

Abstract
Reports of C–H insertions forming six-membered rings containing heteroatoms are rare due to Stevens rearrangements occurring after nucleophilic attack on the carbene by a heteroatom. Using donor/donor carbenes and Rh2(R-PTAD)4 as a catalyst, we have synthesized a collection of isochroman substrates in good yield, with excellent diastereo- and enantioselectivity, and no rearrangement products were observed. Furthermore, we report the first synthesis of six-membered rings containing nitrogen by C–H insertion to form tetrahydroisoquinolines. In one case, a Stevens rearrangement product was isolated at elevated temperature from a carbamate-protected amine substrate and computational evidence suggests formation through a free ylide not bound to rhodium.
Six-membered ring oxygen and nitrogen heterocycles are formed stereoselectively by C–H insertion of donor/donor carbenes.
Insertion reactions of metal carbenes into C–H bonds are useful methods for making C–C bonds.1–6 In most cases, C–H insertion reactions are catalyzed with dirhodium tetracarboxylate complexes, often using chiral ligands to provide enantioselectivity.1,6–8 Asymmetric C–H insertion has been well-documented for carbenes with two electron withdrawing substituents (CO2R, COR, CN, SO2R, acceptor/acceptor)7 or with one electron donating (aryl, alkenyl) and one electron withdrawing group (donor/acceptor).9 Acceptor and acceptor/acceptor carbenes are very reactive and can react indiscriminately with other functionalities on the molecule.7,10,11 Donor/acceptor carbenes are less electrophilic due to the donating substituent, which increases selectivity for C–H insertion (Fig. 1).3,5,9 Recently, we have been interested in donor/donor carbenes,12–14 which have two electron-donating groups and, as a result, are highly selective for C–H insertion while also tolerating many functional groups present in complex substrates.15,16 Although C–H insertion reactions have been used in the synthesis of a wide variety of five-membered ring (1,5-C–H insertion) heterocycles,1,7,10 there are few examples of six-membered ring (1,6-C–H insertion) formation and the substrates are limited to oxygen-containing rings.17–24
Fig. 1. Six-membered ring formation has proven difficult in the past due to the kinetic favorability of five-membered ring insertions and the possibility of Stevens rearrangement products. By utilizing donor/donor carbenes, which are less electrophilic than donor/acceptor or acceptor/acceptor carbenes, the Stevens rearrangement product is not observed.

Accessing 1,6-C–H insertion is difficult due to the kinetic favorability of 1,5-C–H insertion and the potential for rearrangement products when heteroatoms are present (Fig. 1). Previous work to synthesize 1,6-C–H insertion products without heteroatoms consistently observed mixtures of five- and six-membered ring products when a 1,5-C–H insertion site was available.25–29 Installing heteroatoms can eliminate 1,5-C–H insertion however this introduces the possibility of nucleophilic attack on the carbene by the heteroatom.17,19,24 This reaction forms an ylide that undergoes the Stevens rearrangement (or a Stevens-type rearrangement when the heteroatom is oxygen), forming a five-membered ring (5).24,30 Despite these challenges, there are scattered reports of forming tetrahydropyran, chroman, and chromanone cores through 1,6-C–H insertion using donor/acceptor or acceptor carbenes.18,20,22–24,31–33 In 2012, Cossy and coworkers demonstrated the use of donor carbenes generated from cyclopropenes in the synthesis of tetrahydropyrans by C–H insertion,22,31 further expanding the diazo-free insertion work demonstrated by Zhu.34,35 In the cases where chiral catalysts were used, enantioselectivity was often moderate;19,20 it wasn't until 2015 when Hashimoto achieved higher levels of enantioselectivity using Rh2(S-PTTL)4 (up to 97 : 3 er).24 We hypothesized that using donor/donor carbenes would reduce the electrophilicity of the rhodium carbene enough to allow C–H insertion to proceed selectively without interference by the heteroatom.
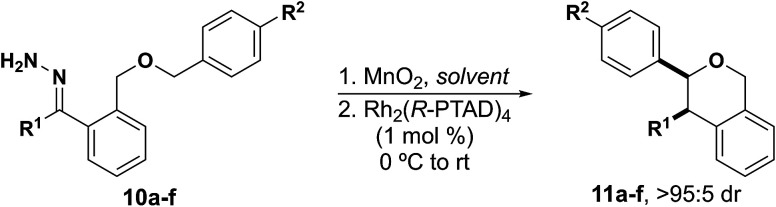
Due to the propensity of C–H insertions to form five-membered rings, we designed substrates that would eliminate the possibility for 1,5-insertion by installing the requisite heteroatom at the 5-position. We started by developing substrates with benzylic insertion sites, as we had found those to be favored with benzodihydrofuran systems.15 We were pleased to find that hydrazone 10a was successfully oxidized to the diazo with eight equivalents of MnO2 and inserted into the benzylic C–H bond with 1 mol percent catalyst loading affording a single diastereomer, with excellent enantioselectivity, and with no observed rearrangement product (Table 1, entry 1).
Insertion into benzylic C–H Bonds.
| ||||||
|---|---|---|---|---|---|---|
| Entry | Product | R1 | R2 | Solvent | Yield (%) | er |
| 1 | 11a | Ph | H | CH2Cl2 | 60 | 97 : 3 |
| 2 | 11b a | Ph | OCH3 | CH2Cl2 | 69 | >99.5 : 0.5 |
| 3 | 11c | 4-CNC6H4 | OCH3 | CH3CN | 97 | 99 : 1 |
| 4b | 11d | 4-H3COC6H4 | OCH3 | CH2Cl2 | 98 | 99 : 1 |
| 5 | 11e | 3-Pyridyl | OCH3 | CH3CN | 82 | 94 : 6 |
| 6 | 11f | CH3 | OCH3 | CH2Cl2 | 56 | 60 : 40 |
With Rh2(S-PTAD)4 as catalyst.
Catalyst added at rt.
With this result we were encouraged to expand the scope of benzylic insertions to form isochromans. We saw a modest increase in yield and enantioselectivity when a methoxy group was installed para to the insertion site (Table 1, entry 2). We then wanted to explore electronic effects at the carbene center and interestingly found no discernible effect when there was a nitrile group versus a methoxy group para to the carbene (Table entries 3, 4). Furthermore, heterocycles with a Lewis basic moiety were also well-tolerated as substituents off of the carbene (Table 1, entry 5). Encouraged by these results we wanted to explore the effect of alkyl substituents on the carbene in combination with an aryl substituent (Table 1, entry 6). The yield was slightly lower, and we saw a significant decrease in enantioselectivity, suggesting that the other aryl ring plays a role in fitting the substrate into the chiral catalyst pocket.
Allylic and propargylic ethers were excellent substrates for C–H insertion (Fig. 2). Although the analogous substrates for the synthesis of benzodihydrofurans can suffer from competing dipolar cycloaddition reactions,36,37 this side reaction was not observed in the reactions leading to isochromans. Stevens-type rearrangement products were also not observed for unsaturated substrates. Notably, 12a did not undergo any Stevens-type rearrangement under reaction conditions, in contrast to a similar substrate derived from a donor/acceptor carbene in work by Hashimoto and co-workers.24 These results appear to support our hypothesis that the reduced electrophilicity of the donor/donor carbene prevents the competing rearrangement reaction. Additionally, while an unsubstituted allyl group underwent insertion with modest yield, the addition of a single substituent resulted in yields of 90% or greater. The crystal structure of 13b demonstrated that the absolute configuration produced with Rh2(R-PTAD)4 is the same for isochromans as was observed with benzodihydrofurans and related heterocycles.15,16,38Cis and trans alkene hydrazones 13d and 13e led to their respective insertion products with no detectable isomerization. A propargyl substituted insertion site proved to be the lowest yielding isochroman (13f). Although the yield was comparatively lower than the other unsaturated substrates, no evidence of dipolar cycloaddition was observed. We hypothesize that a larger ring size helps to limit the dipolar cycloaddition pathway.36,37
Fig. 2. Unsaturated isochroman substrates.

Aliphatic substrates also react with high diastereo- and enantioselectivity. There is a clear trend of decreasing yield from substrates that have two alkyl substituents versus one alkyl substituent at the insertion site. Isochromans 15a–c were synthesized in excellent yield with insertion into a carbon with two alkyl substituents (Fig. 3). When inserting into a carbon with one alkyl substituent the yields decreased significantly (15d–e, 54–62%) indicating some substrate preference for inserting into more highly substituted carbons. Importantly, throughout the isochroman examples, all products were formed as a single diastereomer and no Stevens-type rearrangement products were observed.
Fig. 3. Aliphatic isochroman substrates.a Run with Rh2(S-PTAD)4.

With the formation of six-membered rings comes the opportunity to explore the influence of 1,3-diaxial interactions in the diastereoselective formation of the stereogenic centers that form during insertion (Fig. 4). A single diastereomer (17a) was observed from hydrazone 16a using Rh2(Mes)4 as the catalyst, the configuration of which was determined by a NOE NMR experiment. This indicates that the C–H insertion step occurs in a diastereoselective manner despite having a bulky group close to the rhodium carbene. Furthermore, compounds 17b and 17c were also isolated in good yields with excellent diastereoselectivity when Rh2(R-TCPTTL)4 was used as the catalyst to accelerate the rate of C–H insertion. The chiral phthalimido catalysts generally exhibit higher activity than any of the achiral catalysts used with donor/donor carbenes. The structure of compound 17c was also determined by X-ray diffraction.38
Fig. 4. Diastereoselective insertions to form tri-substituted isochromans.a With Rh2(Mes)4.b With Rh2(R-TCPTTL)4.

Our success in synthesizing isochromans motivated our attempts to synthesize the more challenging nitrogen analogues. To the best of the authors' knowledge, 1,6-C–H insertion on aliphatic systems has never been done to synthesize nitrogen-containing heterocycles. Jefford and coworkers demonstrated related work involving a C(sp2)–H insertion on pyrrole using an acceptor carbene.39–41 Using our prior work to synthesize indolines as inspiration,16 we first developed amine- and aniline-based substrates. These substrates proved unsuccessful as the crude reactions were often complex mixtures with no discernible traces of product. In order to reduce the possibility of side reactions, N-sulfonyl groups were installed to reduce the nucleophilicity of the nitrogen lone pair. No identifiable products were isolated with these substrates.
Computational evidence suggests that donor/donor C–H insertions forming benzodihydrofurans proceed through a stepwise mechanism that has a zwitterionic intermediate in which a carbocation is formed on the insertion carbon.36 Based on this observation we reasoned that a sulfonyl protecting group on nitrogen might be too electron withdrawing, to the detriment of stabilizing a possible carbocation intermediate. We hypothesized that an amide would reduce the nucleophilicity of the nitrogen but to a lesser extent than a sulfonyl protecting group.
We next attempted substrates using lactams and were pleased to synthesize tetrahydroisoquinoline 23a as a single diastereomer in 54% yield and with 99 : 1 er (Fig. 5). We went on to synthesize tetrahydroisoquinoline 23b in good yield and with slightly reduced er. Carbamate derived tetrahydroisoquinoline 23c was also synthesized in moderate yield and with excellent stereoselectivity. X-ray crystallography of 23c showed the same absolute stereochemistry as is observed with isochromans.38 As these insertions occur rapidly, we found that reducing the temperature before the addition of catalyst allowed the reaction to complete within 30 minutes to 3 hours while producing a cleaner reaction mixture.
Fig. 5. Scope of tetrahydroisoquinoline substrates.

In analogy to our work with indolines, in which insertion into methyl C–H bonds was possible,16 we attempted to synthesize a mono-substituted tetrahydroisoquinoline. We synthesized hydrazone 24 with a carbamate protecting group, which we reasoned would provide roughly an equivalent level of electron withdrawing potential as an amide and is easily removed for further functionalization. This substrate reacted much more slowly and eventually required heating to reflux for the diazo to be fully consumed. Upon consumption of diazo no insertion product was observed. The sole identifiable product was isoindoline 26, resulting from an apparent Stevens [1,2]-rearrangement (Fig. 6A).42,43 A similar product involving attack by an amide nitrogen leading to Stevens rearrangement was observed by Padwa.42
Fig. 6. (A) Stevens rearrangement product synthesis. (B) The DFT (uB3LYP/LANL2DZ[6-31G(d)]) computed mechanism suggests that N-attack to the rhodium carbene and the subsequent Stevens rearrangement is energetically feasible at experimental conditions; relative free energies (electronic energies in parentheses) for metal-bound (normal text) and ylide (italics) reactions are reported in kcal mol−1.

After identifying the isoindoline as the primary product of the reaction using Rh2(R-PTAD)4, a second run using Rh2(TFA)4 proceeded in 50% yield. The isolation of Stevens product 26 indicates that the properties of the insertion site are as important as the electron withdrawing effect on the nitrogen, given that tetrahydroisoquinoline 23c was isolated from insertion of hydrazone 22c. Although previous studies propose a stepwise diradical mechanism for the Stevens rearrangement,43 density functional theory (DFT) studies support N-attack to the metal carbene, but then a concerted free ylide Stevens rearrangement (Fig. 6B).44 This model suggests that the metal catalyst is not essential in the Stevens rearrangement step.45
Conclusions
In conclusion, we have used donor/donor rhodium carbenes to synthesize isochromans in good to excellent yields and with overall excellent stereoselectivity. For isochroman substrates, no Stevens-type rearrangement products were observed. Furthermore, we also explored the synthesis of isochromans in a diastereoselective fashion by installing a substituent on the benzylic carbon alpha to oxygen and were gratified to see a single diastereomer formed. Additionally, we synthesized the first six-membered rings containing nitrogen through 1,6-C(sp3)–H insertion in moderate yields and with excellent stereoselectivity. A Stevens rearrangement product was isolated from one substrate using increased temperature and computational evidence suggests that isoindoline formation occurs via a free ylide not bound to rhodium.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health (R01/GM124234). Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R01GM124234. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. LAN and BDB acknowledge UC Davis for financial support in the form of Borge and Corson/DOW fellowships. MRC thanks the California Alliance for Minority Participation (CAMP) for financial support (NSF grant 1826900). We thank the Franz group for providing access to a chiral HPLC and particularly Austin Kelly and Jake Jagannathan (Franz group, UC Davis) for providing assistance with HPLC traces. We also thank the Kurth group, (UC Davis) for use of their IR with assistance from Winston Chow (Olson group, UC Davis). We thank the National Science Foundation (Grant CHE-1531193) for the Dual Source X-ray diffractometer. We gratefully acknowledge support from the XSEDE program via TG-CHE050017N for computational resources.
Electronic supplementary information (ESI) available: Synthetic details, spectroscopic characterization and crystallographic data. CCDC 1921673, 1921674, 1916890 and 1920825. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05111b
Notes and references
- Doyle M. P. Ratnikov M. Liu Y. Org. Biomol. Chem. 2011;9:4007–4016. doi: 10.1039/C0OB00698J. [DOI] [PubMed] [Google Scholar]
- Doyle M. P. Duffy R. Ratnikov M. Zhou L. Chem. Rev. 2010;110:704–724. doi: 10.1021/cr900239n. [DOI] [PubMed] [Google Scholar]
- Davies H. M. L. Denton J. R. Chem. Soc. Rev. 2009;38:3061–3071. doi: 10.1039/B901170F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H. M. L. Manning J. R. Nature. 2008;451:417–424. doi: 10.1038/nature06485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H. M. L. Morton D. Chem. Soc. Rev. 2011;40:1857–1869. doi: 10.1039/C0CS00217H. [DOI] [PubMed] [Google Scholar]
- Davies H. M. L. and Pelphrey P. M., in Organic Reactions, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2011, pp. 75–212 [Google Scholar]
- Davies H. M. L. Beckwith R. E. J. Chem. Rev. 2003;103:2861–2904. doi: 10.1021/cr0200217. [DOI] [PubMed] [Google Scholar]
- Davies H. M. L. Hansen T. J. Am. Chem. Soc. 1997;119:9075–9076. doi: 10.1021/ja971915p. [DOI] [Google Scholar]
- Davies H. M. Loe Ø. Synthesis. 2004;16:2595–2608. doi: 10.1055/s-2004-834876. [DOI] [Google Scholar]
- Sulikowski G. A. Cha K. L. Sulikowski M. M. Tetrahedron: Asymmetry. 1998;9:3145–3169. doi: 10.1016/S0957-4166(98)00342-5. [DOI] [Google Scholar]
- Doyle M. P., McKervey M. A. and Ye T., in Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides, Wiley, 1998, pp. 112–162 [Google Scholar]
- Seidel G. Fürstner A. Angew. Chem., Int. Ed. 2014;53:4807–4811. doi: 10.1002/anie.201402080. [DOI] [PubMed] [Google Scholar]
- Werlé C. Goddard R. Philipps P. Farès C. Fürstner A. J. Am. Chem. Soc. 2016;138:3797–3805. doi: 10.1021/jacs.5b13321. [DOI] [PubMed] [Google Scholar]
- Werlé C. Goddard R. Philipps P. Farès C. Fürstner A. Angew. Chem., Int. Ed. 2016;55:10760–10765. doi: 10.1002/anie.201605502. [DOI] [PubMed] [Google Scholar]
- Soldi C. Lamb K. N. Squitieri R. A. González-López M. Di Maso M. J. Shaw J. T. J. Am. Chem. Soc. 2014;136:15142–15145. doi: 10.1021/ja508586t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza L. W. Squitieri R. A. Dimirjian C. A. Hodur B. M. Nickerson L. A. Penrod C. N. Cordova J. Fettinger J. C. Shaw J. T. Angew. Chem., Int. Ed. 2018;57:15213–15216. doi: 10.1002/anie.201809344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy N. McKervey M. A. Ye T. McCann M. Murphy E. Doyle M. P. Tetrahedron Lett. 1992;33:5983–5986. doi: 10.1016/S0040-4039(00)61106-1. [DOI] [Google Scholar]
- Lee E. Choi I. Song S. Y. J. Chem. Soc., Chem. Commun. 1995:321–322. doi: 10.1039/C39950000321. [DOI] [Google Scholar]
- Ye T. García C. F. McKervey M. A. J. Chem. Soc., Perkin Trans. 1. 1995:1373–1379. doi: 10.1039/P19950001373. [DOI] [Google Scholar]
- Rosales A. Rodríguez-García I. López-Sánchez C. Álvarez-Corral M. Muñoz-Dorado M. Tetrahedron. 2011;67:3071–3075. doi: 10.1016/j.tet.2011.03.004. [DOI] [Google Scholar]
- Archambeau A. Miege F. Meyer C. Cossy J. Angew. Chem., Int. Ed. 2012;51:11540–11544. doi: 10.1002/anie.201205913. [DOI] [PubMed] [Google Scholar]
- Archambeau A. Miege F. Meyer C. Cossy J. Angew. Chem. 2012;124:11540–11544. doi: 10.1002/anie.201205913. [DOI] [PubMed] [Google Scholar]
- Taber D. F. Paquette C. M. Gu P. Tian W. J. Org. Chem. 2013;78:9772–9780. doi: 10.1021/jo4014996. [DOI] [PubMed] [Google Scholar]
- Ito M. Kondo Y. Nambu H. Anada M. Takeda K. Hashimoto S. Tetrahedron Lett. 2015;56:1397–1400. doi: 10.1016/j.tetlet.2015.01.125. [DOI] [Google Scholar]
- Estevan F. Herbst K. Lahuerta P. Barberis M. Pérez-Prieto J. Organometallics. 2001;20:950–957. doi: 10.1021/om0010060. [DOI] [Google Scholar]
- Slattery C. Clarke L.-A. O'Neill S. Ring A. Ford A. Maguire A. Synlett. 2012;23:765–767. doi: 10.1055/s-0031-1290598. [DOI] [Google Scholar]
- Franck-Neumann M. Geoffroy P. Gassmann D. Winling A. Tetrahedron Lett. 2004;45:5407–5410. doi: 10.1016/j.tetlet.2004.05.052. [DOI] [Google Scholar]
- Aburel P. S. Rømming C. Undheim K. J. Chem. Soc., Perkin Trans. 1. 2001:1024–1029. doi: 10.1039/B100179P. [DOI] [Google Scholar]
- West F. G. Eberlein T. H. Tester R. W. J. Chem. Soc., Perkin Trans. 1. 1993:2857. doi: 10.1039/P19930002857. [DOI] [Google Scholar]
- Doyle M. P. Ene D. G. Forbes D. C. Tedrow J. S. Tetrahedron Lett. 1997;38:4367–4370. doi: 10.1016/S0040-4039(97)00930-1. [DOI] [Google Scholar]
- Archambeau A. Miege F. Meyer C. Cossy J. Angew. Chem., Int. Ed. 2012;51:11540–11544. doi: 10.1002/anie.201205913. [DOI] [PubMed] [Google Scholar]
- Ye T. García C. F. McKervey M. A. J. Chem. Soc., Perkin Trans. 1. 1995:1373–1379. doi: 10.1039/P19950001373. [DOI] [Google Scholar]
- McCarthy N. McKervey M. A. Ye T. McCann M. Murphy E. Doyle M. P. Tetrahedron Lett. 1992;33:5983–5986. doi: 10.1016/S0040-4039(00)61106-1. [DOI] [Google Scholar]
- Zhu D. Ma J. Luo K. Fu H. Zhang L. Zhu S. Angew. Chem., Int. Ed. 2016;55:8452–8456. doi: 10.1002/anie.201604211. [DOI] [PubMed] [Google Scholar]
- Zhu D. Chen L. Zhang H. Ma Z. Jiang H. Zhu S. Angew. Chem., Int. Ed. 2018;57:12405–12409. doi: 10.1002/anie.201805676. [DOI] [PubMed] [Google Scholar]
- Lamb K. N. Squitieri R. A. Chintala S. R. Kwong A. J. Balmond E. I. Soldi C. Dmitrenko O. Castiñeira Reis M. Chung R. Addison J. B. Fettinger J. C. Hein J. E. Tantillo D. J. Fox J. M. Shaw J. T. Chem.–Eur. J. 2017;23:11843–11855. doi: 10.1002/chem.201701630. [DOI] [PubMed] [Google Scholar]
- Dimirjian C. A. Castiñeira Reis M. Balmond E. I. Turman N. C. Rodriguez E. P. Di Maso M. J. Fettinger J. C. Tantillo D. J. Shaw J. T. Org. Lett. 2019;21:7209–7212. doi: 10.1021/acs.orglett.9b02124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CCDC 1921674 (11c), 1921673 (13b), 1916890 (17c), and 1920825 (23c) contain the supplementary crystallographic data for this paper.†
- Jefford C. W. Johncock W. Helv. Chim. Acta. 1983;66:2666–2671. doi: 10.1002/hlca.19830660835. [DOI] [Google Scholar]
- Jefford C. W. Zaslona A. Tetrahedron Lett. 1985;26:6035–6038. doi: 10.1016/S0040-4039(00)95118-9. [DOI] [Google Scholar]
- Jefford C. W. Tang Q. Zaslona A. J. Am. Chem. Soc. 1991;113:3513–3518. doi: 10.1021/ja00009a043. [DOI] [Google Scholar]
- Padwa A. Beall L. S. Eidell C. K. Worsencroft K. J. J. Org. Chem. 2001;66:2414–2421. doi: 10.1021/jo001684w. [DOI] [PubMed] [Google Scholar]
- Ghigo G. Cagnina S. Maranzana A. Tonachini G. J. Org. Chem. 2010;75:3608–3617. doi: 10.1021/jo100367z. [DOI] [PubMed] [Google Scholar]
- See ESI† for complete computational work
- Harrison J. G. Gutierrez O. Jana N. Driver T. G. Tantillo D. J. J. Am. Chem. Soc. 2016;138:487–490. doi: 10.1021/jacs.5b11427. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.