

Abstract
PZM21 (1) was recently reported as a biased agonist of the mu-opioid receptor (MOR) with improved antinociceptive effects but reduced side effects than traditional opioid-based analgesics. The original synthesis of PZM21 with the desired (S,S) configuration required the separation of diastereomeric mixture in the final step using chiral HPLC. We have designed a concise synthesis of 1 in the enantiomeric pure form starting with commercially available L-alanine and via a chiral aziridine as a key intermediate. The final product 1 as the (S,S) diastereomer was obtained in 7 steps in 22.5% yield from L-alanine. This synthetic strategy could be readily applied to the development of PZM21 analogs at the thiophenyl position.
Keywords: biased agonist, enantiopure, PZM21, synthesis, opioid
Graphical Abstract
A novel synthesis of the enantiopure PZM21, a mu-opioid receptor biased agonist with improved antinociceptive effects but reduced side effects, has been accomplished starting from L-alanine. The new synthetic route avoids previously used HPLC chiral separation and allows the potential preparation of analogs.
Introduction
Opioid analgesics are the most effective and widely used medications for many pain conditions, particularly for chronic and neuropathic pains.[1] However, the side effects associated with opioids, including abuse liability, physical dependence, respiratory depression, and constipation, have limited their use in pain management.[2] During the last few years studies have been conducted which suggested that biased agonists, agonists that selectively activate certain downstream signaling pathways, could be designed and developed that possessed pain relief with reduced opioid type side effects.[3] One such biased agonist, TRV130 (Oliceridine Olinvo™), is currently in phase 3 clinical studies.[4]
More recently, another mu-opioid receptor (MOR) biased agonist PZM21 (1, Figure 1) was reported where in vitro and animal studies suggest that 1 may show a better separation than TRV130 between pain relief and opioid side effects.[5] Like TRV130, 1 primarily signaled through G protein (Gi/o), but induced no detectable β-arrestin-2 recruitment. As a result, 1 produced antinociceptive effects with decreased respiratory depression and constipation and did not induce conditioned place preference in rodents. This is consistent with the recent notions that opioid analgesia mainly results from MOR signaling through the G protein, whereas signaling through β-arrestin may lead to many of the opioid side effects.[3a] Therefore, biased MOR agonists such as 1 appear as promising analgesics without the typical side effects of opioids, thus resulting in improved safety and tolerability.[6]
Scheme 1.

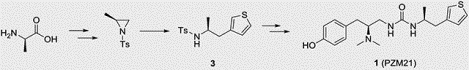
Reported synthetic approach to 1.
1 was initially discovered by computational docking studies of a library of over 3 million commercial lead-like molecules against the MOR, followed by structure-guided synthetic optimization.[5] Chemical synthesis led to all four isomers and it was discovered that the (S,S) stereoisomer was the most potent and efficacious agonist signaling through the Gi/o protein. The published synthetic route to 1 involved the urea coupling of racemic mixture of amine 3 with amine 2 at the final step, and the subsequent separation of the resulting diastereomers by chiral HPLC (Scheme 1). Enantiomeric enrichment of the amine 3 was also attempted with chiral resolution; however, it required repetitive co-crystallization of the racemic mixture of 3 with di-p-anisoyl-D-tartaric acid and was only used to assist in the assignment of the absolute configuration of 3 but not in the final synthesis of 1.[5] In an effort to identify a more amenable synthesis of 1 that avoids chiral HPLC separation or repeated chiral resolution and allows for potential derivatization, we have explored and herein describe a synthetic route that would lead to 1 as the desired (S,S) diastereomer, starting from commercially available L-alanine.
Results and Discussion
Retrosynthetically, 1 can be synthesized from key intermediate 4 either via detosylation of 5 (Method 1, Scheme 2), or via urea formation of enantiomerically pure 3 (Method 2, Scheme 2). The common intermediate 4 could be obtained by the ring opening of aziridine 6 using a thienyl Grignard reagent (Scheme 2). By varying the Grignard reagents employed in this key reaction step, analogs of 1 can be readily prepared with different substituents at the thiophenyl position, which appeared important for its pharmacological activity.[5] Finally, aziridine 6 can be obtained from commercially available L-alanine.
Scheme 2.

Retrosynthetic plan to prepare PZM21 (1) as the (S,S) isomer.
The preparation of the (S) enantiomer of amine 2 was accomplished following modified literature procedures (Scheme 3).[5] Reductive amination of the commercially available L-tyrosinamide with formaldehyde provided the dimethylamine intermediate 7 in 70% yield. However, the subsequent amide reduction was not complete in 48 hours using borane-DMS, with significant amount of unreacted amide 7. Since a low yield of 46% was also reported for this step in the original synthesis,[5] the reduction was then performed using a stronger reductant, LiAlH4. Under these conditions, only a small amount of the unreacted amide 7 was present, as judged by TLC and LC/MS analysis. Since both amide 7 and reduced amine 2 are highly polar and had similar Rf’s, the primary amine and the phenolic hydroxyl group were protected with Boc groups to facilitate separation. The Boc groups of 8 were then removed using HCl in dioxane to give amine 2 as the di-HCl salt. This conversion improved the yield of 2 from 7 to 72% from the reported 46%.
Scheme 3.

Synthesis of amine 2.
The preparation of enantiomerically pure amine (S)-3 required intermediate aziridine 6 (Scheme 2), which can be obtained from commercially available and inexpensive L-alanine.[7] Thus, N-tosylation of L-alanine using tosyl chloride and diisopropylethylamine in acetone gave a 93% yield of the sulfonamide 9. Reduction of the carboxylic acid with lithium aluminum hydride in THF/diethyl ether furnished alcohol 10 in 78% yield. O-Tosylation using tosyl chloride with pyridine in dichloromethane gave the bis-tosylate in 84% yield. Finally, ring closure using potassium hydroxide in methanol gave the aziridine 6 in 51% yield after chromatography (Scheme 4).[8]
Scheme 4.

Two routes of aziridine 6 formation from L-alanine.
Given the modest yield in the last ring closure step of the 4-step sequence, we then attempted a shorter and more efficient route to compound 6 using a one-pot procedure from intermediate L-alaninol 11, which was readily available from L-alanine (Scheme 4). The first attempt of the reduction of L-alanine using lithium aluminum hydride afforded 11 in 61% yield;[9] however, a slightly modified work-up,[10] which involved quenching with saturated aqueous potassium carbonate solution followed by addition of 2% methanol in dichloromethane, improved the yield to 79%. The next one-pot formation of aziridine 6 using an excess of tosyl chloride in dichloromethane with aqueous potassium hydroxide only provided the desired product in 29% yield. After several conditions failed to improve the yield, the best results (65%) were obtained using a 2-stage procedure (Scheme 4), in which the bis-tosylation was first attained and, after an aqueous work-up, the crude product was redissolved in acetone and stirred with potassium carbonate at RT overnight. This sequence gave the desired aziridine 6 in 51% yield in two steps from L-alanine, as compared to the first route that gave a 31% yield in four steps.
Optical rotation of compound 6 (+30.3, c 1, CH2Cl2) matched literature report (+32.1, c 1, CH2Cl2),[11] confirming the stereochemistry.
Conversion of aziridine 6 to 4 was then attempted via ring opening with a 3-thienyl Grignard reagent (Scheme 5). The only commercially available 3-thienyl Grignard reagent, 3-thienylmagnesium iodide (as a 0.3 M solution in THF), reacted with aziridine 6 under the catalysis of copper (I) iodide at −30 °C (Condition A, Figure 5), but afforded the product 4 in only 11% yield. However, in a test reaction, a different Grignard reagent, o-tolylmagnesium bromide, afforded the expected product in high yield under similar conditions. Therefore, conditions with in situ generation of thienylmagnesium bromide or chloride were explored. This transformation has been previously achieved using “Turbo Grignard” (isopropylmagnesium chloride and lithium chloride complex).[12] Thus, the Grignard reagent was pre-formed by mixing 3-bromothiophene with this complex, followed by addition of copper (I) iodide, before aziridine 6 was added and stirred overnight (Condition B). While the initial literature procedure resulted in mostly starting material, together with the ring-opened by-product, chloride 12, elongation of Grignard formation (2 hours) and the total reaction time after aziridine 6 addition (overnight) led to significant improvement in the yield to 93% of 4.
Scheme 5.

Ring opening of aziridine 6 and synthesis of 4.
With the sulfonamide 4 in hand, we then explored the urea formation to furnish the final product 1 via intermediate 5 (Method 1). Thus, activation of sulfonamide 4 with 4-nitrophenyl chloroformate to give 13 and subsequent reaction with amine 2, under the same conditions reported,[5] afforded the N-tosyl analog 5 in 45% yield over 2 steps (Scheme 6). Removal of the tosyl group was achieved using magnesium powder (10 eq.) in methanol under sonication for 60 minutes in 71% yield.[13] However, a competing reaction also took place under these conditions, where the urea was cleaved to the methyl carbamate 14 and sulfonamide 4. Unfortunately, the side reaction became more prevalent when the reaction was performed on larger scales, and no improvement was observed by varying the conditions, particularly the stoichiometry of magnesium.
Scheme 6.

Synthesis of 1 via detosylation of 5 (Method 1).
The other route to 1 would require the removal of the tosyl group of 4, followed by urea coupling between the resulting amine 3 with amine 2 (Method 2, Scheme 2). Removal of the tosyl group of 4 was attempted under several conditions (Scheme 7). Sonication in the presence of magnesium in methanol (Condition A) gave no reaction,[13] whereas magnesium and titanium isopropoxide with chlorotrimethylsilane (Condition B),[14] and samarium iodide and pyrrolidine in THF/water (Condition C),[15] both resulted in decomposition. Finally, we explored a 3-step approach involving the addition of a Boc group to N-tosylamines which, although having two extra steps, would accomplish the removal of the tosyl group in high yields.[13] Thus, Boc protection proceeded in high yield to give 15 and removal of the tosyl group via sonication with magnesium in methanol was considerably more facile than the earlier examples, proceeding in 79% yield over 2 steps to yield Boc-amine 16. This reaction was amenable to scale up with consistently high yields, though the duration required for complete detosylation seemed to lengthen with reaction scales. The Boc group was then easily removed by HCl in dioxane to give the desired free amine 3 in 99% yield. Trifluoroacetic acid was not suitable for this Boc-deprotection as it led to product degradation. This 3-step sequence was accomplished in 78% yield.
Scheme 7.

Synthesis of amine 3 via detosylation of sulfonamide 4. 17 was prepared to confirm the absolute configuration of 3.
In order to confirm the absolute configuration, amine 3 was converted to the corresponding acetamide 17 (Scheme 7). The rotation of 17 (−50.0 (c 0.3, CHCl3)) matched the previously reported value of −46.6 (c 1, CHCl3).[5]
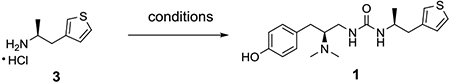
Finally, the optimal conditions for the urea formation between amines 2 and 3 were explored (Method 2). As listed in Table 1, the 2-step procedure described by Manglik,[5] with activation of amine 3 with 4-nitrophenylcarbamate and then reaction with amine 2, gave the product 1 in only 32–39% yields in our hands (Entry 1). Triphosgene in a dichloromethane and saturated sodium bicarbonate solution mixture, furnished the product 28% yield (Entry 2). Using 1,1’-carbonyl diimidazole (CDI) in dichloromethane or chloroform gave better yields (47% and 49%, Entries 3 and 4). Other solvents such as THF and 1,4-dioxane led to lower yields (26–28%, Entries 5–6). Finally, when the reaction was conducted in acetonitrile (Entry 7), 1 was obtained in 60% yield, though reversal of the addition order of reactants provided no final product (Entry 8).
Table 1.
Optimization of conditions to synthesize 1 via amine 3 (Method 2).
 | ||
|---|---|---|
| Entry | Conditions | Yield |
| 1 | (a) 3, 4-NO2PhOCOCl, Et3N, THF, 6 hr; (b) 2, Et3N, DMF, 20 hr | 32–39% |
| 2 | 3, (Cl3CO)2CO, NaHCO3 (aq), CH2Cl2, 10 min, then 2, 16 hr | 28% |
| 3 | 3, CDI, iPr2EtN, CH2Cl2, 15 min then 2, 16 hr | 47% |
| 4 | 3, CDI, iPr2EtN, CHCl3, 15 min then 2, 16 hr | 49% |
| 5 | 3, CDI, iPr2EtN, THF, 15 min then 2, 16 hr | 28% |
| 6 | 3, CDI, iPr2EtN, 1,4-Dioxane, 15 min then 2, 16 hr | 26% |
| 7 | 3, CDI, iPr2EtN, CH3CN, 15 min then 2, 16 hr | 60% |
| 8 | 2, CDI, iPr2EtN, CH3CN, 15 min then 3, 16 hr | 0% |
The stereochemistry of the final product 1 was confirmed by optical rotation (+39.1 (c 0.43, CHCl3) vs. lit. +40.3 (c 1.0, CHCl3)).
A summary of the synthetic route is shown in Figure 8. Under these conditions, 1 was prepared in gram quantities and then tested in animal models. The compound exerted analgesic, reinforcing, and pruritic effects comparable to those of clinically used mu opioid receptor agonists such as morphine and oxycodone in primates.[16]
Scheme 8.

Summary of synthesis of 1 from L-alanine.
Conclusions
We have designed and carried out the synthesis of enantiopure 1, a biased agonist of the MOR, starting from the commercially available L-alanine and L-tyrosinamide. The amine intermediate 2 was prepared from L-tyrosinamide following a slightly modified procedure using Boc protection of the amino groups. The key intermediate sulfonamide 4 with the desired (S) configuration was prepared via the ring opening of a chiral N-tosylaziridine 6, which was synthesized from L-alanine. A Boc group was added to assist the removal of the N-tosyl group and the sequence proceeded in high yields to afford amine 3. Finally, coupling between 2 and 3 was achieved in a single step using CDI in good yield to furnish the final product 1 in the desired (S,S) isomer. The synthesis was accomplished in 7 steps from L-alanine in 22.5% overall yield (Scheme 8). The sequence has been successfully employed to prepare 1 in gram quantities to support animal testing. Similar to other mu opioid receptor agonists, the compound exhibited analgesic effects in primates. Further, this synthetic route provides the flexibility of rapid preparation of potential PZM21 analogs at the thiophenyl position, one appearing important for activity,[5] by employing other Grignard reagents in the reaction with the common intermediate aziridine 6.
Experimental Section
General:
All solvents and chemicals were reagent grade. Unless otherwise mentioned, all were purchased from commercial vendors and used as received. Flash column chromatography was done on a Teledyne ISCO CombiFlash Rf system using prepacked columns. Solvents used were hexane, ethyl acetate (EtOAc), dichloromethane, methanol and chloroform:methanol:ammonium hydroxide (80:18:2) (CMA-80). Purity and characterization of compounds was established by a combination of high pressure liquid chromatography (HPLC), thin layer chromatography (TLC), mass spectrometry (MS) and nuclear magnetic resonance (NMR) analysis. 1H and 13C NMR spectra were recorded on a Bruker Avance DPX-300 (300 MHz) spectrometer and were determined in chloroform-d, methanol-d4 or dimethyl sulfoxide-d6 with tetramethylsilane (TMS) (0.00 ppm) or solvent peaks as the internal reference. Chemical shifts are reported in ppm relative to the reference signal, and coupling constant (J) values are reported in Hz. TLC was done on EMD precoated silica gel 60 F254 plates, and spots were visualized with UV light or iodine staining. HPLC analysis was performed on an Agilent 1100 system using an Agilent Zorbax SB-Phenyl, 2.1 mm × 150 mm, 5 μm column with gradient elution using the mobile phases (A) H2O containing 0.1% CF3COOH and (B) MeCN, with a flow rate of 1.0 mL/min. High resolution mass spectra were obtained on an Agilent 6230 time-of-flight mass spectrometer. Optical rotations were measured on an AutoPol III polarimeter, purchased from Rudolf Research.
(S)-2-(Dimethylamino)-3-(4-hydroxyphenyl)propanamide (7):
7 was synthesized following the literature procedure from L-tyrosinamide. The free base, as opposed to the hydrochloride salt in the literature procedure,[5] was obtained after column chromatography (0–80% CMA-80/CHCl3) in 70% yield. 1H NMR (300 MHz, CDCl3) δ 7.02 – 7.09 (m, 2H, 3-phenyl CHs), 6.69 – 6.76 (m, 2H, 2-phenyl CHs), 3.09 – 3.16 (m, 1H, α-CH), 2.93 – 3.03 (m, 1H, 1 of CH2), 2.84 (dd, J = 5.50, 13.75 Hz, 1H, 1 of CH2), 2.33 – 2.37 (m, 6H, N(CH3)2).
(S)-4-[3-Amino-2-(dimethylamino)propyl]phenol dihydrochloride (2):
Lithium aluminum hydride (8.51 g, 225 mmol) was suspended in THF (100 mL) and amide 7 (8.49 g, 40.766 mmol) in THF (30 mL) was added at 0 °C. After stirring at 0 °C for 15 min, the reaction was brought to room temperature and stirred for another 15 min. The reaction was stirred at reflux for 24 hours and was then cooled to 0 °C before quenching slowly by careful addition of 10 N NaOH (100 mL, 1 mol). Di-tert-butyl dicarbonate (22.3 g, 102 mmol) was added and the mixture was stirred for 1 hour. The reaction was extracted 3 times with EtOAc (3 × 150 mL). The combined organic layers were washed with water and dried over MgSO4. The solvent was removed under reduced pressure and the residue was purified by chromatography on silica (0–100% EtOAc/hexane) to afford pure compound 8. 1H NMR (300 MHz, CDCl3) δ 6.96 – 7.20 (m, 4H, aromatic CHs), 5.15 (br. s., 1H, Boc NH), 3.10 – 3.28 (m, 1H, CHN), 2.61 – 2.99 (m, 4H, 2xCH2), 2.22 – 2.40 (m, 6H, N(CH3)2), 1.55 (s, 9H, Boc CH3s), 1.31 – 1.49 (m, 9H, Boc CH3s).
Compound 8 was subjected to 4 N HCl in dioxane (160 mL, 840 mmol) and then all solvents were removed under reduced pressure to yield 2 as a white solid (7.85g, 72%). The spectroscopic data match with those reported in the literature. [α]D20 −10.6 (c 0.29, H2O) (lit. −10.1 (c 0.47, H2O)).
(2S)-2-(4-Methylbenzenesulfonamido)propanoic acid (9):
This was prepared according to the published procedure in 93% yield.[7] The spectroscopic data match with those reported in the literature.
(2S)-1-Hydroxy-S-(4-methylphenyl)propane-2-sulfonamide (10):
This was prepared according to the published procedure in 78% yield.[7] The spectroscopic data match with those reported in the literature.
(2S)-2-Aminopropan-1-ol (11):
To an ice-cooled suspension of lithium aluminum hydride (10.40 g, 0.274 mol) in anhydrous THF (200 mL) under N2 was added slowly L-alanine (11.0 g, 0.123 mol) in small portions over 2 hours. After complete addition, the reaction mixture was warmed to RT over 1 hour then heated to reflux overnight. The heat was removed and the reaction cooled in an ice bath, and then cautiously quenched with saturated aqueous potassium carbonate. The reaction mixture was then stirred at RT for 1 hr before 2% methanol in dichloromethane was added. The mixture was stirred a further hour, and then the crude product was filtered through Celite, rinsing thoroughly with dichloromethane. The filtrate was dried over MgSO4 then the solvent was removed under reduced pressure (water bath at less than 25 °C) to give the desired alcohol 9 (7.32 g, 79%). The spectroscopic data match with those reported in the literature.[8]
(2S)-2-Methyl-1-(4-methylbenzenesulfonyl)aziridine (6):
From alcohol 10 (as shown in Scheme 4): This was prepared according to the published procedure in 43% over 2 steps.[8]
From L-alaninol 11 (as shown in Scheme 4): 4-Dimethylaminopyridine (0.16 g, 1.33 mmol) was added to a solution of p-toluenesulfonyl chloride (2.79 g, 14.65 mmol) in pyridine (2.11 g, 2.2 mL, 26.63 mmol) and dichloromethane (4 mL) cooled in ice. To this was added slowly a solution of 11 (0.50 g, 6.66 mmol) in dichloromethane (1 mL). The reaction formed a deep purple color during addition. Upon complete addition, the reaction was stirred at RT overnight. It was then poured into cold 1N aqueous HCl. The layers were separated, and the aqueous layer extracted with dichloromethane. The combined organic layers were washed with 5% aqueous CuSO4 solution and brine, back-extracting with dichloromethane after each wash. The organic solution was dried over MgSO4 and the solvent was removed under reduced pressure.
The crude product was redissolved in acetone (35 mL) and potassium carbonate (3.31 g, 23.97 mmol) was added. The reaction was stirred at RT overnight. The solution was filtered through Celite and then the solvent was removed under reduced pressure. The crude material was purified by chromatography on silica (0–20% EtOAc/hexane) to give the aziridine as a white solid (0.92 g, 65%). The spectroscopic data match with those reported in the literature.[7] HRMS (ESI, CH3OH) m/z calcd for C10H14NO2S [M + H]+ 212.0740, m/z found 212.0730. [α]D20 +30.3 (c 1.0, CH2Cl2) (lit. +32.1 (c 1, CH2Cl2)).
4-Methyl-N-[(2S)-1-(thiophen-3-yl)propan-2-yl]benzene-1-sulfonamide (4):
To a solution of 3-bromothiophene (13.63 g, 7.8 mL. 83.60 mmol) in anhydrous THF (80 mL) under N2 in an oven-dried flask at RT was added isopropylmagnesium chloride lithium chloride complex (as a 1.3 M solution in THF, 58.5 mL, 76.0 mmol). The mixture was stirred at RT for 2 hr, then cooled to −30 °C. Copper (I) iodide (2.17 g, 11.4 mmol) was added and the reaction stirred at −30 °C for 2 hr. To this was added dropwise a solution of aziridine 6 (4.01 g, 19.0 mmol) in THF (30 mL). Upon complete addition, the cold bath was removed, and the reaction stirred at RT for 48 hr. The reaction was quenched by the addition of saturated aqueous NH4Cl solution then extracted twice with diethyl ether. The combined extracts were dried over MgSO4 and the solvents were removed under reduced pressure. The crude was absorbed onto Celite and purified by chromatography on silica (0–15% EtOAc/hexane) to give the desired product as a brown oil (5.25 g, 93%). 1H NMR (300 MHz, CDCl3) δ 7.66 (d, J = 8.29 Hz, 2H, 3,5-tosyl CHs), 7.26 (d, J = 7.91 Hz, 2H, 2,6-tosyl CHs), 7.19 (dd, J = 2.92, 4.80 Hz, 1H, 5-thiophene CH), 6.88 (d, J = 1.70 Hz, 1H, 2-thiophene CH), 6.76 (dd, J = 1.13, 4.90 Hz, 1H, 4-thiophene CH), 4.35 (d, J = 7.16 Hz, 1H, NH), 3.53 (td, J = 6.50, 13.37 Hz, 1H, NHCH), 2.71 (d, J = 6.22 Hz, 2H, CH2), 2.42 (s, 3H, Ph-CH3), 1.09 (d, J = 6.59 Hz, 3H, NHCHCH3). 13C NMR (75 MHz, CDCl3) δ 143.2 (aromatic C-CH3), 137.8 (C-SO2), 137.2 (quat thiophene), 129.6 (3,5-tosyl CHs), 128.5 (4-thiophene CH), 127.0 (2,6-tosyl CHs), 125.8 (5-thiophene CH), 122.5 (2-thiophene CH), 50.2 (NH-CH), 37.6 (CH2), 21.5 (tosyl CH3), 21.3 (CH CH3). HRMS (ESI, CH3OH) m/z calcd for C14H18NO2S2 [M + H]+ 296.0773, m/z found 296.0762. [α]D20 −19.7 (c 0.47, CHCl3).
4‐Nitrophenyl N‐(4‐methylbenzenesulfonyl)‐N‐[(2S)‐1‐(thiophen‐3‐yl)propan‐2‐yl]carbamate (13):
To a solution of 4 (0.14 g, 0.46 mmol) in anhydrous dichloromethane (2.3 ml) was added 4-dimethylaminopyridine (0.006 g, 0.05 mmol), triethylamine (0.13 ml, 0.92 mmol) and 4-nitrophenyl chloroformate (0.14 g, 0.69 mmol) at 0 °C. The reaction mixture was brought to room temperature and stirred for 4 h. The reaction mixture was then diluted with dichloromethane and washed with water and brine. The organic layer was dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo. The residue was purified by chromatography on silica gel (0–15% ethyl acetate/hexanes) to afford the desired product as a white solid (0.21 g, quant. yield). 1H NMR (300 MHz, CDCl3) δ 8.22 (d, J = 9.04 Hz, 2H, nitrophenyl CHs), 7.42 (d, J = 8.29 Hz, 2H, 3,5-tosyl CHs), 7.28 – 7.33 (m, 1H, 5-thiophene CH), 7.20 (d, J = 8.29 Hz, 2H, 2,6-tosyl CHs), 7.08 (d, J = 9.04 Hz, 2H, nitrophenyl CHs), 7.00 – 7.04 (m, 2H, 2,4-thiophene CHs), 5.01 (s, 1H, CH), 3.45 (dd, J = 9.51, 14.22 Hz, 1H, 1 of CH2), 3.06 (dd, J = 6.31, 14.22 Hz, 1H, 1 of CH2), 2.41 (s, 3H, tosyl CH3), 1.63 (d, J = 6.78 Hz, 3H, CHCH3). 13C NMR (75MHz, CDCl3) δ 154.3 (carbamate CO), 149.4 (O-C), 145.6 (C-NO2), 144.7 (tosyl C-CH3), 138.7 (C-SO2), 136.7 (quat thiophene), 129.5 (3,5-tosyl CHs), 128.5 (4-thiophene CH), 128.0 (2,6-tosyl CHs), 126.1 (5-thiophene CH), 125.3 (NO2-C-CHs), 122.6 (2-thiophene CH), 122.1 (O-C-CHs), 57.4 (CH), 35.0 (CH2), 21.6 (tosyl CH3), 19.6 (CHCH3). [α]D20 +76.0 (c 0.075, CHCl3).
3‐[(2S)‐2‐(Dimethylamino)‐3‐(4‐methylphenyl)propyl]‐1‐(4‐methylbenzenesulfonyl)‐1‐[(2S)‐1‐(thiophen‐3‐yl)propan‐2‐yl]urea (5):
To a solution of 13 (0.21 g, 0.46 mmol) in anhydrous DMF (2.3 ml) was added amine 2 (0.09 g, 0.46 mmol), triethylamine (0.07 ml, 0.51 mmol) and DMAP (0.006 g, 0.05 mmol). The reaction mixture was stirred at room temperature for 16 h. The reaction mixture was then diluted with ethyl acetate, washed twice with saturated aqueous sodium bicarbonate solution and once with brine. The organic layer was dried over anhydrous magnesium sulfate, filtered and concentrated in vacuo. The residue was purified by chromatography on silica gel to afford the desired product as a white solid (0.11 g, 45%). 1H NMR (300 MHz, CDCL3) δ 7.65 (d, J = 8.29 Hz, 2H, 3,5-phenyl CHs), 7.44 (d, J = 4.52 Hz, 1H, NH), 7.23 – 7.27 (m, 2H2,6-phenyl CHs), 7.21 (dd, J = 2.92, 4.80 Hz, 1H, 5-thiophene CH), 6.92 – 6.99 (m, 3H, 3,5-tosyl CHs, 2-thiophene CH), 6.89 (dd, J = 1.13, 4.90 Hz, 1H, 4-thiophene CH), 6.68 – 6.74 (m, 2H, 2,6-tosyl CHs), 4.46 – 4.60 (m, 1H, CH-N(CH3)2), 3.21 – 3.33 (m, 1H, CH-CH3), 3.12 (d, J = 7.54 Hz, 2H, PhCH2), 2.83 – 3.00 (m, 2H, CH2-thiophene), 2.68 (d, J = 4.52 Hz, 1H, 1 of CH2NH), 2.41 (s, 3H, tosyl CH3), 2.35 (s, 6H, N(CH3)2), 2.18 – 2.30 (m, 1H, 1 of CH2NH), 1.32 (d, J = 6.78 Hz, 3H, CH-CH3). 13C NMR (75MHz, CDCl3) δ 154.4 (C-OH), 152.6 (urea CO), 144.3 (tosyl C-CH3), 139.4 (C-SO2), 137.2 (quat thiophene), 130.7 (phenol C-CH2), 130.0 (CH-C-CH2), 129.7 (3,5-tosyl CHs), 128.5 (4-thiophene CH), 127.3 (2,6-tosyl CHs), 125.3 (5-thiophene CH), 122.1 (2-thiophene CH), 115.5 (CH-C-OH), 64.6 (CH-N(CH3)2), 56.7 (CH-CH3), 41.0 (NH-CH2), 40.0 (N(CH3)2), 36.0 (phenol-CH2), 30.7 (thiophene-CH2), 21.5 (tosyl CH3), 18.9 (CH-CH3). HRMS (ESI, CH3OH) m/z calcd for C26H34N3O4S2 [M + H]+ 516.1985, m/z found 516.1992. [α]D20 +42.8 (c 0.14, CHCl3).
4‐Methyl‐N‐[(2S)‐1‐(thiophen‐3‐yl)propan‐2‐yl]benzene‐1‐sulfonamide (15):
To a solution of 4 (6.29 g, 21.3 mmol) in anhydrous acetonitrile (180 mL) was added di-tert-butyl dicarbonate (5.35 g, 24.5 mmol), 4-dimethylaminopyridine (0.26 g, 2.1 mmol) and N,N-diisopropylethylamine (5.50 g, 7.4 mL, 42.6 mmol). The reaction mixture was stirred at room temperature for 16 h. When the reaction has completed, the reaction mixture was concentrated in vacuo. The crude was purified by chromatography on silica gel (0–20% ethyl acetate/hexanes) to provide the desired product as a colorless liquid (7.31 g, 87%). 1H NMR (300 MHz, CHCl3) δ 7.31 (d, J = 8.29 Hz, 2H, 3,5-tosyl CHs), 7.25 – 7.28 (m, 1H, 5-thiophene CH), 7.15 (d, J = 8.10 Hz, 2H, 2,6-tosyl CHs), 7.00 (d, J = 4.14 Hz, 2H, 2,4-thiophene CHs), 4.85 (td, J = 6.78, 9.23 Hz, 1H, CH), 3.43 (dd, J = 9.32, 14.03 Hz, 1H, 1 of CH2), 2.99 (dd, J = 6.59, 14.13 Hz, 1H, 1 of CH2), 2.39 (s, 3H, tosyl CH3), 1.53 (d, J = 6.78 Hz, 3H, CHCH3), 1.33 (s, 9H, Boc CH3s). 13C NMR (75MHz, CDCl3) δ 150.7 (Boc CO), 143.4 (tosyl C-CH3), 139.4 (C-SO2), 138.1 (quat thiophene), 129.0 (3,5-tosyl CHs), 128.7 (4-thiophene CH), 127.4 (2,6-tosyl CHs), 125.6 (5-thiophene CH), 122.4 (2-thiophene CH), 84.0 (quat tBu), 56.1 (CHCH3), 35.3 (CH2), 27.9 (tBu CH3s), 21.5 (tosyl CH3), 19.8 (CH-CH3). HRMS (ESI, CH3OH) m/z calcd for C19H25NNaO4S2 [M + Na]+ 418.1117, m/z found 418.1108. [α]D20 +44.3 (c 0.16, CHCl3).
tert‐Butyl N‐[(2S)‐1‐(thiophen‐3‐yl)propan‐2‐yl]carbamate (16):
To a solution of 15 (6.82g, 17.2 mmol) in anhydrous methanol (170 mL) was added magnesium powder (2.1 g, 86.2 mmol). The reaction mixture was sonicated at room temperature for 15 min. The solvent was removed in vacuo and the residue was dissolved in dichloromethane. This solution was washed sequentially with 0.5 M HCl solution, saturated sodium bicarbonate solution, and brine. The organic fraction was dried over anhydrous magnesium sulfate, filtered and concentrated in vacuo. The crude was purified by chromatography on silica (0–15% ethyl acetate/hexanes) to give the desired product as a white solid (3.98 g, 91%). 1H NMR (300MHz, CDCl3) δ 7.23 – 7.28 (1H, m, 5-thiophene CH), 6.98 (1H, d, J = 1.7 Hz, 2-thiophene CH), 6.94 (1H, dd, J = 0.9, 4.9 Hz, 4-thiophene CH), 4.36 (1H, br. s., NH), 3.91 (1H, s, CH-CH3), 2.82 (1H, dd, J = 5.8. 14.1 Hz, 1 of CH2), 2.74 (1H, dd, J = 6.8, 13.9 Hz, 1 of CH2), 1.43 (9H, s, Boc CH3s), 1.10 (3H, d, J = 6.8 Hz, CH-CH3). 13C NMR (75 MHz, CDCl3) δ 155.2 (Boc CO), 138.5 (quat thiophene), 128.9 (4-thiophene CH), 125.3 (5-thiophene CH), 121.9 (2-thiophene CH), 79.2 (quat tBu), 46.9 (CH), 37.4 (CH2), 28.4 (tBu CH3s), 20.4 (CH3). [α]D20 −18.1 (c 0.11, CHCl3).
(2S)‐1‐(Thiophen‐3‐yl)propan‐2‐amine hydrochloride (3):
To a solution of 16 (3.98 g, 16.5 mmol) dissolved in a minimum amount of ethyl acetate was added 4N HCl in dioxane (60 mL, 240 mmol) at room temperature and then sonicated at RT for 1 h. Hexane (30 mL) was added, the precipitate was collected by filtration and dried to afford the desired product 3 as the free base as a white solid (2.91 g, 99%). 1H NMR (300 MHz, MeOD) δ 7.43 (dd, J = 2.83, 4.90 Hz, 1H, 5-thiophene CH), 7.24 (dd, J = 0.75, 1.70 Hz, 1H, 2-thiophene CH), 7.04 (dd, J = 1.32, 4.90 Hz, 1H, 4-thiophene CH), 3.47 – 3.60 (m, 1H, CH), 3.02 (dd, J = 6.22, 14.13 Hz, 1H, 1 of CH2), 2.89 (dd, J = 7.91, 13.94 Hz, 1H, 1 of CH2), 1.28 (d, J = 6.59 Hz, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ 136.8 (quat thiophene), 128.5 (4-thiophene CH), 126.4 (5-thiophene CH), 122.9 (2-thiophene CH), 47.3 (CH), 34.6 (CH2), 17.7 (CH3). HRMS (ESI, CH3OH) m/z calcd for C7H12NS [M + H]+ 142.0685, m/z found 142.0682. [α]D20 +10.8 (c 0.19, H2O).
(S)-N-[1-(Thiophen-3-yl)propan-2-yl]acetamide (17):
This was prepared following the literature procedure.[5] [α]D20 −50.0 (c 0.3, CHCl3) (lit. −46.6 (c 1, CHCl3)).
1-[(2S)-2-(Dimethylamino)-3-(4-hydroxyphenyl)propyl]-3-[(2S)-1-(thiophen-3-yl)propan-2-yl]urea (1):
Method 1 (via detosylation of 5 as shown in Scheme 6):
To a solution of urea 5 (0.55 g, 1.06 mmol) in anhydrous methanol (10 ml) was added magnesium powder (0.26 g, 10.6 mmol). After sonicating at room temperature for 1 h, the reaction mixture was adsorbed onto Celite and purified by chromatography on silica gel (0–20% CMA-80/ethyl acetate) to afford the desired product as white solid (0.27 g, 71%).
Method 2 (via CDI coupling of amines 2 and 3 as shown in Entry 7, Table 1):
To a solution of amine hydrochloride 3 (0.53 g, 3.0 mmol) in anhydrous acetonitrile (60 mL) was added diisopropylethylamine (0.78 g, 1.1 mL, 6.0 mmol) followed by 1,1’-carbonyldiimidazole (0.29 g, 1.77 mmol). The reaction mixture was stirred at rt for 15 min. Diamine dihydrochloride 2 (0.80 g, 3.0 mmol) and diisopropylethylamine (1.17 g, 1.6 mL, 9.0 mmol) was added in one portion then the resulting mixture was stirred at RT overnight. The reaction was diluted with EtOAc and then washed with aqueous NaHCO3 solution and brine, dried over MgSO4 and the solvent was removed under reduced pressure. The crude was then purified by chromatography on silica (0–15% CMA-80 in EtOAc) to give the desired product as a white foamy solid (0.64 g, 60%). The spectroscopic data match with those reported in the literature. HRMS (ESI, CH3OH) m/z calcd for C19H28N3O2S [M + H]+ 362.1897, m/z found 362.1881. [α]D20 +39.1 (c 0.43, CHCl3) (lit. +40.3 (c 1.0, CHCl3)).[5] Analytical HPLC tR = 10.0 min (98.4%).
Acknowledgements
This work was supported by National Institute on Drug Abuse, National Institutes of Health, U.S. (Grants DA044775 and DA040693). We also thank Mr. Charles McElhinny for providing technical assistance.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Nuckols TK, Anderson L, Popescu I, Diamant AL, Doyle B, Di Capua P, Chou R, Annals of internal medicine 2014, 160, 38–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a Volkow ND, McLellan AT, N Engl J Med 2016, 374, 1253–1263; [DOI] [PubMed] [Google Scholar]; b Dart RC, Surratt HL, Cicero TJ, Parrino MW, Severtson SG, Bucher-Bartelson B, Green JL, N Engl J Med 2015, 372, 241–248. [DOI] [PubMed] [Google Scholar]
- [3].a Rankovic Z, Brust TF, Bohn LM, Bioorg Med Chem Lett 2016, 26, 241–250; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kenakin T, Expert Opin Drug Discov 2017, 12, 321–333; [DOI] [PubMed] [Google Scholar]; c Siuda ER, Carr R 3rd, Rominger DH, Violin JD, Curr Opin Pharmacol 2017, 32, 77–84. [DOI] [PubMed] [Google Scholar]
- [4].Soergel DG, Subach RA, Burnham N, Lark MW, James IE, Sadler BM, Skobieranda F, Violin JD, Webster LR, Pain 2014, 155, 1829–1835. [DOI] [PubMed] [Google Scholar]
- [5].Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G, Levit A, Kling RC, Bernat V, Hubner H, Huang XP, Sassano MF, Giguere PM, Lober S, Da D, Scherrer G, Kobilka BK, Gmeiner P, Roth BL, Shoichet BK, Nature 2016, 537, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a Luttrell LM, Maudsley S, Bohn LM, Molecular pharmacology 2015, 88, 579–588; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Schmid CL, Bohn LM, Pharmacology & therapeutics 2009, 121, 285–293; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Violin JD, Crombie AL, Soergel DG, Lark MW, Trends in pharmacological sciences 2014, 35, 308–316. [DOI] [PubMed] [Google Scholar]
- [7].Berry MB, Craig D, Synlett 1992, 41–44. [Google Scholar]
- [8].Daub GW, Heerding DA, Overman LE, Tetrahedron 1988, 44, 3919–3930. [Google Scholar]
- [9].Barsanti PA, Aversa RJ, Jin X, Pan Y, Lu Y, Elling R, Jain R, Knapp M, Lan J, Lin X, Rudewicz P, Sim J, Taricani L, Thomas G, Xiao L, Yue Q, ACS Med Chem Lett 2015, 6, 37–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dugar S, MAHAJAN D, HOLLINGER PF, Sharma A, Tripathi V, KUILA B, WO 2014 016849 A2
- [11].Farmer JJ, Schroeder FC, Meinwald J, Helv Chim Acta 2000, 83, 2594–2606. [Google Scholar]
- [12].Krasovskiy A, Knochel P, Angew Chem Int Edit 2004, 43, 3333–3336. [DOI] [PubMed] [Google Scholar]
- [13].Nyasse B, Grehn L, Ragnarsson U, Chem Commun 1997, 1017–1018. [Google Scholar]
- [14].Ankner T, Hilmersson G, Org Lett 2009, 11, 503–506. [DOI] [PubMed] [Google Scholar]
- [15].Xiao X, Hou C, Zhang Z, Ke Z, Lan J, Jiang H, Zeng W, Angew Chem Int Ed Engl 2016, 55, 11897–11901. [DOI] [PubMed] [Google Scholar]
- [16].Ding H, Kiguchi N, Perrey DA, Nguyen T, Czoty PW, Zhang Y, Ko MC, in Exp. Biol, San Diego, CA, USA, 2018. [Google Scholar]