

ABSTRACT
Burn-induced coagulopathy is not well understood, and consensus on diagnosis, prevention, and treatments are lacking. In this review, literature on burn-induced (and associated) coagulopathy is presented along with the current understanding of the effects of burn injury on the interactions among coagulation, fibrinolysis, and inflammation in the acute resuscitative phase and reconstructive phase of care. The role of conventional tests of coagulopathy and functional assays like thromboelastography or thromboelastometry will also be discussed. Finally, reported methods for the prevention and treatment of complications related to burn-induced coagulopathy will be reviewed.
Keywords: Clot dysfunction, coagulopathy, hemostasis, thermal injury, thrombosis
The development of coagulopathy, (i.e., dysfunctional hemostasis resulting in either hemorrhagic or thrombotic events) after burn injury complicates the already intricate level of care required for this patient population. An experienced multidisciplinary team is essential to meet the needs of a severely-burned individual. The typical course of a patient who has survived a critical thermal injury is represented in Figure 1. The dynamic process of recovery is highlighted as patients transition from aggressive interventions immediately after injury and in the phase of acute burn shock to the period of chronic recovery that begins during hospitalization and continues after discharge.
Fig. 1.
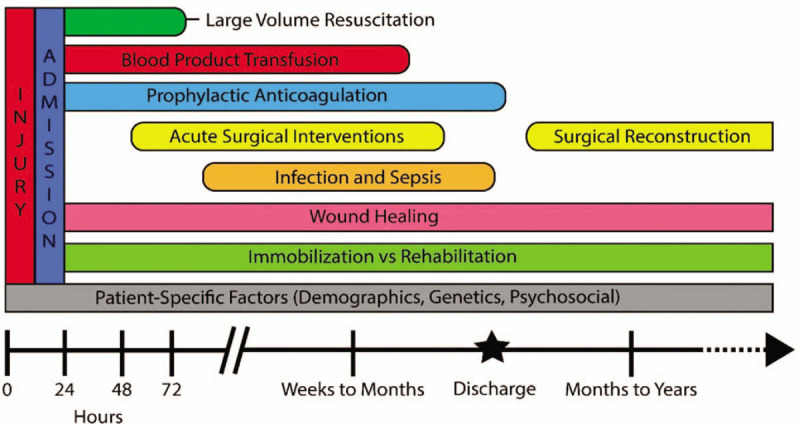
A simplified schematic illustrating a patient's hospital course after burn injury.
At each of these phases, internal and external forces impact coagulation homeostasis. The inflammatory mechanism initiated by the injury itself has profound effects that impact clot formation and can lead to burn induced coagulopathy (BIC). During transport and initial hospital care, patients are subjected to lifesaving measures including large volume resuscitation and surgical interventions that further influence coagulation. After stabilization, hemostasis is altered by care that may include blood product transfusion, extensive excision and grafting, and other surgical interventions.
The coagulation status of patients who survive the acute phase is impacted during recovery by a battery of forces. The development of infection or sepsis remains a leading cause of mortality for severely-burned patients, and the physiologic stress of this event can precipitate coagulopathy. After discharge, patients may encounter events that could trigger coagulopathy, such as incomplete wound healing, surgical reconstructions, and impaired mobility.
Underlying these external forces, patient-specific characteristics must be continually recognized. Age, gender, race, pre-existing conditions, and concomitant injuries may play a role in the physiologic response to thermal injury, and must be considered when treating BIC.
In the past few decades, impressive advances have been made in the understanding of coagulopathy following blunt and penetrating trauma resulting in promising clinical interventions to diagnose, prevent, and treat trauma induced coagulopathy (TIC) (1–5). While the extent of research in the burn population has lagged in comparison with other types of trauma, a growing body of knowledge elucidates many unique characteristics of BIC.
In this review, we examine the history of BIC research, and discuss the current understanding of potential biochemical mechanisms underlying BIC in the contexts of the initial response to injury, the acute care provided during large-volume resuscitation, the recovery during a prolonged hospital stay, and long after discharge. The tools that allow clinicians to recognize BIC and initiate clinical interventions are also be discussed. This review will synthesize basic science and clinical studies by connecting the pathophysiologic mechanisms of BIC with clinical manifestations, and identify areas that merit further investigation.
HISTORY OF BURN-INDUCED COAGULOPATHY RESEARCH
On review of the literature, the first mention of coagulopathy in thermally-injured patients appeared in the Polish journal, Polski Tygodnik Lekarski, by Blonska and Kamienski in 1957 (6). Hemostasis following scald injuries in children and adults was investigated. By measuring clotting time, a hypercoagulable state following scald injury was observed, and this was more pronounced and prolonged in children. A transition to a hypocoagulable state was noted in patients with severe scalds at 3 weeks postinjury. The authors postulated the increased bleeding tendency was a result of liver damage induced by bacterial toxins.
Longitudinal studies of BIC by the French military in the early 1960s examined clot formation and platelet function (7, 8). Thrombocytopenia was described in the initial 24 to 48 h after injury with a rebound thrombocytosis and slow return to normal platelet levels over 2 to 4 months. Thromboelastography (TEG), a viscoelastic hemostatic assay that was first introduced in 1948 in Germany by Hartert (9), which measures the global viscoelastic properties of whole blood clot formation under low shear stress, was performed to supplement platelet counts. Despite initial thrombocytopenia, TEG conducted at 2 days demonstrated hypercoagulability and was attributed to high levels of fibrinogen and thromboplastin due to tissue lysis. The investigators also demonstrated a reduction of this hypercoagulability observed using TEG with the administration of heparin and proposed the use of thromboembolic prophylaxis with heparin in burn patients.
The earliest available English-language publication focusing on coagulopathy in burn patients appeared in Surgery in 1963 (10). Holder et al. (10) noted evidence of thromboembolic events on autopsy of severely-burned patients. They further examined coagulopathy after thermal injury in a mouse model using clotting time measurements and evaluated the effects of heparin and thromboplastin on BIC. A novel method of extracting thromboplastin from burned skin was developed. Subsequently injecting this extract into mice resulted in significant procoagulant states or death (10). Ultimately, the authors concluded that thermal injury induced a hypercoagulable state and hypothesized that thromboplastic material in the injured tissue may be partially responsible.
Investigation of BIC continued in the 1970s with animal models again focusing on platelet function and clot dynamics. In 1974, using a scald-burned rat model, Eurenius and Rothenberg (11) observed an initial period of markedly depressed platelet function in burned animals using ADP-induced platelet aggregation tests. The initial depression was followed by increased platelet aggregation by 24 and 48 h, which the authors presented as a possible mechanism behind the clinically observed hypercoagulability noted in human burn and trauma patients.
In 1975, using a similar scald-burned rat model, Curreri et al. (12) studied the effects of heparin and protamine sulfate on coagulation dynamics. In this study, an initial low-normal platelet level was followed by thrombocytosis after 48 h, echoing the findings of Eurenius and Rothenberg. When fibrin split product concentration was measured after administering heparin to burned rats at 24 h postinjury, no significant effect was observed relative to controls (12). The authors conclude by strongly advocating against prophylactic anticoagulation with heparin, contrary to current practices in many burn institutions. Debate continues on the role of prophylactic anticoagulation in burn care and will be discussed, but it is important to note that here, the effects of heparin were measured at 24 h postinjury.
A more longitudinal and thorough case series on BIC in human patients was published by Bartlett et al.(13) . Numerous coagulation tests were serially conducted on 11 patients with large burns (total body surface area (TBSA) range 30%–68%) with data collected up to 5 weeks postinjury. In addition to tests of platelet counts and function, coagulation function was assessed by activated partial thromboplastin time (aPTT), prothrombin time (PT), and thrombin time (TT), as well as individual clotting factors (fibrinogen, Factor (F) V, and FVII). Again, an initial decrease in platelet function and clotting factors was observed in the first 48 h before returning toward baseline, and platelet function remained depressed even after initial resuscitation (up until 3 or 4 days postinjury) (13). The authors used these findings to propose changes in care plans of their patients that included avoiding surgical interventions until the fourth or fifth day after injury. However, the conclusions of this series must be viewed in the context of the institution-specific treatment protocols and standard practices of the time. Colloid-only resuscitation with albumin was used and average time to excision and grafting was 21 days (13). This is in stark contrast to crystalloid-based resuscitation (14) and early excision and grafting that is the practice of most burn institutions today (15, 16).
More recently, Sherren et al. (17) defined the effects of clotting alterations caused by thermal injury in their retrospective review of acute BIC. Their analyses demonstrated that 39.3% of severely-burned patients met their established criteria for coagulopathy on admission based on elevations of PT (>14.6 s), aPTT (>45 s), or international normalized ratio (INR, > 1.2) on admission labs. They found a significant correlation between PT elevation and burn severity and acute BIC was an independent predictor of 28-day mortality.
In contrast to Sherren et al. (17), in another retrospective study from the same year, Lu et al. (18) concluded that major burn injury was not associated with acute coagulopathy. Acute coagulopathy was defined as an INR of 1.3 or greater, an aPTT of 1.5 or greater times normal, and normal platelet count. Thermally-injured patients needed to meet all three criteria to be classified as having coagulopathy. Of the 102 patients included, none met criteria on admission. Lu et al. highlighted the difference of this 0% frequency with a previously established 20% frequency in blunt and penetrating trauma to assert that TIC is unique to non-burn trauma.
Perceptions regarding existence and relevance of BIC are clearly varied. The difference between time of injury and admission sampling in the retrospective studies above could explain the divergent conclusions. For example, Sherren et al. (17) reported a mean time from burn to hospital arrival of approximately 6 h, while Lu et al. presented a median time of only 2.5 h. Review of the longitudinal data presented by Lu et al. reveals increases in INR to levels above 1.3 in the days following injury for several patients. Furthermore, 68% of the patients in the Lu et al. study suffered burns between 15% and 30% TBSA, whereas all 117 patients included in the Sherren et al. review had burns of at least 30% TBSA. The extent of burn injury has been shown to correlate with development of coagulopathy (19, 20). The relatively small number of patients with burns greater than 30% TBSA in the Lu et al. study weakens their assertion that BIC does not exist.
In 2016, Glas et al. (21) summarized current understanding of BIC, presented outcomes, and highlighted the need for better management strategies for this disease process. The review aimed to clarify definitions to arrive at consensus. Here, acute BIC was defined as abnormal INR (>1.5) and aPTT (>60s) with early onset BIC defined as presence of these abnormalities in the first 24 h after injury (21). Severe burns are defined as greater than 30% TBSA and are associated with a greater coagulopathy with earlier onset. It is noted that early detection of coagulopathy is a challenge and routine tests of coagulation (PT, aPTT, INR) have limited diagnostic value, while the presence of coagulopathy is associated with increased mortality. The pathophysiology of BIC was characterized over time with a discussion of relevant clinical influences and the response of systemic markers of coagulation and inflammatory cytokines. Ultimately, the authors conclude that further work is needed to optimize the diagnosis and subsequent treatment of BIC.
The lack of consensus for monitoring BIC was highlighted in a reported survey of burn care providers conducted by Lavrentieva et al. (22) in 2016. Of the 55 respondents who completed the questionnaire, 46 burn specialists (70.8%) indicated that they only use PT, aPTT, fibrinogen, and platelets as tests to detect BIC. The remaining providers noted the use of measurements of antithrombin (AT), protein C (PC), and D-dimer levels, as well as viscoelastic methods like TEG to supplement traditional measurements. Furthermore, 41 respondents (74.5%) revealed that no specific coagulopathy scoring system was used at their institutions.
Conventional tests of coagulopathy, such as PT, INR, and aPTT, do not reliably detect the presence of coagulopathy in burn patients, and global assay methods (i.e., viscoelastic and thrombin generation assays [TGA]) are more informative. Viscoelastic testing of whole-blood clotting uses technology that has been in existence since it was introduced by Hartert (9) in 1948. Viscoelastic testing is conducted using TEG or rotational thromboelastometry (ROTEM). TGAs are plasma-based assays that can identify phenotypic differences in thrombin generation that can contribute to altered hemostatic states and coagulopathy (23–26) and provide more information beyond the clotting end point. Wiegele et al. (27) showed a procoagulant state in burn patients in the first 2 weeks following injury reflected by TGA and ROTEM, while conventional assays including PT, aPTT, and platelet count remained within reference ranges. The authors concluded that further studies correlating TGA and ROTEM results and clinical outcomes are needed to potentially develop patient-specific clinical interventions. A recent review in 2019 suggests that the use of viscoelastography during intraoperative blood product resuscitation in burn surgery is associated with decreased transfusion requirements (28).
The greater availability and array of laboratory and point of care testinghas led to a deeper understanding of BIC. In turn, greater understanding of BIC informs the selection of diagnostic tests. In recent years user-friendly point-of-care viscoelastic devices have enabled the development of treatment algorithms aimed at identifying and managing coagulopathy (2, 28–31). Nonetheless, there is no consensus on how best to diagnose BIC, potentially related to an incomplete understanding of the pathophysiology.
MECHANISMS CONTROLLING HEMOSTATIC BALANCE: INTERACTIONS BETWEEN COAGULATION, FIBRINOLYSIS, AND INFLAMMATION
There are two fundamental mechanisms for triggering a coagulation, the exposure of blood to functional tissue factor (TF) (extrinsic pathway) and the exposure of blood to materials capable of supporting FXII autoactivation (intrinsic/contact pathway). Functional TF interactions with blood arise through two primary routes: exposure of blood to extravascular cells such as smooth muscle cells and fibroblasts (which constitutively express TF on their surfaces); and intravascular display of functional TF on the surface of white cells (monocytes) and/or endothelial cells which occurs in response to agonists (i.e., inflammatory cytokines). In the first scenario, TF exposure is coincident with the exposure of extracellular matrix proteins that support the adhesion/activation of platelets thus localizing platelets and TF at the injury site. With respect to a coagulant response dependent on FXII activation, the mechanisms include the release into the blood of bacterial products (sepsis) and the presence of intracellular material such as DNA-histone complexes due to physical disruption of cells or the induction of apoptosis. Extravascular TF functions to initiate a localized coagulant response upon vessel injury with the goal of forming and maintaining a physical barrier (fibrin/platelet clot) that restores the separation between the circulatory system and the extravascular space. In contrast, the presence of initiators of coagulation in circulating blood, whether molecules triggering FXII activation or intravascular cells expressing TF, is often a part of a secondary consequence of injury or is associated with disease states.
The enzyme thrombin is the critical product of this response to vascular injury, displaying procoagulant, anticoagulant, antifibrinolytic, and cellular effects (32, 33). Figure 2 summarizes the series of enzymatic events for each of the pathways leading to thrombin generation. Key roles of thrombin in the initial phase of the response to vascular injury include: formation of fibrin from fibrinogen; activation of platelets; activation of FXIII which crosslinks fibrin subunits improving the mechanical stability of the fibrin matrix; and activation of thrombin activatable fibrinolysis inhibitor (TAFI), a carboxypeptidase that modifies fibrin making it more resistant to lysis.
Fig. 2.
Pathways to thrombin generation: Extrinsic pathway: Extravascular TF, exposed as a result of vascular injury, forms a complex with circulating FVIIa forming the extrinsic tenase (“1”) which activates FIX and FX.
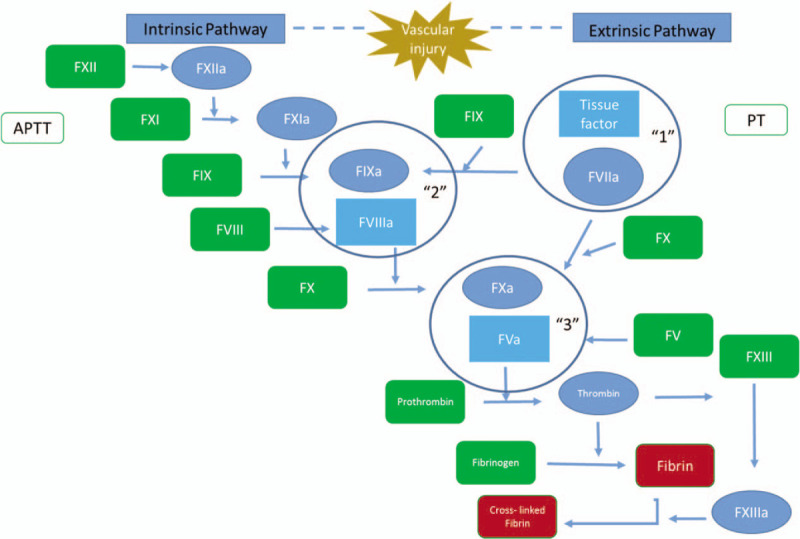
Direct activation of prothrombin by FXa results in trace amounts of thrombin which: rapidly activates the procofacors FVIII and FV to their functional forms, FVIIIa and FVa; and activates platelets Activated platelets provide separate binding sites for the intrinsic tenase (“2”) and the prothrombinase complex (“3”). The intrinsic tenase provides the majority of FXa used to assemble the prothrombinase complex while the prothrombinase complex is the primary source of thrombin needed to propagate the clotting reaction. The PT assay relies on the use of very high concentrations of TF, thus eliminating the need for the intrinsic tenase production of FXa. Intrinsic pathway: Initiation of the intrinsic pathway to thrombin formation requires the exposure of blood to materials capable of supporting FXII autoactivation. FXIIa then activates FXI; FXIa is an extremely efficient activator of FIX. FIXa directly activates FX to FXa which then directly activates prothrombin as described above leading to the formation of both the intrinsic and prothrombinase complexes. Both the PT (utilizing the extrinsic pathway) and the aPTT, (utilizing the intrinsic pathway) assays are ideal for detecting gross coagulation defects. However, as the endpoint of these assays is the fibrin clot, these assays exclude over 95% of the thrombin generated in the complete reaction.
Negative regulation of the coagulant response is primarily accomplished by the activities of tissue factor pathway inhibitor (TFPI), AT, protein S (PS), and the PC pathway (Fig. 3). In combination, these inhibitory processes provide a threshold-limited reaction system in which a stimulus of sufficient magnitude must be provided if the reaction is to proceed. TFPI is a multivalent Kunitz type plasma proteinase inhibitor functioning as the principal inhibitor of the FVIIa-TF complex. TFPI inhibits the FVIIa-TF complex in a FXa-dependent manner and is a primary regulator of the initiation phase of thrombin generation. AT is an abundant member of the serpin proteinase inhibitory family and forms irreversible 1:1 complexes with most of the proteinases participating in the coagulant response including thrombin, FXa, FIXa, TF-FVIIa, and FXIa. AT appears to be the primary inhibitor of thrombin and deficiency is associated with increased thromboembolic events. PS functions as an inhibitory cofactor, improving the efficiency of TFPI inhibition of FVIIa-TF complex and activated PC (APC) inactivation of FVa and FVIIIa. The PC pathway is responsible primarily for limiting the conversion of prothrombin to thrombin. Consistent with these activities, PS and PC deficiency is associated with increased risk of thrombosis (34). Key components of the PC pathway include two proteins expressed on the luminal surface of endothelial cells: thrombomodulin (TM), which binds thrombin to form the PCase complex, an efficient catalyst of PC conversion to APC, and the endothelial cell PC receptor, which delivers PC to the thrombin–TM complex. APC has been shown to proteolytically inactivate FV/FVa, FVIII/FVIIIa. The formation of APC via the PC pathway is the central dynamic anticoagulant mechanism limiting clot formation to the injury site.
Fig. 3.
Pathways linking fibrin formation and fibrinolysis.
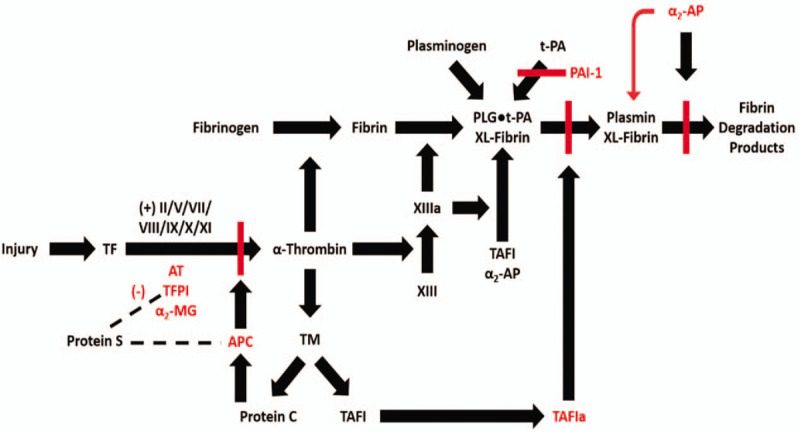
Key proteins and pathways linking the onset of coagulation at an injury site to thrombin formation, the formation of a crosslinked fibrin matrix and the process of fibrin lysis are shown. Key inhibitors and their targeted pathways are shown in red. α2-AP indicates α2-antiplasmin; α2-MG, α2-macroglobulin; PLG, plasminogen; XL-Fibrin, cross-linked fibrin.
The presence of fibrin triggers a complementary pathway, the fibrinolytic system, which plays an important role in down regulating the physical extension of the clot and in the ultimate restoration of vessel structure and integrity (Fig. 3). Key components of the fibrinolytic pathway include: plasminogen, the zymogen precursor of the enzyme plasmin which degrades fibrin, t-PA (tissue plasminogen activator), which catalyzes the conversion of plasminogen to plasmin, PAI-1, (plasminogen activator inhibitor 1), the primary plasma inhibitor of t-PA; alpha-2-antiplasmin (AP), the primary plasma inhibitor of plasmin; and TAFI, which in its activated form works to suppress plasminogen activation on the fibrin surface. The fibrin component of the blood clot acts as a cofactor for the activation of plasminogen by t-PA, leading to efficient production of plasmin only at the site of fibrin deposition.
The complex interplay of inflammation with coagulation through an induced expression of TF has been described (35). Interleukin-6 (IL-6) and interleukin-8 (IL-8) have been shown to increase monocyte TF expression in vitro while other inflammatory cytokines such as IL-1 and tumor necrosis factor-α (TNF-α) have been shown to induce the tethering of TF-bearing micro-particles from monocytes at the endothelial surface (36). This would augment the pre-existing pro-coagulant effects of TF in the blood. A study of endotoxemia in humans reported that TF mRNA expression increased 100-fold following lipopolysaccharide injection (37). A rise in thrombin–antithrombin complex (TAT) and prothrombin fragment 1.2 (F1.2) closely followed the elevation in TF mRNA indicating a resulting increase in thrombin activity (37).
Inflammation also increases coagulation through the inhibition of fibrinolysis (35). PAI-1 is stimulated by IL-6 and TNF-α, contributing to the inhibition of fibrinolysis (38). Furthermore, the down regulation of TM and PC by IL-1 and TNF-α can promote the spread of coagulation beyond directly injured tissue (38).
In subsequent sections, we will examine a more global picture of burn injury to include studies that investigate procoagulant, anticoagulant, fibrinolytic, and inflammatory changes during the acute phase in the first 48 h and beyond.
THE FIRST 48 HOURS: BURN SHOCK AND ACUTE RESUSCITATION PHASE
The first 48 h of care for burn patients differ markedly from that of other trauma patients due to myriad of factors including fluid resuscitation, inhalation injury, and dysfunctional integument (Fig. 1). Derangements in coagulation and fibrinolysis can manifest rapidly after injury, Figure 3 graphically depicts the relationships between many of these factors. Given the acute hyperinflammatory state of individuals with severe burns, the inflammatory cascade likely contributes to coagulopathy.
Procoagulant factors
In the setting of thermal injury, the most commonly studied components used to assess the level of hemostatic dysfunction are platelets, fibrinogen, and two markers of in vivo thrombin generation, the TAT complex and prothrombin activation fragment 1.2, a noncatalytic peptide released during the conversion of prothombin to thrombin (Table 1). More recently thrombin generation potential has been measured using direct (TGA), and indirect (TEG/ROTEM) measurements, as well as mechanism based computational modeling (27, 39).
Table 1.
Effects of burn injury on procoagulants

Overwhelmingly, platelet levels are found to be unchanged by acute thermal injury (40–47). There are a few studies that suggest platelet counts are elevated as early as admission in thermally injured patients (47–49), which may reflect variability in time to measurement following injury. Lawrence and Atac (50) concluded that intravascular hemolysis and fragmentation of erythrocytes can result in pseudothrombocytosis, which may occur during the early resuscitation period. Thrombocytopenia on admission may reflect early disseminated intravascular coagulation (DIC) (47). Growing use of viscoelastic measurements has suggested that in the setting of normal platelet counts, platelet function is diminished (27, 43, 45).
Fibrinogen levels increase in the first 48 h after burn injury (40, 41, 48, 49), and in the preoperative period before excision and grafting (43, 46, 51), although one study notes normal or decreased levels in burn patients exhibiting severe coagulopathy and DIC (47). Levin and Egorihina (49) reported an association between increasing fibrinogen levels in burn patients and a decrease in the degree and velocity of platelet aggregation, which would clearly impact clot formation.
TAT levels increase immediately post burn and peak within 48 h (44, 52–59). The time frame of the onset of this change has been more consistent than for other markers, occurring within the first 24 h (53, 56). Interestingly, TAT levels correlate with burn severity. Two separate studies reported elevated TAT levels in all patients, with significantly higher levels among nonsurvivors (54, 58). Consistent with data for TAT, several studies have found F1.2 levels to be elevated within the first 24 h, often in proportion to TBSA. Burn patients who developed DIC exhibited even higher elevations (45, 47, 58, 59).
Increased F1.2 and TAT levels in the first 48 h post-burn, suggest ongoing systemic thrombin generation. However, the increased free thrombin levels do not appear sufficient to affect the elevated level of fibrinogen. Independent of fibrinogen, thrombin has numerous roles in coagulation homeostasis, inflammation, and in cellular functions such as growth, migration, and regulation of protein synthesis and secretion (60). Thrombin generation assays (27) and computational thrombin modeling (39) have demonstrated a shift toward increased thrombin generation potential following burn injury. This is consistent with the data for the in vivo thrombin generation surrogates F1.2 and TAT. If viewed in context with other hemostatic markers, measuring F1.2 and TAT may help to paint a more comprehensive picture of the progression of coagulopathy in burns.
Several studies have investigated factors within the coagulation cascade in the first 48 h after burn injury, including FV, FVII, and FVIII. FV levels are normal within the first 48 h of injury (40). This suggests that the observed increase in thrombin generation is not substantial enough to deplete FV levels. Additionally, it may point to an underlying difference in the mechanisms underlying hemorrhage risk in thermal versus mechanical trauma. In TIC, reductions in the levels of FV due to APC-mediated proteolysis have been advanced as an important contributor to bleeding risk (61–63).
FVIIa activity appears elevated in the first 48 h after thermal injury (54, 55). FVIIa may have use as a prognostic indicator. Garcia-Avello et al. (54) found FVIIa levels to be higher in non-survivors compared with survivors and controls within 24 h after burn (post-burn day 1, PBD 1). The activity of FVIIa is elevated in burn patients when compared with healthy controls, indicating a hypercoagulable state (55). Increasing levels of FVIIa correspond to the severity of injury in the acute phase (54). Increased levels of FVII activating protease have been reported in trauma patients, thus providing a potential mechanism for the observed higher levels of FVIIa (64).
Lastly, FVIII rapidly increases in the first 48 h and remains elevated above normal values up to 40 days postinjury (40, 45, 48). The extent of FVIII elevation in the acute phase correlates with TBSA (45). FVIII is stored along with von-Willebrand Factor (vWF) within endothelial cell Wiebel-Palade Bodies (WPB) (65). The exocytosis of vWF from WPB into the vascular lumen is an important initiator of primary hemostasis in response to vascular endothelial injury (66). Elevated FVIII levels may be a result of endothelial damage in burned tissue and subsequent WPB exocytosis. In an environment of hemostatic activation, as in burn injury, increasing concentrations of FVIII may work to accelerate production of FXa and thrombin (Fig. 2).
Anticoagulant system
Endogenous anticoagulants have been better characterized in burn injury relative to procoagulants. Factors such as AT, PC, and PS, TM, and TFPI inhibit coagulation by attenuating or suppressing the effects of procoagulant factors (Fig. 3). In burns, three of these factors (AT, PC, and PS) have been shown to decrease during the first 48 h after injury, while the other two (TM and TFPI) increase or remain at normal levels (Table 2).
Table 2.
Effects of burn injury on anticoagulants

AT is the primary inhibitor of key coagulation enzymes including thrombin and FXa. Thus, decreased AT levels reflect reduced capacity to suppress coagulation. Within 48 h of burn injury, there is a marked decrease in AT levels (43–45, 47, 48, 52–55, 58, 59, 67, 68). Consistent with F1.2 and TAT level increases after burn injury, the extent of AT level decrease following burn injury is correlated with severity (52, 55). From a prognostic side, survivors return to near normal levels while non-survivors exhibit persistently depressed levels (55).
PC and PS are key components of the anticoagulant response. The concentrations of PC and PS decrease immediately after thermal injury (45, 54, 55, 58, 59, 69). Loss of both PC and PS following burn injury results in a simultaneous reduction in the capacity to suppress coagulation, with potentially increased thrombotic risk (55), and decreased inhibition of inflammation (35). Levels of PC and PS have been investigated as prognostic indicators; significant lower PC and PS levels are found in non-survivors and with larger burns, as compared with healthy controls (54, 58, 69, 70).
TM is a transmembrane glycoprotein expressed on the luminal surface of endothelial cells (71). It functions as a high affinity receptor for thrombin and the resulting complex is the primary physiologic activator of PC. Assessment of TM following injury generally depends on the measurement of fragments released by the proteolysis of the TM extracellular domain (72), measured as soluble TM. In a rabbit burn model, thermal injury was found to produce shedding of TM from the endothelium (71), effectively removing it from its anticoagulant role in the activation of PC. In human patients, Aoki et al. (53) found TM plasma levels to increase by 48 h after injury. Taken together, these findings suggest endothelial activation or damage following burn, results in shedding of TM from the endothelium and an increase in plasma soluble TM. Burned integument and progression of the zone of stasis may initially contribute to a hypercoagulable state through proteolysis of TM. Elevated levels of soluble TM have also been associated with TIC (61, 73).
TFPI, an inhibitor of the initiation phase of TF-mediated thrombin generation, is present on endothelial cell surfaces and circulates through plasma bound to lipoproteins. Association of TFPI with the endothelial glycocalyx provides localized control to thrombin generation, which may have implications for thrombotic complications in microvasculature adjacent to burns (74). It has been shown in non-burn trauma that despite markedly increased levels of thrombin 24 h after injury, there are no compensatory increases in TFPI levels (75). One of the few studies investigating TFPI levels in burns by Ravindranath et al.(76) found TFPI levels in rats with 30% TBSA wounds to be decreased significantly at 24 h after burn. In contrast, Kowal-Vern et al.(77) found in humans that TFPI levels remain normal during the acute and restorative phases after large burn injury, consistent with observations in non-burn trauma patients. The available data suggests TFPI is not significantly altered by thermal injury in humans. A decrease in TFPI would be cause for concern as it would add to a hypercoagulable state.
Fibrinolysis
After burn injury, pro-fibrinolytic enzymes increase in activity and concentration. The most frequently studied are t-PA and plasminogen. t-PA concentration increases during the acute phase of burn injury. When comparing survivors and non-survivors, both Garcia-Avello et al. and Lavrentieva et al. found t-PA levels to be elevated in both groups, with non-survivors having higher levels immediately after burn injury (54, 58). It is important to note that increased t-PA levels alone are not indicative of increased fibrinolytic potential. Plasminogen levels decrease immediately after burn, proportionately to the severity of the injury (41, 54, 55). While there is a rapid increase in pro-fibrinolytic activity following burn injury, increases in anti-fibrinolytic factors are also observed. PAI-1 and AP are the most widely studied fibrinolytic inhibitors while plasmin-α2-antiplasmin complex (PAP) is measured to infer the amount of plasmin inhibition (Table 3). PAI-1 binds with t-PA and urokinase plasminogen activator (u-PA) to prevent the activation of plasminogen to plasmin. PAI-1 levels increase and peak immediately after burn injury, then proceed to decrease in concentration toward normal or near normal levels (53–55, 58, 59, 77). This trend is similarly exhibited by t-PA levels, several studies have noted that both the fibrinolytic activator (t-PA) and inhibitor (PAI-1) elevate in tandem correlating with burn size and mortality (54, 55, 58). Aoki et al. (53) studied 15 patients in a detailed analysis of PAI-1. An increase in free PAI-1 was observed earlier and of greater magnitude (40 times healthy control) than the elevation in t-PA. The authors suggested the greater increase in PAI-1 compared with t-PA immediately after burn injury may be explained by the behavior of PAI-1 as an acute-phase reactant. The authors go on to speculate that immediate post-burn suppression of fibrinolysis may be necessary to preserve coagulation homeostasis after thermal injury, but that secondary activation of the fibrinolytic system in response to fibrin deposition may lead to a paradoxical state of hypofibrinolysis coexisting with targeted fibrinolysis (53). Several phenotypes of fibrinolysis have been described following traumatic injury. While the hyperfibrinolytic phenotype is associated with a doubling of mortality, the subset of non-burn trauma patients presenting with fibrinolytic shutdown also experience poor outcomes (78, 79). These fibrinolytic phenotypes are less well studied in the burn population.
Table 3.
Effects of burn injury on fibrinolysis

AP is the primary inhibitor of activated plasmin; unlike PAI-1, it is observed to decrease after burn injury (54, 77). From a prognostic standpoint, various authors have found AP levels to be low immediately post-burn, with non-survivors having lower levels compared with survivors. However, AP levels eventually normalized in both groups (54, 77). Interestingly, plasminogen levels follow a similar trend to AP, in that both factors are decreased within the first 48 h after injury (44, 54). This could be explained by a progression of rapid conversion of plasminogen to plasmin resulting in lowered levels of plasminogen. As plasminogen is converted to plasmin, AP quickly complexes and is consumed, resulting in depressed AP levels. This hypothetical progression is further supported by evidence in other studies that PAP complex levels increase immediately post burn (53). However, PAP levels remained within normal physiologic range for both survival and non-survival burn patients, despite a marked increase in PAI and moderate increase in t-PA after injury (58, 59). The lack of PAP increase in this more recent data can be explained by a number of scenarios. Either AP and activated plasmin are not complexing efficiently, AP production is inhibited or depressed, or the higher increase of PAI-1 relative to t-PA is preventing excessive activation of plasmin (80). Another alternative could simply be technical; the rapid degradation or clearance of PAP after complex formation could result in the lack of change observed within this data.
The last metric used to measure fibrinolytic activity is the presence of fibrin degradation products (FDP), the most notable being D-dimer (81, 82). D-Dimer is a marker that reflects activation of coagulation and fibrinolysis, it is a sensitive but not specific marker, with elevated levels found in a myriad of disease states (83). Although various studies report that burn patient's D-dimer levels are elevated upon admission (43–45, 47, 54–59, 77, 84), further investigation is needed to determine its clinical utility. Given local tissue damage and the resulting hyperinflammatory state following burn injury, elevated D-dimer levels are not surprising. As stated earlier, there seems to be a greater increase of PAI-1 relative to t-PA during the acute phase of injury suggesting an initial systemic hypofibrinolytic state (53). In one study, eight of 36 patients showed no clot lysis after 24 h when assessed using the euglobulin clot lysis time assay and had a significantly lower plasma ratio of PAI-1/t-PA complex to free PAI-1 (53). Elevated D-dimer may therefore be due to an excessive amount of fibrin clot formation; the local fibrin degradation at the site of burn injury raising the level of FDPs (53, 54, 85).
INFLAMMATORY FACTORS
Burn injury causes both a local inflammatory response to coagulative necrosis as well as a systemic inflammatory state secondary to circulating cytokines (86). Many pro-inflammatory molecules are elevated in the acute phase in response to burn injury (85, 87, 88). IL-1, IL-6, IL-12, and TNF-α are elevated by 24 h and levels are significantly higher in burn patients who die of their injuries at 48 h (85). Burn injury is associated with a systemic inflammatory response that is associated with injury severity and precedes a hypermetabolic state (89). Cytokine derangements are more severe in adult patients when compared with children and this heightened immunoinflammatory response has been suggested as a contributor to the increased morbidity and mortality observed in adults (87). Matsuura et al. examined a cytokine network including monocyte chemoattractant protein-1 (MCP-1), IL-6, IL-8, and IL-10 and showed that cytokine derangements in burn patients with > 20% TBSA compared with healthy controls were correlated with burn injury severity and prognosis (90). When compared with non-burn trauma patients, patients with severe burns were found to have higher levels of IL-6 and IL-8 as early as 24 h after injury (91). The inflammatory state, specifically IL-6, IL-8, TNF-α, among other cytokines have been shown to shift the hemostatic balance towards coagulation (35). However, the interplay between the inflammatory response to burn injury and its effects on BIC has not been well studied.
Summary
Overall, in the first 48 h thermally-injured patients appear to exhibit a net increase in pro-coagulant potential. Studies of the components of the fibrinolytic cascade provide evidence of both hyper- and hypofibrinolysis. Whether these changes are part of a larger phenomenon unique to burn injury or rather, are part of a general response to trauma is uncertain.
Caution must be taken when trying to define the first 48 h after burn injury, as a hyper- or hypocoagulable state based solely on the levels of individual markers. Indeed, studies have demonstrated induction of both states following burn injury. Global coagulation assays have the potential to better characterize individual differences in the dynamics of clot formation and lysis among patients. In a study on TEG measurements of 65 patients with > 15% TBSA injuries, Huzar et al. (92) found 60% of patients to be hypercoagulable on admission while 24% were hypocoagulable. Identifying potential factors that predispose patients to hyper- or hypocoagulability, such as severity of injury, pre-existing conditions, or demographics, must be further explored in future research.
FORTY-EIGHT HOURS AND BEYOND: POST BURN SHOCK RESUSCITATION
Once a patient survives the acute phase of burn injury, he or she moves into a period wherein infections, protracted hyper-metabolism, and hospital convalescence impact outcomes. During this period, excision and grafting is initiated to mitigate the impact of sustained exposure to thermally damaged tissue (93, 94). During these surgeries burned and devitalized tissue is excised and replaced with auto-, allo-, or xenografts to gain temporary or permanent skin resurfacing. The ability to make and sustain a clot plays a pivotal role in the ability to perform safe operations. Of note, while levels of a few of the components of coagulation discussed in the first 48 h of injury change in this time frame, most remain abnormally elevated or depressed. Longitudinal data studying BIC are limited.
Procoagulant
After 48 h, many studies identify an increase in platelet levels (41, 42, 44, 48, 95). In the presence of infection, Housinger et al. (96) determined falling platelet counts to be an independent predictor of sepsis in severely burned pediatric patients. Cowan et al. (40) noted mean platelet counts were significantly lower in adult burn patients who developed sepsis, and that development of dysfunctional platelet aggregation was more reliably correlated with sepsis than lower count. Burn patients generally present with normal platelet counts and develop a thrombocytosis following the acute phase (Table 1). Dysfunctional platelets at any point after burn injury correlate with poor outcomes. TAT and FVII activity remain elevated in the week following burn injury, suggesting hypercoagulability (54, 55). While overall TAT values in burn patients return toward normal over time, average levels are higher in larger TBSA injury (56) and in non-survivors (54, 58). FVIII activity is elevated in the days following burn injury during excision and grafting while the activities of FII and FX are depressed (43). The differential activity of FVIII has been rationalized by its function as an acute phase reactant. Factor activities were shown to decrease perioperatively reflecting a consumptive process secondary to intraoperative blood loss (42, 43). FVIII activity is higher in non-survivors and FII functional activity is lower with larger burns (85). F1.2 remains elevated for 5 to 7 days but is not significantly higher than on admission (47) or by mortality (58). At 1 to 2 weeks following burn injury, the potential to generate thrombin appears to increase (27). Fibrinogen levels peak within the first week following burn injury and remain elevated (27, 41). Consistent with factor activity, fibrinogen levels fall in the perioperative period with blood loss (42, 43).
Anticoagulant
AT activity is depressed after thermal injury (53, 58). The depression of AT activity tends to be more severe with increasing TBSA (45, 77). Decreased AT activity correlated with poor outcomes including increased length of stay and mortality (67). The perioperative period further exacerbates AT activity depression (43). Kowal-Vern et al. have published case reports on systemic infusion of AT concentrate to reduce microvascular thrombosis associated with burn wound progression (44, 68). PC and PS levels tend to be decreased in the days after burn injury (45, 54, 55, 85) (Table 2). PC and PS normalization following burn injury has been associated with survival. All patients had reductions in PC and PS on admission; survivors’ PC levels were normalized by day 5 and PS by day 7, while PC/PS remained low in non-survivors (58). As discussed, normalization of AT, PC, and PS after 48 h has been associated with survival.
Soluble TM is typically elevated by 48 h and continues to be elevated as far out as 7 days from injury (53). In addition, soluble TM levels in humans have been shown to closely parallel TNF-α increases after burn injury, suggesting TNF-α stimulates TM production (97). Plasma TM levels were found to be significantly higher in non-survivors, as well as those who developed sepsis (97), perhaps reflecting an ongoing state of endothelial activation or damage. TFPI levels are generally within normal range following burns, however, TFPI levels are elevated at later timepoints in non-survivors, and this increase was shown to be significant at hour 60 (85). Again, this clinical finding differs from TFPI activity described in a rat model of sepsis following burn injury. TFPI activity levels were decreased in animals subjected to either burn injury or sepsis alone, with even lower activity when combinatory insults occurred, this decrease was most pronounced at 24 h with a rebound to near normal TFPI activity at 72 h (76). In humans, TFPI levels are found to be increased in several disease states, including sepsis (98, 99). However, interpretation of TFPI levels as a biomarker of coagulopathy can be misleading, given its various isoforms and distribution within the body, and varied methods of measurement (100).
Fibrinolysis
Most studies describe an acute rise in tPA levels within 24 h of burn injury that normalizes within the first week (53–55). Mortality may influence tPA levels, in one study survivors were within normal range and non-survivors had persistently elevated levels (58). This survivor – non-survivor difference was also observed for PAI-1, though not for PAP, which remained normal throughout, or D-Dimer, which remained elevated (58). It has been reported that plasminogen levels gradually increase to normal or near normal physiologic levels by post-burn day 5. When compared by mortality, the drop and subsequent return to normal levels for plasminogen was comparable (54). When plasminogen levels are compared by burn severity, larger burns experienced a sharper decrease and the return to normal levels was slower (55). Overall, it seems plasminogen levels are sensitive to burn severity but are not indicative of mortality (Table 3).
The relationship between plasminogen levels and burn severity may be illustrating a “dose-dependent” activation of fibrinolytic activity: greater severity burns result in more fibrinolysis and drop in plasminogen. If evaluated in tandem with observed t-PA activity, there is evidence that profibrinolytic factors increase their activity, as t-PA levels increase, plasminogen levels fall. Circulating PAI-1 effectively inhibits tPA. tPA is released locally in response to burned tissue as microvasculature thrombosis develops. Relatively greater disturbances in this homeostasis are observed in non-surviving patients (53). It is possible that ineffective fibrinolysis contributes to burn wound progression or organ system dysfunction.
INFLAMMATORY FACTORS
The inflammatory and hypermetabolic state that develops after burn injury persists following the acute phase (90, 101). Nonsurvivors have significantly higher plasma levels of IL-1, IL-6, IL-10, IL-12, and TNF-α at 1 week from burn injury when compared with survivors (85). Septic complications are devastating sources of morbidity and mortality in burn patients. Burn injury alone causes an inflammatory state, infection, and sepsis compound this inflammatory response (102). Lantos et al. (103) observed an acute and sustained elevation in both pro- and anti-inflammatory cytokines in response to burn injury, consistent with other studies. Furthermore, IL-10 which suppresses immune function and predisposes patients to infection was found to be significantly higher at admission and perimortem in nonsurvivors (103). Sepsis has been associated with patterns of gene expression such as increased concentrations of mRNA for IL-10 and TNF-α (104). In pediatric patients, IL-8 levels were correlated with injury severity, infection, and mortality and were suggested as a sensitive and specific prognostic biomarker (105). Sepsis can be associated with DIC characterized by alterations in fibrinolysis that result in microvascular thrombotic complications (106). Burn injury and subsequent injury-specific complications such as infection and sepsis are independently associated with the inflammatory state. Following the acute phase of injury, the complex interplay between these factors and their impact on coagulation homeostasis are not well characterized.
Summary
Based on current studies, the period following acute burn injury is characterized by either maintenance of admission levels of coagulation factors or recovery to normal range values of said factors. In general, a hypercoagulable state persists for days to weeks. Derangements in biomarkers of coagulation are often correlated with size of burn (TBSA), as well as mortality. Excision and grafting impacts factor function, causing a consumptive coagulopathy, which may underlie bleeding complications. Sepsis, a common complication of major burn injury, further impacts coagulation causing platelet dysfunction as well as increases in the release of the endogenous anticoagulant TFPI. The hyperinflammatory state caused by burn injury persists over time, and infection and sepsis further exacerbate inflammation and may impact coagulation. Normalization of fibrinolytic factors seems to be associated with survival, though that data still needs to be explored. The paucity of longitudinal data characterizing BIC outside of the acute phase underscores the need for further research.
CLINICAL COMPLICATIONS OF BURN-INDUCED COAGULOPATHY
The advances in the general understanding of BIC in the last half-century have led to improvements in outcomes for burn patients through targeted thrombotic prophylaxis and hemorrhage control. Applications of the understanding of BIC and future directions that will be discussed in the following sections arose from the foundations built by these pioneers in coagulopathy research and burn care.
Venous thromboembolism
Given the recognized hypercoagulable state and potential for hypofibrinolysis after thermal injury, the development of deep vein thrombosis (DVT) and venous thromboembolism (VTE) in burn patients is a concern. Burn patients often meet all criteria of Virchow's triad for the pathogenesis of VTE—endothelial injury, venous stasis, and hypercoagulability (107). The actual incidence and significance of DVT and VTE after burn injury, however, remains unclear.
In 2015, Meizoso et al. (108) published a comprehensive review of VTE in burn patients. Through their review of over 50 studies, the authors discovered reported incidence of VTE from 0.2% to 25%. The wide range can be explained by varying methodology among studies. Higher incidence is found in those studies with autopsy or with sonographic screening of all patients. Lower incidence is reported when investigating only symptomatic patients. The incidence of potentially fatal consequences of DVT like pulmonary embolism (PE) has been reported to be as low as 0.001%, but this figure was derived from a study that also included patients with minor injuries (109). Others have reported rates of PE as high as 3.3% (110).
Given the wide range of reported incidence of thrombotic events, it is not surprising to find disparate views on the use of chemoprophylaxis for DVT and VTE in burn patients. In a retrospective analysis of a burn registry data, Sikora and Papp (111) determined that although burn severity was associated with VTE complications, chemoprophylaxis did not prevent VTEs. Other authors have proposed that bleeding risks associated with chemoprophylaxis, especially heparin, outweigh the potential benefits (112).
In contrast, Ahuja et al. (113) published a randomized, controlled trial in 2016 of 100 patients with 30% to 60% TBSA burns that examined DVT risk factors and the use of chemoprophylaxis. In the control group of 50 patients that did not receive chemoprophylaxis, four patients (8%) had sonographic evidence of DVT, compared with none of the patients on chemoprophylaxis. One patient in the treatment group developed epistaxis; no other complications associated with treatment were noted. The authors concluded that the benefit of DVT prevention outweighed bleeding risk in burn patients. These findings should be validated in future studies to develop consensus on chemoprophylaxis among providers.
Even when providers opt for chemoprophylaxis, studies have shown that dosing may be inadequate given burn pathophysiology. By monitoring anti-FXa levels, Lin et al. (114) found that standard dosing of enoxaparin in burn patients failed to achieve target anticoagulation. Thus, conflicting evidence on incidence, clinical significance, and prevention of VTE in burn patients obfuscates guidelines on chemoprophylaxis.
Disseminated intravascular coagulation (DIC)
DIC is a consumptive coagulopathy in which excessive, systemic activation of coagulation results in depletion of platelets and coagulation factors (115). Subsequently, diffuse bleeding can occur and lead to poor outcomes. In a 2010 review, Lippi et al. (116) examined the development of DIC in burn patients. As with VTE, there was variability in reported prevalence and recognition of DIC in this population.
The rarity of DIC in burn injury was argued by Barrett and Gomez in a retrospective review of 3,331 consecutive burn patients admitted over 9 years (117). Of all patients, 454 patients (13.6%) sustained burns greater than 20% TBSA. Only three patients were diagnosed with DIC—0.09% of all study patients and 0.66% of patients with TBSA > 20%. The authors used discharge diagnoses of DIC and reviewed clinical/autopsy data to identify other potential patients with DIC.
In contrast to these findings, Lavrentieva et al. (58) reported that in patients with greater than 25% TBSA, the prevalence of a DIC phenotype was 91%. Here, the authors used a scoring system developed by the International Society of Thrombosis and Hemostasis to identify patients with DIC that is based on markers such as platelet count, fibrin split products, prolonged PT, and decreased fibrinogen (118). The wide range of DIC reported in the burn population highlights the need for uniform definitions and large population studies.
INTRAOPERATIVE BLOOD LOSS
Intraoperative blood loss is a challenge in burn surgery and coagulopathy with transfusion requirement is associated with poor outcomes. Niemi et al. (43) studied perioperative hemostatic parameters in burn patients and identified an intraoperative depletion of factors, fibrinogen, and platelets with resulting coagulopathy consistent with a consumptive process. Several studies have described a similar phenomenon, where burn patients typically show preoperative factor, platelet, and fibrinogen levels above reference ranges that fall predictably during excision and grafting (42, 95).
In a recent review, Welling et al. (28) concluded that coagulopathy in burn patients is a heterogenous disease process with a described hypercoagulable state following injury, but that intraoperative hemorrhage, hypothermia, and hemodilution can lead to significant hemorrhage. On review of the data, they suggested that a more protocolized use of viscoelastic assays such as TEG and goal-directed blood product resuscitation can reduce transfusion requirements and improve outcomes (28). In a prospective study randomizing 30 severely burned patients undergoing excision and grafting into a TEG-based resuscitation algorithm versus standard of care, cumulative blood product use was reduced by more than half (46). In this study, the treatment algorithm was based on recommendations for TIC, highlighting the need for burn-specific protocols.
BURN WOUND CONVERSION
The zones of burn wounds were first described by Jackson in 1953. The central zone of coagulation is surrounded by a zone of stasis, and an outermost zone of hyperemia (119). Burn wound conversion is a significant source of morbidity with further tissue loss over time within the zone of stasis, which is characterized by coagulation, inflammation, and resulting ischemia. Investigations into the use of anticoagulant and anti-inflammatory agents to prevent burn wound progression have been undertaken in animal models with varied results (120). One study investigated the administration of t-PA to prevent burn wound progression in a rat model. They found significantly greater percentages of interspace viability (87.8% vs. 31.8%) at 1 week in the t-PA treatment group (121). In a similar rat burn model, Meyerholz et al. (122) studied the utility of APC, and found that it increased burn depth severity, local inflammation, and tissue damage.
Kowal-Vern et al. conducted a prospective trial on administration of human AT concentrate to burn patients with injuries greater than 20% TBSA. AT plasma levels were reduced on admission for all burn patients, treatment with AT was initiated within 24 h of burn injury, and plasma levels improved over the course of treatment in the first 4 days following injury. The treatment group showed shorter time to graft healing in all body regions, and this difference reached significance for the hands. The authors concluded that human AT concentrate infusion is safe and suggested clinical trials to confirm the potential to improve clinical outcomes (77). More recent reviews on the topic of AT therapy in burn care reaffirm that AT has been shown in small human studies and animal models to be safe with a positive effect on wound healing in burns; however, well-designed prospective clinical trials are required to establish its role as clinical adjunct in burn care (123, 124).
CONCLUSIONS AND FUTURE DIRECTIONS
Overall, current understanding of BIC proposes that alterations in the components of the coagulation, fibrinolytic, and inflammatory systems yield functional changes in clot dynamics after burn injury. These alterations differ from those seen in TIC. However, focusing only on changes in the levels of these markers may lead to erroneous conclusions about an individual's clotting dynamics and thus may lead to missed opportunities for identifying (and preventing or treating) thrombotic or hemorrhagic states. Attempts to capture these changes with conventional assays of coagulation (PT, aPTT, platelet counts) have proven to be generally insufficient in the characterization of BIC. Assays that incorporate and combine more information regarding a patient's blood clotting phenotype into a single profile will be more useful.
Many methods of identifying potential BIC have been developed and continue to be optimized. Global and dynamic assays of coagulation such as the viscoelastic assays (TEG/ROTEM) and TGA provide more comprehensive and clinically relevant assessments of coagulation homeostasis (27). The availability of TEG/ROTEM parallels utilization at the point-of-care, TGA, and computational thrombin modeling show promise in characterizing procoagulant potential in burn patients and may experience increased use with increasing availability (27, 39). Research and development of new technologies to advance trauma and critical care is ongoing. Application of these developments to burn patients is promising in regards to diagnosis of BIC and clinical decision-making, such as the management of resuscitation (92). In addition to these laboratory tests, a consensus on an appropriate BIC diagnostic scoring system will clarify the prevalence of this phenomenon across institutions.
The clinical significance and approaches to prevention and treatment of BIC continue to be debated. While this review focused on coagulopathy after thermal injury, burn teams treat injuries from a variety of mechanisms, including chemical and electrical. BIC studies often exclude these groups or include them within the larger thermal injury population without regard for potential disparities. Further studies should investigate potential manifestations of coagulopathy unique to these subgroups.
Severely-burned patients face innumerable challenges on their journey to recovery. After the initial management and resuscitation, these patients face prolonged hospital stays, multiple surgeries, increased risk of infection and sepsis, and a host of psychosocial obstacles. The addition of a progressive or unrecognized coagulopathy can further complicate this journey and lead to devastating consequences. By anticipating and better understanding BIC, providers will be able to optimize the care of their patients with reliable triage and targeted interventions.
Footnotes
The authors report no conflicts of interest.
REFERENCES
- 1.Chang R, Cardenas JC, Wade CE, Holcomb JB. Advances in the understanding of trauma-induced coagulopathy. Blood 128:1043–1049, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kornblith LZ, Moore HB, Cohen MJ. Trauma-induced coagulopathy: the past, present, and future. J Thromb Haemost 17:852–862, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stensballe J, Henriksen HH, Johansson PI. Early haemorrhage control and management of trauma-induced coagulopathy: the importance of goal-directed therapy. Curr Opin Crit Care 23:503–510, 2017. [DOI] [PubMed] [Google Scholar]
- 4.Shupp JW, Brummel-Ziedins KE, Cohen MJ, Freeman K, Hammamieh R, Mudunuri US, Orfeo T, Moffatt LT, Brownstein BH, Mann KG, et al. Military supplement: assessment of coagulation homeostasis in blunt, penetrating, and thermal trauma: guidance for a multi-center systems biology approach. Shock 52: 1S Suppl 1: 84–91, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeeshan M, Hamidi M, Kulvatunyou N, Jehan F, O’Keeffe T, Khan M, Rashdan L, Tang A, Zakaria E-R, Joseph B. 3-factor versus 4-factor pcc in coagulopathy of trauma: four is better than three. Shock 52:23–28, 2019. [DOI] [PubMed] [Google Scholar]
- 6.Blonska T, Kamienski R. [Coagulation disorders in burns]. Pol Tyg Lek (Wars) 12:1151–1153, 1957. [PubMed] [Google Scholar]
- 7.Monteil R, Reynier C. [Evolution of platelets in burns. role of heparin]. Bull Mens Soc Med Mil Fr 57:239–246, 1963. [PubMed] [Google Scholar]
- 8.Monteil R, Reynier C, Vigne J, Leherpeur H, Raby C. [Study of blood platelet variations in coagulation disorders in burned patients]. Hemostase 3:239–249, 1963. [PubMed] [Google Scholar]
- 9.Hartert H. Blutgerinnungsstudien mit der Thrombelastographie; einem neuen Untersuchungs verfahren. Klin Wochenschr 26:577–583, 1948. [DOI] [PubMed] [Google Scholar]
- 10.Holder IA, Malin LL, Fox CL., Jr Hypercoagulability after thermalinjuries: simulation by injection of skin extracts. Surgery 54:316–321, 1963. [PubMed] [Google Scholar]
- 11.Eurenius E, Rothenberg J. Platelet aggregation after thermal injury. J Lab Clin Med 83:355–363, 1974. [PubMed] [Google Scholar]
- 12.Curreri PW, Wilterdink ME, Baxter CR. Coagulation dynamics following thermal injury: effect of heparin and protamine sulfate. Ann Surg 181:161–163, 1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bartlett RH, Fong SW, Marrujo G, Hardeman J, Anderson W. Coagulation and platelet changes after thermal injury in man. Burns 7:370–377, 1981. [Google Scholar]
- 14.Cartotto R, Greenhalgh DG, Cancio C. Burn state of the science: fluid resuscitation. J Burn Care Res 38:e596–e604, 2017. [DOI] [PubMed] [Google Scholar]
- 15.Sterling JP, Heimbach DM. Hemostasis in burn surgery—a review. Burns 37:559–565, 2011. [DOI] [PubMed] [Google Scholar]
- 16.Gacto-Sanchez P. Surgical treatment and management of the severely burn patient: review and update. Med Intensiva 41:356–364, 2017. [DOI] [PubMed] [Google Scholar]
- 17.Sherren PB, Hussey J, Martin R, Kundishora T, Parker M, Emerson B. Acute burn induced coagulopathy. Burns 39:1157–1161, 2013. [DOI] [PubMed] [Google Scholar]
- 18.Lu RP, Ni A, Lin FC, Ortiz-Pujols SM, Adams SD, Monroe DM, 3rd, Whinna HC, Cairns BA, Key NS. Major burn injury is not associated with acute traumatic coagulopathy. J Trauma Acute Care Surg 74:1474–1479, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitra B, Wasiak J, Cameron PA, O’Reilly G, Dobson H, Cleland H. Early coagulopathy of major burns. Injury 44:40–43, 2013. [DOI] [PubMed] [Google Scholar]
- 20.Younan D, Griffin R, Thompson M, Swain T, Honkanen M, Crosby J, Ellis C, Pittet J-F, Kerby J. Early coagulopathy is associated with increased incidence of ventilator-associated events among burn patients. Shock 47:107–110, 2017. [DOI] [PubMed] [Google Scholar]
- 21.Glas GJ, Levi M, Schultz MJ. Coagulopathy and its management in patients with severe burns. J Thromb Haemost 14:865–874, 2016. [DOI] [PubMed] [Google Scholar]
- 22.Lavrentieva A, Depetris N, Kaimakamis E, Berardino M, Stella M. Monitoring and treatment of coagulation abnormalities in burn patients. An international survey on current practices. Ann Burns Fire Disasters 29:172–177, 2016. [PMC free article] [PubMed] [Google Scholar]
- 23.Brummel-Ziedins KE, Wolberg AS. Global assays of hemostasis. Curr Opin Hematol 21:395–403, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tripodi A. Thrombin generation assay and its application in the clinical laboratory. Clinl Chem 62:699–707, 2016. [DOI] [PubMed] [Google Scholar]
- 25.Lecut C, Peters P, Massion PB, Gothot A. Quelle place pour le test de génération de thrombine au sein du laboratoire de biologie clinique ? Ann Biol Clin (Paris) 73:137–149, 2015. [DOI] [PubMed] [Google Scholar]
- 26.Espitia O, Fouassier M, Hardouin J-B, Pistorius M-A, Agard C, Planchon B, Trossaert M, Pottier P. Thrombin generation assay in hospitalized nonsurgical patients: a new tool to assess venous thromboembolism risk? Clin Appl Thromb Hemost 23:45–51, 2017. [DOI] [PubMed] [Google Scholar]
- 27.Wiegele M, Schaden E, Koch S, Bauer D, Krall C, Adelmann D. Thrombin generation in patients with severe thermal injury. Burns 45:54–62, 2019. [DOI] [PubMed] [Google Scholar]
- 28.Welling H, Ostrowski SR, Stensballe J, Vestergaard MR, Partoft S, White J, Johansson PI. Management of bleeding in major burn surgery. Burns 45:755–762, 2019. [DOI] [PubMed] [Google Scholar]
- 29.Harrison PaML. Michelson A. Clinical Tests of Platelet Function. Platelets 3 edLondon, UK: Academic Press; 2013. 519–545. [Google Scholar]
- 30.Wiegele M, Kozek-Langenecker S, Schaden E. Point-of-care testing in burn patients. Semin Thromb Hemost 43:433–438, 2017. [DOI] [PubMed] [Google Scholar]
- 31.Baksaas-Aasen K, Van Dieren S, Balvers K, Juffermans NP, Naess PA, Rourke C, Eaglestone S, Ostrowski SR, Stensballe J, Stanworth S, et al. Data-driven development of ROTEM and TEG algorithms for the management of trauma hemorrhage: a prospective observational multicenter study. Ann Surg 270:1178–1185, 2019. [DOI] [PubMed] [Google Scholar]
- 32.Mann KG, Brummel K, Butenas S. What is all that thrombin for? J Thromb Haemost 1:1504–1514, 2003. [DOI] [PubMed] [Google Scholar]
- 33.Crawley JT, Zanardelli S, Chion CK, Lane DA. The central role of thrombin in hemostasis. J Thromb Haemost 5: Suppl 1: 95–101, 2007. [DOI] [PubMed] [Google Scholar]
- 34.Comp PC, Nixon RR, Cooper MR, Esmon CT. Familial protein S deficiency is associated with recurrent thrombosis. J Clin Invest 74:2082–2088, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Esmon CT. The impact of the inflammatory response on coagulation. Thromb Res 114:321–327, 2004. [DOI] [PubMed] [Google Scholar]
- 36.Herndon DN. Total burn care. Saunders Elsevier, 4th ed.Edinburgh; New York:2012. [Google Scholar]
- 37.Franco RF, de Jonge E, Dekkers PE, Timmerman JJ, Spek CA, van Deventer SJ, van Deursen P, van Kerkhoff L, van Gemen B, ten Cate H, et al. The in vivo kinetics of tissue factor messenger RNA expression during human endotoxemia: relationship with activation of coagulation. Blood 96:554–559, 2000. [PubMed] [Google Scholar]
- 38.Cicala C, Cirino G. Linkage between inflammation and coagulation: an update on the molecular basis of the crosstalk. Life Sci 62:1817–1824, 1998. [DOI] [PubMed] [Google Scholar]
- 39.Bravo MC, Tejiram S, McLawhorn MM, Moffatt LT, Orfeo T, Jett-Tilton M, Pusateri AE, Shupp JW, Brummel-Ziedins KE. Utilizing plasma composition data to help determine procoagulant dynamics in patients with thermal injury: a computational assessment. Mil Med 184:392–399, 2019. [DOI] [PubMed] [Google Scholar]
- 40.Cowan DH, Bowman LS, Fratianne RB, Ahmed F. Platelet aggregation as a sign of septicemia in thermal injury. A prospective study. JAMA 235:1230–1234, 1976. [PubMed] [Google Scholar]
- 41.Caprini JA, Lipp V, Zuckerman L, Vagher JP, Winchester DP. Hematologic changes following burns. J Surg Res 22:626–635, 1977. [DOI] [PubMed] [Google Scholar]
- 42.Chang P, Murray DJ, Olson JD, Pennell BJ, Lewis RW, Kealey GP. Analysis of changes in coagulation factors after postoperative blood loss in burn and non-burn patients. Burns 21:432–436, 1995. [DOI] [PubMed] [Google Scholar]
- 43.Niemi T, Svartling N, Syrjala M, Asko-Seljavaara S, Rosenberg P. Haemostatic disturbances in burned patients during early excision and skin grafting. Blood Coagul Fibrinolysis 9:19–28, 1998. [DOI] [PubMed] [Google Scholar]
- 44.Kowal-Vern A, McGill V, Walenga JM, Gamelli RL. Antithrombin III concentrate in the acute phase of thermal injury. Burns 26:97–101, 2000. [DOI] [PubMed] [Google Scholar]
- 45.King DR, Namias N, Andrews DM. Coagulation abnormalities following thermal injury. Blood Coagul Fibrinolysis 21:666–669, 2010. [DOI] [PubMed] [Google Scholar]
- 46.Schaden E, Kimberger O, Kraincuk P, Baron DM, Metnitz PG, Kozek-Langenecker S. Perioperative treatment algorithm for bleeding burn patients reduces allogeneic blood product requirements. Br J Anaesth 109:376–381, 2012. [DOI] [PubMed] [Google Scholar]
- 47.Kowal-Vern A, Walenga JM, Hoppensteadt D, Gamelli RL. Prothrombin fragment 1.2 and modified antithrombin as predictors of disseminated intravascular coagulation and thrombotic risk in thermal injury. J Burn Care Res 34:459–464, 2013. [DOI] [PubMed] [Google Scholar]
- 48.Alkjaersig N, Fletcher AP, Peden JC, Jr, Monafo WW. Fibrinogen catabolism in burned patients. J Trauma 20:154–159, 1980. [DOI] [PubMed] [Google Scholar]
- 49.Levin GY, Egorihina MN. The role of fibrinogen in aggregation of platelets in burn injury. Burns 36:806–810, 2010. [DOI] [PubMed] [Google Scholar]
- 50.Lawrence C, Atac B. Hematologic changes in massive burn injury. Crit Care Med 20:1284–1288, 1992. [DOI] [PubMed] [Google Scholar]
- 51.Galganski LA, Greenhalgh DG, Sen S, Palmieri TL. Randomized comparison of packed red blood cell-to-fresh frozen plasma transfusion ratio of 4: 1 vs 1: 1 during acute massive burn excision. J Burn Care Res 38:194–201, 2017. [DOI] [PubMed] [Google Scholar]
- 52.Ueyama M, Yamamoto I, Sawada Y. [Disseminated intravascular coagulation in the early stage after severe burn: the role of excessive thrombin generation]. Nihon Geka Gakkai Zasshi 92:907–912, 1991. [PubMed] [Google Scholar]
- 53.Aoki K, Aikawa N, Sekine K, Yamazaki M, Mimura T, Urano T, Takada A. Elevation of plasma free PAI-1 levels as an integrated endothelial response to severe burns. Burns 27:569–575, 2001. [DOI] [PubMed] [Google Scholar]
- 54.Garcia-Avello A, Lorente JA, Cesar-Perez J, Garcia-Frade LJ, Alvarado R, Arevalo JM, Navarro JL, Esteban A. Degree of hypercoagulability and hyperfibrinolysis is related to organ failure and prognosis after burn trauma. Thromb Res 89:59–64, 1998. [DOI] [PubMed] [Google Scholar]
- 55.Kowal-Vern A, Gamelli RL, Walenga JM, Hoppensteadt D, Sharp-Pucci M, Schumacher HR. The effect of burn wound size on hemostasis: a correlation of the hemostatic changes to the clinical state. J Trauma 33:50–56, 1992. [DOI] [PubMed] [Google Scholar]
- 56.Kowal-Vern A, Sharp-Pucci MM, Walenga JM, Dries DJ, Gamelli RL. Trauma and thermal injury: comparison of hemostatic and cytokine changes in the acute phase of injury. J Trauma 44:325–329, 1998. [DOI] [PubMed] [Google Scholar]
- 57.Kowal-Vern A, Walenga JM, McGill V, Gamelli RL. The impact of antithrombin (H) concentrate infusions on pulmonary function in the acute phase of thermal injury. Burns 27:52–60, 2001. [DOI] [PubMed] [Google Scholar]
- 58.Lavrentieva A, Kontakiotis T, Bitzani M, Papaioannou-Gaki G, Parlapani A, Thomareis O, Tsotsolis N, Giala MA. Early coagulation disorders after severe burn injury: impact on mortality. Intensive Care Med 34:700–706, 2008. [DOI] [PubMed] [Google Scholar]
- 59.Lavrentieva A, Kontakiotis T, Bitzani M, Parlapani A, Thomareis O, Scourtis H, Tsotsolis N, Lazaridis L, Giala M-A. The efficacy of antithrombin administration in the acute phase of burn injury. Thromb Haemost 100:286–290, 2008. [PubMed] [Google Scholar]
- 60.Crawley JTB, Zanardelli S, Chion CKNK, Lane DA. The central role of thrombin in hemostasis. J Thromb Haemost 5:95–101, 2007. [DOI] [PubMed] [Google Scholar]
- 61.Brohi K, Cohen MJ, Ganter MT, Schultz MJ, Levi M, Mackersie RC, Pittet JF. Acute coagulopathy of trauma: hypoperfusion induces systemic anticoagulation and hyperfibrinolysis. J Trauma 64:1211–1217, 2008. [DOI] [PubMed] [Google Scholar]
- 62.Cohen MJ, Call M, Nelson M, Calfee CS, Esmon CT, Brohi K, Pittet JF. Critical role of activated protein C in early coagulopathy and later organ failure, infection and death in trauma patients. Ann Surg 255:379–385, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rizoli SB, Scarpelini S, Callum J, Nascimento B, Mann KG, Pinto R, Jansen J, Tien HC. Clotting factor deficiency in early trauma-associated coagulopathy. J Trauma 71:S427–S434, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kanse SM, Gallenmueller A, Zeerleder S, Stephan F, Rannou O, Denk S, Etscheid M, Lochnit G, Krueger M, Huber-Lang M. Factor VII-activating protease is activated in multiple trauma patients and generates anaphylatoxin C5a. J Immunol 188:2858–2865, 2012. [DOI] [PubMed] [Google Scholar]
- 65.Turner NA, Moake JL. Factor VIII is synthesized in human endothelial cells, packaged in weibel-palade bodies and secreted bound to ULVWF strings. PLoS One 10:e0140740–e140750, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nightingale TD, McCormack JJ, Grimes W, Robinson C, Lopes da Silva M, White IJ, Vaughan A, Cramer LP, Cutler DF. Tuning the endothelial response: differential release of exocytic cargos from Weibel-Palade bodies. J Thromb Haemost 16:1873–1886, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Niedermayr M, Schramm W, Kamolz L, Andel D, Romer W, Hoerauf K, Zimpfer M, Andel H. Antithrombin deficiency and its relationship to severe burns. Burns 33:173–178, 2007. [DOI] [PubMed] [Google Scholar]
- 68.Kowal-Vern A, Latenser BA. Antithrombin (human) concentrate infusion in pediatric patients with > 50% TBSA burns. Burns 29:615–618, 2003. [DOI] [PubMed] [Google Scholar]
- 69.Lo SC, Lai WT, Kwok F. Protein C and protein S levels in some burn patients. Burns 20:186, 1994. [DOI] [PubMed] [Google Scholar]
- 70.Lorente JA, Garcia-Frade LJ, Landin L, de Pablo R, Torrado C, Renes E, Garcia-Avello A. Time course of hemostatic abnormalities in sepsis and its relation to outcome. Chest 103:1536–1542, 1993. [DOI] [PubMed] [Google Scholar]
- 71.von Bulow S, Hartmann T, Fuchs PC, Schrimpf C, Pallua N. Endothelial thrombomodulin (CD 141) in a rabbit burn model. Burns 31:459–464, 2005. [DOI] [PubMed] [Google Scholar]
- 72.Ikegami K, Suzuki Y, Yukioka T, Matsuda H, Shimazaki S. Endothelial cell injury, as quantified by the soluble thrombomodulin level, predicts sepsis/multiple organ dysfunction syndrome after blunt trauma. J Trauma 44:789–794, 1998. [DOI] [PubMed] [Google Scholar]
- 73.Cohen MJ, Carles M, Brohi K, Calfee CS, Rahn P, Call MS, Chesebro BB, West MA, Pittet JF. Early release of soluble receptor for advanced glycation endproducts after severe trauma in humans. J Trauma 68:1273–1278, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mann KG. Thrombin generation in hemorrhage control and vascular occlusion. Circulation 124:225–235, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Selby R, Geerts W, Ofosu FA, Craven S, Dewar L, Phillips A, Szalai JP. Hypercoagulability after trauma: hemostatic changes and relationship to venous thromboembolism. Thromb Res 124:281–287, 2009. [DOI] [PubMed] [Google Scholar]
- 76.Ravindranath TM, Goto M, Demir M, Tobu M, Kujawski MF, Hoppensteadt D, Samonte V, Iqbal O, Sayeed MM, Fareed J. Tissue factor pathway inhibitor and thrombin activatable fibrinolytic inhibitor plasma levels following burn and septic injuries in rats. Clin Appl Thromb Hemost 10:379–385, 2004. [DOI] [PubMed] [Google Scholar]
- 77.Kowal-Vern A, McGill V, Walenga JM, Gamelli RL. Antithrombin(H) concentrate infusions are safe and effective in patients with thermal injuries. J Burn Care Rehabil 21:115–127, 2000. [DOI] [PubMed] [Google Scholar]
- 78.Moore HB, Moore EE, Liras IN, Gonzalez E, Harvin JA, Holcomb JB, Sauaia A, Cotton BA. Acute fibrinolysis shutdown after injury occurs frequently and increases mortality: a multicenter evaluation of 2, 540 severely injured patients. J Am Coll Surg 222:347–355, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cardenas JC, Wade CE, Cotton BA, George MJ, Holcomb JB, Schreiber MA, White NJ. Group obotPS. TEG lysis shutdown represents coagulopathy in bleeding trauma patients: analysis of the PROPPR cohort. Shock 51:273–283, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Abdul S, Leebeek FWG, Rijken DC, Uitte de Willige S. Natural heterogeneity of α2-antiplasmin: functional and clinical consequences. Blood 127:538–545, 2016. [DOI] [PubMed] [Google Scholar]
- 81.Elsevier, Olson JD. Makowski GS. Chapter one - D-dimer: an overview of hemostasis and fibrinolysis, assays, and clinical applications. Advances in Clinical Chemistry 2015; 1–46. [DOI] [PubMed] [Google Scholar]
- 82.Hayakawa M, Maekawa K, Kushimoto S, Kato H, Sasaki J, Ogura H, Matauoka T, Uejima T, Morimura N, Ishikura H, et al. High D-Dimer levels predict a poor outcome in patients with severe trauma, even with high fibrinogen levels on arrival: a multicenter retrospective study. Shock 45:308–314, 2016. [DOI] [PubMed] [Google Scholar]
- 83.Weitz JI, Fredenburgh JC, Eikelboom JW. A test in context: D-dimer. J Am Coll Cardiol 70:2411–2420, 2017. [DOI] [PubMed] [Google Scholar]
- 84.Wahl WL, Brandt MM, Ahrns K, Corpron CA, Franklin GA. The utility of D-dimer levels in screening for thromboembolic complications in burn patients. J Burn Care Rehabil 23:439–443, 2002. [DOI] [PubMed] [Google Scholar]
- 85.Tejiram S, Brummel-Ziedins KE, Orfeo T, Mete M, Desale S, Hamilton BN, Moffatt LT, Mann KG, Tracy RP, Shupp JW. In-depth analysis of clotting dynamics in burn patients. J Surg Res 202:341–351, 2016. [DOI] [PubMed] [Google Scholar]
- 86.Ruiz-Castilla M, Roca O, Masclans JR, Barret JP. Recent advances in biomarkers in severe burns. Shock 45:117–125, 2016. [DOI] [PubMed] [Google Scholar]
- 87.Finnerty CC, Jeschke MG, Herndon DN, Gamelli R, Gibran N, Klein M, Silver G, Arnoldo B, Remick D, Tompkins RG. Investigators of the I., the Host Response Glue G. Temporal cytokine profiles in severely burned patients: a comparison of adults and children. Mol Med 14:553–560, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tapking C, Popp D, Herndon D, Branski L, Hundeshagen G, Armenta A, Busch M, Most P, Kinsky M. Cardiac dysfunction in severely burned patients: current understanding of etiology, pathophysiology and treatment. Shock 2019; [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 89.Barayan D, Vinaik R, Auger C, Knuth C, Abdullahi A, Jeschke M. Inhibition of lipolysis with acipimox attenuates post-burn white adipose tissue browning and hepatic fat infiltration. Shock 2019; [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Matsuura H, Matsumoto H, Osuka A. Clinical importance of a cytokine network in major burns. Shock 51:185–193, 2019. [DOI] [PubMed] [Google Scholar]
- 91.Mace JE, Park MS, Mora AG, Chung KK, Martini W, White CE, Holcomb JB, Merrill GA, Dubick MA, Wolf SE, et al. Differential expression of the immunoinflammatory response in trauma patients: burn vs. non-burn. Burns 38:599–606, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Huzar TF, Martinez E, Love J, George TC, Shah J, Baer L, Cross JM, Wade CE, Cotton BA. Admission rapid thrombelastography (rTEG(R)) values predict resuscitation volumes and patient outcomes after thermal injury. J Burn Care Res 39:345–352, 2018. [DOI] [PubMed] [Google Scholar]
- 93.Heimbach DM. Early burn excision and grafting. Surg Clin North Am 67:93–107, 1987. [DOI] [PubMed] [Google Scholar]
- 94.Shirani KZ, Vaughan GM, Mason ADJ, Pruitt BAJ. Update on current therapeutic approaches in burns. Shock 5:4–16, 1996. [PubMed] [Google Scholar]
- 95.Cullen JJ, Murray DJ, Kealey GP. Changes in coagulation factors in patients with burns during acute blood loss. J Burn Care Rehabil 10:517–522, 1989. [DOI] [PubMed] [Google Scholar]
- 96.Housinger TA, Brinkerhoff C, Warden GD. The relationship between platelet count, sepsis, and survival in pediatric burn patients. Arch Surg 128:65–66, 1993. [DOI] [PubMed] [Google Scholar]
- 97.Nakae H, Endo S, Inada K, Yamada Y, Takakuwa T, Yoshida M. Plasma levels of endothelin-1 and thrombomodulin in burn patients. Burns 22:594–597, 1996. [DOI] [PubMed] [Google Scholar]
- 98.Creasey AA, Reinhart K. Tissue factor pathway inhibitor activity in severe sepsis. Crit Care Med 29:S126–S129, 2001. [DOI] [PubMed] [Google Scholar]
- 99.Al Otair HA, Abdel Gader AGM, Khurshid SM, Alzeer AH, Al Momen AK, Al Shaikh M, Al Gahtani F, Al Aseri ZA, Abdelrazik HAH. The levels of tissue factor pathway inhibitor in sepsis patients receiving prophylactic enoxaparin. Turk J Haematol 33:112–118, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Adams M. Tissue factor pathway inhibitor: new insights into an old inhibitor. Semin Thromb Hemost 38:129–134, 2012. [DOI] [PubMed] [Google Scholar]
- 101.Dehne MG, Sablotzki A, Hoffmann A, Mühling J, Dietrich FE, Hempelmann G. Alterations of acute phase reaction and cytokine production in patients following severe burn injury. Burns 28:535–542, 2002. [DOI] [PubMed] [Google Scholar]
- 102.Chipp E, Milner CS, Blackburn AV. Sepsis in burns: a review of current practice and future therapies. Ann Plast Surg 65:228–236, 2010. [DOI] [PubMed] [Google Scholar]
- 103.Lantos J, Földi V, Roth E, Wéber G, Bogár L, Csontos C. Burn trauma induces early hmgb1 release in patients: its correlation with cytokines. Shock 33:562–567, 2010. [DOI] [PubMed] [Google Scholar]
- 104.O’Dwyer MJ, Mankan AK, Stordeur P, O’Connell B, Duggan E, White M, Kelleher DP, McManus R, Ryan T. The occurrence of severe sepsis and septic shock are related to distinct patterns of cytokine gene expression. Shock 26:544–550, 2006. [DOI] [PubMed] [Google Scholar]
- 105.Kraft R, Herndon DN, Finnerty CC, Cox RA, Song J, Jeschke MG. Predictive value of IL-8 for sepsis and severe infections after burn injury: a clinical study. Shock 43:222–227, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Moore HB, Winfield RD, Aibiki M, Neal MD. Is coagulopathy an appropriate therapeutic target during critical illness such as trauma or sepsis? Shock 48:159–167, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chung I, Lip GYH. Virchow's triad revisited: blood constituents. Pathophysiol Haemost Thromb 33:449–454, 2003. [DOI] [PubMed] [Google Scholar]
- 108.Meizoso JP, Ray JJ, Allen CJ, Van Haren RM, Ruiz G, Namias N, Schulman CI, Pizano LR, Proctor KG. Hypercoagulability and venous thromboembolism in burn patients. Semin Thromb Hemost 41:43–48, 2015. [DOI] [PubMed] [Google Scholar]
- 109.Barret JP, Dziewulski PG. Complications of the hypercoagulable status in burn injury. Burns 32:1005–1008, 2006. [DOI] [PubMed] [Google Scholar]
- 110.Wahl WL, Brandt MM, Ahrns KS, Zajkowski PJ, Proctor MC, Wakefield TW, Greenfield LJ. Venous thrombosis incidence in burn patients: preliminary results of a prospective study. J Burn Care Rehabil 23:97–102, 2002. [DOI] [PubMed] [Google Scholar]
- 111.Sikora S, Papp A. Venous thromboembolism in burn patients is not prevented by chemoprophylaxis. Burns 43:1330–1334, 2017. [DOI] [PubMed] [Google Scholar]
- 112.Faucher LD, Conlon KM. Practice guidelines for deep venous thrombosis prophylaxis in burns. J Burn Care Res 28:661–663, 2007. [DOI] [PubMed] [Google Scholar]
- 113.Ahuja RB, Bansal P, Pradhan GS, Subberwal M. An analysis of deep vein thrombosis in burn patients (part II): a randomized and controlled study of thrombo-prophylaxis with low molecular weight heparin. Burns 42:1693–1698, 2016. [DOI] [PubMed] [Google Scholar]
- 114.Lin H, Faraklas I, Cochran A, Saffle J. Enoxaparin and antifactor Xa levels in acute burn patients. J Burn Care Res 32:1–5, 2011. [DOI] [PubMed] [Google Scholar]
- 115.Levi M, van der Poll T. A short contemporary history of disseminated intravascular coagulation. Semin Thromb Hemost 40:874–880, 2014. [DOI] [PubMed] [Google Scholar]
- 116.Lippi G, Ippolito L, Cervellin G. Disseminated intravascular coagulation in burn injury. Semin Thromb Hemost 36:429–436, 2010. [DOI] [PubMed] [Google Scholar]
- 117.Barret JP, Gomez PA. Disseminated intravascular coagulation: a rare entity in burn injury. Burns 31:354–357, 2005. [DOI] [PubMed] [Google Scholar]
- 118.Taylor FB, Jr, Toh CH, Hoots WK, Wada H, Levi M. Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation. Thromb Haemost 86:1327–1330, 2001. [PubMed] [Google Scholar]
- 119.Jackson DM. [The diagnosis of the depth of burning]. Br J Surg 40:588–596, 1953. [DOI] [PubMed] [Google Scholar]
- 120.Schmauss D, Rezaeian F, Finck T, Machens H-G, Wettstein R, Harder Y. Treatment of secondary burn wound progression in contact burns—a systematic review of experimental approaches. J Burn Care Res 36:e176–e189, 2015. [DOI] [PubMed] [Google Scholar]
- 121.Isik S, Sahin U, Ilgan S, Guler M, Gunalp B, Selmanpakoglu N. Saving the zone of stasis in burns with recombinant tissue-type plasminogen activator (r-tPA): an experimental study in rats. Burns 24:217–223, 1998. [DOI] [PubMed] [Google Scholar]
- 122.Meyerholz DK, Piester TL, McNamara AR, Sokolich JC, Jaskille AD, Orion KC, Zamba KD, Light TD. Pharmacologic modification to resuscitation fluid after thermal injury—is drotrecogin alfa the answer to arrest burn depth progression? J Trauma 67:996–1003, 2009. [DOI] [PubMed] [Google Scholar]
- 123.Kowal-Vern A, Orkin BA. Antithrombin in the treatment of burn trauma. World J Crit Care Med 5:17–26, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lavrentieva A. Replacement of specific coagulation factors in patients with burn: a review. Burns 39:543–548, 2013. [DOI] [PubMed] [Google Scholar]