

Abstract
Minimally invasive medical devices are of great interest, with shape memory polymers (SMPs) representing one such possibility for producing these devices. Previous work with low density, highly porous SMPs has demonstrated oxidative degradation, while attempts to incorporate hydrolytic degradation have resulted in rapidly decreasing glass transition temperature (Tg), ultimately preventing strain fixity of the materials at clinically relevant temperatures. Through esterification of the amino alcohol triethanolamine, an alcohol containing network was synthesized and incorporated into SMPs. These ester networks were used to control the bulk morphology of the SMP, with the Tg remaining above 37 °C when 50% of the alcohol was contributed by the ester network. This methodology also yielded SMPs that could degrade through both hydrolysis and oxidation; by oxidation, the SMPs degrade at a similar rate as the control materials (0.2%/day mass) for the first 30 days, at which point the rate changes to 3.5%/day until the samples become too fragile to examine at 80 days. By comparison, control materials have lost approximately 30% of mass by 140 days, at a constant rate of degradation, demonstrating that the ester SMPs are a promising material system for producing more rapidly degradable, soft, porous biomaterials.
Keywords: shape memory polyurethanes, oxidative degradation, resorbable polymers, porous scaffold
Graphical Abstract
1. INTRODUCTION
Many implantable medical devices would benefit from tunable or controllable degradation rates, as the balance between the mechanical support of the scaffold must offset the rate of tissue in-growth and mechanical integrity.1,2 For cerebrovascular aneurysm occlusion, degradable materials could allow healing of the aneurysm to reduce the long-term risk of rupture and hemorrhage. In a pediatric device, the long-term presence of a material may also be detrimental to tissue healing and restoration of functionality; a degradable scaffold could allow for healing alongside native tissue infiltration.3–5 Such biomaterials require compositions that are biocompatible across their entire lifecycles, including degradation products, to minimize host response, are minimally invasive to reduce surgical trauma, and with current trends in green manufacturing, new biomedical materials should be utilizing a resorbable unit.5
Shape memory polymers (SMPs) are a class of material with the capability to respond to an applied external stimulus.5–9 In the case of thermally driven SMPs, this allows for a controlled shape change of the material due to temperature change.5 The shape change, and the temperature threshold about which this may occur are the glass transition temperature (Tg) or melt transition temperature (Tm), and are tunable with the polymer composition.5,6
Porous SMPs have been utilized for aneurysm occlusion, with rapid aneurysm healing, along with other applications, which leverage variations in recovery stresses and strains or recovery rates.10–16 The porous scaffold allows rapid cellular infiltration along with reduced material density.10,11 Polyurethane foams have previously been synthesized from renewable resources such as castor oil polyols and corn sugar-based diols, lactic acid, isosorbides, itaconic acid, and ethylene glycol.17–20,25 The use of poly(ester ureas) based on peptides, synthesized from amino acids, has been explored with good success indicated by cellular compatibility and tunable thermomechanical properties.21,22 While this specific approach is potentially a limiting factor due to cost, renewable resources such as metabolites from cellular cycles may offer cheap cytocompatible monomers as alternative sources.23,24
Here, we demonstrate several improvements over the previous methods in producing degradabel SMPs, including the use of the renewable starting material succinic acid for the synthesis of polyester prepolymers, to produce highly crosslinked ester networks which are then incorporated into porous shape memory polyurethanes. These SMPs demonstrate tunable thermomechanical properties and degradation rates without compromising shape memory behavior. The degradation can occur via two distinct pathways: hydrolytic scission of the ester linkages or oxidative degradation of the amines. The mechanisms and final metabolites are proposed for this material, and a review of toxicology data indicates the SMPs may be ideally suited for further studies as vascular materials.
2. METHODS AND MATERIALS
2.1. Materials.
N,N,N′,N′-Tetrakis(2-hydroxypropyl)-ethylenediamine (HPED, 99%, Sigma-Aldrich), triethanolamine (TEA, 98%, Alfa Aesar), succinic acid (SuA, Sigma-Aldrich, 99%), and 2,2,4-trimethyl hexamethylene diisocyanate (TMHDI, 98%, TCI America, a mixture of 2,2,4 and 2,4,4 isomers) were used as monomers. Hydrochloric acid (HCl, 0.1M), tetrahydrofuran (THF), ethanol (EtOH, 95%), acetic acid (anhydrous, 99%), Nile chloride, sodium hydroxide (NaOH), potassium hydroxide (KOH), isopropyl alcohol (IPA, 95%), phenolphthalein, and dimethylformamide (DMF) were obtained from Sigma-Aldrich and used without further modification.
2.2. Polycondensation of Ester Network.
Esterification of succinic acid into ester networks was performed in solution, using an acid catalyst. Carboxylic acid moieties were added as 1 equiv to approximately 3.5 equiv of alcohol functional groups. Then 0.05 equiv of acid catalyst was added to initiate the reaction, and molecular sieves were used to shift equilibrium toward ester formation by removing water from the solution at 150 °C. The solution was filtered and dried under vacuum overnight.
Initially, the reaction was conducted over a 30-h period to determine the growth kinetics of the reaction; all subsequent ester networks were allowed to form over 10 h. End group analysis was required to determine the relative concentration of alcohol end groups compared to carboxylic acid end groups by analysis of alcohol number and acid number.26,27 Alcohol number was determined by acetylation via titration under atmosphere. Acetic acid in dry pyridine was added to the cleaned ester and heated to 110°C before the addition of Nile chloride solution, followed by titration with NaOH solution, to determine the free alcohol functional groups using eq 1.28 The acid number was determined by titration of the ester with KOH solution with phenolphthalein indicator. The acid number is related to the free carboxylic acid groups in 1 g of material, determined via eq 2:28
| (1) |
| (2) |
The volume of the sample is represented by V, the volume of the blank is Z, the mass of the polymer sample is m, the concentration of the NaOH is CN, and the concentration of KOH is CK.
Carboxylic acid will react with an isocyanate group to produce carbon dioxide and amide linkages, potentially reducing shape memory and altering thermomechanical properties, as well as the rate of degradation. Therefore, the reaction conditions were tailored to maximize the alcohol number and minimize the acid number, to reduce the side reactions during the SMP synthesis.
The model network was synthesized through an overnight reaction of the ester network with stoichiometric amounts of ethyl isocyanate in a sealed vial containing DMF. Afterward, the reaction was quenched with water, isolated, and characterized.
2.3. Synthesis of Shape Memory Polyurethane.
SMP synthesis was carried out as previously described.29–31 Porous SMPs were synthesized containing stoichiometric amounts of the ester network (0%, 15%, 25%, and 50% of the alcohol content as ester), using a traditional two-step polyurethane synthesis method.29–31 An isocyanate premix with approximately 40% of required alcohol groups was mixed with DMF (approximately 20 wt %) in a round-bottom flask for 12 h at 100°C. Viscosity was tailored via adjustment of DMF and reaction time. The isocyanate premix was added to surfactants (DC 5943, 7.5 wt %) and mixed using a high-speed shear mixer. An alcohol premix, containing the remaining alcohols, catalysts, and water, was mixed and added to the isocyanate premix. After mixing both components with a physical blowing agent, the mixture was cured in the oven at 90 °C for 30 min. Foams were cold cured overnight at room temperature and washed using RO water/IPA with sonication. Gel fraction and swelling ratio were calculated for starting materials using previously established protocols.30
2.4. Rheology of Ester Network.
A parallel plate shear test was conducted using 5 to 6 drops of ester network using a Physica MCR 301 rheometer (Anton Paar USA Inc., Ashland, VA) under a sustained 50 s−1 shear rate for 120 s and was evaluated using the RheoCompass software (Anton Paar USA Inc., Ashland, VA).
2.5. Mass Spectrometry.
Voyager DE-STR mass spectrometer (Applied Biosystems, Foster City, CA) in positive linear mode, using a pulsed nitrogen laser at 337 nm and 25 kV, was used to determined molecular weight of the ester network over the course of the 30-h synthesis. trans-2-[3-(4-t-Butylphenyl)-2-methyl-2-propenylidene]-malononitrile (DCTB) and potassium trifluoroacetate (KTFA) were used as matrix and cationization reagents, respectively. The sample and DCTB matrix were mixed with KTFA at a volume ratio of 1:5:1; approximately 0.5 μL of this mixture was deposited on a stainless-steel sample tray for analysis.
2.6. Nuclear Magnetic Resonance Spectroscopy.
Nuclear magnetic resonance (13C NMR 125 MHz and 1H NMR 300 MHz) was performed on a Mercury 300 MHz spectrometer operating in the Fourier transform mode with CDCl3 as the solvent.
Solid state CP (cross-polarization) MAS NMR (13C 100 MHz) was performed using a 4.0 mm 13C probe on an Avance-400 Solids spectrometer (Bruker). A frequency of 123.63 MHz was used, with a spin speed of 3.5 kHz and 4.5 kHz and 90° pulse lengths of 4.5 μs. A recycle delay of 5 s and a contact time of 1.6 ms were the used measurement parameters. Edited CP MAS NMR experiments were performed observe positively phased signals for C and CH carbons, negatively phased CH2, and zero-intensity signals for CH carbons to assist in peak assignments, with an external TMS reference.
2.7. Fourier Transform Infrared Spectroscopy.
Attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR) spectroscopy was used to determine any spectroscopic changes to the bulk material. ATR-FTIR spectra were taken using a Bruker ALPHA infrared spectrometer (Bruker, Billerica, MA) using 32 scans per spectra for both background and samples. Spectra data was collected in absorption mode with a resolution of 4 cm−1. OPUS software was used to examine spectra, identify peaks, and perform baseline and atmospheric corrections. Examinations were performed in triplicate to confirm results.
2.8. Scanning Electron Microscopy.
Foam cell structure was determined by cutting axial and transverse samples that were examined using scanning electron microscopy (SEM). Samples were mounted onto a stage and sputter coated with gold using a Cressington Sputter Coater (Ted Pella, Inc., Redding, CA) for 60 s at 20 mA. Samples were then examined using a Joel NeoScope JCM-5000 SEM (Nikon Instruments Inc., Melville, NY) at 11× magnification and 15 kV under high vacuum.
2.9. Thermal Characterization.
Dynamic mechanical analysis (DMA) of cylindrical samples (6 mm diameter, 5 mm length) was used to conduct thermomechanical analysis using a Q800 TA DMA (TA Instruments, New Castle, DE).30 Dry temperature sweep samples were equilibrated at 20 °C for 15 min and then ramped to 120 °C at a rate of 2 °C/min. The storage modulus (E’) and the loss modulus (E”) were used to determine the peak tan δ (E’/E”), with the maximum value recorded as the dry Tg. Wet samples were submerged for 5 min in 50 °C reverse osmosis (RO) water prior to analysis. Samples were equilibrated for 5 min at 25 °C and then ramped to 70 °C at a rate of 2 °C/min. The temperature of the tan δ maxima was recorded as the wet Tg. Differential scanning calorimetry (DSC) was also used to measure both dry and wet Tg using a Q200 DSC (TA Instruments, Inc., New Castle, DE). Samples of approximately 5.0 mg ±1.0 mg were sealed in TZero aluminum pans and placed in the heating cell. The test profile for dry samples was as follows: equilibration at −40 °C, heating to 120 °C at 10 °C/min, cooling 10 °C/min to −40 °C and holding for 5 min, and a final heating to 120 °C at 10 °C/min. The half-height transition of the final heating cycle was the reported Tg. Wet samples were weighed and sealed in the same manner and were then heated from −40 to 80 °C.9
2.10. Shape Recovery Characterization.
Shape recovery of cylindrical foam samples (6 mm diameter, 10 mm length, six samples per series) crimped over a wire was examined at 50 °C in RO water to determine the volume recovery behavior (strain recovery). Samples were crimped to a minimal diameter (approximately 1.0 mm using a SC150-42 Stent Crimper (Machine Solutions, Flagstaff, AZ)). To crimp, samples were equilibrated at 100 °C for 10 min and then radially compressed and cooled to room temperature. Samples relaxed for 12 h and were tested over the course of 30 min. ImageJ was used for analysis of the change in diameters over time.8
2.11. Gravimetric Analysis of Degradation.
For analysis of degradation rates, cleaned samples were completely immersed in respective solutions of 2% H2O2, PBS, and 0.1 M NaOH, all stored at 37 °C, based upon modeled cerebrovascular aneurysm in vivo conditions.30 Sample solutions were changed every 3 days to ensure a relatively stable ion concentration, and once per week samples were removed, cleaned in EtOH, and dried in at 50 °C under vacuum (30 in Hg), after which sample mass was recorded and samples returned to fresh solution. The effects of ester network concentration and network molecular weight on degradation rates were determined.
Predicted dosing and toxicity risk were determined from gravimetric analysis. Degradation product release was assumed proportional to starting material concentration and corresponding to mass loss. Literature review was used to determine the risk threshold as well as average dimensions of aneurysms for risk calculations, based upon ISO 10993 protocols.
3. RESULTS AND DISCUSSIONS
3.1. Synthesis and Characterization.
The reaction of SuA with TEA resulted in the formation of the ester network over the course of several hours (Figures 1 and 2), with an acid number of 9.5 and an alcohol number of 379. The acid number of the ester (9.5 ± 1.1) compared with the high alcohol number (379 ± 50) indicates that while there is a presence of carboxylic acid groups that can undergo reactions with isocyanate groups, the end groups are prominently alcohols, which will yield the desired urethane linkages. The formation of the product network was noted by a darkening of the solution (clear to brown) and a distinct increase in solution viscosity.
Figure 1.
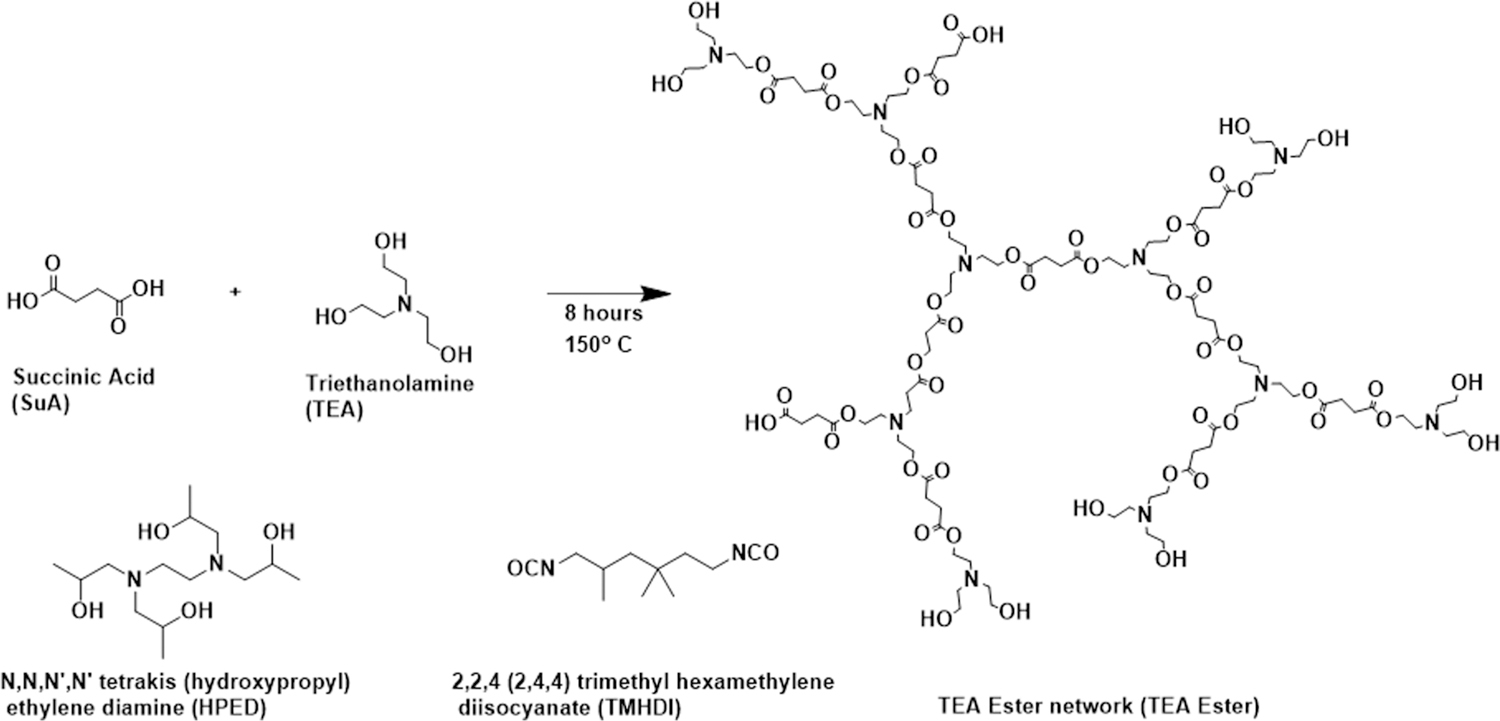
Synthetic scheme for the ester network prepolymer and the urethane-ester SMP network, displaying the idealized network structure.
Figure 2.
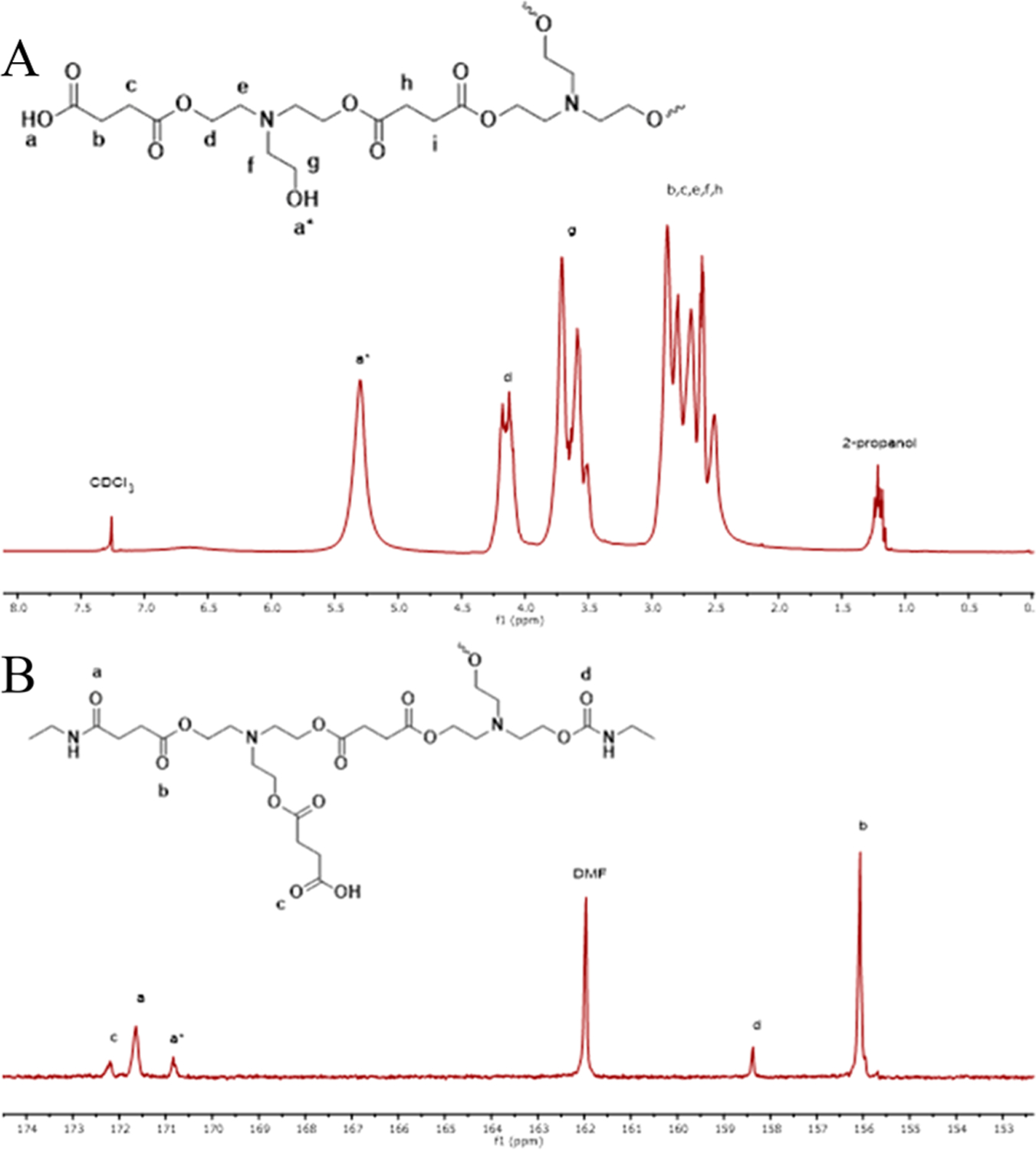
(A) 1H NMR spectra of succinic acid-triethanolamine ester network and (B) 13C NMR of the ester network reacted with ethyl isocyanate to form a model network.
Formation of the ester band at approximately 1700 cm−1 confirmed the formation of ester linkages. 1H and 13C NMR of the ester network was further indicative of ester linkage formation (172.9 ppm), also confirmed by FTIR in Figure 3 (peak at 1730 cm−1) as well as the presence of some carboxylic acid (173.8 ppm) (Figure S1). The alcohol stretch peak is broad (3676 cm−1 to 3011 cm−1), indicating hydrogen bonded hydroxyl groups, and the peak at 804 cm−1 is indicative of out of plane bending of the free hydroxyl.
Figure 3.

(A–C) FTIR spectra of SMP foams containing stoichiometric amounts of (A) 50% ester, (B) 25% ester, (C) 0% ester (control); 13C solid state NMR of (D) 0% ester film, (E) 25% ester foam, (F) 50% ester foam at 4.5 kHz.
The reaction of the diisocyanate with HPED and the ester network utilized DMF for solvation and as a physical blowing agent during foaming. Figure 3 displays the FTIR spectra for the 50%, 25%, and 0% ester SMPs. The carbonyl peak for 0% ester associated with the urethane was found at 1688 cm−1, with the shift to lower wavenumbers occurring due to the hydrogen bonding of the polymer. In the 50% ester, this is shifted to 1692 cm−1, indicating the presence of the ester linkage. The widening of this peak, as well as the slight shift in the peak at 1542 cm−1 (0% ester) to 1528 cm−1, is indicative of amide linkage presence within the polymer.
A carbonyl peak at 155 ppm associated with the urethane and at 169 ppm for the urea appears in all of the examined samples, along with the appearance of a shoulder centered at 164 ppm, which may be associated with amide formation. Additional peaks are found ranging from 216 to 221 ppm, indicating carboxylic acids with various states of hydrogen bonding associated with shape memory behavior as reported elsewhere.32–35
3.2. Morphology of Bulk Materials.
The gel fraction of the reaction decreased with increasing ester (Table 1); the control sample gel fraction was approximately 99%, and this decreased to approximately 92% with 50% ester. This may be due to the presence of carboxylic acid end groups, which may initially react more slowly with isocyanates compared with alcohols during the initial polymerization; the carboxylic acid-isocyanate reactions seem to be result in the more spherical final pore morphology as noted above.40,41 The cross-link density, as expected, was increased with increasing ester concentration, as presented by the dramatic decreasing in swelling ratio (44.7% with 0% ester decreasing to 6.6% with 50% ester).
Table 1.
Density, Swelling Ratio, Gel Fraction, and Average Contact Angle of Ester-SMPs (n = 3)
| control | 15% ester | 25% ester | 50% ester | |
|---|---|---|---|---|
| density (g/mL) | 0.022 ± 0.002 | 0.055 ± 0.006 | 0.047 ± 0.003 | 0.059 ± 0.002 |
| swelling ratio (%) | 44.70 ± 9.09 | 9.96 ± 1.51 | 7.39 ± 1.04 | 6.64 ± 1.20 |
| gel fraction (%) | 0.99 ± 0.01 | 0.97 ± 0.05 | 0.97 ± 0.01 | 0.92 ± 0.01 |
| average contact angle (deg) | 92.5 | 91.7 | 83.1 | 52.2 |
The swelling ratio of the porous SMPs, density, hydrophobicity, and the gel fraction are presented in Figure 4 and Table 1. With increasing ester concentration, the material density was slightly increased, confirmed qualitatively with SEM (Figure 5). As ester replaced HPED, the pores became more homogeneous and spherical, and the pore diameter (measured as the average distance across the pore) decreased; the ester seemingly increases the isotropy of SMP pores and pore wall thickness. Recent work by Quell et al. examined the foaming process of polyHIPEs, determining that surfactant and initiator species and concentration play considerable roles in determining macroscopic morphology. This work suggests that decreased wall thickness in the higher ester samples could be achieved in future studies by increasing surfactant concentration, which might also increase pore interconnectivity.36 Additionally, the shift in the morphology of the pores from polyhedral to spherical seems to be due to the increase in CO2 release due to carboxylic acid-isocyanate reactions.37–39 Interestingly, the amino alcohol-diisocyanate reaction seems to favor polyhedral morphologies, and it only with the addition of the ester network here (with alcohol and carboxylic acid end groups) that the pore morphology changes.30 Diisocyanate species, surfactant species, and alcohol species have not previously impacted the pore shape, including both amino alcohols and ester-containing alcohols.25,30
Figure 4.
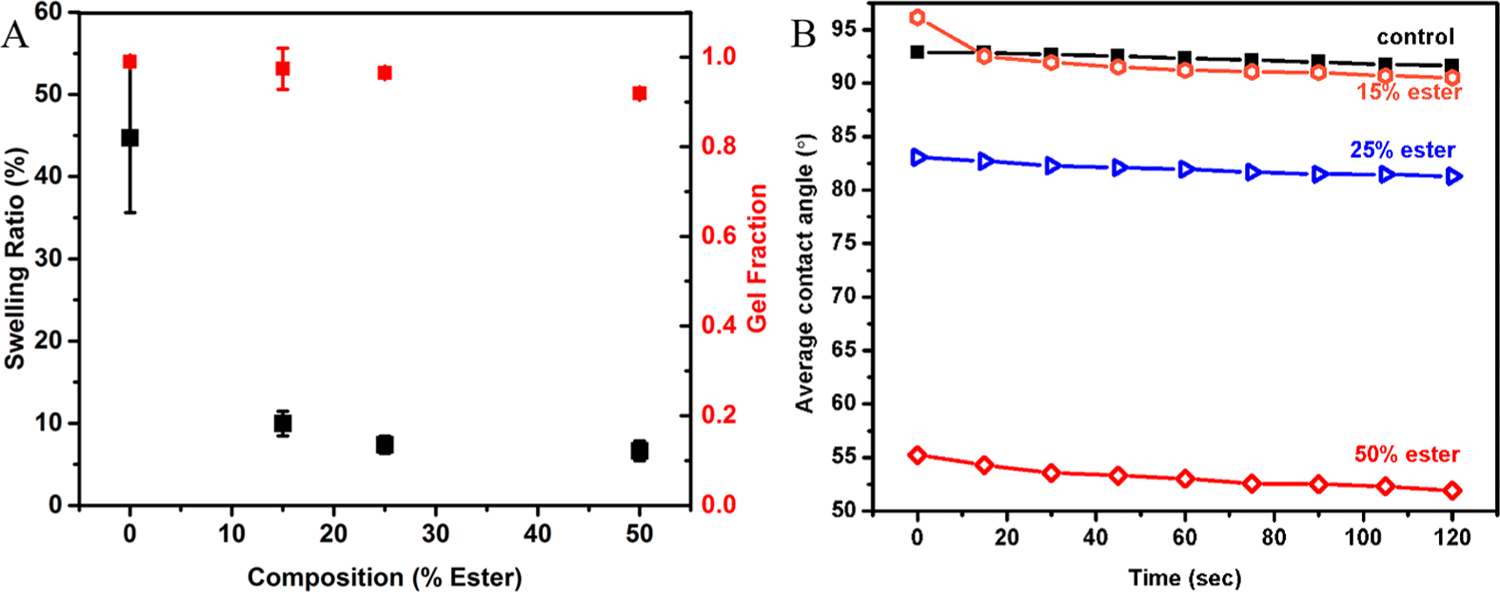
(A) Swelling ratio and gel fraction of the SMPs based on ester concentration (alcohol ratio %). (B) Advancing contact angle measured by DI water dropped onto nonporous SMP surfaces at ambient conditions.
Figure 5.

SEM images of (A, D) 0% ester, (B, E) 25% ester, and (C, F) 50% ester, with (G) average pore diameters for both long and short axis displayed and the (H) anisotropy of the pores (ratio of long to short axis) (n = 75 pores per composition).
3.3. Thermomechanical Analysis.
Previous work with PCL has resulted in drastic decreases in thermal transitions (18.5°C for 50% PCL), preventing strain fixation at room temperature due to decreased Tg, which could ultimately limit clinical use. By comparison, our SMPs display a Tg of ∼65 °C at concentrations of 50% ester (Table 2); the decrease in Tg due to the presence of the ester seems to be offset by the increase in polymer network cross-linking, ultimately resulting in a theoretical lower Tg threshold of approximately 34.6 °C (DSC) or 61.1 °C (DMA) (Figure S2).25
Table 2.
Thermal and Mechanical Characterization of Ester-SMPs
| composition | wet Tg (DSC) (°C) | dry Tg (DSC) (°C) | wet tan δ (DMA) (°C) | dry tan δ (DMA) (°C) | Young’s modulus (MPa) | strain at break (%) | ultimate strength (MPa) | toughness (J m3) |
|---|---|---|---|---|---|---|---|---|
| control | 47.8 | 74.0 | 57.0 | 89.1 | 1.02 ± 0.2 | 26.95 ± 12.2 | 25.06 ± 6.4 | 26.7 ± 5.7 |
| 15% ester | 28.7 | 64.9 | 42.9 | 71.3 | 3.45 ± 0.8 | 8.99 ± 2.3 | 20.22 ± 6.8 | 20.63 ± 7.2 |
| 25% ester | 22.0 | 37.5 | 47.9 | 70.0 | 3.54 ± 1.0 | 11.60 ± 3.3 | 25.48 ± 4.2 | 26.99 ± 6.2 |
| 50% ester | 26.2 | 36.7 | 45.9 | 65.4 | 4.55 ± 1.6 | 7.32 ± 5.1 | 23.39 ± 4.1 | 19.34 ± 8.1 |
SMP tensile testing revealed that with increasing ester concentration, a decrease in strain at break occurred, along with an increase in Young’s modulus.42 Overall, this trade off did not statistically alter the toughness of the materials. Predicted final trends for thermomechanical properties (Figures S2 and S3) indicate that no significant changes will occur with ester concentrations increasing more than 50%.
3.4. Shape Memory Characterization.
The inclusion of the ester network did not alter the shape recovery kinetics compared with the control samples, as presented in Figure 6; the slight reduction in recoverable strain displayed by the 25% ester may be contributable to the material’s macrostructure as opposed to the composition. Itt appears that the 50% ester SMP will recover when implanted in the body (tan δ peak occurring at 37°C) and the DSC measured plasticized Tg occurring at 26°C. Further tuning may be achieved by alternating the diisocyanate species to a less rigid, less methylated species.30 In solvated conditions (plasticized polymer chains), the onset of recovery begins at 45 °C for this system. Extrapolating the relationship between TMHDI-based SMPs and HDI-based SMPs, this gives an approximate wet Tg of 37 °C for the HDI SMPs. Studies by Boyle et al. and others compliment those presented here, indicating that for HDI SMPs even with this Tg, the initiation regime of the shape recovery process for similar material geometries would give up to 120 s before the onset of recovery begins, indicating that this ester-based SMP system is plausible for clinical applications.12 Our TMHDI-based ester SMPs displayed nearly 60 s delays prior to initial volume expansion (strain recovery onset), prior to immersion in 50 °C water; in a clinical setting we envision such a system allowing the clinician to place the SMP, inject a bolus of heated PBS, and have time for device adjustment before shape recovery occurs. This working time delay would allow for repositioning or removal while the SMP is still in its compressed, secondary shape.
Figure 6.
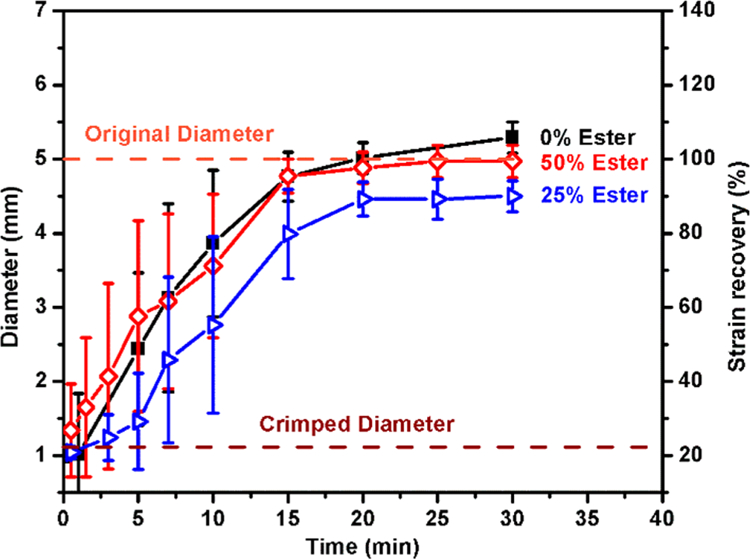
Shape recovery of SMPs, determined through expansion of compressed SMP cylinders when immersed in 50 °C water (n = 3).
3.5. Degradation Analysis.
Gravimetric change profiles are presented in Figure 7 for both hydrolytic and oxidative tests. In hydrolytic basic solution, the 50% ester lost 40% of the mass over 4 months; control has approximately the same mass as at day 0 (0% mass lost), 25% ester retained 75% mass, and 15% ester has approximately 85% of the original mass, which are comparable to other ester-containing polyurethanes, including in porous materials (Figure S4).25,43 Figure S4 SMP hydrolysis was demonstrated by the loss of the ester carbonyl peak (13C NMR) at approximately 170 ppm, as well as a slight shift in the carbonyl IR peak (1698 cm−1 in acidic conditions and 1692 cm−1 in basic conditions).
Figure 7.
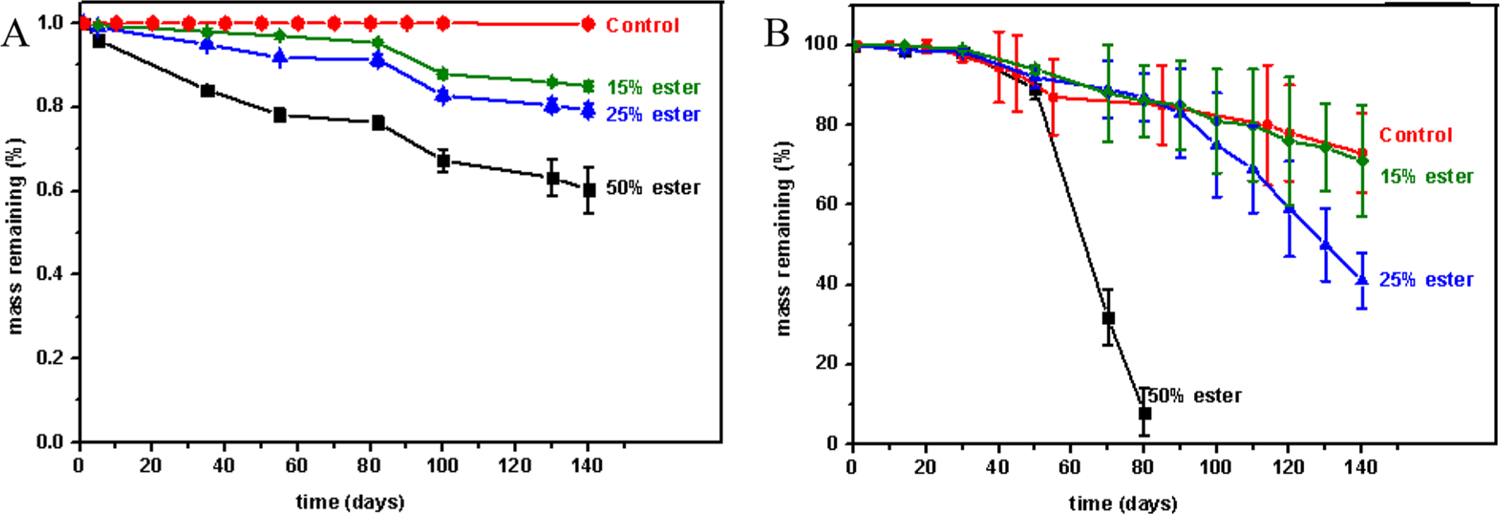
Mass loss profiles for hydrolytic degradation in (A) 0.1 M NaOH and oxidative degradation in (B) 2% H2O2 of porous SMPs at 37 °C (n = 7).
An added benefit of our approach is that the SMPs will undergo oxidatively induced mass loss as well. In these conditions, the 50% ester scaffold displays little gravimetric change until approximately 6 weeks. By 10 weeks, less than 10% of the original mass remains, and total mass loss is expected to occur within 3 months, compared with the 0% ester SMP, which is expected to have mass remaining past 18 months. Oxidation of the SMPs does not alter the ester carbonyl (Figure 8) but the splitting of the urethane carbonyl peak at approximately 160 ppm may correspond to the oxidation of TEA and subsequent carboxylic acid formation.30 TEA will rapidly oxidize to form N-oxides, after which oxidative fragmentation may result in aldehyde and amine formation.30,44,45 Secondary amines may further oxidize to a second aldehyde and a primary amine, and the aldehydes (peak at approximately 210 ppm) may eventually form carboxylic acids.30,44,45 IR spectra displayed a carbonyl shift to 1702 cm−1, indicative of the scission of the tertiary amine and subsequent formation of carboxylic acid and lower amine.30
Figure 8.
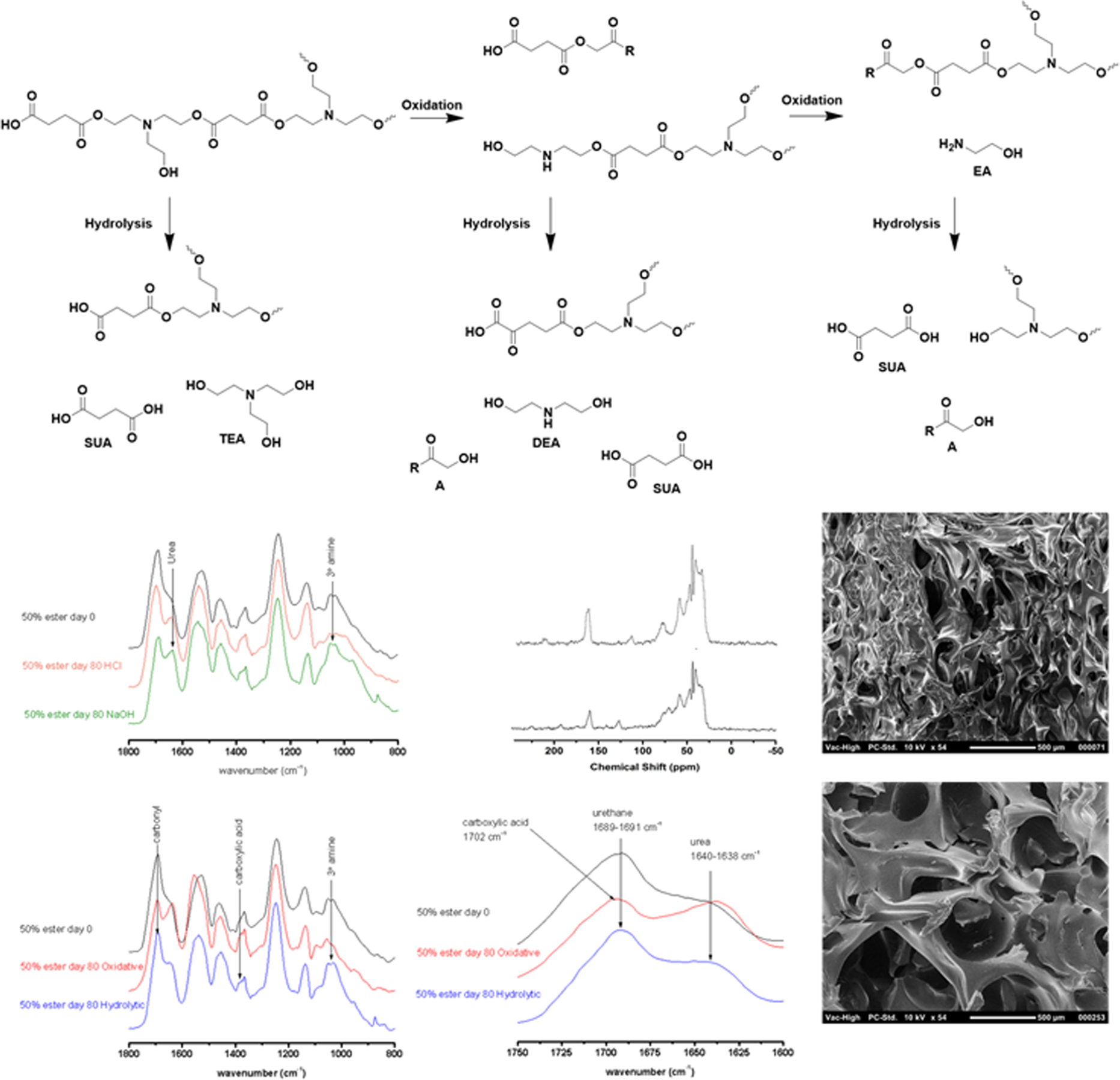
(A) Major degradation hydrolytic and oxidative products. (B) FTIR spectra of 50% ester SMPs comparing acidic and basic degradation signals at 140 days. (C) 13NMR spectra of 50% ester SMP at 80 days oxidation and (D) hydrolysis. (E) 50% ester SMP at 140 days hydrolysis. (F) Comparison of degradation signals comparing basic hydrolysis and oxidation of 50% ester SMPs with virgin material and (G) the expanded spectra of the carbonyl region, displaying the peak shift associated with oxidative degradation. (H) SEM of 50% ester SMP oxidized for 50 days.
The gravimetric analysis was then used to determine toxicity risks to determine maximum possible degradation product doses. The major hydrolytic products are succinic acid and TEA; succinic acid can be taken up by cells and therefore will be resorbed without adverse effects at sufficiently low concentrations.46,47 Intravenous studies of TEA have revealed that it is primarily excreted within hours of introduction.48 The oxidation products of remaining TEA includes diethanolamine (DEA), a secondary amine diol, monoethanolamine (EA), and glycolic acid with its derivatives (A).30 These molecules have varying levels of toxicity; oxalic acid (one such glycolic acid derivative) has a chronic toxicity threshold of approximately 40 mg/day and has been linked to renal failure above this threshold.49 Of diethanolamine and monoethanolamine, which mostly are also excreted, have a lowest no adverse event threshold of 0.162 mg/kg/day.50,51
Oxalic acid, glycoaldehyde, glyoxal, and glycolic acid are products that also may form, with, the primary toxicity concerns being oxalic acid and glycolic acid, with toxicity thresholds of 0.35 mmol/L and 17.0 mmol/L respectively.52,53 These correspond to approximately 40 mg/kg/day for oxalic acid, and more than 1 g/kg/day for glycolic acid.52,53 10 cm3 of SMP corresponds to an approximate mass of 60 mg corresponding to 17.167 mg of oxalic acid in total in an average cerebrovascular aneurysm volume of 0.187 cm3.54 SMPs will undergo oxidative mass loss at a rate of 0.784 mg/day assuming uniform oxidation of the material (calculated only over the period during which mass loss occurs). Assuming the entire mass will be metabolized to either oxalic or glycolic acids, this is still well below the chronic toxicity thresholds mentioned previously (40 mg/day as the most stringent). While translation of this SMP into a medical device requires additional chronic toxicity analysis, based upon the assessment of the degradation products and the limited solubility of larger products, it appears that this material has minimal long-term toxic risk.
Unlike previous degradable SMPs, the Tg of our SMPs was demonstrated to be more suitable for clinical applications (Tg of approximately 37 °C by DSC), and furthermore, the degradation products by hydrolysis include the resorbable monomer succinic acid and the rapidly excreted TEA. SMP oxidation results in primary and secondary amines, and causes substantial mass loss after nearly 60 days, a time period during which the implant site would have undergone endothelialization, collagen deposition, and fibrinogen migration into the polymer as demonstrated in studies by Horn et al.55 The performed oxidative testing was found to be approximately twice as fast for in vitro studies as for in vivo calculations using the control group.30 It appears that this degradable SMP can be used as a porous scaffold with tunable degradation for ensuring the material is losing mass at a very similar rate to that of tissue remodeling.
4. CONCLUSIONS
We present a degradable, highly cross-linked porous SMP intended for cardiovascular applications. SuA cross-linked ester network from TEA and the renewable monomer succinic acid was produced and incorporated into the SMP to yield a porous scaffold displaying clinically relevant Tgs and shape memory behavior. with the SMPs are both oxidatively and hydrolytically degradable with degradation metabolites assessed for their toxicological risk, with the main toxicological risk factors including oxalic acid and glycolic acid. The maximum possible product mass was compared with literature studies that have established acute and chronic toxic thresholds, with the presented ester SMPs producing an order of magnitude less than then most stringent threshold, demonstrating the potential of these SMPs for biomedical materials.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank Dr. Vladimir Bakhmoutov for assistance with NMR experiments, Dr. Lawrence Dangott for technical expertise and advice with LC–MS, and Dr. Elizabeth Cosgriff-Hernandez for scientific discussions. ThermoFisher generously provided the OrbiTrap instrument and time on the experiment. Dr. Sayyeda Hasan provided advice and support for the synthesis of the porous SMPs.
Funding
We would like to acknowledge the NASA Harriett G. Jenkins Fellowship (NNX15AU29H, A.C.W.) and National Institutes of Health/National Institute of Neurological Disorders and Stroke (Grant No. U01-NS089692, D.J.M.) for supporting this work.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsabm.8b00650.
Additional spectroscopic characterization, mechanical behavior quantification, and additional gravimetric analysis (PDF)
The authors declare the following competing financial interest(s): Regarding conflicts, Andrew C. Weems holds stock in Shape Memory Medical, Inc. (SMM), which holds a license for the shape memory polymers developed in Maitlands laboratory. Duncan Maitland owns stock and is on the board of directors at SMM.
REFERENCES
- (1).Song JE; Kim MJ; Yoon H; Lee YJ; Kim NH; Lee D; Yuk SH; Khang G Effect of Hyaluronic Acid in a HA/PLGA Scaffold on Annulus Fibrosus Regeneration: In Vivo Tests. Macromol. Res 2013, 21 (10), 1075–1082. [Google Scholar]
- (2).Huh BK; Kim BH; Kim SN; Park CG; Lee SH; Kim KR; Heo CY; Choy YB Surgical Suture Braided with a Diclofenac-Loaded Strand of Poly(Lactic-co-Glycolic Acid) for Local, Sustained Pain Mitigation. Mater. Sci. Eng., C 2017, 79 (1), 209–215. [DOI] [PubMed] [Google Scholar]
- (3).Sullins VF; Wagner JP; Suwarnasarn AT; Lee SL; Wu BM; Dunn JC Y. A Novel Biodegradable Device for Intestinal Lengthening. J. Ped. Surg 2014, 49 (1), 109–113. [DOI] [PubMed] [Google Scholar]
- (4).Mavrogenis AF; Kanellopoulos AD; Nomikos GN; Papagelopoulos PJ; Soucacos PN Early Experience with Biodegradable Implants in Pediatric Patients. Clin. Orthop. Relat. Res 2009, 467, 1591–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Peterson GI; Dobrynin AV; Becker ML Biodegradable Shape Memory Polymers in Medicine. Adv. Healthcare Mater 2017, 6, 1700694. [DOI] [PubMed] [Google Scholar]
- (6).Behl M; Lendlein A Shape-Memory Polymers. Mater. Today 2007, 10 (4), 20–28. [Google Scholar]
- (7).van Herck N; du Prez FE Fast Healing of Polyurethane Thermosets using Reversible Triazolinedione Chemistry and Shape Memory. Macromolecules 2018, 51 (9), 3405–3414. [Google Scholar]
- (8).Zhu Y; Radlauer MR; Schneiderman DK; Shaffer MSP; Hillmyer MA; Williams CK Multilock Polyesters Demonstrating High Elasticity and Shape Memory Effects. Macromolecules 2018, 51 (7), 2466–2475. [Google Scholar]
- (9).Steelman AC; Weems AC; Traverso AJ; Szafron JM; Maitland DJ; Yakovlev VV Revealing the Glass Transition in Shape Memory Polymers using Brillouin Spectroscopy. Appl. Phys. Lett 2017, 111 (24), 241904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Sokolowski W; Metcalfe A; Hayashi S; Yahia L; Raymond J Medical Applications of Shape Memory Polymers. Biomed. Mater. 2007, 2 (1), 23–27. [DOI] [PubMed] [Google Scholar]
- (11).Metcalfe A; Desfaits AC; Salazkin I; Yahia L; Sokolowski WM; Raymond J Cold Hibernated Elastic Memory Foams for Endovascular Interventions. Biomaterials 2003, 24, 491–497. [DOI] [PubMed] [Google Scholar]
- (12).Boyle AJ; Weems AC; Hasan SM; Nash LD; Monroe MBB; Maitland DJ Solvent Stimulated Actuation of Polyurethane-Based Shape Memory Polymer Foams using Dimethyl Sulfoxide and Ethanol. Smart Mater. Struct 2016, 25 (7), 075014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Li X; Pan Y; Zheng Z; Ding X A Facile and General Approach to Recoverable High-Strain Multishape Shape Memory Polymers. Macromol. Rapid Commun 2018, 39 (6), 1700613–1700618. [DOI] [PubMed] [Google Scholar]
- (14).Raidt T; Schmidt M; Tiller JC; Katzenberg F Crosslinking of Semiaromatic Polyesters toward High-Temperature Shape Memory Polymers with Full Recovery. Macromol. Rapid Commun 2018, 39 (6), 1700768–1700772. [DOI] [PubMed] [Google Scholar]
- (15).de Nardo L; Alberti R; Cigada A; Yahia L; Tanzi MC; Fare S Shape Memory Polymer Foams for Cerebral Aneurysm Reparation: Effects of Plasma Sterilization on Physical Properties and Cytocompatibility. Acta Biomater 2009, 5 (5), 1508–1518. [DOI] [PubMed] [Google Scholar]
- (16).Hampikian JM; Heaton BC; Tong FC; Zhang Z; Wong CP Mechanical and Radiographic Properties of a Shape Memory Polymer Composite for Intracranial Aneurysm Coils. Mater. Sci. Eng., C 2006, 26 (8), 1373–1379. [Google Scholar]
- (17).Ugarte L; Saralegi A; Fernandez R; Martin L; Corcuera MA; Eceiza A Flexible Polyurethane Foams based on 100% Renewably Sourced Polyols. Ind. Crops Prod 2014, 62, 545–551. [Google Scholar]
- (18).Cooper TR; Storey RF Poly(Lactic Acid) and Chain-Extended Poly(Lactic Acid)-Polyurethane Functionalized with Pendent Carboxylic Acid Groups. Macromolecules 2008, 41 (3), 655–662. [Google Scholar]
- (19).Stanzione M; Russo V; Olivero M; Verdolotti L; Sorrentino A; Di Serio M; Tesser R; Iannace S; Lavorgna M Synthesis and Characterization of Sustainable Polyurethane Foams based on Polyhydroxyls with Different Terminal Groups. Polymer 2018, 149 (1), 134–145. [Google Scholar]
- (20).Noordover BAJ; van Stallduien VG; Duchateau R; Koning CE; Mak M; Heise A; Frissen AE; van Haveren J Co-and Terpolyesters based on Isosorbide and Succinic Acid for Coating Applications: Synthesis and Characterization. Biomacromolecules 2006, 7 (12), 3406–3416. [DOI] [PubMed] [Google Scholar]
- (21).Li S; Yu J; Wade MB; Policastro GM; Becker ML Radiopaque, Iodine Functionalized Phenylalanine-based Poly(Ester Urea)s. Biomacromolecules 2015, 16 (2), 615–624. [DOI] [PubMed] [Google Scholar]
- (22).Zhou J; Defante AP; Lin F; Xu Y; Yu J; Gao Y; Childers E; Dhinojwala A; Becker ML Adhesion Properties of Catechol-based Biodegradable Amino Acid-based Poly(Ester Urea) Copolymers Inspired from Mussel Proteins. Biomacromolecules 2015, 16 (1), 266–274. [DOI] [PubMed] [Google Scholar]
- (23).Sheldon RA The E Factor 25 years on: the Rise of Green Chemistry and Sustainability. Green Chem 2017, 19, 18–43. [Google Scholar]
- (24).Becker J; Wittmann C Advanced Biotechnology: Metabolically Engineered Cells for the Bio-based Production of Chemicals and Fuels, Materials, and Health-Care Products. Angew. Chem., Int. Ed 2015, 54, 3328–3350. [DOI] [PubMed] [Google Scholar]
- (25).Singhal P; Small W; Cosgriff-Hernandez E; Maitland DJ; Wilson TS Low Density Biodegradable Shape Memory Polyurethane Foams for Embolic Biomedical Applications. Acta Biomater 2014, 10 (1), 67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Lee S; Yu S; Lee Y Degradable Polyurethanes Containing Poly(Butylene Succinate) and Poly(Ethylene Glycol). Polym. Degrad. Stab 2001, 71 (1), 81–87. [Google Scholar]
- (27).Hu S; Wan C; Li Y Production and Characterization of Biopolyols and Polyurethane Foams from Crude Glycerol-based Liquefaction of Soybean Straw. Bioresour. Technol 2012, 103 (1), 227–233. [DOI] [PubMed] [Google Scholar]
- (28).Polymer Synthesis: Theory and Practice, Fundamentals, Methods, Experiments, 5th ed.; Bruan D, Cherdon H, Rehahn M, Ritter H, Voit B, Eds.; Springer, 2013. [Google Scholar]
- (29).Weems AC; Szafron JM; Easley AD; Herting S; Smolen J; Maitland DJ Shape Memory Polymers with Enhanced Visibility for Magnetic Resonance and X-Ray Imaging Modalities. Acta Biomater 2017, 54, 45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Weems AC; Wacker KT; Carrow JK; Boyle AJ; Maitland DJ Shape Memory Polyurethanes with Oxidation-Induced Degradation: In Vivo and In Vitro Correlations for Endovascular Material Applications. Acta Biomater 2017, 59, 33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Weems AC; Boyle AJ; Maitland DJ Two-Year Performance Study of Porous, Thermoset, Shape Memory Polyurethanes Intended for Vascular Medical Devices. Smart Mater. Struct 2017, 26 (3), 035054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Borsacchi S; Paderni K; Messori M; Toselli M; Pilati F; Geppi M Insights into Shape Memory Poly(Caprolactone) Materials by Solid-State NMR. Macromolecules 2014, 47, 3544–3552. [Google Scholar]
- (33).Ito T; Yamaguchi Y; Watanabe H; Askura T Structural Analysis of Oriented Poly(Caprolactone) Including CaCO3 Particles with 13C Solid State NMR. J. Appl. Polym. Sci 2001, 80, 2376–2382. [Google Scholar]
- (34).Kaji H; Horii F One and Two Dimensional Solid-State 13C NMR Analyses of the Solid Structure and Molecular Motion of Poly(Caprolactone) Isothermally Crystallized from the Melt. Macromolecules 1997, 30, 5791–5798. [Google Scholar]
- (35).Bertmer M; Buda A; Blomenkamp-Hofges I; Kelch S; Lendlein A Biodegradable-Shape Memory Polymer Networks: Characterization with Solid-State NMR. Macromolecules 2005, 38 (9), 3793–3799. [Google Scholar]
- (36).Quell S; de Bergolis B; Drenckhan W; Stubenrauch C How the Locus of Initiation Influences the Morphology and the Pore Connectivity of a Monodisperse Polymer Foam. Macromolecules 2016, 49, 5059–5067. [Google Scholar]
- (37).Viswanathan P; Johnson DW; Hurley C; Cameron NR; Battaglia G 3D Surface Functionalization of Emulsion-Templated Polymeric Foams. Macromolecules 2014, 47, 7091–7098. [Google Scholar]
- (38).Bakir M; Meyer JL; Economy J; Jasiuk I Heat-Induced Polycondensation Reaction with Self-Generated Blowing Agent Forming Aromatic Thermosetting Copolyester Foams. Macromolecules 2016, 49, 6489–6496. [Google Scholar]
- (39).David D; Silverstein MS Porous Polyurethanes Synthesized within High Internal Phase Emulsions. J. Polym. Sci., Part A: Polym. Chem 2009, 47, 5806. [Google Scholar]
- (40).Xiao H; Xiao HX; Frisch KC; Malwitz N Kinetic Studies of the Reactions between Isocyanates and Carboxylic acids. High Perform. Polym 1994, 6, 235–239. [Google Scholar]
- (41).Schieler L Kinetics of the Reaction between Alcohols and Isocyanates Catalyzed by Ferric Acetylacetonate NASA Technical Report No 32-129; NASA, 1961. [Google Scholar]
- (42).Shi S; Wu Q; Gu L; Zhang K; Yu H Bio-based Copolylactide-Urethane Networks with Shape Memory Behavior at Body Temperature. RSC Adv 2016, 6, 79268. [Google Scholar]
- (43).Zhou L; Yu L; Ding M; Li J; Tan H; Wang Z; Fu Q Synthesis and Characterization of pH-Sensitive Biodegradable Polyurethane for Potential Drug Delivery Applications. Macromolecules 2011, 44, 857–864. [Google Scholar]
- (44).Shechter H; Rawalay SS Oxidation of Primary, Secondary, and Tertiary Amines with Neutral Potassium Permanganate. II. J. Am. Chem. Soc 1964, 86 (9), 1706–1709. [Google Scholar]
- (45).Smith JRL; Norman ROC; Rowley AG Amine Oxidation. Pt VIII. Evidence for Intramolecular Hydrogen-Atom Transfer in Amine Radical Cations. J. Chem. Soc., Perkin Trans 1 1973, 565–571. [Google Scholar]
- (46).de castro Fonseca M; Agular CJ; Da Rocha Franco JA; Gingold RN; Leite MF GPR91: Expanding the Frontiers of Krebs Cycle Intermediates. Cell Commun. Signaling 2016, 14, 3 10.1186/s12964-016-0126-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Jager E; Donato RK; Perchacz M; Jager A; Surman F; Hocherl A; Konefal R; Donato KZ; Venturini CG; Bergamo VZ; Schrekker HS; Fuentegria AM; Raucci MG; Ambrosio L; Stepanek P Biocompatible Succinic Acid-based Polyesters for Potential Biomedical Applications: Fungal Biofilm Inhibition and Mesenchymal Stem Cell Growth. RSC Adv 2015, 5, 85756–85766. [Google Scholar]
- (48).Knaak JB; Leung HW; Stott WT; Busch J; Bilsky J Toxicology of Mono-, Di-, and Triethanolamine. Rev. Environ. Contam. Toxicol 1997, 149, 1–86. [DOI] [PubMed] [Google Scholar]
- (49).Mendrala AL; Waechter JM; Bormett GA; Bartels MJ; Stott WT The Pharmacokinetics of Diethanolamine in Sprague-Dawley Rats following Intravenous Administration. Food Chem. Toxicol 2001, 39 (9), 931–939. [DOI] [PubMed] [Google Scholar]
- (50).TOXNET Toxicology Data Network. Diethanolamine; ToxNet, 2014. https://toxnet.nlm.nih.gov/cgi-bin/sis/search/a?dbs+hsdb:@term+@DOCNO+924 (accessed on 4 Nov 2017).
- (51).National research Council (US) Committee on Toxicology. Emergency and Continuous Exposure Limits for Selected Airborne Contaminants - Ethanolamine; National Academies Press (US): Washington D.C., 1984; Vol 2. [Google Scholar]
- (52).Woolf AD; Wynshaw-Boris A; Rinaldo P; Levy HL Intentional Infantile Ethylene Glycol Poisoning Presenting as an Inherited Metabolic Disorder. J. Pediatr 1992, 120 (3), 421–424. [DOI] [PubMed] [Google Scholar]
- (53).Hlozek T; Bursova M; Cabalaa R Fast Determination of Ethylene Glycol, 1,2-Propylene Glycol and Glycolic Acid in Blood Serum and Urine for Emergency and Clinical Toxicology by GC-FID. Talanta 2014, 130 (1), 470–474. [DOI] [PubMed] [Google Scholar]
- (54).Carter BS; Sheth S; Chang E; Sethl M; Ogilvy CS Epidemiology of the Size Distribution of Intracranial Bifurcation Aneurysms: Smaller Size of Distal Aneurysms and Increasing Size of Unruptured Aneurysms with Age. Neurosurg 2006, 58 (2), 217–223. [DOI] [PubMed] [Google Scholar]
- (55).Horn J; Hwang W; Jessen SL; Keller BK; Miller MW; Tuzun E; Hartman J; Clubb FJ; Maitland DJ Comparison of Shape Memory Polymer Foam Versus Bare Metal Coil Treatments in an In Vivo Porcine Sidewall Aneurysm Model. J. Biomed. Mater. Res., Part B 2017, 105 (7), 1892–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.