

Abstract
Here, we describe an extension of our original transformation-associated recombination (TAR) cloning protocol, enabling selective isolation of DNA segments from microbial genomes. The technique is based on the previously described TAR cloning procedure developed for isolation of a desirable region from mammalian genomes that are enriched in autonomously replicating sequence (ARS)-like sequences, elements that function as the origin of replication in yeast. Such sequences are not common in microbial genomes. In this Protocol Extension, an ARS is inserted into the TAR vector along with a counter-selectable marker, allowing for selection of cloning events against vector circularization. Pre-treatment of microbial DNA with CRISPR–Cas9 to generate double-stranded breaks near the targeted sequences greatly increases the yield of region-positive colonies. In comparison to other available methods, this Protocol Extension allows selective isolation of any region from microbial genomes as well as from environmental DNA samples. The entire procedure can be completed in 10 d.
Introduction
At present, transformation-associated recombination (TAR) cloning is undoubtely the most reliable method for selective isolation of large chromosomal regions. This method has enabled the direct cloning of chromosomal fragments up to almost 300 kb from complex mammalian genomes in the yeast Saccharomyces cerevisiae as circular YACs (yeast artificial chromosomes) for their further functional and structural analyses1–3. TAR cloning draws on several features of the yeast. First, yeast cells can take up several DNA molecules during transformation. Second, the method leverages high rates of recombination between homologous DNA sequences during transformation in the yeast Saccharomyces cerevisiae. When both 3′- and 5′-end sequence information is available, a genomic region can be isolated from total genomic DNA by TAR using a vector containing two unique sequences (hooks) homologous to the flanking sequences of a targeted region. After co-transfection of genomic DNA along with vector DNA into yeast cells, recombination between the vector hooks and the ends of a region of interest leads to the establishment of a circular TAR/YAC molecule. A TAR/YAC molecule may be retrofitted into a BAC (bacterial artificial chromosome) form. Alternatively, a TAR vector may contain a BAC cassette that allows YAC/BAC molecules to be transferred directly from yeast into bacterial cells for further DNA isolation, sequencing and analysis.
During the past decade, TAR cloning was successfully applied in multiple studies of mammalian genomes, including the characterization of genome variations such as polymorphic structural rear-rangements, mutation analysis of genes and segmental duplications, closing the gaps between contigs in the human genome sequence, separation of alleles and long-range haplotyping1–3. Moreover, simultaneous isolation of the same genomic region from multiple individuals may be performed only by this method. Thus, at present, TAR technology is a unique tool to create a library of individual human genes and to physically characterize the sites of chromosomal alterations (translocations, copy number variations and inversions) in the human population associated with predisposition to different diseases2. Most homologous regions from different mammals can be selectively cloned by in vivo recombination in yeast using targeting hooks developed from an available human genome sequence, because the level of sequence divergence typically seen between mammalian gene homologs of ~15% does not affect recombination in yeast. This has been demonstrated for several nonhuman primate genes4–6. In TAR cloning, the desired region is typically rescued at a frequency of 1–2%. Recently, we demonstrated that generation of double-stranded breaks (DSBs) near the targeted genomic region by pre-treatment of human genomic DNA with CRISPR–Cas9 endonuclease results in a dramatic increase in the fraction of gene-positive colonies (up to 32%)7 due to much more efficient homologous recombination between TAR vector hooks and targeted genomic sequences.
The original TAR cloning method requires the presence in the cloned genomic DNA fragment of at least one autonomously replicating sequence (ARS) that can function as the origin of replication in yeast. Potential ARS-like sequences can be identified based on the presence of a 17-bp ARS core consensus, WWWWTT TAYRTTTWGTT, in which W = A or T, Y = T or C and R = A or G8. Sequences that function as ARSs in yeast occur at a frequency of ~1/20–40 kb in all of the eukaryotic genomes thus far examined9. This suggests that TAR cloning can readily isolate most chromosomal regions with a vector that lacks an ARS because it relies on the acquisition of an ARS element from the targeted chromosomal DNA. The vector itself cannot propagate in yeast cells. Indeed, >150 different genomic regions were isolated using TAR vectors lacking an ARS1–3.
Isolation of natural product biosynthetic gene clusters (BGCs) from microbial genomes is the most rapidly developing application of the TAR cloning technique. As is well known, microbial natural products and their derivatives are indispensable to modern medicine and are an important source for drug discovery10,11. In some cases, cloning of BGCs from cultured microorganisms is possible using the original TAR protocol12. However, due to the low density of ARS elements in microbial genomes, isolation of BGCs that do not possess ARS-like sequences cannot be carried out by the original protocol. Many of the potentially valuable BGCs have been identified in environmental samples (e.g., in soil or gut microbiota) that are known to contain thousands of bacterial species. Therefore, an individual BGC represents only a tiny fraction of a metagenome compared to a single-copy gene in the mammalian genomes. For such cases, a modified TAR system was established by integrating an ARS element into the TAR vector backbone13 and the counter-selectable marker URA3 that allows background due to vector recircularization by non-homologous end joining to be eliminated13. This Extension to the original TAR protocol enables the isolation of the genomic regions that do not possess ARS-like elements, e.g., centromere and telomere regions from complex mammalian genomes and microbial genomic DNA.
Here, we present a TAR cloning protocol adapted for genomes that have few or no ARS-like sequences, to capture large BGCs with sizes that may be potentially as big as 300 kb. The protocol is also applicable for capturing BGCs from environmental DNA samples, as has been recently demonstrated14–17. This TAR cloning Protocol Extension includes five procedures. The first is construction of the TAR vector containing the targeting hooks (Fig. 1). The second is preparation of genomic DNA in solid agarose plugs (Steps 9–11). The third procedure describes pre-treatment of genomic DNA in agarose plugs by CRISPR–Cas9 nucleases before TAR cloning, which greatly increases the yield of region-positive clones (See Box 1). DNA pre-treatment by CRISPR–Cas9 is especially critical when soil or gut microbiota consisting of thousands of bacterial species is used for cloning of a gene or a gene cluster. The fourth procedure describes preparation of highly competent yeast spheroplasts (Steps 1–8) and transformation of the spheroplasts by a mixture of genomic DNA and a TAR vector adapted for microbial genomes (Steps 12–20). The fifth procedure is identification of gene-positive clones among primary yeast transformants (Steps 21–31). It is worth noting that circular TAR/YACs can be isolated as covalently closed circular DNA molecules directly from yeast cells18.
Fig. 1 |. Construction of the TAR vector with targeting sequences.
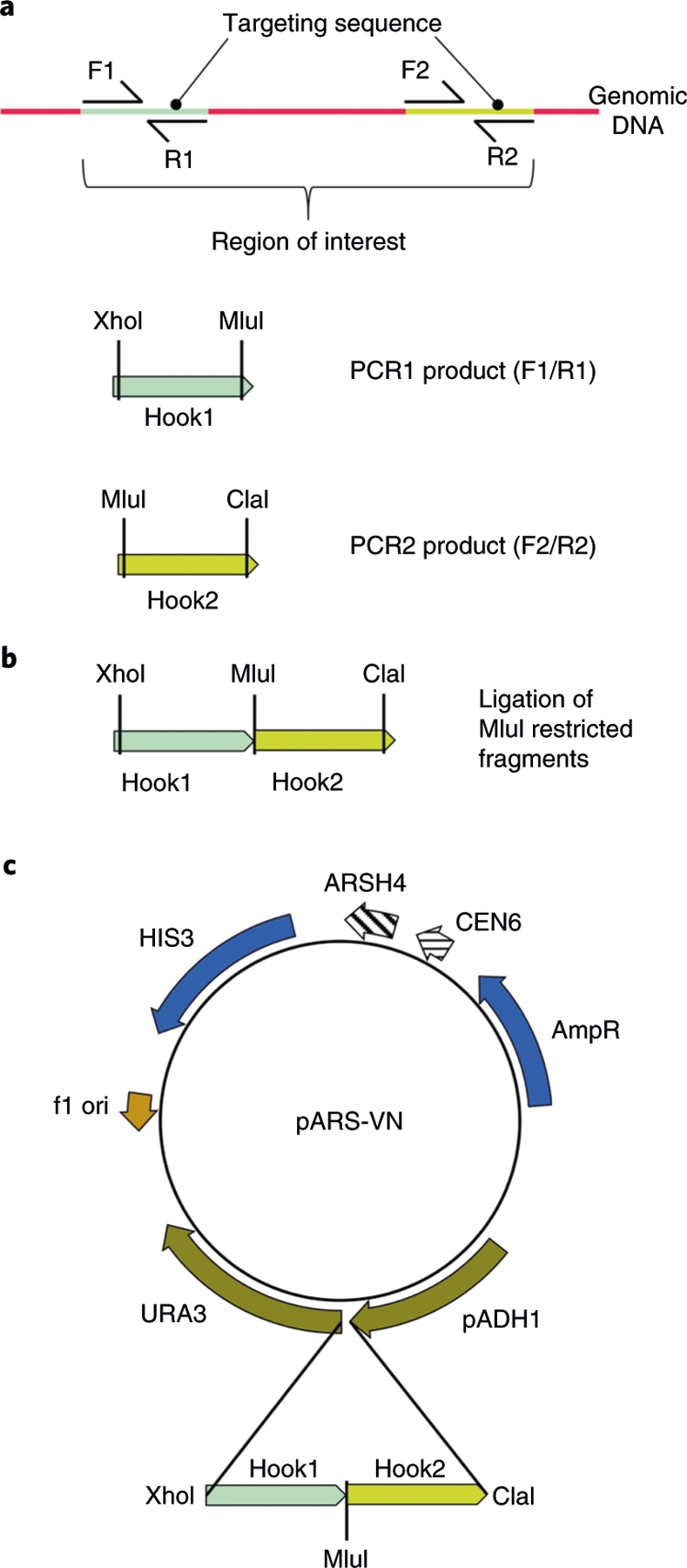
a, Hook1 and Hook2 are targeting sequences designed from the sequence of a region of interest. The Hook1 sequence is PCR amplified from genomic DNA with the primers F1/R1 containing XhoI and MluI sites (the MluI site is not present in the pARS-VN plasmid) (PCR1 product). The Hook2 sequence is PCR amplified from genomic DNA with the primers F2/R2 containing MluI and ClaI sites (PCR2 product). b, PCR1 and PCR2 products are digested with MluI and then ligated using DNA ligase. A second-round PCR reaction is carried out to amplify the ligated hook sequences with primers containing XhoI and ClaI sites, and then the final PCR product is cut using XhoI and ClaI. c, The ligated PCR products (Hook1 and Hook2) are inserted into the pAR-VN plasmid digested by XhoI and ClaI restriction endonucleases. Before TAR cloning, the pAR-VN vector with the hooks should be linearized by MluI to release the targeting sequences.
Box 1 |. Treatment of genomic DNA in agarose plugs by RNA-guided CRISPR–Cas9 endonuclease ● Timing 6 h.
After Step 9 of the Procedure, wash the agarose plugs with 0.1× wash buffer at room temperature for 1 h.
Discard the wash buffer using a 1-ml pipette and add 400 μl of 1× Cas9 nuclease reaction buffer for each plug. Equilibrate the agarose plugs at room temperature for 1 h.
-
During the plug equilibration, assemble Cas9 with guide RNAs (gRNAs) by mixing the following reagents to prepare cleavage solution:
Reagent Volume Final concentration Cas9 nuclease reaction buffer (×10)*Diagnostic primers 60 μl ×1 Cas9 nuclease (20 μM) 20 μl 0.67 μM gRNAI (1 μg/(μl) 20 μl 0.03 μg/μl gRNA2 (1 μg/(μl) 20 μl 0.03 μg/μl RNase-free H2O 480 μl - Total 600 μl - *See Cas9 nuclease kit in Reagents.▲CRITICAL STEP The cleavage solution is prepared for six plugs. For one plug, use 100 μl of the cleavage solution. gRNAs are designed as described in ‘Experimental design’ and synthesized using HiScribe T7 quick high yield RNA synthesis kit as described in refs.7,31,32.
Remove Cas9 nuclease buffer from the tube with the plug in Step 2 using a 1-ml pipette and discard; then add 100 μl of the prepared cleavage solution from Step 3.
Incubate the tube with the plug at 37 °C for 3 h, allowing digestion by Cas9.
After incubation, discard the cleavage solution from the tube using a 1-ml pipette and wash the plug two times with 25 mM NaCl at room temperature for 30 min. To release genomic DNA from the agarose plug, follow Steps 9–11 of the main Procedure.
Applications of the TAR method
This Extension to the original TAR cloning method has found many applications in the postgenomic era. First, TAR cloning was applied to selectively isolate large BGCs from microbial genomes15. Second, recent progress in synthetic genomics was also enabled to a certain extent by the use of TAR cloning. For example, the whole bacterial Mycoplasma genitalium genome (582,970 bp) was assembled using a combination of in vitro enzymatic and in vivo TAR cloning methods19. TAR cloning was applied to assemble eleven ~100-kb DNA segments into a complete 1.1 Mb Mycoplasma mycoides genome20. Cloning of whole genomes or individual chromosomes in yeast was successfully accomplished for species such as M. pneumoniae (0.8 Mb)21, Acholeplasma laidlawii (1.5 Mb)22, Prochlorococcus marinus MED4 (1.6 Mb)23 and eukaryotic algal Phaeodactylum tricornutum (27.4 Mb)24. Recently, the TAR cloning strategy has been applied to assemble four-, five- and six-gene pathways to generate yeast cells synthesizing beta-carotene and violacein25.
TAR cloning is a powerful tool to engineer synthetic viruses with novel properties. A synthetic genome of the Autographa californica multiple nucleopolyhedrovirus was recently constructed by a combination of PCR and TAR cloning26. TAR cloning was used to assemble the fragments into a complete herpes simplex virus type 1 genome27. Recently, TAR cloning was adapted to directly clone the large human cytomegalovirus genomes28. Such cloning facilitates the genetic manipulation of the primary isolates and provides a pathway to design a new generation of HCMV vaccines and vaccine vectors.
To summarize, the adapted TAR method provides a powerful tool for synthetic biology and for isolation of natural product BGCs for biomedicine. It is worth noting that a new TAR protocol can be applied for selective isolation of GC-rich regions lacking ARS-like elements such as clusters of ribosomal DNA from complex mammalian genomes.
Comparison with other methods
Compared with other available methods, this TAR cloning Protocol Extension allows genomic fragments of up to almost 300 kb to be selectively isolated. Such a size is sufficient for successful cloning of most microbial gene clusters. Notably, the size of TAR-isolated regions may be even bigger because, as has been shown previously, some YAC libraries constructed using DNA prepared in agarose blocks (to keep high-molecular-weight DNA) contain YACs with a size ranging from 430 to 1,200 kb29, meaning that there is no definite limitation for the size of the cloned material in yeast. It should also be emphasized that when it is necessary, a gene/region isolated by TAR cloning in yeast may be easily modified using homologous recombination in yeast30.
Recently, an alternative technique, Cas9-assisted targeting of chromosome segments, has been developed for cloning of genomic fragments of up to 100 kb from microorganisms in Escherichia coli31,32. This method is based on cleaving target DNA in vitro from intact bacterial chromosomes using RNA-guided Cas9 nuclease, with its subsequent ligation to a cloning vector through Gibson assembly19. Cas9-assisted targeting of chromosome segments has been successfully applied for isolation of BGCs from microbial genomes31. The method is an alternative to TAR cloning from individual microbes. However, many valuable BGCs have been identified in environmental samples (e.g., in soil or gut microbiota) that are known to contain thousands of bacterial species. Moreover, BGCs have been identified in more complex plant genomes33. For these cases, TAR cloning is the only method to selectively isolate BGCs.
Limitations of the TAR method
Despite being a very reliable and reproducible technology, TAR cloning has several limitations. For example, if a targeted region contains repeated homologous sequences, unwanted recombinant products (e.g., deletions) might occasionally occur. In such cases, the integrity of the recombinant target DNA may be confirmed by contour-clamped homogeneous electric field (CHEF) gel electrophoresis and then sequence analysis that helps to discard aberrant products. At present, this Protocol Extension, based on TAR cloning of genes/gene clusters from mammalian genomes3, has never been applied for capturing genomic fragments bigger than 300 kb. Preparation of high-molecular-weight genomic DNA is critical to clone long-size genomic fragments. Therefore, optimization of the existing methods for DNA preparation and its transfer to yeast spheroplasts would be an important step to overcome this problem. Another limitation is that at least a partial sequence of the targeted region must be available to develop the hooks. The entire genomes of the most commonly used organisms have been sequenced. Therefore, because hook sequences with only 85% homology to the targeted sequence may be used for TAR cloning34, sequence information available for the genomes of similar organisms may be used to design a TAR vector. Finally, when applied to environmental samples, combining the CRISPR–Cas9 endonuclease system with TAR cloning may induce undesirable properties such as off-target effects that affect the cleavage and cloning efficiency, leading to false-positive clones. Such aberrant clones, which have sequences homologous to that of the desirable target region, may be excluded by restriction digestion and sequence analyses.
Experimental design
TAR cloning vector
As in the original TAR protocol, the TAR vector carries two region-of-interest–specific targeting hooks, a yeast centromere (CEN6), and a yeast positive-selectable marker (HIS3). For the TAR protocol adapted for simple genomes, the TAR vector contains a yeast ARS element (ARSH4) and a negative-selectable marker (URA3). URA3 is a hybrid gene containing the open reading frame of the Saccharomyces cerevisiae URA3 gene and the promoter of the Shizosaccharomyces pombe ADH1 gene, which has particular spacing requirements for its function; the distance between the TATA element and the transcription initiation site must be ≤130 bp35,36, as a greater distance causes transcription to initiate at an alternative site, inactivating URA3 expression. The specific spacing requirements allow selection against the recircularized vector. The size of hooks may be as small as 60 bp37. In the TAR vector, the targeting hooks are placed between the promoter and the open reading frame of the URA3 gene. To ensure that the targeting hooks do not disrupt URA3 expression, their combined length should be <130 bp. The hook sequences should not contain ATG codons. The targeting sequences should be cloned into the TAR vector in the same orientation as they occur within the genome. Before transformation, the TAR vector is ‘activated’ by linearization with a unique endonuclease located between the targeting sequences to release the recombinogenic sequences (hooks). The basic vector without hooks was named ‘pARS-VN’13.
Yeast cells
The current protocol has been optimized for the highly transformable Saccharomyces cerevisiae strain VL6–48N (MAT alpha, his3-Δ200, trp1-Δ1, ura3-Δ1, lys2, ade2–101, met14, psi+cir0) that has HIS3, TRP1 and URA3 genes deleted13. The resulting His+Ura− transformants are selected on synthetic, complete histidine-minus plates containing 5-fluoroorotic acid (5-FOA). The ‘recombinational cloning’ part of the protocol has been optimized for processing 1.0 × 109 spheroplasts from early stationary phase cultures grown in 100 ml of yeast extract peptone dextrose (YEPD) media. The time of the cells’ treatment by Zymolyase-20T for each new batch of zymolyase should be experimentally determined.
Genomic DNA
For the isolation of genomic DNA from a single organism in pure culture, standard genomic DNA isolation procedures or commercial isolation kits are generally adequate, but the DNA should be checked by Nanodrop or Qubit to measure DNA purity and concentration, and by agarose gel electrophoresis to confirm integrity. The microbiota composition is highly diverse and complex. Isolation of DNA in sufficient quantity and integrity from metagenomic samples may require comparative analysis of known metagenomic DNA extraction methods to ascertain the appropriate protocol38. Usually, genomic DNA isolated from microbe genomes using commercial DNA kits allows TAR cloning of fragments not bigger than 100 kb. To isolate larger size fragments (bigger than 100 kb), the genomic DNA should be prepared in agarose blocks39,40 or according to CHEF Genomic DNA Plug Kits.
Pre-treatment of genomic DNA with CRISPR–Cas9 to increase the fraction of region-positive colonies
In our early work, we observed that homologous recombination between the TAR cloning vector hooks and targeted genomic sequences is much more efficient if the hooks are located closer to the DNA ends, rather than embedded within the target sequences41. We demonstrated that gene capture efficiency increased ~10–15 times when DSBs were specifically introduced close (<200 bp) to the ends of the desired genomic fragment by rare cutting restriction enzyme(s)41. However, this approach has drawbacks, as it can sometimes be impossible to choose restriction enzymes that cleave near the 5′ and 3′ ends of a targeted region without making additional cuts within the region itself. Therefore, a programmable endonuclease that cleaves at a user-defined sequence is more appropriate.
Recently, we demonstrated that pre-treatment of human genomic DNA with CRISPR–Cas9 endonuclease in vitro to generate DSBs near the ends of the targeted genomic region resulted in a large increase in the fraction of region-positive colonies (from 1% to 32%)7, such that a maximum of 12 yeast transformants needed to be screened to identify a clone with the desired chromosomal region. Therefore, extensive experience with yeast is no longer required. For TAR cloning from an individual microbe, CRISPR–Cas9 pre-treatment is not so important, because the yield of region-positive clones is high enough. This is critical when soil or gut microbiota consisting of thousands of bacterial species is used for cloning.
The CRISPR design tool (http://crispr.mit.edu/) is used to identify CRISPR guide sequences within 1 kb upstream of the 5′ TAR hook and ~1 kb downstream of the 3′ TAR hook sequences. All CRISPR guide sequences that have off-target sites within the gene of interest should be discarded. The off-target sites are given by the CRISPR design tool. If a complete sequence is available, a BLAT search (http://genome.ucsc.edu/cgi-bin/hgBlat) may also be used to further validate that the CRISPR guide candidates do not have other homologies within the gene of interest. CRISPR guides located closest to the TAR hooks are preferred, and a minimum of two CRISPR guides should be selected for each TAR hook position. This provides redundancy, in case one guide does not cleave DNA efficiently7.
Rationale for TAR cloning of genomic regions lacking ARS sequences
Figure 2a shows the basic scheme of the TAR vector pARS-VN. Figure 2b shows co-transfection of the TAR vector containing two targeting sequences (Hook1 and Hook2) linearized between the hooks and genomic DNA cut by CRISPR–Cas9 into yeast spheroplasts. There are two different events involving such a TAR vector. The first one is homologous recombination between the region-specific targeting sequences of the TAR vector and the genomic region of interest that leads to separation of the TATA box from the transcription initiation site in the URA3 gene by the DNA insert. 5-Fluoroorotic acid (5-FOA) is used to counterselect URA3-expressing yeast. The URA3 gene product (orotine-5’-monophosphate decarboxylase) converts 5-FOA to 5-fluorouracil, which is toxic for cells. Because the resulting TAR clones do not express URA3, they can grow on the medium containing 5-FOA. The second event is the nonhomologous end-joining that leads to recircularization of the vector. In this case, the distance between the TATA element and the transcription initiation site is not changed, and URA3 is expressed normally, making the yeast cells sensitive to 5-FOA.
Fig. 2 |. TAR cloning of genomic regions lacking ARS sequences from microbial genomes.
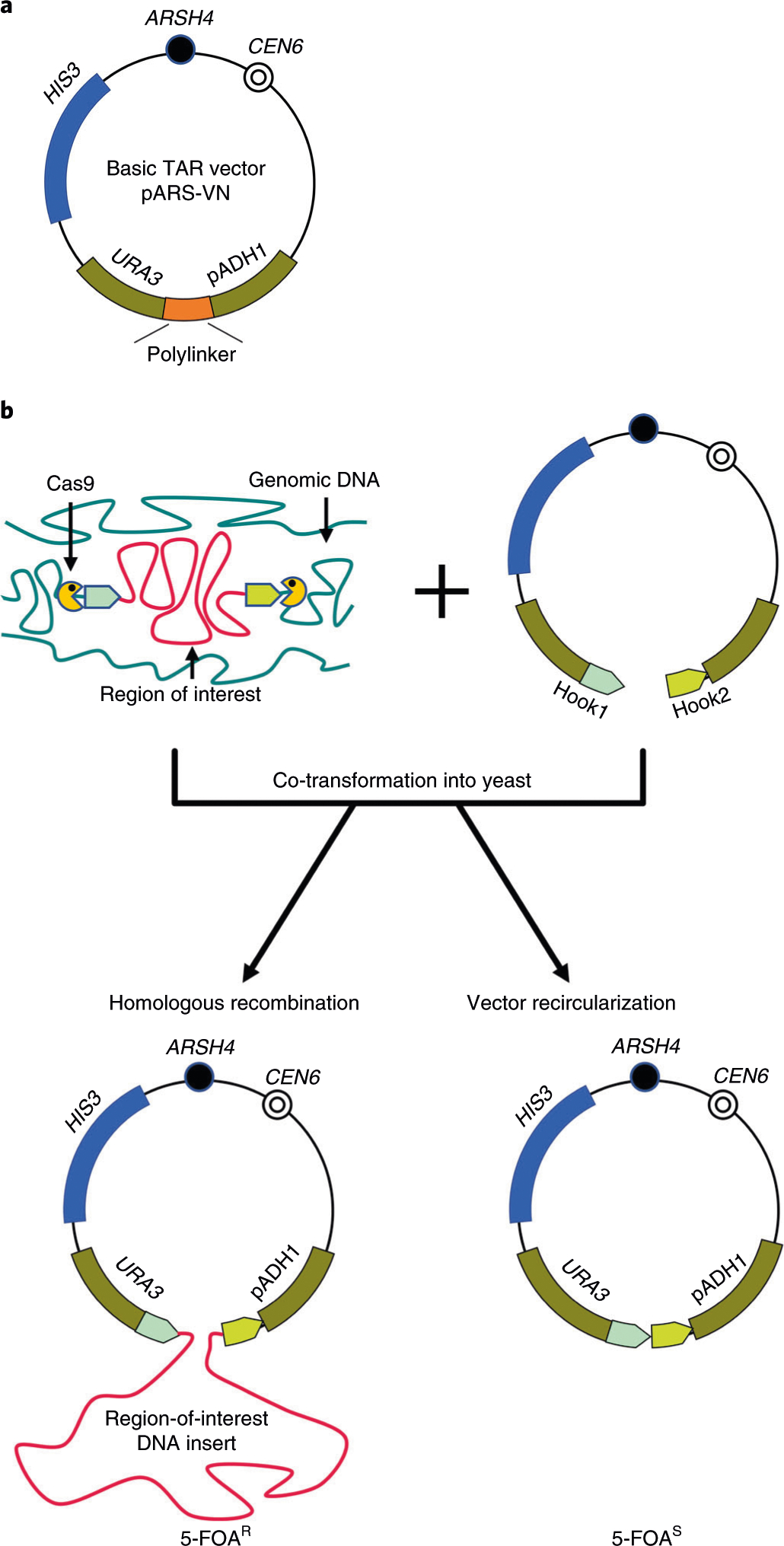
a, The scheme of the basic TAR vector pARS-VN. The plasmid carries a yeast positive-selectable marker (HIS3), a yeast centromere (CEN6), a negative-selectable marker (URA3), a yeast origin of replication (ARSH4) and a polylinker (a multiple cloning site). URA3 is a hybrid gene containing the open reading frame of the Saccharomyces cerevisiae URA3 gene and the promoter of the Shizosaccharomyces pombe pADH1 gene, which tolerates insertion of only up to 130 bp of sequence between the TATA box and the transcription initiation site. Any increase of the distance between the TATA box and the transcription initiation site above this inactivates URA3 expression. The combined length of the targeting hooks that are inserted into the ADH1 promoter should be <130 bp to not disrupt URA3 expression. Such configuration allows selection of TAR cloning events, which inactivate URA3 expression compared to vector recircularization events, which do not13,14. b, A diagram of the components required for TAR cloning of a region of interest from total genomic DNA. A TAR vector contains two unique targeting sequences (Hook1 and Hook2) homologous to the 5′ and 3′ ends of a region of interest inserted into the polylinker. The TAR vector DNA is linearized at the unique endonuclease site located between the hooks to expose targeting sequences. If necessary, genomic DNA may be treated by CRISPR–Cas9 endonuclease before yeast transformation7, which greatly increases the yield of region-positive TAR clones. Recombination between the region-specific unique targeting hooks and the homologous targeted sequences on the genomic fragment containing the region of interest leads to insertion of the cloned material between the TATA box and the transcription initiation site ATG, which abolishes URA3 expression. Such clones can therefore be selected by their ability to grow on media containing 5-FOA. Recircularization of ARS-containing TAR vector with the hooks due to nonhomologous end-joining, however, leads to reconstruction of the functional counter-selectable URA3 marker. In this case, the distance between the TATA element and the transcription initiation site ATG is not changed, and the URA3 marker is normally expressed in these clones; therefore, they do not grow on media containing 5-FOA. 5-FOAR, resistant to 5-FOA,; 5-FOAS, sensitive to 5-FOA.
Diagnostic primers to screen yeast transformants for colonies containing the region of interest
The region-positive colonies are identified by PCR. Two pairs of diagnostic primers should be designed. The first one is for the predicted junction sequences when one PCR primer is designed from the vector part and another one from the genomic insert. This PCR confirms an appropriate homologous recombination between the hooks in the TAR vector and the targeted genomic sequences. Another pair of primers is chosen from the unique internal region sequences. This PCR confirms that the correct genomic region is cloned.
Retrofitting of a TAR/YAC molecule into a BAC form
In this Protocol Extension, we describe the basic TAR vector pARS-VN13. The vector was constructed based on the pUC plasmid. If necessary, there is a simple procedure to retrofit a TAR/YAC into a YAC/BAC molecule with self-replicating broad-host-range elements for heterologous expression39,40. As an alternative, the basic TAR vector can be modified for BGC heterologous expression in various host organisms. All of the vector derivatives should contain the elements for maintenance and selection in yeast, propagation in E. coli and BGC heterologous expression in host organisms. For example, Yamanaka and colleagues designed a TAR cloning vector that consists of three elements, i.e., from yeast, E. coli and actinobacteria15. Thus, the TAR-isolated region can be shuttled between yeast, E. coli and actinobacteria species. The vector also contains oriT and attP-int that allows the targeted region to be transferred by conjugation and insertion of the TAR-isolated region into heterologous host chromosomes. In another work, Zhang and co-authors constructed vectors allowing for expression of TAR-isolated genomic fragments in gram-positive and gram-negative microorganisms14.
Materials
Biological materials
Genomic DNA: see ‘Experimental design’ for details.
Reagents
▲CRITICAL The Reagents list is similar to that described in the original TAR cloning protocol by Kouprina and Larionov12. In this Protocol Extension, we describe the yeast strain, the TAR basic vector, genomic DNA, the media and additional reagents, which differ from the original protocol, but please see ref.12 for the remaining reagents.
Saccharomyces cerevisiae strain VL6–48N. The highly transformable Saccharomyces cerevisiae strain VL6–48N (MAT alpha, his3-Δ200, trp1-Δ1, ura3-Δ1, lys2, ade2–101, met14, psi+cir0) that has HIS3, TRP1 and URA3 genes deleted is used13. The strain is a derivative of VL6–4812 with the ura3–52 gene replaced by the KanMX cassette13, deleting the URA3 gene. The strain is available from our laboratory upon request.
pARS-VN vector containing the targeting hooks (or its derivative carrying a cassette for transfer of a TAR-isolated BGC into a specific microbial host) is propagated in E. coli cells. The basic plasmid pARS-VN (6,441 bp) used for construction of a TAR cloning vector with targeting sequences (hooks) contains a yeast positive-selectable marker (HIS3), a yeast centromeric sequence (CEN6), a yeast ARS element (ARSH4), a negative-selectable marker URA3 under the pADH1 promoter region, an ampicillin resistance gene (AmpR) and colE1 origin of replication. The pARS-VN basic plasmid and its sequence are available from our laboratory upon request.
5-FOA (ThermoFisher Scientific, cat. no. R0811)
SD-His-Ura plates (Teknova, cat. no. C3200)
SORB-TOP-His medium or top agar (Teknova, cat. no. C3743)
CHEF Genomic DNA Plug Kits (Bio-Rad, cat. no. 170–3591, 170–3592, 170–3593)
DNeasy Blood & Tissue Kit (Qiagen, cat. no. 69504).
QIAquick gel extraction kit (Qiagen, cat. no. 28704)
1-kb DNA Ladder (Takara, cat. no. 3426A)
Certified molecular biology agarose (Bio-Rad, cat. no. 1613100)
10× TBE buffer (Tris-borate-EDTA; Thermo Scientific, cat. no. B52)
Cas9 nuclease, Streptococcus pyogenes (NEB, cat. no. MO386M)
HiScribe T7 quick high yield RNA synthesis kit (NEB, cat. no. E2050S)
Diagnostic PCR primers; appropriate diagnostic PCR primers to screen yeast transformants for gene/region-positive clones (Integrated DNA Technologies; see ‘Experimental design’)
Equipment
▲CRITICAL The Equipment list is similar to that described in the original TAR cloning protocol by Kouprina and Larionov12 except for the additional equipment listed below.
0.2-ml PCR tubes (Axygen, cat. no. PCR-02-C)
Thermal cycler (Applied Biosystems, cat. no. 4375786)
Microwave oven
Incubator at 37 °C
Reagent setup
▲CRITICAL The Reagent Setup is similar to that described in the original TAR cloning protocol by Kouprina and Larionov12. In this Protocol Extension, we describe setup for the yeast strain, the TAR basic vector, genomic DNA and the medium for yeast spheroplast transformation, which differ from the original protocol, but please see ref.12 for the remaining Reagent setup information.
The TAR cloning vector
To construct a TAR cloning vector, hooks specific for a region of interest should be inserted between the URA3 open reading frame and the pADH1 promoter region. Such a configuration would allow selection for region-of-interest capture events versus vector recircularization events. Sequences of the hooks should be unique. The combined length of the targeting sequences should be ≤130 bp. Each hook may be as small as 60 bp37. A unique site between the hooks depends on the TAR vector construction (see Fig. 1 and ‘Experimental design’ for further information). The hooks are inserted into XhoI/ClaI sites of the basic pARS-VN plasmid. Before TAR cloning, the TAR vector should be linearized by MluI to release the targeting sequences.
Before cloning, the TAR vector with the hooks should be tested for growth on SD-His-Ura and SD-His-5-FOA media. For this purpose, transformation into yeast Saccharomyces cerevisiae may be carried out by either LiAc or electroporation methods. After transformation of the vector alone into the VL6–48N strain, the transformants should grow on SD-His-Ura medium and should not grow on SD-His-5-FOA medium. This result will indicate the absence of ATG codons in the hooks and correct construction of the TAR vector. Before TAR cloning, the vector should be linearized between the targeting hooks to ‘activate’ the hooks. The linearized vector may be kept at −20 °C for several months. If necessary to transfer a TAR-isolated DNA molecule from yeast to a specific microbial host, a corresponding selectable marker and an ori or attP-int system for chromosome integration can be inserted into a TAR vector17,42,43. Vector DNA may be isolated by using a DNA Maxi kit.
Genomic DNA
High-molecular-weight genomic DNA (bigger than 100 kb) should be prepared in agarose blocks to prevent DNA sharing according to the CHEF Genomic DNA Plug Kits Instruction Manual. For TAR cloning of genomic fragments with sizes not bigger than 100 kb, DNA may be prepared in solution by DNeasy Blood & Tissue Kit.
Petri plates with selection medium SORB-His-5-FOA
(Without histidine and with 5-FOA) 1 M sorbitol, 2% (wt/vol) d-glucose, 0.17% (wt/vol) yeast nitrogen base, 0.5% (wt/vol) (NH4)2SO4, 2% (wt/vol) Bacto agar and 0.1% (wt/vol) 5-FOA supplemented with amino acids as described in SORB-TOP-His. Add 182 g of sorbitol to ~500 ml of distilled/deionized water in a 1000-ml beaker. Stir until dissolved. Make the volume up to 750 ml. Transfer the solution to a 2000-ml glass flask. Add 20 g of agar, mix carefully and autoclave. Separately, mix 20 g of d-glucose, amino acids, 1.7 g of yeast nitrogen base, 5 g of (NH4)2SO4 and 1 g of 5-FOA with 250 ml of double-distilled water. Stir vigorously on a magnetic stirrer at 65 °C. After dissolving, the solution should be filter sterilized. Then, add 250 ml of this solution to 750 ml of melted agar with sorbitol, mix briefly and pour your plates.
Petri plates with selection medium SD-His-Ura
(Without histidine and uracil) 2% (wt/vol) d-glucose, 0.17% (wt/vol) yeast nitrogen base, 0.5% (wt/vol) (NH4)2SO4 and 2% (wt/vol) Bacto agar containing the following supplements: 0.006% (wt/vol) adenine sulfate, 0.005% (wt/vol) l-arginine HCl, 0.008% (wt/vol) l-aspartic acid, 0.01% (wt/vol) l-glutamic acid, 0.005% (wt/vol) l-isoleucine, 0.01% (wt/vol) l-leucine, 0.012% (wt/vol) l-lysine·HCl, 0.002% (wt/vol) l-methionine, 0.005% (wt/vol) l-phenylalanine, 0.0375% (wt/vol) l-serine, 0.01% (wt/vol) l-threonine, 0.005% (wt/vol) l-tryptophan, 0.005% (wt/vol) l-tyrosine, 0.015% (wt/vol) l-valine. Alternatively, synthetic SD-His-Ura medium can be purchased from Teknova (www.teknova.com).
Petri plates with selection medium SD-His-5-FOA
(Without histidine and with 5-FOA) 2% (wt/vol) d-glucose, 0.17% (wt/vol) yeast nitrogen base, 0.5% (wt/vol) (NH4)2SO4, 2% (wt/vol) Bacto agar and 0.1% (wt/vol) 5-FOA supplemented with amino acids as described in SORB-TOP-His without sorbitol. Add 20 g of agar into 750 ml of double-distilled water and autoclave. Separately, mix 20 g of d-glucose, amino acids, 1.7 g of yeast nitrogen base, 5 g of (NH4)2SO4, and 1 g of 5-FOA with 250 ml of double-distilled water. Stir vigorously on a magnetic stirrer at 65 °C. After dissolving, the solution should be filter sterilized. Then, add 250 ml of this solution into 750 ml of melted agar, mix briefly and pour your plates.
Procedure
▲CRITICAL The procedure is similar to that described in the original TAR cloning protocol by Kouprina and Larionov12 except that in this Protocol Extension: (i) the procedure is adapted to microbial genomes, (ii) the VL6–48N strain with the URA3 gene deleted is used, (iii) pARS-VN is the basic TAR vector used with the yeast ARS element and the negative-selectable marker URA3, (iv) new sections ‘Preparation of chromosome-size genomic DNA from agarose plugs’ (Steps 9–11) and ‘Treatment of genomic DNA in agarose plugs by RNA-guided CRISPR–Cas9 endonuclease’ (Box 1) are included. In the case of cloning from metagenomic DNA, the yeast transformants should be combined into pools (see Step 21). DNA isolation from the pools and identification of region-positive pools by PCR with the diagnostic primers are described in Steps 16–32 of the original TAR cloning protocol by Kouprina and Larionov12.
Preparation of yeast cultures ● Timing 15 h, overnight
-
1
1 d before the TAR cloning experiment, make three 100-ml aliquots of YEPD medium in 250-ml Erlenmeyer flasks. Inoculate the flasks with three differently sized single-cell colonies of the host yeast strain VL6–48N that has been freshly grown on a YEPD plate with agar. Grow the cultures overnight (for 12–15 h) at 30 °C with vigorous shaking (220-–240 rpm) to ensure good aeration.
? TROUBLESHOOTING
Preparation of competent yeast spheroplasts ● Timing 2–3 h
-
2
In the morning, measure the optical density of the cultures at 20-min intervals until an OD660 of 2.0–2.5 is achieved. To do so, dilute 0.2 ml of culture 1:10 in water (0.2 ml of culture and 1.8 ml of water), and the density should be between 0.2 and 0.25.
▲CRITICAL STEP Cultures with such an optical density, corresponding to approximately 2 × 107 cells/ml, are ready for the preparation of highly competent spheroplasts.
-
3
Transfer the yeast culture into two 50-ml Corning tubes, and pellet the cells by centrifugation for 5 min at 1,200g and 5 °C. Remove and discard the supernatant.
-
4
Resuspend each cell pellet in 20 ml of 1 M sorbitol solution by vortexing and centrifuge for 5 min at 1,200g and 5 °C. The density should be ~1.1–1.2. Remove and discard the supernatant.
■PAUSE POINT Yeast cells in 1 M sorbitol solution may be kept at 4 °C overnight.
-
5
Resuspend each cell pellet in 20 ml of sorbitol, phosphate buffer and EDTA (SPE) solution. Add into each tube 40 μl of Zymolyase solution and 40 μl of 2-mercaptoethanol (ME), mix well and incubate at 30 °C for ~15 min with slow shaking (50–70 rpm).
▲CRITICAL STEP Note that the treatment time may vary depending on the zymolyase stock. When new stocks of enzyme are made, even if from the same lot number, they should be re-titrated to confirm or adjust the amount of enzyme necessary for the optimal level of spheroplasting, since reduction of enzyme activity during storage at 4 °C can occur.
? TROUBLESHOOTING
-
6
Check the level of spheroplasting by comparing the optical densities of the cell suspension in 1 M sorbitol solution (when spheroplasts are intact) versus in 2% (wt/vol) SDS solution (when spheroplasts are lysed). To measure the OD660 difference, dilute 200-μl aliquots of the zymolyase-treated cell suspension (from Step 5) 10-fold in either 1 M sorbitol solution or 2% (wt/vol) SDS solution. Measure the optical density of both diluted suspensions spectrophotometrically. The spheroplasts are determined to be ready when the difference between the sorbitol and SDS OD660 readings is three- to fivefold.
▲CRITICAL STEP Insufficient or excessive treatment of zymolyase greatly affects transformation efficiency. From this point on, extreme care must be taken to avoid lysing the delicate spheroplasts: very slow, gentle resuspensions are necessary.
? TROUBLESHOOTING
-
7
Centrifuge spheroplasts for 5 min at 1,200g and 5 °C. Decant the supernatant, add 10 ml of 1.0 M sorbitol solution, very gently re-suspend the pellet by pipetting and then add up to 50 ml of 1.0 M sorbitol solution. Pellet the spheroplasts again by centrifugation for 5 min at 1,200g and 5 °C.
-
8
Repeat the wash with 1 M sorbitol solution one more time (as in Step 7) and gently re-suspend the final pellets in 2.0 ml of sorbitol, TRIS and calcium (STC) solution.
■PAUSE POINT The spheroplasts are ready for transformation and are stable at room temperature for ≥1 h.
Preparation of chromosome-size genomic DNA from agarose plugs ● Timing 1.0 h
▲CRITICAL For TAR cloning of large genomic regions (bigger than 100 kb), high-molecular-weight genomic DNA should be prepared in solid low-melting-temperature agarose blocks using the CHEF Genomic DNA Plug Kits or as described previously32,39,40. The resulting agarose plugs are stable for several months when stored at 5 °C in a wash buffer. For TAR isolation of genomic fragments smaller than 200 kb, genomic DNA may be prepared using commercial isolation kits (e.g., DNeasy Blood & Tissue Kit). Isolation of DNA from metagenomic samples may require comparative analysis of known metagenomic DNA extraction methods to ascertain the appropriate protocol38,44,45. The isolated genomic DNA may be kept stable at −20 °C for several months. If genomic DNA for TAR cloning was prepared in solution (not in agarose plugs), Steps 9–11 should be omitted.
-
9
Transfer each agarose plug containing genomic DNA to a 2-ml Eppendorf tube. Remove all the dialysis solution carefully from the tubes using a 1-ml pipette and discard it.
? TROUBLESHOOTING
▲CRITICAL STEP Genomic DNA in the agarose plug may be pre-treated with CRISPR–Cas9 at this stage as described in Box 1, to generate DSBs near the targeted genomic sequences that result in a great increase in the fraction of region-positive colonies (see ‘Experimental design’ for details). This is critical when soil or gut microbiota consisting of thousands of bacterial species is used for cloning.
-
10
Place the tubes in a 70 °C temp block and incubate for 5 min until the agarose is melted completely.
▲CRITICAL STEP Incubation at temperatures higher than 70 °C may denature DNA.
? TROUBLESHOOTING
-
11
Add 1 U of β-agarase to each tube and mix gently by flicking. Incubate tubes for 30 min at 42 °C until the agarose is completely digested. Test for completion by placing the tube on ice for 5 min and examining for solid agarose. If solid agarose remains, re-melt at 70 °C, cool to 42 °C, add an additional 1 U of β-agarase and incubate for 30 min more at 42 °C.
■PAUSE POINT Genomic DNA prepared from agarose plugs may be kept stable at −20 °C for several months.
? TROUBLESHOOTING
Transformation of spheroplasts by genomic DNA along with a TAR vector ● Timing 2.0–2.5 h
-
12Linearize the TAR vector to release the targeting hooks by digestion with MluI (see Fig. 1c), using the following reaction:
Reagent Volume Final concentration TAR cloning vector DNA 25 μl (5 μg) 0.05 μg/μl NEBuffer 3 (×10) 10 μl ×1 MluI (10 U/μl) 5 μl 0.5 U/μl H2O 60 μl Total 100 μl ▲CRITICAL STEP The final vector DNA concentration in the restriction solution should be 50 ng/μl. Use 5–10 μl (0.25–0.5 μg of the TAR cloning vector) of the restriction digestion solution for one agarose plug.
? TROUBLESHOOTING
-
13
Incubate the reaction at 37 °C for 2 h.
-
14
After restriction digestion, inactivate the endonuclease at 65 °C for 10 min.
-
15
Mix 200 μl of spheroplast suspension from Step 8 gently with 1–2 μg of genomic DNA from Step 11 and 0.25–0.5 μg (5–10 μl) of the linearized TAR cloning vector from Step 14 in a 2.0-ml Eppendorf tube. Incubate for 10 min at room temperature.
? TROUBLESHOOTING
▲CRITICAL STEP Each 2.0 ml of spheroplasts in STC solution obtained in Step 8 from 100 ml of the original culture provides enough material for 10 transformation reactions to be set up.
-
16
Add 800 μl of PEG 8000 solution into each Eppendorf tube, gently mix by inverting and incubate for 15 min at room temperature.
? TROUBLESHOOTING
-
17
Pellet the spheroplasts by centrifugation in the Eppendorf microfuge for 5 min at 1,200g and 5 °C. Decant the supernatant and gently resuspend the spheroplasts in each tube with 800 μl of SOS solution using Pipetman.
-
18
Incubate the spheroplasts for 40 min at 30 °C without shaking.
-
19
Transfer the spheroplasts from each transformation reaction into a separate 15-ml Corning tube containing 7.0 ml of melted SORB-TOP-His or top agar selection medium (equilibrated at 50–55 °C) using Pipetman, gently mix and quickly pour each 7.0 ml of medium onto a SORB-His-5-FOA plate (containing 5-FOA).
? TROUBLESHOOTING
-
20
Keep the plates at 30 °C for 5–7 d until all the transformants become visible.
Identification of individual region-positive colonies ● Timing 4–5 h
-
21
Individually streak 30 primary yeast transformants from the plates in Step 20 using a toothpick onto an SD-His-5-FOA plate lacking histidine. Incubate the plates at 30 °C overnight and then use His+Ura− transformants for detection of region-positive individual colonies.
▲CRITICAL STEP Usually 30–50 yeast transformants is enough to streak when genomic DNA isolated from a single microorganism is used for TAR cloning. When metagenomic DNA is used for cloning, the number of colonies streaked should be increased up to 300–400. In this case, the transformants can be combined into the pools, each containing 20–30 transformants, and DNA isolation and identification of region-positive pools can be carried out as described in Steps 18–32 of the original TAR cloning protocol by Kouprina and Larionov12.
-
22
Touch the streak of each His+Ura− transformant with a sterile disposable pipette tip and then rinse the tip thoroughly in a 100-μl mixture of 80 μl of water plus 20 μl of zymolyase solution plus 1 μl of ME.
-
23
Incubate the resulting suspension for 1 h at 30 °C.
-
24
Add 10 μl of 2% (wt/vol) SDS solution. Incubate for 15 min at 70 °C.
-
25
Add 10 μl of 5 M KAc solution and let the tubes sit on ice for 15 min.
-
26
Spin at maximum Eppendorf mini-centrifuge speed (16,000g) for 5 min at room temperature.
-
27
Transfer the supernatants to new Eppendorf tubes and add an equal volume of isopropanol. Precipitate at maximum Eppendorf minifuge speed (16,000g) for 5 min at room temperature.
-
28
Discard the supernatant by pipetting and dissolve the pellets in 50 μl of water.
■PAUSE POINT The DNA samples can be left frozen for up to several months at −20 °C.
-
29Use 1 μl of the DNA solution in a 15-μl PCR reaction with appropriate diagnostic primers to identify region-positive clones, using Taq polymerase manufacturer’s suggested protocols. Conditions for PCR reaction with diagnostic primers are as follows:
Temperature (°C) Time Cycles Initial denaturation 95 2 min 1 Denaturation 95 30 s 30 Annealing 55 30 s - Extension 72 30 s - Final extension 72 1 min 1 Hold 4 1 - -
30
Apply the entire volume of one PCR reaction to one agarose well and examine the lengths of the PCR products by conventional electrophoresis in a 1–1.5% (wt/vol) agarose gel using 1-kb DNA Ladder as a marker. Find the PCR products of the expected length.
? TROUBLESHOOTING
-
31
Extract the PCR products using QIAquick Gel Extraction Kit and then further validate them by sequencing.
■PAUSE POINT The PCR products can be left frozen for up to several months at −20 °C.
▲CRITICAL STEP Although TAR cloning produces a very low level of artifacts, if any, we recommend to physically characterize two to three independently isolated region-positive clones before their use for functional/structural studies. The size of the genomic inserts may be examined by CHEF gel electrophoresis, as previously described46–48. In addition, identity of the inserts in the TAR-isolated clones may be checked by a set of overlapping PCR reactions for the targeted region and comparison of their restriction profiles46–48. The size of isolated DNA fragments in the clones and their restriction profiles should be identical. Such analysis is performed after the transfer of TAR/YACs from yeast to E. coli cells. This is possible to do if the F1 origin of replication is included in the basic TAR cloning vector pARS-VN. Alternatively a TAR/YAC may be retrofitted into a YAC/BAC molecule in yeast, and then a YAC/BAC is moved to E. coli cells, as described previously39.
Troubleshooting
Troubleshooting advice can be found in Table 1.
Table 1 |.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 1 | Poor transformation efficiency | Yeast strain is different | Use only VL6–48N, which exhibits abnormally high transformation efficiency. Also, three genes (HIS3, TRP1 and URA3) are deleted in VL6–48N, allowing their use as markers in TAR cloning |
| 5 | Poor transformation efficiency | Zymolyase solution is >6 mo old | Prepare a fresh zymolyase solution |
| 6 | Poor transformation efficiency | Spheroplasts are not competent for transformation | Make spheroplasts according to the Procedure |
| 9 | No region-positive clones | Degraded genomic DNA | Size of genomic DNA should be checked by pulse gel electrophoresis before its use |
| 10 | Failure to melt agarose plugs | The agarose plugs are not appropriately prepared (either the concentration of agarose is >0.5% or the plugs contain >5 μg of genomic DNA) | Add an equal volume of 25 mM NaCl to the plugs before melting at 70 °C |
| 11 | No region-positive clones | Size of genomic DNA is smaller than the size of a desired region | Mb-size regions require very careful manipulation of genomic DNA. DNA should be prepared in agarose plugs where length of genomic fragments are >1,000 kb20 |
| 12 | No region-positive clones and many 5-FOA-resistant transformants | The hooks are longer than 130 bp | The combined length of the hooks should not be >130 bp |
| Sequences of the hooks in TAR vector contain ATG codons | Verify hook sequences by their sequencing | ||
| No region-positive clones | Wrong orientation of the hooks in the TAR vector | Targeting sequences should be cloned into the pARS-VN vector in the same orientation as they occur within the genome | |
| Very few transformants and a low yield of region-positive clones | Incompleteness of the TAR vector linearization | Check completeness of vector linearization after endonuclease digestion by gel electrophoresis | |
| Poor transformation efficiency | Amount of the TAR vector is <0.5 μg | Check concentration of the vector | |
| 15 | Poor transformation efficiency | Amount of genomic DNA is <1–2 μg, or DNA is contaminated by detergent | Check concentration of genomic DNA or purify DNA again |
| 16 | Poor transformation efficiency | PEG reagent is >3 mo old | Prepare a fresh PEG 8000 solution |
| 19 | No transformants | Yeast cells were plated onto the wrong medium | Ensure that the medium contains all required nutrients. Check the growth of the VL6–48N strain on the synthetic SORB-His-5-FOA medium supplemented with histidine |
| No region-positive clones and many 5-FOA-resistant transformants | Concentration of 5-FOA in the medium is not correct | Prepare new medium according to the Procedure | |
| 30 | No region-positive clones | Diagnostic PCR primers do not work | Check the primers with genomic DNA |
Timing
The time estimations below include reaction time, hands-on time and culture/colony growth time.
Preparation of competent yeast spheroplasts
Step 1, growth of the culture of the yeast strain: 15 h, overnight
Steps 2–8, preparation of competent spheroplasts; depending on the formation of spheroplasts: 2–3 h
Preparation of chromosome-size genomic DNA from agarose plugs
Steps 9–11, preparation of chromosome-size genomic DNA from agarose plugs: 1 h Transformation of spheroplasts by genomic DNA along with a TAR vector
Steps 12–19, transformation of spheroplasts by genomic DNA along with a TAR cloning vector: 2.0–2.5 h
Step 20, growth of His+Ura− yeast transformants on synthetic histidine-minus plates containing 5-FOA (SORB-His-5-FOA). Selection of clones that no longer express URA3: 5–7 d
Identification of individual region-positive colonies
Step 21, grow individually streaked His+Ura− yeast transformants on SD-His-5-FOA medium: 15 h, overnight
Steps 22–28, DNA isolation from the individual colonies: 2 h
Step 29–31, identification of region-positive individual transformants by PCR: 2–3 h
Anticipated results
For transformation conditions described above (i.e., with 0.25–0.5 μg of TAR cloning vector, 1–2 μg of genomic DNA and 1 × 108 spheroplasts), the yield of transformants varies from 20 to 200 colonies on one Petri plate. The yield of region-positive clones from environmental DNA samples varies from 20% to 70% if genomic DNA is pre-treated before spheroplast transformation by CRISPR–Cas9 that is designed in such a way to cut near the targeted sequences7. Approximately the same yield of region-positive clones is expected without CRISPR–Cas9 treatment for DNA samples isolated from individual microbes. With CRISPR–Cas9 treatment, the yield of region-positive clones is >70%.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Acknowledgements
This work was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, USA.
Footnotes
Competing interests
The authors declare no competing interests.
Supplementary information is available for this paper at https://doi.org/10.1038/s41596-019-0280-1.
Reprints and permissions information is available at www.nature.com/reprints.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Kouprina N & Larionov V TAR cloning: insights into gene function, long-range haplotypes and genome structure and evolution. Nat. Rev. Genet 7, 805–812 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Kouprina N & Larionov V Transformation-associated recombination (TAR) cloning for genomics studies and synthetic biology. Chromosoma 125, 621–632 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kouprina N & Larionov V TAR cloning: perspectives for functional genomics, biomedicine, and bio-technology. Mol. Ther. Methods Clin. Dev 14, 16–26 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kouprina N et al. Accelerated evolution of the ASPM gene controlling brain size begins prior to human brain expansion. PLOS Biol. 2, 653–663 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kouprina N et al. Evolutionary diversification of SPANX-N sperm protein gene structure and expression. PLOS One 2, e359(2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pavlicek A et al. Evolution of the tumor suppressor BRCA1 locus in primates: implications for cancer predisposition. Hum. Mol. Genet 13, 1–15 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Lee NCO, Larionov V & Kouprina N Highly efficient CRISPR/Cas9-mediated TAR cloning of genes and chromosomal loci from complex genomes in yeast. Nucleic Acids Res. 43, e55(2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Theis JF & Newlon CS The ARS309 chromosomal replicator of Saccharomyces cerevisiae depends on an exceptional ARS consensus sequence. Proc. Natl Acad. Sci. USA 94, 10786–10791 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stinchcomb DT, Thomas M, Kelly J, Selker E & Davis RW Eukaryotic DNA segments capable of autonomous replication in yeast. Proc. Natl Acad. Sci. USA 77, 4559–4563 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi SS et al. Genome engineering for microbial natural product discovery. Curr. Opin. Microbiol 45, 53–60 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Zhang MM, Qiao Y, Ang EL & Zhao H Using natural products for drug discovery: the impact of the genomics era. Expert Opin. Drug Discov 12, 475–487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kouprina N & Larionov V Selective isolation of genomic loci from complex genomes by transformation-associated recombination cloning in the yeast Saccharomyces cerevisiae. Nat. Protoc 3, 371–377 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Noskov VN et al. A general transformation-associated recombination cloning system to selectively isolate any eukaryotic or prokaryotic genomic region. BMC Genomics 4, 16(2003). Epub 2003 Apr 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang JJ, Yamanaka K, Tang X & Moore BS Direct cloning and heterologous expression of natural product biosynthetic gene clusters by transformation-associated recombination. Methods Enzymol. 621, 87–110 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng Z, Kim JH & Brady SF Fluostatins produced by the heterologous expression of a TAR reassembled environmental DNA derived type II PKS gene cluster. J. Am. Chem. Soc 132, 11902–11913 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim JH et al. Cloning large natural product gene clusters from the environment: piecing environmental DNA gene clusters back together with TAR. Biopolymers 93, 833–844 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamanaka K et al. Direct cloning and refactoring of a silent lipopeptide biosynthetic gene cluster yields the antibiotic taromycin A. Proc. Natl Acad. Sci. USA 111, 1957–1962 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Noskov VN et al. Isolation of circular yeast artificial chromosomes for synthetic biology and functional genomics studies. Nat. Protoc 6, 89–96 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gibson DG et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–347 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Lartigue C et al. Creating bacterial strains from genomes that have been cloned and engineered in yeast. Science 325, 1693–1696 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Benders GA et al. Cloning whole bacterial genomes in yeast. Nucleic Acids Res. 38, 2558–2569 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karas BJ, Tagwerker C, Yonemoto IT, Hutchison CA 3rd & Smith HO Cloning the Acholeplasma laidlawii PG-8A genome in Saccharomyces cerevisiae as a yeast centromeric plasmid. ACS Synth. Biol 1, 22–28 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Tagwerker C et al. Sequence analysis of a complete 1.66 Mb Prochlorococcus marinus MED4 genome cloned in yeast. Nucleic Acids Res 40, 10375–1038 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karas BJ et al. Assembly of eukaryotic algal chromosomes in yeast. J. Biol. Eng 7, 30(2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mitchell LA et al. Versatile genetic assembly system (VEGAS) to assemble pathways for expression in S. cerevisiae. Nucleic Acids Res. 43, 6620–6630 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shang Y et al. Construction and rescue of a functional synthetic baculovirus. ACS Synth. Biol 6, 1393–1402 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Oldfield LM et al. Genome-wide engineering of an infectious clone of herpes simplex virus type 1 using synthetic genomics assembly methods. Proc. Natl Acad. Sci. USA 114, E8885–E8894 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vashee S et al. Cloning, assembly, and modification of the primary human cytomegalovirus isolate Toledo by yeast-based transformation-associated recombination. mSphere 2, pii: e00331–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xie YG et al. Cloning of a novel, anonymous gene from a megabase-range YAC contig in the neurofibromatosis type 2/meningioma region on human chromosome 22q12. Hum. Mol. Genet 2, 1361–1368 (1993). [DOI] [PubMed] [Google Scholar]
- 30.Loots GG Modifying yeast artificial chromosomes to generate Cre/LoxP and FLP/FRT site-specific deletions and inversions. Methods Mol. Biol 349, 75–84 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Jiang W et al. Cas9-Assisted Targeting of CHromosome segments CATCH enables one-step targeted cloning of large gene clusters. Nat. Commun 6, 8101(2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang W & Zhu TF Targeted isolation and cloning of 100-kb microbial genomic sequences by Cas9-assisted targeting of chromosome segments. Nat. Protoc 11, 960–975 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Medema MH & Osbourn A Computational genomic identifcation and functional reconstitution of plant natural product biosynthetic pathways. Nat. Prod. Rep 33, 951–962 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Noskov VN et al. A novel strategy for analysis of gene homologs and segmental genome duplications. J. Mol. Evol 56, 702–710 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Furter-Graves E & Hall BD DNA sequence elements required for transcription initiation of the Shizosaccharomyces pombe ADH gene in Saccharomyces cerevisiae. Mol. Gen. Genet 223, 407–417 (1990). [DOI] [PubMed] [Google Scholar]
- 36.Miret JJ, Pessoa-Brandao L & Lahue RS Orientation-dependent and sequence-specific expansions of CTG/CAG trinucleotide repeats in Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA 95, 12438–12443 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Noskov V et al. Defining the minimal length of sequence homology required for selective gene isolation by TAR cloning. Nucleic Acids Res. 29, E32(2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.D’Rose V, Johny TK & Bhat S Comparative analysis of metagenomic DNA extraction methods from gut microbiota of zebrafish (Danio rerio) for downstream next-generation sequencing. J. Appl. Biol. Biotechnol 7, 1–15 (2019). [Google Scholar]
- 39.Kouprina N, Noskov VN, Koriabine M, Leem SH & Larionov V Exploring transformation-associated recombination cloning for selective isolation of genomic regions. Methods Mol. Biol 255, 69–89 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Kouprina N, Noskov VN & Larionov V Selective isolation of large chromosomal regions by transformation-associated recombination cloning for structural and functional analysis of mammalian genomes. Methods Mol. Biol 349, 85–101 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Leem SH et al. Optimum conditions for selective isolation of genes from complex genomes by transformation-associated recombination cloning. Nucleic Acids Res. 31, e29(2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang X et al. Identification of thiotetronic acid antibiotic biosynthetic pathways by target-directed genome mining. ACS Chem. Biol 10, 2841–2849 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bilyk O, Sekurova ON, Zotchev SB & Luzhetskyy A Cloning and heterologous expression of the grecocycline biosynthetic gene cluster. PLOS One 11, e0158682(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanveer A, Yadav S & Yadav D Comparative assessment of methods for metagenomic DNA isolation from soils of different crop growing fields. 3 Biotech 6, 220(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lazarevic V, Gaïa N, Girard M, François P & Schrenzel J Comparison of DNA extraction methods in analysis of salivary bacterial communities. PLOS One 8, e67699(2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Larionov V, Kouprina N, Solomon G, Barrett JC & Resnick MA Direct isolation of human BRCA2 gene by transformation-associated recombination in yeast. Proc. Natl Acad. Sci. USA 94, 7384–7387 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Annab L et al. Isolation of a functional copy of the human BRCA1 gene by TAR cloning in yeast. Gene 250, 201–208 (2000). [DOI] [PubMed] [Google Scholar]
- 48.Kouprina N et al. Dynamic structure of the SPANX gene cluster mapped to the prostate cancer susceptibility locus HPCX at Xq27. Genome Res. 15, 1477–1486 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]