

Abstract
Well-defined bis(silylene)pyridine cobalt(III) precatalysts for C(sp2)–H borylation have been synthesized and applied to the investigation of the mechanism of the catalytic borylation of furans and pyridines. Specifically, [(ArSiNSi)CoH3]·NaHBEt3 (ArSiNSi = 2,6-[EtNSi(NtBu)2CAr]2C5H3N, Ar = C6H5 (1-H3·NaHBEt3), 4-MeC6H4 (2-H3·NaHBEt3)) and trans-[(ArSiNSi)Co(H)2BPin] (Ar = C6H5 (1-(H)2BPin), 4-MeC6H4 (2-(H)2BPin), Pin = pinacolato) were prepared and employed as single component precatalysts for the C(sp2)–H borylation of 2-methylfuran, benzofuran and 2,6-lutidine. The cobalt(III) precursors, 2-H3·NaHBEt3 and 2-(H)2BPin also promoted C(sp2)–H activation of benzofuran, yielding [(ArSiNSi)CoH(Bf)2] (Ar = 4-MeC6H4, 2-H(Bf)2, Bf = 2-benzofuranyl). Monitoring the catalytic borylation of 2-methylfuran and 2,6-lutidine by 1H NMR spectroscopy established the trans-dihydride cobalt(III) boryl as the catalyst resting state at low substrate conversion. At higher conversion two distinct pincer modification pathways were identified, depending on the substrate and the boron source.
Graphical Abstract

Introduction.
The metal-catalyzed borylation of C–H bonds is one of the most widely applied methods of C–H functionalization, due in part to the versatility and synthetic utility of the resulting aryl- or heteroaryl boronic ester products.1 State-of-the-art methods rely on in situ-generated iridium catalysts derived from an iridium(I) precursor and a bidentate supporting ligand, typically a (bis)phosphine or substituted 2,2’-bipyridines.2 The mechanism of iridium-catalyzed C–H borylation has been studied and it is well-accepted that an Ir(III)-Ir(V) cycle is operative where an iridium(III) trisboryl is responsible for turnover limiting C–H activation.3 Highly active iridium catalysts have been discovered with predictable site-selectivity that is typically sterically driven, where sterically accessible C–H bonds undergo borylation with statistical selectivity. Approaches involving hydrogen-bonding interactions,4 directing group strategies5 and specialized ligand designs6 have been developed to overcome sterically-driven site-selectivity.
Recent interest has focused on the use of catalysts with more Earth-abundant first-row transition metals to promote C–H borylation and examples with Fe,7 Co8 and Ni9 are now known. In addition to possible economic benefits and reduced environmental impact, these catalysts offer the opportunity for selectivity distinct or orthogonal to state-of-the-art iridium.10 With cobalt, the bis(diisopropylphosphine)pyridine pincer, [iPrPNP], imparts a strong ligand field and enables the two-electron oxidative addition of H2, C–H and B–H bonds to cobalt(I)–alkyl precursors.11,12 In catalytic C–H borylation (Scheme 1a), the [(iPrPNP)Co] family of precatalysts offers unique ortho-to-fluorine site selectivity in the borylation of fluorinated arenes, arising from reversible, thermodynamically-controlled oxidative addition where formation of the strongest M–Caryl bond governs borylation selectivity.13 In all cases examined, a Co(I)–Co(III) cycle is favored where a cobalt(I)–boryl intermediate is responsible for cleavage of the C(sp2)–H bond.14,15,16
Scheme 1.

Comparison of cobalt pincer precatalysts for the C(sp2)–H borylation of heterocycles and motivations for the present study.
Our laboratory has explored a variety of tridentate pincer ligands that support active cobalt catalysts for C–H borylation.6,8b Most exhibited inferior activity to the [(iPrPNP)Co] family. In addition, rapid formation of catalytically inactive bis(chelate) metal complexes was also deleterious. Driess, Cui and coworkers reported a notable exception with the efficient C–H borylation of arenes and heteroarenes employing [(PhSiNSi)CoBr2] (PhSiNSi = (2,6-[EtNSi(NtBu)2CPh]2C5H3N)) activated in situ with NaHBEt3 in the presence of excess cyclohexene (Scheme 1b).18 The introduction of silylene donors generates one of the most electron-donating pincers known, and delineation of the consequences of such an electronic effect in catalysis has not been established.19,20,21
Despite their competence in catalytic C–H borylation, little is known about the identity of the active species, the composition of the cobalt complex responsible for C–H activation, the role of the cyclohexene additive and what redox cycle is operative during catalysis. Here we describe the synthesis and characterization of well-defined cobalt(III) complexes supported by [SiNSi]-type pincers (Scheme 1c) and establish key elements of the catalytic cycle including the redox states accessed during catalysis, demonstration of catalyst modification during turnover and insights into the role of the cyclohexene additive. These studies uncover the unique consequences of introduction of such an electron rich pincer on elementary steps relevant to C–H borylation.
RESULTS AND DISCUSSION
Synthesis of Bis(silylene)pyridine Cobalt(III) Complexes.
To gain insight into the mechanism and the redox cycle operative in C–H borylation promoted by [SiNSi]-supported cobalt compounds, well-defined coordination precatalysts were targeted. Furthermore, single component precatalysts eliminate the need for air-sensitive and pyrophoric activators that because of their extreme reactivity, limit synthetic applications. To date, the cobalt(II) dibromide, [(PhSiNSi)CoBr2]18 is the only reported cobalt complex of this pincer also inspiring additional investigations in the coordination chemistry and accessible oxidation states of these complexes.
Inspired by synthetic route used to prepare [(iPrPNP)Co(I)] complexes,8a two equivalents of NaHBEt3 (1.0 M solution in toluene) were added to a frozen toluene solution containing [(ArSiNSi)CoCl2] (Ar = C6H5, 4-MeC6H4) at −95 °C (see Supporting Information). Filtration and subsequent recrystallization resulted in the isolation of pale yellow solids identified as [(ArSiNSi)CoH3]·NaHBEt3 in 40% (Ar = C6H5, 1-H3·NaHBEt3) and 43% (Ar = 4-MeC6H4, 2-H3·NaHBEt3) yield respectively (Scheme 2). The p-tolyl variant of the [SiNSi] ligand was employed to improve the crystallinity of the products.
Scheme 2.

Synthesis of [(ArSiNSi)CoH3]·NaHBEt3 (Ar = C6H5, 1-H3·NaHBEt3; Ar = 4-MeC6H4, 2-H3·NaHBEt3).
The 1H and 13C NMR spectra of 1-H3·NaHBEt3 in benzene-d6 exhibited signals consistent with a C2v symmetric compound. The ethyl groups of the NaHBEt3 ligand were observed as broad signals at 0.86 ppm (CH2) and 1.50 ppm (CH3) that were maintained in the spectrum even after repeated washing with Et2O, supporting an interaction between the cobalt complex and the NaHBEt3 that is preserved in benzene solution. A singlet was observed at −17.19 ppm consistent with rapid exchange of the three hydride ligands on the NMR timescale at 23 °C. Upon cooling a toluene-d8 solution to −75 °C, two broad resonances were observed at −13.32 (2 H) and −24.33 ppm (1 H), corresponding to the apical and equatorial hydrides respectively (Figure S6). Increasing the temperature to −35 °C resulted in coalescence of the signals at −17.19 ppm. A similar dynamic was reported for [(iPrPNP)CoH3] lacking the coordinated NaHBEt3.13 A minimum T1 value of 113 ms was measured at −5°C for 1-H3·NaHBEt3, supporting its formulation as a classical cobalt trihydride, although the impact of the coordinated NaHBEt3 on this value is unknown.13,22
A pathway to rationalize the formation of 1-H3·NaHBEt3 and 2-H3·NaHBEt3 involves initial reduction of the cobalt(II) complex [(ArSiNSi)CoCl2] by NaHBEt3 to the cobalt(I) derivative, [(ArSiNSi)CoCl] with concomitant formation of H2. Generation of the cobalt(I) hydride, [(ArSiNSi)CoH], followed by oxidative addition of the H2 present in the reaction and interaction with a molecule of NaHBEt3 yields the isolated cobalt(III) product.
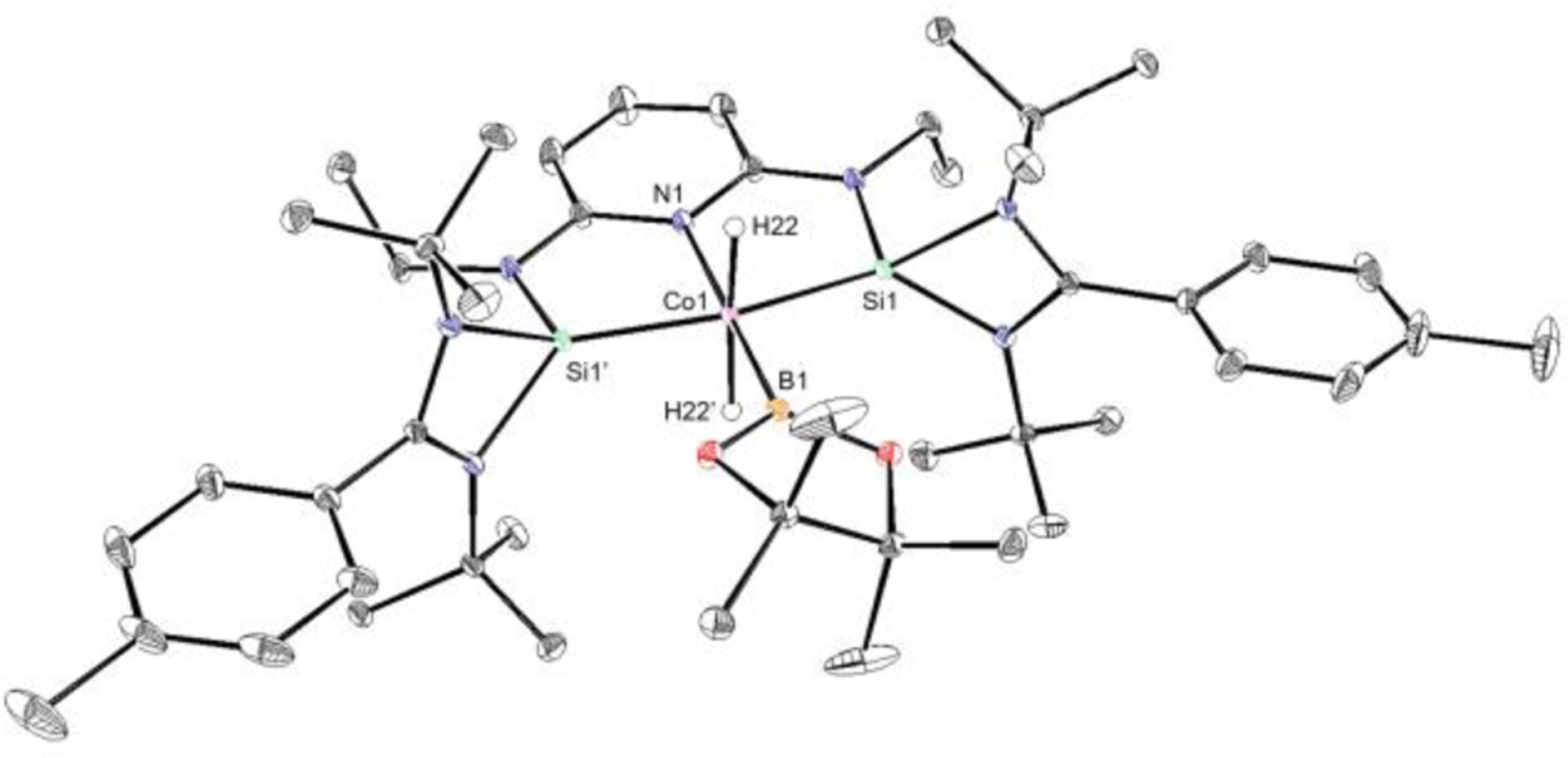
Slow evaporation of a diethyl ether solution of 2-H3·NaHBEt3 at ambient temperature produced yellow crystals suitable for single-crystal X-ray diffraction (Figure 1). A distorted octahedral geometry is observed with the pincer and the three hydride ligands defining the coordination sphere around the cobalt. The sodium of the NaHBEt3 bridges two of the hydride ligands (H(50) and H(51)) and the Co(1)–Si(1) and Co(1)–Si(2) bond distances of 2.1226(5) and 2.1101(5) Å respectively, are consistent with the presence of two silylene donors.21 The dihedral angles between the planes of the p-tolyl substituents (C(11)–C(12)–C(13) and C(19)–C(20)–C(21)) and the plane defined by the pincer ligand (Co(1)–N(4)–C(7)) are 11.9(2)° and 5.2(2)° respectively, supporting a nearly coplanar orientation of the p-tolyl substituents with the SiNSi ligand.
Figure 1.

Solid-state structure of 2-H3·NaHBEt3 at 30% probability ellipsoids. The hydrogen atoms bonded to cobalt (H(50), H(51) and H(52)) have been located and freely refined. All hydrogen atoms except those bonded to cobalt, a B-CH2 group and two of the tert-butyl groups in the pincer have been omitted for clarity.
Cobalt(I) complexes of [(iPrPNP)Co] are useful precatalysts for C–H borylation and are routinely prepared by treatment of [(iPrPNP)CoCl2] with two equivalents of an alkyl lithium reagent, RLi (R = Me, CH2SiMe3).8a,15 Attempts to synthesize analogous cobalt(I) complexes starting from [(ArSiNSi)CoCl2] and MeLi, PhLi or CH2SiMe3Li were unsuccessful, producing decomposition (with MeLi or PhLi) or unidentified mixtures of products (with LiCH2SiMe3). Likewise, treatment of (RPNP)CoCl2 complexes with one equivalent of NaHBEt3 has been used to synthesize the corresponding (RPNP)CoCl derivatives.23 Applying this methodology to [(ArSiNSi)CoCl2] resulted in lower yield and purity of the hydride adducts, 1-H3·NaHBEt3 and 2-H3·NaHBEt3.
The synthesis of a cobalt(III) dihydride boryl was also pursued as these complexes have been identified as catalyst resting states and precatalysts for C–H borylation.8a,10c,13,14,15 Addition of excess (~ 6 equiv) of HBPin to 1-H3·NaHBEt3 or 2-H3·NaHBEt3 in THF and heating to 80 °C for 10 minutes followed by washing with Et2O resulted in isolation of yellow solids identified as trans-[(ArSiNSi)Co(H)2BPin] (Ar = Ph, 1-(H)2BPin; Ar = 4-MeC6H4, 2-(H)2BPin) in 65% and 68% yield, respectively (Scheme 3). The 1H NMR spectrum of 1-(H)2BPin in benzene-d6 exhibited the number of signals consistent with a C2v symmetric molecule. A diagnostic singlet was observed at −10.93 ppm assigned to the two equivalent trans hydrides. No signals were observed between 0.50 to 1.50 ppm attributable to the ethyl groups of NaHBEt3, demonstrating elimination of this salt from the coordination sphere of the cobalt. The 4-Me-iPrPNP (4-methyl-2,6-iPr2P-methylpyridine) analog of 1-(H)2BPin and 2-(H)2BPin, [(4-Me-iPrPNP)Co(H)2BPin], has been previously synthesized by treatment of the corresponding cobalt(II) dichloride complex with two equivalents of NaHBEt3 in the presence of 4 equivalents of HBPin.14 Attempts to apply this method to the synthesis of 1-(H)2BPin produced the desired cobalt(III) product along with other unidentified products as judged by 1H NMR spectroscopy.
Scheme 3.

Synthesis of trans-[(ArSiNSi)Co(H)2BPin] (Ar = C6H5, 1-(H)2BPin; Ar = 4-MeC6H4, 2-(H)2BPin).
Slow evaporation of a diethyl ether solution of 2-(H)2BPin at room temperature furnished yellow crystals suitable for single-crystal X-ray diffraction (Figure 2). As with 2-H3·NaHBEt3, a distorted octahedral geometry about the cobalt is observed in 2-(H)2BPin, with the two hydride ligands in a mutually trans disposition and the BPin ligand trans to the pyridyl ring. The Co(1)–B(1) (1.912(2) Å) and Co(1)–H(22) (1.44(3) Å) bond distances are similar to those in the iPrPNP analog, trans-[(iPrPNP)Co(H)2BPin] (Co–B is 1.926(4) Å and Co–H is 1.49(4) Å).6a In contrast, the Co(1)–Si(1) (2.1128(4) Å) bond distances are shorter than the Co–P distance in [(iPrPNP)Co(H)2BPin] (2.1296(6) Å), and the Co(1)–N(1) bond (2.0378(16) Å) is slightly longer than that in [(iPrPNP)Co(H)2BPin] (2.016(3) Å). Both metrics are in agreement with the silylenes in [SiNSi] being stronger electron-donors than the phosphines in [iPrPNP]. The strong electron-donating ability of the silylene donors is likely the origin of the elongation of the Co-N(pyridine) bond in 2-(H)2BPin, as previously reported for the [(PhSiNSi)FeCl2] complex, where the pyridine ring does not coordinate.21a
Figure 2.
Solid-state structure of 2-(H)2BPin at 30% probability ellipsoids. Hydrogen atoms except those bonded to cobalt have been omitted for clarity.
The conversion of 1-H3·NaHBEt3 to 1-(H)2BPin was monitored by 1H NMR spectroscopy in the presence of excess HBPin at 25 °C, 50 °C and 80 °C with the goal of detecting intermediates. The transformation of 1-H3·NaHBEt3 to 1-(H)2BPin occurred in 7 hours, 40 minutes and 5 minutes respectively and neither the cobalt(I) hydride, [(PhSiNSi)CoH], nor the cis-[(PhSiNSi)Co(H)2BPin] complex were detected (Figure S32).
C(sp2)–H Borylation of Heterocycles with Well-Defined Bis(silylene)pyridine Cobalt(III) Complexes as Precatalysts.
The synthesis of well-defined cobalt(III) dihydride boryl complexes prompted exploration of the performance of these compounds in catalytic C–H borylation. Three representative substrates were selected: 2-methylfuran, benzofuran and 2,6-lutidine (Scheme 4). The borylations of 2-methylfuran and benzofuran proceeded to >98% conversion in 24 hours in THF at 80 °C with one equivalent of HBPin in the presence of 5 mol% of either 1-(H)2BPin, 2-(H)2BPin or 1-H3·NaHBEt3. The borylation of 2,6-lutidine with 1 equiv of HBPin at 100 °C with 5 mol% of 2-(H)2BPin over 24 hours produced only small (<5% conversion) amounts of product. In contrast, when B2Pin2 was used as the borylating agent under the same conditions, 46% conversion of the starting material to borylated product was observed. As with [(iPrPNP)Co] catalysts, B2Pin2 was the preferred boron reagent for the C–H functionalization of 2,6-lutidine.14
Scheme 4.

C(sp2)–H borylation of 2-methylfuran, benzofuran and 2,6-lutidine employing 1-H3·NaHBEt3, 1-(H)2BPin or 2-(H)2BPin as precatalysts. The reported percentages are substrate conversions determined by GC with mesitylene as an internal standard.
The bis(silylene)pyridine cobalt(III) precatalysts are proposed to operate by a redox Co(I)-Co(III) cycle, where reductive elimination of H2 and loss of NaHBEt3 from 1-H3·NaHBEt3 and of H2 or HBPin in 1-(H)2BPin and 2-(H)2BPin generates a Co(I) intermediate responsible for activation of the C(sp2)–H bond. Because only Co(III) complexes have been detected or isolated, a redox-neutral Co(III) cycle where bond activation occurs by σ-bond metathesis is also plausible although seegminly unlikely because of the high electron count and a hexacoordinate cobalt. Pathways involving reversible modification of the pincer assists in oxidative addition, reductive elimination or other elementary steps are also possible and supported by the various unique modifications processes discovered in this study.
Determination of the Catalyst Resting State: Furan Borylation.
With well-defined cobalt(III) precatalysts in hand, the mechanism of 2-methylfuran borylation was studied. Monitoring the catalytic reaction by 1H NMR spectroscopy with either 1 equivalent of HBPin and 5 mol% of 1-H3·NaHBEt3 or 0.5 equivalents of B2Pin2 and 15 mol% of 1-H3·NaHBEt3 at 80 °C in THF-d8 resulted in observation of 1-(H)2BPin as the catalyst-resting state in both experiments (Scheme 5).
Scheme 5.

(a) Catalyst resting-state in the borylation of 2-methylfuran using 1-H3·NaHBEt3 as a precatalyst (b) aromatic region of the 1H NMR spectrum in THF-d8 after 1 hour and 40 minutes of reaction (35% substrate conversion) and after 10 hours of reaction (54% substrate conversion).
When the reaction was carried out with 0.5 equivalents of B2Pin2 in the presence of 15 mol% of 1-H3·NaHBEt3 at 80°C, complete conversion was reached in 40 minutes whereas with 1 equivalent of HBPin and 5 mol% of 1-H3·NaHBEt3, it took 10 hours to reach 54% conversion. In both cases, the initial formation of 1-(H)2BPin (δ Co–H = −11.46 ppm, THF-d8) was accompanied by formation of two other cobalt species (δ Co–H = −12.23 (major) and −12.26 (minor) ppm, THF-d8) whose concentration increased over time at expense of 1-(H)2BPin. Formation of these compounds was slower when the borylation was carried out with 0.5 equivalents of B2Pin2 than with 1 equivalent of HBPin (see Figures S46–S49). When B2Pin2 was employed as the borylating agent, the ratio of the 1-(H)2BPin to the new cobalt hydrides was 1:0.04 at the completion of the reaction, in contrast, when HBPin was used the ratio was 1:1.3 at 54% conversion.
Addition of 4 atm of H2 to THF solutions of 1-H3·NaHBEt3, 2-H3·NaHBEt3 or 1-(H)2BPin and heating at 80 °C for 16 hours produced the same cobalt hydrides present in the catalytic reaction. From the hydrogenation of 1-H3·NaHBEt3, a brown solid was isolated in a 27% yield and was identified as a 4:1 ratio of a mixture of stereoisomers of [(Ar,HSiNSi)CoH2(H2)] (3-H2(H2), Ph,HSiNSi = 2-[EtNSi(NtBu)2CHPh]-6-[EtNSi(NtBu)2CPh]C5H3N). Based on analogy, the tolyl variant of the compound was assigned accordingly as 4-H2(H2) = ptol,HSiNSi = 2-[EtNSi(NtBu)2CH(p-tol)]-6-[EtNSi(NtBu)2C(p-tol)]C5H3N) (Scheme 6a). The products arise from hydride migration from the cobalt to one of the imine groups on the silylene donor along with trapping by σ-H2 coordination.24 As a consequence of this migration, the [Ar,HSiNSi] ligand becomes anionic, as the Si-donor bearing the new C–H group is best described as a silyl donor. As a result of hydride migration, both the incipient carbon (C7) and the cobalt are stereogenic. The oxidation state of the cobalt is maintained at +3 and both 3-H2(H2) and 4-H2(H2) are diamagnetic with two hydride ligands and a σ-H2 ligand. A related cobalt(III) complex has been reported by Heinekey and coworkers.25
Scheme 6.

(a) Independent synthesis of the catalyst resting-state II (at high substrate conversion) in the borylation of 2-methylfuran with HBPin from hydrogenation of 1-H3·NaHBEt3, 2-H3·NaHBEt3 or 1-(H)2BPin. (b) Borylation of benzofuran with 3-H2(H2) as the precatalyst. The reported percentage is the substrate conversion determined by GC using mesitylene as an internal standard.
Integration of the hydride signals in the 1H NMR spectrum of 3-H2(H2) (δ Co–H= −11.57 (major); −11.73 (minor), benzene-d6) established a 4:1 diastereomeric ratio. Because the minor isomer is formed in such small amounts and many of the ligand signals overlap with the major isomer, only the signals for the Co–H and the tBu substituents are resolved. The major isomer exhibits the number of 1H NMR resonances consistent with a Cs symmetric product. Accordingly, a singlet at 5.86 ppm in the 1H NMR spectrum and a signal at 76.8 ppm in the 13C NMR spectrum were assigned to the newly formed C(sp3)–H group. A minimum value of T1 of 94 ms was measured at 0 °C for 4-H2(H2), in the range diagnostic for σ-H2 ligands.28 At −85 °C, the exchange between the hydrides and the dihydrogen ligand in 4-H2(H2) was slowed and the 1H NMR spectrum in toluene-d8 exhibited two broad signals in the hydride region at - 10.05 (3 H) and −22.65 (1 H) ppm that were assigned as the mutually trans hydride and σ-H2 ligands and to the hydride trans to the pyridyl ring, respectively (see Figure S51).
The observation of 3-H2(H2) from hydrogenation of 1-(H)2BPin accounts for its formation during catalytic C–H borylation. The borylation of 2-methylfuran with HBPin generates H2 as the stoichiometric byproduct; capture of the H2 by the 1-(H)2BPin resting state produces the cobalt product with the modified pincer. This also accounts for the higher conversion of 1-(H)2BPin to 3-H2(H2) when HBPin is used in place of B2Pin2. With the latter boron source, HBPin is the stoichiometric byproduct and the concentration of H2 is low and hence little modified product was observed.
The catalytic performance of the modified pincer complex, 3-H2(H2) was evaluated in the borylation of benzofuran. With 1 equivalent of HBPin and 5 mol% of 3-H2(H2), 83% conversion to product was observed at 80 °C over the course of 24 hours (Scheme 6b). The conversion was lower than when 1-H3·NaHBEt3 or 1-(H)2BPin were used, suggesting that 3-H2(H2) is less ative. The catalytic borylation of 2-methylfuran with HBPin and 5 mol % of 3-H2(H2) was monitored by 1H NMR spectroscopy in THF-d8 to gain insight into the catalyst resting state. A Co–H resonance was observed at −11.97 ppm throughout catalytic turnover corresponding to an unidentified cobalt compound, along with those for the 3-H2(H2) mixture (Figure S54). Detectable quantities of 1-(H)2BPin were not observed during in these experiments supporting the irreversibility of the intramolecular hydride migration process and suggesting a different cycle operative than that with the precatalysts with the intact ligand.
Intramolecular hydride or alkyl migration to imine ligands in transition metal complexes has been reported,26 and, more importantly, has been recognized as a deactivation pathway for pyridine(diimine) iron-catalyzed olefin polymerization.27 It is particularly remarkable that in the case of the borylation of 2-methylfuran by bis(silylene)pyridine cobalt complexes, this process does not constitute a deactivation pathway, and 3-H2(H2) is a competent precatalyst for C(sp2)–H borylation. The mechanism of operation, active redox cycle and nature of the C–H activating species are unknown.
C–H Activation of Benzofuran by Bis(silylene) pyridine Cobalt(III) Complexes.
Because cobalt(III) dihydride boryl and cobalt(III) trihydrides complexed with NaHBEt3 containing the bis(silylene)pyridine pincer are efficient precatalysts for the borylation of heterocycles, additional insights into the C–H activation step were sought. Addition of 6 equivalents of benzofuran to a THF solution of 2-H3·NaHBEt3 or 2-(H)2BPin followed by heating (50 °C for 2-H3·NaHBEt3; 80 °C for 2-(H)2BPin) for 1 h and 30 min, yielded a single new cobalt compound that was isolated as an orange powder in moderate (46% and 40% respectively) yield (Scheme 7). The product was identified as cis-[(ptolSiNSi)CoH(Bf)2] (2-H(Bf)2, Bf = 2-benzofuranyl) based on a combination of 1D and 2D NMR characterization. The 1H and 13C NMR spectra of 2-H(Bf)2 in benzene-d6 exhibited the number of signals consistent with a Cs symmetric complex. A singlet at −11.41 (1H) ppm in the 1H NMR spectrum was assigned to the hydride ligand while the C(4)-H groups of each benzofuranyl ligand occurred at 7.51 and 7.48 ppm respectively. Both signals did not show any crosspeak in the 1H, 1H-COSY spectrum supporting activation of the C(2)–H bond of benzofuran. A 1H,13C-HMBC spectrum allowed for the assignment of the 13C NMR signals at 203.7 and 194.6 ppm to the Co-bonded C(5) atoms.
Scheme 7.

Net C–H activation of benzofuran by [(SiNSi)Co(III)].
Complex 2-H(Bf)2 exhibited a distorted octahedral geometry with the two benzofuranyl ligands in a mutually cis arrangement and the hydride ligand trans to one of the benzofuranyl ligands (Figure 3). The benzofuranyl ligand trans to the hydride is tilted 12.2(3)° from the plane defined by Co(1)–N(4)–C(42), whereas the benzofuranyl ligand trans to the pyridyl ring is tilted 45.4(4)° from the plane that contains Co(1)–N(4)–C(1). The Co(1)–C(42) bond distance is longer than the Co(1)–C(50) as a result of an stronger trans influence of the hydride in comparison to the pyridyl ring. Co(1)–Si(1) and Co(1)–Si(2) bond distances (2.1552(10), 2.1371(10) Å respectively) are slightly longer than those found for 2-(H)2BPin (see above), whereas the Co(1)–N(4) bond distance (2.031(3) Å) is similar for both, supporting both as cobalt(III) complexes.
Figure 3.

Solid-state structure of 2-H(Bf)2 at 30% probability ellipsoids. The hydrogen atom bonded to cobalt (H(57)) has been located. All other hydrogen atoms and two of the tert-butyl groups in the pincer have been omitted for clarity.
To gain additional insight into the formation of 2-H(Bf)2, the reaction of 2-H3·NaHBEt3 or 2-(H)2BPin with 6 equivalents of benzofuran was monitored by 1H NMR spectroscopy at 50 °C in THF-d8. Three cobalt complexes were observed prior to formation of 2-H(Bf)2. The diastereomers of the modified pincer complex, 4-H2(H2) were identified as one of the cobalt complexes present and increased over the course of the C–H activation reaction. The other two compounds were identified as cis-[(ptolSiNSi)CoH2(Bf)] (cis-2-H2(Bf)) and trans-[(ptolSiNSi)CoH2(Bf)] (trans-2-H2(Bf)) based on 1D and 2D NMR spectroscopic experiments. Based on these observations, a pathway accounting for formation of cis-2-H2(Bf) and trans-2-H2(Bf) from 2-H3·NaHBEt3 is presented in Scheme 8.
Scheme 8.

Proposed pathway for the formation of 2-H(Bf)2.
Loss of NaHBEt3 and reductive elimination of H2 generates a putative and unobserved cobalt(I)–hydride, [(ptolSiNSi)CoH], that promotes rapid C–H oxidative addition of benzofuran accounting for cis-2-H2(Bf) and trans-2-H2(Bf) depending on the trajectory of the substrate. Reductive elimination from cis-2-H2(Bf) generates a cobalt(I)–benzofuranyl that undergoes oxidative addition of benzofuran to form 2-H(Bf)2. At initial stages of the reaction (from 5 min to 1 h 45 min), when 2-H3·NaHBEt3 has not been fully consumed, the cis:trans ratio of the 2-H2(Bf) complexes was 1:0.9 and remained constant. When all the starting material was consumed (1 h 45 min), the cis:trans ratio gradually changed to 1:2.1, supporting that formation of 2-H(Bf)2 takes place preferentially from the cis isomer. The fact that the cis isomer is present in the reaction until all the trans complex is consumed to form 2-H(Bf)2 supports that trans-2-H2(Bf) isomerizes to cis-2-H2(Bf) which ultimately yields the final product. During the reaction, the amount of 4-H2(H2) gradually increased (cis-2-H(Bf)2:4-H2(H2) 1:0.4 at 45 min, 1:1.4 at 2h 30 min and 1:5.4 at 3 h 5 min) reaching a 2-H(Bf)2:4-H2(H2) ratio of 1:0.2 at the completion of the reaction. The generation of H2 during this process accounts for formation of 4-H2(H2). Because pincer modification is irreversible, this compound accumulates over the course of the C–H activation reaction.
The same three cobalt products were obtained when the reaction was conducted with one rather than six equivalents of benzofuran. Notably, 2-H3·NaHBEt3 remained in solution after all the benzofuran was consumed. Observation of the same product independent of whether 2-H3·NaHBEt3 or 2-H2BPin was used as the starting material demonstrates that the coordinated NaHBEt3 plays little role in the C–H activation of benzofuran. It is important to note that no spectroscopic evidence has been obtained for any [(SiNSi)Co(I)] intermediate, in stark contrast to the PNP cases where such complexes are common precatalysts, highlighting the impact of the increased electron donation of the silylene-based pincer.
To probe the presence of a cobalt(I) hydride in the C–H activation of benzofuran, attempts were made to synthesize a cobalt(I) complex capable of C–H activation. Addition of neohexene to a benzene-d6 solution of 1-H3·NaHBEt3 resulted in decomposition with no evidence for a cobalt(I) alkyl. Addition of π-acidic carbon monoxide was explored in an attempt to stabilize the lower oxidation state. Addition of 1 atm of CO to a benzene-d6 solution of 1-H3·NaHBEt3 or 2-(H)2BPin yielded yellow solids in 75% and 71% yields, respectively. The products were identified as the cobalt(I) dicarbonyl derivatives, [(ArSiNSiH)Co(CO)2] (Ar = C6H5, 5-(CO)2; Ar = 4-MeC6H4, 6-(CO)2) where the pincer has been modified to an anionic silylene-pyridine-silyl ligand formed from cobalt-to-silicon hydride migration (Scheme 9).
Scheme 9.

Reaction of 1-H3·NaHBEt3 or 2-(H)2BPin with CO to yield 5-(CO)2 and 6-(CO)2 respectively.
The 1H NMR spectrum of 5-(CO)2 in benzene-d6 exhibited resonances consistent with an asymmetric SiNSi ligand, e. g., four singlets for the four tBu groups (at 1.54, 1.29, 1.27 and 1.10 ppm, 9 H each) supporting the loss of the horizontal and vertical mirror planes in the molecule. This symmetry contrasts the Cs symmetry observed for 3-H2(H2) or 4-H2(H2), further supporting hydride migration to the amidinate C=N bond in the latter compounds. A singlet at 6.49 ppm was assigned to the silicon-bonded hydrogen atom and was assigned based on a 1H,13C-HSQC experiment, which did not exhibit a crosspeak for that signal, establishing that the hydrogen atom is not bonded to a C atom. The 13C NMR spectrum exhibited two signals at 206.7 and 205.6 ppm that were assigned to the two inequivalent carbonyl ligands. Moreover, the C(25) in the amidinate (see Scheme 10) is upfield shifted (163.0 ppm) in agreement with a localized double bond and contrasts with the chemical shift of C(10) (172.6 ppm) engaged in delocalized N-C=N bonding. The IR spectrum of 5-(CO)2 in KBr showed two bands at 1871 and 1860 cm−1 in the carbonyl region, consistent with the presence of two CO ligands and a band at 1925 cm−1 that is assigned as the Si–H stretch.28
Scheme 10.

(a) Catalyst resting-states for the borylation of 2,6-lutidine. (b) aromatic region of the 1H NMR spectrum after 5 minutes of reaction (5% substrate conversion), showing signals for 1-(H)2BPin, and after 4 hours of reaction (45% substrate conversion), showing signals of a mixture of 1-(H)2BPin and 7-(H)2BPin.
Single crystals of 5-(CO)2 suitable for X-ray diffraction were obtained from a pentane solution stored at −35 °C for three days (Figure 4). An idealized trigonal bipyramidal geometry was observed with a Co(1)–Si(1) bond distance of 2.1734(4) Å contracted from the value of 2.2392(4) Å of Co(1)–Si(2), consistent with Si(1) being a silylene donor and Si(2) being a silyl. In addition, the C(25)–N(6) bond distance of 1.4046(17) Å is consistent with a C–N single bond while the C(25)–N(7) bond length of 1.2769(18) Å is in agreement with a C=N bond. In contrast, the C(10)–N(1) and C(10)–N(2) bond distances on the unmodified amidinate donor are 1.3385(17) and 1.3425(17) Å, supporting a delocalized double bond.
Figure 4.

Solid-state structure of 5-(CO)2 at 30% probability ellipsoids. The hydrogen atom bonded to silicon has been located and freely refined. All other hydrogen atoms have been omitted for clarity.
Formation of 5-(CO)2 from carbonylation of 1-H3·NaHBEt3 likely proceeds by initial H2 reductive elimination and elimination of NaHBEt3 to yield an undetected cobalt(I) hydride complex, [(PhSiNSi)CoH(CO)] (Scheme 9). This intermediate then undergoes cobalt-to-silicon hydride migration promoted by the coordination of a second molecule of CO to yield the final product. During the course of this migration, one of the Si–N bonds breaks and the silylene donor converts to an anionic silyl ligand. A similar pathway is envisioned for the formation of 6-(CO)2 from 2-(H)2BPin, however, in this case 2-(H)2BPin should undergo an initial step of HBPin reductive elimination (instead of H2) to yield the final product.
Determination of the Catalyst Resting State for the Borylation of 2,6-lutidine with Well-Defined Bis(silylene)pyridine Cobalt (III) Precatalysts.
Monitoring the borylation of 2,6-lutidine with one equivalent of B2Pin2 in THF-d8 at 100 °C in the presence of 15 mol% of 1-H3·NaHBEt3 or 1-(H)2BPin resulted in observation of two cobalt complexes by 1H NMR spectroscopy (Scheme 10). At low substrate conversion (5%), 1-(H)2BPin remained as the predominant cobalt species while at higher conversion (45%), a new cobalt compound identified as trans-[(BPinSiNSi)Co(H)2BPin] (BPinSiNSi = 2,6-[EtNSi(NtBu)2C(3-BPinC6H4)]2C5H3N) (7-(H)2BPin) was observed. The latter arises from borylation of the meta positions of both phenyl rings in the [PhSiNSi] pincer during catalysis. A trace amount of 3-H2(H2) was also detected at high (74%) substrate conversion, presumably formed from reaction of 1-(H)2BPin with the H2 formed from the borylation of lutidine with the HBPin byproduct.
The independent synthesis of 7-(H)2BPin was pursued from treatment of 1-H3·NaHBEt3 with excess of B2Pin2 in THF in the absence of substrate (Scheme 11). At 25 °C, the major species was 1-(H)2BPin while raising the temperature to 100 °C afforded 7-(H)2BPin as the major product. Attempts to access pure 7-(H)2BPin have been unsuccessful and the borylation of the pincer did not reach completion even when 20 equivalents of B2Pin2 and longer reaction times of 16 h at 100 °C were used. Nevertheless, diagnostic features of 7-(H)2BPin have been identified that signal its formation. The Co–H appears at −11.42 ppm, close to the value of −11.44 ppm observed for 1-(H)2BPin in THF-d8. A diagnostic singlet at 7.92 ppm also appeared at the expense of the Ph signals of 1-(H)2BPin. The fact that this signal was a singlet enabled the assignment as the ortho-H (Ho) of the Ph ring, supporting selective borylation at the meta position.
Scheme 11.

Attempted independent synthesis of 7-(H)2BPin by treatment of 1-H3·NaHBEt3 with B2Pin2.
To further explore the role of C–H activation under catalytically relevant conditions as well as to gain insight into reductive elimination from cobalt(III), 1-(H)2BPin was treated with excess DBPin in THF at 100 °C. A mixture of isotopologues with the general formula [(Ph-dSiNSi)CoDxH2-x(BPin)] (x = 0, 1, 2) was observed as judged by 2H and 1H NMR spectroscopies. Deuterium incorporation was observed in the Co–H bonds as well as on the Ph rings of the [PhSiNSi] ligand (15 % d-incorporation), demonstrating that oxidative addition of the C(sp2)–H bonds of the Ph groups in the SiNSi pincer is operative. In contrast, addition of excess DBPin to 2-(H)2BPin under the same conditions produced a mixture of isotopologues with the general formula [(ptolSiNSi)CoDxH2-x(BPin)] (x = 0, 1, 2). Notably, deuterium was not observed into the p-tolyl substituents of the pincer ligand, a result of the steric hindrance imposed by the methyl groups in the para position that inhibits oxidative addition.
Competing borylation of the 4-position of the pyridyl ring in the pincer was also observed in the catalytic borylation of 2,6-lutidine with B2Pin2 with [(iPrPNP)CoCH2SiMe3].14 Notably, the borylation of the pincer was faster than the substrate and deactivated the cobalt toward turnover limiting C–H oxidative addition. To determine the effect of pincer borylation on the catalytic performance of (PhSiNSi)-based cobalt catalysts, the borylation of 2,6-lutidine with 2-H3·NaHBEt3 or 2-(H)2BPin was carried out as the para-methyl groups on the aryl rings prevent C–H borylation. Monitoring the catalytic reaction by 1H NMR spectroscopy in THF-d8 at 100 °C established 2-(H)2BPin as the exclusive cobalt resting state confirming that borylation of the phenyl rings did not take place (Scheme 12). This result is also consistent with the lack of observable deuterium incorporation in stoichiometric experiments with DBPin.
Scheme 12.

(a) Catalyst resting-state for the borylation of 2,6-lutidine employing 2-H3·NaHBEt3 as precatalyst. (b) aromatic region of the 1H NMR spectrum after 5 minutes of reaction (5% substrate conversion) and after 11 hours and 30 minutes of reaction (62% substrate conversion) showing signals of 2-(H)2BPin.
Comparing the rate of disappearance of 2,6-lutidine in the borylation with B2Pin2 and either 1-H3·NaHBEt3 or 2-H3·NaHBEt3 established that the reaction was approximately 1.4 times faster with the former (Figure S103). This result demonstrates that formation of 7-(H)2BPin for pincer borylation has little discernable effect on the rate of the catalytic reaction. However, integration of the hydride signal of 2-H2BPin against the mesitylene internal standard revealed that deactivation occurred during turnover (see Figure S102) but the origin of this deactivation process has not been identified.
Having identified pincer borylation with well-defined cobalt precatalysts and obtaining the appropriate spectroscopic signatures raised the question as to whether this process occurs with in-situ generated catalysts. Monitoring the catalytic borylation of 2,6-lutidine with B2Pin2 using 5 mol% of (PhSiNSi)CoCl2 activated with 2 equiv of NaHBEt3 by 1H NMR spectroscopy revealed formation of 1-(H)2BPin and 7-(H)2BPin, supporting access to the same catalytic cycle as with the well-defined 1-H3·NaHBEt3 precatalyst. The same catalyst resting-states were observed when the borylation was initiated from 1-H3·NaHBEt3 in the presence of a stoichiometric amount of cyclohexene under the same conditions again supporting a similar catalytic cycle. However, in this case, the reaction was found to be 2.4 times faster than in the absence of cyclohexene (Figure S106). In addition, a signal at 1.44 ppm (THF-d8) was observed corresponding to cyclohexane that increased as the catalytic reaction progressed. These observations support the role of cyclohexene as a H2 acceptor that likely facilitates generation of the cobalt(I) complex responsible for C–H activation.29 When the borylation of 2,6-lutidine with B2Pin2 was carried out with 2-(H)2BPin and in the presence of one equivalent of cyclohexene, >98% substrate conversion was observed also consistent with reductive elimination from cobalt(III).
To further support the role of cyclohexene as a hydrogen acceptor, excess cyclohexene (6 equivalents) was added to a solution of 1-H3·NaHBEt3 in benzene-d6 and the reaction was monitored by 1H NMR spectroscopy at 25°C with mesitylene as an internal standard. Formation of cyclohexane was observed and the concentration of 1-H3·NaHBEt3 decreased over time consistent with hydrogen transfer from cobalt(III) to form a putative cobalt(I) complex that is unstable preventing observation.
CONCLUSIONS
In summary, well-defined cobalt(III) precatalysts with an electron-donating bis(silylene)pyridine pincer have been synthesized and structurally characterized. These complexes proved effective single-component precatalysts for the borylation of furans and 2,6-lutidine. These compounds were also identified from in-situ catalyst activation procedures, supporting common active species using both procedures. While the strong electron donating properties of the pincer precluded synthesis or observation of cobalt(I) derivatives, a Co(I)-Co(III) redox cycle is plausible for catalysis although steps where the pincer undergoes reversible modification are also possible. Support for this conclusion derives from a modest rate acceleration upon addition of cyclohexene, which serves as a hydrogen acceptor presumably facilitating generation of cobalt(I) intermediates.
Pincer modification processes were also identified that were competitive with catalytic turnover and were observed at higher conversions of substrate. When HBPin was used as the borylating agent for the functionalization of furans, the stoichiometric H2 byproduct proved deleterious, prompting a hydride migration from the cobalt to an imine position of the pincer. The resulting cobalt complex proved less active cobalt σ-H2 complex. For the borylation 2,6-lutidine with B2Pin2, borylation of the phenyl rings on the pincer was observed with little effect on catalytic activity. The results of these studies highlight the consequences of introducing a more electron rich pincer. These insights should prove useful for the design of next generation catalysts with improved performance.
Supplementary Material
ACKNOWLEDGMENTS
Financial support was provided by the NIH (5R01GM121441). R. A. acknowledges the Principado de Asturias-FICYT and the European Union for a Marie Curie Clarín-COFUND fellowship. We thank Dr. Istvan Pelczer and Kenith Conover (Princeton University) for the assistance with the acquisition of T1 values and AllyChem for a gift of B2Pin2.
Footnotes
The authors declare no competing financial interests.
The Supporting Information is available free of charge at, https://doi.org/10.1021/acs.organomet.0c00382.
Accession Codes
CCDC 2008090, 2008091, 2008092 and 2008093, contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.uc.uk/data_request/cif or by emailing data_request@ccdc.cam.ac.uk or by contacting the The Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB2 1EZ UK fax: + 44 1223 336033.
REFERENCES
- 1.(a) Hall DG; Boronic acids; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]; (b) Mkhalid IAI; Barnard JH; Marder TB; Murphy JM; Hartwig JF C−H Activation for the Construction of C−B Bonds. Chem. Rev 2010, 110, 890–931. [DOI] [PubMed] [Google Scholar]; (c) Hartwig JF Borylation and Silylation of C–H Bonds: a Platform for Diverse C–H Bond Functionalizations. Acc. Chem. Res 2012, 45, 864–873. [DOI] [PubMed] [Google Scholar]
- 2.(a) Cho J-Y; Tse MK; Holmes D; Maleczka RE; Smith MR Remarkably Selective Iridium Catalysts for the Elaboration of Aromatic C-H Bonds. Science 2002, 295, 305–308. [DOI] [PubMed] [Google Scholar]; (b) Ishiyama T; Takagi J; Ishida K; Miyaura N; Anastasi NR; Hartwig JF Mild Iridium-Catalyzed Borylation of Arenes. High Turnover Numbers, Room Temperature Reactions, and Isolation of a Potential Intermediate. J. Am. Chem. Soc 2002, 124, 390–391. [DOI] [PubMed] [Google Scholar]
- 3.(a) Boller TM; Murphy JM; Hapke M; Ishiyama T; Miyaura N; Hartwig JF Mechanism of the Mild Functionalization of Arenes by Diboron Reagents Catalyzed by Iridium Complexes. Intermediacy and Chemistry of Bipyridine-Ligated Iridium Trisboryl Complexes. J. Am. Chem. Soc 2005, 127, 14263–14278. [DOI] [PubMed] [Google Scholar]; (b) Tamura H; Yamazaki H; Sato H; Sakaki S Iridium-Catalyzed Borylation of Benzene with Diboron. Theoretical Elucidation of Catalytic Cycle Including Unusual Iridium(v) Intermediate. J. Am. Chem. Soc 2003, 125, 16114–16126. [DOI] [PubMed] [Google Scholar]; (c) Green AG; Liu P; Merlic CA; Houk KN Distortion/Interaction Analysis Reveals the Origins of Selectivities in Iridium-Catalyzed C–H Borylation of Substituted Arenes and 5-Membered Heterocycles. J. Am. Chem. Soc 2014, 136, 4575–4583. [DOI] [PubMed] [Google Scholar]
- 4.(a) Genov GR; Douthwaite JL; Lahdenperä ASK; Gibson DC; Phipps RJ Enantioselective remote C–H activation directed by a chiral cation. Science 2020, 367, 1246–1251. [DOI] [PubMed] [Google Scholar]; (b) Kuninobu Y; Ida H; Nishi M; Kanai M A meta-selective CH borylation directed by a secondary interaction between ligand and substrate. Nat. Chem 2015, 7, 712–717. [DOI] [PubMed] [Google Scholar]; (c) Lu X; Yoshigoe Y; Ida H; Nishi M; Kanai M; Kuninobu Y Hydrogen Bond-Accelerated meta-Selective C-H Borylation of Aromatic Compounds and Expression of Functional Group and Substrate Specificities. ACS Catal. 2019, 9, 1705–1709. [Google Scholar]; (d) Saito Y; Yamanoue K; Segawa Y; Itami K Selective Transfor-mation of Strychnine and 1,2-Disubstituted Benzenes by C–H Borylation. Chem 2020, 6, 1–9. [Google Scholar]
- 5.(a) Rouquet G; Chatani N Catalytic functionalization of C(sp2)–H and C(sp3)–H bonds using bidentate directing groups. Angew. Chem. Int. Ed 2013, 52, 11726–11743. [DOI] [PubMed] [Google Scholar]; (b) Ros A; Fernández R; Lassaletta JM Functional group directed C–H borylation. Chem. Soc. Rev, 2014, 43, 3229–3243. [DOI] [PubMed] [Google Scholar]; (c) Boebel TA; Hartwig JF Silyl-Directed, Iridium-Catalyzed ortho-Borylation of Arenes. A One-Pot ortho-Borylation of Phenols, Arylamines, and Alkylarenes. J. Am. Chem. Soc 2008, 130, 7534–7535. [DOI] [PubMed] [Google Scholar]; (d) Montero Bastidas JR; Oleskey TJ; Miller SL; Smith MR; Maleczka RE Para-Selective, Iridium-Catalyzed C−H Borylations of Sulfated Phenols, Benzyl Alcohols, and Anilines Directed by Ion-Pair Electrostatic Interactions. J. Am. Chem. Soc 2019, 141, 15483–15487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Hoque ME; Bisht R; Haldar C; Chattopadhyay B Noncovalent Interactions in Ir-Catalyzed C−H Activation: L-Shaped Ligand for Para-Selective Borylation of Aromatic Esters. J. Am. Chem. Soc 2017, 139, 7745–7748. [DOI] [PubMed] [Google Scholar]; (b) Smith MR; Bisht R; Haldar C; Pandey G; Dannatt JE; Ghaffari B; Maleczka RE; Chattopadhyay B Noncovalent Interactions in Ir-Catalyzed C−H Activation: L Shaped Ligand for Para-Selective Borylation of Aromatic Esters. ACS Catal. 2018, 8, 6216–6223.30147990 [Google Scholar]; (c) Saito Y; Segawa Y; Itami K para-C–H Borylation of Benzene Derivatives by a Bulky Iridium Catalyst. J. Am. Chem. Soc 2015, 137, 5193–5198. [DOI] [PubMed] [Google Scholar]; (d) Yang L; Semba K; Nakao Y para-Selective C–H Borylation of (Hetero)Arenes by Cooperative Iridium/Aluminum Catalysis. Angew. Chem. Int. Ed 2017, 56, 4853–4857. [DOI] [PubMed] [Google Scholar]
- 7.(a) Dombray T; Werncke CG; Jiang S; Grellier M; Vendier L; Bontemps S; Sortais J-B; Sabo-Etienne S; Darcel C Iron-Catalyzed C–H Borylation of Arenes. J. Am. Chem. Soc 2015, 137, 4062–4065. [DOI] [PubMed] [Google Scholar]; (b) Waltz KM; He X; Muhoro C; Hartwig JF Hydrocarbon Functionalization by Transition Metal Boryls. J. Am. Chem. Soc 1995, 117, 11357–11358. [Google Scholar]; (c) Waltz KM; Hartwig JF Selective Functionalization of Alkanes by Transition-Metal Boryl Complexes. Science 1997, 277, 211–213. [Google Scholar]; (d) Waltz KM; Muhoro CN; Hartwig JF C−H Activation and Functionalization of Unsaturated Hydrocarbons by Transition-Metal Boryl Complexes. Organometallics 1999, 18, 3383–3393. [Google Scholar]; (e) Waltz KM; Hartwig JF Functionalization of Alkanes by Isolated Transition Metal Boryl Complexes. J. Am. Chem. Soc 2000, 122, 11358–11369. [Google Scholar]; (f) Mazzacano TJ; Mankad NP Thermal C-H Borylation Using a CO-Free Iron Boryl Complex. Chem. Commun 2015, 51, 5379–5382. [DOI] [PubMed] [Google Scholar]; (g) Hatanaka T; Ohki Y; Tatsumi K C-H Bond Activation/Borylation of Furans and Thiophenes Catalyzed by a Half-Sandwich Iron N-Heterocyclic Carbene Complex. Chem. Asian J 2010, 5, 1657–1666. [DOI] [PubMed] [Google Scholar]; (h) Mazzacano TJ; Mankad NP Base Metal Catalysts for Photochemical C–H Borylation That Utilize Metal–Metal Cooperativity. J. Am. Chem. Soc 2013, 135, 17258–17261. [DOI] [PubMed] [Google Scholar]; (i) Parmelee SR; Mazzacano TJ; Zhu Y; Mankad NP; Keith JA A Heterobimetallic Mechanism for C–H Borylation Elucidated From Experimental and Computational Data. ACS Catalysis 2015, 5, 3689–3699. [Google Scholar]; (j) Kato T; Kuriyama S; Nakajima K; Nishibayashi Y Catalytic C-H Borylation Using Iron Complexes Bearing 4,5,6,7-Tetrahydroisoindol-2-ide-Based PNP-Type Pincer Ligand. Chem. Asian J 2019, 14, 2097–2101. [DOI] [PubMed] [Google Scholar]
- 8.(a) Obligacion JV; Semproni SP; Chirik PJ Cobalt-Catalyzed C–H Borylation. J. Am. Chem. Soc 2014, 136, 4133–4136. [DOI] [PubMed] [Google Scholar]; (b) Léonard NG; Bezdek MJ; Chirik PJ Cobalt-Catalyzed C(sp2)–H Borylation with an Air-Stable, Readily Prepared Terpyridine Cobalt(II) Bis(Acetate) Precatalyst. Organometallics 2017, 36, 142–150. [Google Scholar]; (c) Palme WN; Obligacion JV; Pappas I; Chirik PJ Cobalt-Catalyzed Benzylic Borylation: Enabling Polyborylation and Functionalization of Remote, Unactivated C(sp3)–H Bonds. J. Am. Chem. Soc 2016, 138, 766–769. [DOI] [PubMed] [Google Scholar]; (d) Jayasundara CRK; Sabasovs D; Staples RJ; Oppenheimer J; Smith MRIII, Maleczka RE Cobalt-catalyzed C-H borylation of alkyl arenes and heteroarenes including the first selective borylations of secondary benzylic C-H bonds. Organometallics 2018, 37, 1567–1574. [Google Scholar]
- 9.(a) Palmer WN; Zarate C; Chirik PJ Benzyltriboronates: Building Blocks for Diastereoselective Carbon–Carbon Bond Formation. J. Am. Chem. Soc 2017, 139, 2589–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Furukawa T; Tobisu M; Chatani N Nickel-Catalyzed Borylation of Arenes and Indoles via C–H Bond Cleavage. Chem. Commun, 2015, 51, 6508–6511. [DOI] [PubMed] [Google Scholar]; (c) Zhang H; Hagihara S; Itami K Aromatic C–H Borylation by Nickel Catalysis Chem. Lett. 2015, 44, 779–781. [Google Scholar]; (d) Das A;Hota PK; Mandal SK Nickel-Catalyzed C(sp2)–H Borylation of Arenes. Organometallics 2019, 38, 3286–3293. [Google Scholar]
- 10.(a) Yu RP; Hesk D; Rivera N; Pelczer I; Chirik PJ Iron-catalyzed tritiation of pharmaceuticals. Nature 2016, 529, 195–199. [DOI] [PubMed] [Google Scholar]; (b) Zarate C; Yang H; Bezdek MJ; Hesk D; Chirik PJ Ni(I)–X Complexes Bearing a Bulky alpha-Diimine Ligand: Synthesis, Structure and Superior Catalytic Performance in the Hydrogen Isotope Exchange of Pharmaceuticals. J. Am. Chem. Soc 2019, 141, 5034–5044. [DOI] [PubMed] [Google Scholar]; (c) Obligacion JV; Bezdek MJ; Chirik PJ C(sp2)–H Borylation of Fluorinated Arenes Using an Air-Stable Cobalt Precatalyst: Electronically Enhanced Site Selectivity Enables Synthetic Opportunities. J. Am. Chem. Soc 2017, 139, 2825–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arevalo R; Chirik PJ Enabling Two-Electron Pathways with Iron and Cobalt: From Ligand Design to Catalytic Applications. J. Am. Chem. Soc 2019, 141, 9106–9123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Semproni SP; Hojilla Atienza CC; Chirik PJ Oxidative Addition and C–H Activation Chemistry with a PNP Pincer-Ligated Cobalt Complex. Chem. Sci 2014, 5, 1956–1960. [Google Scholar]
- 13.Pabst TP; Obligacion JV; Rochette E; Pappas I; Chirik PJ Cobalt-Catalyzed Borylation of Fluorinated Arenes: Thermodynamic Control of C(sp2)-H Oxidative Addition Results in ortho-to-Fluorine Selectivity. J. Am. Chem. Soc 2019, 141, 15378–15389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pabst TP; Obligacion JV; Rochette E; Pappas I; Chirik PJ Cobalt-Catalyzed Borylation of Fluorinated Arenes: Thermodynamic Control of C(sp2)-H Oxidative Addition Results in ortho-to-Fluorine Selectivity. J. Am. Chem. Soc 2019, 141, 15378–15389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Obligacion JV; Chirik PJ Mechanistic Studies of Cobalt-Catalyzed C(sp2)–H Borylation of Five-Membered Heteroarenes with Pinacolborane. ACS Catal. 2017, 7, 4366–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H; Obligacion JV; Chirik PJ; Hall MB Cobalt Pincer Complexes in Catalytic C–H Borylation: the Pincer Ligand Flips Rather Than Dearomatizes. ACS Catal. 2018, 8, 10606–10618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schaefer BA; Margulieux GW; Small BL; Chirik PJ Evaluation of Cobalt Complexes Bearing Tridentate Pincer Ligands for Catalytic C–H Borylation. Organometallics 2015, 34, 1307–1320. [Google Scholar]
- 18.Ren H; Zhou Y-P; Bai Y; Cui C; Driess M Cobalt-Catalyzed Regioselective Borylation of Arenes: N-Heterocyclic Silylene as an Electron Donor in the Metal-Mediated Activation of C−H Bonds. Chem. Eur. J 2017, 23, 5663–5667. [DOI] [PubMed] [Google Scholar]
- 19.(a) Brueck A; Gallego D; Wang W; Irran E; Driess M; Hartwig JF Pushing the σ-donor strength in iridium pincer complexes: bis(silylene) and bis(germylene) ligands are stronger donors than bis(phosphorus(III)) ligands. Angew. Chem. Int. Ed 2012, 51, 11478–11482. [DOI] [PubMed] [Google Scholar]; (b) Yuwen Wang; Arseni Kostenko; Shenglai Yao; Matthias Driess. Divalent Silicon-Assisted Activation of Dihydrogen in a Bis(N-heterocyclic silylene)xanthene Nickel(0) Complex for Efficient Catalytic Hydrogenation of Olefins. J. Am. Chem. Soc 2017, 139, 13499–13506. [DOI] [PubMed] [Google Scholar]; (c) Benedek Z; Szilvasi T Can low-valent silicon compounds be better transition metal ligands than phosphines and NHCs? RSC Adv. 2015, 5, 5077–5086. [Google Scholar]; (d) Inoue S The Next Generation of Silylene Ligands for Better Catalysts Discovering the Future of Molecular Sciences, pp. 243–273. Ed. Pignataro B. Wiley-VCH Verlag GmbH & Co; KGaA Weinheim, Germany, 2014. [Google Scholar]; (e) Luecke M-P; Porwal D; Kostenko A; Zhou Y-P; Yao S; Keck M; Limberg C; Oestreich M; Driess M.Bis(silylenyl)-substituted ferrocene-stabilized η6-arene iron(0) complexes: synthesis, structure and catalytic application. Dalton Trans. 2017, 46, 16412–16418. [DOI] [PubMed] [Google Scholar]; (f) Wang W; Inoue S; Irran E; Driess M Synthesis and Unexpected Coordination of a Silicon(II)-Based SiCSi Pincerlike Arene to Palladium. Angew. Chem. Int. Ed 2012, 51, 3691–3694. [DOI] [PubMed] [Google Scholar]; (g) Wang W; Inoue S; Enthaler S; Driess M Bis(silylenyl)- and Bis(germylenyl)-Substituted Ferrocenes: Synthesis, Structure, and Catalytic Applications of Bidentate Silicon(II)-Cobalt Complexes. Angew. Chem. Int. Ed 2012, 51, 6167–6171. [DOI] [PubMed] [Google Scholar]; (h) Gallego D; Brueck A; Irran E; Meier F; Kaupp M; Driess M; Hartwig JF From Bis(silylene) and Bis(germylene) Pincer-Type Nickel(II) Complexes to Isolable Intermediates of the Nickel-Catalyzed Sonogashira Cross-Coupling Reaction. J. Am. Chem. Soc 2013, 135, 15617–15626. [DOI] [PubMed] [Google Scholar]
- 20.Zhou Y-P; Mo Z; Luecke M-P;Driess M. Stereoselective Transfer Semi-Hydrogenation of Alkynes to E-Olefins with N-Heterocyclic Silylene-Manganese Catalysts. Chem. Eur. J 2018, 24, 4780–4784. [DOI] [PubMed] [Google Scholar]
- 21.(a) Gallego D; Inoue S; Blom B; Driess M Highly electron-rich pincer-type iron complexes bearing innocent bis(metallylene)pyridine ligands: syntheses, structures, and catalytic activity. Organometallics 2014, 33, 6885–6897. [Google Scholar]; (b) Metsaenen TT; Gallego D; Szilvasi T; Driess M; Oestreich M Peripheral mechanism of a carbonyl hydrosilylation catalysed by an SiNSi iron pincer complex. Chem. Sci 2015, 6, 7143–7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tokmic K; Markus CR; Zhu L; Fout AR Well-Defined Cobalt(I) Dihydrogen Catalyst: Experimental Evidence for a Co(I)/Co(III) Redox Process in Olefin Hydrogenation. J. Am. Chem. Soc 2016, 138, 11907–11913. [DOI] [PubMed] [Google Scholar]
- 23.Semproni SP; Milsmann C; Chirik PJ Four-Coordinate Cobalt Pincer Complexes: Electronic Structure Studies and Ligand Modification by Homolytic and Heterolytic Pathways. J. Am. Chem. Soc 2014, 136, 9211–9224. [DOI] [PubMed] [Google Scholar]
- 24.(a) Kubas GJ Metal Dihydrogen and σ-Bond Complexes: Structure, Theory and Reactivity. Springer, New York, 2001. [Google Scholar]; (b) Crabtree RH Dihydrogen complexes & related sigma complexes. Encyclopedia of Inorganic and Bioinorganic Chemistry 2011, 1–6. [Google Scholar]
- 25.Hebden TJ; St. John AJ; Gusev DG; Kaminsky W; Goldberg KI; Heinekey DM Preparation of a Dihydrogen Complex of Cobalt. Angew. Chem. Int. Ed 2011, 50, 1873–1876. [DOI] [PubMed] [Google Scholar]
- 26.(a) Kundu S, Brennessel WW, Jones WD. Synthesis and Reactivity of New Ni, Pd, and Pt 2,6-Bis(di-tert-butylphosphinito)pyridine Pincer Complexes. Inorg. Chem 2011, 50, 9443–9453. [DOI] [PubMed] [Google Scholar]; (b) Berno P, Gambarotta S. Reactivity of a Four-Membered Vanadacycle Ring Supported by Bulky Silazanate. Regioselective Hydrogenation of Pyridine. Organometallics 1994, 13, 2569–2571. [Google Scholar]
- 27.(a) Scott J, Gambarotta S, Korovkov I, Budzelaar PHM Metal versus Ligand Alkylation in the Reactivity of the (Bis-iminopyridinato)Fe Catalyst. J. Am. Chem. Soc 2005, 127, 13019–13029; [DOI] [PubMed] [Google Scholar]; (b) Sugiyama H, Aharonian G, Gambarotta S, Yap GPA, Budzelaar PHM. Participation of the α,α’-Diiminopyridine Ligand System in Reduction of the Metal Center during Alkylation. J. Am. Chem. Soc 2002, 124, 12268–12274; [DOI] [PubMed] [Google Scholar]; (c) Reardon D, Conan F, Gambarotta S, Yap G, Wang Q. Life and Death of an Active Ethylene Polymerization Catalyst. Ligand Involvement in Catalyst Activation and Deactivation. Isolation and Characterization of Two Unprecedented Neutral and Anionic Vanadium(I) Alkyls. J. Am. Chem. Soc 1999, 121, 9318–9325. [Google Scholar]
- 28. The IR νCO and νSiH of 3-(CO)2 are in the range of those reported for the cobalt(I) complex containing an anionic PSiP pincer ligand [(PSiP)Co(CO)2]. See.; Zhang J; Foley BJ; Bhuvanesh N; Zhou J; Janzen DE; Whited MT; Ozerov OV Organometallics 2018, 37, 3956–3962. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.