

Abstract
Combining alignment-free methods for phylogenetic analysis with multi-regional sampling using next-generation sequencing can provide an assessment of intra-patient tumour heterogeneity. From multi-regional sampling divergent branching, we validated two different lesions within a patient’s prostate. Where multi-regional sampling has not been used, a single sample from one of these areas could misguide as to which drugs or therapies would best benefit this patient, due to the fact these tumours appear to be genetically different. This application has the power to render, in a fraction of the time used by other approaches, intra-patient heterogeneity and decipher aberrant biomarkers. Another alignment-free method for calling single-nucleotide variants from raw next-generation sequencing samples has determined possible variants and genomic locations that may be able to characterize the differences between the two main branching patterns. Alignment-free approaches have been applied to relevant clinical multi-regional samples and may be considered as a valuable option for comparing and determining heterogeneity to help deliver personalized medicine through more robust efforts in identifying targetable pathways and therapeutic strategies. Our study highlights the application these tools could have on patient-aligned treatment indications.
INTRODUCTION
Tumour heterogeneity and its complexity can now be explored in more detail, thanks to advances in technology within genomics and sequencing (1). Key functional genetic roles in the progression of prostate cancer can be highly affected by interfocal heterogeneity, where mutational heterogeneity can be high across different sites within the same patient (2). Such tumours that display heterogeneous traits impede, and ultimately fail, in our capabilities to deliver treatment options for optimal clinical care. Studies using exome (3,4) and multi-region sequencing on multiple different types of cancers have revealed intra-tumour heterogeneity within each (3–11). Examples of this include human clear cell renal cell carcinomas’ spatial heterogeneity, where two-thirds of non-synonymous somatic mutations across different regions could not be seen in all biopsies (12). Spatial variations within patient samples could hinder delivering personalized medicine or developing biomarkers in the future. Treatment decisions for metastatic disease are often influenced by information from the original primary tumour, which again highlights the need to overcome the issues that heterogeneity presents.
Other scenarios where tumours are genetically distinct would rely on the need for multi-regional sampling over biopsies. The reconstruction of phylogenetic trees can be achieved through the inclusion of the tumours’ full mutational landscape that can begin to unravel explanations of treatment resistance, relapse and metastatic disease (13). Popular techniques for comparative sequence analysis and phylogenetic tree reconstruction tend to follow alignment-based methods (14,15). These methods are focused towards purpose-built programs that can take sequencing reads from sequencing technologies and align them towards already pre-defined reference genomes to determine which are part of the target genome (16). Alignment-based techniques provide accurate results when a study can be reliably and well aligned to its reference genome; however, drawbacks include diverging sequence or unreliable alignment where vital information may be lost (17). Other issues include tools that lack speed and may not be appropriate for some computer systems or large next-generation sequencing (NGS) studies. Most NGS data are commonly used with short read sequencers, presenting issues in the alignment process. Sequence comparison without the use of alignment methods has the potential to overcome these issues. Alignment-free techniques, in which shared properties of sub-sequences or k-mers are extracted to determine distance matrices, have previously been used in phylogenetic studies (18). Alignment-free analysis has been used across different areas, highlighting its strength within NGS analysis that can be seen across various studies (19).
Alignment-free methods with phylogenetic analysis have shown promising results in the assessment of spatiotemporal heterogeneity in NGS cancer datasets (20). The NUQA (NGS tool for Unsupervised analysis of fastQ using Alignment-free) tool can produce different phylogenetic trees from the same patient data when compared to alignment-based approaches, potentially highlighting key sequence that may have been missed in alignment methods that are influencing the mutational landscape of these tumours. To complement this, other alignment-free techniques can count the number of unique k-mers in raw sequencing to infer genotypes of known variants (21). FastGT identifies variants with speed on basic computer systems.
In this study, we aim to utilize multi-regional sampling from prostate cancer patients following prostatectomy, where their associated whole exome sequencing (WES) profiles were captured (22). From here, we will apply their raw sequencing reads for alignment-free phylogenetic analysis from NUQA to compare intra-tumour heterogeneity, as well as calling variants from FastGT to highlight their locations in order to determine why intra-tumour heterogeneity could potentially be seen for any of these patients. By visualizing these locations using the Integrative Genomics Viewer (IGV) (23) through aligning these FASTQ files with the Burrows–Wheeler Aligner (24), we can demonstrate evidence as differential read build-up using analysis from R Studio. This could potentially allow for the application of alignment-free techniques to translate towards use within clinical practice in the future due to its much faster processes to complete analysis and its highlighted abilities over traditional approaches.
MATERIALS AND METHODS
Details of all the relevant sample collection and sequencing procedures for the prostate cancer patient multi-regional sampling study can be found in the original manuscript (22). Regarding those most relevant to this study, six prostate cancer patients had their whole genetic profile captured from their whole prostate glands resulting in 43 prostate cores taken in total, 22 of these being tumour samples and 21 of these being tumour adjacent samples. Five of these patients also had their circulating free and germline DNA assessed from their blood, which we used as a control normal core for these patients. All raw FASTQ WES samples corresponding to each patient multi-regional sampling core were quality controlled, pre-processed and analysed using the same pipeline. FastQC was used to perform QC on the raw sequence to highlight any potential issues with the data quality. MultiQC created single reports for all the patient cores to better visualize and compare FastQC outputs (25). Trimmomatic ensured the best quality raw sequence input to allow NUQA to produce the most reliable and relevant phylogenetic trees (26). Default paired-end parameters were used when using Trimmomatic: removing Illumina TruSeq3 paired-end adapters, reads below 36 bases long and reads with a phred score below 33 to retain only the high-quality score reads. Clumpify from BBMap was used for deduplication of possible duplicated reads to again allow for the best quality input for NUQA. Again, default paired-end parameters were used to remove normal optical duplicates from Illumina sequencers. All the samples for each core were then merged to create a single FASTQ file for each core. FastQC and MultiQC were again applied for QC following the pre-processing pipeline to validate the removal of any potential issues. Each of the core’s FASTQ files were decompressed and used as an input for NUQA to create a tree of cores for each patient. NUQA’s input parameters used default k-mer length 21 and the Jensen–Shannon divergence distance metric. The shell script generated to perform our pre-processing and analysis pipeline on the raw FASTQ files can be seen in Supplementary Data S1. The resulting NUQA output in Newick tree format is used as input for the Interactive Tree Of Life (iTOL) for visualizing the phylogenetic tree analysis (27).
For patient case 1, eight cores were taken in total, as well as their associated circulating free and germline DNA assessed from their blood as a control normal core. Four of these cores were tumour samples, and the remaining four were tumour adjacent samples. For this patient’s cores, the FASTQ files were also used as input for FastGT to call possible variants on each of these cores using alignment-free methods. We used the exome k-mer database as our input was WES and followed the standard genotyping procedure to count matching k-mers and call their genotype for each possible variant on the database. Using RStudio, we loaded each FastGT calls’ output file corresponding to each of patient case 1’s multi-regional cores into our session and took reference and alternative allele calls to build data frames for each core. From here, we applied variance filtering to keep the top 20% most variant calls across all the cores, using the varFilter function in the genefilter Bioconductor package (28). Scaling of the data frames was performed as a method of normalization among the call counts. Branching groups seen from the phylogenetic tree analysis were created, such as the top and bottom branching groups seen from patient case 1, to allow for t-tests to be performed among these groups for the variant alleles that had been called to determine those that were statistically significant (those with a two-sided P-value of ≤0.05) against these groups. The biomaRt Bioconductor package allowed for these variants to be annotated to their corresponding genes and those without a gene annotation were removed from the data frames (29). For genes with multiple variant results for the given gene, the mean of the variants with annotation duplicates was taken for each core sample to create an averaged single representation of the gene. Hierarchical clustering heat maps were created using the pheatmap R package where the variant call counts were standardized through the mean of counts for each variant against the standard deviation of counts for each variant. The RColorBrewer R package was used to help create the colour palette for visualizing the heat map. These statistically significant calls were finally grouped as a data frame to make comparisons as to how each variant call was made for each given genomic location or single-nucleotide variant (SNV) position for each core in patient case 1. This would allow us to identify interesting locations that had different genotype calls among different cores.
RESULTS
Phylogenetic analysis with NUQA
The NUQA analysis on patient case 1 displayed a tree depicting a divergent pattern. Areas within the prostate, from which the samples were taken (Figure 1A), associate strongly with patient case 1’s phylogenetic tree (Figure 1B and C). The right-hand side of the prostate sample (left-hand side of the image) was defined as having one distinct tumour lesion where tumour cores 1 and 2 and tumour adjacent cores 3 and 4 have been punch biopsied. The left-hand side of the prostate sample (right-hand side of the image) was also defined as having one distinct tumour lesion where tumour cores 5 and 6 and tumour adjacent cores 7 and 8 have been punch biopsied. Tumour cores are punch biopsies from within the tumour lesion, whereas tumour adjacent cores are punch biopsies outside of the tumour lesion. The two branching patterns, stemming from the control blood WES sample, map strongly to the sampling locations and, more subtlety, indicate whether samples were tumour or tumour adjacent. The top branch of NUQA’s phylogenetic tree maps with the four punch biopsies seen on the right-hand side of the prostate (left-hand side of the image), whereas the bottom branch maps with the four punch biopsies seen on the opposite side of the prostate.
Figure 1.
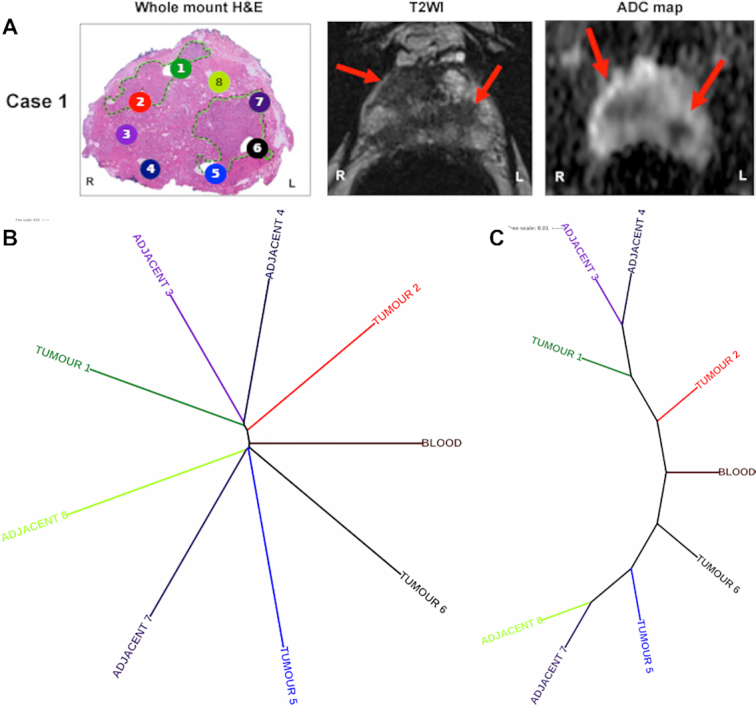
Multi-regional sampling WES to asses intra-patient heterogeneity with NUQA. (A) Patient case 1’s coloured cores in their haematoxylin and eosin whole-mount sections [adapted from (22)], as well as their associated axial T2-weighted image and apparent diffusion coefficient maps that align with the phylogenetic tree from NUQA. The patient’s magnetic resonance imaging (MRI) scans show visible tumour sites on both sides of the prostate, indicated by red arrows as annotations. (B) Phylogenetic tree from NUQA that adheres to branch lengths of the tree to show the calculated distance between samples, and the tree produced when these branch lengths are ignored to highlight their ordering and clustering (C).
Variant calling with FastGT
Using this evidence to employ FastGT for patient case 1, we can identify variant calls against the two main branches seen due to the differential build-up of k-mers (Figure 2). FastGT’s output included a median value of 26 for all the calls on these different k-mer genomic locations, as well as a median value of 33 for all the calls on the top divergent branching pattern and a median value of 20 for all the calls on the opposite divergent branching pattern. Variance filtering resulted in retaining 141,760 SNVs with a median value for all the calls increasing to 41, the median value for all the calls on the top divergent branching pattern cores increasing to 52 and the median value for all the calls on the opposite divergent branching pattern cores increasing to 29. Scaling the whole data set and filtering these k-mer genomic locations using t-tests resulted in 7688 statistically significant SNV positions. Multiple comparisons were not made on these P-values due to the large amount of input SNVs, which made retaining several statistically significant SNVs difficult. This median value for the filtered data set was 0.73, where the top branching pattern cores had a median value of 0.82 and the opposite branching pattern cores had a median value of 0.62. The annotation of SNVs to their associated genes further reduced the data set to 248 statistically significant genomic locations, with a further decreased median value of 0.72 for the whole data set and decreased median values of 0.81 and 0.61 for both the top and bottom branching pattern cores, respectively. Averaging the values of SNVs annotated to the same gene produced a final 183 gene annotations representing the 248 statistically significant genomic locations. The median value increased to 0.72 for the final data set, while also increasing the median values for the top and bottom branching pattern cores to 0.84 and 0.62, respectively.
Figure 2.

Heat map of variant calls from patient case 1’s cores using FastGT. The heat map's dendrogram shows the two main branching patterns from NUQA’s phylogenetic tree, as well as the two tumour and tumour adjacent samples being more related to each other for each of the branches. Gene annotations associated with each of the variant calls in the heat map, in order from top to bottom, can be found in Supplementary Tables S3 and S4. High expression changes are shown in red, low expression changes are shown in blue and little expression changes are shown in yellow.
The vast majority of statistically significant genomic locations had reference genotype calls as genotype AA across all of the eight cores, with the exception that there were calls across the cores that failed to determine the correct genotype for the given genomic location and thus registered as a non-call, as well as three locations that had cores that had alternative genotype calls within them. These three locations were within the area around SNVs rs12917189, rs60000174 and rs6179. These SNVs were related to the genes CDAN1, APOL4 and GHR, respectively. All cores across APOL4 had both a reference and an alternative allele call as genotype AB, whereas all cores across GHR had alternative allele calls as genotype BB. One interesting statistically significant candidate was rs12917189, associated with CDAN1. This would be ranked according to where there is a significant difference in the number of k-mers in one branch in comparison with the other for the same variant call, realizing that one core sample did not call this variant where the other cores had. This was selected as our target example to validate with traditional alignment pipelines.
IGV for tumour adjacent core 4 that is seen in the top branch was able to confirm differential abundances of information in this location when contrasted with that of tumour adjacent core 7 that is seen in the bottom branch (see Figure 1A for site number and Figure 3 for IGV). For the genomic location of rs12917189, there were a total of 46 aligned reads for tumour adjacent core 4 in which 27 of these reads contained the alternative C calls and the remaining 19 of these reads contained the reference T call. In comparison with tumour adjacent core 7 below, while some reads contain the blue alternative C call, this only amounts to 4 reads in total out of a total 25 aligned reads in this location.
Figure 3.

IGV visualization of area around CDAN1 for patient case 1. Zoomed-in visualization for the genomic location where SNV rs12917189 is found showing tumour adjacent core 4 (top track) and tumour adjacent core 7 (bottom track).
DISCUSSION
We used NUQA and highlighted its ability to assess patient heterogeneity in each case where prostate MRI showcased two geographically separate tumour masses. In combination with FastGT, the difference in genotyping calls for the highlighted tumour adjacent cores and the statistical significance of this SNV through our analysis have provided evidence that NUQA can detect genetically distinct samples from within the same patient. This has proved a unique opportunity to validate new approaches on modern drives within clinical practice. It enables rapid assessment of tumour heterogeneity and identification of key drivers within samples in a shorter time frame than traditional approaches. In the future, this may improve treatment selection at diagnosis.
We have applied NUQA towards six prostate cancer patients within the original multi-regional sampling study (22). Here, we have focused on patient case 1 due to the complexity within this patient’s resulting phylogenetic tree and the interest around the two main divergent branching patterns seen that align with the prostate sample locations. Regarding the other cases, one other patient also showed an interesting phylogenetic tree mapping towards the locations on the patient prostate, but only contained five cores, limiting the assessment. This patient case can be seen in Supplementary Figure S2. Out of the remaining four patients, the trees produced were more typical of a singular evolving mass. All of the other five patient cases can be found in Supplementary Figures S1–S5, with their associated discussion found in Supplementary Information S1–S6. Each patient case’s clinical and pathological information is displayed in Supplementary Table S1. Additional information for each tumour core in the patient cohort can be seen in Supplementary Table S2. Intra- and inter-tumour heterogeneity remains a clinical challenge in prostate cancer for diagnosis and treatment (30). When analysed for multifocality across the cases in this study, inter-tumour heterogeneity can be observed in the majority of cases where there was divergence between different tumour foci from the same prostate. However, in some of the patients, including case 1, both inter- and intra-tumour heterogeneity can be observed where samples from the same patient tumour are exhibiting some differences. This reflects the variability of prostate cancer observed in the clinic.
We also used matched low-pass WGS for each of the cores of the six patients. For these results, all the phylogenetic trees produced by NUQA demonstrated no intra-patient tumour heterogeneity for any of the cases, despite the WES phylogenetic trees demonstrating otherwise. We believe the phylogenetic trees were produced in this fashion due to the very low coverage seen across the WGS (0.7× coverage), in comparison to the high coverage seen across the WES (54× and greater coverage).
Other potentially relevant SNVs may also exist from our analysis that were not selected due these locations failing to be annotated by their associated gene. Genes of interest involved in prostate cancer were also selected and parsed from the data frames as a separate analysis and visualization to find variants calls associated with the disease (22). The analysis and visualization of the genes of interest followed the previous RStudio analysis pipeline that has been described. In this analysis, we found that there was little evidence of significant variant calls for SNVs associated with these genes across all the cores for patient case 1.
In the precision medicine era, we will care for our patients over extended time periods utilizing multiple data modalities. This digital ‘data lake’ should adopt tools that can demonstrate new insights into an individual’s tumour profile and ultimately start pointing towards mechanisms of treatment resistance or alternative therapies. Alignment-free applications represent such an opportunity and are perfectly placed to harness applications within accelerative computing, artificial intelligence and machine learning to enable this reform.
DATA AVAILABILITY
NUQA is an open-source software that can be accessed from the GitHub repository (https://github.com/ACRoddy/NUQA/).
FastGT is an open-source software and its source code is available in the GitHub repository (https://github.com/bioinfo-ut/GenomeTester4/).
The prostate cancer multi-regional sampling WES cohort was provided by the original data authors on request and cited in (22).
RStudio is an open-source software that can be accessed from their website (http://www.rstudio.com/).
FastQC is an open-source software that can be accessed from their website (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/).
MultiQC is an open-source software that can be accessed from the GitHub repository (https://github.com/ewels/MultiQC/).
Trimmomatic is an open-source analytical package that be accessed from their website (http://www.usadellab.org/cms/?page=trimmomatic).
BBMap is an open-source analytical package that can be accessed from the SourceForge repository (https://sourceforge.net/projects/bbmap/).
The iTOL is an online tool that can be accessed from their website (https://itol.embl.de/).
Bioconductor is an R package that can be installed using R.
genefilter is a Bioconductor R package that can be installed using R and Bioconductor.
biomaRt is a Bioconductor R package that can be installed using R and Bioconductor.
Pheatmap is an R package that can be installed using R.
RColorBrewer is an R package that can be installed using R.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Richard Marais for his role in data provision of the prostate cancer patient multi-regional sampling cohort.
Contributor Information
Ross G Murphy, Movember FASTMAN Centre of Excellence, Patrick G Johnston Centre for Cancer Research, School of Medicine, Dentistry and Biomedical Sciences, Queen’s University Belfast, Belfast BT9 7AE, UK.
Aideen C Roddy, Movember FASTMAN Centre of Excellence, Patrick G Johnston Centre for Cancer Research, School of Medicine, Dentistry and Biomedical Sciences, Queen’s University Belfast, Belfast BT9 7AE, UK.
Shambhavi Srivastava, Movember FASTMAN Centre of Excellence, Patrick G Johnston Centre for Cancer Research, School of Medicine, Dentistry and Biomedical Sciences, Queen’s University Belfast, Belfast BT9 7AE, UK; Molecular Oncology, Cancer Research UK Manchester Institute, The University of Manchester, Alderley Park SK10 4TG, UK; Belfast–Manchester Movember Centre of Excellence, Cancer Research UK Manchester Institute, The University of Manchester, Alderley Park SK10 4TG, UK.
Esther Baena, Belfast–Manchester Movember Centre of Excellence, Cancer Research UK Manchester Institute, The University of Manchester, Alderley Park SK10 4TG, UK; Prostate Oncobiology, Cancer Research UK Manchester Institute, The University of Manchester, Alderley Park SK10 4TG, UK.
David J Waugh, Movember FASTMAN Centre of Excellence, Patrick G Johnston Centre for Cancer Research, School of Medicine, Dentistry and Biomedical Sciences, Queen’s University Belfast, Belfast BT9 7AE, UK; School of Biomedical Sciences, Faculty of Health, Queensland University of Technology, Brisbane, Queensland, QLD 4000, Australia.
Joe M. O’Sullivan, Movember FASTMAN Centre of Excellence, Patrick G Johnston Centre for Cancer Research, School of Medicine, Dentistry and Biomedical Sciences, Queen’s University Belfast, Belfast BT9 7AE, UK; Northern Ireland Cancer Centre, Belfast Health & Social Care Trust, Belfast BT9 7JL, UK.
Darragh G McArt, Movember FASTMAN Centre of Excellence, Patrick G Johnston Centre for Cancer Research, School of Medicine, Dentistry and Biomedical Sciences, Queen’s University Belfast, Belfast BT9 7AE, UK.
Suneil Jain, Movember FASTMAN Centre of Excellence, Patrick G Johnston Centre for Cancer Research, School of Medicine, Dentistry and Biomedical Sciences, Queen’s University Belfast, Belfast BT9 7AE, UK; Northern Ireland Cancer Centre, Belfast Health & Social Care Trust, Belfast BT9 7JL, UK.
Melissa J LaBonte, Movember FASTMAN Centre of Excellence, Patrick G Johnston Centre for Cancer Research, School of Medicine, Dentistry and Biomedical Sciences, Queen’s University Belfast, Belfast BT9 7AE, UK.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
Belfast–Manchester Movember Prostate Cancer UK Centre of Excellence FASTMAN program [CEO13_2-004]; Brainwaves Northern Ireland Charity [NIC103464]; CRUK Manchester Institute [C5759/A20971 to E.B.]; Friends of the Cancer Centre [to R.G.M.]; Cancer Research UK [C11512/A20877 to A.C.R.]; HSC Research and Development Division of the Public Health Agency in Northern Ireland [COM/4965/14].
Conflict of interest statement. None declared.
REFERENCES
- 1. Caswell D.R., Swanton C.. The role of tumour heterogeneity and clonal cooperativity in metastasis, immune evasion and clinical outcome. BMC Med. 2017; 15:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Suzuki H., Freije D., Nusskern D.R., Okami K., Cairns P., Sidransky D., Isaacs W.B., Bova G.S.. Interfocal heterogeneity of PTEN/MMAC1 gene alterations in multiple metastatic prostate cancer tissues. Cancer Res. 1998; 58:204–209. [PubMed] [Google Scholar]
- 3. Kovac M., Navas C., Horswell S., Salm M., Bardella C., Rowan A., Stares M., Castro-Giner F., Fisher R., de Bruinet E.C. et al.. Recurrent chromosomal gains and heterogeneous driver mutations characterise papillary renal cancer evolution. Nat. Commun. 2015; 6:6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cao W., Wu W., Yan M., Tian F., Ma C., Zhang Q., Li X., Han P., Liu Z., Gu J. et al.. Multiple region whole-exome sequencing reveals dramatically evolving intratumor genomic heterogeneity in esophageal squamous cell carcinoma. Oncogenesis. 2015; 4:e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Campbell P.J., Yachida S., Mudie L.J., Stephens P.J., Pleasance E.D., Stebbings E.D., Morsberger L.A., Latimer C., McLaren S., Lin M.-L. et al.. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010; 467:1109–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shah S.P., Morin R.D., Khattra J., Prentice L., Pugh T., Burleigh A., Delaney A., Gelmon K., Guliany R., Senz J. et al.. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009; 461:809–813. [DOI] [PubMed] [Google Scholar]
- 7. Gerlinger M., Horswell S., Larkin J., Rowan A.J., Salm M.P., Varela I., Fisher R., McGranahan N., Matthews N., Santos C.R. et al.. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat. Genet. 2014; 46:225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yates L.R., Gerstung M., Knappskog S., Desmedt C., Gundem G., Van Loo P., Aas T., Alexandrov L.B., Larsimont D., Davieset H. et al.. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat. Med. 2015; 21:751–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. de Bruin E.C., McGranahan N., Mitter R., Salm M., Wedge D.C., Yates L., Jamal-Hanjani M., Shafi S., Murugaesu N., Rowan A.J. et al.. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science. 2014; 346:251–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nikbakht H., Panditharatna E., Mikael L.G., Li R., Gayden T., Osmond M., Ho C. Y., Kambhampati M., Hwang E.I., Faury D. et al.. Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nat. Commun. 2016; 7:11185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang J., Fujimoto J., Zhang J., Wedge D.C., Song X., Zhang J., Seth S., Chow C. W., Cao Y., Gumbs C. et al.. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science. 2014; 346:256–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Caiado F., Silva-Santos B., Norell H.. Intra-tumour heterogeneity: going beyond genetics. FEBS J. 2016; 283:2245–2258. [DOI] [PubMed] [Google Scholar]
- 13. Gupta R.G., Somer R.A.. Intratumour heterogeneity: novel approaches for resolving genomic architecture and clonal evolution. Mol. Cancer Res. 2017; 15:1127–1137. [DOI] [PubMed] [Google Scholar]
- 14. Felsenstein J. Inferring Phylogenies. 2003; Sunderland: Sinauer Associates. [Google Scholar]
- 15. Leimeister C.A., Boden M., Horwege S., Lindner S., Morgenstern B.. Fast alignment-free sequence comparison using spaced-word frequencies. Bioinformatics. 2014; 30:1991–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Heng L., Durbin R.. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. 2010; 26:589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Song K., Ren J., Reinert G., Deng M., Waterman M.S., Sun F.. New developments of alignment-free sequence comparison: measures, statistics and next-generation sequencing. Brief. Bioinform. 2014; 15:343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chan C.X., Bernard G., Poirion O., Hogan J.M., Ragan M.A.. Inferring phylogenies of evolving sequences without multiple sequence alignment. Sci. Rep. 2014; 4:6504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zielezinski A., Vinga S., Almeida J., Karlowski W.M.. Alignment-free sequence comparison: benefits, applications, and tools. Genome Biol. 2017; 18:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Roddy A.C., Jurek-Loughrey A., Souza J., Gilmore A., O’Reilly P.G., Stupnikov A., Gonzalez de Castro D., Prise K.M., Salto-Tellez M., McArt D.G.. NUQA: estimating cancer spatial and temporal heterogeneity and evolution through alignment-free methods. Mol. Biol. Evol. 2019; 36:2883–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pajuste F.D., Kaplinski L., Möls M., Puurand T., Lepamets M., Remm M.. FastGT: an alignment-free method for calling common SNVs directly from raw sequencing reads. Sci. Rep. 2017; 7:2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Parry M. A., Srivastava S., Ali A., Cannistraci A., Antonello J., Barros-Silva J. D., Ubertini V., Ramani V., Lau M., Shanks J. et al.. Genomic evaluation of multiparametric magnetic resonance imaging-visible and -nonvisible lesions in clinically localised prostate cancer. Eur. Urol. Oncol. 2018; 2:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Robinson J.T., Thorvaldsdóttir H., Wenger A.M., Zehir A., Mesirov J.P.. Variant review with the Integrative Genomics Viewer (IGV). Cancer Res. 2017; 77:31–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li H., Durbin R.. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 2013; arXiv doi:26 May 2013, preprint: not peer reviewedhttps://arxiv.org/abs/1303.3997.
- 25. Ewels P., Magnusson M., Lundin S., Käller M.. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016; 32:3047–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bolger A.M., Lohse M., Usadel B.. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014; 20:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Letunic I., Bork P.. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019; 47:W256–W259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J. et al.. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004; 5:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Durinck S., Spellman P.T., Birney E., Huber W.. Mapping identifiers for the integration of genomic datasets with the R/Biocondictor package biomaRt. Nat. Protoc. 2009; 4:1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Carm K.T., Hoff A.M., Bakken A.C., Axcrona U., Lothe R.A., Skotheim R.I., Løvf M.. Interfocal heterogeneity challenges the clinical usefulness of molecular classification of primary prostate cancer. Sci. Rep. 2019; 9:13579. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
NUQA is an open-source software that can be accessed from the GitHub repository (https://github.com/ACRoddy/NUQA/).
FastGT is an open-source software and its source code is available in the GitHub repository (https://github.com/bioinfo-ut/GenomeTester4/).
The prostate cancer multi-regional sampling WES cohort was provided by the original data authors on request and cited in (22).
RStudio is an open-source software that can be accessed from their website (http://www.rstudio.com/).
FastQC is an open-source software that can be accessed from their website (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/).
MultiQC is an open-source software that can be accessed from the GitHub repository (https://github.com/ewels/MultiQC/).
Trimmomatic is an open-source analytical package that be accessed from their website (http://www.usadellab.org/cms/?page=trimmomatic).
BBMap is an open-source analytical package that can be accessed from the SourceForge repository (https://sourceforge.net/projects/bbmap/).
The iTOL is an online tool that can be accessed from their website (https://itol.embl.de/).
Bioconductor is an R package that can be installed using R.
genefilter is a Bioconductor R package that can be installed using R and Bioconductor.
biomaRt is a Bioconductor R package that can be installed using R and Bioconductor.
Pheatmap is an R package that can be installed using R.
RColorBrewer is an R package that can be installed using R.