

Cells sense the presence of more than one version of mtDNA and select the variant according to its impact on function.
Abstract
Heteroplasmy, multiple variants of mitochondrial DNA (mtDNA) in the same cytoplasm, may be naturally generated by mutations but is counteracted by a genetic mtDNA bottleneck during oocyte development. Engineered heteroplasmic mice with nonpathological mtDNA variants reveal a nonrandom tissue-specific mtDNA segregation pattern, with few tissues that do not show segregation. The driving force for this dynamic complex pattern has remained unexplained for decades, challenging our understanding of this fundamental biological problem and hindering clinical planning for inherited diseases. Here, we demonstrate that the nonrandom mtDNA segregation is an intracellular process based on organelle selection. This cell type–specific decision arises jointly from the impact of mtDNA haplotypes on the oxidative phosphorylation (OXPHOS) system and the cell metabolic requirements and is strongly sensitive to the nuclear context and to environmental cues.
INTRODUCTION
The oxidative phosphorylation (OXPHOS) system is the only structure in animal cells with components encoded by two genomes, maternally transmitted mitochondrial DNA (mtDNA) and biparentally transmitted nuclear DNA (nDNA). In mammals, mtDNA encodes for a reduced number of genes: 13 messenger RNAs (mRNAs), 22 transfer RNAs (tRNAs), and 2 ribosomal RNAs (rRNAs). All proteins encoded in the mtDNA are structural components of the multiprotein mitochondrial respiratory complexes. The mitochondrial-encoded OXPHOS structural proteins have to jointly assemble with up to 70 structural proteins encoded in the nDNA to build functional respiratory complexes. Therefore, the functionality of OXPHOS-encoded genes is limited by a physical matching constraint. This imposes a close-fitting coevolution of both genomes challenged by two very different mechanisms generating variability in nDNA (by sexual reproduction, mutation, and coexistence of two alleles) and in mtDNA (by mutation, polyploidy, and segregation). In addition, nuclear OXPHOS genes have tissue-specific variants (1), which present identical mtDNA-encoded proteins with alternative nDNA variants. These properties have been known for decades, but the mechanisms by which the compatibility of nuclear and mitochondrial structural components is achieved are completely unknown.
One of the more intriguing features of mtDNA is its uniparental transmission, for which no comprehensive explanation exists (2). Because of uniparental transmission, offspring mtDNA arises from the expansion of inherited maternal mtDNA and is substantially clonal (homoplasmic). It has been repeatedly observed in mice that when the mtDNA homoplasmy is artificially disrupted by the generation of animals with more than one mtDNA haplotype (heteroplasmy), an active segregation response is triggered, potentially acting to recover the homoplasmic natural condition (3–5). However, this response does not just promote the recovery of homoplasmy by random segregation. In most tissues, segregation is nonrandom: Some tissues systematically favor one haplotype, while other tissues favor the other, in a markedly consistent pattern (3–5). The driving force underlying this phenomenon remains a mystery. By the same token, it remains unclear why a subset of tissues maintains heteroplasmy, failing to exhibit pronounced segregation bias.
Recently, we reported that mice with identical nuclear genomes but with different mtDNA haplotypes (conplastic mice) generate functionally different OXPHOS systems that shape organismal metabolism (6), and we proposed that this metabolic influence may determine mtDNA segregation patterns. Here, we generated novel heteroplasmic animals combining wild-type mtDNA haplotypes from C57BL/6 and NZB mouse inbred strains. We analyzed the evolution of heteroplasmy in 19 tissues from birth to natural death, shedding new light on the behavior of the different cell types that some tissues contain. In addition, we investigated the impact of genetic, pharmacological, and environmental challenges to mitochondrial function on the evolution of heteroplasmic cells. To complete the picture, we generated and analyzed chimeric mice mixing cells that are homoplasmic for each variant of mtDNA. Our collected, multifaceted, large-scale data allow us to conclude that somatic segregation of heteroplasmy is driven by the intracellular sorting of the organelles due to the impact of mtDNA on the OXPHOS performance and the heterogeneous metabolic roles that the OXPHOS system has in different cell types.
RESULTS
mtDNA heteroplasmy dynamics during postnatal life
We analyzed mtDNA segregation from birth to natural death in 19 different tissues of the BL/6C57-NZB heteroplasmic mice (Fig. 1 and fig. S1). Most tissues progressively select for one of the alternative variants of mtDNA, while in a small number of tissues the heteroplasmy level remains largely stable. During late fetal development, we observed mtDNA segregation only in brown adipose tissue (BAT) and heart (7). After birth, biased segregation continues in BAT, while in heart there is no further segregation bias (Fig. 1, B and C). Thus, we successfully reproduced quantitatively and qualitatively the segregation behavior of mtDNA variants, previously reported by others (3–5). In summary, segregation bias in favor of one or the other mtDNA haplotype is the norm. Only a limited number of tissues do not select postnatally against heteroplasmy (thereby allowing the long-term retention of two distinct mtDNA populations along the life of the animals) (Fig. 1C), and the kinetics of segregation is specific for each tissue.
Fig. 1. Nonrandom segregation of wild-type mtDNA heteroplasmy.
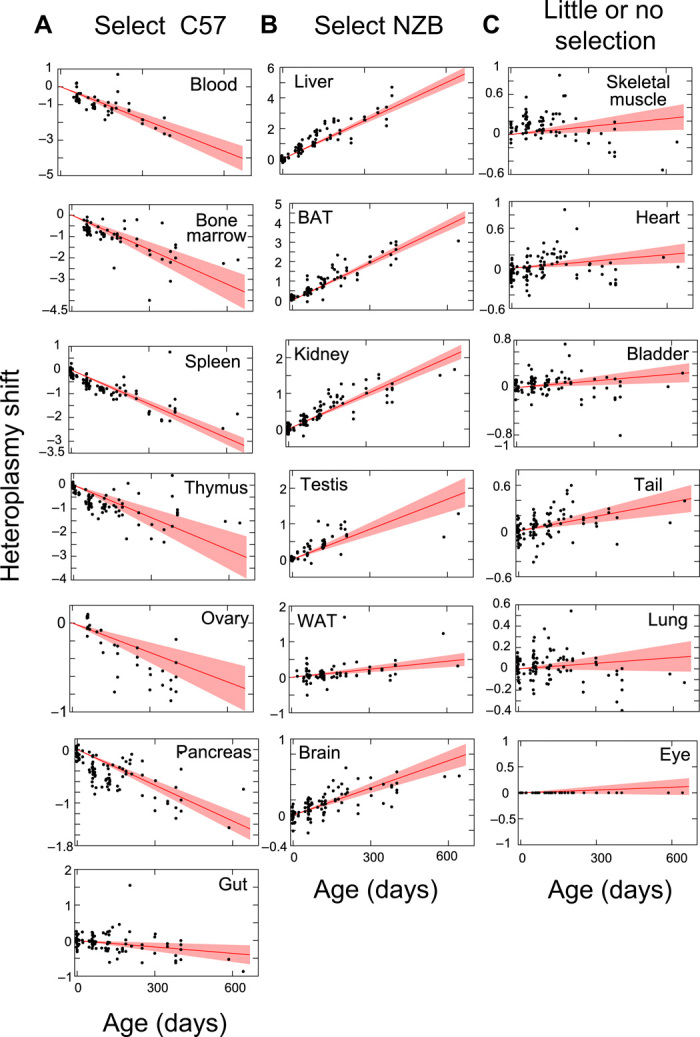
Estimation of heteroplasmy shift for the indicated tissue from birth to more than 600 days old. (A) Tissues that shift toward C57 mtDNA. (B) Tissues that shift toward NZB mtDNA. (C) Tissues that do not resolve heteroplasmy or display very slow shifts. Red lines give inferred mean segregation behavior with 95% confidence intervals (n = 119 BL/6C57-NZB mice). Black dots show heteroplasmy data in the given tissue plotted relative to eye (see Materials and Methods), which is inferred to have a low segregation rate and is hence used as an approximate control tissue. Absolute values for each tissue are also shown in fig. S1.
mtDNA haplotype selection is an intracellular phenomenon
All published studies follow the evolution of somatic mtDNA heteroplasmy in animals by quantifying the proportion of the two mtDNA haplotypes in total tissue. This approach places limitations on the level of mechanistic detail that can be inferred. One of the more relevant constraints is that it cannot distinguish if the segregation behavior requires the interaction of the two variants of mtDNA in the same cytoplasm or if it is due to competition between cells that become homoplasmic by random segregation. To address this question, we generated chimeric animals by aggregation of conplastic morulae BL/6C57 with BL/6NZB to create individuals containing both mtDNA types at homoplasmy in different cells. Then, we tracked the proportion of the mtDNA haplotypes over time to determine whether the observed segregation had any component of cell-to-cell competition. We analyzed the proportion of the two haplotypes in 16 tissues [spleen, thymus, ovary, pancreas, gut, liver, BAT, kidney, testis, white adipose tissue (WAT), brain, skeletal muscle, heart, bladder, lung, and eye] and also the tail of neonates (16 individuals) and of 29- to 279-day-old chimeric mice (38 individuals) (Fig. 2). Because the initial chimeric proportion was unpredictable, we normalized the mtDNA proportion in each tissue as indicated in Materials and Methods. We found no detectable segregation bias in favor of any mtDNA haplotype in any of the tissues analyzed (Fig. 2). Therefore, we conclude that the biased segregation observed in most tissues of heteroplasmic animals requires the interaction of both mtDNAs in the same cytoplasm. In other words, it is an intracellular event.
Fig. 2. Absence of mtDNA-driven cell competition in chimeric mice.
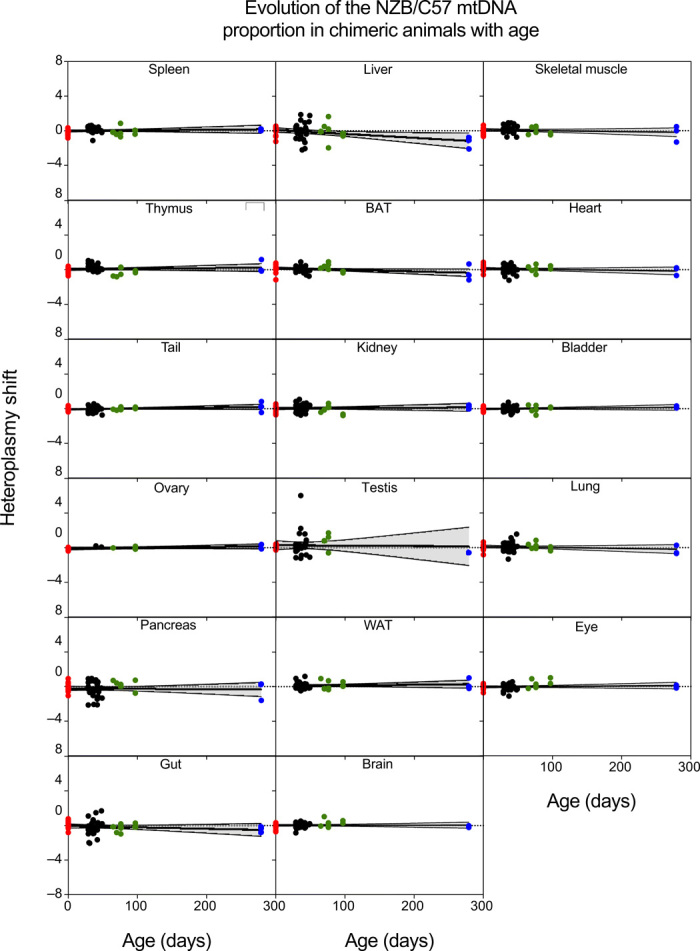
Estimation of mtDNA proportion shift using eye as the reference tissue for the indicated tissue from birth to 279-day-old chimeric mice (n = 54 mice; red dots, newborn pups; black dots, 29-49 days; green dots, 65-97 days old; blue dots, 279 day-old mice) generated by morula aggregation of homoplasmic C57 and homoplasmic NZB embryos. Black lines give inferred mean segregation behavior with 95% confidence intervals (shadowed areas). No statistically significant P values were observed after correction for multiple testing.
mtDNA preference is cell type–specific rather than tissue specific
A second major limitation of the previous studies on mtDNA segregation is that with the analysis of mtDNA content from total tissue, it is not possible to distinguish whether different cell types within the same tissue behave differently, and thus whether segregation bias is tissue or cell type specific. To address this, we worked with the observation that all tissues that produce or store blood cells showed segregation bias toward C57 mtDNA (Fig. 1A and fig. S1). Separation of different populations of blood cells is straightforward (see Materials and Methods) and confirmed the generalized preference for C57 mtDNA of all cell types regardless of their lymphoid or myeloid origin (fig. S2, A to D). Nevertheless, there are evident kinetic differences in this tendency. Lymphocytes showed a progressive selection, while monocytes and neutrophils produced the shift at a very early age (fig. S2, B to D).
The fact that all blood cell types manifest identical preference for C57 mtDNA may suggest that this is a tissue rather than a cell type decision. This raised the question whether this is also the case in solid tissues, which have higher variety in the metabolic and functional specialization of their cell types. This specialization is particularly relevant in the case of the brain, which has an enormous complexity, with several cell types and with intricate metabolic interrelationship between them (8, 9). We found that when the proportion of mtDNA heteroplasmy was measured in brain (specifically in cerebral cortex) along the whole life of the animals, NZB mtDNA is preferentially selected (Fig. 1B). The brain enables us the opportunity to study a complete system to evaluate the driving forces that trigger mtDNA segregation bias by specific isolation of different cell types from the same heteroplasmic individual. Particularly interesting are the differences in metabolism between neurons and astrocytes (10). To explore that, astrocytes (left brain hemisphere) and neurons (right brain hemisphere) were isolated and mtDNA heteroplasmy was estimated at different ages. Astrocytes did not manifest any detectable preference for a particular mtDNA haplotype (Fig. 3A). However, neurons selected for NZB mtDNA (Fig. 3B). When animals were young (8-week-old females), the amount of NZB mtDNA was higher in astrocytes than in neurons, while neurons presented higher amount of NZB mtDNA than astrocytes with age. Neurons rely more than astrocytes on OXPHOS to fulfill their energetic demands (11). Contrary to our observation of generalized C57 mtDNA selection in blood cells, the results in brain reinforce the interpretation that mtDNA segregation is cell type specific rather than tissue specific, which may be determined by OXPHOS demands.
Fig. 3. Analysis of heteroplasmy in brain and cardiac cell populations.
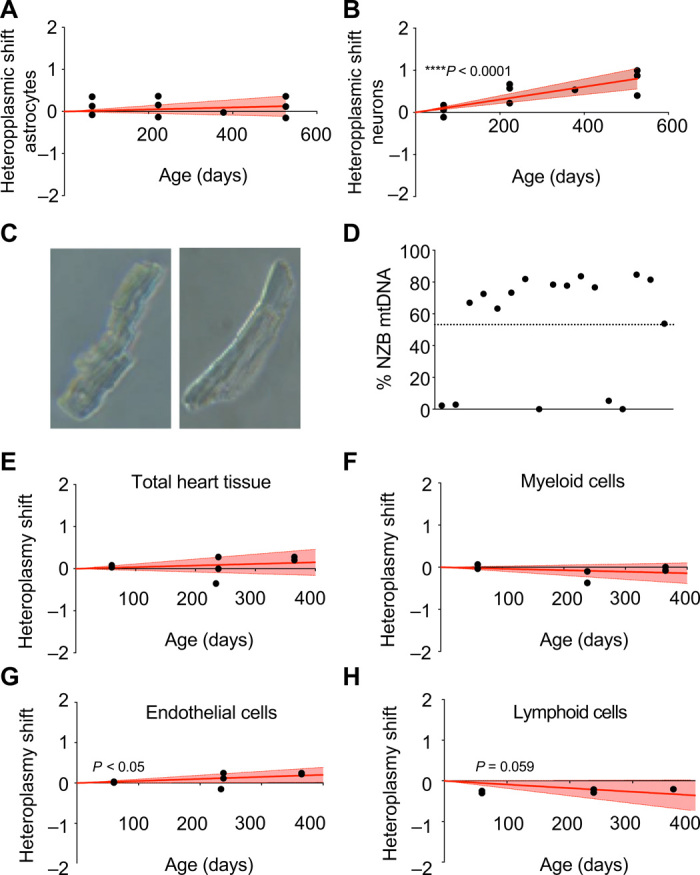
(A and B) Transformed heteroplasmy shift in isolated astrocytes (A) and neurons (B) using tail as reference tissue. In (A) and (B), red lines correspond to the mean and discontinuous lines show 95% confidence intervals of inferred segregation trajectories. Each dot corresponds to a different animal at the indicated age. (C) Bright-field microscopy of single isolated adult cardiomyocytes. (D) Evaluation of intracellular mtDNA heteroplasmy in single cardiomyocytes from 20-week-old heteroplasmic mice (n = 4) Each dot corresponds to a different cardiomyocyte. Dotted line: average of entire tissue. (E to H) Evaluation of transformed heteroplasmy shift using tail as the reference tissue in isolated cardiac cell populations from heteroplasmic hearts at different ages. (E) Total cardiac tissue, (F) myeloid cells, (G) endothelial cells, and (H) lymphoid cells. Each dot corresponds to a different animal. *P < 0.05 and ****P < 0.0001, linear regression coefficient. (A, B, E, F, G, and H): positive toward mtDNA NZB and negative toward mtDNA C57.
We next investigate, at a cellular level, the fate of heteroplasmy in a tissue that does not manifest biased segregation: the heart. Cardiomyocytes are the dominant cells in the heart, with high mitochondrial content. Thus, overall heart mtDNA heteroplasmy likely represents the mtDNA of cardiomyocytes. However, the estimation of the proportion of heteroplasmy in total heart tissue does not allow us to determine between-cell variability. To investigate this important heterogeneity, we isolated individual adult cardiomyocytes (Fig. 3, C and D) from heteroplasmic hearts. The average of the heteroplasmy was 53.2% of NZB mtDNA, in the range of the whole tissue. Most cardiomyocytes showed the coexistence of both mtDNAs in a single cell (Fig. 3D). We found two cardiomyocytes that had only C57 mtDNA and another three that shifted toward C57 mtDNA (Fig. 3D). The rest of the analyzed cells remained heteroplasmic with a slight increase in NZB mtDNA over tissue average (Fig. 3D).
Are other cardiac cell populations different from cardiomyocytes, and able to segregate mtDNA? We performed cell sorting from heteroplasmic hearts at different ages and estimated the proportion of heteroplasmy in whole heart and in heart-derived myeloid, endothelial, and lymphoid cells (Fig. 3, E to H). Thus, while the heteroplasmy proportion in neither the whole heart nor myeloid cells shifted with age (Fig. 3, E and F), we could observe that lymphoid cells selected for C57 mtDNA, while endothelial cells selected for NZB mtDNA (Fig. 3, G and H). Comparison to our previous observations in blood showed that lymphoid cells either from blood or from the heart manifest an identical preference for C57 mtDNA. In summary, the analysis of the heteroplasmy evolution in three different tissues, which selected for C57 mtDNA (blood) or for NZB mtDNA (brain) or showed no selection (heart), allows us to conclude that segregation bias is a cell type–specific phenomenon.
Postnatal mtDNA segregation depends on OXPHOS function
The unanswered question is: Why do most cell types segregate mtDNA variants? To evaluate whether postnatal segregation depends on the impact of heteroplasmy on OXPHOS function, we challenged the heteroplasmic animals with a variety of environmental and pharmacological interventions altering the mitochondrial metabolism and followed the subsequent mtDNA segregation behavior. Environmental alterations in heteroplasmic animals also promoted changes in segregation. Per-oral N-acetylcysteine (NAC) administration in drinking water reduced the overall magnitudes of segregation bias, with a particularly prominent effect in BAT (Fig. 4A and fig. S2E). Even more acute changes in the pattern of segregation were observed when mice were fed with a hypercaloric diet with high-fat content. High-fat diet (HFD) promoted a marked modification in the segregation rate and direction of mtDNA variants in gut (Fig. 4A and fig. S2E). Last, we treated the animals with dichloroacetate (DCA) to activate mitochondrial pyruvate oxidation and enhance OXPHOS activity. DCA considerably exacerbated the mtDNA segregation pattern and also inverted the selected mtDNA haplotype in the case of gut (Fig. 4A and fig. S2E). Therefore, we find that metabolic interventions can modulate OXPHOS performance–driven haplotype selection.
Fig. 4. mtDNA segregation is sensitive to modulation of organismal metabolism.
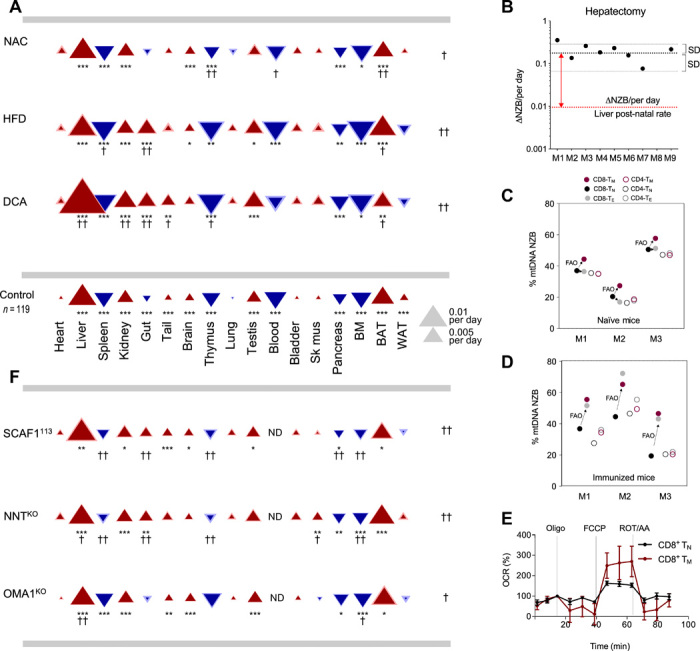
(A) Tissue-specific segregation patterns in basal and intervention conditions. Triangles indicate magnitude (size), s.e.m. (thickness of lighter border), and direction of the segregation toward NZB mtDNA (red, upward) or C57 mtDNA (blue, downward). Control: Segregation with no intervention. Experimental: Segregation resulting from chemical (A) and genetic (F) perturbations. DCA, n = 22 (age 57 to 159); NAC, n = 16 (age 96 to 273); HFD, n = 25 (age 59 to 188). (B) Rate of segregation of mtDNA in control (red line) and hepatectomized (black line) mice. (C and D) Heteroplasmy in different T cell subsets in 15-week-old (C) naïve and (D) immunized mice. Circles indicate CD8+ (filled) or CD4+ (open) T cells, and the colors indicate the different T cell subsets [naïve (TN), memory (TM), or effector (TE) T cells]. FAO, fatty acid oxidation; M, mouse. Data are representative of two independent experiments. (E) Oxygen consumption rate (OCR) Seahorse profiles for indicated cells (n = 5 BL/6C57-NZB). Oligo, oligomycin; FCCP, carbonilcyanide p-triflouromethoxyphenylhydrazone; ROT, rotenone; AA, antimycin. (F) Tissue-specific segregation patterns in heteroplasmic mouse with the indicated nuclear genetic variations. ND, not described. BL/6C57-NZB:NNTKO: n = 27 ovaries, n = 16 testes, and n = 25 rest of the tissues. BL/6C57-NZB:SCAF1113: n = 11 ovaries, n = 10 testes, and n = 20 to 21 for the rest of the tissues. BL/6C57-NZB:OMA1KO: n = 11 ovaries, n = 11 testes, and n = 14 to 16 for the rest. In (A) and (F), †P < 0.05 and ††P < 0.01, at the side for significance affecting the overall pattern of segregation and under triangles for significance at individual tissues. Asterisks: Significant differences from zero segregation (bootstrap percentile method with Bonferroni correction; *P < 0.05, **P < 0.01, and ***P < 0.001).
Preferred mtDNA haplotype is determined by the cellular metabolic program
Comparing the tissue mtDNA segregation bias, those tissues showing preference for C57 are highly proliferative (Fig. 1A), while those that choose NZB mtDNA haplotype are more quiescent (Fig. 1B). Thus, the metabolic requirements of an elevated cellular proliferation rate might determine mtDNA haplotype preference. If this is the case, a switch toward a proliferative phenotype should then change the preferred mtDNA haplotype. Alternatively, the mtDNA haplotype preference may be dictated by the cell type–specific OXPHOS performance that determines its preferred metabolic “fuel” or combination of “fuels.” In this sense, in the C57BL/6JOlaHsd nuclear background, the NZB mtDNA promotes lipid metabolism, while C57 mtDNA promotes glucose utilization (7). Therefore, the cell-intrinsic metabolic architecture would define the preferred mtDNA haplotype. To test between these two alternatives, we chose liver and T cell lymphocytes as representative of NZB or C57 mtDNA preference, respectively. In one side, we induced in vivo the regression of adult liver to a highly proliferative status by hepatectomy, and in the other side, we investigated the impact of proliferation and differentiation of T cell populations, as well of activation, on the mtDNA haplotype segregation.
Partial hepatectomy (removing 75% of the liver) induces cell proliferation until the original mass of the liver is restored in few days. Most of the changes in metabolism occur during the first 24 hours (12). One of the short-term effects of hepatectomy is the overall reduction of blood glucose, with a marked increase in hepatic triglyceride content causing the induction of the adipogenic transcriptional program. Therefore, after hepatectomy, cell proliferation occurs in a context of low use of glucose as fuel (13).
After these changes, a replicative phase in which DNA synthesis is very active is triggered (12). Hepatectomy surgery was performed in 10-week-old males. Fifteen days after the tissue removal, mice were euthanized and total liver regeneration and mtDNA haplotypes were measured. Total liver weight revealed that livers from all mice are able to recover (fig. S2F). To evaluate mtDNA segregation, we compared the excised tissue during the surgery (day 0) with regenerated livers at day 15. The analysis revealed that (i) the rate of mtDNA segregation does not correlate with the total recovery of liver weight (fig. S2G) and (ii) 9 of 10 regenerated livers had an increase in NZB mtDNA compared to day 0 (Fig. 4B and fig. S2G). A reproducible percentage of each haplotype was found independently of the hepatic region analyzed. Moreover, the rate per day of NZB positive selection during regeneration was one order of magnitude faster than that of liver in the absence of surgery. In conclusion, mtDNA segregation bias toward NZB haplotype is an intrinsic property of the hepatocytes that does not change qualitatively, although it is substantially accelerated upon an increase in their proliferation rate.
To test whether the direction of segregation in favor of C57 mtDNA in blood cells was also determined by the cell metabolic or replicative status, we followed the heteroplasmic proportion in different subsets of lymphocytes. Lymphocytes are highly responsive cells that shift their proliferative and metabolic programs upon natural stimulus. Lymphocytes express a variety of differentiation programs that diversify them into T cell populations that perform specific physiological roles. CD4+ or CD8+ antigens respectively defined the helper and cytotoxic classes of lymphocytes. Each class can be subdivided in naïve (TN), effector (TE), or memory (TM) T cells (14). The transition from the quiescent phase of naïve T cells to growth and proliferation requires an overall increase in mitochondrial biogenesis. However, CD4+ T cell mitochondria are enriched in one-carbon metabolism enzymes but not in OXPHOS, TCA (tricarboxylic acid) cycle, or β-oxidation components (15). On the other hand, CD8+ TN cells rely on oxidative metabolism, but their mitochondrial content and activity is low (16). CD8+ TE and TM cells increase mitochondrial mass and CD8+ TM cells significantly activate their β-oxidation metabolism (17). We therefore investigated the segregation of mtDNA between these types of lymphocytes in control (Fig. 4C) and immunized (Fig. 4D) animals (see also fig. S2, H and I). Mice were intravenously injected with the model antigen ovalbumin (OVA) combined with the invariant Natural Killer T (NKT) cell agonist IMM60 and the TLR4 agonist monophosphoryl lipid A (MPLA) (18). Seven days after vaccination, naïve and immunized mice were euthanized and TM, TE, and TN cells were sorted according to the expression of CD62L and CD44 (19), and the proportion of heteroplasmy was measured in each population (fig. S2, H and I). We confirmed that spare respiration capacity of CD8+ TM was substantially higher than that of CD8+ TN (fig. 4E). In addition, we found no differences in the proportion of heteroplasmy for CD4+ T cells from naïve to effector or memory, regardless of the immunization status. CD8+ TM in naïve and immunized mice, which mostly rely on fatty acid oxidation, systematically shifted their mtDNA content toward NZB mtDNA when compared with the CD8+ TN (Fig. 4, C and D, and fig. S2, H and I). On the other hand, CD8+ TE cells manifested an intermediate situation in naïve animals and behave closer to TM in immunized mice (Fig. 4, C and D, and fig. S2, H and I).
The maintenance of the preferred NZB mtDNA haplotype by liver upon hepatectomy, and the reversion of the segregation tendency in lymphocytes toward NZB (both shift their metabolic program to a more active fatty acid oxidation), indicate that a major determinant of mtDNA haplotype segregation is the characteristic cellular metabolic program instead of cellular proliferation rate.
mtDNA selection is strongly modulated by the nucleus-mitochondrial interaction
Thus far, we have established that the functional role of OXPHOS and its particular modulation in different cell types and cell metabolic context influence mtDNA segregation. However, most of the studies performed to date have analyzed the segregation phenomenon in a homogeneous nuclear genetic context of an inbred strain. We have previously observed several nuclear features that affect mtDNA germline segregation. Specifically, the mild alteration of the mitochondrial reactive oxygen species (ROS) signaling/detoxification due to the absence of NNT (nicotinamide nucleotide transhydrogenase), impairment of superassembly of the respiratory complexes by the lack of a functional SCAF1 (supercomplex assembly factor 1)/COX7A2L, and mild modification of the mitochondrial quality control system under stress by the ablation of OMA1 all affect mtDNA germ-line segregation (7).
To explore the role of these nuclear features on mtDNA segregation, we used three heteroplasmic mouse models: (i) mice without NNT (BL/6C57-NZB:NNTKO), (ii) mice harboring the wild-type form of SCAF1 (BL/6C57-NZB:SCAF1113), or (iii) mice without OMA1 (BL/6C57-NZB:OMA1KO) (20).
NNT is a mitochondrial inner membrane protein that catalyzes the transhydrogenation between NAD and NADP coupled to proton pumping (21). BL/6C57-NZB:NNTKO mice promoted the segregation toward NZB mtDNA while reducing the advantage of C57 mtDNA (Fig. 4F). These patterns involved a shift in the segregation in gut from C57 toward NZB mtDNA while eliminating or reducing the segregation in favor of C57 mtDNA in BAT, thymus, and spleen.
SCAF1 (also named COX7A2L) is required for the superassembly between complex III and complex IV (22, 23). We found that wild-type C57BL/6 mice carry a naturally occurring mutation in the SCAF1 gene that renders the protein nonfunctional (named SCAF1111) (22, 23). We have generated heteroplasmic individuals harboring the wild-type version of SCAF1 (SCAF1113) in the C57BL/6 nuclear background, in which its function is restored. The expression of SCAF1113 had a notable impact on mtDNA segregation behavior. In this system, heart, lung, bladder, and skeletal muscle do not show significant selection for one of the two mtDNA haplotypes (Fig. 4F). We found that WAT lost its original segregation behavior toward NZB mtDNA. Moreover, spleen, thymus, pancreas, bone marrow (BM), and gut, which segregated toward C57 mtDNA in control heteroplasmic mice, lose or strongly reduced its rate of segregation (Fig. 4F).
Last, OMA1 is a mitochondrial inner membrane metalloprotease activated upon stress that participates in the regulation of the mitochondrial cristae dynamics by the proteolytic regulation of OPA1 (24). Mitochondrial dynamics, specifically mitochondrial fission, are critical for mitochondrial quality control and mitophagy. Loss of OPA1 cleavage by OMA1 protein was demonstrated to mildly reduce mitophagy without causing an overt disease phenotype (25). This mild reduction in mitophagy is sufficient to prevent the selective degradation of impaired mitochondria (25). Our previous results in mouse embryonic fibroblasts (MEFs) demonstrated that heteroplasmy promotes a loss in mitochondria quality associated to changes in metabolism (7). We hypothesized that removal of the suboptimal mitochondria in heteroplasmy by mitophagy may be responsible for an efficient segregation. If this is the case, segregation should be sensitive to the ablation of OMA1. We focused on the OPA1 cleavage by OMA1 in liver as a tissue that strongly selects one of the mtDNA variants and in heart as tissue that cannot segregate. OPA1 presented an accumulation of cleaved short OPA1 in liver of heteroplasmic mice (fig. S2J). On the other hand, OPA1 cleavage was reduced in heteroplasmic hearts when compared to homoplasmic ones (fig. S2K). Moreover, the overall expression of OPA1 in heteroplasmic hearts was reduced and the OPA1 unprocessed band more affected was the one cleavage by OMA1 (fig. S2, J and K). Those observations strongly suggested an active OMA1-dependent mitophagy in liver, while it is impaired in heart. Thus, we studied the behavior of segregation of the different tissues and confirmed our hypothesis: The ablation of OMA1 reduces the rate of segregation in the tissues that strongly selected one mtDNA (toward NZB mtDNA in liver and C57 mtDNA in BM) (Fig. 4F), tissues that presented the strongest segregation. These experiments demonstrate that the overall mtDNA haplotype segregation behavior is strongly modulated by mild genetic differences affecting mitochondrial dynamics, ROS handling, or respiratory chain performance.
mtDNA selection is mediated by carbon source adaptation
To explore, in a more controlled situation, the segregation behavior of mtDNA haplotypes, mouse adult fibroblasts (MAFs) were generated and immortalized from the different heteroplasmic strains. BL/6C57-NZB maintained their percentage of heteroplasmy at least until the 12th passage after immortalization using 25 mM glucose as a carbon source. Basal and maximum respiratory capacity were diminished in BL/6C57-NZB MAFs regardless of whether cells were cultured under high (25 mM) or low (5 mM) glucose concentrations (Fig. 5, A and B). The production of ROS by homoplasmic cells is higher when carrying NZB mtDNA. The decrease in respiration in heteroplasmic MAFs was accompanied by an increase in mitochondrial membrane potential (MMP) (Fig. 5C) and ROS production, despite their lower respiration rate (Fig. 5D). Consequently, the extracellular acidification rate (ECAR), an indirect measurement of lactic fermentation, per oxygen consumption rate (OCR) was significantly elevated in heteroplasmic MAFs (Fig. 5E). This is a strong indicator of an enhanced glycolytic activity induction by heteroplasmy. Therefore, the presence of mtDNA heteroplasmy strongly affects the mitochondrial metabolic capacity of MAFs in culture, favoring a reduction in the oxidative metabolism.
Fig. 5. Impact of heteroplasmy on MAF metabolic performance.
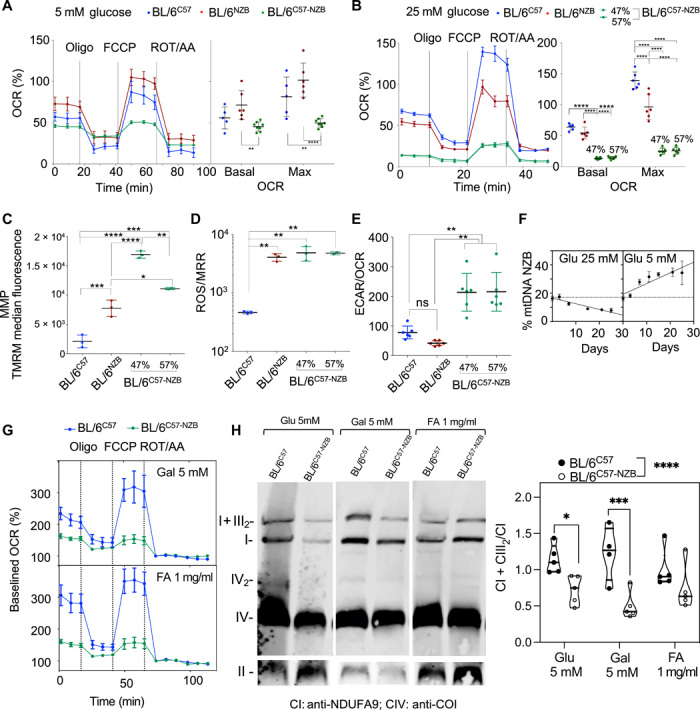
Estimation of the respiratory performance of homoplasmic and heteroplasmic MAFs at low (A) or high (B) glucose concentration. Left panels show representative profiles of OCRs determined by Seahorse analysis, and right panels show the summary of five to six independent assays per clone. Data are means ± SEM. (C) Assessment of the mitochondrial membrane potential (MMP) of the indicated cell clones. (D) ROS production normalized by the maximum respiration rate (MRR) for the indicated clone. (E) Extracellular acidification rate (ECAR) per oxygen consumption rate (OCR) as an index of glycolytic versus OXPHOS metabolism of the indicated cell clones. Data are means ± SD. Significant differences were assessed by ordinary two-way ANOVA and for multiple comparisons of the means. (F) Dynamics of mtDNA segregation in heteroplasmic MAFs under different nutritional conditions: 25 mM glucose displays linear segregation toward C57 mtDNA, and 5 mM glucose displays linear segregation toward NZB mtDNA. Data are means ± SD. (G) Evaluation of the respiratory performance of control homoplasmic and heteroplasmic MAFs under different carbon source media conditions: 5 mM glucose, 5 mM galactose, and Albumax lipid-rich BSA [fatty acids (FA), 1 mg/ml]. Data are means ± SEM. (H) Representative BNE showing the assembly and superassembly status of respiratory complexes in BL/6C57 and heteroplasmic MAFs under different nutritional conditions (left) and quantification of the proportion of free versus super-assembled complex I (right) (n = 5 in all cases except BL/6C57 (n = 4); significance assessed by two-way ANOVA with Fisher’s least significant difference post hoc test for multiple comparisons: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
We next asked whether forcing the metabolism of the cells by controlling fuel availability has the reciprocal impact in the dynamics of heteroplasmy. We found that while growing under 25 mM glucose promotes segregation bias toward C57 mtDNA, the direction shifted toward NZB mtDNA when the availability of glucose was reduced to 5 mM (Fig. 5F). Heteroplasmic cells were unable to grow for more than 72 hours in galactose or Albumax™, likely due to glucose deprivation–induced ER (endoplasmic reticulum) stress combined with the lack of functional SCAF1 (26). While homoplasmic cells efficiently respired with both substrates, heteroplasmic ones did not (Fig. 5G), which correlated with a reduction in the relative proportion of complex I in SC:I+III2 observed by blue native electrophoresis after 24 hours of treatment (Fig. 5H).
Autophagy stalling and mitochondrial network disorganization in heteroplasmic cells are linked to segregation behavior
The fact that biased mtDNA haplotype segregation is an intracellular event dependent on OXPHOS impact of the competing variants strongly suggests that mitochondrial sorting by the autophagy machinery (mitophagy) may play a relevant role in determining the “winning” mtDNA. This hypothesis was supported by analyzing the steady-state levels of a variety of autophagy protein components (fig. S3A). Thus, heteroplasmic cells have a marked reduction in the amount of LC3, and especially in the LC3I/LC3II ratio, together with an accumulation of several ATG proteins, which suggest that autophagy is stalled. This is confirmed by in situ immunodetection of LC3 (fig. S3, B and C). To determine whether the segregation phenotype was causally connected with the autophagy phenotype, we induced autophagy through different drugs—the inhibition of mTOR (mammalian target of rapamycin) with rapamycin and the activation of PERK (protein kinase R-like endoplasmic reticulum kinase) with CCT020312—and we induced the removal of mitochondria specifically by collapsing the membrane potential with CCCP (carbonyl cyanide m-chlorophenylhydrazone). PERK activation partially restores LC3 levels in heteroplasmic cells (fig. S3C). Moreover, damaging membrane potential alone did not affect the segregation behavior, while both PERK activation and mTOR inhibition abolished the segregation in favor of C57 mtDNA (fig. S3D). In agreement with that, PERK activation enhanced oxygen consumption of heteroplasmic cells, while PERK inhibition reduced it (fig. S3E). To obtain information about the mitochondrial network and dynamic status of the heteroplasmic MAFs, we performed immunodetection of TOM20 for the staining of the mitochondrial outer membrane and phalloidin for staining the F-actin (phalloidin) (fig. S3F). First, we noticed that cell shape and size varied between different heteroplasmic models. Control homoplasmic BL/6C57 MAFs showed the most elongated mitochondrial networks (fig. S3F). In BL/6C57 MAFs, mitochondria were spread through the whole cytoplasm, with a high concentration of them in the perinuclear region (fig. S3G). This distribution was disrupted in control BL/6C57-NZB heteroplasmic MAFs, where mitochondrial content was diminished, with an increase in the fragmentation and showing a more uniform distribution within the cell. Furthermore, lower clustering around the perinuclear region was observed. As with basal respiration, the activation of PERK pathway in heteroplasmic MAFs partially restored the homoplasmic features as mitochondrial content and the higher density of mitochondria surrounding the nucleus (fig. S3G). The concentration of mitochondria in these positions, assessed by the relative fluorescence intensities of TOM20, was highest than in any area of the cytoplasm (fig. S3G).
The analysis in cultured cells suggests that mtDNA segregation can be modulated by the activation of autophagy by rapamycin or PERK activation. To determine whether this would also be the case in animals, we treated heteroplasmic mice with those drugs (Fig. 6). Both drugs, in general, favored the tendency to accumulate NZB mtDNA. This confirms that modulation of autophagy could be a strategy to orientate the direction of the heteroplasmic shift.
Fig. 6. Evaluation of mtDNA dynamics using in vivo modulation of autophagy.
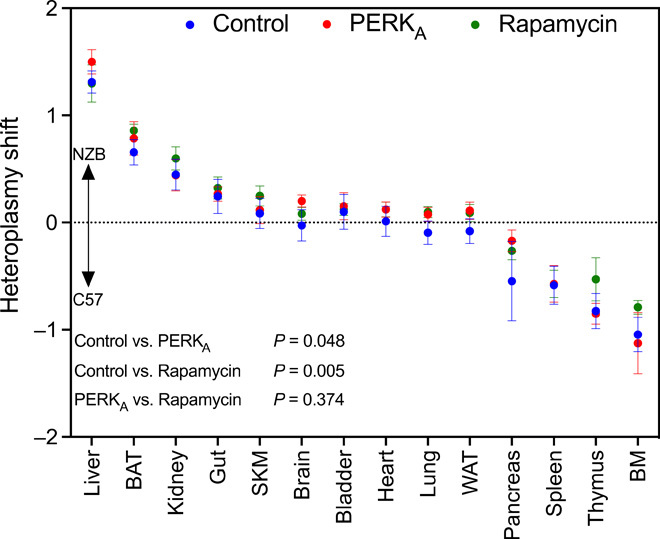
The graphic represents the heteroplasmy shift calculated using eye as reference tissue (positive toward mtDNA NZB and negative toward mtDNA C57) after 30 days of treatment with PERK activator (in red) or rapamycin (in green). Blue dots represent control heteroplasmic mice (injected with the vehicle) (n = 9 to 10 12-week-old mice per treatment). Data expressed as means ± SEM. Differences between cases were analyzed by fitting a mixed model with correction for multiple comparisons by controlling false discovery rate.
DISCUSSION
mtDNA is limited in the size and content of genetic information. Its very existence, physically separated from the rest of the genome and genetically regulated by unique rules, imposes exceptional and costly demands to the cells (27). Therefore, understanding its biology is necessary to decipher the transition from health to disease. The general instability of mtDNA heteroplasmy in most tissues, which select progressively and nonrandomly for one of the alternative variants of mtDNA, was described more than 20 years ago (3) and confirmed repeatedly (4, 5, 28, 29). However, the explanation of this robust phenomenon has remained elusive.
Here, we advance substantially in our understanding of this behavior by demonstrating that (i) biased mtDNA segregation is an intracellular event (Fig. 2); (ii) the haplotype selection is determined by the cell type (Fig. 3); (iii) the segregation phenotype can be strongly modulated by pharmacological, nutritional, and genetic determinants (Fig. 4); (iv) the driving force for nonrandom segregation relies on the different OXPHOS performance associated with each mtDNA phenotype and cell type (Fig. 4); (v) the heteroplasmic status markedly alters the metabolism of the cell (Fig. 5); (vi) heteroplasmy imposes a chronic stress situation that affects the autophagic flux (fig. S2); and (vii) in vivo and in vitro genetic and/or pharmacological modulation of autophagy can drive the direction of segregation by selective removal of mitochondria (Figs. 4 and 6 and fig. S3). Therefore, the combination of the interaction between mtDNA-encoded genes, genetic interaction with the nuclear context, and the different metabolic requirements of each cell type as well as environmental cues defines the segregation phenotype. All the collected data point to a mechanism of segregation able to sample the entire organelle rather than mtDNA molecules. The description that GIMAP3, a protein of unknown function located at the endoplasmic reticulum, contributes to mtDNA segregation in blood cells but not in other tissues (30) also suggests that it is the entire organelle that is selected.
Somatic mtDNA haplotype selection thus depends on the impact of the mtDNA haplotype on the OXPHOS system performance, with metabolic fuel preference and differential mitochondrial ROS handling playing a substantial role in the selection process. A plausible scenario has to connect fuel use, ROS signaling, mitochondrial fragmentation, and labeling for mitophagy of the organelle containing the mtDNA that has to be eliminated. Different carbon sources generate a variable proportion of NADH [reduced form of nicotinamide adenine dinucleotide (NAD+)]/FADH2 [reduced form of flavin adenine dinucleotide (FAD)] electrons to feed the mitochondrial electron transport chain (ETC). NADH gives electrons to complex I, while FADH2 circumvents complex I. The mitochondrial ETC modifies its configuration when confronted with carbon sources that generate different proportions of NADH/FADH2 electrons. This phenomenon is mediated by alterations in the CoQH2/CoQ ratio, which is elevated when the proportion of FADH2 electrons was raised, triggering ROS production by reverse electron transfer (31). This elevation in ROS induces the degradation of complex I and the release of complex III to enhance the oxidation of FADH2-derived electrons (31). This regulatory feedback between mitochondria and nucleus allows the adaptation between the demands of a given mtDNA haplotype and the requirements of the cell, minimizing the mitochondrial ROS production and maximizing OXPHOS efficiency to the predetermined metabolic program. A mechanism compatible with our observation is then as follows. The heteroplasmic status compromises this regulatory feedback mechanism, because reaching the minimum ROS per maximum performance for the organelles with NZB mtDNA would happen at the expense of losing the efficiency in the organelles with C57 mtDNA, and vice versa. Therefore, the metabolic program of different cell types offers transcriptional programs closer to the “minimum ROS/optimal OXPHOS” of either one or the other mtDNA haplotype. As a consequence, organelles with the haplotype closer to the minimum ROS/optimal OXPHOS would generate less ROS than the organelles with the alternative haplotype. Last, the less fitted organelle would have a higher likelihood to be preferentially selected for mitophagy. In agreement with this model, the ablation of NNT compromises mitochondrial ROS handling and the reexpression of functional SCAF1 enhances supercomplex formation and decreases ROS production (32), and both contexts substantially affect the haplotype segregation.
Understanding the driving forces that determine mtDNA segregation is vital to explain why heteroplasmy remains largely stable in critical tissues (i.e., skeletal muscle, heart, lung, and eye) or cell types (i.e., astrocytes and cardiomyocytes). Those are some of the most sensitive tissues to OXPHOS dysfunction. A common general feature of abundant cell populations in these tissues (myocytes, rods and cones, astrocytes, or type I pneumocytes) is the highly structured cytoplasm, which may compromise mitochondrial mobility and hence mtDNA segregation. In agreement with this, analyses of specific cell types showed that cardiomyocytes and astrocytes with a highly structured cytoplasm were unable to segregate, while lymphocytes, neutrophils, neurons, or endothelial cells did segregate. The genetic and OXPHOS functions are both coupled with the physical dynamics of mitochondria (33, 34).
Our analysis of the MAFs in culture and lymphocytes in vivo demonstrate that heteroplasmic status significantly affects the bioenergetic metabolism of the cells. Given the physiological relevance of the tissues that are unable to segregate, a deeper analysis of the metabolism and functionality of them will be of major interest. This is particularly relevant in view of the potential generation of heteroplasmy between common haplotypes in humans as a result of novel medical technologies. For example, the proposal of improving human oocyte fertility capability by injection of cytoplasm from younger donor oocytes unavoidably generates mtDNA heteroplasmy (35). A recent controversial report described three independent families where the mechanism of elimination of paternal mtDNA upon fertilization failed, generating mtDNA heteroplasmy (36). These families allowed the identification of heteroplasmic individuals where two common mtDNA variants reached proportions in blood of around 60/40, 70/30, and 50/50 heteroplasmy ratios (36). Those proportions are similar to those studied in our animal models. These examples of heteroplasmy, both naturally occurring and synthetically generated by medical interventions in humans, stress the necessity to understand heteroplasmy dynamics from a clinical perspective.
Although we are confronting nonpathological mtDNA haplotypes in heteroplasmy, our observations may also have relevance regarding mtDNA-linked diseases. Evidence obtained in cultured cells with patient-derived mtDNA mutations in heteroplasmy also found alteration in autophagy/mitophagy (37, 38), and the use of rapamycin to drive selection against mutant mtDNA in human cybrids has been reported (39). The fact that rapamycin and a number of other interventions can modulate the segregation of mtDNA heteroplasmy in animals opens the possibility to explore those modulators as potential treatments of mtDNA causing diseases.
MATERIALS AND METHODS
Experimental design
Mouse experimentation
All animal procedures conformed to EU Directive 86/609/EEC and Recommendation 2007/526/EC regarding the protection of animals used for experimental and other scientific purposes, enforced in Spanish law under Real Decreto 1201/2005. Approval of the different experimental protocols requires the estimation of the adequate sample size as well as the definition of the randomization and blinding criteria. The mice were fed a standard chow diet (5K67 LabDiet) or, when indicated, a HFD (D12492, OpenSource Diets).
Generation of heteroplasmic mice
Heteroplasmic mice were generated by electrofusing cytoplasts from conplastic BL/6NZB zygotes to recipient C57BL/6JOlaHsd (BL/6C57) one-cell embryos, cultured overnight, and transplanted as two-cell embryos into pseudo-pregnant Hsd:ICR (CD-1) females to complete development to term as previously described (3). To the best of our knowledge, no consensus rule exists for naming heteroplasmic mouse strains. Here, we propose the following designation to name heteroplasmic mouse strains: NUCLEAR GENOMEmtGENOME #1-CYTOPLASMIC GENOME #2 [i.e., CBL/6C57-NZB, a strain with the nuclear genome of C57BL/6JOlaHsd and the cytoplasmic (mitochondrial) genomes C57BL/6JOlaHsd and NZB/OlaHsd]. To simplify this term, we have called this strain BL/6C57-NZB throughout this report. The female heteroplasmic offspring (named BL/6C57-NZB) were mated with C57BL/6JOlaHsd males to prevent nuclear genetic drift in our particular mouse lines. Only the offspring of the established heteroplasmic mouse were used.
Molecular confirmation and quantification of heteroplasmy
All mice were genotyped at postnatal day 21 using DNA extracted from tail tip. Total genomic DNA was isolated from tail using REDExtract-N-Amp Tissue PCR (polymerase chain reaction) (Sigma-Aldrich). To determine the heteroplasmy levels of each tissue or cell culture, total genomic DNA was isolated using the DNeasy Blood & Tissue kit (Qiagen) from liver, lung, kidney, ovary, testis, heart (left ventricle), brain (cerebral cortex), skeletal muscle (gastrocnemius muscle), bladder, gut (duodenum), thymus, eye, pancreas, spleen, BM, BAT, WAT (gonadal adipose tissue), and MAFs.
The polymorphic G4276A nucleotide was used to genotype individual animals and tissues. This polymorphism in C57 mtDNA formed part of a Bam H1 restriction site, which is absent in NZB mtDNA. Total genomic DNA was PCR-amplified using standard conditions with the 5Prime Master Mix Kit and the following primers: 5′-AAGCTATCGGGCCCATACCCCG-3′ (3862-3884) and 5′-GTTGAGTAGAGTGAGGGATGGG-3′ (4503-4525), as follows: 95°C, 30 s; 58°C, 30 s; 72°C, 45 s for 30 cycles. A 15 μl aliquot of PCR was digested with 20 U of Bam H1 (New England Biolabs) at 37°C for 2 hours. After agarose gel electrophoresis, DNA was visualized with the Gel Doc XR+ System (Bio-Rad), and band intensity was quantified with Quantity One 1-D Analysis Software. The proportion of NZB mtDNA was calculated using the intensities of the undigested 664-bp Bam H1 fragments and was divided by the sum of the intensities of the undigested 664-bp fragments and the 414-and 250-bp Bam H1–digested fragments. To correct for heteroduplex formation, a standard curve was generated by mixing pure C57 and NZB mtDNAs. In addition, this technique was confirmed by solid-phase mini-sequencing as previously described (41).
Heteroplasmy statistical analysis
Heteroplasmy dynamics were modeled as h(t) = 1 / (1 + (1 − h0)/h0 e−βt), where h is heteroplasmy, t is time after conception, h0 is initial heteroplasmy at conception, and β is segregation rate (40). When reporting a change in heteroplasmy h with respect to a reference value h0 (for example, a previous measurement or a reference tissue), we therefore use the transformation Δh = βt = log ((h(h0–1))/(h0(h − 1))), motivated by the above model as in (40). To determine segregation rates for each dataset, the values of h0 for each sample and of β for each tissue were simultaneously inferred using a maximum likelihood approach implemented with Markov chain Monte Carlo, assuming normally distributed measurement noise, with confidence intervals derived using bootstrapping with the percentile method. To assess the influence of chemical and genetic perturbations, we took h0 to be the heteroplasmy value in eye tissue and used the linear model Δh = β0 t for control mice and Δh = β0 τ + β1 (t − τ), where β0 and β1 are rates of heteroplasmy shift before and after treatment, and treatment begins at time τ. In these cases, a significant difference between β0 and β1 implies a difference in segregation behavior between treatment and control cases. Model selection using the likelihood ratio test was performed to distinguish the most supported model. Normalization between chimeric mice values was performed using multiple testing analysis, and the Benjamini-Hochberg procedure was applied to estimate the adjusted P value.
In vivo mtDNA segregation intervention
To produce heteroplasmic mice OMA1KO, BL/6C57-NZB female were outcrossed to male OMA1KO (C57BL/6JOlaHsd background) to generate F1 heterozygote mice, which were then intercrossed to obtain a F2 OMA1KO heteroplasmic mice. OMA1 mice were genotyped as previously described (20). To produce heteroplasmic mice NNTKO, BL/6C57-NZB females were outcrossed to NNTKO males (C57BL/6J background) to generate the F1 heterozygote mice, which were then intercrossed to obtain the F2 NNTKO heteroplasmic mice. Heteroplasmic strains were maintained by outcrossing heteroplasmic females (BL/6C57-NZB: NNTKO) with BL/6C57:NNTKO males. Nnt was genotyped as previously described (7). To produce heteroplasmic mice SCAF1113, BL/6C57-NZB females were outcrossed to SCAF1113 males (C57BL/6JOlaHsd nuclear background) to generate the F1 heterozygote mice, which were then intercrossed to obtain the F2 SCAF1113 heteroplasmic mice. Heteroplasmic strains were maintained by outcrossing heteroplasmic females (BL/6C57-NZB:SCAF1113) with BL/6C57:SCAF1113 males. SCAF1 was genotyped as previously described (7). Experimental mice were fed with HFD (D12492, OpenSource Diets) from 28 days of age until being sacrificed. Mice treated with NAC or DCA were given 61.2 mM (1% g/vol) of NAC (Sigma-Aldrich) or 66.2 mM (1% g/vol) of DCA (Sigma-Aldrich) in drinking water from 28 days of age until being sacrificed. For in vivo modulation of autophagy-related pathways, heteroplasmic mice were intraperitoneally injected with vehicle, CCT020312 (0.5 mg/kg per day), or rapamycin (1.3 mg/kg per day) for 30 days until being sacrificed.
Generation of chimeric mice
Chimeric mice were generated by aggregation of eight-cell embryos (E8C) BL/6C57 and BL/6NZB. Fifteen drops of KSOM (potassium simplex optimized medium) were placed in 35-mm tissue culture dish, and all the drops were covered with a mineral oil and the dishes were stored at 37°C and 5% CO2. E8C was extracted and zona pellucida was removed using an acid Tyrode’s solution. Embryos were washed with Hepes-KSOM, placed in the KSOM drop, and incubated overnight at 37°C and 5% CO2. Those aggregations that develop to blastocyst were further transferred into pseudo-pregnant CD-1 females.
Immunomagnetic purification of neurons and astrocytes from adult brain
Brains from mice were dissociated using the Adult Mouse Brain Dissociation Kit (Miltenyi Biotec). Debris- and red blood cell (RBC)–free dissociated cells were separated using either the astrocyte-specific anti–ACSA-2 Microbead Kit (mouse) or the neuron-specific Neuron Isolation Kit (mouse), according to the manufacturer’s protocol [MACS (magnetic-activated cell sorting) technology].
Adult cardiomyocyte isolation
Animals were heparinized (1000 U per mouse) and euthanized. The hearts were surgically excised and immediately immersed in Ca2+-free ice-cold buffer containing 10 mM of the contractile inhibitor 2,3-butanedione monoxime, 13 mM NaCl, 4.7 mM KCl, 0.6 mM KH2PO4, 0.6 mM Na2HPO4, 1.2 mM MgSO4, 0.032 mM phenol red (to verify perfusion of the coronaries), 12 mM NaHCO3, 10 mM KHCO3, 10 mM Hepes, 30 mM taurine, 10 mM 2,3-butanedione monoxime, and 5 mM glucose; the pH was adjusted to 7.46 with NaOH. The heart was then cannulated and connected to a Langendorff apparatus for retrograde perfusion, with the flow rate adjusted to 3 ml/min. The cannulated heart was first perfused with perfusion buffer at 37°C for 4 min and then digested at the same temperature for 22 to 24 min with the above perfusion buffer supplemented with liberase (0.2 mg/ml; Sigma-Aldrich), trypsin (0.14 mg/ml), and Ca2+ (12.5 μM). To stop digestion, the heart was removed from the Langendorff apparatus, mechanically minced with forceps, and progressively introduced to a series of “digestion stop” solutions, which are based on the above perfusion buffer with 12.5 μM Ca2+ added and additionally contain variable percentages of fetal bovine serum (FBS): first, 10%, and then, 5%. Cells were mechanically dissociated by gentle agitation of the heart debris with a Pasteur pipette. Physiological Ca2+ levels were slowly reintroduced by applying several steps of increasing Ca2+ concentration. Heteroplasmy was assessed in single cardiomyocytes.
Quantification of heteroplasmy in cardiac cell populations
Hearts from heteroplasmic mice of different ages were collected, cut, and digested in Hanks’ balanced salt solution (HBSS) with liberase (1 U/ml, Roche) and deoxyribonuclease (DNase) I (10 mU/ml, Sigma-Aldrich) for 40 min at 37°C. After digestion, single-cell suspensions were obtained by gentle pipetting and mechanical dissociation of the remaining tissue through cell strainers (BD Falcon). Single-cell suspensions were incubated with the fluorochrome-conjugated antibodies for 15 min at 4°C. Sorting panel was designed using the following antibodies: anti-CD11b [phycoerythrin (PE), AFS98, eBioscience], CD45 [peridinin chlorophyll protein (PerCP) Cy5.5, 30-F11, eBioscience], and CD31 [allophycocyanin (APC), 390, eBioscience]. Samples were acquired using the digital flow sorter FACSAria SORP (Becton Dickinson). The FlowJo software (FlowJo LLC, Ashland, OR) was used to analyze the data. The different cardiac cell populations were sorted (myeloid cells, lymphoid cells, and endothelial cells) and collected into 1.5-ml Eppendorf tubes. Cell suspensions were centrifuged for 7 min at 1500 rpm. DNA was extracted to assess the heteroplasmy proportion using 10 μl of 50 mM NaOH, incubation of 10 min at 95°C, and neutralization with 2 μl of 1 M tris-HCl (pH 7.5).
Partial hepatectomy surgery
For partial hepatectomy, adult mice (10-week-old males) were anaesthetized using a mixture of isoflurane/oxygen. Seventy-five percent of the liver was excised, which involves removal of the medial and left lateral lobes (used for initial estimation of heteroplasmy). Liver proliferation, regeneration, and mtDNA analysis were performed at 15 days after partial hepatectomy. Transformed heteroplasmy was calculated to analyze the change in NZB mtDNA proportion before and after liver regeneration.
Assessment of heteroplasmy in immune cell populations
Adult heteroplasmic mice (15 weeks old) were euthanized, and blood and spleens were collected for analysis. Cell suspensions were obtained after chopping the spleens and incubated them with collagenase D (1 mg/ml, Sigma-Aldrich) and DNase (0.5 mg/ml, Sigma-Aldrich) for 15 min at 37°C. Enzymatic activity was inactivated with R10 media (RPMI medium, R5878, Sigma-Aldrich), 10% FBS, 2 mM l-glutamine, and penicillin/streptomycin (100 U/ml) (P0781, Sigma-Aldrich). The resulting cell suspension was filtered (70-μm strainer), resuspended in RBC lysis buffer (Qiagen), and washed before staining with the specific antibodies. Sorting panel was designed using the following antibodies: anti-Ly6G (AF647, 1A8, BioLegend), anti-CD115 (PE, M1/70, Tonbo), anti-B220 (APC-Cy7, RA36B2, BioLegend), anti-CD3 (BV510, 17A2, BioLegend), anti-CD4 (BV421, RMA4-5, BioLegend), anti-CD8 (PE-Cy7, 53-6.7, BioLegend), anti-CD62L [fluorescein isothiocyanate (FITC), MEL-14, eBioscience], and anti-CD44 (APC, IM7, BioLegend). LIVE/DEAD Near-IR (Invitrogen) was used to discard dead cells according to the manufacturer’s instructions. The concentrations of the antibodies in the final solution were specified in the manufacturer instructions. B cells, myeloid cells, monocytes, neutrophils, CD4+ T cells (naïve, memory, and effector), and CD8+ T cells (naïve, memory, and effector) were sorted and collected (20,000 to 25,000 cells per population). DNA was extracted to assess the heteroplasmy proportion using 10 μl of 50 mM NaOH, incubation of 10 min at 95°C, and neutralization with 2 μl of 1 M tris-HCl (pH 7.5). Transformed heteroplasmy was calculated using tail as the tissue of reference.
Mice immunization
Adult heteroplasmic mice (12 to 15 weeks of age) received a boost immunization intravenously using OVA, Sigma-Aldrich (800 μg per mouse), IMM60 (1 μg per mouse), and MPLA (25 μg per mouse). Mice were sacrificed and spleens were excised for analysis after 7 days of the immunization.
MAF cell lines generation and experimental conditions
Fibroblasts from homoplasmic and heteroplasmic mice were isolated from mouse ear and immortalized by transfection with pLOX-Ttag-iresTK (Addgene). Cell lines were grown in Dulbecco’s modified Eagle’s medium (Gibco) supplemented with 5% FBS, 1% penicillin-streptomycin (Lonza), and 1 mM sodium pyruvate (Sigma-Aldrich). Where indicated, the carbon source was 5 mM glucose (Sigma-Aldrich) or 5 mM galactose (Sigma-Aldrich) or Albumax lipid-rich bovine serum albumin (BSA; 1 mg/ml). For in vitro assessment of mtDNA segregation using modulators of the autophagy flux, we used 1 μM final concentration of the uncoupling agent CCCP (Sigma-Aldrich), mTOR inhibitor rapamycin (LC Laboratories), PERK activator (CCT020312, Millipore), and PERK inhibitor (GSK2606414, Millipore).
Seahorse analysis
Oxygen consumption in MAFs was measured using the XF96 MitoStress Test (Seahorse Bioscience). OCRs were normalized to cell number using CyQuant (Molecular Probes).
MMP and ROS assessment in MAFs
ROS and MMP were measured in MAFs using 2′-7′dichlorofluorescein diacetate (H2DCFDA; 0.4 μM final concentration, Thermo Fisher Scientific) and tetramethylrhodamine methyl ester perchlorate (TMRM; 100 nM final concentration, Sigma-Aldrich). A total of 10,000 events were recorded for each sample using the FACSCanto II System (BD Biosciences). All experiments were performed in triplicate. Samples were analyzed with BD FACSDiva Software, and an average of the medians and standard deviations was calculated using FlowJo Software (v10).
Blue native gel electrophoresis
Supercomplex levels and compositions were analyzed in isolated mitochondria from cells by BNE. Mitochondrial proteins were solubilized with 10% digitonin (4 g/g) (D5628, Sigma-Aldrich) and run on a 3 to 13% gradient blue native gel. The gradient gel was prepared in 1.5-mm glass plates using a gradient former connected to a peristaltic pump. Proteins were electroblotted onto polyvinylidene difluoride (PVDF) transfer membrane (Immobilon-FL, 0.45 μm, Merck Millipore, IPFL00010) for 1 hour at 100 V in transfer buffer (48 mM tris, 39 mM glycine, 20% EtOH). A Mini Trans-Blot Cell system (Bio-Rad) was used. Sea Blocking buffer (37527, Thermo Fisher Scientific) or phosphate-buffered saline (PBS) with 5% BSA was used for 1 hour at room temperature (RT) to avoid nonspecific binding of antibodies. For protein detection, antibodies were incubated with the membrane for 2 hours at RT. Secondary antibodies were incubated for 45 min at RT. The membrane was washed with PBS–0.1% Tween 20 for 5 min three times between primary and secondary antibodies, and after secondary antibodies, the last wash was only PBS. To study supercomplex assembly, the PVDF membrane was sequentially probed with complex I (anti-NDUFA9, Abcam), complex IV (anti-COI, Invitrogen), and complex II (anti-SDHA, Thermo Fisher Scientific) antibodies.
Western blot
Proteins in whole lysates were separated using SDS–polyacrylamide gel electrophoresis and transferred to PVDF membranes (Bio-Rad), which were then blocked with 5% BSA–0.1% PBS Tween 20 for 1 hour at RT. The membranes were then probed using anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (mouse monoclonal, Ab8245, Abcam) and different autophagy-related antibodies (Autophagy Antibody Sampler Kit, Cell Signaling Technology) and revealed using DyLight 680/800 secondary antibodies (Thermo Fisher Scientific, Rockland, MA). The images were acquired with the Odyssey Infrared Imaging System (LI-COR).
Immunocytochemistry
For in vitro immunofluorescence assays, cells were seeded on coverslips, fixed (4% formaldehyde) for 15 min at RT, and permeabilized in PBS containing 0.1% Triton X-100. Cells were then stained with F-actin marker (phalloidin, Life Technologies), LC3 primary antibody (Cell Signaling Technology), and secondary Alexa Fluor antibodies (Life Technologies) in blocking solution (5% BSA in PBS). Coverslips were mounted in ProLong Gold Antifade Reagent, and images were acquired using a Leica TCS SP5 confocal system. Quantitative analyses were performed using LAS AF software (Leica Microsystems, Germany) and ImageJ 1.48v (National Institutes of Health, USA).
Statistical analysis
Sample size for each measurement was based on previously reported results and varied depending on the type of data and/or study. The experiments were not randomized. The investigators were blinded to animal genotype and strain in the analysis. Unless specified, statistical analyses and graphics were produced with GraphPad Prism 8 software. Datasets were compared by t test, analysis of variance (ANOVA), or nonparametric analysis, as corresponding to the structure of the data, with P values adjusted for multiple tests. Differences were considered statistically significant at P values below 0.05. Unless otherwise stated, *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. All results are presented as means ± SD or means ± SEM as stated.
Supplementary Material
Acknowledgments
We thank C. López-Otín for the gift of the OMA1KO mice; M. M. Muñoz-Hernandez, A. Gonzalez-Guerra, I. Martinez-Carrascoso, C. Jimenez, and E. R. Martínez-Jimenez for technical assistance; the WIMM and CNIC Flow Cytometry Facility for technical support; and M. Cueva and R. Alvarez for mouse work. Funding: This study was supported by MINECO SAF2015-65633-R to JAE, SAF2016-78114-R to J.P.B. and SAF2017-84494-C2-1-R to J.R.-C. G.S. thanks MINECO-FEDER SAF2016-79126-R and Comunidad de Madrid IMMUNOTHERCAN-CM B2017/BMD-3733. N.S.J. thanks EP/N014529/1. CIBERFES (CB16/10/00282) to J.P.B. and J.A.E., and H2020 European Commission (BatCure grant 666918) to J.P.B. A.V.L.-V. was supported by SOFPI-fellowship from the MINECO. V.C. is supported by the UK Medical Research Council, Cancer Research UK (CRUK) (C399/A2291), and the Oxford Biomedical Research Centre. I.G.J. thanks the European Research Council (ERC- No. 805046-EvoConBiO). The CNIC is supported by MINECO and Pro-CNIC Foundation and is a SO-MINECO (award SEV-2015-0505). CIC biomaGUNE is supported by the Maria de Maeztu Units of Excellence Program from the Spanish State Research Agency (grant no. MDM-2017-0720), ELKARTEK Program (grant no. KK-2019/bmG19), and BBVA Foundation (Ayudas a Equipos de investigación científica Biomedicina 2018). Author contributions: A.V.L.-V. and A.L.-P. performed most of the mouse and cellular experimental work with R.J.-M. and R.M.-d.-M. I.G.J. and N.S.J. performed the data analysis and mathematical modeling. J.M.F.-T. and L.M.C. generated the heteroplasmic and chimeric animals. G.P. and U.G. performed the immune cell analysis in naïve and immunized mice. A.M. and G.S. performed the hepatectomy analysis. D.J.-B. and J.P.B. performed brain cell acute separation from the mice for DNA analysis. S.G.P. and D.J.S. performed the isolation of individual cardiomyocytes. J.A.N.-A. performed heart cellular sorting. R.A.-P. performed Seahorse analysis of the MAFs. J.A.E., A.V.L.-V., A.L.-P., V.C., and J.R.-C. participated in the design of the experimental work and the integrated analysis of the results and the writing of the manuscript. J.A.E. directed and designed the research. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data are present in the paper and/or the Supplementary Materials. Cell and mouse lines generated in this work can be requested to the corresponding author and will be delivered through a materials transfer agreement.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/31/eaba5345/DC1
REFERENCES AND NOTES
- 1.Lomax M. I., Grossman L. I., Tissue-specific genes for respiratory proteins. Trends Biochem. Sci. 14, 501–503 (1989). [DOI] [PubMed] [Google Scholar]
- 2.Greiner S., Sobanski J., Bock R., Why are most organelle genomes transmitted maternally? Bioessays 37, 80–94 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jenuth J., Peterson A., Shoubridge E. A., Tissue-specific selection for different mtDNA genotypes in heteroplasmic mice. Nat. Genet. 16, 93–95 (1997). [DOI] [PubMed] [Google Scholar]
- 4.Sharpley M. S., Marciniak C., Eckel-Mahan K., McManus M., Crimi M., Waymire K., Lin C., Masubuchi S., Friend N., Koike M., Chalkia D., MacGregor G., Sassone-Corsi P., Wallace D. C., Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition. Cell 151, 333–343 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burgstaller J., Johnston I. G., Jones N. S., Albrechtová J., Kolbe T., Vogl C., Futschik A., Mayrhofer C., Klein D., Sabitzer S., Blattner M., Gülly C., Poulton J., Rülicke T., Pialek J., Steinborn R., Brem G., mtDNA segregation in heteroplasmic tissues is common in vivo and modulated by haplotype differences and developmental stage. Cell Rep. 7, 2031–2041 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Latorre-Pellicer A., Moreno-Loshuertos R., Sánchez-Cabo F., Acín-Peréz R., Calvo E., Logan A., Cruz R., Cogliati S., Carracedo A., Pérez-Martos A., Fernández-Silva P., Murphy M. P., Flores I., Vázquez J., Enriquez J. A., Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 535, 561–565 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Latorre-Pellicer A., Lechuga-Vieco A., Johnston I. G., Hämäläinen R. H., Pellico J., Justo R., Fernandez-Toro J., Clavería C., Guaras A. M., Llop J., Torres M., Criado L., Suomalainen A., Jones N. S., Ruiz-Cabello J., Enriquez J. A., Regulation of mother-to-offspring transmission of mtDNA heteroplasmy. Cell Metab. 30, 1120–1130.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bélanger M., Allaman I., Magistretti P. J., Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 14, 724–738 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Bolaños J., Bioenergetics and redox adaptations of astrocytes to neuronal activity. J. Neurochem. 139, 115–125 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herrero-Mendez A., Almeida A., Fernández E., Maestre C., Moncada S., Bolaños J., The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C–Cdh1. Nat. Cell Biol. 11, 747–752 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Lopez-Fabuel I., Douce J. L., Logan A., James A. M., Bonvento G., Murphy M. P., Almeida A., Bolaños J. P., Complex I assembly into supercomplexes determines differential mitochondrial ROS production in neurons and astrocytes. Proc. Natl. Acad. Sci. U.S.A. 113, 13063–13068 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferri D., Moro L., Mastrodonato M., Capuano F., Marra E., Liquori G. E., Greco M., Ultrastructural zonal heterogeneity of hepatocytes and mitochondria within the hepatic acinus during liver regeneration after partial hepatectomy. Biol. Cell 97, 277–288 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Huang J., Rudnick D. A., Elucidating the metabolic regulation of liver regeneration. Am. J. Pathol. 184, 309–321 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geltink R. I. K., O’Sullivan D., Corrado M., Bremser A., Buck M. D., Buescher J. M., Firat E., Zhu X., Niedermann G., Caputa G., Kelly B., Warthorst U., Rensing-Ehl A., Kyle R. L., Vandersarren L., Curtis J. D., Patterson A. E., Lawless S., Grzes K., Qiu J., Sanin D. E., Kretz O., Huber T. B., Janssens S., Lambrecht B. N., Rambold A. S., Pearce E. J., Pearce E. L., Mitochondrial priming by CD28. Cell 171, 385–397.e11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ron-Harel N., Santos D., Ghergurovich J. M., Sage P. T., Reddy A., Lovitch S. B., Dephoure N., Satterstrom K. F., Sheffer M., Spinelli J. B., Gygi S., Rabinowitz J. D., Sharpe A. H., Haigis M. C., Mitochondrial biogenesis and proteome remodeling promote one-carbon metabolism for T cell activation. Cell Metab. 24, 104–117 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cao Y., Rathmell J. C., Macintyre A. N., Metabolic reprogramming towards aerobic glycolysis correlates with greater proliferative ability and resistance to metabolic inhibition in CD8 versus CD4 T cells. PLOS ONE 9, e104104 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van der Windt G. J. W., Everts B., Chang C.-H., Curtis J. D., Freitas T. C., Amiel E., Pearce E. J., Pearce E. L., Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 36, 68–78 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jukes J.-P., Gileadi U., Ghadbane H., Yu T.-F., Shepherd D., Cox L. R., Besra G. S., Cerundolo V., Non-glycosidic compounds can stimulate both human and mouse i NKT cells. Eur. J. Immunol. 46, 1224–1234 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaech S., Hemby S., Kersh E., Ahmed R., Molecular and functional profiling of memory CD8 T cell differentiation. Cell 111, 837–851 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Quirós P. M., Ramsay A. J., Sala D., Fernández-Vizarra E., Rodríguez F., Peinado J. R., Fernández-García M., Vega J. A., Enríquez J. A., Zorzano A., López-Otín C., Loss of mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective thermogenesis in mice. EMBO J. 31, 2117–2133 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoek J. B., Rydström J., Physiological roles of nicotinamide nucleotide transhydrogenase. J. Biochem. 254, 1–10 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lapuente-Brun E., Moreno-Loshuertos R., Acín-Pérez R., Latorre-Pellicer A., Colás C., Balsa E., Perales-Clemente E., Quirós P. M., Calvo E., Rodríguez-Hernández M. A., Navas P., Cruz R., Carracedo Á., López-Otín C., Pérez-Martos A., Fernández-Silva P., Fernández-Vizarra E., Enríquez J. A., Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 340, 1567–1570 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Cogliati S., Calvo E., Loureiro M., Guaras A. M., Nieto-Arellano R., Garcia-Poyatos C., Ezkurdia I., Mercader N., Vázquez J., Enriquez J. A., Mechanism of super-assembly of respiratory complexes III and IV. Nature 539, 579–582 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Käser M., Kambacheld M., Kisters-Woike B., Langer T., Oma1, a novel membrane-bound metallopeptidase in mitochondria with activities overlapping with the m-AAA Protease. J. Biol. Chem. 278, 46414–46423 (2003). [DOI] [PubMed] [Google Scholar]
- 25.MacVicar T. D. B., Lane J. D., Impaired OMA1-dependent cleavage of OPA1 and reduced DRP1 fission activity combine to prevent mitophagy in cells that are dependent on oxidative phosphorylation. J. Cell Sci. 127, 2313–2325 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balsa E., Soustek M. S., Thomas A., Cogliati S., García-Poyatos C., Martín-García E., Jedrychowski M., Gygi S. P., Enriquez J., Puigserver P., ER and nutrient stress promote assembly of respiratory chain supercomplexes through the PERK-eIF2α axis. Mol. Cell 74, 877–890.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoitzing H., Gammage P. A., Van Haute L., Minczuk M., Johnston I. G., Jones N. S., Energetic costs of cellular and therapeutic control of stochastic mitochondrial DNA populations. PLOS Comput. Biol. 15, e1007023 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meirelles F. V., Smith L. C., Mitochondrial genotype segregation during preimplantation development in mouse heteroplasmic embryos. Genetics 148, 877–883 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takeda K., Takahashi S., Onishi A., Hanada H., Imai H., Replicative advantage and tissue-specific segregation of RR mitochondrial DNA between C57BL/6 and RR heteroplasmic mice. Genetics 155, 777–783 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Battersby B. J., Loredo-Osti J. C., Shoubridge E. A., Nuclear genetic control of mitochondrial DNA segregation. Nat. Genet. 33, 183–186 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Guarás A., Perales-Clemente E., Calvo E., Acín-Peréz R., Loureiro-Lopez M., Pujol C., Martínez-Carrascoso I., Nuñez E., García-Marqués F., Rodríguez-Hernández M., Cortés A., Diaz F., Pérez-Martos A., Moraes C. T., Fernández-Silva P., Trifunovic A., Navas P., Vázquez J., Enriquez J. A., The CoQH2/CoQ ratio serves as a sensor of respiratory chain. Cell Rep. 15, 197–209 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Maranzana E., Barbero G., Falasca A. I., Lenaz G., Genova M. L., Mitochondrial respiratory supercomplex association limits production of reactive oxygen species from complex I. Antioxid. Redox Signal. 19, 1469–1480 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnston I. G., Tension and resolution: Dynamic, evolving populations of organelle genomes within plant cells. Mol. Plant 12, 764–783 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Cogliati S., Frezza C., Soriano M. E., Varanita T., Quintana-Cabrera R., Corrado M., Cipolat S., Costa V., Casarin A., Gomes L. C., Perales-Clemente E., Salviati L., Fernández-Silva P., Enriquez J. A., Scorrano L., Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155, 160–171 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barritt J. A., Willadsen S., Brenner C., Cohen J., Cytoplasmic transfer in assisted reproduction. Hum. Reprod. Update 7, 428–435 (2001). [DOI] [PubMed] [Google Scholar]
- 36.Luo S., Valencia C. A., Zhang J., Lee N.-C., Slone J., Gui B., Wang X., Li Z., Dell S., Brown J., Chen S. M., Chien Y.-H., Hwu W.-L., Fan P.-C., Wong L.-J., Atwal P. S., Huang T., Biparental inheritance of mitochondrial DNA in humans. Proc. Natl. Acad. Sci. U.S.A. 115, 13039–13044 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gilkerson R. W., De Vries R. L. A., Lebot P., Wikstrom J. D., Torgyekes E., Shirihai O. S., Przedborski S., Schon E. A., Mitochondrial autophagy in cells with mtDNA mutations results from synergistic loss of transmembrane potential and mTORC1 inhibition. Hum. Mol. Genet. 21, 978–990 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suen D.-F., Narendra D. P., Tanaka A., Manfredi G., Youle R. J., Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc. Natl. Acad. Sci. U.S.A. 107, 11835–11840 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dai Y., Zheng K., Clark J., Swerdlow R. H., Pulst S. M., Sutton J. P., Shinobu L. A., Simon D. K., Rapamycin drives selection against a pathogenic heteroplasmic mitochondrial DNA mutation. Hum. Mol. Genet. 23, 637–647 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnston I. G., Jones N. S., Evolution of cell-to-cell variability in stochastic, controlled, Heteroplasmic mtDNA Populations. Am. J. Hum. Genet. 99, 1150–1162 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wartiovaara A., Syvänen A. C., Analysis of nucleotide sequence variations by solid-phase minisequencing. Methods Mol. Biol. 187, 57–63 (2002). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/31/eaba5345/DC1