice treated with SB431542 had lower metastases to the other lung lobes and liver, with no liver metastasis detected in the SB431542 treated group (Figure 2F).
CMT-luc cells undergo EMT and increase migration in response to TGF-β1
Because TGF-β1 signaling inhibition decreased primary tumor size and metastasis, we sought to further delineate the effects TGF-β1 signaling has on CMT-luc cells that could be contributing to inhibition of tumor progression. Since TGF-β1 is known to affect both proliferation and migration of cancer cells [27–29], we examined the effect of TGF-β1 treatment on these processes in CMT-luc cells in vitro. TGF-β1 treatment decreased proliferation of CMT-luc parental and control shRNA cells, but had no effect on CMT-luc cells expressing TGFβRII shRNA 602. CMT-luc cells expressing TGFβRII shRNA 531, which resulted in a smaller degree of TGFβRII knockdown compared to shRNA 602 (Figure 2A), still exhibited decreased proliferation in response to TGF-β1 as assessed by luciferase activity (Figure 3A). These data would suggest that loss of TGF-β1 signaling in vivo should increase primary tumor size, which is contrary to the observed results that TGFβRII knockdown decreased primary tumor size of CMT-luc in vivo (Figure 2C). This would suggest that TGF-β1 is regulating additional biological processes critical for progression in these cells.
Figure 3: Response of CMT-luc cells treated with TGF-β1 in vitro.
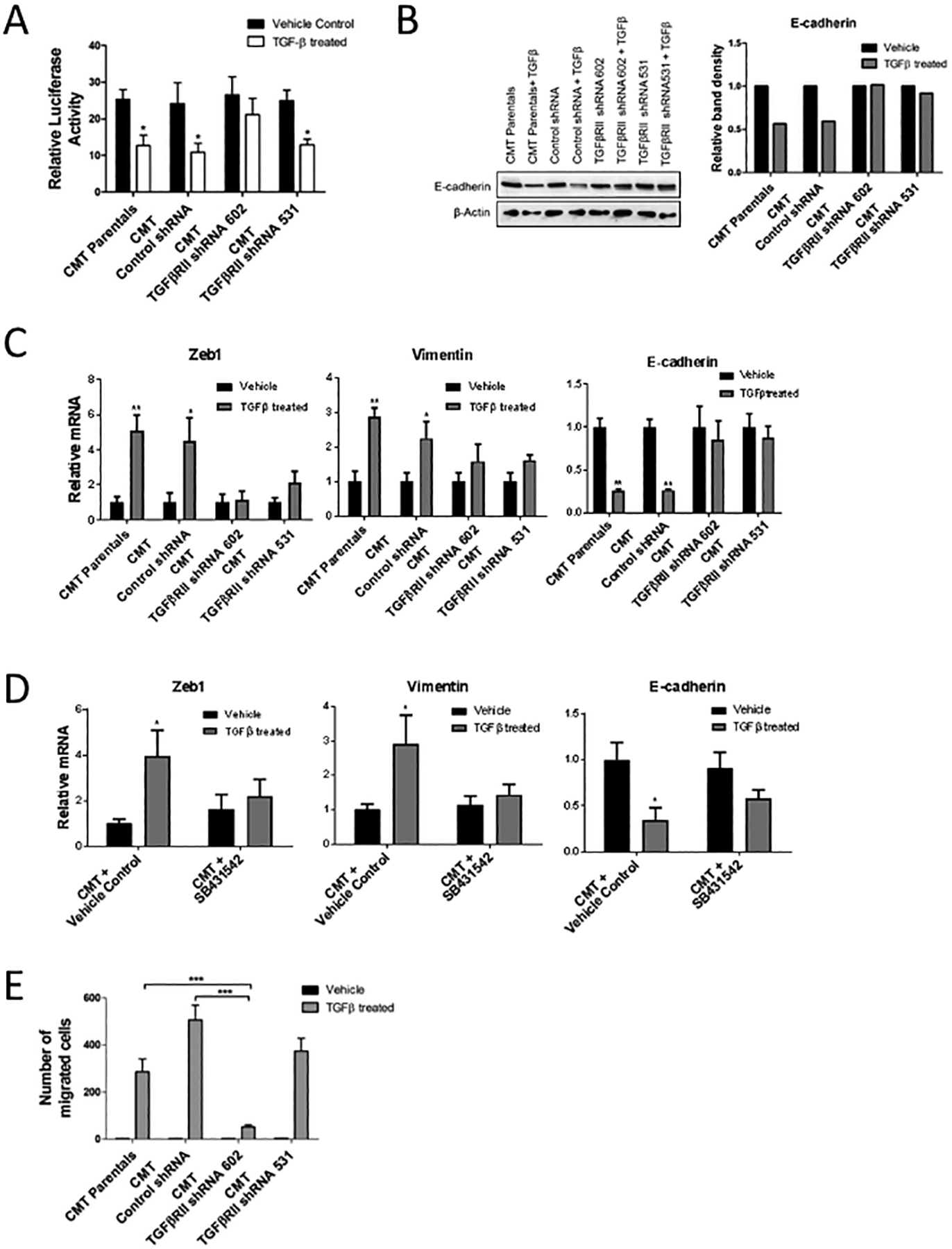
(A) Proliferation of cells treated with TGF-β1 or vehicle control in vitro, measured as change in total luminescence (*p<0.05 for TGF-β1 treated compared to respective vehicle treated controls). (B) Representative western blot for E-cadherin in CMT-luc cells treated with TGF-β1 compared to control with corresponding densitometry analysis reported as relative band intensity. (C) qRT-PCR for Zeb1, Vimentin and E-cadherin in CMT-luc cells post TGF-β1 treatment compared to control. (D) qRT-PCR for E-cadherin, Zeb1, and Vimentin in CMT-luc cells post TGF-β1 treatment combined with the TGFβRI inhibitor SB431542 or vehicle control (DMSO). (E) Number of migrating cells in transwell migration assays using CMT-luc cells treated with TGF-β1 or vehicle control (**p<0.01, ***p<0.001).
When treated with TGF-β1 in vitro, CMT-luc cells underwent a morphologic change to a more spindle-like morphology, consistent with EMT (Supplemental Figure S3B). This change was associated with decreased expression of E-cadherin and increased expression of mesenchymal markers, Zeb1 and vimentin. These changes were not observed in either of the CMT-luc cells with TGFβRII knockdown (Figure 3B, 3C). In addition, the TGFβRI inhibitor SB431542 inhibited induction of EMT in response to TGF-β1 in CMT-luc cells, blunting the inhibition of E-cadherin expression, and decreasing the induction of Zeb1 and vimentin (Figure 3D). Since TGF-β1 is known to promote a more invasive phenotype [26, 30] we examined the effects of TGF-β1 on cell migration in vitro using transwell migration assays. CMT-luc cells had low levels of migration at baseline, but readily increased migration in response to TGF-β1 treatment. In CMT-luc TGFβRII knockdown shRNA 602 cell migration in response to TGF-β1 was almost completely eliminated, whereas shRNA531 showed little effect (Figure 3E). Collectively, these data demonstrate that in response to TGF-β1, CMT-luc cells undergo EMT and increase migration in vitro, which are two key events associated with tumor progression.
LLC-luc tumors are not dependent on TGF-β1 signaling for progression
We examined this pathway in a second mouse lung cancer cell line, LLC-luc, which is more mesenchymal (Supplemental Figure S1). We sought to determine whether deletion of PPARγ in myeloid cells would affect progression of LLC-luc tumors, as we have observed in CMT167 tumors [8]. Similar to CMT-luc cells, we found no difference in primary tumor size when LLC-luc cells were injected into the lung of PPARγ-WT or PPARγ-MKO mice (Supplemental Figure S4A). There was also no difference in the extent of metastasis, which was different from what we observed with CMT-luc cells (Supplemental Figure S4B, S4C).
To determine if LLC-luc tumors have PPARγ dependent TGF-β1 production, we examined the levels of TGF-β1 of LLC tumors in PPARγ-WT and PPARγ-MKO mice by IHC (Figure 4A, 4B). Similar to the CMT-luc tumors, TGF-β1 expression was detected in LLC-luc tumors and in the surrounding lung tissue of PPARγ-WT mice. TGF-β1 expression was markedly decreased in the tumors in PPARγ-MKO, but not in the surrounding lung. To investigate if LLC-luc tumors would also induce TGF-β1 production by macrophages in a PPARγ dependent manner we tested the ability of LLC-luc tumor homogenate to induce TGF-β1 production in PPARγ-WT and PPARγ-MKO BMDM. LLC-luc tumor homogenate also induced TGF-β1 production by BMDM, similar to what we observed using CMT-luc tumor homogenate. This was dependent on PPARγ activation, as LLC-luc tumor homogenate did not increase TGF-β1 production in PPARγ-MKO BMDM or WT BMDM treated with the PPARγ antagonist, T007 (Figure 4C). This suggests that both murine lung cancer cells lines are able to produce factors that activate PPARγ and induce TGF-β1 production within macrophages of the TME.
Figure 4: LLC-luc induce PPARγ dependent myeloid cell TGF-β1 production but do not require TGF-β1 signaling for progression.
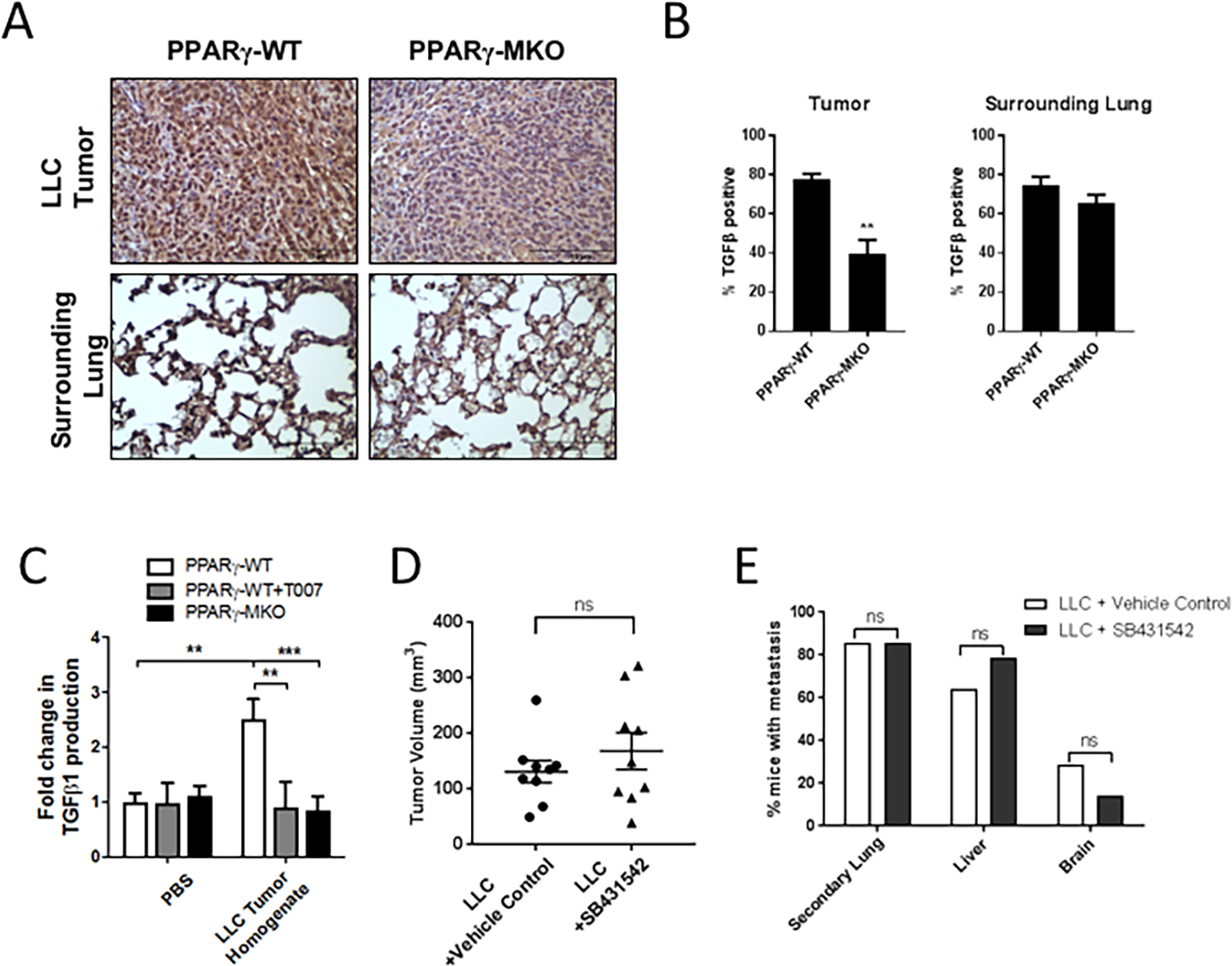
(A) PPARγ-WT or PPARγ-MKO mice were orthotopically injected with LLC-luc cells. Tumor bearing lungs were collected 2.5 weeks later. IHC for TGF-β1 was performed on tissue sections and representative images at 40X magnification are shown. (B) Quantification of TGF-β1 positive cells within tumor and surrounding lung tissue in IHC sections, reported as a percent of the total. (C) TGF-β1 released from PPARγ-WT or PPARγ-MKO BMDM treated with media supplemented with LLC-luc tumor homogenate. (D) WT mice were orthotopically injected with LLC-luc cells and treated with the TGFβRI inhibitor SB431542 or vehicle control (1:1 DMSO:PBS) starting at the time of injection and continuing 5 days a week for 2.5 weeks. There were 8 mice per group injected with LLC-luc cells. Primary tumor size was measured by caliper and reported as the tumor volume in mm3. (E) Incidence of metastasis reported as the percent of mice with metastasis to the other lobes of the lung (secondary lung), liver, or brain as measured by bioluminescence (**p<0.01, ***p<0.001).
Since both CMT-luc and LLC-luc tumors induce activation of PPARγ to produce TGF-β1, we examined the responsiveness of LLC-luc cells to TGFβ receptor signaling. We compared tumor progression of LLC treated with the TGFβRII inhibitor. We observed no difference in primary tumor size or the extent of metastasis (Figure 4D, 4E). To correlate effects of TGF-β1 on LLC-luc tumors in vivo with in vitro responsiveness, we treated LLC-luc cells with TGF-β1 in vitro. Similar to CMT-luc cells, TGF-β1 exposure inhibited LLC-luc cell proliferation (Supplemental Figure S5A). LLC-luc cells had high basal rates of migration, which was not affected by TGF-β1 treatment (Supplemental Figure S5B). Similarly, when LLC-luc cells were treated with TGF-β1 or with TGFβRI inhibitor SB431542 in vitro there was no change in expression of EMT markers (Supplemental Figure S5C).
Overexpression of ZEB1 in CMT cells results in a less differentiated phenotype and resistance to TGF-β inhibition
Together these data indicate that while TGF-β1 decreases proliferation of both cell lines in vitro, these two cell lines show very different responses to TGFβRI inhibition in vivo. While there are potentially many differences between CMT-luc and LLC-luc cells, one distinguishing feature is their state of differentiation. To test if this difference affects their response TGFβRI inhibition, we overexpressed ZEB1, a promoter of EMT, in CMT-luc cells to generate cells that were less differentiated (CMT-ZEB1OE). Morphologically, ZEB1 overexpressing cells showed a more mesenchymal morphology compared to control cells transfected with empty vector (Figure 5A). Overexpression of ZEB1 resulted in increased mRNA and protein expression of the mesenchymal marker vimentin, and decreased protein levels of E-cadherin (Figure 5B, 5C); levels of E-cadherin mRNA were not significantly altered, suggesting that this is not a complete reversal of the epithelilal phenotype. Importantly, we assessed the effect of altering the state of differentiation on response to TGFβRII inhibition in vivo. Primary tumor growth was strongly inhibited in the empty vector controls, similar to what we observed in parental CMT-luc cells. However, CMT-ZEB1OE cells were resistant to this treatment (Figure 5D), similar to what we observed in LLC tumors. In addition, ZEB1 overexpression also reversed the effect of the inhibitor on the incidence of secondary lung metastasis (Figure 5E). Thus, converting epithelial CMT cells to a more mesenchymal phenotype via overexpression of ZEB1 induced resistant to TGF-β1 inhibition.
Figure 5: Overexpression of ZEB1 in CMT cells results in a less differentiated phenotype and resistance to TGF-β inhibition.
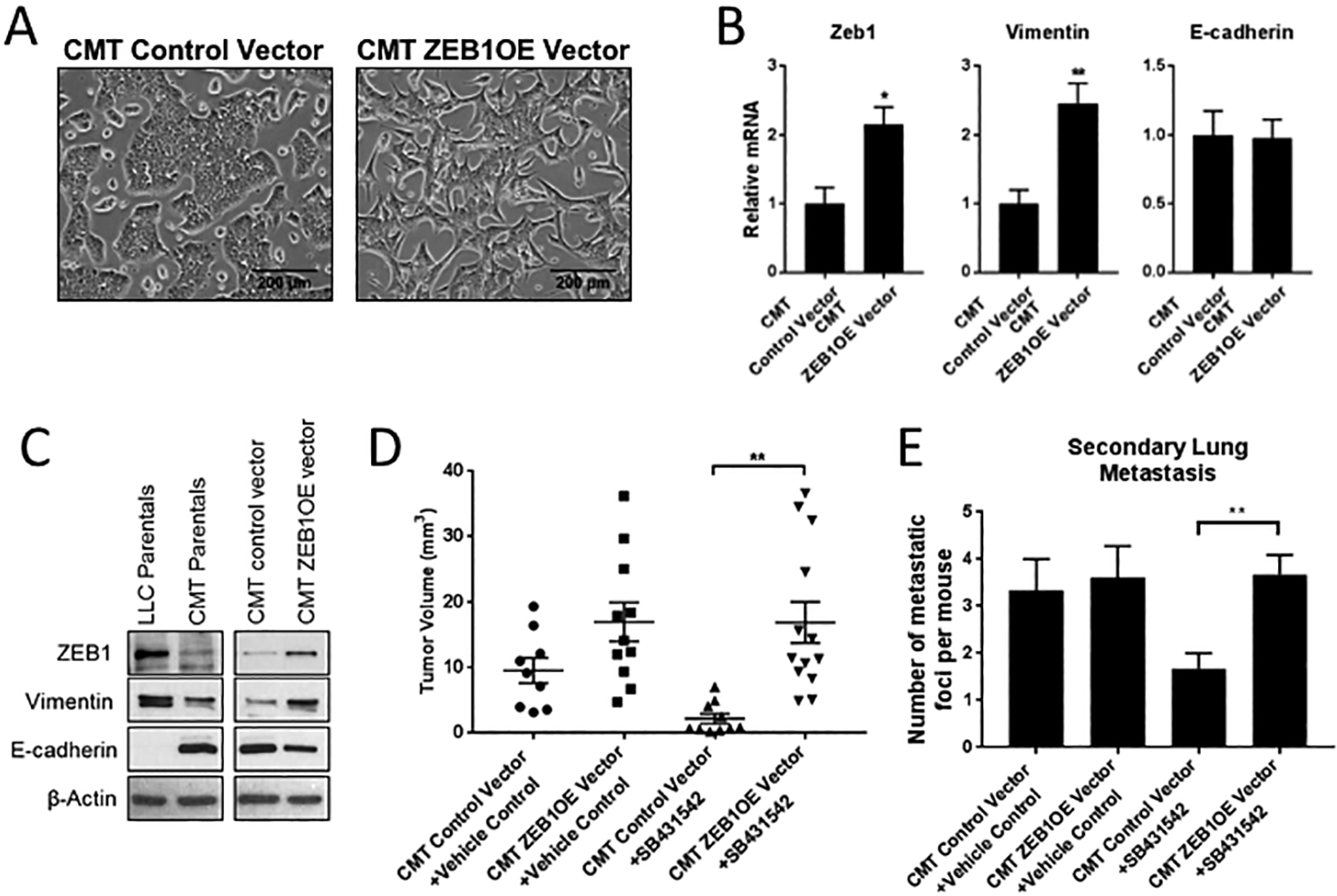
(A) Light microscope images of CMT-luc control vector and CMT-ZEBOE vector in vitro. (B) qRT-PCR of Zeb1, Vimentin, and E-cadherin in CMT-luc control vector and CMT-ZEBOE vector cells. (C) Western blot of Zeb1, Vimentin, and E-cadherin in LLC-luc, CMT-luc parentas, CMT-luc control vector and CMT-ZEBOE vector cells. (D,E) WT mice were orthotopically injected with CMT control vector or CMT-luc ZEBOE and treated with TGFβRI inhibitor SB431542 or vehicle control (1:1 DMSO:PBS) starting 10 days after injection and continuing 5 days a week for 5 weeks. There were five mice per group in the vehicle treated and 6 mice per group in the SB431542 treated. (D) Primary tumor size was measured by caliper and reported as the tumor volume in mm3. (E) Number of secondary lung metastases was counted on H&E stained lung tissue sections. (*p<0.05, **p<0.01)
DISCUSSION
The role that the microenvironment plays in tumor progression has become increasingly apparent. In terms of cell composition, myeloid cells are large contributors to the TME in many tumor types and have been associated with poorer prognosis in patients with increased densities, including lung cancer [31, 32]. For lung cancer, this correlation has been controversial in the past, although further characterization of tumor associated macrophages (TAMs) in terms of location and phenotype have been more reliable at predicting outcomes in patients (reviewed in [33, 34]). While myeloid cells have mostly been studied in terms of their ability to suppress the adaptive immune system, how these cells regulate other cells in the tumor, including cancer cells is not well understood. Myeloid cells induce angiogenesis, influence matrix deposition and remodeling, and promote migration and invasion of cancer cells. In terms of metastasis, the most well-defined mechanism by which myeloid cells are involved is through the production of epidermal growth factor (EGF) in response to CSF-1 production by the tumor. EGF then acts on the cancer cells to promote their migration (reviewed in [35]).
Though it has been previously demonstrated that TAMs produce TGF-β within the TME, the consequence of its secretion from myeloid cells has mostly been implicated in immunosuppression. We have shown that TGF-β1 expression within the TME is mediated, at least in part, through PPARγ-dependent production in myeloid cells. Blocking this pathway, either through deletion of PPARγ in myeloid cells [8] or silencing TGFβRII on cancer cells, results in inhibition of primary tumor growth and lower incidence of metastases in the mouse lung cancer cell line CMT-luc, suggesting a direct effect of TGF-β1 released from macrophages on the cancer cells themselves. This is similar to a recent study that demonstrated macrophage production of TGF-β1 leads to EMT and acquisition of cancer stem cell like properties in the mouse hepatocellular carcinoma cell line Hepa1–6 cells [36]. Importantly, another mouse lung cancer cell line, LLC-luc, also induced TGF-β production by myeloid cells in a PPARγ dependent manner, but did not require TGF-β signaling for progression and metastasis. Our data indicate that the importance of specific pathways activated in the TME will be related to the nature of the cancer cell. It should also be noted that other cells, including the cancer cell, can produce TGF-β. However, since the production of active TGF-β involves release from the matrix [37], it is likely that production by distinct components of the tumor have different effects on progression.
Recent studies have subdivided human lung cancers into separate categories based on the identification of dominant oncogenic drivers. Therapeutics that target cancer cells are based on identifying these drivers and personalizing treatment to target these pathways [3]. In general, studies focusing on the TME have not sufficiently considered the drivers and the properties of the cancer cells, assuming a “one size fits all” approach in targeting the TME. Both CMT-luc and LLC-luc cells express oncogenic KRAS [9]. Perhaps more importantly, the phenotype of these cancer cells is clearly different; illustrating that despite having the same oncogenic driver, tumors are very heterogeneous. In fact, converting epithelial CMT cells to a more mesenchymal phenotype by overexpression of ZEB1 made them resistant to inhibition of TGF-β1, strongly suggesting that the state of differentiation is a major determinant of this response. We propose that activation of PPARγ in the TME, specifically in myeloid cells, will accelerate progression in more epithelial cancer cells, such as CMT167, where EMT is critical for progression, but will have lesser effects in more mesenchymal tumors. A recent study analyzed the TCGA dataset for the expression of classic epithelial or mesenchymal markers and found that the majority of human lung adenocarcinomas are epithelial in nature [38], demonstrating the applicability of finding targets to prevent EMT in these tumors.
Targeting TGF-β signaling in cancer has been a desirable achievement for the treatment of many solid tumors. The most significant problem with this approach is delineating the differential effects of TGF-β signaling in cancer cells versus cells of the tumor microenvironment. For some cancer types the predominant role of TGF-β within the cancer cell is tumor suppressive, while TGF-β signaling in cells of the TME is mostly tumor promoting [29]. Before targeting TGF-β signaling in patients, it is important to be able to identify those that will benefit from the inhibition rather than those that will not respond or will respond negatively. In NSCLC, increased levels of TGF-β1 have been associated with tumor progression and metastasis in both human patients and mouse models [39–42], while other studies have demonstrated a tumor suppressive role of TGFβRII in NSCLC [43–45]. Similarly, we have shown that our mouse lung cancer cell lines have differential responses to inhibition of TGF-β1 signaling, therefore highlighting the necessity of delineating tumors that will be sensitive to TGF-β1 inhibition. While a recent review by Colak and Dijke suggested sensitivity to TGF-β targeting in other cancer types is associated with a mesenchymal phenotype [29], we have found the opposite to be true in our mouse model of lung cancer. Further analysis to confirm the link between epithelial differentiation state and the responsiveness to TGFβR inhibition will need to be performed. The results of this study provide promising preclinical data to support this hypothesis and advocate for targeting TGF-β signaling to prevent progression of epithelial lung cancers.
Supplementary Material
IMPLICATIONS.
This study suggests that TGFβ receptor inhibitors may be an effective therapy in a subset of KRAS mutant NSCLC patients which show an epithelial phenotype.
ACKNOWLEDGMENTS
We wish to thank Lynn Heasley for helpful discussion.
Funding: These studies were supported by grants from the NCI (R01 CA162226, and CA108610 to R. Nemenoff), the University of Colorado Cancer Center SPORE in Lung Cancer (P50 CA058187 to R. Nemenoff), and the VA Career Development Program (CDA IK2BX001282 to H. Li). T. Sippel and A. Johnson were supported by a Ruth L. Kirschstein National Research Service Award T32CA17468. The University of Colorado Cancer Center flow cytometry core and the University of Colorado Functional Genomics Facility are supported by funding from the National Cancer Institute through the Cancer Center Support Grant (P30CA046934). The contents of the study are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Footnotes
Disclosure Statement: Nothing to disclose.
REFERENCES:
- 1.Yoda S, Dagogo-Jack I, and Hata AN, Targeting oncogenic drivers in lung cancer: Recent progress, current challenges and future opportunities. Pharmacol Ther, 2018. [DOI] [PubMed] [Google Scholar]
- 2.Chirieac LR and Dacic S, Targeted Therapies in Lung Cancer. Surg Pathol Clin, 2010. 3(1): p. 71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reungwetwattana T and Dy GK, Targeted therapies in development for non-small cell lung cancer. J Carcinog, 2013. 12: p. 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cho WC, et al. , The role of inflammation in the pathogenesis of lung cancer. Expert Opin Ther Targets, 2011. 15(9): p. 1127–37. [DOI] [PubMed] [Google Scholar]
- 5.Heinrich EL, et al. , The inflammatory tumor microenvironment, epithelial mesenchymal transition and lung carcinogenesis. Cancer Microenviron, 2012. 5(1): p. 5–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whiteside TL, The tumor microenvironment and its role in promoting tumor growth. Oncogene, 2008. 27(45): p. 5904–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kwak JW, et al. , Complement Activation via a C3a Receptor Pathway Alters CD4(+) T Lymphocytes and Mediates Lung Cancer Progression. Cancer Res, 2018. 78(1): p. 143–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li H, et al. , Activation of PPARgamma in myeloid cells promotes lung cancer progression and metastasis. PLoS One, 2011. 6(12): p. e28133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li HY, et al. , The Tumor Microenvironment Regulates Sensitivity of Murine Lung Tumors to PD-1/PD-L1 Antibody Blockade. Cancer Immunol Res, 2017. 5(9): p. 767–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poczobutt JM, et al. , Expression Profiling of Macrophages Reveals Multiple Populations with Distinct Biological Roles in an Immunocompetent Orthotopic Model of Lung Cancer. J Immunol, 2016. 196(6): p. 2847–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poczobutt JM, et al. , Deletion of 5-Lipoxygenase in the Tumor Microenvironment Promotes Lung Cancer Progression and Metastasis through Regulating T Cell Recruitment. J Immunol, 2016. 196(2): p. 891–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weiser-Evans MC, et al. , Depletion of cytosolic phospholipase A2 in bone marrow-derived macrophages protects against lung cancer progression and metastasis. Cancer Res, 2009. 69(5): p. 1733–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nemenoff RA, Peroxisome proliferator-activated receptor-gamma in lung cancer: defining specific versus “off-target” effectors. J Thorac Oncol, 2007. 2(11): p. 989–92. [DOI] [PubMed] [Google Scholar]
- 14.Panigrahy D, et al. , PPARgamma as a therapeutic target for tumor angiogenesis and metastasis. Cancer Biol Ther, 2005. 4(7): p. 687–93. [DOI] [PubMed] [Google Scholar]
- 15.Hazra S and Dubinett SM, Ciglitazone mediates COX-2 dependent suppression of PGE2 in human non-small cell lung cancer cells. Prostaglandins Leukot Essent Fatty Acids, 2007. 77(1): p. 51–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Srivastava N, et al. , Inhibition of Cancer Cell Proliferation by PPARgamma Is Mediated by a Metabolic Switch that Increases Reactive Oxygen Species Levels. Cell Metab, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bren-Mattison Y, et al. , Antitumorigenic effects of peroxisome proliferator-activated receptor-gamma in non-small-cell lung cancer cells are mediated by suppression of cyclooxygenase-2 via inhibition of nuclear factor-kappaB. Mol Pharmacol, 2008. 73(3): p. 709–17. [DOI] [PubMed] [Google Scholar]
- 18.Bren-Mattison Y, et al. , Peroxisome proliferator-activated receptor-gamma (PPAR(gamma)) inhibits tumorigenesis by reversing the undifferentiated phenotype of metastatic non-small-cell lung cancer cells (NSCLC). Oncogene, 2005. 24(8): p. 1412–22. [DOI] [PubMed] [Google Scholar]
- 19.Layton MG and Franks LM, Heterogeneity in a spontaneous mouse lung carcinoma: selection and characterisation of stable metastatic variants. Br J Cancer, 1984. 49(4): p. 415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poczobutt JM, et al. , Eicosanoid profiling in an orthotopic model of lung cancer progression by mass spectrometry demonstrates selective production of leukotrienes by inflammatory cells of the microenvironment. PLoS One, 2013. 8(11): p. e79633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franks LM, et al. , Metastasizing tumors from serum-supplemented and serum-free cell lines from a C57BL mouse lung tumor. Cancer Res, 1976. 36(3): p. 1049–55. [PubMed] [Google Scholar]
- 22.Justilien V, et al. , Matrix metalloproteinase-10 is required for lung cancer stem cell maintenance, tumor initiation and metastatic potential. PLoS One, 2012. 7(4): p. e35040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schliekelman MJ, et al. , Molecular portraits of epithelial, mesenchymal, and hybrid States in lung adenocarcinoma and their relevance to survival. Cancer Res, 2015. 75(9): p. 1789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bouhlel MA, et al. , PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab, 2007. 6(2): p. 137–43. [DOI] [PubMed] [Google Scholar]
- 25.Noy R and Pollard JW, Tumor-associated macrophages: from mechanisms to therapy. Immunity, 2014. 41(1): p. 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nawshad A, et al. , Transforming growth factor-beta signaling during epithelial-mesenchymal transformation: implications for embryogenesis and tumor metastasis. Cells Tissues Organs, 2005. 179(1–2): p. 11–23. [DOI] [PubMed] [Google Scholar]
- 27.Massague J, TGFbeta in Cancer. Cell, 2008. 134(2): p. 215–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen JJ, et al. , Up-regulation of tumor interleukin-8 expression by infiltrating macrophages: its correlation with tumor angiogenesis and patient survival in non-small cell lung cancer. Clin Cancer Res, 2003. 9(2): p. 729–37. [PubMed] [Google Scholar]
- 29.Colak S and Ten Dijke P, Targeting TGF-beta Signaling in Cancer. Trends Cancer, 2017. 3(1): p. 56–71. [DOI] [PubMed] [Google Scholar]
- 30.Nakamura S, et al. , TGF-beta1 in tumor microenvironments induces immunosuppression in the tumors and sentinel lymph nodes and promotes tumor progression. J Immunother, 2014. 37(2): p. 63–72. [DOI] [PubMed] [Google Scholar]
- 31.Bingle L, Brown NJ, and Lewis CE, The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol, 2002. 196(3): p. 254–65. [DOI] [PubMed] [Google Scholar]
- 32.Mei J, et al. , Prognostic impact of tumor-associated macrophage infiltration in non-small cell lung cancer: A systemic review and meta-analysis. Oncotarget, 2016. 7(23): p. 34217–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Conway EM, et al. , Macrophages, Inflammation, and Lung Cancer. Am J Respir Crit Care Med, 2016. 193(2): p. 116–30. [DOI] [PubMed] [Google Scholar]
- 34.Quatromoni JG and Eruslanov E, Tumor-associated macrophages: function, phenotype, and link to prognosis in human lung cancer. Am J Transl Res, 2012. 4(4): p. 376–89. [PMC free article] [PubMed] [Google Scholar]
- 35.Qian BZ and Pollard JW, Macrophage diversity enhances tumor progression and metastasis. Cell, 2010. 141(1): p. 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin CY, et al. , Macrophage activation increases the invasive properties of hepatoma cells by destabilization of the adherens junction. FEBS Lett, 2006. 580(13): p. 3042–50. [DOI] [PubMed] [Google Scholar]
- 37.David CJ and Massague J, Contextual determinants of TGFbeta action in development, immunity and cancer. Nat Rev Mol Cell Biol, 2018. 19(7): p. 419–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen L, et al. , Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun, 2014. 5: p. 5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vazquez PF, et al. , TGF-beta specifically enhances the metastatic attributes of murine lung adenocarcinoma: implications for human non-small cell lung cancer. Clin Exp Metastasis, 2013. 30(8): p. 993–1007. [DOI] [PubMed] [Google Scholar]
- 40.Yu JR, et al. , TGF-beta/Smad signaling through DOCK4 facilitates lung adenocarcinoma metastasis. Genes Dev, 2015. 29(3): p. 250–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salvo E, et al. , Combined targeting of TGF-beta1 and integrin beta3 impairs lymph node metastasis in a mouse model of non-small-cell lung cancer. Mol Cancer, 2014. 13: p. 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hasegawa Y, et al. , Transforming growth factor-beta1 level correlates with angiogenesis, tumor progression, and prognosis in patients with nonsmall cell lung carcinoma. Cancer, 2001. 91(5): p. 964–71. [PubMed] [Google Scholar]
- 43.Malkoski SP, et al. , Loss of transforming growth factor beta type II receptor increases aggressive tumor behavior and reduces survival in lung adenocarcinoma and squamous cell carcinoma. Clin Cancer Res, 2012. 18(8): p. 2173–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anumanthan G, et al. , Restoration of TGF-beta signalling reduces tumorigenicity in human lung cancer cells. Br J Cancer, 2005. 93(10): p. 1157–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Borczuk AC, et al. , Progression of human bronchioloalveolar carcinoma to invasive adenocarcinoma is modeled in a transgenic mouse model of K-ras-induced lung cancer by loss of the TGF-beta type II receptor. Cancer Res, 2011. 71(21): p. 6665–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.