

ARID1A is a tumor suppressor in neuroblastoma, whose loss promotes the adrenergic-to-mesenchymal transition.
Abstract
Mutations in genes encoding SWI/SNF chromatin remodeling complexes are found in approximately 20% of all human cancers, with ARID1A being the most frequently mutated subunit. Here, we show that disruption of ARID1A homologs in a zebrafish model accelerates the onset and increases the penetrance of MYCN-driven neuroblastoma by increasing cell proliferation in the sympathoadrenal lineage. Depletion of ARID1A in human NGP neuroblastoma cells promoted the adrenergic-to-mesenchymal transition with changes in enhancer-mediated gene expression due to alterations in the genomic occupancies of distinct SWI/SNF assemblies, BAF and PBAF. Our findings indicate that ARID1A is a haploinsufficient tumor suppressor in MYCN-driven neuroblastoma, whose depletion enhances tumor development and promotes the emergence of the more drug-resistant mesenchymal cell state.
INTRODUCTION
Neuroblastoma, a solid tumor of the peripheral sympathetic nervous system (PSNS) in children, can represent difficult treatment challenges and, as a result, accounts for 15% of all childhood cancer deaths (1, 2). Neuroblastoma is derived from the sympathoadrenal lineage of neural crest cells (NCCs) and typically arises in the adrenal medulla or sympathetic ganglia (3). MYCN gene amplification and overexpression define approximately 25% of neuroblastomas, making it one of the most common high-risk genetic alterations in these tumors (4). The vast majority of neuroblastomas with MYCN gene amplification also harbor large deletions of chromosome band 1p36 (5–8), and a recent study has implicated the gene encoding AT-rich interacting domain–containing protein 1A (ARID1A; also known as BAF250A and SMARCF1) as the gene on chromosome 1p whose loss collaborates with oncogenic MYCN in neuroblastoma pathogenesis (5). This work showed in studies of primary mouse NCCs in vitro that MYCN-driven tumorigenesis selects for NCCs with ARID1A deletions from a pool of NCCs (5).
ARID1A is present exclusively in the canonical BAF complexes within the mammalian SWI/SNF (SWItch/Sucrose Non-Fermentable) (mSWI/SNF) family of chromatin remodeling complexes that modulate chromatin structure and, therefore, gene transcription (9–11). Whole-genome and exome sequencing studies have implicated mSWI/SNF complexes as the most frequently mutated chromatin remodelers in human tumors, with ARID1A being the most highly mutated component among mSWI/SNF subunits (12,13). Inactivating mutations in ARID1A have been identified in a range of tumor types, including neuroblastoma, colon cancer, ovarian clear cell carcinomas, and endometrioid carcinomas (14–19). Furthermore, immunohistochemistry-based studies have demonstrated that most of these mutations are associated with the loss of detectable ARID1A protein expression (20–22).
Sequence alterations of ARID1A have been identified in 6% of neuroblastomas and shown to be associated with early treatment failure and an unfavorable outcome overall (17). ARID1A is deleted on one allele in at least 87% of cases with loss of chromosome 1p, which is almost always deleted in neuroblastomas with MYCN gene amplification and is the most common deletion in high-risk neuroblastomas. The ARID1A gene does not lie within the smallest common region of deletion on 1p, but the vast majority of these abnormalities are very large and include ARID1A within the deleted region (5–8). As a result, at least 30% of all primary high-risk human neuroblastomas have haploinsufficiency for ARID1A (8, 23).
Given the recent studies implicating ARID1A as the critical haploinsufficient tumor suppressor in MYCN-amplified neuroblastoma (5) and the paucity of information regarding the consequences of ARID1A loss in neuroblastoma, we sought to clarify the pathogenic role of this chromatin regulator in our MYCN zebrafish model of high-risk neuroblastoma (24). Here, we show that disruption of either ARID1A homolog, arid1aa or arid1ab, accelerates the onset and increases the penetrance of MYCN-induced neuroblastoma by increasing the proliferation of cells in the sympathoadrenal lineage. ARID1A depletion in human neuroblastoma cells promotes cell invasion and the adrenergic-to-mesenchymal transition by regulating enhancer-mediated gene expression through alteration of the binding sites of both BAF and PBAF (polybromo-containing BAF) complexes (distinct mSWI/SNF assemblies) in a genome-wide manner. These results support the role of ARID1A as a bona fide tumor suppressor in neuroblastoma, whose loss promotes the transition of committed adrenergic neuroblast cells to undifferentiated mesenchymal cells that drive a more aggressive phenotype.
RESULTS
Zebrafish arid1aa or arid1ab deficiency increases the penetrance of MYCN-induced neuroblastoma in vivo
Analysis of gene expression data of human tumors (the R2 database; https://hgserver1.amc.nl/cgi-bin/r2/main.cgi) revealed that low ARID1A expression is strongly associated with lower overall survival probability in neuroblastoma patients and that ARID1A expression levels are inversely correlated with MYCN expression in human primary neuroblastomas (Fig. 1, A and B). To examine the relevance of ARID1A as a tumor suppressor gene in vivo, we used a CRISPR-Cas9–mediated knockout strategy (25) to disrupt its homologs in a zebrafish neuroblastoma model Tg (dβh:EGFP-MYCN) (designated MYCN) (24) and in wild-type (strain AB) fish. Zebrafish have duplicated ARID1A genes, namely, arid1aa and arid1ab. To eliminate potential off-target effects, we designed two unique guide RNAs (gRNAs) targeting the coding sequence of each gene. After injection, randomly selected embryos were evaluated for double-stranded cleavage efficiency of these gRNAs at 1 day postfertilization (dpf). Disruption of the target site for each gRNA was observed in more than 70% of the embryos (table S1). Observation of these mosaic primary injected MYCN fish revealed a substantially accelerated tumor onset and an increased penetrance of neuroblastoma for each gRNA, as compared with uninjected controls (fig. S1). Neuroblastomas arising in these mosaic fish were dissected and subjected to Sanger sequencing to identify gRNA target sites. Mutations in either arid1aa or arid1ab were found in early-onset tumors [5 and 13 weeks postfertilization (wpf)] but not those with late onset (15 and 27 wpf) (table S2), suggesting that the accelerated tumor onset was attributed to CRISPR-Cas–mediated arid1aa or arid1ab gene mutations.
Fig. 1. Zebrafish arid1aa or arid1ab deficiency increases the penetrance of MYCN-induced neuroblastoma.
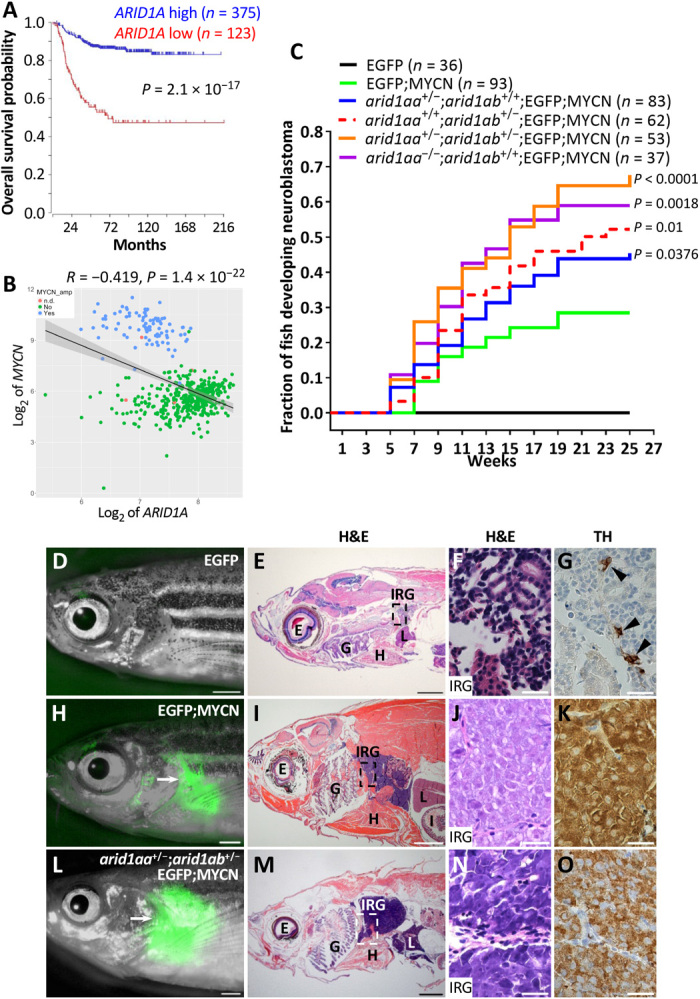
(A) Kaplan-Meier survival analysis according to ARID1A expression with chi-square test. The cutoff value of ARID1A expression levels was determined by the Kaplan scanner tool in R2 web application. (B) Correlation analysis between ARID1A and MYCN in human neuroblastomas. Tumors are categorized as MYCN-amplified (blue), MYCN-nonamplified (green), and MYCN status not determined (n.d.) (pink). Correlation coefficients (r) and statistical significance levels were determined by linear regression with lm method in R, and data were plotted using ggplot2 in R. Shaded area indicates 95% confidence interval. Data used in (A) and (B) were obtained from Tumor Neuroblastoma SEQC-498-RPM-seqcnb1 in R2 database. (C) Cumulative frequency of neuroblastoma onset among stable transgenic and mutant lines by Kaplan-Meier analysis. The overall differences between the curves for various mutant lines versus EGFP;MYCN line are compared by the log-rank test. (D to O) Lack of detectable EGFP expression in the IRG of a 17-wpf EGFP transgenic fish (D) and EGFP expression in the tumors (arrows) arising in the IRG of a 45-wpf EGFP;MYCN fish (H) and a 26-wpf arid1aa+/−;arid1ab+/−;EGFP;MYCN fish (L). Hematoxylin and eosin (H&E) staining and immunohistochemical staining of tyrosine hydroxylase (TH) of the sagittal sections through the IRG of each fish are shown on the right. E, eye; G, gill; H, heart; I, intestine; IRG, interrenal gland; L, liver. Scale bars, 1 mm (D, E, H, I, L, and M) and 20 μm (F, G, J, K, N, and O). Photo credit: Ting Tao, Dana-Farber Cancer Institute.
Wild-type fish injected with arid1aa or arid1ab gRNA and Cas9 mRNA were grown to fertility, and stable zebrafish mutant lines arid1aaΔ1 and arid1abΔ19 (arid1aa−/− and arid1ab−/− hereafter) were established by outcrossing (fig. S2, A and B). Each of these mutations included a deletion and/or insertion within a coding region that created a premature stop codon, resulting in a truncation of the Arid1aa or Arid1ab protein before any functional domains, including the DNA binding ARID domain (fig. S2, B to D). Western blotting confirmed the absence of Arid1aa or Arid1ab protein expression in homozygous mutant embryos at 3 dpf (fig. S2E). Zebrafish arid1aa-/-, but not arid1ab-/-, mutants were observed in the adult population. To investigate this result further, we performed quantitative survival studies. While the arid1aa−/− larvae exhibited similar survival rates as wild-type larvae, the arid1ab−/− larvae survival began decreasing at 13 dpf, with no surviving embryos observed beyond 18 dpf (fig. S2F). The arid1ab−/− larvae also displayed a body curvature morphology and a lack of swim bladder indicative of abnormal development (fig. S2G).
To address whether arid1aa or arid1ab deficiency collaborates with MYCN overexpression during neuroblastoma tumorigenesis in the context of stable lines, we incrossed compound heterozygous mutant arid1aa+/−;arid1ab+/− in the background of Tg (dβh:EGFP;dβh:EGFP-MYCN) fish [designated enhanced green fluorescent protein (EGFP); MYCN]. The presence of the dβh:EGFP transgene enhanced fluorescence intensity of the MYCN-fused EGFP, which is more conducive to visualization of tumor development. Zebrafish arid1ab−/− and arid1aa−/−;arid1ab+/− mutants were not detected in the tumor watch population starting at 5 wpf. While fish transgenic for EGFP alone did not develop neuroblastoma regardless of arid1aa and arid1ab genotype (Fig. 1C), both arid1aa or arid1ab deficiency markedly increased the penetrance of MYCN-driven tumors induced in the interrenal gland (IRG; zebrafish counterpart of the human adrenal gland) of EGFP;MYCN fish, with the arid1aa+/−;arid1ab+/−;EGFP;MYCN line showing the highest penetrance: 67.6% in the arid1aa+/−;arid1ab+/−;EGFP;MYCN line versus 28.4% in the control EGFP;MYCN line by 25 wpf (P < 0.0001; Fig. 1C). The mature IRG of EGFP control fish consists mainly of tyrosine hydroxylase (TH)–expressing chromaffin cells (Fig. 1, D to G). By contrast, all MYCN-transformed neuroblastoma cell populations, with or without mutations in the arid1aa or arid1ab gene, included TH-expressing, undifferentiated, small round tumor cells with hyperchromatic nuclei (Fig. 1, H to O), similar to findings in human neuroblastoma (26). Together, these findings demonstrate synergism between arid1aa or arid1ab deficiency and MYCN overexpression in both mosaic and stable zebrafish lines, thus strengthening the case for a tumor suppressor role of arid1aa and arid1ab during neuroblastoma tumorigenesis.
Haploinsufficiency of arid1aa and arid1ab enhances MYCN-induced hyperplasia by increasing cell proliferation in the sympathoadrenal lineage
During normal development, a subset of sympathoadrenal precursor cells migrate ventrally from the neural crest to the anterior-ventral edge of head kidney and differentiate into adrenal chromaffin cells. Sympathoadrenal precursor cells express TH and the panneuronal marker HuC, while chromaffin cells lose HuC expression as they undergo lineage specification, indicating a loss of the neuronal cell identity (24, 27, 28). To clarify the mechanisms responsible for accelerated tumor growth in arid1aa- or arid1ab-deficient MYCN fish, we examined the numbers of TH+HuC+ neuroblasts in sections of the IRG in (i) EGFP, (ii) arid1aa+/−;arid1ab+/−;EGFP, (iii) EGFP;MYCN, and (iv) arid1aa+/−;arid1ab+/−;EGFP;MYCN lines. At 5 wpf, the numbers of TH+HuC+ neuroblasts in the IRG of normal EGFP fish were quite low and the numbers of these PSNS neuroblasts were identical in arid1aa+/−;arid1ab+/−;EGFP fish (Fig. 2A). By contrast, both EGFP;MYCN and arid1aa+/−;arid1ab+/−;EGFP;MYCN fish showed significantly increased numbers of TH+HuC+ neuroblasts in the IRG region, as compared to controls without the MYCN transgene (P = 0.0003; Fig. 2A). The numbers of TH+HuC+ neuroblasts were similar between EGFP;MYCN and arid1aa+/−;arid1ab+/−;EGFP;MYCN fish at 5 wpf (Fig. 2A). At 7 wpf, however, 9 of 10 EGFP;MYCN transgenic fish lacked notable TH+HuC+ neuroblast populations in the IRG region (Fig. 2B) because of a previously reported, developmentally timed apoptotic event (24). By contrast, arid1aa+/−;arid1ab+/−;EGFP;MYCN line continued to display expansion of TH+HuC+ neuroblasts in the IRG at this time point (P = 0.0276; Fig. 2B and fig. S3, G to L), indicating either decreased cell apoptosis or increased cellular proliferation in the sympathoadrenal lineage. arid1aa+/−;arid1ab+/−;EGFP fish showed a slightly increased number of TH+HuC+ neuroblasts in the IRG, as compared to the EGFP control fish at 7 wpf (P = 0.0478; Fig. 2B and fig. S3, A to F), although neuroblastomas were not evident in the lines lacking the MYCN transgene.
Fig. 2. arid1aa and arid1ab haploinsufficiency enhances MYCN-induced sympathoadrenal lineage hyperplasia by increasing cell proliferation in the IRG.
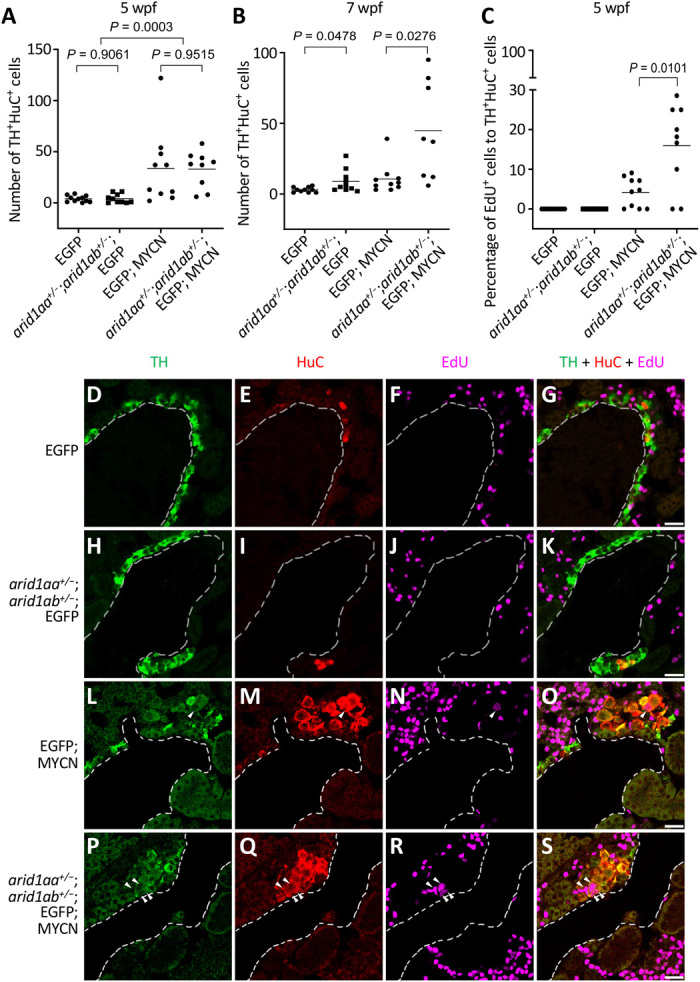
(A and B) Numbers of TH+HuC+ sympathoadrenal cells in the IRG of EGFP, arid1aa+/−;arid1ab+/−;EGFP, EGFP;MYCN, and arid1aa+/−;arid1ab+/−;EGFP;MYCN lines at both 5 wpf (A) and 7 wpf (B). (C) Percentage of EdU-positive TH+HuC+ sympathoadrenal cells in the IRG of the indicated fish lines at 5 wpf. In all plots, each symbol represents the value for an individual fish. Horizontal bars indicate mean values that were compared with the two-tailed unpaired Welch t test. (D to S) Representative coimmunostaining images of TH (green), HuC (red), and EdU (magenta) of the sagittal sections through IRG of EdU-labeled EGFP (D to G), arid1aa+/−;arid1ab+/−;EGFP (H to K), EGFP;MYCN (L to O), and arid1aa+/−;arid1ab+/−;EGFP;MYCN (P to S) lines at 5 wpf. Arrowheads indicate the EdU-labeled proliferating sympathoadrenal cells. Dotted lines indicate the head kidney boundary. Scale bars, 20 μm.
To explore this mechanism further, we performed experiments based on activated caspase-3 immunofluorescence and 5-ethynyl-2ʹ-deoxyuridine (EdU) pulse labeling. Apoptotic HuC+ neuroblasts with positive staining for activated caspase-3 were detected in both EGFP;MYCN and arid1aa+/−;arid1ab+/−;EGFP;MYCN fish (P = 1) but not in EGFP and arid1aa+/−;arid1ab+/−;EGFP fish (fig. S3, M to Y). EdU labeling of the proliferating fraction of sympathoadrenal lineage cells in the IRG region of the EGFP;MYCN fish revealed that 4.16% of the TH+HuC+ neuroblasts showed incorporation of EdU after 2 hours of pulse labeling (Fig. 2, C and L to O). By contrast, the percentage of EdU-positive TH+HuC+ neuroblasts was significantly increased to 15.9% in the arid1aa+/−;arid1ab+/−;EGFP;MYCN fish (P = 0.0101; Fig. 2, C and L to S), consistent with the sympathoadrenal hyperplasia observed in this line at 7 wpf (Fig. 2B). No EdU-positive TH+HuC+ neuroblasts were detected in either EGFP or arid1aa+/−;arid1ab+/−;EGFP fish under the same pulse labeling condition (Fig. 2, C to K). Thus, arid1aa+/−;arid1ab+/− haploinsufficiency did not affect the levels of MYCN-induced developmentally timed apoptosis of PSNS neuroblasts but rather increased the numbers of PSNS neuroblasts by increasing the rate of cell proliferation, leading to a higher penetrance of tumors in MYCN fish with arid1aa+/−;arid1ab+/− haploinsufficiency.
ARID1A loss affects enhancer-mediated gene regulation by altering both BAF and PBAF complexes binding in neuroblastoma cells
To assess the tumor-suppressive role of ARID1A in human neuroblastoma cells, we generated two ARID1A homozygous mutant clones of the MYCN-amplified NGP neuroblastoma cell line (designated ARID1A−/− #1 and ARID1A−/− #2) using CRISPR-Cas9 genome editing system with two unique gRNAs (gRNA #1 and #2) targeting human ARID1A (fig. S4). Each of these mutations created a premature stop codon and a truncation of the ARID1A protein product (fig. S4, C and D). An ARID1A heterozygous mutant clone of NGP (designated ARID1A+/−) was also generated using gRNA #2 to assess the consequences of ARID1A haploinsufficiency (fig. S4). A control NGP cell line was generated by transfection with a nontargeting control gRNA. ARID1A protein expression was lower in ARID1A+/− cells and was not detected in either ARID1A−/− #1 or ARID1A−/− #2 cells, when compared to control NGP cells by western blotting (Fig. 3A). Proliferation was unchanged in each of the ARID1A heterozygous and homozygous mutant NGP cells (Fig. 3B). However, transwell invasion and migration assays demonstrated that remarkably more ARID1A−/− #1 and ARID1A−/− #2 NGP cells passed through collagen-coated permeable membranes than did control NGP cells (P < 0.001; Fig. 3, C and D). ARID1A haploinsufficiency also enhanced cell invasion and migration, with an intermediate number of ARID1A+/− NGP cells passing through the membranes compared to control NGP cells (P < 0.01; Fig. 3, C and D). ARID1B is mutually exclusive with ARID1A in BAF complexes and is a specific vulnerability in ARID1A-mutant cancers (29). To test whether these findings extend to neuroblastoma, we first performed an immunoprecipitation experiment with an anti-ARID1B antibody and found that ARID1B-containing BAF complexes were intact in ARID1A knockout NGP cells (Fig. 3E). While knockdown of ARID1B expression with two short hairpin RNAs (shRNAs) did not affect the growth of control NGP cells, it slightly inhibited the growth of ARID1A+/− heterozygous mutant clone and effectively inhibited the growth of both ARID1A−/− #1 and ARID1A−/− #2 homozygous mutant clones (Fig. 3, F and G). Together, these data indicate that ARID1A loss promotes the invasion and migration of human neuroblastoma cells and support the finding of others that ARID1B is synthetic lethal with ARID1A (29), and thus, ARID1B could serve as a therapeutic target in ARID1A-mutant neuroblastomas.
Fig. 3. Loss of ARID1A promotes NGP cell invasion and migration.
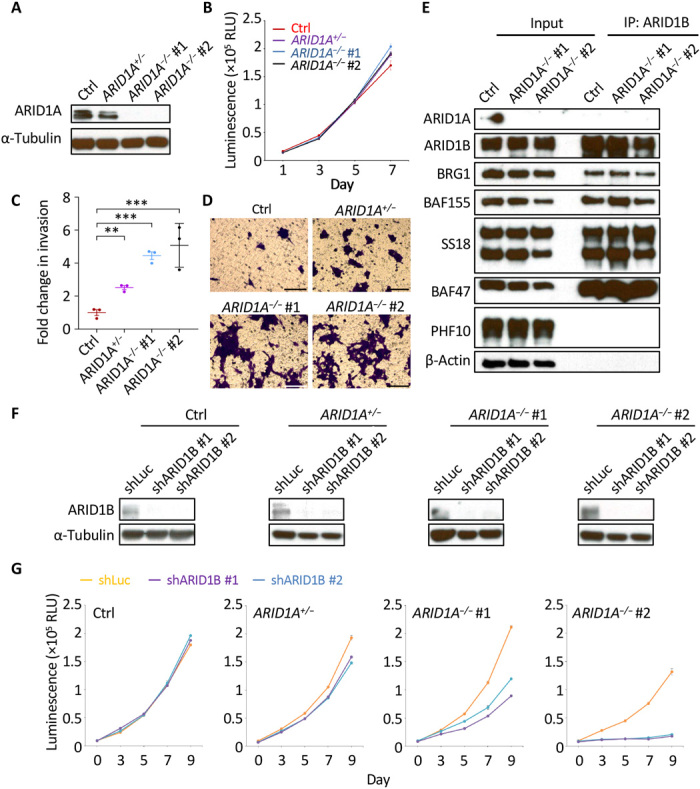
(A) Western blot analysis of ARID1A protein expression in control (Ctrl), ARID1A+/−, ARID1A−/− #1, and ARID1A−/− #2 NGP cells. α-Tubulin was used as a loading control. (B) Cell growth was measured at days 1, 3, 5, and 7. Values are means ± SEM of triplicate experiments. RLU, relative light unit. (C and D) Transwell invasion and migration assay of the indicated cells. The fold change of the number of migrated cells passing through the membrane per field was relative to the mean for control NGP cells and was compared with the two-tailed unpaired t test (C). **P < 0.01; ***P < 0.001. Cells were stained with 0.1% crystal violet (D). Scale bars, 100 μm. (E) Immunoprecipitation (IP) of the components of the BAF complexes using an anti-ARID1B antibody. PHF10 is present exclusively in the PBAF complexes and served as a negative control in this experiment; β-actin was used as a loading control. (F) Control luciferase shRNA (shLuc) or each of the ARID1B-targeting shRNAs (shARID1B #1 and #2) was induced by doxycycline (1.5 μg/ml) for 6 days in the indicated cells. The expression of ARID1B was detected by western blot analysis. α-Tubulin was used as a loading control. (G) Cell growth was measured at days 0, 3, 5, 7, and 9 after pretreatment with doxycycline (1.5 μg/ml) for 3 days to induce the expression of shLuc, shARID1B #1, or shARID1B #2. Values are means ± SEM of triplicate experiments.
mSWI/SNF family proteins are components of multiple complexes, including the BAF complexes (canonical BAF and recently defined noncanonical BAF complexes) and PBAF complexes (9, 11, 30). Because the genome-wide occupancy of these complexes has not been reported in neuroblastoma, we performed chromatin immunoprecipitation coupled with massively parallel DNA sequencing (ChIP-seq) for BRG1 (shared core subunit in both BAF and PBAF complexes), SS18 (specific in BAF complexes), and ARID2 (BAF200; specific in PBAF complexes) to identify their chromatin localization in control NGP cells. A total of 4346 BAF binding sites defined by overlapping SS18 and BRG1 peaks were identified, while 4017 PBAF binding sites defined by overlapping ARID2 and BRG1 peaks were identified (data file S1 and Materials and Methods). More than half of the PBAF complex binding sites were located at promoter regions, while the BAF complexes preferentially bound to distal intergenic regions (fig. S5A) annotated by ChIPseeker (31). Further analysis showed that about 40% of BAF and PBAF complex binding sites overlapped with each other, and about 60% of the BAF complex target genes were also PBAF complex targets (fig. S5, B and C). Gene ontology (GO) analysis using Enrichr (32) revealed that BAF complex–bound genes in NGP cells were significantly enriched in the categories of nervous system development, cell adhesion, and transcriptional regulation, while PBAF complex–bound genes were mainly enriched for genes involved in transcriptional regulation (fig. S5, D and E).
To investigate the effects of ARID1A loss on the genomic occupancy of BAF and PBAF complexes, we also performed BRG1, SS18, and ARID2 ChIP-seq on ARID1A−/− #1 and ARID1A−/− #2 NGP cells. The BAF or PBAF complex binding sites in control NGP cells were compared to the two homozygous mutant NGP cells, which were computationally merged for downstream analysis (see Materials and Methods for details). A large fraction of BAF binding sites (33.18%) showed weakened SS18 binding in both ARID1A−/− #1 and ARID1A−/− #2 cells compared to control NGP cells, while only a few sites (5.25%) showed strengthened SS18 binding in both ARID1A homozygous mutant cells (Fig. 4A). The genomic occupancy of PBAF complexes was also affected by ARID1A depletion: 11.26% sites gained ARID2-binding capacity, while 4.53% sites lost ARID2 binding in both ARID1A homozygous mutant cells compared to control NGP cells (fig. S6A). Thus, loss of ARID1A not only alters BAF complex targeting of genomic sites but also promotes retargeting of PBAF complexes genome-wide.
Fig. 4. Loss of ARID1A affects BAF complex targeting and regulation of enhancers.
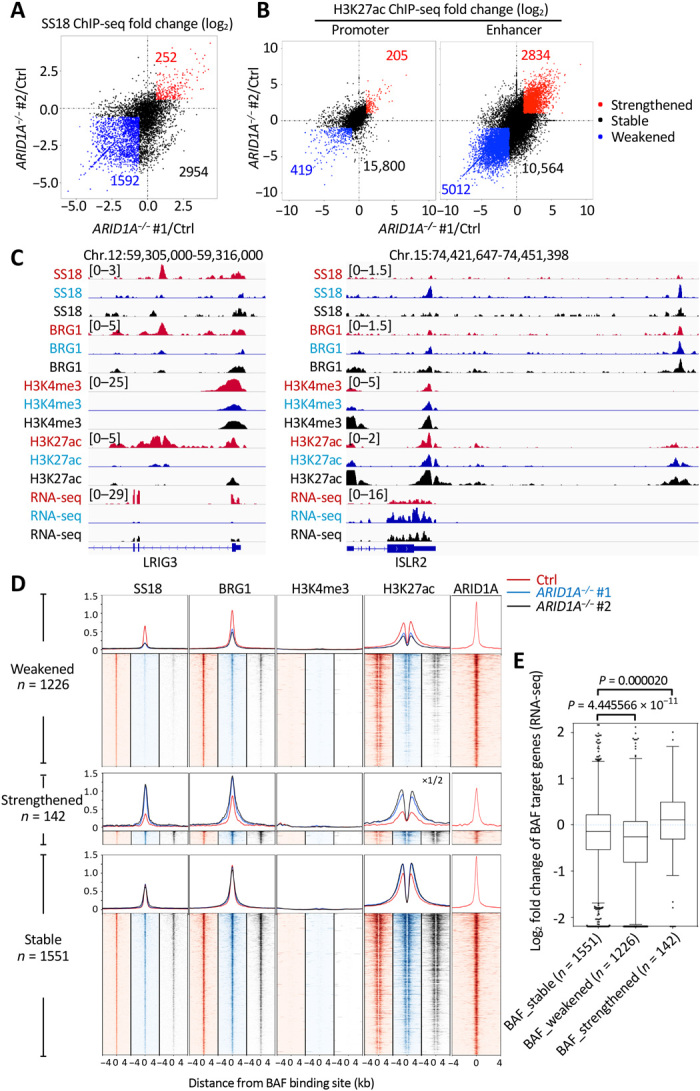
(A) Fold change (log2) in SS18 ChIP-seq signals in ARID1A−/− #1 (x value) or ARID1A−/− #2 (y value) relative to control (Ctrl) NGP cells at each BAF binding site. Sites with weakened (<2/3×) signals in both ARID1A−/− #1 and ARID1A−/− #2 are indicated by blue, while sites with strengthened (>3/2×) signals in both ARID1A−/− #1 and ARID1A−/− #2 are indicated by red; others are indicated by black. (B) Fold change (log2) in H3K27ac ChIP-seq signals in ARID1A−/− #1 (x value) or ARID1A−/− #2 (y value) relative to control (Ctrl) NGP cells at each promoter and enhancer. Promoters or enhancers with weakened (<1/2×) signals in both ARID1A−/− #1 and ARID1A−/− #2 are indicated by blue, while sites with strengthened (>2×) signals in both ARID1A−/− #1 and ARID1A−/− #2 are indicated by red; others are indicated by black. (C) Example of SS18, BRG1, H3K4me3, and H3K27ac ChIP-seq tracks and RNA sequencing (RNA-seq) tracks at TSS-distal BAF weakened leucine rich repeats and immunoglobulin like domains 3 (LRIG3) locus (left) and strengthened immunoglobulin superfamily containing leucine rich repeat 2 (ISLR2) locus (right) in control (red), ARID1A−/− #1 (blue), and ARID1A−/− #2 (black) NGP cells. (D) ChIP-seq profiles and heat maps of SS18, BRG1, H3K4me3, and H3K27ac in control (red), ARID1A−/− #1 (blue), and ARID1A−/− #2 (black) NGP cells and of ARID1A in control NGP cells (red), around all TSS-distal BAF binding sites. (E) Log2 fold change of RPKM in RNA-seq between ARID1A mutants and control NGP cells for TSS-distal BAF target genes. Boxes indicate the median (horizontal line), 25th percentile, and 75th percentile; whiskers, distances from the largest and smallest value to each end of the box that are within 1.5× box length; dots, outliers. Data were analyzed with the Mann-Whitney test.
To further resolve altered binding activities of the BAF and PBAF complexes and to evaluate the effect of ARID1A deficiency on promoter and enhancer activities, we performed ChIP-seq for H3K4me3 and H3K27ac in all the conditions. Transcription start site (TSS)–proximal/distal BAF and PBAF complex binding sites were classified by distances to the active TSS marked by H3K4me3 (16). We defined TSS-proximal H3K27ac-enriched regions as promoters and TSS-distal H3K27ac-enriched regions as enhancers (see Materials and Methods for details). Thus, we observed widespread changes in H3K27ac levels at enhancers (15.39% up and 27.22% down) but only a few changes at promoters (1.25% up and 2.55% down) upon loss of ARID1A (Fig. 4B). Although a small set of TSS-proximal regions showed altered BAF or PBAF binding, the H3K27ac signal at these sites did not appear to relate to BAF/PBAF binding strength (fig. S7). However, in TSS-distal regions, H3K27ac signal was reduced in both ARID1A−/− #1 and ARID1A−/− #2 homozygous mutant cells at sites with weakened BAF or PBAF binding and was markedly increased in strengthened TSS-distal BAF or PBAF binding sites (Fig. 4, C and D, and fig. S6, B and C). NGP cells with ARID1A haploinsufficiency showed a small effect on BAF but not PBAF binding across TSS-distal regions genome-wide (fig. S8, A and B). Because H3K27ac is indicative of active enhancers and associated with high levels of transcription, we assessed enhancer activities by mRNA expression levels of the nearest genes to BAF/PBAF binding sites. With this approach, changes in the mRNA expression levels of the nearest genes correlated with changes of TSS-distal BAF/PBAF binding upon homozygous loss of ARID1A (Fig. 4E, fig. S6D, and data file S2). GO analysis using Enrichr showed that weakened BAF complex–bound genes upon homozygous loss of ARID1A were selectively enriched in the categories of nervous system development and cell adhesion (fig. S9A), likely indicating these as key pathways involved in ARID1A-mediated tumor suppression. Weakened BAF complex binding sites were selectively enriched for unique transcription factor binding motifs including E-box, aristaless-like homeobox (ALX homeobox), and GATA (fig. S9B). Collectively, these results suggest that ARID1A can both activate and repress enhancer-mediated gene expression in neuroblastoma cells by altering the binding activities of the BAF complexes not only directly but also indirectly because of the retargeting of non-ARID1A containing BAF and PBAF complexes that affect enhancer activity.
ARID1A loss alters the differentiation status of neuroblastoma cells
Neuroblastoma tumors are thought to harbor two unique cell states, adrenergic and mesenchymal, and transcriptional enhancers forming feed-forward autoregulatory or core regulatory circuitry have an important role in programming and reinforcing each cell identity (33–37). We therefore asked whether ARID1A loss would alter the differentiation status of neuroblastoma cells. Our ChIP-seq data showed that paired like homeobox 2B (PHOX2B, an adrenergic-associated transcription factor) gene locus exhibited reduced H3K27ac and BRG1 binding, with fibronectin 1 (FN1, a mesenchymal cell adhesion protein)gene locus showing enrichment with these factors in both ARID1A−/− #1 and ARID1A−/− #2 cells, compared to control NGP cells. Correspondingly, whole-transcriptome mRNA sequencing (RNA-seq) data demonstrated decreased PHOX2B but increased FN1 expression in both ARID1A homozygous mutant cells (Fig. 5A). Western blotting showed that PHOX2B protein expression was completely ablated, while the mesenchymal-associated markers FN1, snail family transcriptional repressor 2 (SNAI2), and vimentin (VIM) were markedly up-regulated in both ARID1A−/− #1 and ARID1A−/− #2 cells compared to control NGP cells (Fig. 5B). RNA-seq analysis of neuroblastoma super-enhancer associated transcription factors (SE-TFs) (37) revealed that 6 of 18 adrenergic SE-TFs had reduced mRNA expression, while 6 of 20 mesenchymal SE-TFs were induced in both ARID1A homozygous mutant cell clones (Fig. 5C). ARID1A+/− heterozygous NGP cells expressed intermediate levels of PHOX2B, FN1, and SNAI2 proteins (fig. S8C). RNA-seq analysis of the same SE-TFs revealed that PHOX2B and GATA2, two major adrenergic SE-TFs, and most of the mesenchymal SE-TFs were also expressed at the intermediate levels in ARID1A+/− heterozygous NGP cells compared to control and ARID1A−/− NGP cells (fig. S8D). Gene set enrichment analysis (GSEA) revealed that a generic gene signature for mesenchymal tumor (38) was significantly enriched in ARID1A homozygous mutant NGP cells (Fig. 5D). Consistent with prior reports of mesenchymal cells being more resistant to conventional chemotherapeutic agents, compared with noradrenergic cells (37), we found that ARID1A homozygous mutant cells were more resistant to cisplatin (Fig. 5E), a standard drug in neuroblastoma therapy. These results provide further evidence that ARID1A loss promotes the transition of NGP cells from an adrenergic to a mesenchymal cell state by regulating enhancer-mediated gene expression.
Fig. 5. Loss of ARID1A promotes adrenergic-to-mesenchymal transition and increases cisplatin resistance in NGP cells.
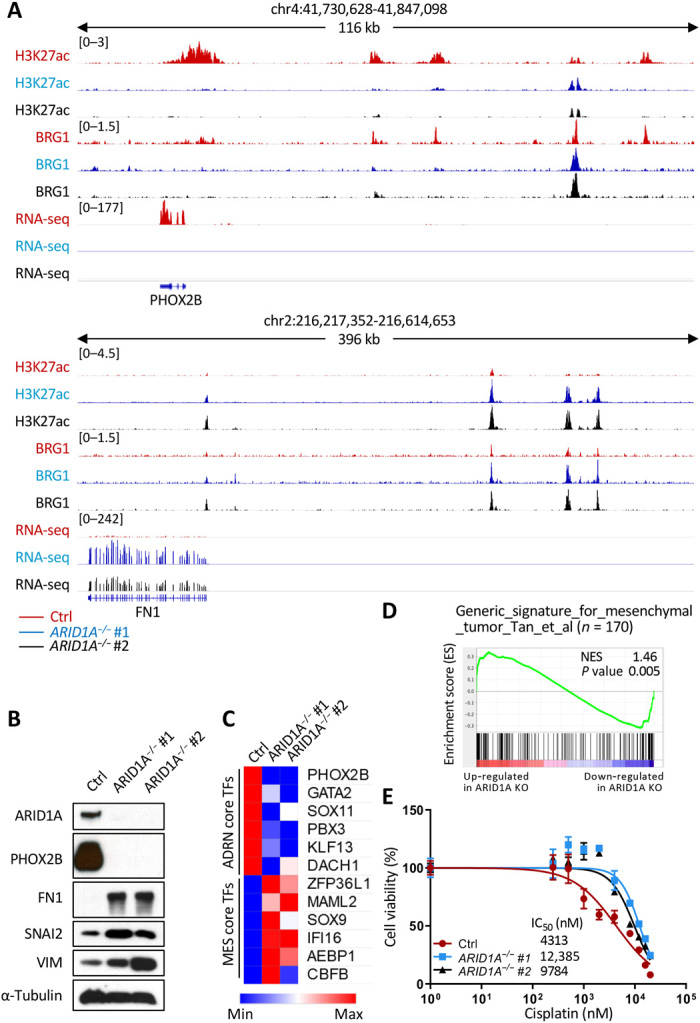
(A) H3K27ac and BRG1 ChIP-seq tracks and RNA-seq tracks at PHOX2B and FN1 loci in control (red), ARID1A−/− #1 (blue), and ARID1A−/− #2 (black) NGP cells. (B) Western blot analysis of ARID1A, an adrenergic maker (PHOX2B), and three mesenchymal markers (FN1, SNAI2, and VIM) in control (Ctrl), ARID1A−/− #1, and ARID1A−/− #2 NGP cells. α-Tubulin was used as a loading control. (C) mRNA expression (RPKM of RNA-seq) of core regulatory circuitries of transcription factors specific for adrenergic (ADRN core TFs) or mesenchymal (MES core TFs) cells in control (Ctrl), ARID1A−/− #1, and ARID1A−/− #2 NGP cells. (D) GSEA to determine the enrichment of a generic gene signature for mesenchymal tumor in ARID1A mutant NGP cells. Genes are ranked by score and plotted along the x axis as vertical black bars. NES, normalized enrichment score; KO, knockout. (E) Cell viability [% relative to N,N′-dimethylformamide (DMF)–treated cells, y value] of control (red), ARID1A−/− #1 (blue), and ARID1A−/− #2 (black). NGP cells were measured after treatment with various concentrations of cisplatin (0 to 20,000 nM, x value) for 72 hours, and half-maximal inhibitory concentration (IC50) values are indicated. Values are means ± SD of triplicate experiments.
DISCUSSION
The ARID1A gene is the most frequently mutated subunit in SWI/SNF complexes in human cancers (12), is deleted on one allele in at least 30% of high-risk neuroblastomas (8, 23), and is mutated in 6% of neuroblastomas overall (17). In this study, we established the first in vivo neuroblastoma model incorporating arid1aa and arid1ab deficiency using zebrafish. Our results demonstrate that loss of just one allele of arid1aa or arid1ab was sufficient to significantly accelerate the onset and increase the penetrance of MYCN-driven neuroblastoma, indicating that loss of even one of the four copies of arid1aa or arid1ab in zebrafish acts to promote the initiation of this tumor. Loss of additional copies of arid1aa or arid1ab further accelerated tumor onset, with the most rapid onset seen in compound heterozygotes of both genes, corresponding to a haploinsufficient tumor suppressor in human cells with only two copies of ARID1A. Our studies emphasize the highly dose-dependent nature of the tumor suppressor role of ARID1A in neuroblastoma tumorigenesis. Although MYCN-induced developmentally timed apoptosis (24, 39) still occurred in the sympathoadrenal lineage cells of arid1aa and arid1ab compound heterozygotes, the increased cell proliferation induced by loss of arid1aa and arid1ab was sufficient to overcome the effects of apoptosis and increase the penetrance of these tumors. Moreover, in studies to further understand the tumor suppressor role of ARID1A in human neuroblastoma, we generated two ARID1A homozygous mutant NGP cell lines, both of which exhibited unchanged cell growth rates (Fig. 3B) but showed increased invasiveness (Fig. 3C) compared to control NGP cells. These results indicate that loss of ARID1A enhances the initiation and migratory properties of neuroblastoma in our model system.
SWI/SNF chromatin remodeling complexes have been shown to be required for the maintenance of lineage-specific enhancers in mouse embryonic fibroblast and rhabdoid tumor cell lines, and SWI/SNF-dependent distal enhancers are essential for controlling expression of genes linked to developmental processes (40). Specifically, SMARCB1 is required for the integrity of SWI/SNF complexes, and its loss alters enhancer activity in rhabdoid tumors (41). Furthermore, ARID1A loss has been reported to impair enhancer-mediated gene regulation in colon cancer (16, 42); similarly, we found that loss of ARID1A reduced genome-wide H3K27ac at enhancer regions in neuroblastoma cells, indicating that they recapitulate this conserved function of SWI/SNF complexes in enhancer regulation. In their study, Mathur and colleagues defined BRG1 and BAF155 cobinding sites as SWI/SNF complexes binding sites, as these factors are shared by both BAF and PBAF complexes (16). By contrast, we considered BAF and PBAF binding sites separately. In wild-type NGP neuroblastoma cells, we found that more than half of all PBAF complex binding sites were located at promoter regions, while BAF complexes preferred to bind at distal intergenic regions (fig. S5A). Our results show that BAF and PBAF complexes colocalize at ~40% of the binding sites and ~60% of the target genes (fig. S5, B and C). Although ARID1A is an exclusive component of the canonical BAF complexes, loss of ARID1A not only impaired BAF complex localization but also affected PBAF complex genomic occupancies of chromatin (Fig. 4 and fig. S6).
SWI/SNF chromatin remodeling complexes are required by leukemia cells for survival and aberrant self-renewal through the maintenance of MYC transcription (43), while the complexes activate p21WAF1/CIP1 through the repression of MYC transcription in differentiation-associated cell cycle arrest (44). These data indicate that the function of SWI/SNF complexes is context dependent. NGP is a MYCN-amplified neuroblastoma cell line, in which the MYCN oncogene is abundantly expressed. Knock out of ARID1A did not significantly alter the expression levels of MYC and MYCN in NGP cells (based on RNA-seq data), indicating that MYC and MYCN are not major downstream effectors of SWI/SNF complexes in the NGP neuroblastoma cell line.
Neuroblastomas include tumor cells exhibiting two different cell states: committed adrenergic cells and undifferentiated mesenchymal or neural crest-like cells, which are more motile and more resistant to chemotherapy. These cells coexist in individual tumor cell populations and exhibit plasticity with the ability to interconvert from one cell state into the other (33, 36, 37). Expression of the mesenchymal transcription factor paired related homeobox 1 (PRRX1) or notch receptor 3 (NOTCH3) intracellular domain can reprogram adrenergic cells toward a mesenchymal state, while PHOX2B, heart and neural crest derivatives expressed 2 (HAND2), and GATA3 work in concert to promote mesenchymal cells to adopt an adrenergic identity (33, 35–37). We found that loss of ARID1A increases the expression of specific mesenchymal markers (FN1, SNAI2, and VIM) and abolishes the expression of an important adrenergic transcription factor dependency gene (PHOX2B), suggesting that ARID1A deficiency promotes the adrenergic-to-mesenchymal transition in neuroblastoma by weakening the influence of BAF complexes that regulate adrenergic enhancer-mediated gene expression (Fig. 5, B and C). Previous studies have shown that increased cell migration and cisplatin resistance in neuroblastoma are accompanied by epithelial-to-mesenchymal transition (37, 45). Thus, the increased invasiveness (Fig. 3C) and cisplatin resistance (Fig. 5E) characteristic of ARID1A mutant NGP cells reflects mesenchymal cell properties acquired upon ARID1A loss. Recent studies to identify and functionally characterize the core regulatory circuitry and signaling feedback loops that reprogram neuroblastoma cells from the adrenergic to the mesenchymal cell state are important (36), as they illuminate pathways that operate in the context of haploinsufficiency for ARID1A to drive the tumor’s metastatic potential and provide a framework for experiments to reveal the most effective therapy.
In conclusion, our work provides compelling in vivo evidence that ARID1A acts as a haploinsufficient tumor suppressor in MYCN-driven neuroblastoma, a prominent and often lethal high-risk tumor in children. Mechanistically, loss of ARID1A has a major impact on the localization of both BAF and PBAF complexes in neuroblastoma cells, with accompanying global effects on transcriptional control. Effects primarily on enhancers of genes are crucial for the determination of cell state and promote adrenergic-to-mesenchymal transition within tumor cell populations of individual tumors. Collectively, these gene expression changes enhance the rate of onset of malignancy and the invasiveness and chemotherapy resistance of the affected tumor cells. Last, our zebrafish model provides an in vivo platform for the identification of small-molecule inhibitors that kill ARID1A-deficient neuroblastoma cells with only minimal toxicity to normal tissues.
MATERIALS AND METHODS
Fish lines and maintenance
Zebrafish were raised and maintained according to the standard procedure. Zebrafish were all AB background strain. All zebrafish studies and maintenance were performed in accord with Dana-Farber Cancer Institute Institutional Animal Care and Use Committee–approved protocol #02-107.
Zebrafish lines Tg(dβh:EGFP) and Tg (dβh:EGFP-MYCN) (24) were described previously and were designated EGFP and MYCN in the text, respectively. The zebrafish arid1aa and arid1ab mutant lines were generated by the CRISPR-Cas genome editing system using the previously described protocol (25). The plasmid constructs pDR274 (#42250) and pMLM3613 (#42251) were purchased from Addgene. Oligonucleotides listed in table S3 were annealed and cloned into the pDR274 vector to generate gRNAs targeting arid1aa and arid1ab. All gRNAs were transcribed in vitro from Dra I linearized pDR274-gRNA plasmid DNA using the MAXIscript T7 Transcription Kit (Ambion, #AM1312). Cas9 mRNA was transcribed in vitro from Pme I linearized pMLM3613 DNA, using the mMESSAGE mMACHINE SP6 Transcription Kit (Ambion, #AM1340). Each embryo was injected with 1 nl of solution containing gene-specific gRNA (25 ng/μl) and Cas9 mRNA (600 ng/μl) at the one-cell stage. Mosaic F0 fish with germline mutation were identified, and stable mutant lines for arid1aa E1-2 gRNA and arid1ab E1-1 gRNA were established by outcrossing. To genotype the arid1aa or arid1ab mutant line, DNA fragment containing the mutated site was amplified with genomic DNA extracted from fin clips serving as template using the primers listed in table S4. The DNA product was either sent for Sanger sequencing with the primers listed in table S4 or digested with the specific restriction enzymes listed in table S4. The DNA product amplified from wild-type allele will be cut into two fragments, while the one amplified from mutant allele will be resistant to digestion.
Tumor watch
Mosaic primary injected or stable fish lines were observed biweekly for evidence of EGFP fluorescent tumors starting at 5 wpf. Fish with tumors were separated, genotyped, and euthanized after anesthesia and characterized by histopathological hematoxylin and eosin (H&E) staining and molecular analysis. The cumulative frequency of neuroblastoma development was analyzed by the Kaplan-Meier method with the log-rank test used to determine statistical significance.
Protein extraction, western blot analysis, and immunoprecipitation
Protein was extracted from cell lines or zebrafish samples using radioimmunoprecipitation assay buffer. Western blotting was performed as described previously (46). Protein samples were separated by an 8% polyacrylamide gel and transferred to a polyvinylidene difluoride membrane (Thermo Fisher Scientific, #PI88518). After incubation with antibodies, the results were visualized on autoradiography films with SuperSignal West Pico Chemiluminescent Substrate (Pierce, #PI34080) or SuperSignal West Dura Extended Duration Substrate (Pierce, #34075). Zebrafish Arid1ab was detected with an antibody produced by Josman LLC. that recognizes a region between residue 1770 and 1845 of zebrafish Arid1ab. Anti-ARID1A rabbit polyclonal antibody was purchased from Novus Biologicals (#NB100-55334) and used to detect both human ARID1A and zebrafish Arid1aa. Other antibodies and their sources were anti–α-tubulin mouse monoclonal (B-5-1-2) antibody (Sigma-Aldrich, #T6074), anti-PHOX2B mouse monoclonal (C-3) antibody (Santa Cruz Biotechnology, #SC-376993), anti-SNAI2 rabbit monoclonal (C19G7) antibody and anti-VIM rabbit monoclonal (D21H3) antibody (Cell Signaling Technology, #9585S and #5741S), and anti-FN1 sheep polyclonal antibody (R&D Systems, #AF1918).
Immunoprecipitation was performed as described previously (12). Cells were first lysed in buffer A [10 mM Hepes (pH 7.6), 25 mM KCl, 1 mM EDTA, and 10% glycerol, supplemented with fresh 1 mM dithiothreitol (DTT) and complete protease inhibitors (Roche, #04693132001)] to eliminate cytoplasmic protein. Nuclei were resuspended in buffer C [10 mM Hepes (pH 7.6), 3 mM Mg2Cl, 100 mM KCl, 0.1 mM EDTA, and 10% glycerol, supplemented with fresh 1 mM DTT and complete protease inhibitors] and lysed for 30 min under rotation after the addition of ammonium sulfate to a final concentration of 0.3 M. Soluble nuclear proteins were separated from insoluble chromatin fraction by ultracentrifugation (100,000g) followed by precipitation with ammonium sulfate (0.3 mg/ml). After ultracentrifugation (100,000g), protein precipitate was resuspended in immunoprecipitation buffer [150 mM NaCl, 50 mM tris-HCl (pH 8.0), 1% NP-40, and 0.5% deoxycholate, supplemented with fresh 1 mM DTT and complete protease inhibitors] for immunoprecipitation. The anti-ARID1B mouse monoclonal (KMN1) antibody (Santa Cruz Biotechnology, #sc-32762 X) coupled with Dynabeads M-270 Epoxy (Life Technologies, #14301) was used for immunoprecipitation. Proteins were detected by the anti-ARID1A rabbit polyclonal antibody (Novus Biologicals, #NB100-55334), anti-ARID1B mouse monoclonal antibody (Abcam, #ab57461), anti-BRG1 mouse monoclonal (EPNCIR111A) antibody (Abcam, #ab110641), anti-BAF155 rabbit monoclonal (D7F8S) antibody (Cell Signaling Technology, #11956), anti-SS18 rabbit monoclonal (D6I4Z) antibody (Cell Signaling Technology, #21792), anti-BAF47 mouse monoclonal (F-4) antibody (Santa Cruz Biotechnology, #sc-166164), anti-plant homeodomain finger protein 10 (PHF10) rabbit polyclonal antibody (GeneTex, #GTX116314), and anti–β-actin rabbit polyclonal antibody (Abcam, #ab8227).
EdU pulse labeling, cryosectioning, paraffin sectioning, and immunostaining
For EdU pulse labeling experiment in zebrafish, Edu from the Click-iT Alexa Fluor 647 imaging kit (Life Technologies, #C10340) was diluted to 2.5 mg/ml for retro-orbital injection. Each euthanized fish examined at 5 wpf was injected with 1 μl of EdU solution. Fish were fixed 2 hours after injection for cryosectioning; EdU detection was performed according to the manufacturer’s protocol.
Zebrafish cryosectioning and immunofluorescence staining were performed as previously described (39). Fish at indicated stage were fixed in 4% paraformaldehyde at 4°C overnight, followed by washing three times with phosphate-buffered saline (PBS). The samples were embedded in 1.5% agarose, melted in 30% sucrose PBS solution, and equilibrated in 30% sucrose PBS solution overnight at 4°C. Sagittal sections were cut serially at a 14-μm thickness and then collected on CLIP CRNR Excell slides (Thermo Fisher Scientific, #22-037-247). For immunofluorescence staining, slides were incubated with a primary antibody at 4°C overnight, washed with PBS-Tween, and then incubated with a secondary antibody for 2 hours at room temperature. Antibodies against TH (Pel-Freez, #P40101), HuC (Life Technologies, #A-21271), and activated caspase-3 (BD Biosciences, #559565) were used as primary antibodies. Secondary antibodies were conjugated with Alexa Fluor 488, Alexa Fluor 568, and Alexa Fluor 647 (Life Technologies). 4′,6-Diamidino-2-phenylindole (Life Technologies, #S36973) was used for nuclear staining. Fluorescent images were taken with a Leica SP5X scanning confocal microscope at the Confocal and Light Microscopy core facility at Dana-Farber Cancer Institute.
Zebrafish paraffin sectioning, H&E staining, and immunohistochemistry with primary antibodies against TH (Pel-Freez, #P40101) were performed at the DF/HCC Research Pathology Core.
Cell lines and cell culture
The NGP neuroblastoma cell line was stocked in our laboratory and maintained in RPMI 1640 medium (Gibco) supplemented with 10% fetal bovine serum (Sigma-Aldrich) and 1× penicillin-streptomycin (Gibco). The absence of mycoplasma contamination was confirmed using the MycoAlert Mycoplasma Detection Kit (Lonza, #LT07-418).
CRISPR-Cas9 technology was used to knock out ARID1A in NGP cells. A control gRNA (Ctrl_gRNA, 5′-GAC CGG AAC GAT CTC GCG TA-3′) or each of huARID1A-specific gRNAs (ARID1A_gRNA #1m, 5′-GCT GCT GTG TCG TCG ACT GC-3′ and ARID1A_gRNA #2, 5′-GCC TGC TGG GAG AGC GTC GA-3′; designed by CRISPR Design web tool; http://crispr.mit.edu) was cloned into lentiCRISPR v2 vector (Addgene, #52961) (47, 48). Each of these vectors was cotransfected with pMD2.G and psPAX2 into human embryonic kidney (HEK) 293T cells using FuGENE 6 transfection reagent (Promega, #PRE2693). Cell culture medium containing the lentivirus was filtered by 0.45-μm nitrocellulose membrane and infected with neuroblastoma cells in the presence of polybrene (8 μg/ml). The cells were treated with puromycin (Sigma-Aldrich, #P8833) at 48 hours after infection to eliminate the noninfected cells. Single cell–derived ARID1A homozygous mutant clones were selected by limiting dilution, and the mutations were confirmed by Sanger sequencing.
To generate ARID1A+/− cells, ARID1A_gRNA #2 was cloned into the pSpCas9(BB)-2A-GFP (PX458) vector (Addgene, #48138) (49) and transfected into NGP cells using FuGENE HD transfection reagent (Promega, #PRE2311). Cells were incubated for 72 hours, and GFP-positive single cells were sorted by flow cytometry and genotyped by Sanger sequencing.
Cell growth assay
Cells were seeded in a 96-well plate at a density of 2500 cells per well. Relative cell growth at days 1, 3, 5, and 7 was evaluated by CellTiter-Glo Luminescent Cell Viability Assay (Promega, #G7571).
For cell growth assay with ARID1B knockdown in control, ARID1A+/−, ARID1A−/− #1, and ARID1A−/− #2 NGP cells, a control luciferase shRNA (shLuc, 5′-CGT ACG CGG AAT ACT TCG A-3′) (50) or each of ARID1B-specific shRNAs (shARID1B#1, 5′-GCC GAA TTA CAA ACG CCA TAT-3′ and shARID1B#2, 5′-GGG TTT GGC CCA GGT TAA TAA-3′; https://portals.broadinstitute.org/gpp/public/) was cloned into the CS-RfA-ETV lentiviral expression vector (a gift from H. Miyoshi at RIKEN Center for Computational Science, Japan). Each of the cell line was cotransfected with the doxycycline-inducible shLuc, shARID1B #1, or shARID1B #2, together with pMD2.G and psPAX2 into HEK293T cells using the FuGENE HD transfection reagent (Promega, #PRE2311). Cell culture medium containing the lentivirus was filtered by 0.45-μm nitrocellulose membrane and infected with neuroblastoma cells in the presence of polybrene (8 μg/ml). The infected cells were sorted by the coexpressed EGFP at 72 hours after infection. Cells were pretreated with doxycycline (1.5 μg/ml) for 3 days to induce the knockout of ARID1B before the regular cell growth assay, which is described above.
The half-maximal inhibitory concentration assay
Cells were seeded into a 96-well plate at a density of 10,000 cells per well 1 day before treatment. After 3 days of treatment with vehicle [N,N′-dimethylformamide (DMF)] or cisplatin at various concentrations, relative cell growth was evaluated by CellTiter-Glo Luminescent Cell Viability Assay (Promega, #G7571) as a percentage of the vehicle (DMF) control. The half-maximal inhibitory concentration value was determined with GraphPad Prism 7.01.
Cell transwell invasion and migration assay
A transwell insert with 8.0-μm pore polycarbonate membrane (Corning, #3403) was coated with collagen. Cells were seeded into the upper compartment with serum-free media. Media containing 10% fetal bovine serum were added to the lower compartment. After incubation for 48 hours, the cells were visualized by staining with 0.1% crystal violet. The cells remaining on the upper membrane were removed by cotton wool. The number of migrated cells in five representative fields per membrane was counted. Experiments were performed in triplicate, with statistical significance determined by two-tailed unpaired t test.
Chromatin immunoprecipitation coupled with massively parallel DNA sequencing
ChIP-seq was performed as described previously (35). Human neuroblastoma cells were cross-linked in 11% formaldehyde at room temperature for 15 min, followed by tris-HCl buffer (pH 7.5) quenching. Nuclear extracts were generated with the Nuclei Isolation Kit: Nuclei EZ Prep (Sigma-Aldrich, #NUC101), and chromatin was fragmented by sonication. Anti-BRG1 rabbit monoclonal (EPNCIR111A) antibody (Abcam, #ab110641), anti-SS18 rabbit monoclonal (D6I4Z) antibody (Cell Signaling Technology, #21792), anti-ARID2 mouse monoclonal (E-3) antibody (Santa Cruz Biotechnology, #sc166117X), anti-H3K4me3 rabbit polyclonal antibody (Abcam, #ab8580), anti-H3K27ac rabbit polyclonal antibody (Abcam, #ab4729), and anti-ARID1A mouse monoclonal (PSG3) antibody (EMD Millipore, #04-080) coupled with Dynabeads M-270 Epoxy (Life Technologies, #14301) were used for immunoprecipitation. ChIP DNA was purified and subjected to library preparation for Illumina NextSeq 500 Next-Generation Sequencing at the Dana-Farber Cancer institute Molecular Biology Core Facilities. ChIP-seq data are available from Gene Expression Omnibus (GEO) (series GSE134632).
ChIP-seq data analysis and display
ChIP-seq reads were aligned to human reference genome (hg19) using Bowtie2/2.2.9 (51) with default parameters. SAMtools/1.3.1 (52) was used to remove duplicated reads and reads with low mapping quality (mapQ < 20). SS18 and ARID2 peaks were called against input reads using MACS2/2.1.1 (53) with default parameters. BRG1 and H3K4me3 peaks were called against input reads using MACS2/2.1.1 at q = 1 × 10−3. H3K27ac peaks were called twice using MACS (53) with parameter sets –keep-dup=auto –p 1e-9 and –keep-dup=all –p 1e-9 and then collapsed together for downstream analysis. Peaks overlapping with MYCN amplified regions (chr2: 16004820-17229328) and ENCODE blacklisted regions were discarded for downstream analysis.
An SS18 peak with at least a 50% overlap with a BRG1 peak was identified as a BAF binding site, while an ARID2 peak with at least a 50% overlap with a BRG1 peak was identified as a PBAF binding site. BAF or PBAF binding sites were determined in each condition (control, ARID1A−/− #1 and ARID1A−/− #2). The BAF and PBAF binding sites in control NGP cells were used for annotation with R Bioconductor package ChIPseeker (31). For this annotation, genes with the closest TSS were considered to be BAF/PBAF complex target genes.
The regions of BAF or PBAF binding sites in all three conditions were then merged together by bedtools 2.26.0 (54) as BAF or PBAF binding sites. To define the alterations of BAF/PBAF binding upon ARID1A loss, ChIP-seq read counts were generated by bedtools at each site, and read counts for each binding site were normalized to per million mapped reads (RPM). Input RPM values for each BAF/PBAF binding site in each condition were subtracted from SS18/ARID2 RPM values in that condition, and all the negative values gained at this step were set to 0. Fold changes were evaluated at each BAF/PBAF binding site using the normalized RPM values mentioned above, with a pseudocount of 0.1. If the fold change of BAF/PBAF binding at a site was greater than 1.5-fold in both ARID1A mutants, then the site was called a strengthened BAF/PBAF binding site. If, on the other hand, the fold change was less than two/threefold in both ARID1A mutants, the site was called a weakened BAF/PBAF binding site. Other sites were designated stable BAF/PBAF binding sites.
TSS-proximal/distal sites were classified as previously described (16). Active TSS was defined as all the TSS annotated in the UCSC hg19 gene track that overlap with H3K4me3 peaks in any of the three conditions. BAF/PBAF binding sites overlapping with both an H3K4me3 peak and an active TSS were designated TSS proximal. Those more than 2 kb away from an active TSS and more than 1 kb away from an H3K4me3 peak were considered TSS distal. Other ambiguous sites were removed for TSS-proximal/distal studies. H3K27ac peaks in all the conditions were merged together. Similar to BAF/PBAF binding sites, H3K27ac peaks overlapping with both an H3K4me3 peak and an active TSS were designated promoters, while those more than 2 kb away from an active TSS and more than 1 kb away from an H3K4me3 peak were considered enhancers.
Bigwig files for display were made RPM using MACS2/2.1.1 with input subtracted. Heat maps for BAF/PBAF binding were generated by deepTools 3.0.2 (55). Every line of the heat maps shows the input-subtracted values with negative values set to 0.
RNA-seq data analysis
RNA-seq reads were aligned to hg19 using HISAT2 2.1.0 (56) with default parameters. GFold v1.1.2 (57) was used to generate reads per kilobase of exon per million reads mapped (RPKM) values and calculate log2 fold change between ARID1A mutants and control cells from RNA-seq data. RNA-seq data are available from GEO (series GSE134632).
Each TSS-distal BAF or PBAF binding site was annotated to the gene with the closest active TSS and classified as strengthened, stable, or weakened site as defined above by ChIP-seq signals. The log2 fold change between ARID1A mutants and control cells was plotted for each category. Statistical significance was determined with two-sided Mann-Whitney test (http://astatsa.com/WilcoxonTest/).
GSEA and GO analysis
Normalized expression values for individual samples of RNA-seq were obtained from DESeq2 (58) using the variance-stabilizing transformation of the raw counts and were used for GSEA. Genes from whole transcriptomic data were ranked on the basis of the stat from DESeq2 in ARID1A mutant versus control NGP cells. The preranked option of GSEA (59) was run with 2500 permutations for statistical evaluation. GSEA was performed with signatures from version 6.0 of the Molecular Signatures Database (www.broadinstitute.org/gsea/msigdb/index.jsp). A generic gene signature for mesenchymal tumor (38) was also used as a gene set for GSEA. The GO analysis was performed using the Enrichr program (http://amp.pharm.mssm.edu/Enrichr/) (32), and the top 10 GO terms ranked by combined score were shown.
Motif analysis
The genomic sequence within 100 base pairs of the center of each weakened BAF binding site in ARID1A mutant NGP cells was generated from the UCSC website with repeats masked with “N.” Motif analysis was performed by MEME-ChIP v4.12.0 (http://meme-suite.org/tools/meme-chip) (60).
Statistical analysis
To analyze the hyperplastic, apoptotic, and proliferative status of the sympathoadrenal cells before tumor onset, we scanned all sections from each fish by confocal z-stack. A single representative section containing the largest number of sympathoadrenal cells or activated caspase-3–positive sympathoadrenal cells or the largest percentage of EdU+ sympathoadrenal cells in the IRG was selected and quantified for each fish. Data were compared with either the two-tailed unpaired Welch t test (hyperplasia analysis) or the two-tailed Fisher’s exact test (apoptosis analysis).
To analyze the proliferative capacity of the sympathoadrenal cells before tumor onset, all sections from each individual fish were scanned by confocal z-stack. A single representative section containing the largest percentage of EdU+ sympathoadrenal cells to the total number of sympathoadrenal cells in the IRG was selected and quantified for each individual fish. The data were analyzed by two-tailed unpaired Welch t test.
Supplementary Material
Acknowledgments
We thank J. R. Gilbert for review of the manuscript and comments; G. Thurston, D. Debiasi, M. T. Coute, and M. Alves for care of the zebrafish; Z. Herbert of the Dana-Farber Molecular Biology Core Facility for genomics support; and Y. Zhou and S. Yang of Boston Children’s Hospital for comments. Funding: This work was supported by an NIH grant R01-CA180692 (A.T.L. and J. M. Maris co-PI grant). T.T. was supported by a research grant from Children’s Neuroblastoma Cancer Foundation, a Friends of DFCI award, and a Friends for Life Fellowship from Dana-Farber Cancer Institute. H.S. was supported by a Young Investigator Grant from Alex’s Lemonade Stand Foundation, a Rally Foundation for Childhood Cancer Research, and a Friends for Life Neuroblastoma Fellowship from Dana-Farber Cancer Institute. M.W.Z. was supported by a Young Investigator Grant from Alex’s Lemonade Stand Foundation, a postdoctoral fellowship from the Charles A. King Trust, and the Claudia Adams Barr Innovative Basic Science research program. A.D.D. was supported by funding from the Damon-Runyon Cancer Research Foundation (DRSG-24-18), the Alex’s Lemonade Stand Foundation, and the CureSearch for Children’s Cancer Foundation. Author contributions: H.S., T.T., C.K., and A.T.L. designed the study and experiments. H.S. and T.T. performed the experiments. H.S., T.T., and B.J.A. performed the bioinformatics analysis. H.S., T.T., B.J.A., A.D.D., M.W.Z., C.K., and A.T.L. analyzed the data. H.S., T.T., and A.T.L. drafted and finalized the manuscript with inputs from all other authors. Competing interests: C.K. is the scientific founder, a fiduciary board of directors member, a scientific advisory board member, a shareholder, and a consultant for Foghorn Therapeutics Inc. (Cambridge, MA). B.J.A is a shareholder in Syros Pharmaceuticals Inc. The other authors declare that they have no potential conflicts of interest. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. ChIP-seq and RNA-seq data are available from GEO (series GSE134632). Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/29/eaaz3440/DC1
REFERENCES AND NOTES
- 1.Cheung N.-K., Dyer M. A., Neuroblastoma: Developmental biology, cancer genomics and immunotherapy. Nat. Rev. Cancer 13, 397–411 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schleiermacher G., Janoueix-Lerosey I., Delattre O., Recent insights into the biology of neuroblastoma. Int. J. Cancer 135, 2249–2261 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Maris J. M., Recent advances in neuroblastoma. N. Engl. J. Med. 362, 2202–2211 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang M., Weiss W. A., Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 3, a014415 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.García-López J., Wallace K., Otero J. H., Olsen R., Wang Y. D., Finkelstein D., Gudenas B. L., Rehg J. E., Northcott P., Davidoff A. M., Freeman K. W., Large 1p36 deletions affecting Arid1a locus facilitate Mycn-driven oncogenesis in neuroblastoma. Cell Rep. 30, 454–464 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henrich K.-O., Schwab M., Westermann F., 1p36 tumor suppression—A matter of dosage? Cancer Res. 72, 6079–6088 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Okawa E. R., Gotoh T., Manne J., Igarashi J., Fujita T., Silverman K. A., Xhao H., Mosse Y. P., White P. S., Brodeur G. M., Expression and sequence analysis of candidates for the 1p36.31 tumor suppressor gene deleted in neuroblastomas. Oncogene 27, 803–810 (2008). [DOI] [PubMed] [Google Scholar]
- 8.White P. S., Thompson P. M., Gotoh T., Okawa E. R., Igarashi J., Kok M., Winter C., Gregory S. G., Hogarty M. D., Maris J. M., Brodeur G. M., Definition and characterization of a region of 1p36.3 consistently deleted in neuroblastoma. Oncogene 24, 2684–2694 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Mashtalir N., D’Avino A. R., Michel B. C., Luo J., Pan J., Otto J. E., Zullow H. J., McKenzie Z. M., Kubiak R. L., Pierre R. S., Valencia A. M., Poynter S. J., Cassel S. H., Ranish J. A., Kadoch C., Modular organization and assembly of SWI/SNF family chromatin remodeling complexes. Cell 175, 1272–1288.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valencia A. M., Kadoch C., Chromatin regulatory mechanisms and therapeutic opportunities in cancer. Nat. Cell Biol. 21, 152–161 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang X., Wang S., Troisi E. C., Howard T. P., Haswell J. R., Wolf B. K., Hawk W. H., Ramos P., Oberlick E. M., Tzvetkov E. P., Vazquez F., Hahn W. C., Park P. J., Roberts C. W. M., BRD9 defines a SWI/SNF sub-complex and constitutes a specific vulnerability in malignant rhabdoid tumors. Nat. Commun. 10, 1881 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kadoch C., Hargreaves D. C., Hodges C., Elias L., Ho L., Ranish J., Crabtree G. R., Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 45, 592–601 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kadoch C., Crabtree G. R., Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci. Adv. 1, e1500447 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones S., Wang T.-L., Shih l.-M., Mao T.-L., Nakayama K., Roden R., Glas R., Slamon D., Diaz L. A. Jr., Vogelstein B., Kinzler K. W., Velculescu V. E., Papadopoulos N., Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 330, 228–231 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones S., Li M., Parsons D. W., Zhang X., Wesseling J., Kristel P., Schmidt M. K., Markowitz S., Yan H., Bigner D., Hruban R. H., Eshleman J. R., Iacobuzio-Donahue C. A., Goggins M., Maitra A., Malek S. N., Powell S., Vogelstein B., Kinzler K. W., Velculescu V. E., Papadopoulos N., Somatic mutations in the chromatin remodeling gene ARID1A occur in several tumor types. Hum. Mutat. 33, 100–103 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mathur R., Alver B. H., San Roman A. K., Wilson B. G., Wang X., Agoston A. T., Park P. J., Shivdasani R. A., Roberts C. W., ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nat. Genet. 49, 296–302 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sausen M., Leary R. J., Jones S., Wu J., Reynolds C. P., Liu X., Blackford A., Parmigiani G., Diaz L. A. Jr., Papadopoulos N., Vogelstein B., Kinzler K. W., Velculescu V. E., Hogarty M. D., Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat. Genet. 45, 12–17 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wiegand K. C., Shah S. P., Al-Agha O. M., Zhao Y., Tse K., Zeng T., Senz J., McConechy M. K., Anglesio M. S., Kalloger S. E., Yang W., Heravi-Moussavi A., Giuliany R., Chow C., Fee J., Zayed A., Prentice L., Melnyk N., Turashvili G., Delaney A. D., Madore J., Yip S., McPherson A. W., Ha G., Bell L., Fereday S., Tam A., Galletta L., Tonin P. N., Provencher D., Miller D., Jones S. J., Moore R. A., Morin G. B., Oloumi A., Boyd N., Aparicio S. A., Shih M., Mes-Masson A.-M., Bowtell D. D., Hirst M., Gilks B., Marra M. A., Huntsman D. G., ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 363, 1532–1543 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu J. N., Roberts C. W. M., ARID1A mutations in cancer: Another epigenetic tumor suppressor? Cancer Discov. 3, 35–43 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guan B., Mao T.-L., Panuganti P. K., Kuhn E., Kurman R. J., Maeda D., Chen E., Jeng Y.-M., Wang T.-L., Shih l.-M., Mutation and loss of expression of ARID1A in uterine low-grade endometrioid carcinoma. Am. J. Surg. Pathol. 35, 625–632 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guan B., Wang T.-L., Shih l.-M., ARID1A, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res. 71, 6718–6727 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wiegand K. C., Lee A. F., Al-Agha O. M., Chow C., Kalloger S. E., Scott D. W., Steidl C., Wiseman S. M., Gascoyne R. D., Gilks B., Huntsman D. G., Loss of BAF250a (ARID1A) is frequent in high-grade endometrial carcinomas. J. Pathol. 224, 328–333 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Maris J. M., Guo C., Blake D., White P. S., Hogarty M. D., Thompson P. M., Rajalingam V., Gerbing R., Stram D. O., Matthay K. K., Seeger R. C., Brodeur G. M., Comprehensive analysis of chromosome 1p deletions in neuroblastoma. Med. Pediatr. Oncol. 36, 32–36 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Zhu S., Lee J.-S., Guo F., Shin J., Perez-Atayde A. R., Kutok J. L., Rodig S. J., Neuberg D. S., Helman D., Feng H., Stewart R. A., Wang W., George R. E., Kanki J. P., Look A. T., Activated ALK collaborates with MYCN in neuroblastoma pathogenesis. Cancer Cell 21, 362–373 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hwang W. Y., Fu Y., Reyon D., Maeder M. L., Tsai S. Q., Sander J. D., Peterson R. T., Yeh J.-R., Joung J. K., Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 31, 227–229 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tornoczky T., Semjen D., Shimada H., Ambros I. M., Pathology of peripheral neuroblastic tumors: Significance of prominent nucleoli in undifferentiated/poorly differentiated neuroblastoma. Pathol. Oncol. Res. 13, 269–275 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Huber K., The sympathoadrenal cell lineage: Specification, diversification, and new perspectives. Dev. Biol. 298, 335–343 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Stewart R. A., Lee J. S., Lachnit M., Look A. T., Kanki J. P., Henion P. D., Studying peripheral sympathetic nervous system development and neuroblastoma in zebrafish. Methods Cell Biol. 100, 127–152 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Helming K. C., Wang X., Wilson B. G., Vazquez F., Haswell J. R., Manchester H. E., Kim Y., Kryukov G. V., Ghandi M., Aguirre A. J., Jagani Z., Wang Z., Garraway L. A., Hahn W. C., Roberts C. W. M., ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat. Med. 20, 251–254 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Michel B. C., D’Avino A. R., Cassel S. H., Mashtalir N., McKenzie Z. M., McBride M. J., Valencia A. M., Zhou Q., Bocker M., Soares L. M. M., Pan J., Remillard D. I., Lareau C. A., Zullow H. J., Fortoul N., Gray N. S., Bradner J. E., Chan H. M., Kadoch C., A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat. Cell Biol. 20, 1410–1420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu G., Wang L.-G., He Q.-Y., ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Kuleshov M. V., Jones M. R., Rouillard A. D., Fernandez N. F., Duan Q., Wang Z., Koplev S., Jenkins S. L., Jagodnik K. M., Lachmann A., McDermott M. G., Monteiro C. D., Gundersen G. W., Ma’ayan A., Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boeva V., Louis-Brennetot C., Peltier A., Durand S., Pierre-Eugene C., Raynal V., Etchevers H. C., Thomas S., Lermine A., Daudigeos-Dubus E., Geoerger B., Orth M. F., Grunewald T. G. P., Diaz E., Ducos B., Surdez D., Carcaboso A. M., Medvedeva I., Deller T., Combaret V., Lapouble E., Pierron G., Grossetete-Lalami S., Baulande S., Schleiermacher G., Barillot E., Rohrer H., Delattre O., Janoueix-Lerosey I., Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat. Genet. 49, 1408–1413 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Decaesteker B., Denecker G., Van Neste C., Dolman E. M., Van Loocke W., Gartlgruber M., Nunes C., De Vloed F., Depuydt P., Verboom K., Rombaut D., Loontiens S., De Wyn J., Kholosy W. M., Koopmans B., Essing A. H. W., Herrmann C., Dreidax D., Durinck K., Deforce D., van Nieuwerburgh F., Henssen A., Versteeg R., Boeva V., Schleiermacher G., van Nes J., Mestdagh P., Vanhauwaert S., Schulte J. H., Westermann F., Molenaar J. J., De Preter K., Speleman F., TBX2 is a neuroblastoma core regulatory circuitry component enhancing MYCN/FOXM1 reactivation of DREAM targets. Nat. Commun. 9, 4866 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Durbin A. D., Zimmerman M. W., Dharia N. V., Abraham B. J., Iniguez A. B., Weichert-Leahey N., He S., Krill-Burger J. M., Root D. E., Vazquez F., Tsherniak A., Hahn W. C., Golub T. R., Young R. A., Look A. T., Stegmaier K., Selective gene dependencies in MYCN-amplified neuroblastoma include the core transcriptional regulatory circuitry. Nat. Genet. 50, 1240–1246 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Groningen T., Akogul N., Westerhout E. M., Chan A., Hasselt N. E., Zwijnenburg D. A., Broekmans M., Stroeken P., Haneveld F., Hooijer G. K. J., Savci-Heijink C. D., Lakeman A., Volckmann R., van Sluis P., Valentijn L. J., Koster J., Versteeg R., van Nes J., A NOTCH feed-forward loop drives reprogramming from adrenergic to mesenchymal state in neuroblastoma. Nat. Commun. 10, 1530 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Groningen T., Koster J., Valentijn L. J., Zwijnenburg D. A., Akogul N., Hasselt N. E., Broekmans M., Haneveld F., Nowakowska N. E., Bras J., van Noesel C. J. M., Jongejan A., van Kampen A. H., Koster L., Baas F., van Dijk-Kerkhoven L., Huizer-Smit M., Lecca M. C., Chan A., Lakeman A., Molenaar P., Volckmann R., Westerhout E. M., Hamdi M., van Sluis P. G., Ebus M. E., Molenaar J. J., Tytgat G. A., Westerman B. A., van Nes J., Versteeg R., Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat. Genet. 49, 1261–1266 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Tan T. Z., Miow Q. H., Miki Y., Noda T., Mori S., Huang R. Y.-J., Thiery J. P., Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol. Med. 6, 1279–1293 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tao T., Sondalle S. B., Shi H., Zhu S., Perez-Atayde A. R., Peng J., Baserga S. J., Look A. T., The pre-rRNA processing factor DEF is rate limiting for the pathogenesis of MYCN-driven neuroblastoma. Oncogene 36, 3852–3867 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alver B. H., Kim K. H., Lu P., Wang X., Manchester H. E., Wang W., Haswell J. R., Park P. J., Roberts C. W., The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat. Commun. 8, 14648 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang X., Lee R. S., Alver B. H., Haswell J. R., Wang S., Mieczkowski J., Drier Y., Gillespie S. M., Archer T. C., Wu J. N., Tzvetkov E. P., Troisi E. C., Pomeroy S. L., Biegel J. A., Tolstorukov M. Y., Bernstein B. E., Park P. J., Roberts C. W. M., SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat. Genet. 49, 289–295 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kelso T. W. R., Porter D. K., Amaral M. L., Shokhirev M. N., Benner C., Hargreaves D. C., Chromatin accessibility underlies synthetic lethality of SWI/SNF subunits in ARID1A-mutant cancers. eLife 6, e30506 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi J., Whyte W. A., Zepeda-Mendoza C. J., Milazzo J. P., Shen C., Roe J. S., Minder J. L., Mercan F., Wang E., Eckersley-Maslin M. A., Campbell A. E., Kawaoka S., Shareef S., Zhu Z., Kendall J., Muhar M., Haslinger C., Yu M., Roeder R. G., Wigler M. H., Blobel G. A., Zuber J., Spector D. L., Young R. A., Vakoc C. R., Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 27, 2648–2662 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagl N. G. Jr., Zweitzig D. R., Thimmapaya B., Beck G. R. Jr., Moran E., The c-myc gene is a direct target of mammalian SWI/SNF-related complexes during differentiation-associated cell cycle arrest. Cancer Res. 66, 1289–1293 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Piskareva O., Harvey H., Nolan J., Conlon R., Alcock L., Buckley P., Dowling P., Henry M., O’Sullivan F., Bray I., Stallings R. L., The development of cisplatin resistance in neuroblastoma is accompanied by epithelial to mesenchymal transition in vitro. Cancer Lett. 364, 142–155 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Tao T., Shi H., Guan Y., Huang D., Chen Y., Lane D. P., Chen J., Peng J., Def defines a conserved nucleolar pathway that leads p53 to proteasome-independent degradation. Cell Res. 23, 620–634 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hsu P. D., Scott D. A., Weinstein J. A., Ran F. A., Konermann S., Agarwala V., Li Y., Fine E. J., Wu X., Shalem O., Cradick T. J., Marraffini L. A., Bao G., Zhang F., DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sanjana N. E., Shalem O., Zhang F., Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., Zhang F., Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morita K., Suzuki K., Maeda S., Matsuo A., Mitsuda Y., Tokushige C., Kashiwazaki G., Taniguchi J., Maeda R., Noura M., Hirata M., Kataoka T., Yano A., Yamada Y., Kiyose H., Tokumasu M., Matsuo H., Tanaka S., Okuno Y., Muto M., Naka K., Ito K., Kitamura T., Kaneda Y., Liu P. P., Bando T., Adachi S., Sugiyama H., Kamikubo Y., Genetic regulation of the RUNX transcription factor family has antitumor effects. J. Clin. Invest. 127, 2815–2828 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; 1000 Genome Project Data Processing Subgroup , The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nusbaum C., Myers R. M., Brown M., Li W., Liu X. S., Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Quinlan A. R., Hall I. M., BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ramirez F., Ryan D. P., Grüning B., Bhardwaj V., Kilpert F., Richter A. S., Heyne S., Dündar F., Manke T., deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim D., Langmead B., Salzberg S. L., HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Feng J., Meyer C. A., Wang Q., Liu J. S., Shirley Liu X., Zhang Y., GFOLD: A generalized fold change for ranking differentially expressed genes from RNA-seq data. Bioinformatics 28, 2782–2788 (2012). [DOI] [PubMed] [Google Scholar]
- 58.Anders S., Huber W., Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., Mesirov J. P., Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Machanick P., Bailey T. L., MEME-ChIP: Motif analysis of large DNA datasets. Bioinformatics 27, 1696–1697 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/29/eaaz3440/DC1