

Supplemental Digital Content is available in the text
Keywords: biomarker, comprehensive analysis, prognosis, prostate cancer, WGCNA
Abstract
Background:
Prostate cancer (PCa) is one of the leading causes of cancer-related death. In the present research, we adopted a comprehensive bioinformatics method to identify some biomarkers associated with the tumor progression and prognosis of PCa.
Methods:
Differentially expressed genes (DEGs) analysis and weighted gene co-expression network analysis (WGCNA) were applied for exploring gene modules correlative with tumor progression and prognosis of PCa. Clinically Significant Modules were distinguished, and Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis were used to Annotation, Visualization and Integrated Discovery (DAVID). Protein–protein interaction (PPI) networks were used in selecting potential hub genes. RNA-Seq data and clinical materials of prostate cancer from The Cancer Genome Atlas (TCGA) database were used for the identification and validation of hub genes. The significance of these genes was confirmed via survival analysis and immunohistochemistry.
Results:
2688 DEGs were filtered. Weighted gene co-expression network was constructed, and DEGs were divided into 6 modules. Two modules were selected as hub modules which were highly associated with the tumor grades. Functional enrichment analysis was performed on genes in hub modules. Thirteen hub genes in these hub modules were identified through PPT networks. Based on TCGA data, 4 of them (CCNB1, TTK, CNN1, and ACTG2) were correlated with prognosis. The protein levels of CCNB1, TTK, and ACTG2 had a degree of differences between tumor tissues and normal tissues.
Conclusion:
Four hub genes were identified as candidate biomarkers and potential therapeutic targets for further studies of exploring molecular mechanisms and individual therapy on PCa.
1. Introduction
Prostate cancer (PCa) is the foremost regularly non-cutaneous malignancy among males and the second primary cause of cancer-related deaths in the Western world, the occurrence rate of which keeps increasing.[1,2] Although plenty of PCas are characterized as slow-growing and indolent, some PCa patients are at high risk of developing local recurrence and/or distant metastases and die of their disease. The universal diagnosis approaches of PCa contain digital examination of the rectum, prostate-specific antigen (PSA) test, and subsequential biopsies for histopathological staging.[3] It is of great benefit but also huge challenges to make the stratification of high-risk PCa patients after diagnosing. At present, management modes of PCa are principally guided by risk degrees on the strength of Gleason grade, clinical stage, as well as serum PSA level. A better understanding of the potential mechanisms and molecular pathways contributing to tumorigenesis and tumor progression is quite essential to enhance our current diagnostic, prognostic, and therapeutic abilities.
Benefited from the development of high-throughput technology, such as whole genome[4] and whole exome sequencing,[5] researchers could explore and discover the full molecular landscapes of tumors at a variety of levels including DNA structural changes in the PCa genome. In recent years, gene expression profiles, noncoding RNA profiles and DNA methylation profiles are promising not only for serving as prognosis biomarkers but also therapy targets. For example, cell cycle genes are proved as potential markers that could predict the risk of clinicopathological outcomes such as biochemical recurrence (BCR) rate after radical prostatectomy.[6] More recently, new research figured out that up-regulated KIF20A expression level was significantly associated with higher BCR, serum PSA value, Gleason score, pathological T stage, and lymph node metastasis in PCa.[7] Besides, the down-regulation of miR-24 was reported to promote cell cycle, proliferation, migration, and clonogenicity potential of PCa cells through regulating the expression of p16 and p27, which supported that it may act as a tumor suppressor in PCa.[8] Particularly, in human prostate adenocarcinomas, 128 CpG sites were found to have high accuracy and sensitivity in differentiating samples, which could help us identify lymphatic metastases at DNA methylation level and also may inspire novel therapeutic methods.[9] Thus, there is a significant clinical need for finding and identifying united novel biomarkers related to clinicopathological parameters, guiding the surveillance of tumorous progression, assessment of prognosis and improvement of personalized treatment programs, especially at gene expression status.
Nevertheless, many researches to date just focused on the screening of differentially expressed genes, and the high degree of interconnection among genes has been largely neglected, where genes with semblable expression patterns might be functionally correlated. Weighted gene co-expression network analysis (WGCNA), a capable approach widely applied for constructing free-scale gene co-expression networks and exploring the connections between different gene sets or between gene sets and interesting phenotypic traits.[10] WGCNA has been used successfully in various biological processes, which vastly favorable for the identification of candidate biomarkers or potential treatment targets.[11] Moreover, it is of great success to identify biomarkers for castration-resistant prostate cancer by using WGCNA.[12] Thus, in the current research, we attempt to construct a free-scale gene co-expression network of correlation patterns among genes through a comprehensive bioinformatics method in the light of WGCNA and identified network-centric genes relative to clinical traits of PCa.
2. Materials and methods
2.1. Microarray data
Figure S1 showed the workflow of our research. We downloaded the gene expression profiles of GSE69223[13] (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE69223) from the Gene Expression Omnibus (GEO) database. Data set GSE69223, performed on Affymetrix Human Genome U133 Plus 2.0 Array, contained malignant and matched normal adjacent tissue samples of 15 prostate cancer patients with prostatectomy. It included microarray data of these patients and detailed clinical variables, such as patients age, tumor stage, and grade, which could be used for constructing co-expression networks, identifying clinical features related modules, and screening hub genes. Ethical approval is not necessary because the data of this study is derived from gene database.
2.2. Data preprocessing and screening of DEGs
The gene expression profile data were preprocessed through Robust Multi-array Average (RMA) algorithm in affy package within Bioconductor (http://www.bioconductor.org) in R, and quantile normalization was used to normalize their processed signals. Affymetrix annotation files were applied for annotating the probes. If different probes linked to the same gene, the median value was chosen as the gene expression value. DEGs between prostate cancer and matched normal adjacent tissue were analyzed by the “limma” R package in R. |Log2-fold change (LFC)| >0.5 and Pfalsediscoveryrate(FDR) < 0.05 and were considered as the thresholds for DEGs screening.
2.3. Construction of co-expression network
Firstly, DEGs were used to construct scale-free co-expression networks. Secondly, the correlation matrix between 2 genes was performed using Pearson correlation matrices and the major connecting rod. Next, the correlation matrix was transformed into a weighted adjacency matrix through a power function axy=|cxy|β (amn=adjacency between gene x and gene y; cmn=Pearson correlation between gene x and gene y). β was the most appropriate soft-thresholding parameter which could raise the co-expression similarity and achieve consistent scale-free topology in multiple datasets. Here, we chose the power of β = 10 (scale-free R2 = 0.85) to ensure a scale-free network (Fig. S2). After that, with the chosen power value, we performed automatic network construction and module detection with the following major parameters: maxBlockSize of 4000, minModuleSize of 100, deepSplit of 2, and mergeCutHeight of 0.5. This procedure comprised the calculation of network adjacencies and topological overlap dissimilarities, followed by scaling of topological overlap matrices (TOM) and calculation of consensus topological overlap. And then, we established an average linkage hierarchical clustering dendrogram of gene expression data for the dataset to cluster genes that have similar expression profiles into 1 gene module and performed adaptive branch cutting to identify shared functional modules. The Above steps were implemented by using the “WGCNA” package in R.
2.4. Identification of clinically significant modules
Module eigengenes (MEs) were characterized as the first principal component of each co-expression module and the expression of MEs was regarded as a representative of entire genes in a given module. Additionally, we recognize the clinically significant module (hub module) by calculating the correlation between MEs and clinical data.
2.5. Gene ontology and KEGG enrichment analysis
For further understanding the function of DEGs in the hub module, all genes in the hub module were subjected to GO and KEGG pathway analysis through the Database for DAVID database (version 6.8) (http://david.abcc.ncifcrf.gov/). The ontology includes 3 categories: biological process (BP), cellular component (CC), and molecular function (MF). P value < .05 was regarded as the cut-off criterion for identifying the enriched GO terms and KEGG pathways.
2.6. Hub gene analysis and construction of PPI network
In this study, after the hub module was confirmed, we selected the candidate hub genes by module connectivity (cor.geneModuleMembership (MM) > 0.6 or >0.8) and clinical feature relationship (cor.geneTraitSignificance (GS) > 0.2), both measured by absolute value of the Pearson correlation. After that, we uploaded these genes to the STRING database (https://string-db.org/) for constructing protein–protein interaction (PPI) networks and screening out hub nodes. Next, Cytoscape software (version 3.7.1) (http://www.cytoscape.org) was used to visualize the interaction relationships between genes in these hub nodes. Finally, a subnetwork was extracted from the PPI network using the Molecular Complex Detection (MCODE) application,[14] a Cytoscape plugin that detected densely connected regions in networks that may represent molecular complexes. The parameters were set as follows. Degree cutoff: 2, node score cutoff: 0.2, cut style: haircut, k-core: 2, and max. Depth: 100. Genes in the PPI subnetwork were considered as hub genes for further analysis.
2.7. Hub gene identification and validation using TCGA data
The RNA-seq data and clinical information of PCa were downloaded from The Cancer Genome Atlas Project database (TCGA, https://cancergenome.nih.gov/). A total of 551 samples were included. The samples in the data were divided into different groups according to the clinical features of hub modules which we were interested in. Spearman correlation analysis was used to estimate the filtered gene expression in differential clinical traits. Then, event-free survival (EFS) estimate and statistical significance were performed using the Kaplan–Meier method, two-sided log-rank test, and univariate Cox regression analysis. A hazard ratio (HR) P value of <.05 was considered significant statistically. The Human Protein Atlas (http://www.proteinatlas.org) was applied for validating the immunohistochemistry (IHC) of latent hub genes.
3. Results
3.1. DEGs screening and WGCNA analysis
Totally 2688 DEGs (1539 up-regulated and 1149 down-regulated) were identified between PCa and matched normal adjacent tissue (Fig. 1A). The WGCNA analysis was performed on these DEGs. The tumor samples of GSE69223 were clustered using Pearson correlation method and the average linkage method (Fig. 1B). Meanwhile, a scale-free network was ensured with the power of β = 10. Totally 6 modules were identified via the average linkage hierarchical clustering (Fig. 1C). Both brown module (r = 0.58, P value = .02) and turquoise module (r = −0.61, P value = .02) were found to have the most conspicuous association with tumor grade (Fig. 1D). The expression levels of genes in the brown module were positively associated with tumor grade of PCa, while those of the turquoise module had the contrary relationship. These 2 modules were selected as hub modules for subsequent analysis.
Figure 1.
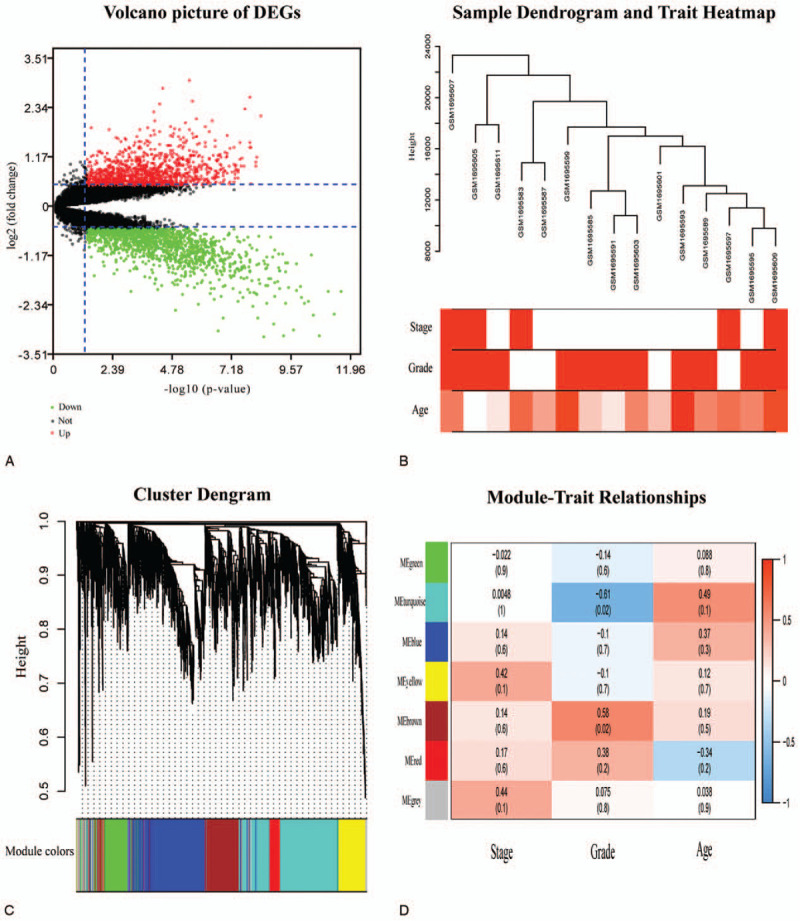
DEGs screening and WGCNA analysis. (A) The volcano picture for DEGs. Green dots represented the downregulated genes. Black dots represented genes that were not differentially expressed, and the red dots represented the upregulated genes. (B) Clustering dendrogram of 15 tumor samples as well as the clinical features. The color intensity was proportional to higher tumor grade, higher tumor stage, and older age. (C) Dendrogram of all DEGs clustered based on a dissimilarity measure (1-TOM). (D) Heatmap of the correlation values between MEs and different clinical features of PCa (tumor stage, tumor grade, and age). Red for positive correlation and Blue for the negative correlation. P values were printed below the correlations. DEGs, differentially expressed genes. MEs = module eigengenes, PCa = prostate cancer, WGCNA = weighted gene co-expression network analysis.
3.2. Gene ontology and pathway enrichment analysis
With the similarly prominent connection of tumor grade, we combined the genes in these 2 modules for enrichment analysis. The genes were categorized into 3 functional groups (BP, CC, and MF). The results showed that DEGs in the BP group was mainly involved in oxidation–reduction process, cell adhesion, extracellular matrix organization, Wnt signaling pathway, and cytoskeleton organization; DEGs in the CC group were enriched primarily on cytoplasm, extracellular space, extracellular exosome, membrane especially plasma membrane; DEGs in the MF group were significantly improved in protein homodimerization activity, GTP binding, structural molecule activity, GTPase activity, and transferase activity. According to KEGG enrichment analysis, our findings indicated that DEGs mainly took part in focal adhesion, proteoglycans in cancer, vascular smooth muscle contraction, cGMP−PKG signaling pathway and ECM−receptor interaction (Fig. S3).
3.3. Hub gene analysis and construction of PPI network
Based the cut-off criteria (showed in Fig. S4. Brown module:|MM| > 0.6 and |GS| > 0.2;Turquoise module: |MM| > 0.8 and |GS| > 0.2), totally 570 genes (286 genes in the brown module and 284 genes in the turquoise module) with high connectivity were identified as candidate hub genes. The number of highly correlated genes between the 2 modules would be so far apart that it might affect the eventual screening of hub genes if we used |MM| > 0.8 as cut-off criteria for brown module. Therefore, we used a lower cutoff for it. Figure 2 showed the 2 PPI networks created by importing these candidate hub genes of 2 hub modules into the STRING database, respectively. The 13 highest-scoring nodes were screened out by MCODE application: CCNB1, TTK, DBF4, WEE1, MCM3, MCM6, ACTG2, CNN1, TAGLN, MYL9, ACTA2, LMOD1, and FLNA (6 genes in the brown module and 7 genes in the turquoise module). These 13 genes were considered as hub genes.
Figure 2.
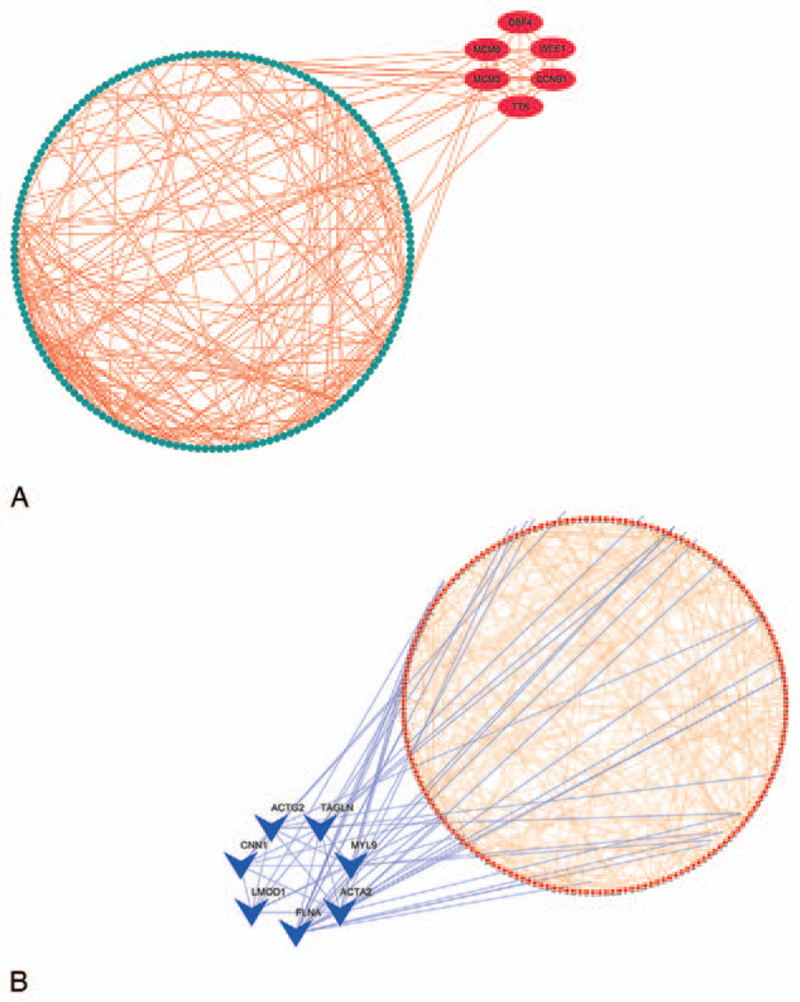
Visualization of PPI networks with candidate hub genes in the brown and turquoise modules, respectively (286 genes in the brown module and 284 genes in the turquoise module). PPI = protein-protein interaction.
3.4. Hub gene identification and validation using TCGA data
Based on the samples in TCGA data which were divided into 5 groups according to the tumor grades, Spearman correlation analysis showed that 2 up-regulated genes (CCNB1, TTK) and 2 down-regulated genes (ACTG2, CNN1) in PCa had the most significant correlation coefficient (|cor|>0.25), which meant there was a highly consistent variation tendency between the expression levels of these genes and the grades of PCa (Fig. 3). Meanwhile, Kaplan–Meier curves and the two-sided log-rank test revealed that lower expression of CCNB1, TTK, and higher expression of ACTG2, CNN1 were associated with better EFS (Fig. 4). Besides, the univariate Cox proportional hazards regression analysis revealed that these 4 genes were also correlative with EFS of PCa (Table 1). Moreover, based on IHC data, the protein levels of CCNB1 and TTK were slightly higher in tumor tissues than those in normal tissues, while that of ACTG2 in tumor tissues was significantly lower than those in the control group (Fig. S5).
Figure 3.
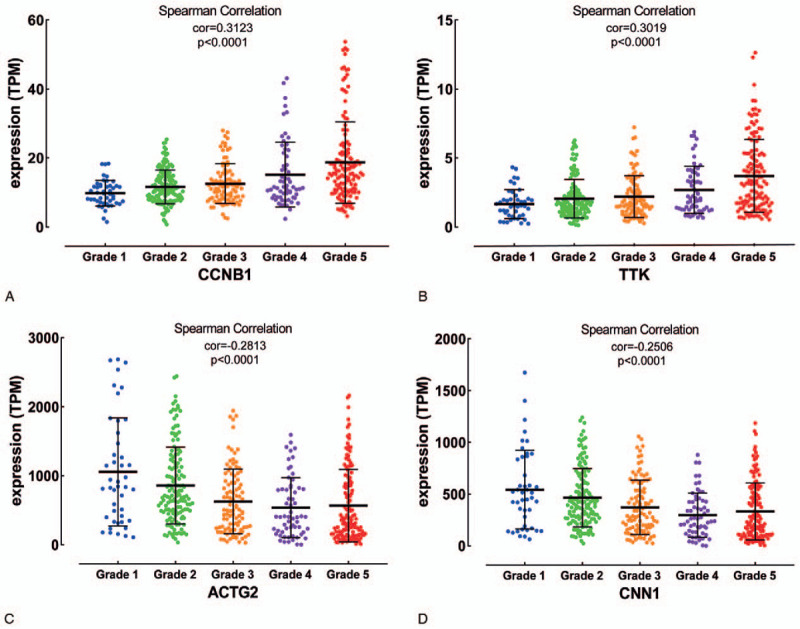
The expression levels of (A) CCNB1, (B) TTK, (C) CNN1, and (D) ACTG2 in different tumor grades of PCa. PCa, prostate cancer.
Figure 4.
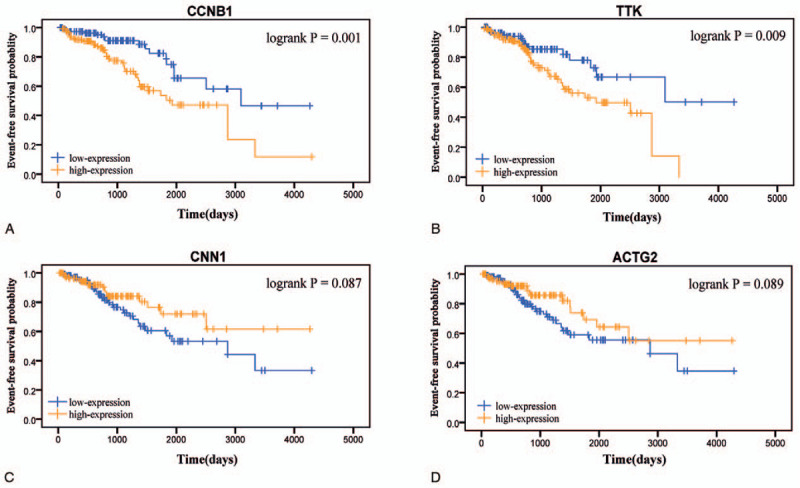
Event-free survival curve of the four hub genes in PCa based on Kaplan–Meier analysis and log-rank test. Patients were divided into the high expression level group and the low expression level group based on quartile cutoff method (cutoff-high:75% and cutoff-low:25%). (A) CCNB1. (B) TTK. (C) CNN1. (D) ACTG2. PCa, prostate cancer.
Table 1.
Univariate Cox analysis of the 4 hub genes using TCGA dataset.

4. Discussion
PCa is one of the most common malignant tumors in men, which seriously harms men's health. Current diagnostic methods such as PSA testing, digital rectal examination, and prostate biopsy are defective and might be rising in a number of non-malignant clinical situations. Thus, it is vital to search for novel non-invasive tumor markers with high sensitivity and high specificity. Many studies have shown that the development and application of multi-omics-based approaches including genomics, proteomics, and metabolomics in malignant tumors provide the possibility for this idea, which is conducive to improving the accuracy of early diagnosis and prognosis evaluation for PCa and reducing the number of unnecessary invasive examinations.[15] For instance, the PCA3 (also called DD3) gene was reported remarkably increased for PCa tissue compared with the normal control, and PCA3 score of 20 could be regarded as the cutoff of avoiding unnecessary biopsies.[16] Besides, 1 research figured out that the levels of Periostin, Cathepsin D, Lysosome-associated membrane glycoprotein 2 and Adipocyte plasma membrane-associated protein were detected highly specific in PCa serum with respect to the normal group, which were proved by a fast protocol based on highly sensitive mass spectrometric analysis by Selected Reaction Monitoring and TiO2 enrichment.[17] What is more, another research summarized that 2 major metabolomics characteristics of PCa were an increased expression of genes in the tricarboxylic acid cycle and the accumulation of metabolic intermediates, which might guide us in exploring novel therapeutic targets for PCa.[18]
It is worth noting that genomics, as a relatively mature omics technology, has shown unique advantages in the exploration of biomarkers for malignant tumors. In the study, we performed an integrated bioinformatics analysis based on WGCNA towards PCa patients gene expression profiling, finally highlighting some key hub genes which could act as candidate biomarkers associated with the tumor progression and prognosis of PCa. We also made use of the RNA-seq data and clinical traits of PCa from the TCGA database, as well as IHC data from the Human Protein Atlas database for validation.
Both GO and KEGG pathway analysis in the combination of identified modules highly enriched in cell adhesion. Interestingly, numerous studies figured out that changes of adhesion-related proteins were related to high Gleason score and metastasis, such as E-cadherin[19] and CD44 standard (CD44s).[20]
Additionally, another research revealed that glucose-regulated protein 78 was up-regulated in PCa and served as a modulator of cell adhesion markers, which contributes to metastatic spread.[21] GO terms are also enriched in the Wnt signaling pathway that is of great interest to the PCa community. Wnt family contains several secreted glycoproteins, which could combine with some Frizzled transmembrane receptors to activating one of 2 principal Wnt pathways (i.e., “non-canonical” or “canonical”).[22] Several studies demonstrated that WNT5a might serve as a tumor-inhibiting factor because its expression could induce apoptosis in PCa cell lines[23] and predict for increased biochemical recurrence-free survival.[24,25] Similarly, the expression level of WNT11 seems to be upregulated by androgen deprivation treatment,[26] which contribute to neuroendocrine differentiation in PCa.[27]
Furthermore, we screened out 4 hub genes (CCNB1, TTK, CNN1, and ACTG2) with high connectivity from the identified modules through PPI networks. The expression patterns of these genes were correlated with the tumor grade (WGCNA), and the gene expression status was associated with prognosis. Our findings suggested that the mentioned genes might play a crucial role in the occurrence and development of PCa.
CCNB1, also named cyclin B1, regulates the G2 to M phase transition in mitosis. Its expression and clinical value have been examined in a variety of cancers. For example, overexpression of CCNB1 played an essential role in cell proliferation and tumor growth of colorectal cancer, which suggested that CCNB1 may be an effective biomarker in colorectal cancer.[28] What is more, another bioinformatics analysis revealed that CCNB1 could serve as a potential therapeutic target in hepatocellular carcinoma.[29] As for PCa, according to one previous research, androgen receptor could increase the expression level of CCNB1 via Akt phosphorylation, which contributes to the progression of PCa regulated by Jagged1.[30] Besides, the high ratio of cyclin A and B to the proliferation marker Ki67 was proved as the most potent predictor of time to recurrence of PCa.[31] Higher levels of CCNB1 sensitized PCa cells to apoptosis initiated by chemotherapy, supporting that overexpression of CCNB1 in PCa cells may be an effective prognostic biomarker for chemotherapy.[32] In the present study, our results suggested that a high CCNB1 expression level could act as a good predictor of worse EFS.
TTK, also called Monopolar spindle one kinase (MPS1), is another checkpoint in mitosis and correlative with cell proliferation and cell invasion. Currently, overexpression of TTK was confirmed to occur in various cancers that harbor copy number gains (CNG) of chromosome 9p involving PD-L1.[33] Furthermore, TTK was overexpressed within the incredible lions share of esophageal squamous cell carcinoma tissues but barely expressed in ordinary tissues,[34] while another study demonstrated that high expression status of TTK corresponded positively with tumor grade and negatively with prognosis in glioblastoma.[35] Nowadays, there are few types of researches regarding the role of TTK in PCa. Our study discovered that overexpression of TTK was associated with higher Gleason grade and worse prognosis of PCa, which was the first study revealing the relationship between TTK expression level and clinical outcome of PCa, as far as we know.
Functionally, the binding of CNN1 contributed to the stabilization of the 3D structure of actin filaments, which effectively resulted in the decrease of filament turnover and suppression of cytoskeleton remodeling.[36] Unlike CCNB1 and TTK, CNN1 may play a tumor-suppressive role in many kinds of malignancy tumorsr.[37,38] Loss of CNN1 also led to tumor progression and anoikis resistance in high-grade serous carcinoma, which originated from the fallopian tube epithelium.[39] Although the functional importance of CNN1 on many other cancers has been proved, few studies have performed the specific analysis of CNN1 on its tumor-suppressive function in PCa. Based on the present study, we detected that the expression of CNN1 in PCa was down-regulated and related with high tumor grade and terrible clinical outcome, supporting CNN1 may be a candidate biomarker for progression and prognosis of PCa.
Recently, aberrant expression of ACTG2 (gamma smooth muscle 2) has been described in several different types of malignancy tumors. According to 1 previous study, a lower expression level of ACTG2 was correlative with worse disease-specific survival in leiomyosarcoma.[40] On the contrary, it was also reported that ACTG2 was up-regulated in hepatocellular carcinoma, involved as a promoter gene in cell migration and distant metastasis.[41] Higher expression status of ACTG2 was also found in colon tissue compared with colon carcinoma.[42] Its expressions and effects seem to vary in different kinds of cancers. Although the molecular mechanism by which ACTG2 functions in tumors is not very clear and needs further research, it is reasonable to believe that ACTG2 could be a novel prognostic biomarker and candidate therapeutic target for PCa due to the results of our study.
Nevertheless, the limitations should be acknowledged for our research. Firstly, it is required further studies in vivo and in vitro for illuminating the accurate molecular mechanisms that affected the tumorigenesis and the progression of PCa. Secondly, to making our findings more convincing, a larger number of clinical samples were needed, which we will concentrate on in our future research.
Taken together, we identified 4 potential molecular biomarkers (CCNB1, TTK, CNN1, and ACTG2) for prognosing outcome and target therapy of PCa by applying a comprehensive bioinformatics analysis based on WGCNA, but more detailed functional exploration and validation of the molecular mechanisms associated with these genes are indispensable. Our findings might furnish some meaningful insights into individual therapy of PCa patients.
Author contributions
Conceptualization: Xuan Chen, Jingyao Wang, Yongqing Lai.
Data curation: Xiqi Peng.
Formal analysis: Xuan Chen, Jingyao Wang, Chunduo Zhang.
Funding acquisition: Yongqing Lai.
Investigation: Xuan Chen, Jingyao Wang, Xiqi Peng, Chunduo Zhang.
Methodology: Xuan Chen, Jingyao Wang, Xiqi Peng, Kaihao Liu, Xingzhen Zeng.
Project administration: Xingzhen Zeng, Yongqing Lai.
Resources: Xingzhen Zeng, Yongqing Lai.
Software: Xuan Chen, Jingyao Wang, Chunduo Zhang.
Supervision: Xingzhen Zeng, Yongqing Lai.
Validation: Xuan Chen, Xiqi Peng, Kaihao Liu.
Visualization: Xuan Chen, Kaihao Liu.
Writing – original draft: Xuan Chen.
Writing – review & editing: Jingyao Wang, Yongqing Lai.
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Footnotes
Abbreviations: BCR = biochemical recurrence, BP= biological process, CC= cellular component, CNG = copy number gains, DAVID = Annotation, Visualization and Integrated Discovery, DEGs = differentially expressed genes, EFS = event-free survival, FDR = false discovery rate, GEO = gene expression omnibus, GO = gene ontology, GS = coefficient of gene trait significance, HR = hazard ratio, IHC = immunohistochemistry, KEGG = Kyoto Encyclopedia of Genes and Genomes, LFC = Log2-fold change, MCODE = Molecular Complex Detection, MEs = module eigengenes, MF = molecular function, MM = coefficient of module membership, MS = module significance, PCa = prostate cancer, PPI = protein–protein interaction, PSA = prostate-specific antigen, RMA = Robust Multi-array Average TCGA = The Cancer Genome Atlas, TOM = topological overlap measure, WGCNA = weighted gene co-expression network analysis.
How to cite this article: Chen X, Wang J, Peng X, Liu K, Zhang C, Zeng X, Lai Y. Comprehensive analysis of biomarkers for prostate cancer based on weighted gene co-expression network analysis. Medicine. 2020;99:14(e19628).
The datasets generated during and/or analyzed during the current study are publicly available.
XC and JW contributed equally to the study.
This study was supported by Basic Research Project of Peking University Shenzhen Hospital (JCYJ2017001, JCYJ2017004, JCYJ2017005, JCYJ2017006, JCYJ2017007, JCYJ2017012), Clinical Research Project of Peking University Shenzhen Hospital (LCYJ2017001), Science and Technology Development Fund Project of Shenzhen (no. JCYJ20180507183102747) and Clinical Research Project of Shenzhen Health Commission (no. SZLY2018023).
The authors report no conflicts of interest.
Supplemental Digital Content is available for this article.
References
- [1].Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin 2011;61:69–90. [DOI] [PubMed] [Google Scholar]
- [2].Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30. [DOI] [PubMed] [Google Scholar]
- [3].Formosa A, Markert EK, Lena AM, et al. MicroRNAs, miR-154, miR-299-5p, miR-376a, miR-376c, miR-377, miR-381, miR-487b, miR-485-3p, miR-495 and miR-654-3p, mapped to the 14q32.31 locus, regulate proliferation, apoptosis, migration and invasion in metastatic prostate cancer cells. Oncogene 2014;33:5173–82. [DOI] [PubMed] [Google Scholar]
- [4].Berger MF, Lawrence MS, Demichelis F, et al. The genomic complexity of primary human prostate cancer. Nature 2011;470:214–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Grasso CS, Wu YM, Robinson DR, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012;487:239–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Freedland SJ, deGregorio F, Sacoolidge JC, et al. Preoperative p27 status is an independent predictor of prostate specific antigen failure following radical prostatectomy. J Urol 2003;169:1325–30. [DOI] [PubMed] [Google Scholar]
- [7].Song Z, Huang Y, Zhao Y, et al. The identification of potential biomarkers and biological pathways in prostate cancer. J Cancer 2019;10:1398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lynch SM, McKenna MM, Walsh CP, et al. miR-24 regulates CDKN1B/p27 expression in prostate cancer. Prostate 2016;76:637–48. [DOI] [PubMed] [Google Scholar]
- [9].Peng D, Ge G, Xu Z, et al. Diagnostic and prognostic biomarkers of common urological cancers based on aberrant DNA methylation. Epigenomics 2018;10:1189–99. [DOI] [PubMed] [Google Scholar]
- [10].Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Shi K, Bing ZT, Cao GQ, et al. Identify the signature genes for diagnose of uveal melanoma by weight gene co-expression network analysis. Int J Ophthalmol 2015;8:269–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wang L, Gong Y, Chippada-Venkata U, et al. A robust blood gene expression-based prognostic model for castration-resistant prostate cancer. BMC Med 2015;13:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Meller S, Meyer HA, Bethan B, et al. Integration of tissue metabolomics, transcriptomics and immunohistochemistry reveals ERG- and gleason score-specific metabolomic alterations in prostate cancer. Oncotarget 2016;7:1421–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 2003;4:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lucarelli G, Rutigliano M, Galleggiante V, et al. Metabolomic profiling for the identification of novel diagnostic markers in prostate cancer. Expert Rev Mol Diagn 2015;15:1211–24. [DOI] [PubMed] [Google Scholar]
- [16].Ferro M, Buonerba C, Terracciano D, et al. Biomarkers in localized prostate cancer. Future Oncol 2016;12:399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gabriele C, Cantiello F, Nicastri A, et al. High-throughput detection of low abundance sialylated glycoproteins in human serum by TiO2 enrichment and targeted LC-MS/MS analysis: application to a prostate cancer sample set. Anal Bioanal Chem 2019;411:755–63. [DOI] [PubMed] [Google Scholar]
- [18].Lucarelli G, Loizzo D, Ferro M, et al. Metabolomic profiling for the identification of novel diagnostic markers and therapeutic targets in prostate cancer: an update. Expert Rev Mol Diagn 2019;19:377–87. [DOI] [PubMed] [Google Scholar]
- [19].Morton RA, Ewing CM, Nagafuchi A, et al. Reduction of E-cadherin levels and deletion of the alpha-catenin gene in human prostate cancer cells. Cancer Res 1993;53:3585–90. [PubMed] [Google Scholar]
- [20].Aaltomaa S, Lipponen P, Ala-Opas M, et al. Expression and prognostic value of CD44 standard and variant v3 and v6 isoforms in prostate cancer. Eur Urol 2001;39:138–44. [DOI] [PubMed] [Google Scholar]
- [21].Cultrara CN, Kozuch SD, Ramasundaram P, et al. GRP78 modulates cell adhesion markers in prostate Cancer and multiple myeloma cell lines. BMC Cancer 2018;18:1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Murillo-Garzon V, Kypta R. WNT signalling in prostate cancer. Nat Rev Urol 2017;14:683–96. [DOI] [PubMed] [Google Scholar]
- [23].Thiele S, Gobel A, Rachner TD, et al. WNT5A has anti-prostate cancer effects in vitro and reduces tumor growth in the skeleton in vivo. J Bone Miner Res 2015;30:471–80. [DOI] [PubMed] [Google Scholar]
- [24].Khaja AS, Egevad L, Helczynski L, et al. Emphasizing the role of Wnt5a protein expression to predict favorable outcome after radical prostatectomy in patients with low-grade prostate cancer. Cancer Med 2012;1:96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Syed Khaja AS, Helczynski L, Edsjo A, et al. Elevated level of Wnt5a protein in localized prostate cancer tissue is associated with better outcome. PLoS One 2011;6:e26539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Volante M, Tota D, Giorcelli J, et al. Androgen deprivation modulates gene expression profile along prostate cancer progression. Hum Pathol 2016;56:81–8. [DOI] [PubMed] [Google Scholar]
- [27].Uysal-Onganer P, Kawano Y, Caro M, et al. Wnt-11 promotes neuroendocrine-like differentiation, survival and migration of prostate cancer cells. Mol Cancer 2010;9:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Fang Y, Yu H, Liang X, et al. Chk1-induced CCNB1 overexpression promotes cell proliferation and tumor growth in human colorectal cancer. Cancer Biol Ther 2014;15:1268–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhou L, Du Y, Kong L, et al. Identification of molecular target genes and key pathways in hepatocellular carcinoma by bioinformatics analysis. Onco Targets Ther 2018;11:1861–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yu Y, Zhang Y, Guan W, et al. Androgen receptor promotes the oncogenic function of overexpressed Jagged1 in prostate cancer by enhancing cyclin B1 expression via Akt phosphorylation. Mol Cancer Res 2014;12:830–42. [DOI] [PubMed] [Google Scholar]
- [31].Bubendorf L, Sauter G, Moch H, et al. Ki67 labelling index: an independent predictor of progression in prostate cancer treated by radical prostatectomy. J Pathol 1996;178:437–41. [DOI] [PubMed] [Google Scholar]
- [32].Gomez LA, de Las Pozas A, Reiner T, et al. Increased expression of cyclin B1 sensitizes prostate cancer cells to apoptosis induced by chemotherapy. Mol Cancer Ther 2007;6:1534–43. [DOI] [PubMed] [Google Scholar]
- [33].Budczies J, Denkert C, Gyorffy B, et al. Chromosome 9p copy number gains involving PD-L1 are associated with a specific proliferation and immune-modulating gene expression program active across major cancer types. BMC Med Genomics 2017;10:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhang Y, Zhang Y, Zhang L. Expression of cancer-testis antigens in esophageal cancer and their progress in immunotherapy. J Cancer Res Clin Oncol 2019;145:281–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tannous BA, Kerami M, Van der Stoop PM, et al. Effects of the selective MPS1 inhibitor MPS1-IN-3 on glioblastoma sensitivity to antimitotic drugs. J Natl Cancer Inst 2013;105:1322–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gimona M, Kaverina I, Resch GP, et al. Calponin repeats regulate actin filament stability and formation of podosomes in smooth muscle cells. Mol Biol Cell 2003;14:2482–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Liu Y, Wu X, Wang G, et al. CALD1, CNN1, and TAGLN identified as potential prognostic molecular markers of bladder cancer by bioinformatics analysis. Medicine (Baltimore) 2019;98:e13847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yamane T, Asanoma K, Kobayashi H, et al. Identification of the critical site of calponin 1 for suppression of ovarian cancer properties. Anticancer Res 2015;35:5993–9. [PubMed] [Google Scholar]
- [39].Wang KH, Chu SC, Chu TY. Loss of calponin h1 confers anoikis resistance and tumor progression in the development of high-grade serous carcinoma originating from the fallopian tube epithelium. Oncotarget 2017;8:61133–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Beck AH, Lee CH, Witten DM, et al. Discovery of molecular subtypes in leiomyosarcoma through integrative molecular profiling. Oncogene 2010;29:845–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wu Y, Liu ZG, Shi MQ, et al. Identification of ACTG2 functions as a promoter gene in hepatocellular carcinoma cells migration and tumor metastasis. Biochem Biophys Res Commun 2017;491:537–44. [DOI] [PubMed] [Google Scholar]
- [42].Drew JE, Farquharson AJ, Mayer CD, et al. Predictive gene signatures: molecular markers distinguishing colon adenomatous polyp and carcinoma. PLoS One 2014;9:e113071. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.