

Abstract
Despite decades of clinical use, mechanisms of glucocorticoid resistance are poorly understood. We treated primary murine T lineage acute lymphoblastic leukemias (T-ALLs) with the glucocorticoid dexamethasone (DEX) alone and in combination with the pan-PI3 kinase inhibitor GDC-0941 and observed a robust response to DEX that was modestly enhanced by GDC-0941. Continuous in vivo treatment invariably resulted in outgrowth of drug-resistant clones, ~30% of which showed low glucocorticoid receptor (GR) protein expression. A similar proportion of relapsed human T-ALLs also exhibited markedly reduced GR protein levels. De novo or pre-existing mutations in the gene encoding GR (Nr3c1) occurred in relapsed clones derived from multiple independent parental leukemias. CRISPR/Cas9 gene editing confirmed that loss of GR expression confers DEX resistance. Exposing drug-sensitive T-ALLs to DEX in vivo altered transcript levels of multiple genes, and this response was attenuated in relapsed T-ALLs. These data implicate reduced GR protein expression as a frequent cause of glucocorticoid resistance in T-ALL.
Introduction
Glucocorticoids are a cornerstone of therapeutic protocols for T-ALL and other lymphoid cancers and a poor initial response to glucocorticoids remains one of the strongest predictors of relapse in ALL (1). Studies performed in the 1980s revealed low GR protein levels in some diagnostic and relapsed pediatric ALL samples, but uncovered no clear association with treatment response (2–5). In T-ALL cell lines exposed to glucocorticoids in vitro, resistance was associated with somatic NR3C1 mutations resulting in impaired GR activity, reduced receptor expression, and failure to up-regulate GR in response to glucocorticoid exposure (6–12). However, NR3C1 alterations were found only rarely in relapsed human B and T lineage ALLs (13–17) with the exception of relapsed B-cell precursor ALLs, which harbor frequent copy number alterations resulting in NR3C1 deletions (18–21). Thus, the molecular mechanisms contributing to glucocorticoid resistance and relapse in ALL remain poorly understood.
Transplanting primary mouse T-ALLs generated via retroviral insertional mutagenesis (RIM) in wild-type (WT) and KrasG12D mice and treating them in vivo is an unbiased strategy for elucidating resistance mechanisms (22). In a previous study, the pan-PI3 kinase (PI3K) inhibitor GDC-0941 exhibited anti-leukemia activity as a single agent and in combination with the MEK inhibition PD0325901 in this system (REF). These data and a report that identified GR as a substrate for phosphorylation and degradation by the PI3K effector AKT (23) raised the possibility that PI3K inhibition might be synergistic with glucocorticoid treatment in T-ALL. To address this question, we treated 10 independent T-ALLs with the glucocorticoid dexamethasone (DEX) alone and in combination with GDC-0941. These studies revealed frequent down-regulation of GR protein expression in drug-resistant leukemias isolated at relapse, including clones with either pre-existing or acquired Nr3c1 mutations that followed different evolutionary trajectories. Directly inactivating NR3C1 induced profound glucocorticoid resistance in drug-sensitive human T-ALL cells, and primary murine leukemias with reduced GR expression showed impaired regulation of glucocorticoid response genes upon in vivo DEX exposure. These data support the existence of diverse molecular mechanisms that converge on reduced GR expression as a frequent cause of glucocorticoid resistance in T-ALL and potentially other lymphoid cancers.
Results
Dexamethasone and GDC-0941 show in vivo efficacy in T-ALL
Treating healthy mice with 15 mg/kg/day of DEX alone and in combination with 125 mg/kg/day of GDC-0941 (24) caused lymphopenia and splenic and thymic atrophy without other toxicities (data not shown). T-ALLs generated by performing RIM with MOL4070 (25) in wild-type mice (KrasWT) and congenic animals harboring a Kras oncogene (KrasG12D) (22, 26) contain frequent insertions and/or somatic mutations in known human T-ALL genes (26, 27). Ten independent T-ALLs (n=5 each of KrasWT and KrasG12D) were transplanted into 14 mice each and recipients were assigned to treatment with vehicle (n=4), DEX (n=5), or DEX/GDC-0941 (n=5) (Fig. 1a). Mice were treated for eight weeks or until disease progression, and bone marrow was obtained at euthanasia.
Figure 1. RIM-induced T-ALLs respond to dexamethasone alone or in combination with GDC-0941 and exhibit intrinsic glucocorticoid resistance upon re-treatment.
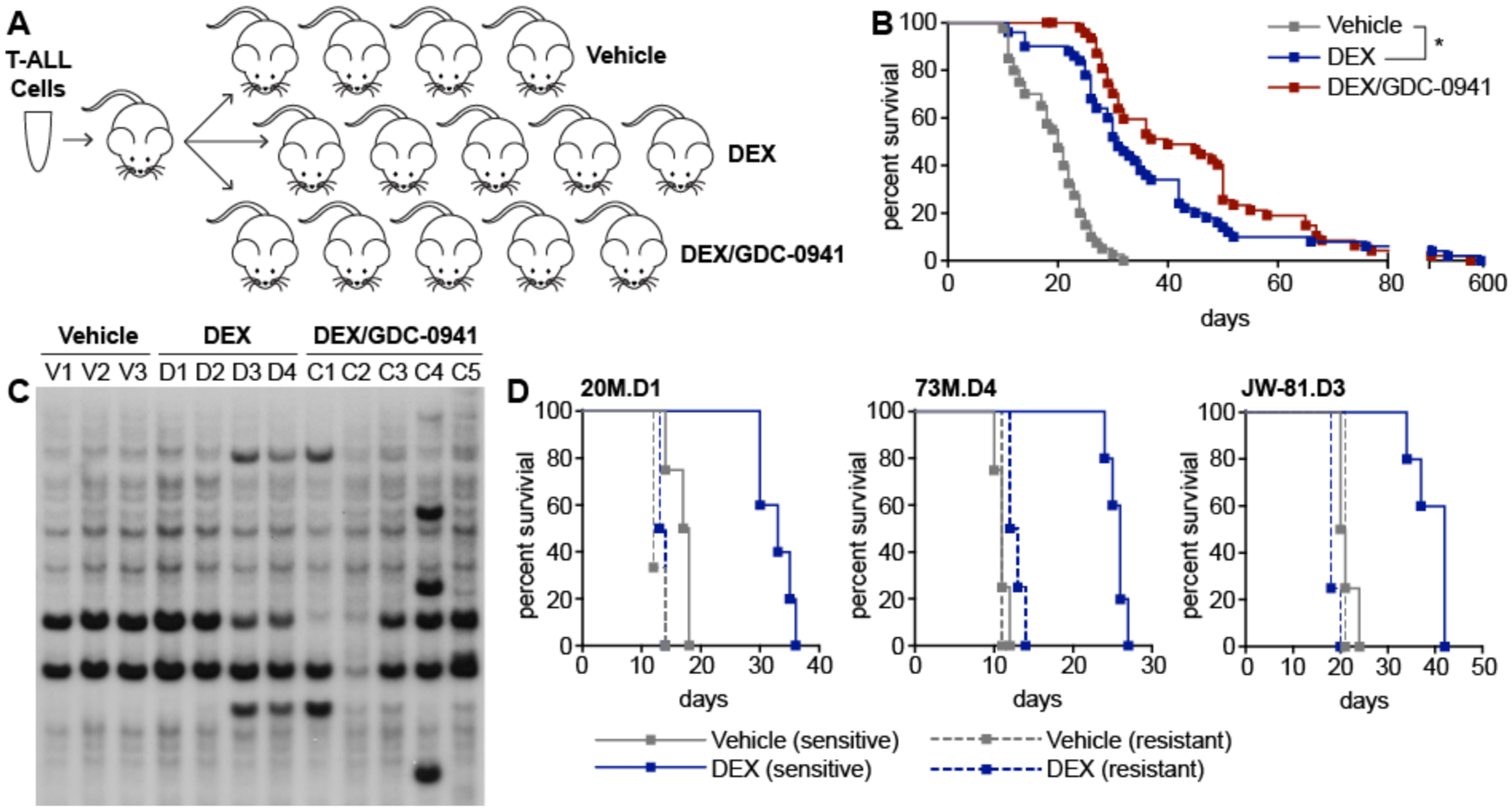
(A) Primary T-ALLs were first expanded in a single recipient. After euthanasia, 2×106 bone marrow cells were harvested and transplanted into 14 recipient mice, which were then randomly assigned to one of the three indicated treatment groups. (B) Kaplan-Meier survival analysis of recipient mice transplanted with 10 independent primary T-ALLs (five KrasWT and five KrasG12D) and treated with vehicle (n=40), DEX (n=50), or DEX/GDC-0941 (n=50) indicated that DEX significantly extended survival (p<0.0001, Log-rank test), which was modestly but not significantly enhanced by GDC-0941 (p=0.0805, Log-rank test). (C) Southern blot analysis with a probe for the MOL4070 virus of DNA extracted from leukemia cells harvested from individual moribund recipients of T-ALL 20M that were treated with vehicle (V1-V3), DEX (D1-D4), or the DEX/GDC-0941 combination (C1-C5). Novel restriction fragments indicative of retroviral integrations that are not present in the dominant parental clone are visible in relapsed leukemias D3, D4, C1, and C4. (D) Kaplan-Meier survival analysis of resistant T-ALLs (dotted lines) treated with vehicle (n=3) or DEX (n=4) compared to the corresponding drug-sensitive parental T-ALLs (solid lines) treated with vehicle (n=4) or DEX (n=5). Resistant T-ALLs demonstrated significantly shorter survival after DEX treatment versus the corresponding parental leukemias; 20M.D1 (p=0.0039, Log-rank test), 73M.D4 (p=0.0027, Log-rank test), and JW-81.D3 (p=0.0025, Log-rank test).
DEX-treated recipient mice showed a significant survival advantage compared to vehicle-treated controls (median 31 versus 20 days; p<0.0001, Log-rank test) that was modestly enhanced by the addition of GDC-0941 (median 31 versus 40 days; p=0.0805, Log-rank test) (Fig. 1b). Examining the 10 individual leukemias revealed variable sensitivity to DEX and DEX/GDC-0941 (Table S1) that was independent of KrasG12D mutation status (Fig. S1a, b). To characterize genetic differences between parental and relapsed T-ALLs, we isolated leukemia cells from 71 mice that progressed after a significant (p<0.005) response to DEX or DEX/GDC-0941 treatment and performed Southern blotting to detect retroviral insertion sites. Twenty-eight relapsed leukemias (39%) exhibited one or more novel integrations compared to the corresponding parental T-ALL (Fig. 1c). Parental and relapsed T-ALLs invariably shared multiple retroviral integrations indicating evolution from a common founder clone, consistent with studies of relapsed human ALLs (13). Moreover, T-ALLs that relapsed after an initial response to DEX acquired intrinsic drug resistance, confirmed by transplantation into secondary recipients and in vivo re-treatment with DEX (Fig. 1d and Fig. S1c). Relapsed T-ALLs with and without clonal evolution as visualized by Southern blotting exhibited DEX resistance, suggesting the existence of both insertional and non-insertional mechanisms.
Reduced GR protein expression is frequent in relapsed mouse and human T-ALLs
We performed Western blotting to assess Notch1 and Ras-regulated signaling molecules. These studies did not uncover consistent differences in the levels of cleaved Notch1, PTEN, phosphorylated ERK, phosphorylated Akt or phosphorylated S6 between parental and relapsed leukemias (Fig. S2). However, we observed markedly reduced or absent GR protein expression in multiple independent relapsed T-ALLs compared to the corresponding DEX-sensitive parental leukemia, which was independent of Kras mutation status (Fig. 2a). We next performed immunohistochemistry (IHC) to assess GR protein levels in a cohort of 102 diagnosic and relapsed T-ALL patient bone marrow biposies in order to determine if human leukemias also exhibited loss of receptor expression at relapse (Fig. 2b). Analysis of Western blotting data in our murine leukemias demonstrated high levels of GR in 80% of parental T-ALLs (n=10) versus 32% of relapsed leukemias (n=65), with the remaining samples exhibiting medium (34%) or low (34%) GR protein expression (Fig. 2c; p-value=0.0109, Fisher’s test). Strikingly, blinded IHC analysis of patient samples with comparable disease burden (Table S2) revealed robust GR protein expression in 90% of diagnostic T-ALL samples (n=72), whereas 40% of the relapsed leukemias (n=30) were either weakly positive (27%) or negative (13%) for GR staining (Fig. 2d; p-value=0.0013, Fisher’s test). Together, these data demonstrate a significant increase in the proportion of T-ALLs with reduced or absent GR protein expression at relapse in large cohorts of mouse and human T-ALLs.
Figure 2. Relapsed mouse and human T-ALLs show reduced glucocorticoid receptor protein expression which confers dexamethasone resistance in vitro.
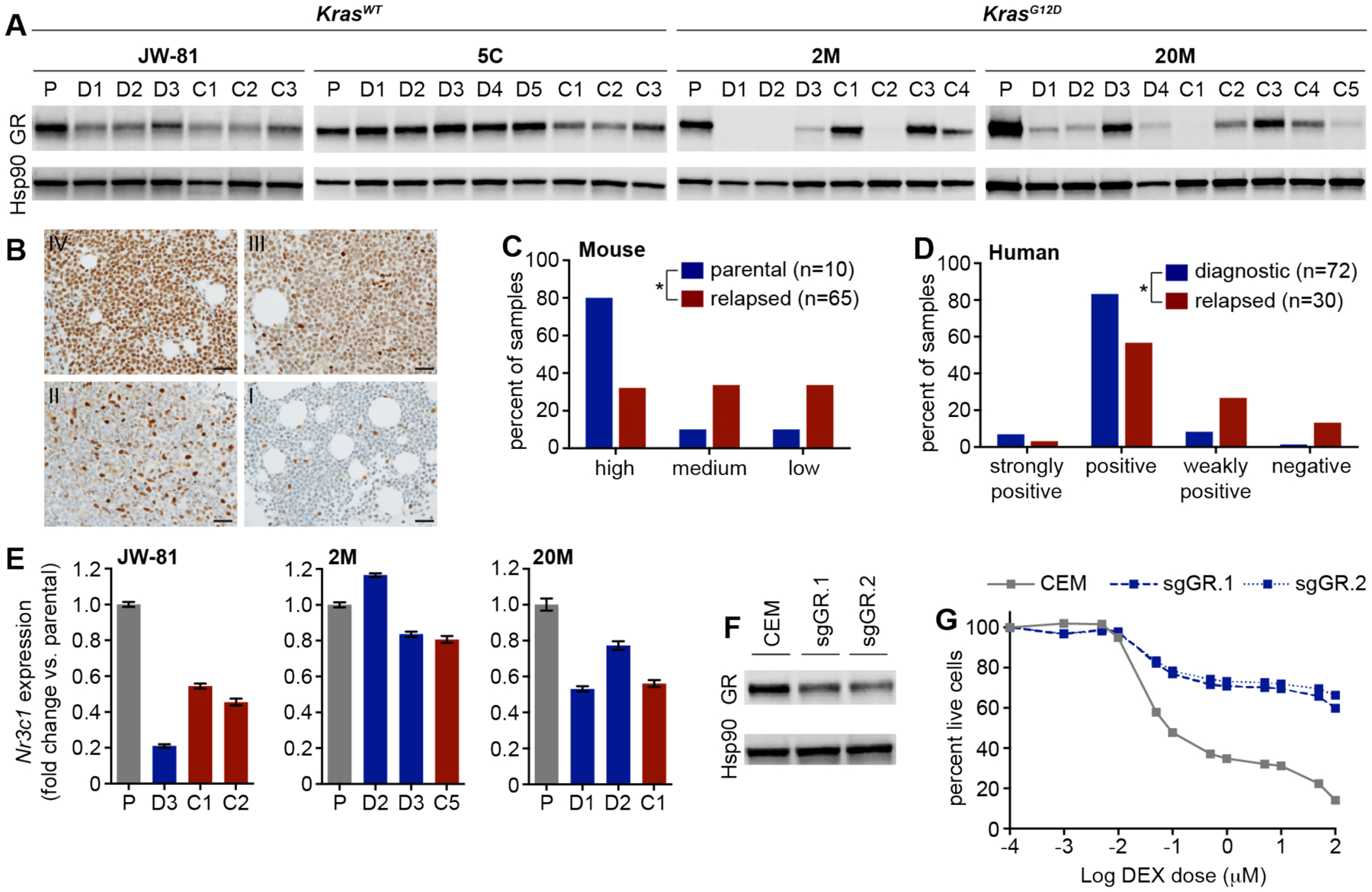
(A) Western blot analysis of bone marrow lysates from KrasWT and KrasG12D T-ALLs in parental (P), DEX treated (D) and DEX/GDC-0941 combination treated (C) mice at relapse. (B) Human bone marrow from T-ALL patients was subjected to immunohistochemistry analysis of GR protein expression and scored in a blinded fashion independently by two hematopathologists to classify samples as strongly positive (IV), positive (III), weakly positive (II) or negative (I). Scale bar represents 30 microns. Graphical representation of GR expression levels assessed by Western blot in primary murine T-ALLs (C) and by immunohistochemistry in human T-ALL patient samples (D). GR expression in murine T-ALLs was classified as low (examples in panel A include 2M.D1, 2M.C2, 20M.D4, 20M.C5), medium (examples include JW-81.C2, 5C.C2, 2M.C4, 20M.C4), or high (examples include 5C.D1, 2M.C1, 2M.C3, 20M.D3). Asterisk in panel C denotes a significant difference between high and medium/low classifications in parental versus relapsed murine T-ALLs (p-value=0.0109, Fisher’s test). Asterisk in panel D denotes a significant difference between strongly positive/positive and weakly positive/negative classifications in diagnostic versus relapsed human T-ALLs (p-value=0.0013, Fisher’s test). (E) Gene expression analysis using a TaqMan assay to measure Nr3c1 transcript levels in three independent parental T-ALLs (P), and corresponding DEX (D) or DEX/GDC-0941 combination (C) treated relapsed T-ALLs. Error bars represent standard error of the mean for technical replicates. (F) Western blot analysis of GR expression in lysates from the CCRF-CEM human T-ALL cell line edited using nucleofection with Cas9 ribonuleoproteins containing two independent sgRNAs targeting the Nr3c1 gene. (G) Analysis of viability in the CCRF-CEM cells CRISPR edited at the GR locus and exposed to increasing doses of DEX. Error bars represent standard deviation of technical replicates.
In order to assess the relationship between GR protein expression and Nr3c1 transcript levels, we performed quantitative RT-PCR (qPCR) analysis of bone marrow harvested from paired parental and relapsed murine T-ALLs. These studies revealed variably decreased Nr3c1 mRNA levels at relapse (Fig. 2e), suggesting GR protein levels may reflect reduced Nr3c1 transcription or result from post-transcriptional or post-translational mechanisms. We next performed CRISPR-mediated gene editing of Nr3c1 in CCRF-CEM, an established DEX-sensitive human T-ALL cell line (28). CRISPR editing using ribonucleoproteins (RNPs) containing one of two independent sgRNA molecules targeting the Nr3c1 locus with high efficiency (58.9% for sgGR.1, 64.7% for sgGR.2) resulted in reduced GR protein expression (Fig. 2f) and also rendered CCRF-CEM cells resistant to DEX-induced cell death (Fig. 2g).
Relapsed T-ALLs harbor pre-existing or acquired Nr3c1 mutations
To identify genomic alterations that might cause DEX resistance, we performed exome sequencing of 25 relapsed murine T-ALLs, including 22 with reduced or absent GR protein expression. This analysis revealed distinct point mutations or small insertions/deletions (indels) within Nr3c1 coding exons in 18% of relapsed T-ALLs analyzed that emerged after treatment with DEX or DEX/GDC-0941 (n=22, including four samples from different recipients transplanted with KrasG12D T-ALL 78A that harbored the same indel). Nr3c1 mutations were also detected in mice transplanted with KrasWT T-ALL 5C and KrasG12D T-ALLs 2M and 20M (Fig. 3a and Table S3). Targeted Sanger sequencing of additional drug-treated relapsed clones isolated from recipients of parental T-ALL 78A revealed the same Nr3c1 L282fs indel at a variant allele frequency (VAF) that correlated with GR protein levels (Fig. 3b and Fig. S3a), and ultra-deep sequencing at approximately 30,000x coverage identified the L282fs indel at 1.1% VAF in parental T-ALL 78A (Fig. S3b). By contrast, the Nr3c1 mutations in T-ALLs 5C.C3, 2M.C4, and 20M.D1 were each detected at relapse in only one recipient mouse (n=8 relapsed clones analyzed for T-ALL 5C; n=8 for T-ALL 2M; n=9 for T-ALL 20M). Accordingly, targeted sequencing did not identify pre-existing Nr3c1 mutations Q517X and I513fs in parental T-ALLs 5C and 2M, respectively at a minimum sensitivity of <0.1% VAF. As in human T-ALL (29), somatic Notch1 mutations represent late genetic events in this model (26, 27). T-ALLs 2M and 20M contain somatic Notch1 PEST domain mutations that were retained in resistant clones with or without Nr3c1 mutations, strongly suggesting that the Nr3c1 mutation was acquired later. Together, these data indicate T-ALL cells with Nr3c1 mutations are selected by DEX treatment, either as rare pre-existing subclones or after de novo acquisition of this alteration, consistent with data in human ALL demonstrating both selection of pre-existing relapse clones during treatment and development of new mutations that drive drug resistance (13, 16, 30–32).
Figure 3. Nr3c1 mutations in relapsed T-ALLs drive leukemia outgrowth in vivo and confer dexamethasone resistance in vitro.

(A) Mutant allele frequencies for mutations identified in the parental (P, x-axis) and the corresponding DEX-treated (D) or DEX/GDC-0941 combination-treated (C) relapsed T-ALLs (y-axis) highlighting the specific Nr3c1 mutations or indels. (B) Box and whiskers plot showing allele frequency of the Nr3c1 indel in each relapsed sample as compared to parental T-ALL 78A as determined from Sanger sequencing data (top), and corresponding Western blot data showing GR protein expression (bottom). Whiskers represent 10–90 percentile. (C) Sanger sequencing traces showing the site of the Nr3c1 point mutation (arrow) in parental and relapsed 20M T-ALLs. Note that representation of the mutant allele is stable after transplantation in the absence of treatment, but increases markedly in secondary recipients that received DEX. (D) Western blot showing GR protein expression in the parental (P), relapsed (D) and re-treated 20M T-ALLs shown in panel C. (E) Viability of independent CRISPR-edited CCRF-CEM cell clones nucleofected with a scramble guide (S1, S2, S3) or with a guide targeting the region of exon 4 in which the mutation in T-ALL 20M.D was identified (GR1, GR2, GR3) in response to increasing doses of DEX. Error bars represent standard deviation of technical replicates. (F) Western blot analysis showing GR expression in lysates from the CCRF-CEM cell clones edited with scramble or Nr3c1-targeting guides.
Loss of Nr3c1 enhances clonal fitness and confers dexamethasone resistance
To determine if Nr3c1 mutations identified in relapsed T-ALLs are sufficient to induce DEX resistance, we performed functional studies in both relapsed T-ALL 20M.D1 and CCRF-CEM cells. Relapsed T-ALL 20M.D1 harbors a truncating Nr3c1 K514X stop-gain mutation at approximately 70% VAF, and exhibited phenotypic resistance upon DEX re-treatment in secondary recipients (Fig. 1d). In addition, in vivo re-treatment of relapsed clone 20M.D1 with DEX, but not the control vehicle, strongly selected for outgrowth of Nr3c1-mutant T-ALL cells (Fig. 3c) that exhibited profoundly reduced GR protein expression (Fig. 3d). We also performed nucleofection in CCRF-CEM cells with RNPs containing sgRNAs targeting the homologous region of Nr3c1 harboring the K514X mutation identified in T-ALL 20M.D1, then used limiting dilution to isolate multiple independent single-cell clones that exhibited bi-allelic editing. Whereas three CRISPR-edited clones exhibited loss of GR protein expression and resistance to DEX, three clones isolated after nucleofection with a control RNP containing a scrambled sgRNA showed no change in GR levels and remained drug-sensitive (Fig. 3e, f). Together, these studies indicate the Nr3c1 K514X stop-gain mutation identified in T-ALL 20M.D1 causes loss of GR protein expression and enhances the fitness of primary leukemia cells exposed to DEX in vivo, and also show that targeted disruption of Nr3c1 is sufficient to induce glucocorticoid resistance in vitro.
Relapsed T-ALLs with pre-existing or acquired Nr3c1 mutations follow distinct evolutionary trajectories
The experimental strategy of transplanting primary T-ALLs into multiple independent recipient mice and treating them in vivo (Fig. 1a) provided a unique opportunity to explore the genetic relatedness of individual relapsed clones with either a pre-existing or acquired Nr3c1 mutation. We analyzed retroviral integration and exome sequencing data from relapsed clones isolated from different recipients transplanted with parental T-ALLs 78A (n=4) or 20M (n=6). Consistent with rare pre-existing T-ALL 78A cells bearing an Nr3c1 L282fs indel that conferred DEX resistance, Southern blotting revealed an identical pattern of MOL4070 integrations in the parental leukemia and all of the relapsed subclones (data not shown). The majority of mutations identified in T-ALL 78A, including the engineered KrasG12D mutation and a Notch1 indel, were also present at similar VAFs in all four relapsed clones analyzed by exome sequencing (78A.D1, 78A.D4, 78A.C2, 78A.C3) (Fig. 4a). The subclone harboring an Nr3c1 L282fs indel was positively selected during treatment and emerged in all four recipients analyzed with relatively few additional relapse-specific mutations identified (Fig. 4b and Table S3). The limited genetic diversity of T-ALL 78A relapsed clones is fully consistent with the existence of a pre-existing Nr3c1 mutant subclone identified in the parental leukemia.
Figure 4. Relapsed T-ALLs with pre-existing or acquired Nr3c1 mutations exhibit different evolutionary trajectories in response to dexamethasone treatment.
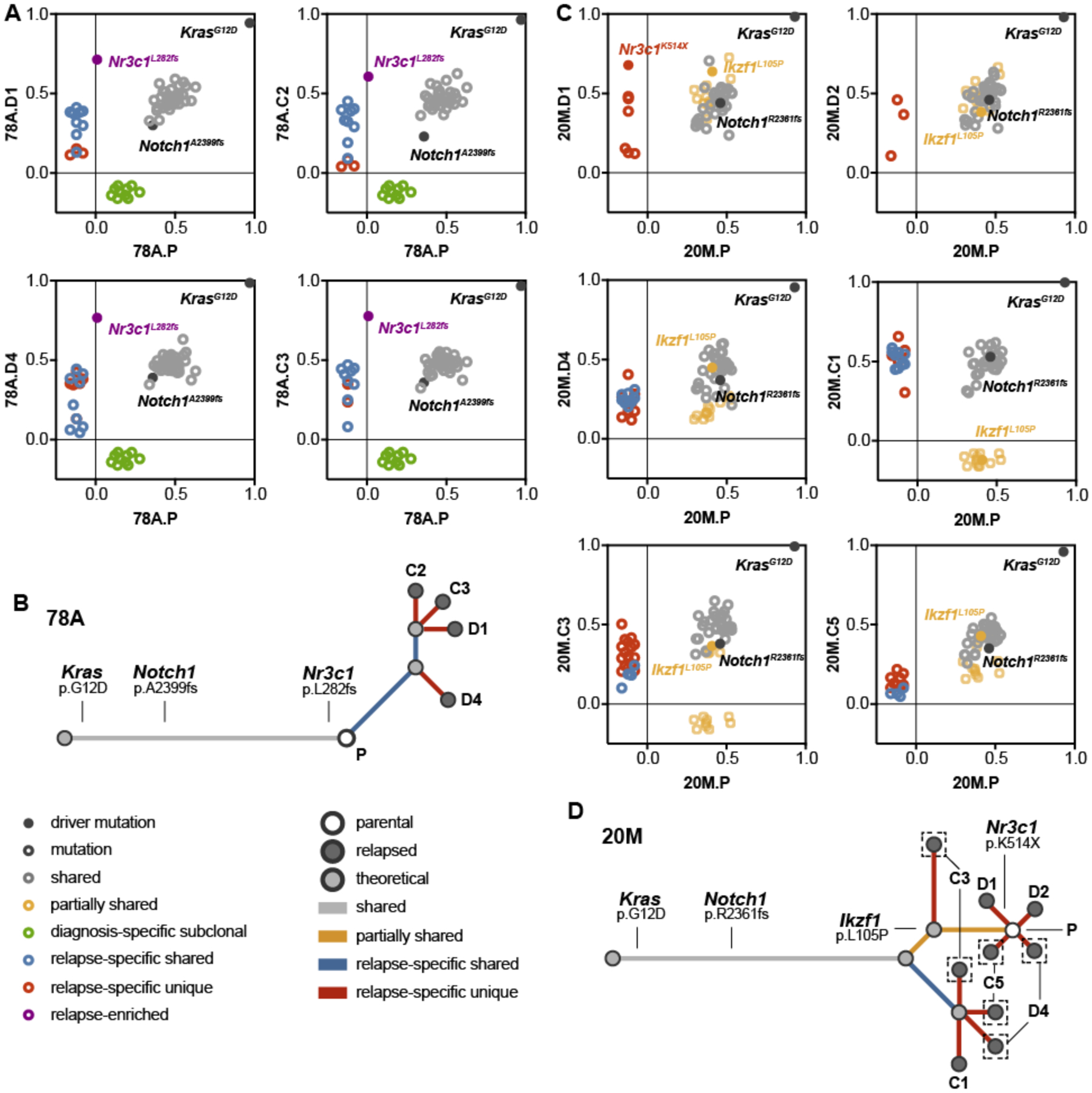
(A) Graphical representation of the mutant allele frequencies of missense mutations and indels shared between the parental T-ALL 78A (x-axis) and four independent relapsed leukemias (y-axis; grey), present only in the parental leukemia (green), shared only with other relapsed leukemias (blue), significantly enriched in the relapsed leukemia (purple), or specific to one of the relapsed leukemias (red). (B) Analysis of the evolutionary relationships among genetic alterations identified by WES in T-ALL 78A indicated a pattern of linear clonal evolution. (C) Graphical representation of the mutant allele frequencies shared between the parental T-ALL 20M (x-axis) and six independent relapsed leukemias (y-axis; grey), present in the parental and a subset of relapsed leukemias (yellow), shared only with other relapsed leukemias (blue), or specific to one of the relapsed leukemias (red). (D) Evolutionary analysis of the genetic alterations identified by WES in T-ALL 20M indicates a branching pattern of clonal dynamics involving multiple ancestral populations. Legends indicate the type of mutation represented by each dot shown in panels A and B (left), and each dot and branch in panels C and D. The same color scheme is used in the evolutionary analysis of panels A through D.
In contrast to T-ALL 78A, the Nr3c1 K514X mutation was only detected in relapsed T-ALL 20M.D1. Interestingly, leukemia cells isolated from four other recipient mice that relapsed during treatment with DEX or DEX/GDC-0941 exhibited clonal evolution by Southern blot. T-ALLs 20M.D3 and 20M.D4 harbored two novel insertions also present in 20M.C1 (Fig. 1c), indicating evolution from common pre-existing founder clone. Exome sequencing of relapsed T-ALLs with (20M.D4, 20M.C1) or without (20M.C3, 20M.C5, 20M.D1, 20M.D2) novel retroviral integrations (Fig. 1c) revealed multiple mutations present at similar VAFs as in the parental leukemia (Fig. 4c, gray circles). In addition, relapsed clones 20M.D4, 20M.C1, 20M.C3, and 20M.C5 shared mutations that were absent in the parental leukemia (Fig. 4c; blue circles and Table S3), indicating derivation from a common ancestral clone. Within this group, relapsed clones 20M.C1 and 20M.C3 exhibited complete or partial loss of numerous mutations identified in the parental T-ALL (Fig. 4c, yellow circles), and thus arose prior to their acquisition. Additionally, 20M.D4, 20M.C3, and 20M.C5 represent multi-lineage relapses harboring at least two subclones based on relapse-specific and partially shared VAFs lower than shared VAFs. The Nr3c1 K514X mutation is among the alterations found in a single relapsed clone (20M.D1), with additional unique mutations identified in the other relapsed leukemias analyzed (Fig. 4d and Table S3). Together, these data indicate DEX resistance in T-ALL 20M is characterized by complex clonal dynamics with outgrowth from distinct but related pre-existing subclones that undergo further genetic evolution upon in vivo drug exposure. These data are most consistent with the existence of an ancestral drug-tolerant clone that persists during treatment, but must acquire additional somatic mutations to cause relapse.
Relapsed T-ALLs show attenuated transcriptional responses to in vivo dexamethasone exposure
We sought to define common transcriptional changes within genetically diverse drug-sensitive T-ALLs following glucocorticoid treatment, which has pleiotropic and cell context-dependent effects (33). Each parental leukemia was first transplanted into a cohort of recipient mice, which received a single dose of DEX (15 mg/kg) or control vehicle when they became moribund. The mice were euthanized four hours later and bone marrow was harvested for RNA sequencing (RNA-seq). We observed modulation of known GR transcriptional targets after short-term DEX exposure in drug-sensitive leukemias. Notably, Myc and several of its target genes were consistently down-regulated upon DEX exposure, while primary GR target genes (Fkbp5, Tsc22d3, Nr3c1) and pro-apoptotic genes (Bcl2l11, Txnip, Bmf) were consistently up-regulated (Fig. 5a). We used qPCR to validate a subset of the transcriptional changes identified by RNA-seq, and then assessed whether short-term in vivo DEX treatment also modulated expression of GR-responsive genes in relapsed T-ALLs with low GR protein levels (Fig. 5b and Fig. S4a, b). Analysis of T-ALL 20M revealed a blunted transcriptional response to DEX in the relapsed sample harboring the Nr3c1 K514X mutation (20M.D1) as compared to the corresponding parental leukemia. Interestingly, four additional relapsed samples from T-ALL 20M that express low GR protein levels but lack Nr3c1 mutations (20M.D2 , 20M.D4, 20M.C1, 20M.C5) also showed a blunted transcriptional response to DEX (Fig. 5b). Similarly, DEX treatment of relapsed subclones from T-ALLs 2M and 73A induced a pattern of target gene expression distinct from that observed in the corresponding parental T-ALL (Fig. S4a, b). These data demonstrate a lack of the expected transcriptional response to in vivo DEX treatment in relapsed T-ALLs with reduced GR protein levels that either harbor or lack a Nr3c1 mutation.
Figure 5. Blunted transcriptional responses of relapsed T-ALLs to dexamethasone treatment in vivo.

(A) Heatmap showing fold increased (p-value<0.001, FDR=0.002) or decreased (p-value<0.001, FDR= 0.050) expression of selected GR-responsive genes after short-term in vivo DEX treatment measured by RNA-sequencing. (B) Gene expression analysis using TaqMan assays for each indicated gene in parental (P, grey bars) and relapsed (D) 20M T-ALLs with low GR protein expression with (blue bars) or without (red bars) an Nr3c1 mutation showing transcript levels of GC response genes as compared to vehicle-treated controls. Error bars represent standard error of the mean for technical replicates.
Discussion
Delineating how individual drugs contribute to resistance and relapse is challenging in the context of multi-agent chemotherapy protocols used in human ALL. To overcome this limitation, we transplanted and treated genetically heterogeneous primary T-ALLs with DEX as a single agent or in combination with a PI3K inhibitor and performed molecular and functional analyses of relapsed leukemias. Of 65 T-ALLs that relapsed after initially responding to glucocorticoid treatment in vivo, 68% showed reduced or absent GR protein expression. Importantly, GR protein levels were also reduced in 40% of relapsed human T-ALL samples, implicating loss of GR expression as a major cause of glucocorticoid resistance. This observation is consistent with clinical data correlating poor response to glucocorticoid treatment during induction with an elevated risk of relapse (1). Although previous studies examining ALL patient samples have generated conflicting data regarding the correlation between GR expression level and glucocorticoid resistance (34–36), functional studies in human T-ALL cell lines have demonstrated that modulation of GR expression level affects glucocorticoid response and resistance in vitro (37) and in xenograft models (38). Consistent with these data, our unbiased forward genetic screen identified loss of GR expression as a mechanism of glucocorticoid resistance in vivo, which we validated using CRISPR/Cas9 gene editing as a functional approach in vitro.
We identified Nr3c1 mutations in 18% of relapsed murine T-ALLs analyzed, and showed those with reduced or absent GR protein expression with or without an Nr3c1 mutation were resistant to DEX upon re-transplant and re-treatment. Whereas previous studies including relatively small numbers of relapsed ALLs uncovered NR3C1 mutations or deletions either rarely (16) or not at all (13, 15), a very recent study in which comprehensive serial sequencing was performed on diagnostic and relapsed samples from 103 Chinese pediatric ALL patients identified NR3C1 mutations as the most frequent relapse-specific alteration, occurring in 14% of cases (39). Copy number alterations leading to deletion of NR3C1 have also been identified in 9% of relapsed B-cell precursor ALLs (21), with one study reporting NR3C1 deletions in 28% of the ETV6/RUNX1-positive subgroup (18). Importantly, NR3C1 deletions were consistently associated with inferior treatment response and adverse outcome in B-cell precursor ALL (18–21). Non-mutational mechanisms have also been implicated in causing reduced GR expression and glucocorticoid resistance in ALL. Paugh and colleagues identified caspase-induced cleavage of GR due to epigenetic up-regulation of the NALP inflammasome as a glucocorticoid resistance mechanism, and showed this reduction in GR protein levels induced substantial DEX resistance using cell line models (40). More recently, a preliminary report showed T-ALL cells with a mutation in the histone methyl transferase NSD2 exhibit both reduced GR expression and impaired up-regulation after glucocorticoid treatment, resulting in a lack of GR binding to DNA and a profound reduction in the subsequent transcriptional response (41). Together with our studies of human and mouse leukemias, these emerging data indicate reduced GR expression is common in relapsed T-ALL and identify Nr3c1/NR3C1 mutations as one mechanism of receptor down-regulation and glucocorticoid resistance.
Recent serial targeted ultra-deep sequencing in a large human ALL cohort of diagnosis/relapse pairs revealed most NR3C1 mutations detected at relapse are likely acquired during treatment and arise in patients that relapse later than those harboring a pre-existing drug resistance mutation (39). Similarly, three of the four Nr3c1 mutations we identified in relapsed mouse T-ALLs were not detected in the corresponding parental leukemia, and present in a single recipient mouse that relapsed later than recipients of the T-ALL harboring a pre-existing Nr3c1 mutation. Gain-of-function PRPS1 and NT5C2 mutations that drive thiopurine resistance in ALL were also not detected at diagnosis by high-depth sequencing (30, 42). A recent study showing NT5C2 mutations reduce fitness of ALL cells in the absence of drug treatment provided an elegant mechanistic explanation for this observation (32). The ability of NR3C1 mutations to alter the competitive fitness of leukemia-initiating cells may be context-dependent, particularly given the diverse physiologic roles of glucocorticoid signaling at different stages of lymphoid development (43).
Several sequencing studies comparing diagnostic and relapsed ALL patient samples have identified pre-existing drug-resistant cells that are selected during treatment (13, 16, 31). An alternative mechanism in which drug-tolerant “persister” cells survive treatment and then acquire additional alterations rendering them fully resistant was first described in solid tumor cell lines (44–48), and has been observed in γ-secretase inhibitor resistant T-ALL cell lines in which drug-tolerant cells with an altered chromatin state exhibit sensitivity to inhibition of the epigenetic reader protein BRD4 (49). However, the role of drug-tolerant persister cells in primary cancers and in the context of treatment with conventional chemotherapy agents remains unclear. Our analysis of evolutionary trajectories in murine T-ALLs uncovered remarkable genetic diversity in a relapsed leukemia harboring an acquired Nr3c1 mutation as compared to the T-ALL with a pre-existing Nr3c1 mutation. These data support a two-step model of glucocorticoid resistance whereby pre-existing drug-tolerant clones survive induction, but must acquire additional alterations that confer full resistance to DEX and drive relapse. Furthermore, our data indicating the majority of Nr3c1 mutations were acquired during treatment suggest this resistance mechanism may be more common than selection of a pre-existing resistant clone in ALL. Similarly, deep genomic analysis of relapsed pediatric ALLs supports acquired mutations in genes involved in drug action within a drug-tolerant cell population as a frequent mechanism of resistance, and demonstrated a comparable evolutionary trajectory as we observed in relapsed mouse leukemias (50).
Our data provide a strong rationale for implementing treatment strategies to eradicate drug-tolerant clones that survive induction and ultimately give rise to resistance in patients with sub-optimal responses to front-line therapies. Although the addition of a PI3K inhibitor markedly enhanced survival in some T-ALLs as compared to treatment with DEX alone, this combination therapy was not sufficient to overcome DEX resistance across our cohort of primary murine leukemias. Newer compounds targeting specific PI3K isoforms (51) or inhibitors of downstream effectors (52) may provide more effective suppression of the pathway and enhance survival in combination with glucocorticoids. In addition, recent studies suggest that targeting interleukin 7 receptor signaling (53, 54) and developing methods for antagonizing aberrant JDP2 expression through MCL1 inhibition (55) are potential strategies for overcoming intrinsic glucocorticoid resistance in select patients. As relapsed/refractory T-ALLs with NR3C1 mutations or loss of GR protein expression are unlikely to respond to glucocorticoids, this knowledge may identify patients who will not benefit from additional glucocorticoid therapy and could thereby avoid potential toxicities. Similarly, developing clinical tests to identify NR3C1 mutations and/or low GR protein expression in the leukemic blasts of patients with detectable minimal residual disease during treatment may identify those at high risk for relapse who could benefit from alternative or novel therapies. Intriguingly, restoring glucocorticoid sensitivity may prove feasible with combination therapy in some leukemias that exhibit reduced GR protein expression due to non-mutational mechanisms, potentially through the use of drugs targeting epigenetic modification (56, 57).
Materials and methods
Preclinical trials.
Retroviral insertional mutagenesis was performed using MOL4070LTR in wild-type (KrasWT) or Mx1-Cre, Lox-STOP-Lox (LSL)-KrasG12D (KrasG12D) mice on a C57BL/6 × 129Sv/Jae (F1) strain background as described previously (26). Cryopreserved primary T-ALL cells were transplanted into sublethally irradiated (450 rads) recipient mice for in vivo expansion, then (2×106) bone marrow cells from these mice were intravenously or retro-orbitally injected into sublethally irradiated 8–12 week old male F1 recipient mice for the preclinical trial. Starting four days post-transplant, GDC-0941 (125 mg/kg/day) or control vehicle (0.5% hydroxypropyl methylcellulose, 0.2% Tween 80) was administered by oral gavage, and DEX (15 mg/kg/day) or control vehicle (phosphate buffered saline) was administered by intraperitoneal injection without blinding at any stage of the study. For the trial cohort, 14 mice were randomly assigned to receive vehicle (n=4), DEX (n=5), or DEX/GDC-0941 (n=5). To confirm drug resistance in relapsed T-ALLs, seven secondary recipients were randomly assigned to receive vehicle (n=3) or DEX (n=4). Mice were euthanized when they appeared moribund and survival was calculated from day of transplant. The time to death for vehicle-treated mice transplanted with individual T-ALLs was highly consistent, allowing determination of significant differences in the survival of drug-treated mice. GraphPad Prism Software was used generate Kaplan-Meier survival curves. Statistical significance was calculated by comparing individual trial arms using a two-tailed Log-rank test. Data analysis from previous studies has verified the statistical power of these cohort sizes to reliably detect significant differences (22, 26, 58–60). All animals assigned to treatment groups were included in the survival analysis. Very rarely (n=3 mice in the original preclinical trial cohort), animals were censored due to a mortality that was not caused by leukemia but rather an unrelated event including complications during drug administration or lack of leukemia engraftment leading to bone marrow failure. Dexamethasone sodium phosphate for injection (NDC 63323-516-10) was purchased through the University of California, San Francisco (UCSF) pharmacy. GDC-0941 was obtained through a Materials Transfer Agreement with Genentech, Inc. All animal studies were approved by the UCSF Institutional Animal Care and Use Committee (AN179357).
Southern blot analysis and MOL4070LTR integration cloning.
Restriction enzyme digestion with HindIII or BamHI of genomic DNA from primary mouse T-ALLs was followed by gel electrophoresis and hybridization with a 32P-radiolabeled MOLO4070LTR-specific probe as described previously (58). Retroviral junction fragments were cloned and sequenced using a linker-based approach (61).
Western blot analysis and immunohistochemistry.
Bone marrow from moribund T-ALL recipients was subjected to red blood cell lysis, then resuspended in RIPA buffer to generate lysates. Protein concentration was determined using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific), then equivalent amounts of protein were mixed with Laemmli Sample Buffer (Bio-Rad Laboratories) followed by separation on a 8–20% Criterion TGX Precast Gel (Bio-Rad Laboratories). Immunoblots were assayed using primary antibodies listed in Table S4. IRDye secondary antibodies were used for detection and Western blots were imaged using an Odyssey Imaging System (LI-COR Biosciences). GR expression level for each leukemia was assigned an unblinded score of high (comparable to the corresponding parental T-ALL), medium (reduced as compared to the corresponding parental T-ALL), or low (nearly or completely absent). The level of GR protein expression for each T-ALL was assessed in bone marrow from at least two independent recipient mice with comparable results. Immunohistochemistry was performed on de-identified human bone marrow samples from children and adults of both sexes diagnosed with T-cell acute lymphoblastic leukemia using GR antibody at 1:400. Diagnostic and relapsed samples were unpaired, with the exception of two patients. T-ALL samples subjected to IHC analysis were scored blindly by two hematopathologists. A detailed protocol for immunohistochemistry and image acquisition in human T-ALLs is provided in the Supplemental Materials and Methods. GraphPad Prism Software was used for graphical representation of the data. The two-tailed Fisher’s exact test was used to determine the significance of association with respect to GR expression levels in parental/diagnostic versus relapsed mouse and human T-ALLs. De-identified human tissue samples were obtained with Institutional Review Board approval UCSF (10–01080), Brigham and Women’s Hospital (2010P002736) and Boston Children’s Hospital (P00025328).
CRISPR/Cas9 modification of CCRF-CEM cells.
CCRF-CEM cells were purchased through the UCSF Cell Culture Facility (ATCC, CCL-119) and cultured in RPMI media supplemented with 10% fetal bovine serum and glutamine. All cell lines described in this study were authenticated through the UC Berkeley DNA Sequencing Facility and tested negative for mycoplasma. Cas9 protein was purchased from the University of California, Berkeley. tracrRNA, sgRNAs, and the HDR template were purchased from Dharmacon, and sequences are listed in Table S5. Transfection was performed using Amaxa Cell Line Nucleofector Kit C (Lonza, VACA-1004) in an Amaxa Nucleofector II Device. Transfection efficiencies were determined using TIDE analysis software (62). CRISPR-edited clones were generated using limiting dilution in conjunction with TIDE analysis to confirm editing of all alleles and Sanger sequencing to confirm HDR template incorporation.
In vitro dexamethasone dose response assays.
Unperturbed or CRISPR-edited CCRF-CEM cells were seeded into 96-well round-bottom tissue culture plates in triplicate (50,000 cells/well) and exposed to increasing concentrations of DEX (Sigma; D4902) or vehicle (DMSO). Cells were resuspended in Hoechst viability dye after 72 hours and analyzed on a BD FACSVerse 8 color Flow Cytometer. FlowJo software was used to determine the percentage of live cells at each DEX concentration. GraphPad Prism Software was used for graphical representation of the data. Each dose response assay was repeated three times with comparable results.
Exome and targeted sequencing.
Genomic DNA was isolated from the bone marrow of moribund T-ALL recipient mice after prolonged in vivo drug exposure using a Puregene DNA Isolation Kit (Qiagen), quantified with PicoGreen and quality controlled by Agilent BioAnalyzer, and 264–500 ng of DNA were used to prepare libraries using the KAPA Hyper Prep Kit (Kapa Biosystems KK8504) with 8 PCR cycles. After sample barcoding, 500–2100 ng of library were captured by hybridization using SureSelectXT Mouse All Exon (Agilent #5190–4641) according to the manufacturer’s protocol. In total, 8–10 cycles of PCR amplification of the post-capture libraries was undertaken after which samples were sequenced on either HiSeq 4000 or HiSeq 2500 instrumentation in 100 bp paired end format using the HiSeq 3000/4000 SBS Kit or HiSeq Rapid SBS Kit v2 (Illumina). Normal and tumor samples were covered to an average of 86- and 173-fold, respectively. Detailed descriptions of the targeted sequencing approach, exome sequencing analysis, and Sanger sequencing validation methods are described in the Supplementary Materials and Methods. Primers used to validate Nr3c1 mutations identified by exome sequencing are listed in Table S6. The exome sequencing data presented in Figures 3 and 4 of this manuscript was deposited into the DNA Data Bank for Japan (https://www.ddbj.nig.ac.jp/dra/index-e.html) under accession code .
Short-term dexamethasone treatment and RNA-seq.
Moribund T-ALL recipient mice received a single dose of vehicle (phosphate buffered saline) or DEX (15 mg/kg), and were euthanized four hours later. Bone marrow cells from these animals were subjected to red blood cell lysis, then frozen in Trizol. Total RNA was extracted and assessed for quality and quantity using the Eukaryote Total RNA Nano Bioanalyzer chip (Agilent Technologies, Inc) and Qubit fluorometer (Thermo Fisher), respectively. RNA-Seq libraries were prepared on the Sciclone liquid handling workstation (PerkinElmer) from 1 μg of total RNA per sample using the TruSeq Stranded Total RNA kit (Illumina, Inc.) following the manufacturer’s recommendations. RNA-Seq libraries were clustered on the cBot and sequenced on HiSeq 2000 and HiSeq 2500 sequencers at 2×100 bp using HiSeq v4 sequencing reagents (Illumina, Inc.). RNA-seq was performed on bone marrow from one vehicle- or DEX-treated mouse per T-ALL indicated. Detailed analysis methods are described in the Supplementary Materials and Methods. False discovery rate and p-values were calculated from RNA-seq data to assess the significance of gene expression changes. RNA-sequencing data presented in Figure 5 of this manuscript was deposited into the National Center for Biotechnology Information Gene Expression Omnibus (NCBI Geo; https://www.ncbi.nlm.nih.gov/geo/) under accession code GSE141967.
Quantitative RT-PCR validation of transcriptome analysis.
Bone marrow cells collected after short-term DEX treatment as described above were subjected to red blood cell lysis, then total RNA was isolated using the RNeasy Mini Kit (Qiagen) and used to generate cDNA using Superscript III (Thermo Fisher Scientific). TaqMan Gene Expression Assays (Applied Biosystems) are listed in Table S7, and qPCR was performed on a QuantStudio 5 Real-Time PCR Instrument (Applied Biosystems). qPCR was performed on bone marrow from one vehicle- or DEX-treated mouse per T-ALL as indicated using technical triplicates. GraphPad Prism Software was used for graphical representation of the data.
Statistical analysis.
The two-tailed Log-rank test was used to perform pairwise comparisons of Kaplan-Meier survival analyses in order to determine significance. The two-tailed Fisher’s exact test was used to determine the significance of association with respect to GR expression levels in parental/diagnostic versus relapsed mouse and human T-ALLs. False discovery rate and p-values were calculated to assess the significance of gene expression changes as determined by RNA-sequencing analysis of murine T-ALLs. Standard deviation was calculated using technical triplicates for in vitro assessment of DEX sensitivity in human T-ALL cell lines. Standard error of the mean was calculated using technical triplicates for qPCR in primary murine T-ALLs.
Supplementary Material
Acknowledgements.
We thank members of the Integrated Genomics Operation in the Marie-Josee and Henry R. Kravis Center for Molecular Oncology at the Memorial Sloan-Kettering Cancer Center, and the Genome Sequencing Lab at St. Jude Children’s Research Hospital for assistance with sequencing; the UCSF Biorepository and Tissue Biomarker Technology Core and the DF/HCC Research Pathology Cores at Harvard Medical School for performing immunohistochemistry analysis. This study was supported in part by the UCSF Helen Diller Family Comprehensive Cancer Center Laboratory for Cell Analysis Shared Resource Facility through a grant from the NIH (P30CA082103). We obtained KrasG12D mice from David Tuveson and Tyler Jacks, and Linda Wolff provided the MOL4070LTR retrovirus. This work was supported in part by National Institutes of Health awards R37 CA72614 (K.S.), R01 CA180037 (K.S.); P30 CA008748, U54 OD020355 (B.S.T.), R01 CA204749 (B.S.T.), and awards from the St. Baldrick’s Foundation (K.S.), the American Cancer Society (RSG-15-067-01-TBG) (K.S.), the Anna Fuller Fund, and the Robertson Foundation (B.S.T.). A.M.W was supported by a Postdoctoral Fellowship (PF-14-070-01-TBG) from the American Cancer Society including a supplement from the Hillcrest Committee, and an Alex’s Lemonade Stand Foundation Young Investigator Grant including support from Northwestern Mutual. K.S. is an American Cancer Society Research Professor (RP 10-078-01-TBE).
Footnotes
Competing Interests: Deepak Sampath and Monique Dail were employees and shareholders of Genentech, Inc. when this work was performed. The remaining authors of this manuscript declare no financial interest related to this work.
References
- 1.Gao J & Liu WJ (2018) Prognostic value of the response to prednisone for children with acute lymphoblastic leukemia: a meta-analysis. European review for medical and pharmacological sciences 22(22):7858–7866. [DOI] [PubMed] [Google Scholar]
- 2.Pui CH, Dahl GV, Rivera G, Murphy SB, & Costlow ME (1984) The relationship of blast cell glucocorticoid receptor levels to response to single-agent steroid trial and remission response in children with acute lymphoblastic leukemia. Leukemia research 8(4):579–585. [DOI] [PubMed] [Google Scholar]
- 3.Pui CH, Ochs J, Kalwinsky DK, & Costlow ME (1984) Impact of treatment efficacy on the prognostic value of glucocorticoid receptor levels in childhood acute lymphoblastic leukemia. Leukemia research 8(3):345–350. [DOI] [PubMed] [Google Scholar]
- 4.Pui CH & Costlow ME (1986) Sequential studies of lymphoblast glucocorticoid receptor levels at diagnosis and relapse in childhood leukemia: an update. Leukemia research 10(2):227–229. [DOI] [PubMed] [Google Scholar]
- 5.Lauten M, Cario G, Asgedom G, Welte K, & Schrappe M (2003) Protein expression of the glucocorticoid receptor in childhood acute lymphoblastic leukemia. Haematologica 88(11):1253–1258. [PubMed] [Google Scholar]
- 6.Ashraf J & Thompson EB (1993) Identification of the activation-labile gene: a single point mutation in the human glucocorticoid receptor presents as two distinct receptor phenotypes. Molecular endocrinology 7(5):631–642. [DOI] [PubMed] [Google Scholar]
- 7.Powers JH, Hillmann AG, Tang DC, & Harmon JM (1993) Cloning and expression of mutant glucocorticoid receptors from glucocorticoid-sensitive and -resistant human leukemic cells. Cancer research 53(17):4059–4065. [PubMed] [Google Scholar]
- 8.Strasser-Wozak EM, et al. (1995) Splice site mutation in the glucocorticoid receptor gene causes resistance to glucocorticoid-induced apoptosis in a human acute leukemic cell line. Cancer research 55(2):348–353. [PubMed] [Google Scholar]
- 9.Geley S, et al. (1996) Resistance to glucocorticoid-induced apoptosis in human T-cell acute lymphoblastic leukemia CEM-C1 cells is due to insufficient glucocorticoid receptor expression. Cancer research 56(21):5033–5038. [PubMed] [Google Scholar]
- 10.Hala M, Hartmann BL, Bock G, Geley S, & Kofler R (1996) Glucocorticoid-receptor-gene defects and resistance to glucocorticoid-induced apoptosis in human leukemic cell lines. International journal of cancer 68(5):663–668. [DOI] [PubMed] [Google Scholar]
- 11.Riml S, Schmidt S, Ausserlechner MJ, Geley S, & Kofler R (2004) Glucocorticoid receptor heterozygosity combined with lack of receptor auto-induction causes glucocorticoid resistance in Jurkat acute lymphoblastic leukemia cells. Cell death and differentiation 11 Suppl 1:S65–72. [DOI] [PubMed] [Google Scholar]
- 12.Mullighan CG, et al. (2008) Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science 322(5906):1377–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hogan LE, et al. (2011) Integrated genomic analysis of relapsed childhood acute lymphoblastic leukemia reveals therapeutic strategies. Blood 118(19):5218–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kunz JB, et al. (2015) Pediatric T-cell lymphoblastic leukemia evolves into relapse by clonal selection, acquisition of mutations and promoter hypomethylation. Haematologica 100(11):1442–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oshima K, et al. (2016) Mutational landscape, clonal evolution patterns, and role of RAS mutations in relapsed acute lymphoblastic leukemia. Proceedings of the National Academy of Sciences of the United States of America 113(40):11306–11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spinella JF, et al. (2018) Mutational dynamics of early and late relapsed childhood ALL: rapid clonal expansion and long-term dormancy. Blood advances 2(3):177–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuster L, et al. (2011) ETV6/RUNX1-positive relapses evolve from an ancestral clone and frequently acquire deletions of genes implicated in glucocorticoid signaling. Blood 117(9):2658–2667. [DOI] [PubMed] [Google Scholar]
- 18.Grausenburger R, et al. (2016) Genetic alterations in glucocorticoid signaling pathway components are associated with adverse prognosis in children with relapsed ETV6/RUNX1-positive acute lymphoblastic leukemia. Leukemia & lymphoma 57(5):1163–1173. [DOI] [PubMed] [Google Scholar]
- 19.Piovan E, et al. (2013) Direct reversal of glucocorticoid resistance by AKT inhibition in acute lymphoblastic leukemia. Cancer cell 24(6):766–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paugh SW, et al. (2015) NALP3 inflammasome upregulation and CASP1 cleavage of the glucocorticoid receptor cause glucocorticoid resistance in leukemia cells. Nature genetics 47(6):607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, et al. (2018) A gain of function mutation in the NSD2 histone methyltransferase drives glucocorticoid resistance of acute lymphoblastic leukemia. Blood 132(Suppl 1):653. [Google Scholar]
- 22.Wolff L, Koller R, Hu X, & Anver MR (2003) A Moloney murine leukemia virus-based retrovirus with 4070A long terminal repeat sequences induces a high incidence of myeloid as well as lymphoid neoplasms. J Virol 77(8):4965–4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lauchle JO, et al. (2009) Response and resistance to MEK inhibition in leukaemias initiated by hyperactive Ras. Nature 461(7262):411–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dail M, et al. (2010) Mutant Ikzf1, KrasG12D, and Notch1 cooperate in T lineage leukemogenesis and modulate responses to targeted agents. Proceedings of the National Academy of Sciences of the United States of America 107(11):5106–5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dail M, et al. (2014) Loss of oncogenic Notch1 with resistance to a PI3K inhibitor in T-cell leukaemia. Nature 513(7519):512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burgess MR, et al. (2017) KRAS Allelic Imbalance Enhances Fitness and Modulates MAP Kinase Dependence in Cancer. Cell 168(5):817–829 e815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Folkes AJ, et al. (2008) The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin −4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem 51(18):5522–5532. [DOI] [PubMed] [Google Scholar]
- 28.Norman MR & Thompson EB (1977) Characterization of a glucocorticoid-sensitive human lymphoid cell line. Cancer research 37(10):3785–3791. [PubMed] [Google Scholar]
- 29.Mansour MR, et al. (2007) Notch-1 mutations are secondary events in some patients with T-cell acute lymphoblastic leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research 13(23):6964–6969. [DOI] [PubMed] [Google Scholar]
- 30.Tzoneva G, et al. (2013) Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nature medicine 19(3):368–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma X, et al. (2015) Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia. Nature communications 6:6604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tzoneva G, et al. (2018) Clonal evolution mechanisms in NT5C2 mutant-relapsed acute lymphoblastic leukaemia. Nature 553(7689):511–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reddy TE, et al. (2009) Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome research 19(12):2163–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li B, et al. (2018) Mutational Landscape and Temporal Evolution during Treatment of Relapsed Acute Lymphoblastic Leukemia. Blood 132(Suppl 1):1.29976776 [Google Scholar]
- 35.Li B, et al. (2015) Negative feedback-defective PRPS1 mutants drive thiopurine resistance in relapsed childhood ALL. Nature medicine 21(6):563–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jamieson CA & Yamamoto KR (2000) Crosstalk pathway for inhibition of glucocorticoid-induced apoptosis by T cell receptor signaling. Proceedings of the National Academy of Sciences of the United States of America 97(13):7319–7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharma SV, et al. (2010) A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141(1):69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramirez M, et al. (2016) Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nature communications 7:10690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Raha D, et al. (2014) The cancer stem cell marker aldehyde dehydrogenase is required to maintain a drug-tolerant tumor cell subpopulation. Cancer research 74(13):3579–3590. [DOI] [PubMed] [Google Scholar]
- 40.Hata AN, et al. (2016) Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nature medicine 22(3):262–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hangauer MJ, et al. (2017) Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551(7679):247–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Knoechel B, et al. (2014) An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nature genetics 46(4):364–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delgado-Martin C, et al. (2017) JAK/STAT pathway inhibition overcomes IL7-induced glucocorticoid resistance in a subset of human T-cell acute lymphoblastic leukemias. Leukemia 31(12):2568–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mansour MR, et al. (2018) JDP2: An oncogenic bZIP transcription factor in T cell acute lymphoblastic leukemia. The Journal of experimental medicine 215(7):1929–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mansour MR, et al. (2014) Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 346(6215):1373–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jing D, et al. (2018) Lymphocyte-Specific Chromatin Accessibility Pre-determines Glucocorticoid Resistance in Acute Lymphoblastic Leukemia. Cancer cell 34(6):906–921 e908. [DOI] [PubMed] [Google Scholar]
- 47.Uren AG, et al. (2009) A high-throughput splinkerette-PCR method for the isolation and sequencing of retroviral insertion sites. Nature protocols 4(5):789–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brinkman EK, Chen T, Amendola M, & van Steensel B (2014) Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic acids research 42(22):e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pronier E, et al. (2018) Genetic and epigenetic evolution as a contributor to WT1-mutant leukemogenesis. Blood 132(12):1265–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iyer G, et al. (2012) Genome sequencing identifies a basis for everolimus sensitivity. Science 338(6104):221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Al-Ahmadie H, et al. (2014) Synthetic lethality in ATM-deficient RAD50-mutant tumors underlies outlier response to cancer therapy. Cancer discovery 4(9):1014–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li H & Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14):1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.DePristo MA, et al. (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature genetics 43(5):491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cibulskis K, et al. (2013) Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature biotechnology 31(3):213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ye K, Schulz MH, Long Q, Apweiler R, & Ning Z (2009) Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25(21):2865–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Robinson JT, et al. (2011) Integrative genomics viewer. Nature biotechnology 29(1):24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Amarasinghe KC, et al. (2014) Inferring copy number and genotype in tumour exome data. BMC genomics 15:732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bolger AM, Lohse M, & Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Langmead B & Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nature methods 9(4):357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li B & Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinformatics 12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robinson MD, McCarthy DJ, & Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26(1):139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Subramanian A, et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102(43):15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.