

Abstract
Fifty years have passed since the discovery of glial fibrillary acidic protein (GFAP) by Lawrence Eng and colleagues. Now recognized as a member of the intermediate filament family of proteins, it has become a subject for study in fields as diverse as structural biology, cell biology, gene expression, basic neuroscience, clinical genetics and gene therapy. This review covers each of these areas, presenting an overview of current understanding and controversies regarding GFAP with the goal of stimulating continued study of this fascinating protein.
Keywords: GFAP, astrocyte, intermediate filament, Alexander disease
Glial fibrillary acidic protein, typically referred to as GFAP, is an intermediate filament protein that arose early in vertebrate evolution, coinciding with the development of different forms of glial cells in the central nervous systems of primitive fish (Wicht et al., 1994; Kálmán and Gould, 2001); reviewed in (Appel, 2013). Intermediate filaments are key components of the cytoplasmic cytoskeleton, with multiple roles encompassing structural support, scaffolding for enzymes and organelles, and mechanosensing of the extracellular environment (Lowery et al., 2015). Based on sequence homology, GFAP is classified as a type III intermediate filament, along with vimentin (expressed in multiple cell types), desmin (skeletal and cardiac muscle), and peripherin (neurons) (Geisler and Weber, 1983; Eriksson et al., 2009). Its discovery was reported by Lawrence Eng at a meeting of the International Society of Neurochemistry in September 1969. GFAP was quickly adopted as a marker of astrocytes in the central nervous system (CNS); and subsequently its elevated expression recognized as an indicator of gliosis associated with brain injury or disease. Since then, there has been a steady increase in our understanding of GFAP’s role in health and disease. Control of GFAP’s expression and measurement of its levels is now pursued in both basic and clinical neuroscience, with its influence now extending to genetic medicine and gene therapy.
In 2000, Eng et al. published a broad overview of research on GFAP, covering its first 31 years in the scientific literature (Eng et al., 2000). A count of publications per year that mention GFAP finds the trend noted by Eng in 2000 continuing to the present (Figure 1). As we now pass the 50th anniversary of GFAP’s discovery, we consider it appropriate to provide an updated account, concentrating on the most recent 20 years. Some may find this survey of current knowledge helpful, some may find the battle over controversies irritating, but hopefully everyone will find the unknowns an irresistible draw to future research. Much remains to be learned.
Figure 1.

GFAP Publications. Number of publications listed in PubMed each year retrieved by a search consisting of “GFAP” or “glial fibrillary acidic protein.”
Discovery
The initial publication describing what would later be called GFAP derived from protein analysis of three different samples of gliotic scars in the CNS—plaques from individuals with multiple sclerosis, postsurgical scars, and the periventricular region from aged individuals with hydrocephalus ex vacuo (causes not further specified) (Eng et al., 1971; Figure 2). With assistance from the laboratory of Eric Shooter, the amino acid composition of the major band isolated by gel electrophoresis was defined, which along with its solubility properties clearly distinguished this protein from epithelial keratins and other filamentous proteins abundant in the CNS, such as microtubules and neurofilaments (then called “filarin”). The terms “GFA protein”, or more simply “GFAP”, came into common use after the publication of an immunological study by Uyeda et al. (1972).
Figure 2.

Western Blot Detection of GFAP. Samples shown in the figure are partial purifications of proteins from brain tissue containing a multiple sclerosis plaque (Lane 5), leukotomy scar (Lane 6), multiple sclerosis plaque + leukotomy scar (Lane 7), and periventricular layer in hydrocephalus ex vacuo (Lane 8). The common, major band marked by an X by the authors was identified by amino acid analysis as a novel acidic protein, now known as GFAP. Figure reprinted from Eng et al. (1971), with permission from Elsevier.
Structure
In 1984, Lewis et al. reported the first cloning of a Gfap transcript by screening a mouse brain cDNA expression library with a polyclonal antiserum against bovine GFAP (Lewis et al., 1984). One nearly full-length clone was isolated, which permitted prediction of 97% of the amino acid sequence for this species. Brenner et al. (1990) subsequently cloned cDNA and genomic sequences for human GFAP and corrected the identification of the protein start site and the initial amino acid sequence. The human cDNA predicted a GFAP 432 amino acids in length. Comparison of its sequence to that of other intermediate filaments suggested that it shared the common property of having a central α-helical rod domain flanked by more variable head and tail domains (Figure 3). GFAP exhibits extremely high conservation among species, having 90% identity at the protein level among human, mouse, and rat (Brenner, 1994), and even 67% between human and zebrafish (Nielsen and Jørgensen, 2003). Much remains to be learned about the 3-dimensional structure of intermediate filaments, with most progress made with another type III filament, vimentin (Chernyatina et al., 2012). The structure of the rod 1B domain of GFAP was analyzed by Kim et al. (2018a), but their speculation about the effects of amino acid variants failed to distinguish between pathogenic and benign mutations (see the Danger section).
Figure 3.
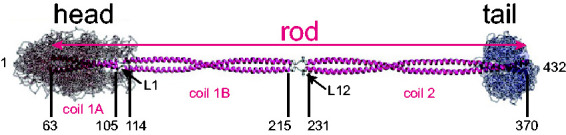
Model of the GFAP Dimer. Shown is a model of the GFAP coiled-coil dimer, based on that of vimentin, showing the α-helical central rod domain flanked by disordered head and tail domains (adapted from Figure 2(E) of Chernyatina et al., 2015). The coordinates for the boundaries of each coil domain are based on amino acid homology between human GFAP and vimentin. Note that GFAP coil 2 may extend further than shown, to position 378 (Roy Quinlan, personal communication, June 4, 2020).
Under normal circumstances, the type III intermediate filaments assemble in a multistep process that begins with parallel binding of monomers to form dimers, which then associate in an antiparallel fashion to form tetramers, with succeeding steps of lateral associations to produce octamers, oligomers, and finally the structures visible by electron microscopy termed unit length filaments that contain anywhere from 30 to 59 monomers in cross section (depending on the filament) (Herrmann et al., 1999). The unit length filaments anneal in a nonpolar fashion (unlike microtubules and actin) to generate mature intermediate filaments (for review, see Herrmann and Aebi, 2004). The association of dimers to form tetramers is primarily mediated through the rod domains. The roles of the head and tail domains appear to vary considerably among the different groups of intermediate filaments and are poorly understood. Using truncation mutants and cell culture as well as cell-free systems, both the head and tail domains of GFAP or its close relative, vimentin, were found important for proper assembly into mature 10-nm filaments (Chen and Liem, 1994a; Ralton et al., 1994; Herrmann et al., 1996). However, these studies did not did not consider the role of binding partners such as αB-crystallin (Nicholl and Quinlan, 1994).
Posttranslational modifications occur at multiple sites in GFAP and may impact its properties and function (Figure 4; for a general review of posttranslational modifications and intermediate filament biology, see Snider and Omary, 2014). Phosphorylation has received the most attention. It is performed by several kinases, including protein kinase A, protein kinase C, calcium/calmodulin-dependent protein kinase II, Rho-kinase, and Cdc2 kinase (summarized in Sullivan et al., 2012; Battaglia et al., 2019). Nothing is known about the relevant phosphatases that reverse these modifications. Phosphorylation of Ser8 and Ser13 (human sequence numbers) in particular has been implicated in regulating assembly and disassembly of GFAP (Inagaki et al., 1990), which might be important during mitosis (Nakamura et al., 1992). Phosphorylation of Ser8 also promotes binding to 14-3-3γ (Li et al., 2006), although the functional significance of this binding is not known. Increased phosphorylation of Ser13 occurs in response to hypoxic injury (Sullivan et al., 2012), and in the conditions of frontotemporal lobar dementia (Herskowitz et al., 2010) and Alexander disease (Battaglia et al., 2019; discussed in the Danger section). Phosphorylation of an abundant protein such as GFAP may also impact the intracellular pools of phosphate, thereby regulating diverse signaling pathways, as has been suggested for keratins as the “sponge hypothesis” (Ku and Omary, 2006).
Figure 4.
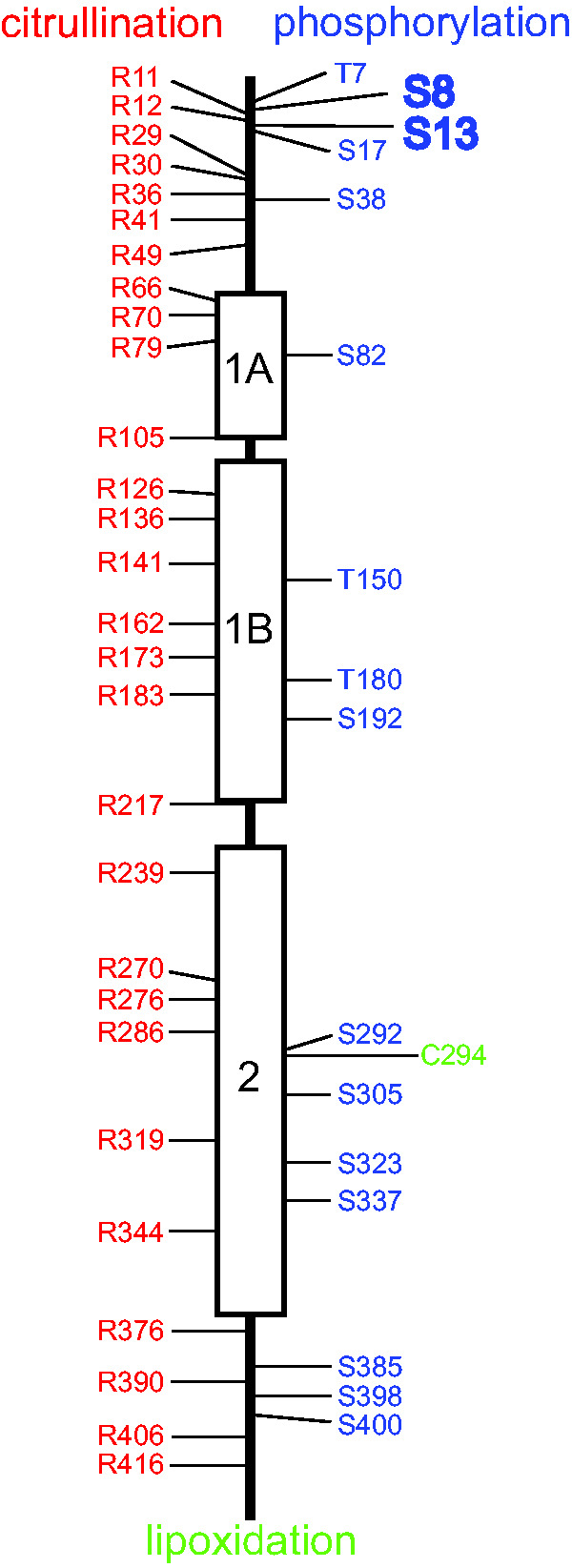
Posttranslational Modifications of GFAP. The three α-helical regions of the GFAP central rod domain are indicated by boxes separated by nonhelical linker regions, and the N-terminal head and C-terminal tail domains are indicated by straight lines. Amino acids identified as sites of citrullination are shown in red (Jin et al., 2013; Brenner and Nicholas, 2017; Faigle et al., 2019), sites of phosphorylation are shown in blue (Inagaki et al., 1994; Battaglia et al., 2019), and the single site for lipoxidation is shown in green (Viedma-Poyatos et al., 2018). Evidence also exists for N-glycosylation and O-glycosylation of GFAP, but none of the specific sites for these modifications is known (Kanninen et al., 2004; Korolainen et al., 2005). Phosphorylation of Ser8 and Ser13 are displayed in larger font because multiple studies have implicated them in regulation of filament assembly or responses to injury or disease (Sullivan et al., 2012; Battaglia et al., 2019). The functional significance of the other modifications has yet to be established.
Citrullination, the enzymatic conversion of peptidyl-arginine to peptidyl-citrulline through deimination, is another posttranslational modification of potential significance (reviewed in Brenner and Nicholas, 2017). Citrullination has been observed at multiple arginine residues in neurologically normal individuals (Jin et al., 2013; Faigle et al., 2019); both the number of sites and/or amount increases in several diseases, including multiple sclerosis (Nicholas et al., 2004; Faigle et al., 2019), experimental allergic encephalomyelitis (Nicholas et al., 2005), Alzheimer’s disease (Ishigami et al., 2005, 2015), and hepatic fibrosis (Kim et al., 2018b). In vitro studies have suggested that citrullination interferes with GFAP polymerization (Inagaki et al., 1989). Finally, the sole cysteine in human GFAP, present at position 294 and conserved in animals ranging from goldfish to mice, is susceptible to lipoxidation in cultured cells via prostaglandins. Mutating this cysteine to serine interferes with the ability of the GFAP to assemble into the cytoskeletal network, suggesting that its lipoxidation would have a similar effect (Viedma-Poyatos et al., 2018). Whether lipoxidation occurs in vivo, and if so, has disease relevance, has not yet been determined.
The predominant isoform in astrocytes is GFAPα, which is the 432 amino acid (for the human sequence) protein that is the subject of most publications. Two other GFAP isoforms, GFAPβ and GFAPγ, derive from alternative RNA start sites (Figure 5). The start site for GFAPβ mRNA is upstream of that of GFAPα, being reported as 169 or 79 nucleotides further 5′ (Feinstein et al., 1992; Lim et al., 2008). In contrast, transcription of GFAPγ commences in GFAPα intron 1, about 130 nucleotides from the end of this intron (Zelenika et al., 1995). Both the GFAPβ and the GFAPγ transcripts contain the GFAPα exons downstream of their start points. GFAPβ is the primary GFAP transcript in the rodent peripheral nervous system (PNS) and also is present in rodent brain and hepatic stellate cells, and in human gliomas and lymphocytes (Galea et al., 1995; Riol et al., 1997). The ability of certain monoclonal antibodies to stain GFAP in the CNS but not in the PNS suggests a difference between the GFAPα and GFAPβ proteins, but it has yet to be established whether this difference is in the primary sequence or posttranslational modifications. This question and other properties of GFAPβ are discussed in detail in the accompanying supplemental file, “GFAPβ.” GFAPγ transcripts are present in mouse and human brain, and in mouse bone marrow and spleen. The longest open reading frame starts in exon 4, which is near the end of rod domain 1B and would thus be expected to produce a polymerization-incompetent protein. The single attempt to detect a protein encoded by the GFAPγ mRNA failed (Zelenika et al., 1995).
Figure 5.

GFAP Isoforms. Diagram of GFAP mRNA structure indicating major isoforms generated by different transcriptional start sites (small arrows) and alternative splicing events. The boxes indicate the translated regions, and the thick lines indicate the untranslated regions (not drawn to scale). For GFAPβ, uncertainty remains about where translation begins (for details, see supplemental file “GFAPβ”). For GFAPγ, transcription begins within intron 1, but the longest open reading frame does not start until exon 4 where it is in phase with GFAPα. Figure modified from Helman et al. (2020).
Additional GFAP isoforms are produced by alternative splicing, a process unique to GFAP among cytoplasmic intermediate filaments other than synemin (reviewed in Hol and Pekny, 2015; Figure 5). In 1999, Condorelli et al. reported a novel transcript in rat hippocampus, designated GFAPδ (Condorelli et al., 1999). Sequencing of the GFAPδ transcript revealed that it contained a previously undetected exon within GFAPα intron 7, and so termed exon 7a, whose presence in GFAPδ replaces exons 8 and 9, resulting in a different sequence for the C-terminal tail domain. This exon, defined by a new splice acceptor site and polyadenylation signal, is present in mammals but not lower vertebrates such as birds and fish (Singh et al., 2003). Nielsen et al. (2002) independently detected this isoform through a yeast two-hybrid assay as a binding partner for presenilin, and gave it the name GFAPε. Despite efforts to settle on only GFAPδ for terminology (Roelofs et al., 2005), its designation as GFAPε continues in the clinical literature (here we will use GFAPδ). Another isoform of interest, also generated by alternative splicing in intron 7 but different from that of GFAPδ, is GFAPκ (Blechingberg et al., 2007). While GFAPα can assemble into mature intermediate filaments on its own, GFAPδ and GFAPκ cannot achieve this feat but can coassemble with GFAPα or vimentin (Nielsen and Jørgensen, 2004; Blechingberg et al., 2007). By varying the proportion of α and δ in cell-free assays or transfected cell lines, both Nielsen and Jørgensen (2004) and Perng et al. (2008) found that GFAPδ by itself is prone to aggregation and prevents normal filament assembly if above a threshold concentration (10%–30% of total GFAP, depending on the assay). Based on the relative level of its mRNA, it constitutes about 4% of the total GFAP in normal mouse brain (Thomsen et al., 2013). Similar coassembly titrations have not been performed for GFAPκ, which is present at less than 1% of total GFAP in normal mouse brain (Blechingberg et al., 2007).
Unlike GFAPα, the C-terminal sequences of both GFAPδ and GFAPκ vary considerably among species. Singh et al. (2003) observed that the sequence of exon 7a is strongly conserved among higher primates (e.g., human, chimpanzee, gorilla, orangutan, baboon), but not among other mammals (e.g., pig, rat, mouse). In addition, the conserved sequence among higher primates contains an unusually high number of nonsynonymous sequence changes compared with other species. This led them to suggest that there has been positive selection for a novel (though as yet unspecified) function in the higher primates. Another isoform that contains sequence from exon 7a is GFAPλ, which is a hybrid between GFAPα and GFAPδ (Helman et al., 2020). It includes nearly all of exon 7a, but just before the last 7a codon, it is spliced to include GFAPα exons 8 and 9 (Figure 5). Point mutations in exon 7a that result in overexpression of this isoform are associated with Alexander disease (see later).
Expression
GFAP is predominantly but not exclusively expressed in astrocytes of the CNS (Figure 6). Early evidence for this specificity came from standard immunostaining of both cultured cells and tissues (Bignami et al., 1972; Raff et al., 1979). It is widely accepted that GFAP is much higher in subpial and white matter astrocytes than gray matter astrocytes (Lundgaard et al., 2013; Olabarria and Goldman, 2017), a pattern noted in early studies from the Eng lab (Ludwin et al., 1976). It also coincides with analysis of mRNA by in situ hybridization (Lewis and Cowan, 1985) as well as ultrastructural observations that intermediate filaments are more abundant in astrocytes of these regions compared with most of those in the gray matter (Peters et al., 1991). However, immunocytochemistry must be interpreted with caution, as it can be highly sensitive to the details of processing and fixation (Shehab et al., 1990).
Figure 6.

Tissue-Specific Expression of GFAP. GFAP expression in different tissues reflected by mRNA levels, illustrated in the Consensus Dataset by the Human Protein Atlas (data available from v19.3.proteinatlas.org; https://www.proteinatlas.org/ENSG00000131095-GFAP/tissue; Uhlén et al., 2015).
PBMC = peripheral blood mononuclear cell; NK = natural killer.
Developmentally, GFAP first appears in radial glia (Levitt and Rakic, 1980), which are progenitors for both astrocytes and neurons. The subsequent rise in GFAP expression as astrocytes differentiate is often viewed as a defining feature of astrocyte maturation. Mouse cortical astrocytes grown in primary culture display monoallelic rather than the more typical biallelic expression of GFAP (Takizawa et al., 2008). Monoallelic expression was not found for mouse hippocampal astrocytes and has not yet been verified to occur in vivo or to take place in astrocytes from other species.
In addition to its baseline expression, GFAP levels increase as a nearly universal response to injury and disease, when astrocytes enter a different state (or states) of “reactive gliosis” that itself defies simplistic definition and likely varies depending on the insult (for recent reviews, see Anderson et al., 2014; Pekny et al., 2016; Liddelow and Barres, 2017; Escartin et al., 2019). Single-cell expression analysis both in vivo and in vitro show marked variation in the levels of expression of GFAP in astrocytes that becomes even more exaggerated after injury (Wilhelmsson et al., 2017; Zeisel et al., 2018; Pekny et al., 2019). The significance of this wide variation in GFAP expression is not known, and indeed, some cells that fulfill many other criteria for being considered “astrocytes” may not express any detectable GFAP (Bradley et al., 2019).
Among the most convincing examples of GFAP expression in nonastrocytic cells are nonmyelinating Schwann cells, enteric glia, and the neurogenic stem cells of the subgranular zone in the hippocampus and the subventricular zone surrounding the lateral ventricles. Other sites of GFAP expression, some still speculative, include stellate cells of the liver, pancreas, and vocal fold, Leydig and Sertoli cells in the testis, lens epithelium, lymphocytes, and vertebral and tracheal cartilage (for review, see Messing, 2018b). In the setting of cancer, several surprising examples occur, such as salivary gland tumors and myoepithelial tumors of soft tissue (Huang et al., 1996; Jo and Fletcher, 2015). Clearly, the utility of GFAP as a marker for astrocytes is context-dependent.
The role of GFAP expression as a marker for astrocytes and for their differentiation and response to injury has prompted multiple studies of its transcriptional regulation (Besnard et al., 1991; Mucke et al., 1991; Brenner et al., 1994; Johnson et al., 1995; Bernardos and Raymond, 2006; Yeo et al., 2013; Brenner et al., 2019). More than a dozen transcription factors have been proposed to directly affect GFAP expression. Paramount among these are STAT3, which is critically important for the developmental onset of expression and for its maintenance in the resting state, and AP-1, which appears required for GFAP upregulation following injury (an extensive discussion of regulation of GFAP expression will be published separately. An important outcome of these promoter studies has been development of cassettes to target expression to astrocytes. These have been extensively used both to create transgenic mice and to direct cell-specific viral activity for both basic research and therapeutics (von Jonquieres et al., 2013; Vagner et al., 2016). Descriptions of the more commonly used GFAP promoters are provided in the accompanying supplemental file, “GFAP Transgenes.”
Functions
Because GFAP arose early in vertebrate evolution and has remained relatively unchanged over an extraordinarily long period of time, one might surmise that it is necessary for one or more critical functions (an extensive review of putative functions for GFAP can be found in Brenner, 2014). The expectation that clarity on this topic would come from generation of mouse knockouts was upended by the results from four groups that independently produced such animals, with essentially the same result. Mice rendered completely deficient for GFAP were born in normal numbers, grew to adulthood, and reproduced well (reflecting a complex behavior) (Gomi et al., 1995; Pekny et al., 1995; Liedtke et al., 1996; McCall et al., 1996). Similar vitality has been observed by us for a GFAP knockout rat (Hagemann, unpublished data). However, although the rodent knockouts appear overtly normal, aspects of both hippocampal and cerebellar physiology, such as long-term potentiation and long-term depression, may be defective (McCall et al., 1996; Shibuki et al., 1996).
More significant deficiencies become apparent following injury. As one example, based on the general concept that intermediate filaments in other cell types provide tensile strength and support intercellular connections, Nawashiro et al. (1998) studied the effects of closed head injury using a weight-drop device with anesthetized animals. In this model, most of the GFAP-null mice suffered mortality, whereas all wild-type mice survived. Death was likely due to whiplash injury to the cervical spinal cord that occurred when the foam cushioning allowed head movement following impact (Figure 7). When the heads were fixed in position during impact, no mortality occurred. In another injury model, regeneration of axons in the PNS was impaired in the GFAP-null mice after crush injury to the sciatic nerve (Triolo et al., 2006), although it is not clear whether this reflects changes in interactions between neurons and astrocytes within the CNS, or between axons and nonmyelinating Schwann cells in the PNS. GFAP-null mice are also impaired in their response to both inflammation (experimental allergic encephalomyelitis) and infection (Staphylococcus aureus, Toxoplasma gondii) (Liedtke et al., 1998; Stenzel et al., 2004). On the other hand, the GFAP-null state does not change susceptibility to scrapie infection (Gomi et al., 1995), and viral infections have not yet been studied. An initial claim that GFAP-null mice have a late-onset myelin deficiency (Liedtke et al., 1996) was not reported by the other three groups that generated GFAP-null mice.
Figure 7.

Hypersensitivity of GFAP-Null Mice to Traumatic Cerebrospinal Injury. Neuropathology following trauma from closed head injury is concentrated in the cervical spinal cord of GFAP-null mice (–/–), whereas wild-type mice (+/+) subjected to the same injury are relatively unaffected. Figure reproduced from Nawashiro et al. (1998), with permission.
The generally mild phenotype of GFAP-null animals in the absence of injury may derive from the continued presence of vimentin. When both GFAP and vimentin are absent, several functional deficits are observed not seen for either of the single knockouts, such as release of taurine from cultured astrocytes exposed to hypotonic stress (Ding et al., 1998), regeneration following lesioning of the entorhinal cortex (Wilhelmsson et al., 2004), and infarct volume after ischemia induced by transection of the middle cerebral artery (Li et al., 2008). Although the double knockout experiments suggest participation of GFAP in these additional functions, further studies are required for establishing causality. Other claims related to GFAP functions have been based on studies of cultured astrocytes, but the validity of these are questionable because these cells deviate considerably from the phenotype of astrocytes in vivo (Berger and Hediger, 2000; Wilhelm et al., 2004; Cahoy et al., 2008; Carter et al., 2012). Pekny and Pekna (2014) have proposed a dual role for GFAP in the context of the reactive response of astrocytes, with beneficial acute effects following injuries such as sequestration of ischemic or inflammatory lesions, but increasingly detrimental effects in some chronic states such as interference with regeneration.
The early era of knockout experiments suffered from important limitations, particularly related to the lack of temporal control. To date, all published studies on GFAP knockouts have used one of the four strains generated in the 1990s, none of which employed targeting vector designs that allow for inducible deletion after administration or withdrawal of drugs such as tetracycline or tamoxifen. Hence, all existing GFAP knockouts are GFAP deficient from the moment of conception, raising the possibility that compensatory changes mask the consequences of GFAP deficiency. Nevertheless, the obvious candidates for compensation (other intermediate filaments such as vimentin and nestin) do not change their levels of mRNA and/or protein (McCall et al., 1996; Triolo et al., 2006; Kamphuis et al., 2015). One study compared gene expression profiles of nulls versus wild types, finding changes in expression of 392 genes. None of the changes was identified as possibly being compensatory for the loss of GFAP. In addition, some of these differences could derive from the mixed genetic backgrounds of the mice used, and it was not stated if the animals were sex-matched (Kamphuis et al., 2015). Removal of GFAP in an adult, after the initial period of development has already taken place, might reveal functions of which we are currently unaware. Recent progress in the development of antisense methods for suppression of gene expression, described below in the Alexander disease section, will allow a new set of approaches for addressing these questions.
It is interesting that some vertebrate species have naturally undergone GFAP gene deletion during their evolution (Martinez-De Luna et al., 2017). These include frogs, toads, and some caecilians, whereas salamanders and newts retain the gene. Whether functional knockouts of the GFAP gene occur in the human population is not known. In a collection of 2,026 individuals at the Children’s Hospital of Philadelphia, 4 were heterozygous for GFAP deletions (Shaikh et al., 2009). If this deletion frequency generally applies to the U.S. population and absence of GFAP is benign, it predicts that approximately 4 individuals per million will lack a functional GFAP, corresponding to about 1,200 people in the United States (or 30,000 worldwide). As genome sequencing becomes more routine, it will be of interest to find if such individuals exist, and if so, the consequence of absence of GFAP.
The subcellular localization of GFAP mRNA and protein might be relevant for function, but few studies have examined this topic, and none has established a causal relationship. A general finding for GFAP mRNA is that it colocalizes with the protein, being particularly present within processes (Sarthy et al., 1989; Erickson et al., 1992; Medrano and Steward, 2001; Thomsen et al., 2013). This might minimize the need for transport of the protein to its assembly site. A subplasmalemmal network of GFAP filaments, including association with hemidesmosomes, has been described in the end feet of subpial and perivascular astrocytes of the rat by Nakazawa and Ishikawa (1998). Extrapolating from a similar arrangement for keratins in epithelial cells, and the possibility that links extend from the plasma membrane to the nuclear membrane, Quinlan et al. (2017) proposed a general model in which the network of intermediate filaments has a role in mechanotransduction. The effect on tissue mechanics of conditions that change GFAP levels has been mixed. In both in vitro (stretch injury) and in vivo (cortical stab wound) models of gliosis, which produce increased GFAP levels, astrocytes and tissues become softer as measured by atomic force microscopy (Miller et al., 2009; Moeendarbary et al., 2017). However, when GFAP increases in the context of heterozygous missense mutations, as occurs in Alexander disease (discussed in the Danger section), tissue stiffening occurs (Wang et al., 2018). Resolution of these apparent contradictory results, including whether the GFAP concentration changes are causal or just epiphenomena, awaits further investigation. Studies of GFAP-null mice or astrocytes could be informative.
A novel role for GFAP as a regulator of chaperone-mediated autophagy (CMA) by lysosomes was reported by Bandyopadhyay et al. (2010), who found that GFAP binds to the lysosome-associated membrane protein type 2A (LAMP-2A), a protein involved in CMA substrate transport into lysosomes. In a follow-up study, phosphorylation of GFAP was proposed as a switch to temporally inactivate CMA activation (Arias et al., 2015). These studies were performed in mouse and rat fibroblasts and hepatocytes with GFAP being identified by multiple methods. There are several curiosities about these results, in addition to the studies being confined to two cell types that are only minor sites of expression. Contrary to expectation, CMA was increased by both the addition of purified GFAP to isolated lysosomes in vitro and by the depletion of GFAP by knockdown or knockout in vivo. In addition, mutation of the predicted serine 8 phosphorylation site to mimic phosphorylation (S to E), or to prevent phosphorylation (S to A), both eliminated the effect of GFAP on CMA. Whether the proposed regulation of CMA activity by GFAP has biological consequences is uncertain. It is not known if the proposed mechanism is present in other species or cell types, including astrocytes, and no fibroblast or hepatic phenotype has been reported in GFAP-null mice.
Most of the preceding section reflects focus on the major isoform, GFAPα. The functions of the minor isoforms, as for GFAPα, are poorly understood. Speculation has focused on potential roles for GFAPδ in stem cells (Roelofs et al., 2005; van den Berge et al., 2010) and gliomas (Moeton et al., 2016; Stassen et al., 2017; van Bodegraven et al., 2019b). No animal models have yet been made to selectively express or delete a specific isoform. Possibly complicating such an approach in animal models is evidence that GFAPδ is expressed in different populations of cells in humans versus mouse (Kamphuis et al., 2012; Mamber et al., 2012); and as noted earlier, the marked divergence between lower and higher mammalian species in the amino acid sequence encoded by exon 7a may correspond to functional differences as well.
Degradation
Although there is a sizable literature on synthesis of GFAP and the factors that regulate this process, relatively little attention has been given to pathways for its degradation. Early studies from the Eng lab found that GFAP was particularly susceptible to degradation by a calcium-mediated protease (later called “calpain”) (DeArmond et al., 1983), with predominant cleavage sites after residues Asn59 and Thr383, yielding a central 37-kDa fragment consisting of residues 60–383 that essentially consists of just the rod domain (Dahl and Bignami, 1975; Zhang et al., 2014). These cleavages may be responsible for the degradation product of about 37 kDa often seen in immunoblots of GFAP isolated from tissue (discussed in Heaven et al., 2019). Subsequently, cleavage sites have been identified for caspase 3 after Asp266 (Mouser et al., 2006; Acarin et al., 2007) and caspase 6 after Asp142 (Jonesco et al., 2019) and D225 (Chen et al., 2013). These cleavage sites are shown diagrammatically in Figure 8. It is not yet known whether any of the degradation products acquires new and deleterious functions, as has been suggested for the huntingtin protein (Orr et al., 2008) and β-amyloid (Yankner et al., 1989). None of these degradation fragments appears likely to be capable of self-assembly, although the 1–225 fragment that is one product caspase 6 cleavage is aggregation prone (Chen et al., 2013). Whether any of these cleavages occur at the level of the filament, and if so, causes disassembly, is not known. In addition to sequence-specific proteases, evidence exists for the involvement of both the proteasome and autophagy in GFAP degradation (Tang et al., 2008; Middeldorp et al., 2009; Tang et al., 2010).
Figure 8.
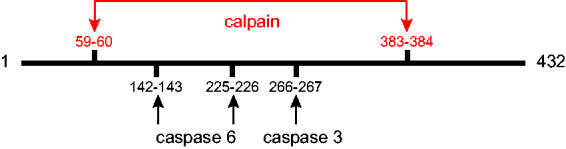
Protease Cleavage Sites in GFAP. Diagram showing the major cleavage sites by proteases that contribute to degradation of GFAP. Calpain cleavage produces a ∼37-kDa product (Zhang et al., 2014). Caspase 3 has one cleavage site, producing 31- and 19-kDa products (Mouser et al., 2006). Caspase 6 has two alternative cleavage sites, producing either 26- and 24-kDa bands (Chen et al., 2013) or 16- and 34-kDa bands (Jonesco et al., 2019).
However GFAP degradation is achieved, it is a slow process; similar to other intermediate filaments, GFAP has a relatively long half-life in vivo. Using S-35-labeled arginine, DeArmond et al. (1986) showed its half-life in mouse spinal cord was approximately 9 weeks. More recently, two groups used nonradioactive methods of isotope labeling with N-15 amino acids administered in the diet to determine the turnover rate for GFAP in adult mice. Under the experimental conditions used, where GFAP levels were essentially at steady state, turnover rate is equivalent to half-life and was measured as 28 days (Price et al., 2010; Moody et al., 2017). What regulates degradation is still poorly understood, although at least one important factor is gigaxonin, an E3 ubiquitin ligase adaptor protein that targets its substrates to the proteasome and that fosters degradation of several intermediate filament proteins, including GFAP, vimentin, peripherin, and neurofilaments (Mahammad et al., 2013; Lin et al., 2016).
Applications
As noted earlier, GFAP expression was predominantly found in astrocytes of the CNS, and thus GFAP was quickly adopted by the neuroscience community as a convenient marker for cell identification (Bignami et al., 1972; Raff et al., 1979; Levitt and Rakic, 1980). Subsequently, the value of GFAP detection in tumor diagnosis quickly emerged. The presence of GFAP complemented previous morphological assessments that related the neoplastic cells in astrocytomas to their presumed normal counterparts and provided a very helpful tool for distinguishing astrocytomas from other gliomas and tumors involving the CNS (Duffy et al., 1977; reviewed by Eng and Rubinstein, 1978). While the presence of GFAP supported the astrocytic nature of such cells, it has since proven difficult to relate the levels of expression with degree of malignancy (recently reviewed by van Bodegraven et al., 2019a). Tumor diagnosis and classification has now moved into a new era that adds more specific immunohistochemical tests and molecular and genetic phenotyping to traditional morphological characterization for subtyping and grading of CNS neoplasms to predict outcomes and guide treatment (Louis et al., 2016; DeWitt et al., 2017; Yeaney and Brat, 2019).
Although GFAP continues to be used as a marker of astrocytes, especially in their reactive state, this use should be tempered by findings that not all astrocytes express detectable levels of GFAP, whereas many nonastrocytic cells both inside and outside of the CNS do (see the earlier section on Expression). In addition, although standard light microscopic immunostaining for GFAP is commonly used to identify gross astrocyte morphology, intracellular dye filling reveals a much more elaborate architecture of fine processes (Bushong et al., 2002; Figure 9).
Figure 9.

GFAP Immunolabeling and Astrocyte Morphology. Astrocytes in the CA1 region (stratum radiatum) of rat hippocampus, stained for GFAP by immunolabeling and dye-filled with Lucifer yellow to reveal the complexity of fine processes. Figure 2(B) from Bushong et al. (2002)(Copyright 2002 Society for Neuroscience).
In addition to serving as a marker for astrocytes, the presence of GFAP in cerebrospinal fluid (CSF) and blood has been receiving increasing attention as a diagnostic marker of brain injury. One can imagine several mechanisms by which GFAP escapes its intracellular location, especially cell death with subsequent release of normal cytoplasmic contents. Halford et al. (2017) have proposed that the presence of 18- to 25-kDa degradation products in the CSF appear late after traumatic brain injury, and signify cell death, but did not specify from which parts of the protein these fragments derive. GFAP may also be contained within exosomes, whose release is under active control (Lee et al., 2016; Willis et al., 2017; Manek et al., 2018). In the setting of injury or disease, GFAP levels in CSF and blood rise, sometimes to striking levels, particularly in traumatic brain injury, stroke, subarachnoid hemorrhage, and neuromyelitis optica (for reviews, see Liem and Messing, 2009; Petzold, 2015). Monitoring of CSF and blood levels could have particular value in Alexander disease (discussed later), where GFAP is the instigator of the disease process and is also elevated (Kyllerman et al., 2005; Jany et al., 2013).
Danger
Alexander Disease
Surprisingly, although the complete absence of GFAP produces only subtle effects (at least in laboratory rodents), mutations that change only a single amino acid can be lethal. This was revealed through investigation of the devastating leukodystrophy, Alexander disease. The disease occurs in all ethnic groups, affects both sexes, and has ages of onset ranging from fetal through the eighth decade (Prust et al., 2011). Most patients have deficits in white matter (thus the classification among the leukodystrophies), but the degree and distribution of these lesions vary by age. Depending on age of onset and locations of pathology, symptoms include seizures, developmental delays, intellectual disability, difficulty in speaking and swallowing, vomiting, pain, abnormal gait, and autonomic dysfunction. Prevalence was calculated at 1 in 2.7 million for the population of Japan, although this is certain to be an underestimate (Yoshida et al., 2011). Extensive reviews have appeared that are comprehensive (Brenner et al., 2009; Flint and Brenner, 2011; Messing, 2018b) or limited to particular aspects of the disease, such as clinical features (Pareyson et al., 2008; Balbi et al., 2010), neuroinflammation (Olabarria and Goldman, 2017), or genetics (Messing, 2018a). Here, we focus on the most recent findings, which mainly represent attempts to determine mechanisms of the disease.
Variants in GFAP are found in more than 90% of patients clinically diagnosed with Alexander disease; the cause in the other ∼10% is currently unknown (Figure 10). One of the striking features of the genetics is that pathogenic variants have been identified in 87 of the 432 amino acids of the major isoform, GFAPα, and variants in only 5 amino acids are currently considered benign (Pro47Leu, Thr110Ser, Glu223Gln, Asp157Asn, and Asp295Asn) (Messing, 2018a). All known pathogenic variants are genetically dominant and appear to cause a toxic gain of function. Thus, Alexander disease is unlikely to result from loss of any of the “normal” GFAP functions, a conclusion consistent with the absence of Alexander disease-related symptoms in GFAP-null animals. A recent report of putative recessive inheritance has appeared (Fu et al., 2020), but given that the same mutation had previously been shown to be dominant in several patients, the results are more plausibly explained by dominant inheritance along with gene dosage effects. Pathogenic variants are distributed throughout the protein (though few in the nonhelical head domain), suggesting that the disorder is caused by a global property of the protein rather than alteration of a local interaction site. One variant within the head domain has been described, Gly18Val, for which pathogenicity is strongly supported by segregation analysis within the family, although with variable expressivity (Casasnovas et al., 2019). Approximately two thirds of the published mutations are “private” (i.e., found in only one patient or family), thus limiting the potential for genotype–phenotype correlations. Because cases are generally not published if the mutation has already been described, this number for private mutations must be an overestimate; we cannot judge by how much without population-based studies.
Figure 10.

GFAP Mutations in Alexander Disease. Shown is a compilation of GFAP mutations in Alexander disease, prepared in 2012, with their locations in relation to protein structure. The wild-type amino acid is indicated next to the structure, and amino acid replacements within symbols on either side. Early-onset cases (first symptom before the age of 2 years) are on the left, shown as blue circles, and late-onset cases (first symptom after the age of 2 years) are on the right, shown as red circles. Each symbol represents a single patient, except that familial cases, including identical twins, are represented by a single symbol coded for the onset type of the proband. A continually updated list of published mutations is maintained at https://alexander-disease.waisman.wisc.edu. Reprinted with permission from (Messing et al., 2012).
The hallmark pathologic feature, required for the diagnosis, is the presence of cytoplasmic protein aggregates within astrocytes known as “Rosenthal fibers,” named after Werner Rosenthal, the 19th century neuropathologist who first described them as part of a case report on another condition (Rosenthal, 1898; for review, see Wippold et al., 2006; Figure 11). Although Rosenthal fibers are present in several neurological conditions, they are particularly abundant in Alexander disease. These aggregates are complex mixtures of GFAP, other intermediate filaments including vimentin and synemin, stress proteins including αB-crystallin and heat shock protein 27, and other recently described components such as cyclin D2 (Perng et al., 2006; Heaven et al., 2016; Messing, 2018b). The early stages of Rosenthal fiber formation are not yet clear, but they may arise from stress granules (Heaven et al., 2016) or involve focal deposits on existing intermediate filaments along with αB-crystallin (Sosunov et al., 2017). Initially, Rosenthal fibers were viewed as static, and perhaps resistant to degradation, but studies in cultured cells transfected with fluorescently tagged GFAP revealed them as dynamic structures that can disappear over the course of 1 to 2 days (Mignot et al., 2007). Loss of Rosenthal fibers has also been demonstrated in vivo in an Alexander disease mouse model treated with GFAP antisense oligonucleotides (ASOs; see the description given in the Relief section) (Hagemann et al., 2018). Considerable evidence supports the hypothesis that Rosenthal fibers result from accumulation of GFAP above a certain threshold (Messing et al., 1998; Tanaka et al., 2007), although what that threshold is remains uncertain. Whether the Rosenthal fibers are themselves the toxic species, or simply a recognizable byproduct of the entire disease cascade, is not known. There is no evidence for either increased GFAP or Rosenthal fibers in any cell type other than astrocytes, except for neural stem cells in the subgranular zone of the hippocampus and perhaps subventricular zone (Hagemann et al., 2013).
Figure 11.

Rosenthal Fibers. Rosenthal fibers in humans with Alexander disease as seen by (A) light microscopy (bright red globular structures surrounding a vein [v]) and (B) electron microscopy (arrow). Figure reprinted from (Messing et al., 2012), used with permission.
The level of GFAP accumulation in Alexander disease roughly correlates with disease severity (Jany et al., 2013; Walker et al., 2014; reviewed in Messing, 2019), but the mechanisms leading to that accumulation are the subject of debate. At least one contributing factor is an increase in GFAP synthesis, prompted by activation of the GFAP promoter that occurs after onset of expression of the mutant GFAP (Jany et al., 2013). Precisely what pathway mediates this activation is not known. Whether decreased degradation also contributes to accumulation of GFAP is even less clear. On the one hand, evidence exists for mutant GFAP impairing proteasome activity (Tang et al., 2006), for which the most likely culprits are soluble GFAP oligomers rather than Rosenthal fibers (Tang et al., 2010). Yet at the same time there is an increase in autophagy (Tang et al., 2008) and activation of caspases 3 and 6 (Chen et al., 2011, 2013). When the knockin mouse model was studied during adulthood using methods of isotope turnover, the half-life of GFAP was observed to be actually decreased (Moody et al., 2017). Furthermore, recent studies on autopsy tissues from three patients found that the mutant form of the protein is not the majority of the GFAP present in the brain, suggesting that it is less stable than the wild type (Heaven et al., 2019). Cleavage of GFAP by caspase 6 has recently been linked to increased phosphorylation of Ser13, with the latter change being most prominent in the more severe, early-onset forms of disease (Battaglia et al., 2019).
How mutant GFAP impacts astrocyte function is not understood. Plausible suspects include decreased levels of glutamate transporters (specifically Glt-1) (Hagemann et al., 2009; Tian et al., 2010), perhaps mediated through caspase 3 (Boston-Howes et al., 2006). Another candidate is change in the potassium channel Kir4.1. Its levels decrease substantially in the caudal CNS and spinal cord of Alexander disease model mice but actually increase in rostral regions (Sosunov et al., 2013; Minkel et al., 2015; Canals et al., 2018). Loss of function mutations in both Glt-1 and Kir4.1 have been linked to epilepsy, ataxia, and other neurological phenotypes in mice and humans (Tanaka et al., 1997; Neusch et al., 2001; Djukic et al., 2007; Bockenhauer et al., 2009; Scholl et al., 2009). Other astrocyte proteins that may be relevant and are the subject of current investigation include aquaporin-4 and connexin 43, given their involvement in other white matter disorders (Hinson, 2008; Lutz et al., 2009; Magnotti et al., 2011; Masaki et al., 2013). However, other changes have been noted that themselves could have wide-ranging downstream effects, including mislocalization and phosphorylation of TAR DNA binding protein of 43 kDa (TDP-43) (Walker et al., 2014) and upregulation of the nuclear lamins A/C (Wang et al., 2018). Studies originating from the Drosophila model implicate the nitric oxide pathway as mediating a toxic effect of glia on neurons, as well as a reverse pathway by which dysfunctional neurons induce death of glia (Wang et al., 2015).
Model systems using Drosophila and rodents have significant limitations for studying the role of GFAP in disease. Flies do have glia, suitable for studying glial-neuronal interactions, but have neither true astrocytes nor myelin. Rodent astrocytes are smaller and less complex than their human counterparts (Oberheim et al., 2009). To more closely model the human disease, several laboratories have investigated use of patient-derived pluripotent stem cells (iPS cells). Using astrocytes differentiated from iPS cells with the Arg88Cys or Arg416Trp variants, Jones et al. (2018) found alterations in astrocytic intracellular vesicle trafficking, calcium dynamics, and ATP release. It is intriguing that defects in these pathways were previously found in a transgenic mouse model that expressed the human Arg239His variant from a cDNA, though ATP release was changed in the opposite direction predicted by the human iPS cell results (Lee et al., 2013; Saito et al., 2018). Canals et al. (2018) introduced the common Arg239Cys variant into human embryonic stem cells, which were then induced toward astrocyte differentiation, and found decreases in both Kir4.1 and the Na+/K+ ATPase ATP1B2. Again using patient-derived iPS cells, Li et al. (2018) observed an increase in secretion of a factor (YKL-40) that inhibited the differentiation of oligodendrocyte progenitor cells. However, an effect at the level of oligodendrocyte progenitor cells does not explain the wide variations in age of onset of Alexander disease, and why some patients have relatively normal white matter.
Minor Isoforms and Disease
Until recently, the question of whether any of the minor isoforms have a role in disease has been unclear. GFAPκ is the major isoform in enteric glia, where it is differentially upregulated in patients with Parkinson’s disease, but a direct connection of GFAP to the gut phenotypes associated with this disorder has yet to be established (Clairembault et al., 2014). Although GFAPδ is usually coordinately upregulated with GFAPα in the CNS, as demonstrated in a mouse model of Alzheimer’s disease with prominent gliosis (Kamphuis et al., 2014), in two neurological diseases, the ratio of GFAPδ to GFAPα rises. One of these is vanishing white matter disease, which like Alexander disease is a primary disorder of astrocytes (Bugiani et al., 2011; Huyghe et al., 2012; Dooves et al., 2016; Zhou et al., 2019). A second is a subset of astrocytoma patients with poor outcome (van Bodegraven et al., 2019b). The authors suggest that the poor prognosis is due to the elevated GFAPδ/GFAPα ratio causing upregulation of the phosphatase dual-specificity phosphatase-4 (DUSP4), with speculation involving mitogen-activated protein kinase signaling pathways and interactions with the extracellular matrix. A sequence variant present in GFAPδ but not in GFAPα that predicts an Arg430His amino acid change has been causally linked to Alexander disease (Melchionda et al., 2013; Karp et al., 2018). However, Helman et al. (2020) recently reported that this variant (as well as a silent R430R variant found in a different family) causes disease not through the predicted amino acid change but by altering splicing to increase the level of the GFAPλ isoform (described earlier in the Structure section; Figure 5).
Autoimmunity to GFAP
GFAP may also pose danger by acting as a target in autoimmune disease. Autoantibodies to a variety of neural proteins, including GFAP, occur in many different neurological diseases but are likely nonspecific responses that have little bearing on the course of disease. Examples where presumably incidental GFAP autoantibodies are seen include traumatic brain or spinal cord injury (Zhang et al., 2014), lead poisoning (Moneim et al., 1999), Alzheimer’s disease (Tanaka et al., 1989), and gliomas (Wei et al., 2013). GFAP autoantibodies also occur in a small percentage of patients with multiple sclerosis and in rodent experimental allergic encephalomyelitis, both clearly immune-mediated diseases; however, here also, whether GFAP is necessary or sufficient as a target for disease is not known (Pekovic et al., 1990; Kaiser et al., 1997; Lambracht-Washington et al., 2007).
The best case for a pathogenic immune response aimed at GFAP exists for a condition first described in 2016 and generally referred to as “autoimmune GFAP astrocytopathy” (for review, see Kunchok et al., 2019). These patients suffer from a steroid-responsive meningoencephalitis, sometimes including myelitis, and display a broad range of symptoms affecting vision, gait, and cognition, but not seizures (Fang et al., 2016). One third also have some form of neoplasia, suggesting classification of autoimmune GFAP astrocytopathy among the paraneoplastic syndromes. Studies using mouse models implicate cytotoxic T cells as having a key role in pathogenesis (Sasaki et al., 2014). GFAP autoantibodies are also prominent in a canine disorder, especially seen in Pugs, referred to as necrotizing meningoencephalitis (Uchida et al., 1999). Like the human disease, this canine version displays GFAP autoantibodies in blood and CSF, where their levels may serve as useful biomarkers of severity and progression (Shibuya et al., 2007; Miyake et al., 2013). What exactly initiates this autoimmune attack, and the significance of the GFAP-targeted immune response in driving disease progression, is not yet clear. Most patients have an immune response directed against the GFAPα isoform (Flanagan et al., 2017), though a subset shows specificity for the minor GFAPδ isoform (Long et al., 2018).
Two additional examples of potential GFAP autoimmunity, surprisingly outside the nervous system, are type I diabetes and arthritis. In the pancreas, peri-islet stellate cells express GFAP, similar to the perisinusoidal stellate cells in the liver (Buniatian et al., 1996; Apte et al., 1998). Both T- and B-cell responses to GFAP have been reported as potential early events in the evolution of diabetes in the mouse and human (Winer et al., 2003), with responsible epitopes at several sites within the protein (Standifer et al., 2006; Tsui et al., 2008; Pang et al., 2017b). In the nonobese diabetic mouse model of diabetes, vaccination against GFAP at 9 weeks of age with the goal of inducing tolerance reduced the number of mice with hyperglycemia at 1 year (Pang et al., 2017a). In rheumatoid arthritis, patients may have elevated levels of GFAP in their synovial fluid and antibodies to GFAP in their plasma (Biswas et al., 2013). This finding may relate to reports from the early literature of expression of GFAP in cartilage of mouse and human, although these initial conclusions were based on limited immunohistochemical data (Viale et al., 1988; Kasper and Stosiek, 1990).
Relief
If we focus on Alexander disease, can the danger posed by mutant GFAP be addressed? Genetic studies in both mouse and Drosophila demonstrated that forced overexpression of stress response genes such as Cryab and Nrf2 could mitigate the effects of pathogenic variants in GFAP. However, whether these findings can be translated into clinical application is an open question. Pharmacologic approaches have targeted other pathways downstream of mutant GFAP, such as use of ceftriaxone to increase expression of glutamate transporters (Bachetti et al., 2010; Sechi et al., 2013). Based on the evidence described earlier that Alexander disease is associated with reduced expression of Glt-1, increasing its levels could be beneficial. However, the literature on ceftriaxone induction of Glt-1 is mixed, and we found it had no beneficial effect in our mouse models (unpublished observations). Using the Drosophila model, Wang et al. (2016) conducted an unbiased screen of a Food and Drug Administration-approved drug library for compounds that reduced the neuronal toxicity induced by expression of mutant GFAP in fly glia. Major hits included compounds that target muscarinic, cholinergic, and serotonergic pathways, but whether these compounds can rescue disease phenotypes has not been tested in other model systems.
The early finding that elevated levels of even wild-type GFAP could lead to both Rosenthal fibers and death in mice led to the suggestion that reducing the expression of GFAP could be a treatment for Alexander disease (Messing et al., 1998). As an initial (and unbiased with respect to mechanism) attempt to reduce GFAP, Cho et al. (2010) screened a library of Food and Drug Administration-approved and other compounds for their ability to reduce expression of GFAP in primary cultures of mouse astrocytes. Compounds identified in this screen, such as clomipramine, reduced GFAP levels in both cultured cells and wild-type mice, but subsequent attempts to extend these findings to mice expressing mutant GFAP failed (unpublished observations). Lithium was administered to mice via the oral route with the goal of reducing GFAP by stimulating autophagy (as in Sarkar et al., 2005). This did achieve significant reductions in GFAP in multiple areas of the nervous system (although likely through effects on Stat3 rather than autophagy) (LaPash Daniels et al., 2015). However, lithium had a very narrow window of safety and is not being pursued for clinical use.
Antisense suppression as a strategy to reduce GFAP was tried in cell culture models decades ago with either the broad goal of reducing gliosis (Weinstein et al., 1991; Yu et al., 1991, 1993; Ghirnikar et al., 1994; Lefrançois et al., 1997) or to alter migration and proliferation of gliomas (Chen and Liem, 1994b; Rutka et al., 1994). Others have used lentiviral vectors to deliver GFAP-targeted antisense in vivo, in the context of spinal cord injury, but the effects were modest, and no follow-up has appeared (Desclaux et al., 2015).
Most recently, Hagemann et al. (2018) reported the dramatic ability of ASOs to suppress expression of GFAP and change the course of Alexander disease in a mouse model (Figure 12). Single intracerebroventricular injections of GFAP-targeted ASOs reduced mRNA and protein throughout the CNS to essentially the null levels found in genetic knockouts of the gene for at least 8 weeks duration (how long the suppression lasts is the subject of current investigation). Even in the Alexander disease mouse models that express mutant GFAP and have elevated GFAP levels, nearly all the GFAP was eliminated by 8 weeks after injection. Rosenthal fibers disappeared, and other downstream markers of disease, such as Lcn2, Ccl2, Cxcl1, and ceruloplasmin, were normalized. The elimination of GFAP in brain parenchyma was also tracked by a drop in the CSF to undetectable levels. Of particular interest for clinical application of GFAP-targeted ASOs, treatment of these mice achieved both prevention and reversal of disease based on both cellular and clinical phenotypes (e.g., Rosenthal fibers disappeared, and body weight was restored). Attempts are now underway to extend these findings to humans in a formal clinical trial.
Figure 12.

ASO Suppression of GFAP Expression. A single injection of antisense oligonucleotides into the lateral ventricle of adult mice leads to nearly complete elimination of GFAP throughout the CNS (GFAP quantitation by ELISA). Mice given saline as a control show the natural elevation of GFAP that occurs in the mutant mice. Mutants (red), wild type (black), 8 weeks posttreatment. GFAP levels were measured in three biochemically defined pools of intermediate filaments, defined by solubility in Triton-X-100 (left and middle, representing monomers through small oligomers and mature filaments, respectively) and SDS (right, representing an enrichment for Rosenthal fibers). ****p<0.0001, unpaired 2-tailed t test. Figure reprinted from Hagemann et al. (2018). GFAP = glial fibrillary acidic protein;
ASO = antisense oligonucleotide; RF = Rosenthal fiber.
Future
We anticipate fruitful future investigations into a number of topics, including the role of minor isoforms, the question of whether variants in the gene influence normal biology or cause pathologies other than Alexander disease, and the question of whether GFAP has currently unknown functions, particularly in higher primates, given that they have more extensive astrocyte networks than lower vertebrates. The full collection of binding partners for GFAP has yet to be identified, and even potential structural relationships remain to be discovered, such as with the nuclear lamins and the recently described subplasmalemmal rim (Quinlan et al., 2017). A previous review by Brenner (2014) cites instances in which there is uncertainty about multiple possible functions of GFAP because of conflicting reports in the literature. It points out that in many instances, these apparent conflicts could be due to GFAP performing different functions in different brain regions. Pursuing comparative regional studies of GFAP function could thus also be a fruitful area for future research. The past 50 years of research on GFAP has yielded many insights over a continuously changing landscape of areas in modern biology and medicine. We expect no less of the next 50.
Supplemental Material
Supplemental material, sj-pdf-1-asn-10.1177_1759091420949680 for GFAP at 50 by Albee Messing and Michael Brenner in ASN Neuro
Supplemental material, sj-pdf-2-asn-10.1177_1759091420949680 for GFAP at 50 by Albee Messing and Michael Brenner in ASN Neuro
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Work in our laboratories has been funded by the National Institutes of Health (HD076892, HD090256, NS110719, NS42803, NS39055, and NS060120), the United Leukodystrophy Foundation, Children Living with Inherited Metabolic Diseases, the Palamaro Fund, the Juanma Fund, the Jelte Rijkaart Fund, and Elise’s Corner.
ORCID iD
Albee Messing https://orcid.org/0000-0001-6049-0521
Supplemental Material
Supplemental material for this article is available online.
References
- Acarin L., Villapol S., Faiz M., Rohn T. T., Castellano B., González B. (2007). Caspase-3 activation in astrocytes following postnatal excitotoxic damage correlates with cytoskeletal remodeling but not with cell death or proliferation. Glia, 55, 954–965. [DOI] [PubMed] [Google Scholar]
- Anderson M. A., Ao Y., Sofroniew M. V. (2014). Heterogeneity of reactive astrocytes. Neurosci Lett, 565, 23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel B. (2013). Nonmammalian vertebrate glia In: Kettenmann H., Ransom B. R. (Eds.), Neuroglia (pp. 24–31). Oxford University Press. [Google Scholar]
- Apte M. V., Haber P. S., Applegate T. L., Norton I. D., McCaughan G. W., Korsten M. A., Pirola R. C., Wilson J. .S. (1998). Periacinar stellate shaped cells in rat pancreas – Identification, isolation, and culture. GUT, 43, 128–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias E., Koga H., Diaz A., Mocholi E., Patel B., Cuervo A. M. (2015). Lysosomal mTORC2/PHLPP1/Akt regulate chaperone-mediated autophagy. Mol Cell, 59, 270–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachetti T., Di Zanni E., Balbi P., Bocca P., Prigione I., Deiana G. A., Rezzani A., Ceccherini I., Sechi G, (2010). In vitro treatments with ceftriaxone promote elimination of mutant glial fibrillary acidic protein and transcription down-regulation. Exp Cell Res, 316, 2152–2165. [DOI] [PubMed] [Google Scholar]
- Balbi P., Salvini S., Fundarò C., Frazzitta G., Maestri R., Mosah D., Uggetti C., Sechi G. (2010). The clinical spectrum of late-onset Alexander disease: A systematic literature review. J Neurol, 257, 1955–1962. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay U., Sridhar S., Kaushik S., Kiffin R., Cuervo AM. (2010) Identification of regulators of chaperone-mediated autophagy. Mol Cell, 39, 535–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia R. A., Beltran A. S., Delic S., Dumitru R., Robinson J. A., Kabiraj P., Herring L. E., Madden V. J., Ravinder N., Willems E., Newman R. A., Quinlan R. A., Goldman J. E., Perng M. D., Inagaki M., Snider N. T. (2019). Site-specific phosphorylation and caspase cleavage of GFAP are new markers of Alexander Disease severity. Elife, 8, e47789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger U. V., Hediger M. A. (2000). The vitamin C transporter SVCT2 is expressed by astrocytes in culture but not in situ. NeuroReport, 11, 1395–1399. [DOI] [PubMed] [Google Scholar]
- Bernardos R. L., Raymond P. A. (2006). GFAP transgenic zebrafish. Gene Expr Patterns, 6, 1007–1013. [DOI] [PubMed] [Google Scholar]
- Besnard F., Brenner M., Nakatani Y., Chao R., Purohit H. J., Freese E. (1991). Multiple interacting sites regulate astrocyte-specific transcription of the human gene for glial fibrillary acidic protein. J Biol Chem, 266, 18877–18883. [PubMed] [Google Scholar]
- Bignami A., Eng L. F., Dahl D., Uyeda C. T. (1972). Localization of the glial fibrillary acidic protein in astrocytes by immunofluorescence. Brain Res, 43, 429–435. [DOI] [PubMed] [Google Scholar]
- Biswas S., Sharma S., Saroha A., Bhakuni D. S., Malhotra R., Zahur M., Oellerich M., Das H. R., Asif A. R. (2013). Identification of novel autoantigen in the synovial fluid of rheumatoid arthritis patients using an immunoproteomics approach. PLoS One, 8, e56246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blechingberg J., Holm I. E., Nielsen K. B., Jensen T. H., Jørgensen A. L., Nielsen A. L. (2007). Identification and characterization of GFAPk, a novel glial fibrillary acidic protein isoform. Glia, 55, 497–507. [DOI] [PubMed] [Google Scholar]
- Bockenhauer D., et al. (2009). Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med, 360, 1960–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boston-Howes W., Gibb S. L., Williams E. O., Pasinelli P., Brown R. H., Jr., Trotti D. (2006). Caspase-3 cleaves and inactivates the glutamate transporter EAAT2. J Biol Chem, 281, 14076–14084. [DOI] [PubMed] [Google Scholar]
- Bradley R. A., Shireman J., McFalls C., Choi J., Canfield S. G., Dong Y., Liu K., Lisota B., Jones J. R., Petersen A., Bhattacharyya A., Palecek S. P., Shusta E. V., Kendziorski C., Zhang S. C. (2019). Regionally specified human pluripotent stem cell-derived astrocytes exhibit different molecular signatures and functional properties. Development, 146(13), dev170910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner M. (2014). Role of GFAP in CNS injuries. Neurosci Lett, 565, 7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner M., Goldman J. E., Quinlan R. A., Messing A. (2009). Alexander disease: A genetic disorder of astrocytes In: Parpura V., Haydon P. G. (Eds.), Astrocytes in (patho)physiology of the nervous system (pp.591–648). Springer. [Google Scholar]
- Brenner M., Kisseberth W. C., Su Y., Besnard F., Messing A. (1994). GFAP promoter directs astrocyte-specific expression in transgenic mice. J Neurosci, 14, 1030–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner M., Lampel K., Nakatani Y., Mill J., Banner C., Mearow K., Dohadwala M., Lipsky R., Freese E. (1990). Characterization of human cDNA and genomic clones for glial fibrillary acidic protein. Brain Res Mol Brain Res, 7, 277–286. [DOI] [PubMed] [Google Scholar]
- Brenner M., Messing A., Olsen M. L. (2019). AP-1 and the injury response of the GFAP gene. J Neurosci Res, 97, 149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner M., Nicholas A. P. (2017). The significance of deiminated GFAP in neurodegenerative diseases with special emphasis on Alexander disease In Nicholas A. P., Bhattacharya S. K., Thompson P. R. (Eds.), Protein deimination in human health and disease (pp. 391–412). Springer-Verlag. [Google Scholar]
- Bugiani M., Boor I., van Kollenburg B., Postma N., Polder E., van Berkel C., van Kesteren R. E., Windrem M. S., Hol E. M., Scheper G. C., Goldman S. A., van der Knaap M. S. (2011). Defective glial maturation in vanishing white matter disease. J Neuropathol Exp Neurol, 70, 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buniatian G., Hamprecht B., Gebhardt R. (1996). Glial fibrillary acidic protein as a marker of perisinusoidal stellate cells that can distinguish between the normal and myofibroblast-like phenotypes. Biol Cell, 87, 65–73. [PubMed] [Google Scholar]
- Bushong E. A., Martone M. E., Jones Y. Z., Ellisman M. H. (2002). Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci, 22, 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy J. D., Emery B., Kaushai A., Foo L. C., Zamanian J. L., Christopherson K. S., Xing Y., Lubischer J. L., Krieg P. A., Krupenko S. A., Thompson W. J., Barres B. A. (2008). A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J Neurosci, 28, 264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canals I., Ginisty A., Quist E., Timmerman R., Fritze J., Miskinyte G., Monni E., Hansen M. G., Hidalgo I., Bryder D., Bengzon J., Ahlenius H. (2018). Rapid and efficient induction of functional astrocytes from human pluripotent stem cells. Nat Methods, 15, 693–696. [DOI] [PubMed] [Google Scholar]
- Carter B. S., Meng F., Thompson R. C. (2012). Glucocorticoid treatment of astrocytes results in temporally dynamic transcriptome regulation and astrocyte-enriched mRNA changes in vitro. Physiological Genomics, 44, 1188–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casasnovas C., Verdura E., Vélez V., Schlüter A., Pons-Escoda A., Homedes C., Ruiz M., Fourcade S., Launay N., Pujol A. (2019). A novel mutation in the GFAP gene expands the phenotype of Alexander disease. J Med Genet, 56, 846–849. [DOI] [PubMed] [Google Scholar]
- Chen M.-H., Hagemann T. L., Quinlan R. A., Messing A., Der Perng M. (2013). Caspase cleavage of GFAP produces an assembly-compromised proteolytic fragment that promotes filament aggregation. ASN Neuro, 5, Art: e00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W. J., Liem R. K. H. (1994. a). The endless story of the glial fibrillary acidic protein. J Cell Sci, 107, 2299–2311. [DOI] [PubMed] [Google Scholar]
- Chen W. J., Liem R. K. H. (1994. b). Reexpression of glial fibrillary acidic protein rescues the ability of astrocytoma cells to form processes in response to neurons. J Cell Biol, 127, 813–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. S., Lim S. C., Chen M. H., Quinlan R. A., Perng M. D. (2011). Alexander disease causing mutations in the C-terminal domain of GFAP are deleterious both to assembly and network formation with the potential to both activate caspase 3 and decrease cell viability. Exp Cell Res, 317, 2252–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernyatina A. A., Guzenko D., Strelkov S. V. (2015). Intermediate filament structure: The bottom-up approach. Curr Opin Cell Biol, 32, 65–72. [DOI] [PubMed] [Google Scholar]
- Chernyatina A. A., Nicolet S., Aebi U., Herrmann H., Strelkov S. V. (2012). Atomic structure of the vimentin central alpha-helical domain and its implications for intermediate filament assembly. Proc Natl Acad Sci U S A, 109, 13620–13625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho W., Brenner M., Peters N., Messing A. (2010). Drug screening to identify suppressors of GFAP expression. Hum Mol Genet, 19, 3169–3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clairembault T., Kamphuis W., Leclair-Visonneau L., Rolli-Derkinderen M., Coron E., Neunlist M., Hol E. M., Derkinderen P. (2014). Enteric GFAP expression and phosphorylation in Parkinson's disease. J Neurochem, 130, 805–815. [DOI] [PubMed] [Google Scholar]
- Condorelli D. F., Nicoletti V. G., Barresi V., Conticello S. G., Caruso A., Tendi E. A., Stella A. M. G. (1999). Structural features of the rat GFAP gene and identification of a novel alternative transcript. J Neurosci Res, 56, 219–228. [DOI] [PubMed] [Google Scholar]
- Dahl D., Bignami A. (1975). Glial fibrillary acidic protein from normal and gliosed human brain. Demonstration of multiple related polypeptides. Biochim Biophys Acta, 386, 41–51. [DOI] [PubMed] [Google Scholar]
- DeArmond S. J., Fajardo M., Naughton S. A., Eng L. F. (1983). Degradation of glial fibrillary acidic protein by a calcium dependent proteinase: An electroblot study. Brain Res, 262, 275–282. [DOI] [PubMed] [Google Scholar]
- DeArmond S. J., Lee Y. L., Kretzschmar H. A., Eng L. F. (1986). Turnover of glial filaments in mouse spinal cord. J Neurochem, 47, 1749–1753. [DOI] [PubMed] [Google Scholar]
- Desclaux M., Perrin F. E., Do-Thi A., Prieto-Cappellini M., Gimenez y Ribotta M., Mallet J., Privat A. (2015). Lentiviral-mediated silencing of glial fibrillary acidic protein and vimentin promotes anatomical plasticity and functional recovery after spinal cord injury. J Neurosci Res, 93, 43–55. [DOI] [PubMed] [Google Scholar]
- DeWitt J. C., Mock A., Louis D. N. (2017). The 2016 WHO classification of central nervous system tumors: What neurologists need to know. Curr Opin Neurol, 30, 643–649. [DOI] [PubMed] [Google Scholar]
- Ding M., Eliasson C., Betsholtz C., Hamberger A., Pekny M. (1998). Altered taurine release following hypotonic stress in astrocytes from mice deficient for GFAP and vimentin. Mol Brain Res, 62, 77–81. [DOI] [PubMed] [Google Scholar]
- Djukic B., Casper K. B., Philpot B. D., Chin L. S., McCarthy K. D. (2007). Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci, 27, 11354–11365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooves S., Bugiani M., Postma N. L., Polder E., Land N., Horan S. T., van Deijk A. L. F., van de Kreeke A., Jacobs G., Vuong C., Klooster J., Kamermans M., Wortel J., Loos M., Wisse L. E., Scheper G. C., Abbink T. E. M., Heine V. M., van der Knaap M. S. (2016). Astrocytes are central in the pathomechanisms of vanishing white matter. J Clin Invest, 126, 1512–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy P. E., Graf L., Rapport M. M. (1977). Identification of glial fibrillary acidic protein by the immunoperoxidase method in human brain tumors. J Neuropathol Exp Neurol, 36, 645–652. [DOI] [PubMed] [Google Scholar]
- Eng L. F., Ghirnikar R. S., Lee Y. L. (2000). Glial fibrillary acidic protein: GFAP-thirty-one years (1969-2000). Neurochem Res, 25, 1439–1451. [DOI] [PubMed] [Google Scholar]
- Eng L. F., Rubinstein L. J. (1978). Contribution of immunohistochemistry to diagnostic problems of human cerebral tumors. J Histochem Cytochem, 26, 513–522. [DOI] [PubMed] [Google Scholar]
- Eng L. F., Vanderhaeghen J. J., Bignami A., Gerstl B. (1971). An acidic protein isolated from fibrous astrocytes. Brain Res, 28, 351–354. [DOI] [PubMed] [Google Scholar]
- Erickson P. A., Feinstein S. C., Lewis G. P., Fisher S. K. (1992). Glial fibrillary acidic protein and its mRNA: Ultrastructural detection and determination of changes after CNS injury. J Struct Biol, 108, 148–161. [DOI] [PubMed] [Google Scholar]
- Eriksson J. E., Dechat T., Grin B., Helfand B., Mendez M., Pallari H.-M., Goldman R. D. (2009). Introducing intermediate filaments: From discovery to disease. J Clin Invest, 119, 1763–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escartin C., Guillemaud O., Carrillo-de Sauvage M. A. (2019). Questions and (some) answers on reactive astrocytes. Glia, 67, 2221–2247. [DOI] [PubMed] [Google Scholar]
- Faigle W., Cruciani C., Wolski W., Roschitzki B., Puthenparampil M., Tomas-Ojer P., Sellés-Moreno C., Zeis T., Jelcic I., Schaeren-Wiemers N., Sospedra M., Martin R. (2019). Brain citrullination patterns and T cell ceactivity of cerebrospinal fluid-derived CD4+ T cells in multiple sclerosis. Front Immunol, 10, 540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang B. Y., McKeon A., Hinson S. R., Kryzer T. J., Pittock S. J., Aksamit A. J., Lennon V. A. (2016). Autoimmune glial fibrillary acidic protein astrocytopathy: A novel meningoencephalomyelitis. JAMA Neuro, 73, 1297–1307. [DOI] [PubMed] [Google Scholar]
- Feinstein D. L., Weinmaster G. A., Milner R. J. (1992). Isolation of cDNA clones encoding rat glial fibrillary acidic protein: Expression in astrocytes and in Schwann cells. J Neurosci Res, 32, 1–14. [DOI] [PubMed] [Google Scholar]
- Flanagan E. P., Hinson S. R., Lennon V. A., Fang B. Y., Aksamit A. J., Morris P. P., Basal E., Honorat J. A., Alfugham N. B., Linnoila J. J., Weinshenker B. G., Pittock S. J., McKeon A. (2017). Glial fibrillary acidic protein immunoglobulin G as biomarker of autoimmune astrocytopathy: Analysis of 102 patients. Ann Neurol, 81, 298–309. [DOI] [PubMed] [Google Scholar]
- Flint D., Brenner M. (2011). Alexander disease In: Raymond G. V., Eichler F., Fatemi A., Naidu S. (Eds.), Leukodystrophies (pp. 106–129). MacKeith Press. [Google Scholar]
- Fu M. H., Chang Y. Y., Lin N. H., Yang A. W., Chang C. C., Liu J. S., Peng C. H., Wu K. L. H., Perng M. D., Lan M. Y. (2020). Recessively-inherited adult-onset Alexander disease caused by a homozygous mutation in the GFAP gene. Mov Disord. Advance online publication. 10.1002/mds.28099 [DOI] [PubMed]
- Galea E., Dupouey P., Feinstein D. L. (1995). Glial fibrillary acidic protein mRNA isotypes: Expression in vitro and in vivo. J Neurosci Res, 41, 452–461. [DOI] [PubMed] [Google Scholar]
- Geisler N., Weber K. (1983). Amino acid sequence data on glial fibrillary acidic protein (GFA); implications for the subdivision of intermediate filaments into epithelial and non-epithelial members. EMBO J, 2, 2059–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghirnikar R. S., Yu A. C. H., Eng L. F. (1994). Astrogliosis in culture: III. Effect of recombinant retrovirus expressing antisense glial fibrillary acidic protein RNA. J Neurosci Res, 38, 376–385. [DOI] [PubMed] [Google Scholar]
- Gomi H., Yokoyama T., Fujimoto K., Ideka T., Katoh A., Itoh T., Itohara S. (1995). Mice devoid of the glial fibrillary acidic protein develop normally and are susceptible to scrapie prions. Neuron, 14, 29–41. [DOI] [PubMed] [Google Scholar]
- Hagemann T. L., Boelens W., Wawrousek E., Messing A. (2009). Suppression of GFAP toxicity by αB-crystallin in mouse models of Alexander disease. Hum Mol Genet, 18, 1190–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemann T. L., Paylor R., Messing A. (2013). Deficits in adult neurogenesis, contextual fear conditioning and spatial learning in a Gfap mutant mouse model of Alexander disease. J Neurosci, 33, 18698–18706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemann T. L., Powers B., Mazur C., Kim A., Wheeler S., Hung G., Swayze E., Messing A. (2018). Antisense suppression of GFAP as a treatment for Alexander disease. Ann Neurol, 83, 27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halford J., Shen S., Itamura K., Levine J., Chong A. C., Czerwieniec G., Glenn T. C., Hovda D. A., Vespa P., Bullock R., Dietrich W. D., Mondello S., Loo J. A., Wanner I. B. (2017). New astroglial injury-defined biomarkers for neurotrauma assessment. J Cereb Blood Flow Metab 37, 3278–3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaven M. R., Flint D., Randall S. M., Sosunov A. A., Wilson L., Barnes S., Goldman J. E., Muddiman D. C., Brenner M. (2016). The composition of Rosenthal fibers, the protein aggregate hallmark of Alexander disease. J Proteome Res, 15, 2265–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaven M. R., Wilson L., Barnes S., Brenner M. (2019). Relative stabilities of wild type and mutant glial fibrillary acidic protein in patients with Alexander disease. J Biol Chem, 294, 15604–15612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helman G., Takanohashi A., Hagemann T. L., Perng M. D., Walkiewicz M., Woidill S., Sase S., Cross Z., Du Y., Zhao L., Waldman A., Haake B. C., Fatemi A., Brenner M., Sherbini O., Messing A., Vanderver A., Simons C. (2020). Type II Alexander disease caused by splicing errors and aberrant overexpression of a GFAP isoform. Hum Mutat, 41, 1131–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann H., Aebi U. (2004). Intermediate filaments: Molecular structure, assembly mechanism, and integration into functionally distinct intracellular scaffolds. Annu Rev Biochem, 73, 749–789. [DOI] [PubMed] [Google Scholar]
- Herrmann H., Häner M., Brettel M., Ku N. O., Aebi U. (1999). Characterization of distinct early assembly units of different intermediate filament proteins. J Mol Biol, 286, 1403–1420. [DOI] [PubMed] [Google Scholar]
- Herrmann H., Häner M., Brettel M., Müller S. A., Goldie K. N., Fedtke B., Lustig A., Franke W. W., Aebi U. (1996). Structure and assembly properties of the intermediate filament protein vimentin: The role of its head, rod and tail domains. J Mol Biol, 264, 933–953. [DOI] [PubMed] [Google Scholar]
- Herskowitz J. H., Seyfried N. T., Duong D. M., Xia Q. W., Rees H. D., Gearing M., Peng J. M., Lah J. J., Levey A. I. (2010). Phosphoproteomic analysis reveals site-specific changes in GFAP and NDRG2 phosphorylation in Frontotemporal Lobar Degeneration. J Proteome Res, 9, 6368–6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinson S. R. (2008). Aquaporin-4-binding autoantibodies in patients with neuromyelitis optica impair glutamate transport by down-regulating EAAT2. J Exp Med, 205, 2473–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hol E. M., Pekny M. (2015). Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the central nervous system. Curr Opin Cell Biol, 32, 121–130. [DOI] [PubMed] [Google Scholar]
- Huang J. W., Shrestha P., Takai Y., Mori M. (1996). Immunohistochemical evaluation of GFAP in salivary gland tumors. Acta Histochem Cytochem, 29, 85–92. [Google Scholar]
- Huyghe A., Horzinski L., Henaut A., Gaillard M., Bertini E., Schiffmann R., Rodriguez D., Dantal Y., Boespflug-Tanguy O., Fogli A. (2012). Developmental splicing deregulation in leukodystrophies related to EIF2B mutations. PLoS One, 7, e38264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki M., Gonda Y., Nishizawa K., Kitamura S., Sato C., Ando S., Tanabe K., Kikuchi K., Tsuiki S., Nishi Y. (1990). Phosphorylation sites linked to glial filament disassembly in vitro locate in a non-α-helical head domain. J Biol Chem, 265, 4722–4729. [PubMed] [Google Scholar]
- Inagaki M., Nakamura Y., Takeda M., Nishimura T., Inagaki N. (1994). Glial fibrillary acidic protein: Dynamic property and regulation by phosphorylation. Brain Pathol, 4, 239–243. [DOI] [PubMed] [Google Scholar]
- Inagaki M., Takahara H., Nishi Y., Sugawara K., Sato C. (1989). Ca2+-dependent deimination-induced disassembly of intermediate filaments involves specific modification of the amino-terminal head domain. J Biol Chem, 264, 18119–18127. [PubMed] [Google Scholar]
- Ishigami A., Masutomi H., Handa S., Nakamura M., Nakaya S., Uchida Y., Saito Y., Murayama S., Jang B., Jeon Y. C., Choi E. K., Kim Y. S., Kasahara Y., Maruyama N., Toda T. (2015). Mass spectrometric identification of citrullination sites and immunohistochemical detection of citrullinated glial fibrillary acidic protein in Alzheimer's disease brains. J Neurosci Res, 93, 1664–1674. [DOI] [PubMed] [Google Scholar]
- Ishigami A., Ohsawa T., Hiratsuka M., Taguchi H., Kobayashi S., Saito Y., Murayama S., Asaga H., Toda T., Kimura N., Maruyama N. (2005). Abnormal accumulation of citrullinated proteins catalyzed by peptidylarginine deiminase in hippocampal extracts from patients with Alzheimer's disease. J Neurosci Res, 80, 120–128. [DOI] [PubMed] [Google Scholar]
- Jany P. L., Hagemann T. L., Messing A. (2013). GFAP expression as an indicator of disease severity in mouse models of Alexander disease. ASN Neuro, 5, Art: e00109 10.1042/AN20130003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z., Fu Z., Yang J., Troncosco J., Everett A. D., Van Eyk J. E. (2013). Identification and characterization of citrulline-modified brain proteins by combining HCD and CID fragmentation. Proteomics, 13, 2682–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo V. Y., Fletcher C. D. (2015). Myoepithelial neoplasms of soft tissue: An updated review of the clinicopathologic, immunophenotypic, and genetic features. Head Neck Pathol, 9, 32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson W. B., Ruppe M. D., Rockenstein E. M., Price J., Sarthy V. P., Verderber L. C., Mucke L. (1995). Indicator expression directed by regulatory sequences of the glial fibrillary acidic protein (GFAP) gene: In vivo comparison of distinct GFAP-LacZ transgenes. Glia, 13, 174–184. [DOI] [PubMed] [Google Scholar]
- Jones J. R., Kong L., Hanna M. G., IV, Hoffmann B., Krencik R., Bradley R., Hagemann T., Choi J., Doers M., Dubovis M., Sherafat M. A., Bhattacharyya A., Kendziorski C., Audhya A., Messing A., Zhang S. C. (2018). Mutations in GFAP disrupt the distribution and function of organelles in human astrocytes. Cell Rep, 25, 947–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonesco D. S., Hassager C., Frydland M., Kjærgaard J., Karsdal M., Henriksen K. (2019). A caspase-6-cleaved fragment of glial fibrillary acidic protein as a potential serological biomarker of CNS injury after cardiac arrest. PLoS One, 14, e0224633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser R., Obert M., Kaufmann R., Czygan M. (1997). IgG-antibodies to CNS proteins in patients with multiple sclerosis. Eur J Med Res, 2, 169–172. [PubMed] [Google Scholar]
- Kálmán M., Gould R. M. (2001). GFAP-immunopositive structures in spiny dogfish, Squalus acanthias, and little skate, Raia erinacea, brains: Differences have evolutionary implications. Anat Embryol, 204, 59–80. [DOI] [PubMed] [Google Scholar]
- Kamphuis W., Kooijman L., Orre M., Stassen O., Pekny M., Hol E. M. (2015). GFAP and vimentin deficiency alters gene expression in astrocytes and microglia in wild-type mice and changes the transcriptional response of reactive glia in mouse model for Alzheimer’s disease. Glia, 63, 1036–1056. [DOI] [PubMed] [Google Scholar]
- Kamphuis W., Mamber C., Moeton M., Kooijman L., Sluijs J. A., Jansen A. H., Verveer M., de Groot L. R., Smith V. D., Rangarajan S., Rodríguez J. J., Orre M., Hol E. M. (2012). GFAP isoforms in adult mouse brain with a focus on neurogenic astrocytes and reactive astrogliosis in mouse models of Alzheimer disease. PLoS One, 7, e42823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphuis W., Middeldorp J., Kooijman L., Sluijs J. A., Kooi E. J., Moeton M., Freriks M., Mizee M. R., Hol E. M. (2014). Glial fibrillary acidic protein isoform expression in plaque related astrogliosis in Alzheimer's disease. Neurobiol Aging, 35, 492–510. [DOI] [PubMed] [Google Scholar]
- Kanninen K., Goldsteins G., Auriola S., Alafuzoff I., Koistinaho J. (2004). Glycosylation changes in Alzheimer's disease as revealed by a proteomic approach. Neurosci Lett, 367, 235–240. [DOI] [PubMed] [Google Scholar]
- Karp N., Lee D., Shickh S., Jenkins M. E. (2018). c.1289G>A (p.Arg430His) variant in the epsilon isoform of the GFAP gene in a patient with adult onset Alexander disease. Eur J Med Genet, 62, 235–238. [DOI] [PubMed] [Google Scholar]
- Kasper M., Stosiek P. (1990). Detection of GFAP in vertebral fibrocartilage in human fetal and newborn tissue. Acta Histochem(Jena), 88, 25–27. [DOI] [PubMed] [Google Scholar]
- Kim B., Kim S., Jin M. S. (2018. a). Crystal structure of the human glial fibrillary acidic protein 1B domain. Biochem Biophys Res Commun, 503, 2899–2905. [DOI] [PubMed] [Google Scholar]
- Kim S. E., Park J. W., Kim M. J., Jang B., Jeon Y. C., Kim H. J., Ishigami A., Kim H. S., Suk K. T., Kim D. J., Park C. K., Choi E. K., Jang M. K. (2018. b). Accumulation of citrullinated glial fibrillary acidic protein in a mouse model of bile duct ligation-induced hepatic fibrosis. PLoS One, 13, e0201744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korolainen M. A., Auriola S., Nyman T. A., Alafuzoff I., Pirttilä T. (2005). Proteomic analysis of glial fibrillary acidic protein in Alzheimer's disease and aging brain. Neurobiol Dis, 20, 858–870. [DOI] [PubMed] [Google Scholar]
- Ku N. O., Omary M. B. (2006). A disease- and phosphorylation-related nonmechanical function for keratin 8. J Cell Biol, 174, 115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunchok A., Zekeridou A., McKeon A. (2019). Autoimmune glial fibrillary acidic protein astrocytopathy. Curr Opin Neurol, 32, 452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyllerman M., Rosengren L., Wiklund L. M., Holmberg E. (2005). Increased levels of GFAP in the cerebrospinal fluid in three subtypes of genetically confirmed Alexander disease. Neuropediatrics, 36, 319–323. [DOI] [PubMed] [Google Scholar]
- Lambracht-Washington D., O'Connor K. C., Cameron E. M., Jowdry A., Ward E. S., Frohman E., Racke M. K., Monson N. L. (2007). Antigen specificity of clonally expanded and receptor edited cerebrospinal fluid B cells from patients with relapsing remitting MS. J Neuroimmunol, 186, 164–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPash Daniels C. M., Paffenroth E., Austin E. V., Glebov K., Lewis D., Walter J., Messing A. (2015). Lithium decreases glial fibrillary acidic protein in a mouse model of Alexander disease. PLoS One, 10, e0138132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. U., Yamazaki Y., Tanaka K. F., Furuya K., Sokabe M., Hida H., Takao K., Miyakawa T., Fujii S., Ikenaka K. (2013). Increased astrocytic ATP release results in enhanced excitability of the hippocampus. Glia, 61, 210–224. [DOI] [PubMed] [Google Scholar]
- Lee J., McKinney K. Q., Pavlopoulos A. J., Han M. H., Kim S. H., Kim H. J., Hwang S. (2016). Exosomal proteome analysis of cerebrospinal fluid detects biosignatures of neuromyelitis optica and multiple sclerosis. Clin Chim Acta, 462, 118–126. [DOI] [PubMed] [Google Scholar]
- Lefrançois T., Fages C., Peschanski M., Tardy M. (1997). Neuritic outgrowth associated with astroglial phenotypic changes induced by antisense glial fibrillary acidic protein (GFAP) mRNA in injured neuron-astrocyte cocultures. J Neurosci, 17, 4121–4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt P., Rakic P. (1980). Immunoperoxidase localization of glial fibrillary acidic protein in radial glial cells and astrocytes of the developing rhesus monkey brain. J Comp Neurol, 193, 815–840. [DOI] [PubMed] [Google Scholar]
- Lewis S. A., Balcarek J. M., Krek V., Shelanski M., Cowan N. J. (1984). Sequence of a cDNA clone encoding mouse glial fibrillary acidic protein: Structural conservation of intermediate filaments. Proc Natl Acad Sci U S A, 81, 2743–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis S. A., Cowan N. J. (1985). Temporal expression of mouse glial fibrillary acidic protein mRNA studied by a rapid in situ hybridization procedure. J Neurochem, 45, 913–919. [DOI] [PubMed] [Google Scholar]
- Li H. H., Guo Y., Teng J. L., Ding M. X., Yu A. C. H., Chen J. G. (2006). 14-3-3γ affects dynamics and integrity of glial filaments by binding to phosphorylated GFAP. J Cell Sci, 119, 4452–4461. [DOI] [PubMed] [Google Scholar]
- Li L., et al. (2008). Protective role of reactive astrocytes in brain ischemia. J Cereb Blood Flow Metab, 28, 468–481. [DOI] [PubMed] [Google Scholar]
- Li L., Tian E., Chen X., Chao J., Klein J., Qu Q., Sun G., Sun G., Huang Y., Warden C. D., Ye P., Feng L., Li X., Cui Q., Sultan A., Douvaras P., Fossati V., Sanjana N. E., Riggs A. D., Shi Y. (2018). GFAP mutations in astrocytes impair oligodendrocyte progenitor proliferation and myelination in an hiPSC model of Alexander disease. Cell Stem Cell, 23, 239–251.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow S. A., Barres B. A. (2017). Reactive astrocytes: Production, function, and therapeutic potential. Immunity, 46, 957–967. [DOI] [PubMed] [Google Scholar]
- Liedtke W., Edelmann W., Bieri P. L., Chiu F. C., Cowan N. J., Kucherlapati R., Raine C. S. (1996). GFAP is necessary for the integrity of CNS white matter architecture and long-term maintenance of myelination. Neuron, 17, 607–615. [DOI] [PubMed] [Google Scholar]
- Liedtke W., Edelmann W., Chiu F. C., Kucherlapati R., Raine C. S. (1998). Experimental autoimmune encephalomyelitis in mice lacking glial fibrillary acidic protein is characterized by a more severe clinical course and an infiltrative central nervous system lesion. Am J Pathol, 152, 251–259. [PMC free article] [PubMed] [Google Scholar]
- Liem R. K. H., Messing A. (2009). Dysfunctions of neuronal and glial intermediate filaments in disease. J Clin Invest, 119, 1814–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim M. C., Maubach G., Zhuo L. (2008). Glial fibrillary acidic protein splice variants in hepatic stellate cells – Expression and regulation. Mol Cells, 25, 376–384. [PubMed] [Google Scholar]
- Lin N. H., Huang Y. S., Opal P., Goldman R. D., Messing A., Perng M. D. (2016). The role of gigaxonin in the degradation of the glial-specific intermediate filament protein GFAP. Mol Biol Cell, 27, 3980–3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long Y., Liang J., Xu H., Huang Q., Yang J., Gao C., Qiu W., Lin S., Chen X. (2018). Autoimmune glial fibrillary acidic protein astrocytopathy in Chinese patients: A retrospective study. Eur J Neurol, 25, 477–483. [DOI] [PubMed] [Google Scholar]
- Louis D. N., Perry A., Reifenberger G., von Deimling A., Figarella-Branger D., Cavenee W. K., Ohgaki H., Wiestler O. D., Kleihues P., Ellison D. W. (2016). The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol, 131, 803–820. [DOI] [PubMed] [Google Scholar]
- Lowery J., Kuczmarski E. R., Herrmann H., Goldman R. D. (2015). Intermediate filaments play a pivotal role in regulating cell architecture and function. J Biol Chem, 290, 17145–17153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwin S. K., Kosek J. C., Eng L. F. (1976). The topographical distribution of S-100 and GFA proteins in the adult rat brain: An immunohistochemical study using horseradish peroxidase-labelled antibodies. J Comp Neurol, 165, 197–207. [DOI] [PubMed] [Google Scholar]
- Lundgaard I., Osório M. J., Kress B., Sanggaard S., Nedergaard M. (2013). White matter astrocytes in health and disease. Neuroscience, 276, 161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz S. E., Zhao Y., Gulinello M., Lee S. C., Raine C. S., Brosnan C. F. (2009). Deletion of astrocyte connexins 43 and 30 leads to a dysmyelinating phenotype and hippocampal CA1 vacuolation. J Neurosci, 29, 7743–7752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnotti L. M., Goodenough D. A., Paul D. L. (2011). Deletion of oligodendrocyte Cx32 and astrocyte Cx43 causes white matter vacuolation, astrocyte loss and early mortality. Glia, 59, 1064–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahammad S., Murthy S. N., Didonna A., Grin B., Israeli E., Perrot R., Bomont P., Julien J. P., Kuczmarski E., Opal P., Goldman R. D. (2013). Giant axonal neuropathy-associated gigaxonin mutations impair intermediate filament protein degradation. J Clin Invest, 123, 1964–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamber C., Kamphuis W., Haring N. L., Peprah N., Middeldorp J., Hol E. M. (2012). GFAPδ expression in glia of the developmental and adolescent mouse brain. PLoS One, 7, e52659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manek R., Moghieb A., Yang Z., Kumar D., Kobessiy F., Sarkis G. A., Raghavan V., Wang K. K. W. (2018). Protein biomarkers and neuroproteomics characterization of microvesicles/exosomes from human cerebrospinal fluid following traumatic brain injury. Mol Neurobiol, 55, 6112–6128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-De Luna R. I., Ku R. Y., Aruck A. M., Santiago F., Viczian A. S., San Mauro D., Zuber M. E. (2017). Muller glia reactivity follows retinal injury despite the absence of the glial fibrillary acidic protein gene in Xenopus. Dev Biol, 426, 219–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masaki K., Suzuki S. O., Matsushita T., Matsuoka T., Imamura S., Yamasaki R., Suzuki M., Suenaga T., Iwaki T., Kira J. I. (2013). Connexin 43 astrocytopathy linked to rapidly progressive multiple sclerosis and neuromyelitis optica. PLoS One, 8, e72919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall M. A., Gregg R. G., Behringer R. R., Brenner M., Delaney C. L., Galbreath E. J., Zhang C. L., Pearce R. A., Chiu S. Y., Messing A. (1996). Targeted deletion in astrocyte intermediate filament (Gfap) alters neuronal physiology. Proc Natl Acad Sci U S A, 93, 6361–6366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medrano S., Steward O. (2001). Differential mRNA localization in astroglial cells in culture. J Comp Neurol, 430, 56–71. [DOI] [PubMed] [Google Scholar]
- Melchionda L., Fang M., Wang H., Fugnanesi V., Morbin M., Liu X., Li W., Ceccherini I., Farina L., Savoiardo M., D'Adamo P., Zhang J., Costa A., Ravaglia S., Ghezzi D., Zeviani M. (2013). Adult-onset Alexander disease, associated with a mutation in an alternative GFAP transcript, may be phenotypically modulated by a non-neutral HDAC6 variant. Orphanet J Rare Dis, 8, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messing A. (2018. a) Alexander disease. Handb Clin Neurol, 148, 693–700. [DOI] [PubMed] [Google Scholar]
- Messing A. (2018. b). Alexander disease: A guide for patients and families (106 p). Morgan & Claypool Publishers. [Google Scholar]
- Messing A. (2019). Refining the concept of GFAP toxicity in Alexander disease. J Neurodev Disord, 11, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messing A., Brenner M., Feany M. B., Nedergaard M., Goldman J. E. (2012). Alexander disease. J Neurosci, 32, 5017–5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messing A., Head M. W., Galles K., Galbreath E. J., Goldman J. E., Brenner M. (1998). Fatal encephalopathy with astrocyte inclusions in GFAP transgenic mice. Am J Pathol, 152, 391–398. [PMC free article] [PubMed] [Google Scholar]
- Middeldorp J., Kamphuis W., Sluijs J. A., Achoui D., Leenaars C. H. C., Feenstra M. G. P., van Tijn P., Fischer D. F., Berkers C., Ovaa H., Quinlan R. A., Hol E. M. (2009). Intermediate filament transcription in astrocytes is repressed by proteasome inhibition. FASEB J, 23, 2710–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignot C., Delarasse C., Escaich S., della Gaspera B., Noé E., Colucci-Guyon E., Babinet C., Pekny M., Vicart P., Boespflug-Tanguy O., Dautigny A., Rodriguez D., Pham-Dinh D. (2007). Dynamics of mutated GFAP aggregates revealed by real-time imaging of an astrocyte model of Alexander disease. Exp Cell Res, 313, 2766–2779. [DOI] [PubMed] [Google Scholar]
- Miller W. J., Leventhal I., Scarsella D., Haydon P. G., Janmey P., Meaney D. F. (2009). Mechanically induced reactive gliosis causes ATP-mediated alterations in astrocyte stiffness. J Neurotrauma, 26, 789–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minkel H. R., Anwer T. Z., Arps K. M., Brenner M., Olsen M. L. (2015). Elevated GFAP induces astrocyte dysfunction in caudal brain regions: A potential mechanism for hindbrain involved symptoms in type II Alexander disease. Glia, 63, 2285–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake H., Inoue A., Tanaka M., Matsuki N. (2013). Serum glial fibrillary acidic protein as a specific marker for necrotizing meningoencephalitis in Pug dogs. J Vet Med Sci, 75, 1543–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeendarbary E., Weber I. P., Sheridan G. K., Koser D. E., Soleman S., Haenzi B., Bradbury E. J., Fawcett J., Franze K. (2017). The soft mechanical signature of glial scars in the central nervous system. Nat Commun, 8, 14787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeton M., Stassen O. M., Sluijs J. A., van der Meer V. W., Kluivers L. J., van Hoorn H., Schmidt T., Reits E. A., van Strien M. E., Hol E. M. (2016). GFAP isoforms control intermediate filament network dynamics, cell morphology, and focal adhesions. Cell Mol Life Sci, 73, 401–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moneim I. A., Shamy M. Y., el-Gazzar R. M., el-Fawal H. A. (1999). Autoantibodies to neurofilaments (NF), glial fibrillary acidic protein (GFAP) and myelin basic protein (MBP) in workers exposed to lead. J Egypt Public Health Assoc, 74, 121–138. [PubMed] [Google Scholar]
- Moody L. R., Barrett-Wilt G. A., Sussman M. R., Messing A. (2017). Glial fibrillary acidic protein exhibits altered turnover kinetics in a mouse model of Alexander disease. J Biol Chem, 292, 5814–5824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouser P. E., Head E., Ha K. H., Rohn T. T. (2006). Caspase-mediated cleavage of glial fibrillary acidic protein within degenerating astrocytes of the Alzheimer's disease brain. Am J Pathol, 168, 936–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L., Oldstone M. B. A., Morris J. C., Nerenberg M. I. (1991). Rapid activation of astrocyte-specific expression of GFAP-lacZ transgene by focal injury. New Biol, 3, 465–474. [PubMed] [Google Scholar]
- Nakamura Y., Takeda M., Aimoto S., Hojo H., Takao T., Shimonishi Y., Hariguchi S., Nishimura T. (1992). Assembly regulatory domain of glial fibrillary acidic protein. A single phosphorylation diminishes its assembly-accelerating property. J Biol Chem, 267, 23269–23274. [PubMed] [Google Scholar]
- Nakazawa E., Ishikawa H. (1998). Ultrastructural observations of astrocyte end-feet in the rat central nervous system. J Neurocytol, 27, 431–440. [DOI] [PubMed] [Google Scholar]
- Nawashiro H., Messing A., Azzam N., Brenner M. (1998). Mice lacking glial fibrillary acidic protein are hypersensitive to traumatic cerebrospinal injury. NeuroReport, 9, 1691–1696. [DOI] [PubMed] [Google Scholar]
- Neusch C., Rozengurt N., Jacobs R. E., Lester H. A., Kofuji P. (2001). Kir4.1 potassium channel subunit is crucial for oligodendrocyte development and in vivo myelination. J Neurosci, 21, 5429–5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas A. P., Sambandam T., Echols J. D., Barnum S. R. (2005). Expression of citrullinated proteins in murine experimental autoimmune encephalomyelitis. J Comp Neurol, 486, 254–266. [DOI] [PubMed] [Google Scholar]
- Nicholas A. P., Sambandam T., Echols J. D., Tourtellotte W. W. (2004). Increased citrullinated glial fibrillary acidic protein in secondary progressive multiple sclerosis. J Comp Neurol, 473, 128–136. [DOI] [PubMed] [Google Scholar]
- Nicholl I. D., Quinlan R. A. (1994). Chaperone activity of α-crystallins modulates intermediate filament assembly. EMBO J, 13, 945–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen A. L., Holm I. E., Johansen M., Bonven B., Jørgensen P., Jørgensen A. L. (2002). A new splice variant of glial fibrillary acidic protein, GFAP-ε, interacts with the presenilin proteins. J Biol Chem, 277, 29983–29991. [DOI] [PubMed] [Google Scholar]
- Nielsen A. L., Jørgensen A. L. (2003). Structural and functional characterization of the zebrafish gene for glial fibrillary acidic protein, GFAP. Gene, 310, 123–132. [DOI] [PubMed] [Google Scholar]
- Nielsen A. L., Jørgensen A. L. (2004). Self-assembly of the cytoskeletal glial fibrillary acidic protein is inhibited by an isoform-specific C-terminus. J Biol Chem, 279, 41537–41545. [DOI] [PubMed] [Google Scholar]
- Oberheim N. A., Takano T., Han X., He W., Lin J. H., Wang F., Xu Q., Wyatt J. D., Pilcher W., Ojemann J. G., Ransom B. R., Goldman S. A., Nedergaard M. (2009). Uniquely hominid features of adult human astrocytes. J Neurosci, 29, 3276–3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olabarria M., Goldman J. E. (2017). Disorders of astrocytes: Alexander disease as a model. Annu Rev Pathol, 12, 131–152. [DOI] [PubMed] [Google Scholar]
- Orr A. L., Li S., Wang C.-E., Li H., Wang J., Rong J. G., Xu X., Mastroberardino P. G., Greenamyre J. T., Li X.-J. (2008). N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J Neurosci, 28, 2783–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang Z., Higuchi M., Koriyama H., Yoshida S., Kurinami H., Shimamura M., Takami Y., Rakugi H., Morishita R., Nakagami H. (2017. a). Evaluating the potential of the GFAP-KLH immune-tolerizing vaccine for type 1 diabetes in mice. FEBS Lett, 591, 129–136. [DOI] [PubMed] [Google Scholar]
- Pang Z., Kushiyama A., Sun J., Kikuchi T., Yamazaki H., Iwamoto Y., Koriyama H., Yoshida S., Shimamura M., Higuchi M., Kawano T., Takami Y., Rakugi H., Morishita R., Nakagami H. (2017. b). Glial fibrillary acidic protein (GFAP) is a novel biomarker for the prediction of autoimmune diabetes. FASEB J, 31, 4053–4063. [DOI] [PubMed] [Google Scholar]
- Pareyson D., Fancellu R., Mariotti C., Romano S., Salmaggi A., Carella F., Girotti F., Gattellaro G., Carriero M. R., Farina L., Ceccherini I., Savoiardo M. (2008). Adult-onset Alexander disease: A series of eleven unrelated cases with review of the literature. Brain, 131, 2321–2331. [DOI] [PubMed] [Google Scholar]
- Pekny M., Levéen P., Pekna M., Eliasson C., Berthold C. H., Westermark B., Betsholtz C. (1995). Mice lacking glial fibrillary acidic protein display astrocytes devoid of intermediate filaments but develop and reproduce normally. EMBO J, 14, 1590–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny M., Pekna M. (2014). Astrocyte reactivity and reactive astrogliosis: Costs and benefits. Physiol Rev, 94, 1077–1098. [DOI] [PubMed] [Google Scholar]
- Pekny M., Pekna M., Messing A., Steinhäuser C., Lee J. M., Parpura V., Hol E. M., Sofroniew M. V., Verkhratsky A. (2016). Astrocytes: A central element in neurological diseases. Acta Neuropathol, 131, 323–345. [DOI] [PubMed] [Google Scholar]
- Pekny M., Wilhelmsson U., Tatlisumak T., Pekna M. (2019). Astrocyte activation and reactive gliosis – A new target in stroke? Neurosci Lett, 689, 45–55. [DOI] [PubMed] [Google Scholar]
- Pekovic D., Raine C. S., Traugott U. (1990). Increase in anti-astrocyte antibodies in the serum of guinea pigs during active stages of experimental autoimmune encephalomyelitis. J Neuroimmunol, 26, 251–259. [DOI] [PubMed] [Google Scholar]
- Perng M. D., Su M., Wen S. F., Li R., Gibbon T., Prescott A. R., Brenner M., Quinlan R. A. (2006). The Alexander disease-causing GFAP mutant, R416W, accumulates into Rosenthal fibers by a pathway involving filament aggregation and the association of αB-crystallin and HSP27. Am J Hum Genet, 79, 197–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perng M. D., Wen S. F., Gibbon T., Middeldorp J., Sluijs J., Hol E. M., Quinlan R. A. (2008). Glial fibrillary acidic protein filaments can tolerate the incorporation of assembly-compromised GFAP-δ, but with consequences for filament organization and αB-crystallin association. Mol Biol Cell, 19, 4521–4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A., Palay S. L., Webster . H. D. (1991). The fine structure of the nervous system: Neurons and their supporting cells. Oxford University Press. [Google Scholar]
- Petzold A. (2015). Glial fibrillary acidic protein is a body fluid biomarker for glial pathology in human disease. Brain Res, 1600, 17–31. [DOI] [PubMed] [Google Scholar]
- Price J. C., Guan S., Burlingame A., Prusiner S. B., Ghaemmaghami S. (2010). Analysis of proteome dynamics in the mouse brain. Proc Natl Acad Sci U S A, 107, 14508–14513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prust M., et al. (2011). GFAP mutations, age of onset, and clinical sub-types in Alexander disease. Neurology, 77, 1287–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan R. A., Schwarz N., Windoffer R., Richardson C., Hawkins T., Broussard J. A., Green K. J., Leube R. E. (2017). A rim-and-spoke hypothesis to explain the biomechanical roles for cytoplasmic intermediate filament networks. J Cell Sci, 130, 3437–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff M. C., Fields K. L., Hakomori S. I., Mirsky R., Pruss R. M., Winter J. (1979). Cell-type-specific markers for distinguishing and studying neurons and the major classes of glial cells in culture. Brain Res, 174, 283–308. [DOI] [PubMed] [Google Scholar]
- Ralton J. E., Lu X., Hutcheson A. M., Quinlan R. A. (1994). Identification of two N-terminal non-alpha-helical domain motifs important in the assembly of glial fibrillary acidic protein. J Cell Sci, 107, 1935–1948. [DOI] [PubMed] [Google Scholar]
- Riol H., Tardy M., Rolland B., Lévesque G., Ven Murthy M. R. (1997). Detection of the peripheral nervous system (PNS)-type glial fibrillary acidic protein (GFAP) and its mRNA in human lymphocytes. J Neurosci Res, 48, 53–62. [PubMed] [Google Scholar]
- Roelofs R. F., Fischer D. F., Houtman S. H., Sluijs J. A., Van Haren W., van Leeuwen F. W., Hol E. M. (2005). Adult human subventricular, subgranular, and subpial zones contain astrocytes with a specialized intermediate filament cytoskeleton. Glia, 52, 289–300. [DOI] [PubMed] [Google Scholar]
- Rosenthal W. (1898). Über eine eigenthümliche, mit syringomyelie complicirte geschwulst des rückenmarks. Bietr Pathol Anat, 23, 111–143. [Google Scholar]
- Rutka J. T., Hubbard S. L., Fukuyama K., Matsuzawa K., Dirks P. B., Becker L. E. (1994). Effects of antisense glial fibrillary acidic protein complementary DNA on the growth, invasion, and adhesion of human astrocytoma cells. Cancer Res, 54, 3267–3272. [PubMed] [Google Scholar]
- Saito K., Shigetomi E., Yasuda R., Sato R., Nakano M., Tashiro K., Tanaka K. F., Ikenaka K., Mikoshiba K., Mizuta I., Yoshida T., Nakagawa M., Mizuno T., Koizumi S. (2018). Aberrant astrocyte Ca2+ signals “AxCa signals” exacerbate pathological alterations in an alexander disease model. Glia, 66, 1053–1067. [DOI] [PubMed] [Google Scholar]
- Sarkar S., Floto R. A., Berger Z., Imarisio S., Cordenier A., Pasco M., Cook L. J., Rubinsztein D. C. (2005). Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol, 17, 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarthy P. V., Fu M., Huang J. (1989). Subcellular localization of an intermediate filament protein and its mRNA in glial cells. Mol Cell Biol, 9, 4556–4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki K., Bean A., Shah S., Schutten E., Huseby P. G., Peters B., Shen Z. T., Vanguri V., Liggitt D., Huseby E. S. (2014). Relapsing-remitting central nervous system autoimmunity mediated by GFAP-specific CD8 T cells. J Immunol, 192, 3029–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl U. I., Choi M., Liu T., Ramaekers V. T., Häusler M. G., Grimmer J., Tobe S. W., Farhi A., Nelson-Williams C., Lifton R. P. (2009). Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci U S A, 106, 5842–5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sechi G., Ceccherini I., Bachetti T., Deiana G. A., Sechi E., Balbi P. (2013). Ceftriaxone for Alexander's disease: A four-year follow-up. JIMD Rep, 9, 67–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh T. H., et al. (2009). High-resolution mapping and analysis of copy number variations in the human genome: A data resource for clinical and research applications. Genome Res, 19, 1682–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shehab S. A., Cronly-Dillon J. R., Nona S. N., Stafford C. A. (1990). Preferential histochemical staining of protoplasmic and fibrous astrocytes in rat CNS with GFAP antibodies using different fixatives. Brain Res, 518, 347–352. [DOI] [PubMed] [Google Scholar]
- Shibuki K., Gomi H., Chen L., Bao S. W., Kim J. S. K., Wakatsuki H., Fujisaki T., Fujimoto J., Katoh A., Ikeda T., Chen C., Thompson R. F., Itohara S. (1996). Deficient cerebellar long-term depression, impaired eyeblink conditioning, and normal motor coordination in GFAP mutant mice. Neuron, 16, 587–599. [DOI] [PubMed] [Google Scholar]
- Shibuya M., Matsuki N., Fujiwara K., Imajoh-Ohmi S., Fukuda H., Pham N. T., Tamahara S., Ono K. (2007). Autoantibodies against glial fibrillary acidic protein (GFAP) in cerebrospinal fluids from pug dogs with necrotizing meningoencephalitis. J Vet Med Sci, 69, 241–245. [DOI] [PubMed] [Google Scholar]
- Singh R., Nielsen A. L., Johansen M. G., Jørgensen A. L. (2003). Genetic polymorphism and sequence evolution of an alternatively spliced exon of the glial fibrillary acidic protein gene, GFAP⋆. Genomics, 82, 185–193. [DOI] [PubMed] [Google Scholar]
- Snider N. T., Omary M. B. (2014). Post-translational modifications of intermediate filament proteins: Mechanisms and functions. Nat Rev Mol Cell Biol, 15, 163–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosunov A. A., Guilfoyle E., Wu X., McKhann G. M., Goldman J. E. (2013). Phenotypic conversions of “protoplasmic” to “reactive” astrocytes in Alexander disease. J Neurosci, 2013, 7439–7450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosunov A. A., McKhann G. M., 2nd, Goldman J. E. (2017). The origin of Rosenthal fibers and their contributions to astrocyte pathology in Alexander disease. Acta Neuropathol Commun, 5, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standifer N. E., Ouyang Q., Panagiotopoulos C., Verchere C. B., Tan R., Greenbaum C. J., Pihoker C., Nepom G. T. (2006). Identification of Novel HLA-A*0201-restricted epitopes in recent-onset type 1 diabetic subjects and antibody-positive relatives. Diabetes, 55, 3061–3067. [DOI] [PubMed] [Google Scholar]
- Stassen O. M. J. A., van Bodegraven E. J., Giuliani F., Moeton M., Kanski R., Sluijs J. A., van Strien M. E., Kamphuis W., Robe P. A. J., Hol E. M. (2017). GFAPdelta/GFAPα ratio directs astrocytoma gene expression towards a more malignant profile. Oncotarget, 8, 88104–88121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenzel W., Soltek S., Schluter D., Deckert M. (2004). The intermediate filament GFAP is important for the control of experimental murine Staphylococcus aureus-induced brain abscess and Toxoplasma encephalitis. J Neuropathol Exp Neurol, 63, 631–640. [DOI] [PubMed] [Google Scholar]
- Sullivan S. M., Sullivan R. K., Miller S. M., Ireland Z., Bjorkman S. T., Pow D. V., Colditz P. B. (2012). Phosphorylation of GFAP is associated with injury in the neonatal pig hypoxic-ischemic brain. Neurochem Res, 37, 2364–2378. [DOI] [PubMed] [Google Scholar]
- Takizawa T., Gudla P. R., Guo L. Y., Lockett S., Misteli T. (2008). Allele-specific nuclear positioning of the monoallelically expressed astrocyte marker GFAP. Genes Dev, 22, 489–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka J., Nakamura K., Takeda M., Tada K., Suzuki H., Morita H., Okado T., Hariguchi S., Nishimura T. (1989). Enzyme-linked immunosorbent-assay for human autoantibody to glial fibrillary acidic protein – Higher titer of the antibody is detected in serum of patients with Alzheimer's disease. Acta Neurol Scand, 80, 554–560. [DOI] [PubMed] [Google Scholar]
- Tanaka K., Watase K., Manabe T., Yamada K., Watanabe M., Takahashi K., Iwama H., Nishikawa T., Ichihara N., Kikuchi T., Okuyama S., Kawashima N., Hori S., Takimoto M., Wada K. (1997). Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science, 276, 1699–1702. [DOI] [PubMed] [Google Scholar]
- Tanaka K. F., Takebayashi H., Yamazaki Y., Ono K., Naruse M., Iwasato T., Itohara S., Kato H., Ikenaka K. (2007). The murine model of Alexander disease: Analysis of GFAP aggregate formation and its pathological significance. Glia, 55, 617–631. [DOI] [PubMed] [Google Scholar]
- Tang G., Perng M. D., Wilk S., Quinlan R., Goldman J. E. (2010). Oligomers of mutant glial fibrillary acidic protein (GFAP) inhibit the proteasome system in Alexander disease astrocytes, and the small heat shock protein, αB-crystallin, reverses the inhibition. J Biol Chem, 285, 10527–10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G., Xu Z., Goldman J. E. (2006). Synergistic effects of the SAPK/JNK and the proteasome pathway on glial fibrillary acidic protein (GFAP) accumulation in Alexander disease. J Biol Chem, 281, 38634–38643. [DOI] [PubMed] [Google Scholar]
- Tang G., Yue Z., Talloczy Z., Hagemann T., Cho W., Messing A., Sulzer D. L., Goldman J. E. (2008). Autophagy induced by Alexander disease-mutant GFAP accumulation is regulated by p38/MAPK and mTOR signaling pathways. Hum Mol Genet, 17, 1540–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen R., Daugaard T. F., Holm I. E., Nielsen A. L. (2013). Alternative mRNA splicing from the glial fibrillary acidic protein (GFAP) gene generates isoforms with distinct subcellular mRNA localization patterns in astrocytes. PLoS One, 8, e72110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian R., Wu X., Hagemann T. L., Sosunov A. A., Messing A., McKhann G. M., Goldman J. E. (2010). Alexander disease mutant GFAP compromises glutamate transport in astrocytes. J Neuropathol Exp Neurol, 69, 335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triolo D., Dina G., Lorenzetti I., Malaguti M. C., Morana P., Del Carro U., Comi G., Messing A., Quattrini A., Previtali S. C. (2006). Loss of glial fibrillary acidic protein (GFAP) impairs Schwann cell proliferation and delays nerve regeneration after damage. J Cell Sci, 119, 3981–3993. [DOI] [PubMed] [Google Scholar]
- Tsui H., Chan Y., Tang L., Winer S., Cheung R. K., Paltser G., Selvanantham T., Elford A. R., Ellis J. R., Becker D. J., Ohashi P. S., Dosch H.-M. (2008). Targeting of pancreatic glia in type 1 diabetes. Diabetes, 57, 918–928. [DOI] [PubMed] [Google Scholar]
- Uchida K., Hasegawa T., Ikeda M., Yamaguchi R., Tateyama S. (1999). Detection of an autoantibody from Pug dogs with necrotizing encephalitis (Pug dog encephalitis). Vet Pathol, 36, 301–307. [DOI] [PubMed] [Google Scholar]
- Uhlén M., et al. (2015). Tissue-based map of the human proteome. Science, 347, 1260419. [DOI] [PubMed] [Google Scholar]
- Uyeda C. T., Eng L. F., Bignami A. (1972). Immunological study of the glial fibrillary acidic protein. Brain Res, 37, 81–89. [DOI] [PubMed] [Google Scholar]
- Vagner T., Dvorzhak A., Wójtowicz A. M., Harms C., Grantyn R. (2016). Systemic application of AAV vectors targeting GFAP-expressing astrocytes in Z-Q175-KI Huntington's disease mice. Mol Cell Neurosci, 77, 76–86. [DOI] [PubMed] [Google Scholar]
- van Bodegraven E. J., van Asperen J. V., Robe P. A. J., Hol E. M. (2019. a). Importance of GFAP isoform-specific analyses in astrocytoma. Glia, 67, 1417–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bodegraven E. J., van Asperen J. V., Sluijs J. A., van Deursen C. B. J., van Strien M. E., Stassen O., Robe P. A. J., Hol E. M. (2019. b). GFAP alternative splicing regulates glioma cell-ECM interaction in a DUSP4-dependent manner. FASEB J, 33, 12941–12959. [DOI] [PubMed] [Google Scholar]
- van den Berge S. A., Middeldorp J., Zhang C. E., Curtis M. A., Leonard B. W., Mastroeni D., Voorn P., van de Berg W. D. J., Huitinga I., Hol E. M. (2010). Longterm quiescent cells in the aged human subventricular neurogenic system specifically express GFAP-δ. Aging Cell, 9, 313–326. [DOI] [PubMed] [Google Scholar]
- Viale G., Doglioni C., Dell'Orto P., Zanetti G., Iuzzolino P., Bontempini L., Coggi G. (1988). Glial fibrillary acidic protein immunoreactivity in human respiratory tract cartilages and pulmonary chondromatous hamartomas. Am J Pathol, 133, 363–373. [PMC free article] [PubMed] [Google Scholar]
- Viedma-Poyatos A., de Pablo Y., Pekny M., Pérez-Sala D. (2018). The cysteine residue of glial fibrillary acidic protein is a critical target for lipoxidation and required for efficient network organization. Free Radic Biol Med, 120, 380–394. [DOI] [PubMed] [Google Scholar]
- von Jonquieres G., Mersmann N., Klugmann C. B., Harasta A. E., Lutz B., Teahan O., Housley G. D., Fröhlich D., Krämer-Albers E. M., Klugmann M. (2013). Glial promoter selectivity following AAV-delivery to the immature brain. PLoS One, 8, e65646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker A. K., LaPash Daniels C. M., Goldman J. E., Trojanowski J. Q., Lee V. M.-Y., Messing A. (2014). Astrocytic TDP-43 pathology in Alexander disease. J Neurosci, 34, 6448–6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Hagemann T. L., Kalwa H., Michel T., Messing A., Feany M. B. (2015). Nitric oxide mediates glial-induced neurodegeneration in Alexander disease. Nat Commun, 6, 8966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Hagemann T. L., Messing A., Feany M. B. (2016). An in vivo pharmacological screen identifies cholinergic signaling as a therapeutic target in glial-based nervous system disease. J Neurosci, 36, 1445–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Xia J., Li J., Hagemann T. L., Jones J. R., Fraenkel E., Weitz D. A., Zhang S. C., Messing A., Feany M. B. (2018). Tissue and cellular rigidity and mechanosensitive signaling activation in Alexander disease. Nat Commun, 9, 1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei P., Zhang W., Yang L. S., Zhang H. S., Xu X. E., Jiang Y. H., Huang F. P., Shi Q. (2013). Serum GFAP autoantibody as an ELISA-detectable glioma marker. Tumor Biol, 34, 2283–2292. [DOI] [PubMed] [Google Scholar]
- Weinstein D. E., Shelanski M. L., Liem R. K. H. (1991). Suppression by antisense mRNA demonstrates a requirement for the glial fibrillary acidic protein in the formation of stable astrocytic processes in response to neurons. J Cell Biol, 112, 1205–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicht H., Derouiche A., Korf H. W. (1994). An immunocytochemical investigation of glial morphology in the Pacific hagfish: Radial and astrocyte-like glia have the same phylogenetic age. J Neurocytol, 23, 565–576. [DOI] [PubMed] [Google Scholar]
- Wilhelm A., Volknandt W., Langer D., Nolte C., Kettenmann H., Zimmermann H. (2004). Localization of SNARE proteins and secretory organelle proteins in astrocytes in vitro and in situ. Neurosci Res, 48, 249–257. [DOI] [PubMed] [Google Scholar]
- Wilhelmsson U., Andersson D., de Pablo Y., Pekny R., Ståhlberg A., Mulder J., Mitsios N., Hortobágyi T., Pekny M., Pekna M. (2017). Injury leads to the appearance of cells with characteristics of both microglia and astrocytes in mouse and human brain. Cereb Cortex, 27, 3360–3377. [DOI] [PubMed] [Google Scholar]
- Wilhelmsson U., Li L. Z., Pekna M., Berthold C. H., Blom S., Eliasson C., Renner O., Bushong E., Ellisman M., Morgan T. E., Pekny M. (2004). Absence of glial fibrillary acidic protein and vimentin prevents hypertrophy of astrocytic processes and improves post-traumatic regeneration. J Neurosci, 24, 5016–5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis C. M., Ménoret A., Jellison E. R., Nicaise A. M., Vella A. T., Crocker S. J. (2017). A refined bead-free method to identify astrocytic exosomes in primary glial cultures and blood plasma. Front Neurosci, 11, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winer S., Tsui H., Lau A., Song A., Li X., Cheung R. K., Sampson A., Afifiyan F., Elford A., Jackowski G., Becker D. J., Santamaria P., Ohashi P., Dosch H. M. (2003). Autoimmune islet destruction in spontaneous type 1 diabetes is not β-cell exclusive. Nat Med, 9, 198–205. [DOI] [PubMed] [Google Scholar]
- Wippold F. J., Perry A., Lennerz J. (2006). Neuropathology for the neuroradiologist: Rosenthal fibers. Am J Neuroradiol, 27, 958–961. [PMC free article] [PubMed] [Google Scholar]
- Yankner B. A., Dawes L. R., Fisher S., Villa-Komaroff L., Oster-Granite M. L., Neve R. L. (1989). Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer's disease. Science, 245, 417–420. [DOI] [PubMed] [Google Scholar]
- Yeaney G. A., Brat D. J. (2019). What every neuropathologist needs to know: Update on cIMPACT-NOW. J Neuropathol Exp Neurol, 78, 294–296. [DOI] [PubMed] [Google Scholar]
- Yeo S., Bandyopadhyay S., Messing A., Brenner M. (2013). Transgenic analysis of GFAP promoter elements. Glia, 61, 1488–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T., Sasaki M., Yoshida M., Namekawa M., Okamoto Y., Tsujino S., Sasayama H., Mizuta I., Nakagawa M. (2011). Nationwide survey of Alexander disease in Japan and proposed new guidelines for diagnosis. J Neurol, 258, 1998–2008. [DOI] [PubMed] [Google Scholar]
- Yu A. C. H., Lee Y. L., Eng L. F. (1991). Inhibition of GFAP synthesis by antisense RNA in astrocytes. J Neurosci Res, 30, 72–79. [DOI] [PubMed] [Google Scholar]
- Yu A. C. H., Lee Y. L., Eng L. F. (1993). Astrogliosis in culture: I. The model and the effect of antisense oligonucleotides on glial fibrillary acidic protein synthesis. J Neurosci Res, 34, 295–303. [DOI] [PubMed] [Google Scholar]
- Zeisel A., Hochgerner H., Lönnerberg P., Johnsson A., Memic F., van der Zwan J., Häring M., Braun E., Borm L. E., La Manno G., Codeluppi S., Furlan A., Lee K., Skene N., Harris K. D., Hjerling-Leffler J., Arenas E., Ernfors P., Marklund U., Linnarsson S. (2018). Molecular architecture of the mouse nervous system. Cell, 174, 999–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelenika D., Grima B., Brenner M., Pessac B. (1995). A novel glial fibrillary acidic protein mRNA lacking exon 1. Mol Brain Res, 30, 251–258. [DOI] [PubMed] [Google Scholar]
- Zhang Z. Q., et al. (2014). Human traumatic brain injury induces autoantibody response against glial fibrillary acidic protein and its breakdown products. PLoS One, 9, e101712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L., Li P., Chen N., Dai L. F., Gao K., Liu Y. N., Shen L., Wang J. M., Jiang Y. W., Wu Y. (2019). Modeling vanishing white matter disease with patient-derived induced pluripotent stem cells reveals astrocytic dysfunction. CNS Neurosci Ther, 25, 759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-pdf-1-asn-10.1177_1759091420949680 for GFAP at 50 by Albee Messing and Michael Brenner in ASN Neuro
Supplemental material, sj-pdf-2-asn-10.1177_1759091420949680 for GFAP at 50 by Albee Messing and Michael Brenner in ASN Neuro