

 This work is licensed under a
This work is licensed under a Abstract
The phosphoinositide 3-kinase (PI3K) pathway is a major mediator of growth factor signaling, cell proliferation and metabolism. Somatic gain-of-function mutations in PIK3CA, the catalytic subunit of PI3K, have recently been discovered in a number of vascular anomalies. The timing and origin of these mutations remain unclear although they are believed to occur during embryogenesis. The cellular origin of these lesions likely involves endothelial cells or an early endothelial cell lineage. This review will cover the diseases and syndromes associated with PIK3CA mutations and discuss the cellular origin, pathways and mechanisms. Activating PIK3CA ‘hot spot’ mutations have long been associated with a multitude of cancers allowing the development of targeted pharmacological inhibitors that are FDA-approved or in clinical trials. Current and future therapeutic approaches for PIK3CA-related vascular anomalies are discussed.
Keywords: vascular malformation, vascular anomalies
The phosphoinositide 3-kinase pathway
The phosphoinositide 3-kinase (PI3K) signaling pathway is a critical regulator of the angiogenic process by controlling endothelial cell (EC) proliferation, migration and survival (1). Growth factor stimulation of receptor tyrosine kinases activates PI3K that in turn catalyzes the conversion of phosphatidylinositol (3,4)‐bisphosphate (PIP2) to phosphatidylinositol (3,4,5)‐triphosphate (PIP3). PIP3 triggers phosphorylation of Pyruvate Dehydrogenase Kinase 1 (PDK1), which phosphorylates the serine and threonine kinase AKT (also known as Protein Kinase B, PKB) (2, 3). This activation of AKT leads to increased cellular proliferation through the mTOR pathway (4, 5). The catalytic subunit of PI3K (p110α) is encoded by the PIK3CA gene. PIK3CA ‘hot spot’ mutations p.E542K (E, glutamic acid replaced by K, lysine at amino acid position 542) and p.E545K (amino acid position 545) in exon 9 (α-helical domain), and p.H1047R/L (H, histidine replaced by R, arginine or L, leucine at amino acid position 1047) in exon 20 (kinase domain) are common in cancer (6), and more recently, have also been identified in pediatric vascular anomalies and overgrowth syndromes. All of the PIK3CA mutations associated with vascular anomalies and cancer are non-inherited and lead to sequestration of the p110α subunit to the plasma membrane thereby generating constitutive hyper-activation of the PI3K‐AKT pathway (7). Of interest, sustained AKT activation induces pathological angiogenesis and increased blood vessel diameter (8). Germline transmission of the PIK3CA oncogenic ‘hotspot’ mutations is not compatible with life. In mouse, ubiquitous or Tie2-driven heterozygous expression of Pik3ca H1047R resulted in disrupted vascular remodeling in the embryonic and extraembryonic tissues and lethality prior to E11.5 (9). Conversely, ubiquitous expression of Pik3ca H1047R in 6–8-week-old mice caused increased body weight, organomegaly and metabolic defects (10).
PIK3CA mutations in vascular anomalies
While vascular malformations are primarily present at birth, they can also become apparent during childhood, and they persist throughout adulthood. The International Society for the Study of Vascular Anomalies (ISSVA) created a classification system to aid the diagnosis of these anomalies (11). A number of genes and gene mutations have been implicated with vascular anomalies, in this review we will focus on the disease categories associated with PIK3CA mutations (Table 1).
Table 1.
Genetic mutations in vascular anomalies.
| Vascular anomalies | Gene mutations | References |
|---|---|---|
| Vascular tumors | KRAS, NRAS, HRAS, BRAF, GNAQ, GNA11, GNA14 | (72, 73, 74, 75) |
| Vascular malformations | ||
| Simple | ||
| Capillary malformation | GNAQ, GNA11, KRAS | (76, 77, 78) |
| Lymphatic malformation | PIK3CA | (18, 19, 20, 21) |
| Venous malformation | TEK, PIK3CA | (29, 31, 32, 33, 34, 35) |
| Arteriovenous malformation | KRAS, MAP2K1, BRAF | (76, 79, 80) |
| Combined | ||
| Lymphatic-Venous malformation | PIK3CA | (81) |
| Capillary-Lymphatic-Venous malformation/KTS, CLOVES | PIK3CA | (18, 46, 47, 48, 49) |
| Capillary malformation-ArterioVenous malformation | RASA1, EPHB4 | (82, 83) |
| Others | ||
| Fibro-adipose vascular anomaly | PIK3CA | (18) |
| Megalencephaly-capillary malformation | PIK3CA | (53) |
Simple vascular anomalies
Lymphatic malformations (LM)
Lymphatic malformations (LM) are characterized by massively dilated and dysmorphic low-flow lymphatic channels (12, 13, 14, 15, 16) lined with flattened EC. Often, vessels are filled with blood or thrombi as result of intralesional trauma and/or improper communication with the venous system. Expansion of LM is caused by vascular distension generated by fluid accumulation and cellular hyperplasia. LM expands during adolescence, and lesions can cause severe disfigurement, affect vital organs and contribute to infections. Histological markers of LM are Prox-1 and VEGFR3, while Podoplanin levels can be variable (17). Somatic activating PIK3CA mutations have been identified in affected tissue resected from the majority (94%) of LM patients (18) and in cells isolated from LM tissue (19, 20, 21). Patient-derived lymphatic endothelial cells (LM-LEC) exhibit pro-angiogenic properties such as increased proliferation, sprout formation, and VEGF-C and VEGFR-3 overexpression. AKT that is downstream of PI3K is constitutively active in the mutant LM-LEC, while ERK1/2 phosphorylation levels are variable (17, 19, 20, 22). A few animal models of LM have been reported and consist of xenografts generated by injection of LM-LEC (17) or LEC progenitor cells (23). In these studies, it is unclear if implanted cells expressed PIK3CA mutations. Another model of thoracic LM was generated by overexpression of VEGF-C in mouse airway epithelial cells resulting in lymphangectasia and mTOR pathway activation (24), although the specific activation status of AKT was not assessed. In the future, improved PIK3CA-based models of LM would be very beneficial to investigate therapeutic targets.
Generalized lymphatic anomaly (GLA)
Generalized lymphatic anomaly (GLA) is a severe form of LM that is diffuse or multifocal. Patients have increased risk of pulmonary involvement and the dysmorphic lymphatics can infiltrate and compromise the medullary bone causing pain and compromised mobility (25, 26). A high-throughput sequencing effort led to the discovery of PIK3CA mutations in affected tissue and LEC in 55% of GLA patients (27). Postnatal transgenic expression of PIK3CA p.H1047R in the LEC (Prox1-CreERT2) caused lymphatic hyperplasia and dysfunction, modeling important aspects of GLA (27).
Venous malformation (VM)
Venous malformation (VM) manifests as unifocal or multifocal bluish compressible lesions and may affect any tissue or organ. Patients with VM can suffer severe disfigurement, painful swelling, chronic recurrent thrombi and increased risk for pulmonary thromboembolism (13, 28). The histological hallmarks of VM are slow-flow thin-walled ectatic veins with scant smooth muscle cells. Pioneering work by Miikka Vikkula’s group identified germline or sporadic somatic activating TIE2 (TEK) mutations in about 60% of VM cases (29, 30, 31, 32). More recently, three independent studies described PIK3CA mutations in about 25% of VM patients (33, 34, 35). PIK3CA mutations are more frequent in intramuscular VM that do not involve the skin (35). TIE2 and PIK3CA mutations are typically mutually exclusive but in few patients both occurred (33, 34, 35, 36). The result of PIK3CA or TIE2 mutation is constitutively active PI3K signaling and downstream AKT activation. While ERK1/2 and STAT1 are activated in TIE2-mutant EC they are not in PIK3CA-mutant EC. Furthermore, AKT activation is higher in PIK3CA-mutated EC compared to TIE2-mutant EC. Our studies suggest this stronger AKT phosphorylation is linked to increased vessel density in a PIK3CA-mutated patient-derived xenograft model (35, 36, 37, 38). PIK3CA mutation-based models include a patient-derived EC xenograft and murine models with transgenic expression of PIK3CA p.H1047R in the embryonic mesoderm or in VE-Cadherin+ cells (33, 34, 39).
Combined vascular anomalies (two or more vascular malformations found in one lesion)
Capillary lymphatic venous malformation (CLVM)
Capillary lymphatic venous malformation (CLVM) includes complex lesions that involve overgrowth of multiple vascular components such as capillaries, lymphatics and veins. CLVM appears at birth and enlarge with time (12, 40). Two syndromes having these complex mixed vascular overgrowth lesions are Klippel-Trenaunay syndrome (KTS) (41) and Congenital Lipomatous Overgrowth, Vascular malformations, Epidermal nevi, Scoliosis/skeletal and spinal syndrome (CLOVES) (42). KTS diagnosis depends on three key features, cutaneous capillary malformations, venous and lymphatic malformations, and asymmetric hypertrophy of bones and overlying soft tissue (43, 44). The presentation of KTS is heterogeneous and there is substantial crossover in presentation of KTS with PIK3CA-Related Overgrowth Syndromes (PROS) (40). CLOVES syndrome is characterized by a combination of vascular, skin, lipomatous overgrowth and musculoskeletal abnormalities (45). PIK3CA somatic mutations have been identified in a high percentage (~90%) of KTS and CLOVES patients (18, 46, 47, 48). An interesting distinctive feature of KTS is that the PIK3CA variant p.C420R, relatively frequent in CLOVES, has not been reported in KTS patients (18, 46, 48, 49). A postnatal murine model of CLOVES was recently generated through ubiquitous expression of a dominant active PIK3CA transgene (P110*) (50, 51, 52).
Other vascular anomalies
A range of overgrowth syndromes combined with vascular anomalies have been associated with PI3KCA mutations. Fibro-adipose vascular anomaly (FAVA) (18) is a disease characterized by fibrous and fatty tissue infiltrating the skeletal muscle and abnormalities in the veins or lymphatic vessels. In a recent study, affected tissue from around 60% of FAVA patients had PIK3CA mutations. Mosaic post-zygotic or rare germline PIK3CA mutations have also been detected in the affected tissue in patients with brain overgrowth or megalencephaly-capillary malformation syndrome (MCAP) (53).
Cellular origin of mutations
In all of the vascular anomalies discussed here, monoallelic expression of gain-of-function PIK3CA mutations was initially identified in the affected tissue from patients while not present in blood samples. The allelic frequency of PIK3CA mutations in LM tissue ranged from 0.8 to 10% in digital droplet (dd) PCR sequencing analysis (18) suggesting only a distinct cell population within the lesion express the genetic variant. Work from our group and others demonstrated that PIK3CA mutations are present in the LM-LEC population, while the non-EC isolated from the affected tissue are mutation-negative (19, 20, 21). In GLA patient tissue, PIK3CA variant allelic frequency ranged from 28–33% in EC (27). Similarly, we recently reported that EC isolated from 9/9 patient-derived VM tissue or lesional blood expressed TIE2 or/and PIK3CA mutations while non-EC were negative (36). In this study, the allelic frequency of 46.9% (TIE2 p.L914F) and 59.6% (PIK3CA p.C420R) was observed in VM-EC populations from two patients. One VM-EC population contained both a TIE2 and a PIK3CA mutation and single-cell-derived clonal expansion revealed that every clone expressed both mutations. Conversely, few studies suggest that in CLVM and in overgrowth syndromes the PIK3CA mutations are expressed in the fibroblast population (49, 54, 55), although in these investigations purified EC were not analyzed. Our group have distinct preliminary data that in 7/7 CLVM patients (CLOVES and KTS) PIK3CA mutations are present solely in the EC purified from the tissue by CD31 expression (Le Cras T., unpublished) while the remaining cell populations (no CD31 expression detected) were negative. Correlations between any particular PIK3CA mutation and the clinical phenotype have not been demonstrated to date. The existence of the same PIK3CA mutations in simple (LM, VM) and combined (CLVM) vascular malformations suggest that it is not the type of mutation that induces different types of vascular anomalies. The type, location and/or severity of the vascular anomaly are likely dependent on when and/or what EC lineage or progenitor cell the mutational event occurs in during embryonic development. PIK3CA mutations that affect an early progenitor cell(s) may affect multiple EC lineages descended from those progenitor cell(s) and therefore affect more types of vessels than mutations in a committed cell lineage, although this has yet to be proven. More studies are needed to determine the exact cellular origin of CLVM and pinpoint the effect of the mutation distribution on the disease phenotype and severity.
Therapeutic targets and future treatments
Rapamycin
Rapamycin (Sirolimus) was introduced in 2011 as the first pharmacologic treatment for complicated vascular anomalies and has completely revolutionized the field (56). Prior to its introduction, targeted molecular therapies were not available for most of the anomalies discussed in this review and medical care consisted primarily of symptomatic treatment through surgery and sclerotherapy. Rapamycin is an immunosuppressant agent with efficacy in treating patients with Lymphangioleiomyomatosis (LAM), a lung disease with lymphatic hyperplasia (57). Rapamycin is an mTOR inhibitor that efficiently suppresses mTORC2-dependent AKT signaling in PIK3CA-mutant EC (Fig. 1). Rapamycin prevented VM lesion growth in different murine models (33, 37, 58) and restored lymphatic function in a GLA pre-clinical model (27). Rapamycin treatment significantly improved the quality of life of patients with complicated vascular anomalies with tolerable side-effects, although not always associated with lesion size reduction (37, 59, 60). Topical rapamycin was efficacious in patients with superficial microcystic LM lesions (61, 62). Mouse studies demonstrated that the mechanism of action of topical rapamycin is diminished phospho-AKT and phospho-S6 levels (63). Future studies should address the efficacy of rapamycin treatment in relation to the type of PIK3CA mutation and other genetic variants (60).
Figure 1.
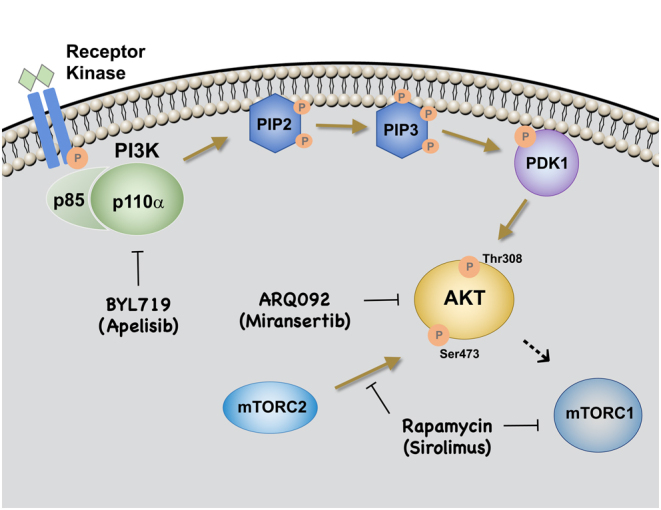
Targeted molecular therapies affecting PI3K pathway activation.
ARQ092
ARQ092 (Miransertib) is an allosteric pan-AKT inhibitor. Several clinical trials for the use of ARQ092 in adult cancer are ongoing (https://clinicaltrials.gov:NCT02476955, NCT01473095). A low dose of ARQ092 (5 mg/m2/day) reduced the levels of phospho-AKT by about half in 83% of biopsies from Proteus patients. Proteus is an overgrowth syndrome caused by somatic activating AKT mutations (p.E17K) (64). Studies using fibroblasts from patients with PIK3CA-related overgrowth syndromes showed that ARQ092 has potent antiproliferative activity (54, 55, 65, 66). Together, these findings lead to a phase I/II clinical trial for the use of ARQ092 for treatment of PIK3CA-related overgrowth syndromes and vascular anomalies (NCT03094832). Expanded access to this drug was recently granted by the FDA (NCT03317366).
BYL719
BYL719 (Apelisib) is a PI3K p110α specific inhibitor. It has a half-maximum inhibitory concentration of 4.6nM for the α isoform, while 55–251-fold higher doses are needed to inhibit the β, γ and δ isoforms (67). BYL719 has a tolerable safety profile and encouraging preliminary activity in patients with PIK3CA-altered solid tumors (68). Promising results were also obtained in eight CLOVES patients in a study conducted in France where treatment with BYL719 was granted under compassionate use (52). In pre-clinical studies, BYL719 effectively prevented organ dysfunction and death in transgenic dominant active PIK3CA mice. In this study and in an independent in vitro investigation (35), BYL719 was more effective than rapamycin as it more efficiently blocked AKT phosphorylation at both Serine 473 and Threonine 308. Interestingly, BYL719 treatment also decreased VM lesion size and promoted apoptosis in a transgenic PIK3CA H1047R VM murine model (33). A topical formulation of BYL719 was also efficacious in VM superficial lesions (33).
Whether the benefits of long-term treatment with these inhibitors will be limited by side-effects is still unclear although early results seem promising. In cancer, it has been shown that inhibition of the PI3K pathway leads to proliferative arrest rather than cell death, and combination of BYL719 with mTOR inhibitor enhanced the antitumoral activity (69, 70, 71). The combination of PI3K/AKT inhibitors with rapamycin for the treatment of PIK3CA-related vascular anomaly patients could enhance clinical response. Much work still needs to be done in pre-clinical models to explore the efficacy of these therapeutic strategies.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of this review.
Funding
Research reported in this manuscript was supported by the National Heart, Lung, and Blood Institute, under Award Number R01 HL117952 (E B), part of the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Funding for this project was also provided by the Lymphatic Malformation Institute (LMI) (T D L) and the Center for Pediatric Genomics (CpG) (CCHMC) (T D L and E B).
Acknowledgements
We thank Jillian Goines and Sandra Schrenk for critical revision.
References
- 1.Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nature Reviews: Molecular Cell Biology 2012. 13 . ( 10.1038/nrm3290) [DOI] [PubMed] [Google Scholar]
- 2.Hiles ID, Otsu M, Volinia S, Fry MJ, Gout I, Dhand R, Panayotou G, Ruiz-Larrea F, Thompson A, Totty NF. Phosphatidylinositol 3-kinase: structure and expression of the 110 kd catalytic subunit. Cell 1992. 70 . ( 10.1016/0092-8674(92)90166-A) [DOI] [PubMed] [Google Scholar]
- 3.Stephens L, Anderson K, Stokoe D, Erdjument-Bromage H, Painter GF, Holmes AB, Gaffney PR, Reese CB, McCormick F, Tempst P, et al Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science 1998. 279 . ( 10.1126/science.279.5351.710) [DOI] [PubMed] [Google Scholar]
- 4.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nature Cell Biology 2002. 4 . ( 10.1038/ncb839) [DOI] [PubMed] [Google Scholar]
- 5.Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nature Cell Biology 2002. 4 . ( 10.1038/ncb840) [DOI] [PubMed] [Google Scholar]
- 6.Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, et al High frequency of mutations of the PIK3CA gene in human cancers. Science 2004. 304 554 ( 10.1126/science.1096502) [DOI] [PubMed] [Google Scholar]
- 7.Burke JE, Perisic O, Masson GR, Vadas O, Williams RL. Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110alpha (PIK3CA). PNAS 2012. 109 . ( 10.1073/pnas.1205508109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Phung TL, Ziv K, Dabydeen D, Eyiah-Mensah G, Riveros M, Perruzzi C, Sun J, Monahan-Earley RA, Shiojima I, Nagy JA, et al Pathological angiogenesis is induced by sustained Akt signaling and inhibited by rapamycin. Cancer Cell 2006. 10 . ( 10.1016/j.ccr.2006.07.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hare LM, Schwarz Q, Wiszniak S, Gurung R, Montgomery KG, Mitchell CA, Phillips WA. Heterozygous expression of the oncogenic Pik3ca(H1047R) mutation during murine development results in fatal embryonic and extraembryonic defects. Developmental Biology 2015. 404 . ( 10.1016/j.ydbio.2015.04.022) [DOI] [PubMed] [Google Scholar]
- 10.Kinross KM, Montgomery KG, Mangiafico SP, Hare LM, Kleinschmidt M, Bywater MJ, Poulton IJ, Vrahnas C, Henneicke H, Malaterre J, et al Ubiquitous expression of the Pik3caH1047R mutation promotes hypoglycemia, hypoinsulinemia, and organomegaly. FASEB Journal 2015. 29 . ( 10.1096/fj.14-262782) [DOI] [PubMed] [Google Scholar]
- 11.Wassef M, Blei F, Adams D, Alomari A, Baselga E, Berenstein A, Burrows P, Frieden IJ, Garzon MC, Lopez-Gutierrez JC, et al Vascular anomalies classification: recommendations from the International Society for the study of vascular anomalies. Pediatrics 2015. 136 e203–e214. ( 10.1542/peds.2014-3673) [DOI] [PubMed] [Google Scholar]
- 12.Brouillard P, Vikkula M. Vascular malformations: localized defects in vascular morphogenesis. Clinical Genetics 2003. 63 . ( 10.1034/j.1399-0004.2003.00092.x) [DOI] [PubMed] [Google Scholar]
- 13.Garzon MC, Huang JT, Enjolras O, Frieden IJ. Vascular malformations: Part I. Journal of the American Academy of Dermatology 2007. 56 ; quiz . ( 10.1016/j.jaad.2006.05.069) [DOI] [PubMed] [Google Scholar]
- 14.Mulliken JB, Glowacki J. Hemangiomas and vascular malformations in infants and children: a classification based on endothelial characteristics. Plastic and Reconstructive Surgery 1982. 69 . ( 10.1097/00006534-198203000-00002) [DOI] [PubMed] [Google Scholar]
- 15.Padwa BL, Hayward PG, Ferraro NF, Mulliken JB. Cervicofacial lymphatic malformation: clinical course, surgical intervention, and pathogenesis of skeletal hypertrophy. Plastic and Reconstructive Surgery 1995. 95 . ( 10.1097/00006534-199505000-00001) [DOI] [PubMed] [Google Scholar]
- 16.Whimster IW. The pathology of lymphangioma circumscriptum. British Journal of Dermatology 1976. 94 . ( 10.1111/j.1365-2133.1976.tb05134.x) [DOI] [PubMed] [Google Scholar]
- 17.Lokmic Z, Mitchell GM, Koh Wee Chong N, Bastiaanse J, Gerrand YW, Zeng Y, Williams ED, Penington AJ. Isolation of human lymphatic malformation endothelial cells, their in vitro characterization and in vivo survival in a mouse xenograft model. Angiogenesis 2014. 17 . ( 10.1007/s10456-013-9371-8) [DOI] [PubMed] [Google Scholar]
- 18.Luks VL, Kamitaki N, Vivero MP, Uller W, Rab R, Bovee JV, Rialon KL, Guevara CJ, Alomari AI, Greene AK, et al Lymphatic and other vascular malformative/overgrowth disorders are caused by somatic mutations in PIK3CA. Journal of Pediatrics 2015. 166 1048.e1–1054.e5. ( 10.1016/j.jpeds.2014.12.069) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blesinger H, Kaulfuß S, Aung T, Schwoch S, Prantl L, Rößler J, Wilting J, Becker J. PIK3CA mutations are specifically localized to lymphatic endothelial cells of lymphatic malformations. PLoS ONE 2018. 13 e0200343 ( 10.1371/journal.pone.0200343) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boscolo E, Coma S, Luks VL, Greene AK, Klagsbrun M, Warman ML, Bischoff J. AKT hyper-phosphorylation associated with PI3K mutations in lymphatic endothelial cells from a patient with lymphatic malformation. Angiogenesis 2015. 18 . ( 10.1007/s10456-014-9453-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Osborn AJ, Dickie P, Neilson DE, Glaser K, Lynch KA, Gupta A, Dickie BH. Activating PIK3CA alleles and lymphangiogenic phenotype of lymphatic endothelial cells isolated from lymphatic malformations. Human Molecular Genetics 2015. 24 . ( 10.1093/hmg/ddu505) [DOI] [PubMed] [Google Scholar]
- 22.Partanen TA, Vuola P, Jauhiainen S, Lohi J, Salminen P, Pitkaranta A, Hakkinen SK, Honkonen K, Alitalo K, Yla-Herttuala S. Neuropilin-2 and vascular endothelial growth factor receptor-3 are up-regulated in human vascular malformations. Angiogenesis 2013. 16 . ( 10.1007/s10456-012-9305-x) [DOI] [PubMed] [Google Scholar]
- 23.Wu JK, Kitajewski C, Reiley M, Keung CH, Monteagudo J, Andrews JP, Liou P, Thirumoorthi A, Wong A, Kandel JJ, et al Aberrant lymphatic endothelial progenitors in lymphatic malformation development. PLoS ONE 2015. 10 e0117352 ( 10.1371/journal.pone.0117352) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baluk P, Yao LC, Flores JC, Choi D, Hong YK, McDonald DM. Rapamycin reversal of VEGF-C-driven lymphatic anomalies in the respiratory tract. JCI Insight 2017. 2 ( 10.1172/jci.insight.90103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lala S, Mulliken JB, Alomari AI, Fishman SJ, Kozakewich HP, Chaudry G. Gorham-Stout disease and generalized lymphatic anomaly – clinical, radiologic, and histologic differentiation. Skeletal Radiology 2013. 42 . ( 10.1007/s00256-012-1565-4) [DOI] [PubMed] [Google Scholar]
- 26.Trenor CC, 3rd, Chaudry G. Complex lymphatic anomalies. Seminars in Pediatric Surgery 2014. 23 . ( 10.1053/j.sempedsurg.2014.07.006) [DOI] [PubMed] [Google Scholar]
- 27.Rodriguez-Laguna L, Agra N, Ibanez K, Oliva-Molina G, Gordo G, Khurana N, Hominick D, Beato M, Colmenero I, Herranz G, et al Somatic activating mutations in PIK3CA cause generalized lymphatic anomaly. Journal of Experimental Medicine 2019. 216 . ( 10.1084/jem.20181353) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boon LM, Mulliken JB, Enjolras O, Vikkula M. Glomuvenous malformation (glomangioma) and venous malformation: distinct clinicopathologic and genetic entities. Archives of Dermatology 2004. 140 . ( 10.1001/archderm.140.8.971) [DOI] [PubMed] [Google Scholar]
- 29.Limaye N, Wouters V, Uebelhoer M, Tuominen M, Wirkkala R, Mulliken JB, Eklund L, Boon LM, Vikkula M. Somatic mutations in angiopoietin receptor gene TEK cause solitary and multiple sporadic venous malformations. Nature Genetics 2009. 41 . ( 10.1038/ng.272) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soblet J, Limaye N, Uebelhoer M, Boon LM, Vikkula M. Variable somatic TIE2 mutations in half of sporadic venous malformations. Mol Syndromol 2013. 4 . ( 10.1159/000348327) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vikkula M, Boon LM, Carraway KL, 3rd, Calvert JT, Diamonti AJ, Goumnerov B, Pasyk KA, Marchuk DA, Warman ML, Cantley LC, et al Vascular dysmorphogenesis caused by an activating mutation in the receptor tyrosine kinase TIE2. Cell 1996. 87 . ( 10.1016/S0092-8674(00)81814-0) [DOI] [PubMed] [Google Scholar]
- 32.Wouters V, Limaye N, Uebelhoer M, Irrthum A, Boon LM, Mulliken JB, Enjolras O, Baselga E, Berg J, Dompmartin A, et al Hereditary cutaneomucosal venous malformations are caused by TIE2 mutations with widely variable hyper-phosphorylating effects. European Journal of Human Genetics 2010. 18 . ( 10.1038/ejhg.2009.193) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Castel P, Carmona FJ, Grego-Bessa J, Berger MF, Viale A, Anderson KV, Bague S, Scaltriti M, Antonescu CR, Baselga E, et al Somatic PIK3CA mutations as a driver of sporadic venous malformations. Science Translational Medicine 2016. 8 332ra42 ( 10.1126/scitranslmed.aaf1164) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Castillo SD, Tzouanacou E, Zaw-Thin M, Berenjeno IM, Parker VE, Chivite I, Mila-Guasch M, Pearce W, Solomon I, Angulo-Urarte A, et al Somatic activating mutations in Pik3ca cause sporadic venous malformations in mice and humans. Science Translational Medicine 2016. 8 332ra43 ( 10.1126/scitranslmed.aad9982) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Limaye N, Kangas J, Mendola A, Godfraind C, Schlogel MJ, Helaers R, Eklund L, Boon LM, Vikkula M. Somatic activating PIK3CA mutations cause venous malformation. American Journal of Human Genetics 2015. 97 . ( 10.1016/j.ajhg.2015.11.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goines J, Li X, Cai Y, Mobberley-Schuman P, Metcalf M, Fishman SJ, Adams DM, Hammill AM, Boscolo E. A xenograft model for venous malformation. Angiogenesis 2018. 21 . ( 10.1007/s10456-018-9624-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boscolo E, Limaye N, Huang L, Kang KT, Soblet J, Uebelhoer M, Mendola A, Natynki M, Seront E, Dupont S, et al Rapamycin improves TIE2-mutated venous malformation in murine model and human subjects. Journal of Clinical Investigation 2015. 125 . ( 10.1172/JCI76004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uebelhoer M, Natynki M, Kangas J, Mendola A, Nguyen HL, Soblet J, Godfraind C, Boon LM, Eklund L, Limaye N, et al Venous malformation-causative TIE2 mutations mediate an AKT-dependent decrease in PDGFB. Human Molecular Genetics 2013. 22 . ( 10.1093/hmg/ddt198) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.di Blasio L, Puliafito A, Gagliardi PA, Comunanza V, Somale D, Chiaverina G, Bussolino F, Primo L. PI3K/mTOR inhibition promotes the regression of experimental vascular malformations driven by PIK3CA-activating mutations. Cell Death and Disease 2018. 9 45 ( 10.1038/s41419-017-0064-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boon LM, Ballieux F, Vikkula M. Pathogenesis of vascular anomalies. Clinics in Plastic Surgery 2011. 38 . ( 10.1016/j.cps.2010.08.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berry SA, Peterson C, Mize W, Bloom K, Zachary C, Blasco P, Hunter D. Klippel-Trenaunay syndrome. American Journal of Medical Genetics 1998. 79 . () [DOI] [PubMed] [Google Scholar]
- 42.Martinez-Lopez A, Blasco-Morente G, Perez-Lopez I, Herrera-Garcia JD, Luque-Valenzuela M, Sanchez-Cano D, Lopez-Gutierrez JC, Ruiz-Villaverde R, Tercedor-Sanchez J. CLOVES syndrome: review of a PIK3CA-related overgrowth spectrum (PROS). Clinical Genetics 2017. 91 . ( 10.1111/cge.12832) [DOI] [PubMed] [Google Scholar]
- 43.Klippel M, Trenaunay P. Du naevus variqueux osteo-hypertrophique. Archives of General Internal Medicine 1900. 185 . [Google Scholar]
- 44.Wang SK, Drucker NA, Gupta AK, Marshalleck FE, Dalsing MC. Diagnosis and management of the venous malformations of Klippel-Trenaunay syndrome. Journal of Vascular Surgery: Venous and Lymphatic Disorders 2017. 5 . ( 10.1016/j.jvsv.2016.10.084) [DOI] [PubMed] [Google Scholar]
- 45.Alomari AI. CLOVE(S) syndrome: expanding the acronym. American Journal of Medical Genetics: A 2009. 149A 294; author reply 295. [DOI] [PubMed] [Google Scholar]
- 46.Keppler-Noreuil KM, Rios JJ, Parker VE, Semple RK, Lindhurst MJ, Sapp JC, Alomari A, Ezaki M, Dobyns W, Biesecker LG. PIK3CA-related overgrowth spectrum (PROS): diagnostic and testing eligibility criteria, differential diagnosis, and evaluation. American Journal of Medical Genetics: Part A 2015. 167A . ( 10.1002/ajmg.a.36836) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Keppler-Noreuil KM, Sapp JC, Lindhurst MJ, Parker VE, Blumhorst C, Darling T, Tosi LL, Huson SM, Whitehouse RW, Jakkula E, et al Clinical delineation and natural history of the PIK3CA-related overgrowth spectrum. American Journal of Medical Genetics: Part A 2014. 164A . ( 10.1002/ajmg.a.36552) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kurek KC, Luks VL, Ayturk UM, Alomari AI, Fishman SJ, Spencer SA, Mulliken JB, Bowen ME, Yamamoto GL, Kozakewich HP, et al Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. American Journal of Human Genetics 2012. 90 . ( 10.1016/j.ajhg.2012.05.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuentz P, St-Onge J, Duffourd Y, Courcet JB, Carmignac V, Jouan T, Sorlin A, Abasq-Thomas C, Albuisson J, Amiel J, et al Molecular diagnosis of PIK3CA-related overgrowth spectrum (PROS) in 162 patients and recommendations for genetic testing. Genetics in Medicine 2017. 19 . ( 10.1038/gim.2016.220) [DOI] [PubMed] [Google Scholar]
- 50.Hu Q, Klippel A, Muslin AJ, Fantl WJ, Williams LT. Ras-dependent induction of cellular responses by constitutively active phosphatidylinositol-3 kinase. Science 1995. 268 . ( 10.1126/science.7701328) [DOI] [PubMed] [Google Scholar]
- 51.Klippel A, Reinhard C, Kavanaugh WM, Apell G, Escobedo MA, Williams LT. Membrane localization of phosphatidylinositol 3-kinase is sufficient to activate multiple signal-transducing kinase pathways. Molecular and Cellular Biology 1996. 16 . ( 10.1128/mcb.16.8.4117) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Venot Q, Blanc T, Rabia SH, Berteloot L, Ladraa S, Duong JP, Blanc E, Johnson SC, Hoguin C, Boccara O, et al Targeted therapy in patients with PIK3CA-related overgrowth syndrome. Nature 2018. 558 . ( 10.1038/s41586-018-0217-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Riviere JB, Mirzaa GM, O'Roak BJ, Beddaoui M, Alcantara D, Conway RL, St-Onge J, Schwartzentruber JA, Gripp KW, Nikkel SM, et al De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nature Genetics 2012. 44 . ( 10.1038/ng.2331) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ranieri C, Di Tommaso S, Loconte DC, Grossi V, Sanese P, Bagnulo R, Susca FC, Forte G, Peserico A, De Luisi A, et al In vitro efficacy of ARQ 092, an allosteric AKT inhibitor, on primary fibroblast cells derived from patients with PIK3CA-related overgrowth spectrum (PROS). Neurogenetics 2018. 19 . ( 10.1007/s10048-018-0540-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Suzuki Y, Enokido Y, Yamada K, Inaba M, Kuwata K, Hanada N, Morishita T, Mizuno S, Wakamatsu N. The effect of rapamycin, NVP-BEZ235, aspirin, and metformin on PI3K/AKT/mTOR signaling pathway of PIK3CA-related overgrowth spectrum (PROS). Oncotarget 2017. 8 . ( 10.18632/oncotarget.17566) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hammill AM, Wentzel M, Gupta A, Nelson S, Lucky A, Elluru R, Dasgupta R, Azizkhan RG, Adams DM. Sirolimus for the treatment of complicated vascular anomalies in children. Pediatric Blood and Cancer 2011. 57 . ( 10.1002/pbc.23124) [DOI] [PubMed] [Google Scholar]
- 57.McCormack FX, Inoue Y, Moss J, Singer LG, Strange C, Nakata K, Barker AF, Chapman JT, Brantly ML, Stocks JM, et al Efficacy and safety of sirolimus in lymphangioleiomyomatosis. New England Journal of Medicine 2011. 364 . ( 10.1056/NEJMoa1100391) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Molecular Cell 2006. 22 . ( 10.1016/j.molcel.2006.03.029) [DOI] [PubMed] [Google Scholar]
- 59.Adams DM, Trenor CC, 3rd, Hammill AM, Vinks AA, Patel MN, Chaudry G, Wentzel MS, Mobberley-Schuman PS, Campbell LM, Brookbank C, et al Efficacy and safety of sirolimus in the treatment of complicated vascular anomalies. Pediatrics 2016. 137 e20153257 ( 10.1542/peds.2015-3257) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hammer J, Seront E, Duez S, Dupont S, Van Damme A, Schmitz S, Hoyoux C, Chopinet C, Clapuyt P, Hammer F, et al Sirolimus is efficacious in treatment for extensive and/or complex slow-flow vascular malformations: a monocentric prospective phase II study. Orphanet Journal of Rare Diseases 2018. 13 191 ( 10.1186/s13023-018-0934-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Garcia-Montero P, Del Boz J, Sanchez-Martinez M, Escudero Santos IM, Baselga E. Microcystic lymphatic malformation successfully treated with topical rapamycin. Pediatrics 2017. 139 e20162105 ( 10.1542/peds.2016-2105) [DOI] [PubMed] [Google Scholar]
- 62.Ivars M, Redondo P. Efficacy of topical sirolimus (rapamycin) for the treatment of microcystic lymphatic malformations. JAMA Dermatology 2017. 153 . ( 10.1001/jamadermatol.2016.3697) [DOI] [PubMed] [Google Scholar]
- 63.Du W, Gerald D, Perruzzi CA, Rodriguez-Waitkus P, Enayati L, Krishnan B, Edmonds J, Hochman ML, Lev DC, Phung TL. Vascular tumors have increased p70 S6-kinase activation and are inhibited by topical rapamycin. Laboratory Investigation 2013. 93 . ( 10.1038/labinvest.2013.98) [DOI] [PubMed] [Google Scholar]
- 64.Keppler-Noreuil KM, Sapp JC, Lindhurst MJ, Darling TN, Burton-Akright J, Bagheri M, Dombi E, Gruber A, Jarosinski PF, Martin S, et al Pharmacodynamic study of Miransertib in individuals with Proteus syndrome. American Journal of Human Genetics 2019. 104 . ( 10.1016/j.ajhg.2019.01.015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lindhurst MJ, Yourick MR, Yu Y, Savage RE, Ferrari D, Biesecker LG. Repression of AKT signaling by ARQ 092 in cells and tissues from patients with Proteus syndrome. Scientific Reports 2015. 5 17162 ( 10.1038/srep17162) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yu Y, Savage RE, Eathiraj S, Meade J, Wick MJ, Hall T, Abbadessa G, Schwartz B. Targeting AKT1-E17K and the PI3K/AKT pathway with an allosteric AKT inhibitor, ARQ 092. PLoS ONE 2015. 10 e0140479 ( 10.1371/journal.pone.0140479) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fritsch C, Huang A, Chatenay-Rivauday C, Schnell C, Reddy A, Liu M, Kauffmann A, Guthy D, Erdmann D, De Pover A, et al Characterization of the novel and specific PI3Kalpha inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Molecular Cancer Therapeutics 2014. 13 . ( 10.1158/1535-7163.MCT-13-0865) [DOI] [PubMed] [Google Scholar]
- 68.Juric D, Rodon J, Tabernero J, Janku F, Burris HA, Schellens JHM, Middleton MR, Berlin J, Schuler M, Gil-Martin M, et al Phosphatidylinositol 3-kinase alpha-selective inhibition with alpelisib (BYL719) in PIK3CA-altered solid tumors: results from the first-in-human study. Journal of Clinical Oncology 2018. 36 . ( 10.1200/JCO.2017.72.7107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Elkabets M, Vora S, Juric D, Morse N, Mino-Kenudson M, Muranen T, Tao J, Campos AB, Rodon J, Ibrahim YH, et al mTORC1 inhibition is required for sensitivity to PI3K p110alpha inhibitors in PIK3CA-mutant breast cancer. Science Translational Medicine 2013. 5 196ra99 ( 10.1126/scitranslmed.3005747) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M, Botero ML, Llonch E, Atzori F, Di Cosimo S, et al NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Research 2008. 68 . ( 10.1158/0008-5472.CAN-08-1385) [DOI] [PubMed] [Google Scholar]
- 71.Zwang Y, Jonas O, Chen C, Rinne ML, Doench JG, Piccioni F, Tan L, Huang HT, Wang J, Ham YJ, et al Synergistic interactions with PI3K inhibition that induce apoptosis. eLife 2017. 6 e24523 ( 10.7554/eLife.24523) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ayturk UM, Couto JA, Hann S, Mulliken JB, Williams KL, Huang AY, Fishman SJ, Boyd TK, Kozakewich HP, Bischoff J, et al Somatic activating mutations in GNAQ and GNA11 are associated with congenital hemangioma. American Journal of Human Genetics 2016. 98 . ( 10.1016/j.ajhg.2016.03.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Groesser L, Peterhof E, Evert M, Landthaler M, Berneburg M, Hafner C. BRAF and RAS mutations in sporadic and secondary pyogenic granuloma. Journal of Investigative Dermatology 2016. 136 . ( 10.1038/JID.2015.376) [DOI] [PubMed] [Google Scholar]
- 74.Lim YH, Bacchiocchi A, Qiu J, Straub R, Bruckner A, Bercovitch L, Narayan D, McNiff J, Ko C, et al GNA14 somatic mutation causes congenital and sporadic vascular tumors by MAPK activation. American Journal of Human Genetics 2016. 99 ( 10.1016/j.ajhg.2016.06.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sabater Marco V, Escutia Munoz B, Morera Faet A, Mata Roig M, Botella Estrada R. Pseudogranulomatous Spitz nevus: a variant of Spitz nevus with heavy inflammatory infiltrate mimicking a granulomatous dermatitis. Journal of Cutaneous Pathology 2013. 40 . ( 10.1111/cup.12011) [DOI] [PubMed] [Google Scholar]
- 76.Al-Olabi L, Polubothu S, Dowsett K, Andrews KA, Stadnik P, Joseph AP, Knox R, Pittman A, Clark G, Baird W, et al Mosaic RAS/MAPK variants cause sporadic vascular malformations which respond to targeted therapy. Journal of Clinical Investigation 2018. 128 . ( 10.1172/JCI98589) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Couto JA, Ayturk UM, Konczyk DJ, Goss JA, Huang AY, Hann S, Reeve JL, Liang MG, Bischoff J, Warman ML, et al A somatic GNA11 mutation is associated with extremity capillary malformation and overgrowth. Angiogenesis 2017. 20 . ( 10.1007/s10456-016-9538-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shirley MD, Tang H, Gallione CJ, Baugher JD, Frelin LP, Cohen B, North PE, Marchuk DA, Comi AM, Pevsner J. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. New England Journal of Medicine 2013. 368 . ( 10.1056/NEJMoa1213507) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Couto JA, Huang AY, Konczyk DJ, Goss JA, Fishman SJ, Mulliken JB, Warman ML, Greene AK. Somatic MAP2K1 mutations are associated with extracranial arteriovenous malformation. American Journal of Human Genetics 2017. 100 . ( 10.1016/j.ajhg.2017.01.018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nikolaev SI, Vetiska S, Bonilla X, Boudreau E, Jauhiainen S, Rezai Jahromi B, Khyzha N, DiStefano PV, Suutarinen S, Kiehl TR, et al Somatic activating KRAS mutations in arteriovenous malformations of the brain. New England Journal of Medicine 2018. 378 . ( 10.1056/NEJMoa1709449) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Glaser K, Dickie P, Neilson D, Osborn A, Dickie BH. Linkage of metabolic defects to activated PIK3CA alleles in endothelial cells derived from lymphatic malformation. Lymphatic Research and Biology 2018. 16 . ( 10.1089/lrb.2017.0033) [DOI] [PubMed] [Google Scholar]
- 82.Amyere M, Revencu N, Helaers R, Pairet E, Baselga E, Cordisco M, Chung W, Dubois J, Lacour JP, Martorell L, et al Germline loss-of-function mutations in EPHB4 cause a second form of capillary malformation-arteriovenous malformation (CM-AVM2) deregulating RAS-MAPK signaling. Circulation 2017. 136 . ( 10.1161/CIRCULATIONAHA.116.026886) [DOI] [PubMed] [Google Scholar]
- 83.Revencu N, Boon LM, Mulliken JB, Enjolras O, Cordisco MR, Burrows PE, Clapuyt P, Hammer F, Dubois J, Baselga E, et al Parkes Weber syndrome, vein of Galen aneurysmal malformation, and other fast-flow vascular anomalies are caused by RASA1 mutations. Human Mutation 2008. 29 . ( 10.1002/humu.20746) [DOI] [PubMed] [Google Scholar]