

Abstract
Microbiomes are vast communities of microorganisms and viruses that populate all natural ecosystems. Viruses have been considered to be the most variable component of microbiomes, as supported by virome surveys and examples of high genomic mosaicism. However, recent evidence suggests that the human gut virome is remarkably stable compared with that of other environments. Here, we investigate the origin, evolution and epidemiology of crAssphage, a widespread human gut virus. Through a global collaboration, we obtained DNA sequences of crAssphage from more than one-third of the world’s countries and showed that the phylogeography of crAssphage is locally clustered within countries, cities and individuals. We also found fully colinear crAssphage-like genomes in both Old-World and New-World primates, suggesting that the association of crAssphage with primates may be millions of years old. Finally, by exploiting a large cohort of more than 1,000 individuals, we tested whether crAssphage is associated with bacterial taxonomic groups of the gut microbiome, diverse human health parameters and a wide range of dietary factors. We identified strong correlations with different clades of bacteria that are related to Bacteroidetes and weak associations with several diet categories, but no significant association with health or disease. We conclude that crAssphage is a benign cosmopolitan virus that may have coevolved with the human lineage and is an integral part of the normal human gut virome.
Phages form the vast majority of the human gut virome in healthy individuals, with an estimated 5 × 109 phages per gram of human faeces versus 9 × 1010 bacteria1,2. Phages are critical for the control of bacterial populations and vary widely between individuals3–5. Evolutionary and genomic studies have suggested that dynamic phage–host interactions are reflected in phage genomes, which show high sequence diversity and mosaicism6,7. In marine aquatic ecosystems, phages only persist in the environment for one to two days8–10, but those dynamics may be substantially different in the human gut virome, where phages can persist for more than a year in individual people3,11. The origin, evolution and epidemiology of the human virome remains uninvestigated at the scale of the global human population. To circumvent the interindividual variations in the gut virome, we investigated the widespread virus crAssphage12, the global ubiquity of which allows these questions to be addressed at the global scale. We found that crAssphage is stable in individuals, common throughout the human population and not associated with any health or disease phenotypes. These results support a model of a highly stable benign association of the human virome with the primate lineage that may be millions of years old.
crAssphage populations in the human gut
We assessed the origin, evolution and epidemiology of crAssphage—one of the most ubiquitous human gut viruses—to understand the stability of the human gut virome. We previously recovered the crAssphage sequence from more than half of 466 faecal metagenomic datasets12 and the first member of the expansive crAss-like family to be cultured13 was recently reported14. We screened the crAssphage genome for regions that were present in many different datasets—regions in which variable segments were flanked by conserved regions suitable for targeting by PCR primers—and we identified three amplicon regions of approximately 1.3 kilobases (kb; see Methods). We tested faecal samples of 45 healthy individuals from four cities in two different continents and found that almost half of these volunteers (21 individuals) were crAss-positive, as determined by gel electrophoresis. We followed six individuals over two months to assess the stability of crAssphage populations (Fig. 1). Two individuals were consistently crAss-negative, whereas others displayed more variable dynamics. Notably, DNA sequencing revealed that crAssphage strains from each individual tended to be phylogenetically clustered, although the sequences are not phylogenetically clustered by date (Fig. 1g–i). This suggests that multiple closely related crAssphage populations may coexist within one individual in whom the abundance—and thus detection—of these populations varies in time. Such ecological dynamics may also explain the fact that Male 1 was intermittently crAss-negative, although his sequences were still clustered in the phylogenetic trees.
Fig. 1 |. crAssphage presence or absence status over time in the human gut.
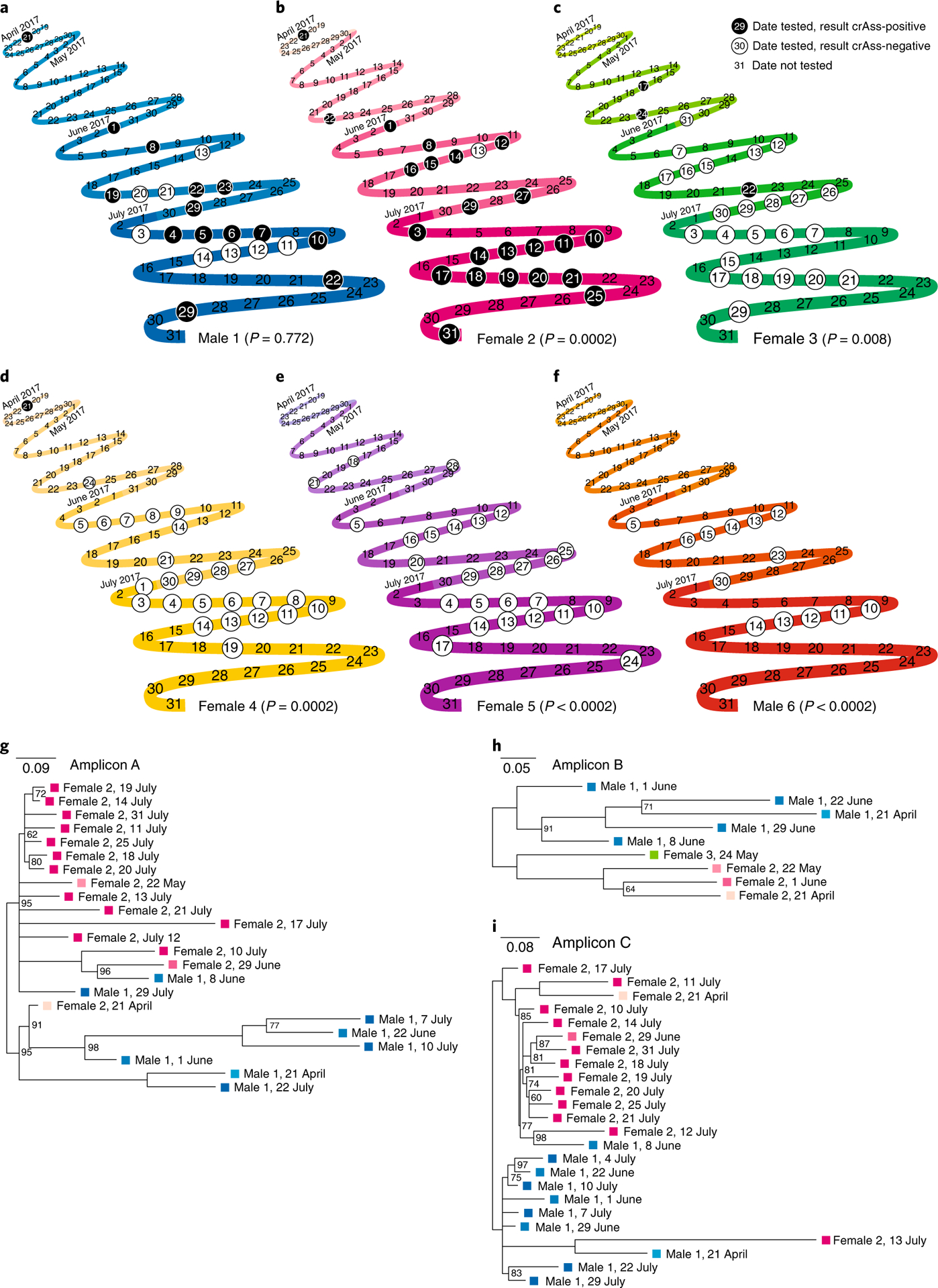
a–f, Timelines of the crAssphage status of six volunteers (a–f) between April and July 2017, in which each limb of the curve represents a week from Monday to Sunday, and subsequent months are indicated by increasingly intense colours per individual. On the circled dates, individuals were tested for crAssphage using PCR analysis of amplicons A–C; gel electrophoresis of the three amplicons was always consistent for each sample. The black and white circles indicate crAss-positive and crAss-negative samples, respectively. P values indicate the fraction of cases in which crAss status within individuals was more consistently crAss-positive or crAss-negative in 5,000 random permutations of the status labels across all individuals than those that were observed; in cases in which P < 0.0002, none of the random permutations were more consistent. g–i, Unrooted maximum-likelihood phylogenies of amplicons A (g), B (h) and C (i) show clustering of the sequences by volunteer; note that not all crAss-positive samples could be sequenced. Branches with less than 60% bootstrap support were collapsed; values of less than 100% are displayed. Scale bars indicate the average number of mutations per alignment position. Colours correspond to the individual and the month in which the sample was taken.
To confirm the intraindividual evolution that we observed, we recovered 20 different crAssphage genomes from the faecal viromes of 3 adult female twin pairs and their mothers, using the same datasets that we originally used to discover crAssphage3,12, and built a phylogenomic tree. Genomes that were sampled up to one year apart from the same individual clustered together in the tree (Supplementary Fig. 1), consistent with a model that describes the intraindividual evolution of these dominant gut virome populations that are generally acquired once, but may diverge into several different—albeit related—subpopulations over time3,11.
crAssphage is globally distributed and locally clustered
The phylogenies shown in Fig. 1g–i and Supplementary Fig. 1 suggested that individuals have a dominant and stable crAssphage population in their gut microbiome; however, these results might be skewed by PCR amplification or metagenome assembly. Although higher order groups, including species and genera, remain controversial in viral taxonomy and depend on complete genome sequences15,16, here we defined strains as unique sequences (that is, 100% identical)17. To analyse the number of strains that could co-occur within a single sample, we downloaded 95,552 metagenomic datasets for all environments from the Sequence Read Archive18. Using a strain-resolved bioinformatics pipeline that was developed for this analysis19 (see Methods), we extracted the three amplicon regions from 2,216 datasets, most of which contained only a single crAssphage strain (Fig. 2). Although 95% of all recovered strains were only found in a single sample, one strain of amplicon C was identified 104 times in different datasets (Supplementary Table 1), showing the exceptional ubiquity of some strains around the world. It has been previously suggested that crAssphage is not acquired early in life20; by contrast, our global analysis identified crAssphage in at least 134 infant samples (26 with locality information; Supplementary File 1), confirming recent incidental findings that crAssphage can be found in infants20,21. Sixteen metagenomes contain more than 100 strains. Phylogenetic trees containing these sequences showed that—as in Fig. 1 and Supplementary Fig. 1—strains within a single individual tend to be recently diverged, although different co-occurring clusters could be observed in some cases (Supplementary Fig. 2). Interestingly, the two samples with the most diverse crAssphage populations are from young individuals, including a sample from a healthy child from the United States22 that contained up to 1,409 strains, and a sample from a one-year-old infant from Finland23 that contained up to 748 strains (Fig. 2, Supplementary File 2).
Fig. 2 |. Diversity of crAssphage strains in metagenomic samples.
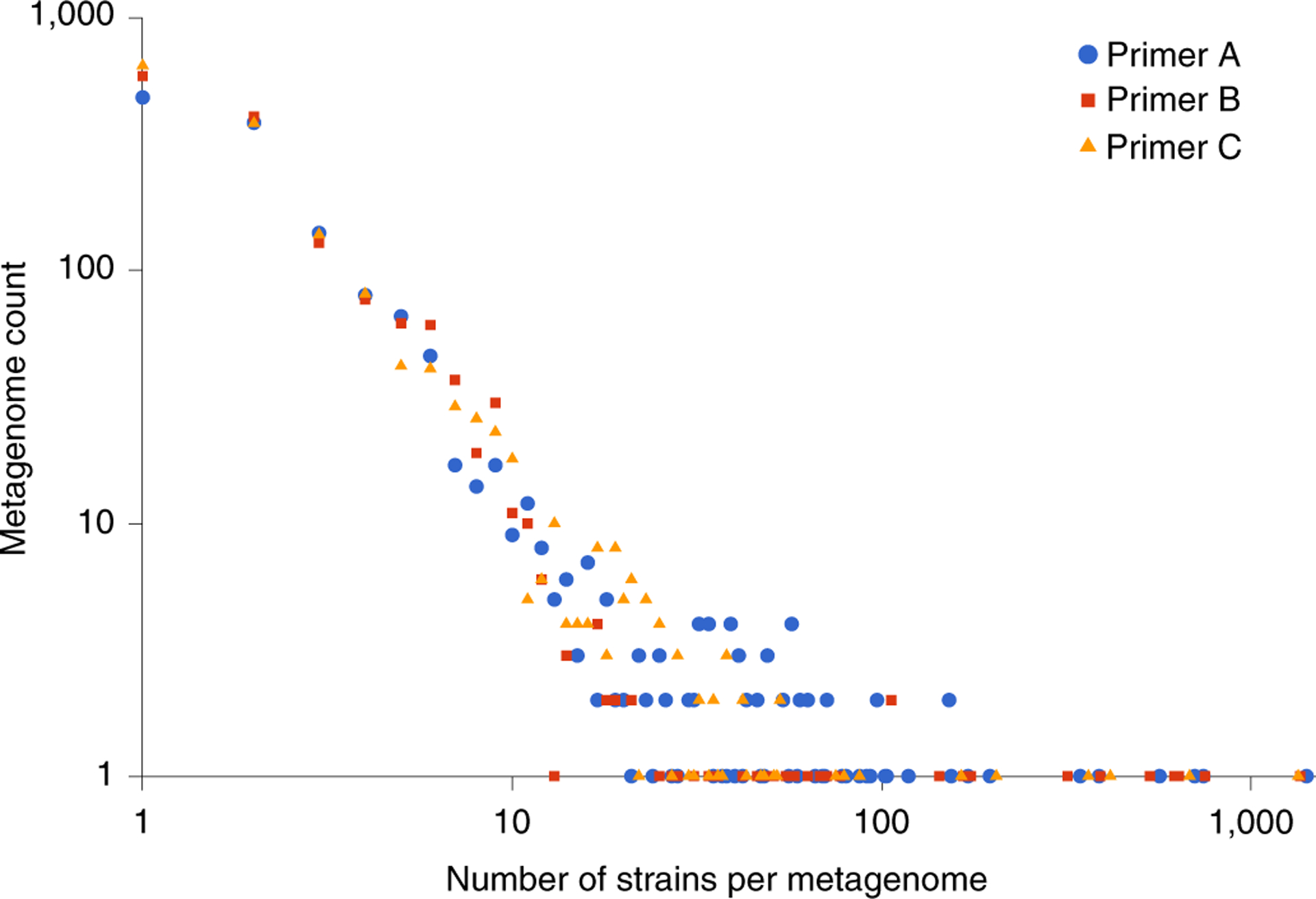
Strains for three amplicon regions A–C were detected in 2,216 metagenomes using Gretel19 (Supplementary File 3).
To investigate the global phylogeography of crAssphage, we collected data about the three amplicon regions from various sources and combined them in a large-scale phylogenetic analysis, providing a worldwide overview of the evolution of an epitome of the human gut virome (Supplementary Table 2). We launched a global collaboration to amplify and sequence the three regions of the crAssphage genome from local sites. To obtain the highest expected rate of detection, collaborators sampled wastewater treatment plants. We combined these sequences with data from the COMPARE sewage sampling project (http://www.compare-europe.eu/) and the sequences from our metagenomics searches and individual volunteers found above. Together, we analysed 32,273 different crAssphage sequences from at least 67 countries in six continents (34% of the countries in the world; Fig. 3, Supplementary Fig. 3, Supplementary File 2). We reconstructed phylogenetic trees for the subset of strains with locality information and used permutation statistics to assess the distribution of the associated sampling metadata24. Sequences from the same country, location and sampling date are significantly clustered in the phylogeny (P < 0.001; Supplementary Fig. 4). Moreover, strains that are genetically most similar tend to be geographically close in most cases (Fig. 3). Thus, crAssphage is a cosmopolitan inhabitant of the human gut throughout the world, with a geographically and temporally local sequence signature that may prove useful in future forensic applications of faecal-contamination identification and detection25–28.
Fig. 3 |. Global locations of 2,424 crAssphage strains for amplicon A.
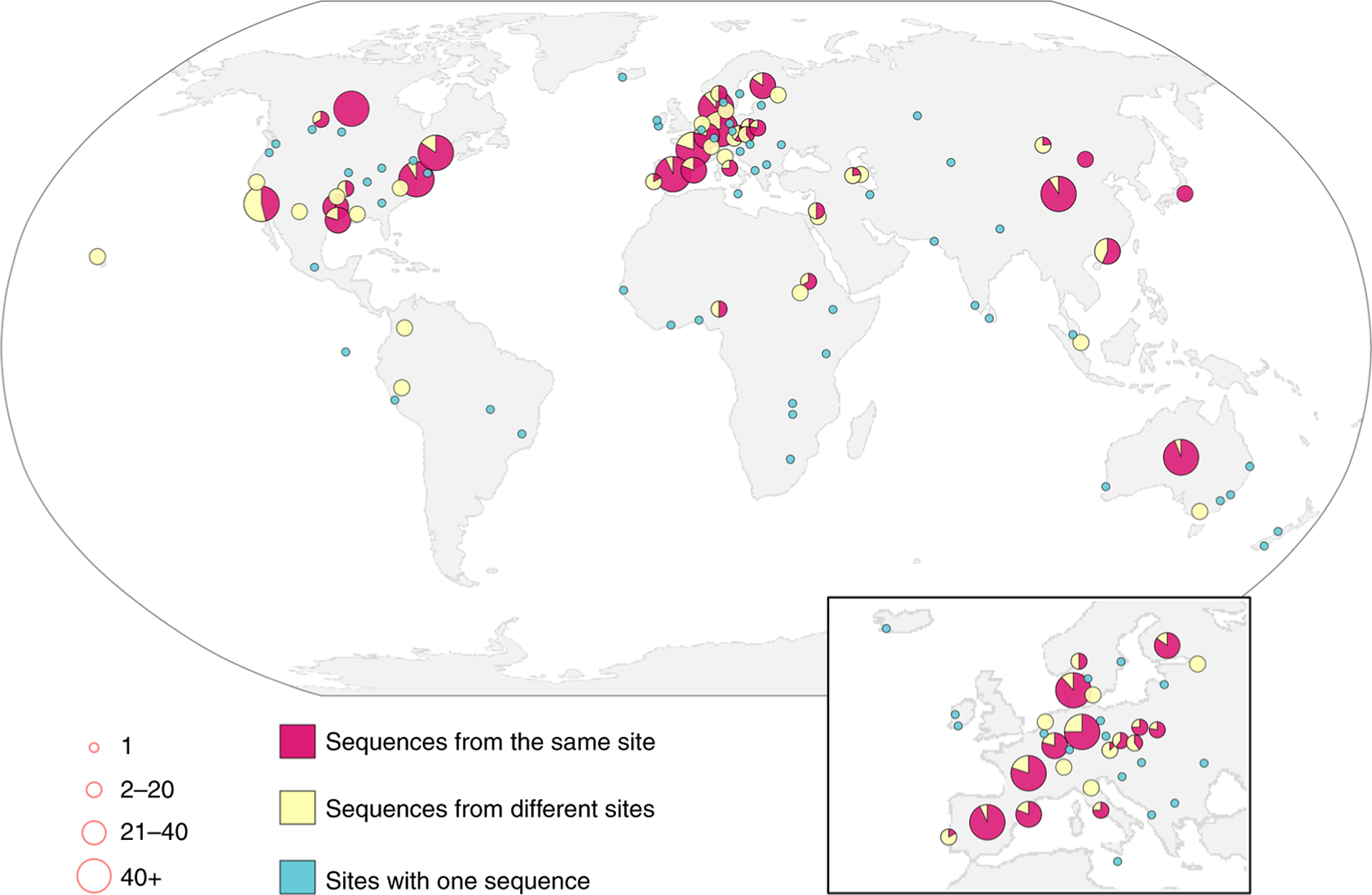
Pie diagrams indicate the fraction of genetically most-similar strains identified at the same site (less than 150 km apart) and at a different site. The number of strains at each location is indicated by the size of the pie diagram. The inset shows a magnification of Europe. See Supplementary Fig. 3 for amplicons B and C.
crAssphage has evolved with humans
The global distribution of crAssphage led us to investigate whether this virus was present in early humans and whether it evolved with us as we spread out and colonized the planet. Alternatively, and consistent with the view of viruses as rapidly evolving entities, it is possible that crAssphage emerged recently—perhaps through recombination of other viruses—and spread around the world either due to factors that relate to the human host—for example, the global food supply chain or international travel—or through the epidemiology of our intestinal bacteria.
To assess the possible ancient association of crAssphage-like phages with the human lineage, we screened the datasets from our global data survey for remote human populations. We found a few crAssphage-like sequences in faecal samples from rural Malawi and from the Amazonas of Venezuela29 (Supplementary Table 3). By contrast, mummified gut samples from three pre-Columbian Andean mummies30 and the European iceman31 were all crAss-negative. Although this could suggest that these individuals were crAss-negative, it is also possible that the DNA of any crAssphage that these individuals may have carried has degraded over thousands of years.
Next, we sequenced and assembled 15 faecal metagenomes from 5 species of non-human primates to search for crAssphage in our distant primate relatives. None of the assembled nucleotide sequences matched the amplicon regions used above; only short stretches of nucleotide homology were identified to the crAssphage genome32. Interestingly, many short homologous regions were found in several long sequences of around 90,000 nucleotides that—when displayed as a dot plot—revealed a range of near-complete genomes of distant crAssphage relatives in apes, Old-World monkeys and New-World monkeys (Fig. 4). Although those genomes were distantly related to crAssphage, they were clearly colinear, showing the long-term genomic stability of this widespread gut virus. These results are consistent with a recent study that identified ten candidate crAss-like phage genera, the genomes of which were also colinear33. To investigate the phylogenetic relationships between those genomes and the ones identified in the non-human primates, we created a concatenated alignment phylogeny of 15 proteins. The sequences from non-human primates are related to candidate genera III and IX, two candidate genera of the Alphacrassvirinae subfamily to which the prototypical crAssphage candidate genus I also belongs33 (Supplementary Fig. 5). Although most sequences from non-human primates form deep clades, two sequences obtained from Gorilla 1 are closely related to the human strains CDZH01002743 (from a 25-year-old man from Canada34) and FDYN_MS_11 (from a healthy individual from Ireland33). We hypothesize that this strain may have been transmitted from humans, as this gorilla has had human contact (http://gracegorillas.org/2017/12/29/pinga/). She also contains a further strain that clusters among the other sequences from non-human primates in candidate genus IX. Notably, this tree does not reflect the phylogeny of the hominids, instead it reflects the presence of multiple crAssphage-like species in the gut virome of non-human primates. This observation is consistent with the higher gut-microbiome diversity of non-human primates35 and may also be explained by the fact that multiple lineages of the probable crAssphage hosts—Bacteroidaceae—coexist in the primate gut microbiome36.
Fig. 4 |. Maximum likelihood phylogeny and dot plot showing full genomic colinearity between crAssphage and ten long contigs that were assembled from faecal metagenomes of different non-human primates.
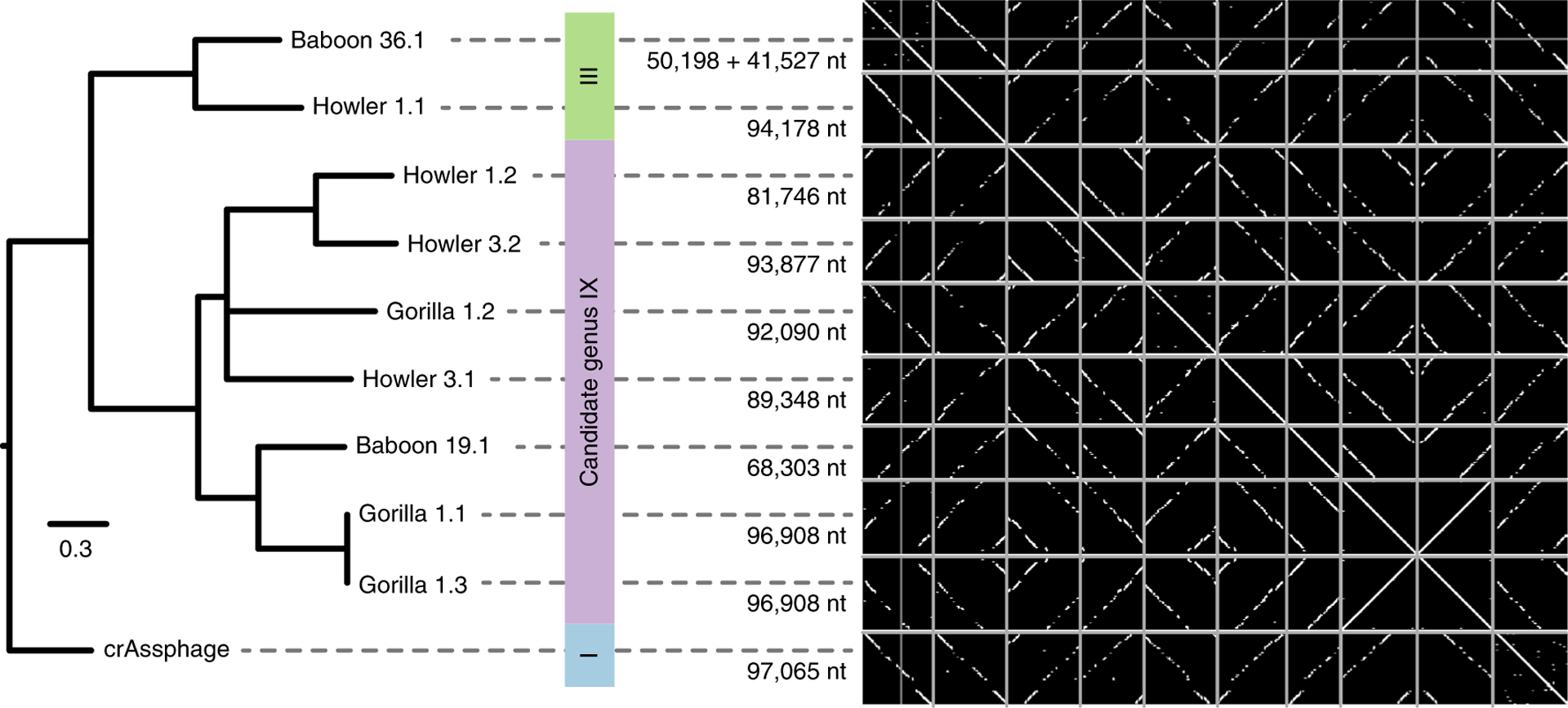
Phylogeny based on a concatenated trimmed-protein alignment of 15 homologous open reading frames (ORFs). The tree is rooted as described previously33, and candidate genera are indicated by coloured blocks. All of the branches had 100% bootstrap support, with one exception of <50%, which was collapsed. The scale bar indicates 0.3 mutations per site. nt, nucleotide. For a phylogeny of all 119 crAssphage-like genomes from Alphacrassvirinae and non-human primates, see Supplementary Fig. 5. For the dot plots, all genomes or contigs are shown in separate boxes along the x and y axes of the plot, and regions of similarity between genomes are displayed in white. Similarity is based on high-scoring segment pairs (BLASTn E < 0.001) between all contigs. The figure is to scale, numbers to the left of the dot plot indicate genome or contig lengths. Note that reverse complement sequences result in diagonals from top-right to bottom-left and circular permutation of some genomes leads to apparently broken diagonals in some dot plots.
crAssphage belongs to the normal human virome
To investigate the association between crAssphage and the characteristics of the human host and its microbiome, we studied the correlation between faecal crAssphage abundance and a range of host factors and microbial taxa. By exploiting shotgun metagenomes and host metadata from the LifeLines-DEEP cohort37,38, we correlated the abundance of crAssphage across 1,135 individuals with 207 exogenous and intrinsic human variables, including 78 dietary factors, 41 intrinsic factors, 39 diseases, 44 drug groups, 5 smoking categories (Supplementary File 3) and 490 microbial taxa (Supplementary File 4). We found significant, albeit weak, correlations with several diet categories (Benjamini–Hochberg adjusted P values using a false discovery rate of <5%), including protein, carbohydrates and caloric intake, basic food groups that are probably related to the dietary preferences of the crAssphage host bacteria12,37,39–41. The most significant correlations of crAssphage with microbial taxa in the LifeLines-DEEP cohort included the family Prevotellaceae, which is consistent with our previous prediction that crAssphage infects bacteria of the Bacteroidetes phylum12. Diverse dietary associations have been observed for different Bacteroidetes members, including the genus Bacteroides that was linked to a long-term western diet rich in animal protein and sugars42, whereas Prevotella and Paraprevotella were linked to low protein and high fibre43. The most reliable computational phage–host signal to date44 is a 100% matching CRISPR spacer in Porphyromonas sp. 31_2 isolated from human faeces13, another species within the Bacteroidetes phylum, and the first cultured crAss-like phage was recently isolated through the use of Bacteroides intestinalis as an isolation host14. Given the potentially family-scale taxonomic diversity of crAssphages13, it is probable that they infect a range of hosts throughout the Bacteroidetes phylum, leading to poor abundance correlations between crAssphage and specific host taxa. The LifeLines-DEEP cohort did not reveal a significant relationship between crAssphage and any human health or disease parameters, which is consistent with a previous study that showed no association between crAssphage and diarrhoea21. As crAssphage abundance is not related to any health-related variables, we conclude that it is a part of the normal human virome45.
Conclusions
The human gut virome consists of mainly phages that infect the abundant and diverse bacteria that live in our gut. Phages are generally thought of as transient entities in the environment, the fast infection cycle and relatively error-prone replication machinery of which enables rapid coevolution with their hosts, which—in turn—should be reflected in highly diverse viral metagenome sequences6,7. Indeed, we found thousands of crAssphage strains throughout human faeces-associated environments around the world. These strains are geographically and temporally clustered, consistent with rapid evolution and local dispersion. However, we also identified one exceptionally widespread strain in up to 104 different samples from, for example, Denmark, France, Germany, Israel, Italy, Japan and United States (Supplementary File 1). We suggest that this conservation primarily reflects recent spread by human global migration, although a crAssphage strain with potentially high fitness or environmental stability cannot be ruled out. Moreover, we identified highly divergent, but fully colinear, genome sequences from the crAss-like candidate genera III and IX33 in all major groups of primates, suggesting that crAssphage has had a stable genome structure for millions of years, and a stable association with the primate lineage and its microbiome36 since our early ancestors began their great migration out of Africa.
Recently, the recombination rate of phages has been estimated to be between 10−3 and 10−4.5 rearrangements per year46. Considering that New-World monkeys diverged from the human lineage 35 to 40 million years ago47, the genomic colinearity observed between their gut viruses implies a strong selective pressure and a highly optimized genomic architecture. Our results challenge high genomic mosaicism in viruses, showing that the genome structure of phages can be remarkably conserved in the stable environment provided by the human gut. The stability of the primate gut also limits the ability of its specialized microorganisms and viruses to escape to other environments. Indeed, this specificity makes crAssphage one of the strongest human faecal contamination markers to date25–27. Taken together, our results provide a global overview of the phylogeography of one of the most abundant and widespread viruses in the human gut, with evidence of both ancient evolution and ongoing local dispersion.
Methods
Phylogenomic tree of crAssphages from the twin study.
To assess the evolution of the intraindividual crAssphage population and its within-family relations, we assembled 20 different near-complete crAssphage genomes from the faecal viromes of three female twin pairs and their mothers3 using SPAdes v.3.11.0 with its default metagenomics settings48. Contigs related to crAssphage were identified by querying the contigs against the crAssphage reference genome sequence (RefSeq identifier NC_024711.1) using BLASTn32 v.2.5.0+ (E < 0.001). Next, the bitscore (which is independent of the database size) was summed for each SPAdes contig, and contigs with a total summed bitscore of at least 4,000 were selected. Note that shorter contigs with homology to crAssphage existed in the datasets, but we limited our analysis to the longest contigs with the strongest similarity signal to the crAssphage genome.
A phylogenomic tree (Supplementary Fig. 1) was created on the basis of the near-complete crAssphage genomes from the gut viromes of twins. ORFs were identified in all contigs using Prodigal v.2.6.349 and were queried against the crAssphage genome using BLASTp32 v.2.5.0+ (E < 0.001). Proteins that were missing from more than three genomes were excluded. This resulted in a dataset of 68 proteins that were aligned using Clustal Omega50 v.1.2.0 with default parameters (crAssphage proteins orf00003, orf00007, orf00009, orf00010, orf00011, orf00012, orf00013, orf00014, orf00015, orf00016, orf00017, orf00018, orf00020, orf00022, orf00023, orf00024, orf00025, orf00026, orf00027, orf00029, orf00031, orf00032, orf00033, orf00035, orf00037, orf00038, orf00040, orf00041, orf00042, orf00044, orf00045, orf00046, orf00047, orf00053, orf00054, orf00055, orf00056, orf00057, orf00059, orf00060, orf00062, orf00063, orf00065, orf00066, orf00067, orf00068, orf00070, orf00071, orf00072, orf00074, orf00075, orf00078, orf00079, orf00080, orf00081, orf00082, orf00084, orf00086, orf00088, orf00091, orf00092, orf00093, orf00094, orf00095, orf00096, orf00097, orf00098 and orf00099). The aligned proteins were concatenated to form a superalignment of 25,066 residues that was converted to an approximate maximum likelihood tree using IQ-tree51,52 v.1.5.5 (options -alrt 1000 -bb 1000), which was shown to be the most robust phylogenetic method53,54.
PCR primer design.
PCR primers were designed to facilitate the identification of crAssphage in a diverse range of sampling sites around the world and amplify a variable region of the genome for phylogenetic analysis (Supplementary Tables 4–6). Several studies designed primers for selected crAssphage proteins25,27,28,55,56 but we took a data-driven approach by identifying regions of the crAssphage genome that are suitable for phylogeographical analysis, that is, variable regions that were flanked by conserved regions that might be targeted by the primers. We identified these regions by determining the consensus sequence of the full crAssphage genome in 148 datasets in which at least 10,000 reads were aligned to crAssphage in our previous study12. We used Bowtie 2 v.2.3.4.357 to map metagenomic sequencing reads against the crAssphage reference genome, and called the consensus using Samtools v.1.858, which yielded 148 aligned consensus sequences. Next, we analysed the genome for suitable regions according to the following criteria: (1) high diversity flanked by conserved regions; (2) present in at least 90% of all sequences (less than 10% gaps); (3) a length of 1,000–1,400 nucleotides. From the resulting candidate regions, we identified potential PCR primer sites for further analysis. We defined three primer regions, which we call A, B and C, that amplify the following regions: primer A, 25,634–26,964 bp; primer B, 33,709–35,062 bp; and primer C, 43,820–45,057 bp in the canonical crAssphage genome (RefSeq ID NC_024711.1)12.
PCR amplification and sequencing of primer regions.
The metagenomics-guided primer design outlined above yielded 11 promising primer regions of the crAssphage genome, and—following testing using raw sewage influent (the raw sewage entering into the wastewater treatment plant) from four sewage plants in southern California (see below)—a standard protocol was developed for three regions of the crAssphage genome. To prepare the DNA template, the raw sewage influent was centrifuged briefly to remove the solids and passed through a 0.2 μm or 0.22 μm filter. Subsequently, 7 μl of supernatant was used in a 50 μl PCR reaction (Supplementary Table 5) with 30 cycles of amplification (Supplementary Table 6).
The crAss status was determined by the identification of a gel electrophoresis band, and sequencing was performed with Sanger sequencing using commercial providers. Bases were identified from the .ab1 files using phred59,60 v.0.071220.b, and overlapping reads were merged using merger from the EMBOSS suite (v.6.5.7.0) with default alignment parameters61. Sequences were then formatted so that the sequence identifier contained metadata about the sequences. Specifically, we recorded the collection-location address, latitude, longitude, country and altitude, the date of collection, the source of the sample (including raw sewage and faeces) and other notes about the sample (Supplementary File 1).
Global crAssphage collaboration.
We initiated a global and local survey of crAssphage using an open science collaboration framework. Scientists were asked to donate their time, expertise and resources to collect samples from local sewage treatment plants, PCR amplify three regions of the crAssphage genome and sequence those regions. To avoid potential contamination that could arise from the use of central reagent stocks, each laboratory was responsible for ordering their own primers, performing the PCR and sequencing the end products. Primers were only provided to researchers involved in the project in a few cases, usually when ordering primers was too financially onerous. For those cases the primers were ordered from Integrated DNA Technologies and provided to the researchers before the tubes had been opened. Note that the sequences resulting from those cases did not show any clustering in our resulting analyses, thus ruling out the possibility of cross-contamination.
Our global survey of crAssphage showed direct evidence of a globally distributed phage associated with humans and wastewater treatment plants. We generated 544 crAssphage sequences (184, amplicon A; 158, amplicon B; and 202, amplicon C) from 70 different locations in 23 countries across five continents. In most cases, a single pure sequence was obtained from each PCR amplification. This suggests that either there is a single dominant crAssphage strain in the environment, or that the PCR amplification resulted in one genotype being dominantly amplified at the expense of other sequences.
We tested for PCR amplification bias in two different ways. First, we started with three sewage samples from different wastewater treatment plants. We extracted five aliquots from each sample and amplified each in a separate reaction. All 15 products were sequenced, and we recovered identical DNA sequences within wastewater plants but not between different wastewater plants. Second, 30 different PCR fragments were cloned into the vector pTZ57R/T (Thermo Scientific) and sequenced independently (see below). Some sewage samples yielded mixed populations of sequences, whereas other sewage samples generated identical sequences.
Two wastewater treatment sites (Greymouth, New Zealand and Leuven, Belgium) identified mixed samples (Supplementary Fig. 6). In these cases, the sequences were identical at the beginning but abruptly degraded, possibly caused by amplification of two different genotypes that differ by a small insertion or deletion. Such an indel would place the sequence out of register, prohibiting the resolution of a single sequence from the trace data.
Only a few sites were unable to amplify crAssphage from the sewage using any of the primers. It is not clear whether that was due to a lack of crAssphage DNA in the sample or potential contaminants in the sample that inhibited the PCR reaction. We deliberately did not provide a positive control for crAssphage to avoid cross-contamination of samples. We sampled a single phage genome across almost the entire globe, demonstrating the ubiquitous spread of this phage. Thus, although care was taken to provide the same protocol to all collaborators and crAssphage was identified at many sites, we focus on only the positive results in this study because negative results could still represent a problem with the experiments rather than a lack of the phage in the sample.
Sampling of volunteers.
The volunteer sampling was conducted under IRB Approved Protocol Number HS-2016-0056 and BUA Protocol 17-02-003E from San Diego State University. In San Diego, we tested 12 American individuals and 1 British individual, 11 from San Diego and 2 from Irvine, between 21 April and 25 May 2017; 4 out of 7 males and 2 out of 6 females were crAss-positive, as determined by PCR and gel-electrophoresis. Three volunteers who were crAss-positive and three who were crAss-negative were followed weekly until 31 July 2017. On two separate weeks, each volunteer was followed daily. After gaining informed consent, each volunteer was provided with swubes (Becton Dickenson) to collect faecal samples immediately after defecation. Samples were processed by adding approximately 0.5 ml of sterile phosphate-buffered saline to the swube and placing the suspended material in an Eppendorf tube. DNA extraction using the QIAamp PowerFecal DNA Kit (Qiagen) was performed according to the manufacturer’s protocol. Samples were tested by PCR, and all of the PCR products were sequenced by Eton Bioscience. Significance of the consistency in crAss status was calculated by randomly reshuffling the labels across all individuals (in total, 41 crAss-positive and 97 crAss-negative cases) 5,000 times. P values indicate the fraction of cases that crAss status within individuals was equally or more extreme (that is, crAss-positive or crAss-negative) than observed; P < 0.0002 when none of the random permutations were more consistent. Another 32 volunteers from Wageningen and Utrecht were tested, for which amplicon C was used for initial detection and amplicon B was used for confirmation. The test showed that 15 people were crAss-positive (11 male, 4 female) and 17 were crAss-negative (14 male, 3 female). The nationalities of the volunteers included 1 American (crAss-negative), 1 Australian (crAss-negative), 1 Chinese (crAss-negative), 1 Colombian (crAss-positive), 23 Dutch (11 crAss-positive, 12 crAss-negative), 1 German (crAss-positive), 1 Greek (crAss-positive), 1 Portuguese (crAss-positive) and 2 Spanish (crAss-negative). Interestingly, at least one couple living together for several years had a discordant crAss status.
COMPARE global sewage sampling.
Samples from 81 sewage plants in 63 countries were taken within the COMPARE project (http://www.compare-europe.eu/) for strain-resolved metagenomic sequencing. Samples were centrifuged and DNA was isolated using the QIAamp Fast DNA Stool protocol62. DNA sequencing was performed at the Oklahoma Medical Research Foundation where the DNA was sheared to around 300 bp and library preparation was performed using the NEXTflex PCR-free DNA-seq library preparation kit. The multiplexed samples were sequenced on a HiSeq3000 using 2 × 150 bp paired-end reads. Subsequently, data were quality trimmed and assembled with SPAdes v.3.9.0 using the -meta flag. Contigs of crAssphage were identified as explained below for the faecal metagenomes of primates. Amplicon regions were identified using BLASTn v.2.5.0+, and hits included when they overlapped >50% with the amplicon regions. Note that 158 out of 179 hits overlapped ≥99% with the amplicon regions. All E values were equal to 0.0 in this small database that comprised only the COMPARE crAssphage contigs.
Strain-resolved metagenomics.
The Sequence Read Archive63 (SRA) contains approximately ten petabases of DNA sequence (1016 bp), including data from many metagenomes. We developed a pipeline to search the SRA using the Jetstream platform64–66. Initially, we screened the 95,552 metagenomes identified by PARTIE67 for the presence of crAssphage by comparing 100,000 reads from each metagenome with the crAssphage reference genome sequence using Bowtie 257. Metagenomes that had one or more matching reads in this initial screen were compared with the crAssphage genome to identify any sequencing reads in those metagenome libraries that match crAssphage. All metagenomic sequences were cleaned using our parallel version of Prinseq, called Prinseq++ v.1.2 (https://github.com/Adrian-Cantu/PRINSEQ-plus-plus)68,69. Sequences were trimmed to ensure that the mean quality score was at least 20, no nucleotides annotated as N were included in the sequences, all sequences were dereplicated, the ends were trimmed on the basis of the quality score cut-off and each read was required to be a minimum 30 nucleotides long. The sequences were mapped and indexed using Bowtie 257 to generate a BAM file. Details of the screening procedure66,70 are provided at https://github.com/linsalrob/SearchSRA. A total of 10,260 metagenomes had matches to crAssphage over 1 kb (Supplementary Fig. 7a, Supplementary File 5). The variability of the three amplicon regions is shown in Supplementary Fig. 7b–d.
Entire amplicon regions were recovered from 2,216 metagenomes derived from 121 SRA Bioprojects, and those were used as input to Gretel v.0.0.819 for probabilistic haplotype recovery. Initially, single-nucleotide polymorphisms were identified from the BAM files using snpper from Gretel-Test (https://github.com/SamStudio8/gretel-test) for each of the three regions used in the PCR. Variants predicted by Gretel were combined into a single FASTA file for downstream analysis19. As in the global collaboration, we focused only on the crAssphage-positive samples. Owing to persistent inconsistencies in the metadata of metagenomes submitted to SRA, we avoided an extensive search of, for example, all human faecal samples and/or all sewage or wastewater samples. Instead, we identified crAssphage in all metagenomic datasets—regardless of their environmental origin—and we refrained from making statements about the percentage of crAss-positive individuals on the basis of this analysis. We observed a weak (r2 = 0.66) but statistically significant correlation (P < 0.01) between the depth of coverage of the three amplicon regions in the metagenomes and the number of strains recovered from each of the three amplicon regions (Supplementary Fig. 8). This may be expected because further rare variants may be detected using deeper sequencing.
Global phylogenetic trees of three amplicon regions.
Using the methods outlined above, we collected sequencing data for each of the three amplicon regions from several different sources (Supplementary Fig. 8, Supplementary Table 2, Supplementary File 1). As shown in Supplementary Table 2, only a subset of the sequences contained locality information and could thus be included in the global phylogeographical analysis.
All sequences were then processed through a pipeline that is provided as a Makefile71 in the GitHub repository (https://github.com/linsalrob/crassphage). The trees can be built using the GNU Make program. After alignment with MUSCLE72 v.3.8.31 (using a maximum of two iterations and with the -diags option to find diagonals), alignments were trimmed to remove any columns that contained gaps in more than 10% of the sequences using a custom-written Python program, which deleted some of the sequences (Supplementary Fig. 3). Phylogenetic trees were constructed using IQ-tree51 (default settings) with ModelFinder52. The MUSCLE alignments and IQ-tree analysis were performed on a 540 node compute cluster in the Edwards Bioinformatics Laboratory. Trees were visualized using iTOL v.473.
Assessment of metadata clustering.
We assessed geographical clustering of crAssphage in the phylogeny, and clustering by sampling date. To obtain meaningful statistics, we developed a permutation approach, as described previously74, that retained the branching structure of the phylogeny and reshuffled the leaf labels n times, each time asking whether the geography and sampling date were more clustered in the randomly permuted tree than in the original tree. To account for phylogenetic noise in the tree topology, we collapsed branches with low bootstrap values. The statistics for the global phylogenies are presented in Supplementary Fig. 4 and empirical P values for the clustering were calculated as explained below, resulting in P < 0.001 for all statistics at all bootstrap levels.
To assess the extent of geographical clustering of crAssphage in the global phylogeny, we measured three different statistics. (1) For each branch in the tree, we measured the frequency of the most frequently annotated country or locality, and averaged across all of the branches to generate a single consistency clustering statistic for the whole tree. (2) We counted the number of branches where all of the leaves have the same country or locality annotation, yielding a ‘perfect branches’ statistic. (3) We calculated the standard deviation of all of the pairwise geographical distances between leaves in a branch on the basis of latitude and longitude coordinates, and averaged across all branches of the tree.
To assess phylogenetic clustering of the sampling dates, we calculated the standard deviation of all dates within a branch in an eight-digit numerical format YYYYMMDD and generated an average across all branches of the tree. Moreover, we calculated the consistency and perfect branches statistics, as above, by treating each date as a categorical rather than a numerical value.
Rural Malawi and Amazonas of Venezuela.
To investigate the presence of crAssphage in the faecal microbiota of human populations that were relatively remote from western culture, we used metagenomic sequencing data from samples of people from rural Malawi and the Amazonas of Venezuela29. The datasets were retrieved from MG-RAST75 and compared with the crAssphage reference genome sequence using Bowtie 257. As shown in Supplementary Table 3, a few reads mapped from both the Malawi and Venezuelan samples. As these hits did not cover the amplicon regions sampled in our global analysis, the sequences were not included in the global phylogeny.
Mummies.
To investigate the presence of crAssphage in the mummified faecal remains of ancient humans, we used metagenomic sequencing data from three pre-Columbian Andean mummies30 and the 5,300-year-old intestinal content of the Tyrollean glacier mummy, Ötzi31. For the three pre-Columbian Andean mummies, sequences were downloaded from MG-RAST75 (MG-RAST project identifier mgp13354; 12 samples, 115,174,154 reads and 11,488,857,080 bp). For Ötzi31, sequences were downloaded from the SRA (ENA project identifier ERP012908; 43 samples, 2,797,498,968 reads and 282,547,395,768 bp). The datasets were compared with the crAssphage reference genome sequence using Bowtie 257 (nucleotide search) and RAPsearch2 v.2.2276 with an E-value threshold of <10−5 (protein search). No hits were found.
Candidate crAss-like genera.
Recently, ten proposed crAssphage genera by reconstructing genomes from metagenomes were identified by Guerin et al.33. In total, 249 genomes were identified and 63 genomes were ascribed to candidate genus I, the genus that contains the prototypical crAssphage. Each of those 63 genomes contained the three amplicon regions described here—as detected using BLASTn—whereas none of the 186 genomes that belonged to other candidate genera contained any sequence similarity to those regions. Thus, here we identify only members of candidate genus I that infect bacteria in the human intestine.
Primates.
Faecal samples were collected from five species of primates in their natural habitats or in rehabilitation and conservation centres as described previously77. Sampling and analysis was approved by the Institutional Animal Care and Use Committees of Dartmouth College (protocol number 11-05-05AT), the University of Colorado Boulder (protocol number 1311.01) and the University of Illinois at Urbana-Champaign (protocol numbers 08044, 11046 and 14098). The collection and export of faecal samples was approved by the Ethiopian Wildlife Conservation Authority (permit numbers DA12/26/03 and DA12/27/11), whereas the import of samples was approved by the US Department of Agriculture, Animal and Plant Health Inspection Service. Samples were obtained from adults but the sex was not recorded. Three baboon samples were collected from wild specimens (Old-World monkeys). Baboon 19 and Baboon 22 are Papio hamadryas–Papio anubis hybrids from Awash, Ethiopia, and Baboon 36 is a Papio hamadryas from Filwoha, Ethiopia. Three black and gold howler monkey samples were collected from wild Alouatta caraya in Argentina (New-World monkeys). Three lemur (sifakas) samples were collected from wild Propithecus diadema in Tsinjoarivo, Madagascar. Three eastern lowland gorilla (Gorilla beringei graueri) samples were taken at the Mountain Gorilla Veterinary Project. The apes were originally wild from Rwanda but were being cared for in the Mountain Gorilla sanctuary when samples were taken. They have therefore had some close contact with humans. Three chimpanzee samples were collected from Pan troglodytes schweinfurthii in Ngamba Island, Uganda. These apes were also sanctuary animals rescued from Congo and Uganda and have relatively close contact with humans. Metagenomic DNA libraries were constructed using the TruSeq DNA Sample Prep kit (Illumina). All of the sequencing was performed at the Roy J. Carver Biotechnology Center’s High-Throughput Sequencing and Genotyping Unit at the University of Illinois, Urbana-Champaign.
Metagenomic sequences were assembled and contigs related to crAssphage were identified as described for the crAssphage genomes from the twin study (above). Shorter contigs with homology to crAssphage existed in the primate faecal metagenomic datasets, but we limited our analysis to the longest contigs with the strongest similarity signal to the crAssphage genome (total summed BLASTn bitscore of ≥4,000) to validate the existence of ancient relatives of crAssphage in primate faeces. The 10 selected primate contigs contained a strong colinearity signal with the crAssphage genome, suggesting that they were near-complete genomes. Two contigs from Baboon 36 were merged because they shared 66 overlapping nucleotides at the end of the contigs, had very similar assembly depth (7.7× and 7.4×) and were homologous to two non-overlapping sections of the crAssphage genome. Gorilla 1.1 and Gorilla 1.3 represent two near-identical strains (one polymorphism in 96,908 nucleotides) that were independently recovered from the same ape, showing the robustness of the metagenomic sequencing and assembly approach. The sequences from Baboon 36 and Howler 1 were most similar to candidate genera III described by Guerin et al.33, whereas the other sequences were most similar to candidate genera IX, measured by the fraction of the genomes aligned with BLASTn and as shown in Fig. 4.
After identifying ORFs in all contigs with Phanotate v.1.0.178, homologous groups were identified by querying against proteins from the crAssphage genome using BLASTp v.2.7.1+ (E ≤ 10−5); the 15 protein homologues that were identified from crAssphage—the non-human primate and the Alphacrassvirinae—were separately aligned using MAFFT79 v.7.407, and concatenated. Positions with gaps in more than 5% of the sequences, and positions at which every amino acid was identical, were removed. A maximum likelihood phylogenomic tree was created on the basis of concatenated protein alignment using IQ-tree51 v.1.5.5 with ModelFinder52, tree search, 1,000 ultrafast bootstraps and a SH-aLRT test (that is, the IQ-tree options -alrt 1000 -bb 1000).
Correlations with host factors and intestinal microorganisms.
To explore the association between crAssphage abundance in the gut and a broad range of exogenous and intrinsic human phenotypes, as well as intestinal microbial taxa, we used data from the LifeLines-DEEP study37. The LifeLines-DEEP cohort is a population-representative cohort of citizens from the northern Netherlands that comprises 1,135 individuals. Methods of sample collection, DNA extraction and sequencing, and phenotype selection were previously described37. To estimate the abundance of crAssphage in the LifeLines-DEEP samples, metagenomic sequencing data were mapped to the reference crAssphage genome using BWA v.0.7.15-r1140 with default parameters. The relative abundance of crAssphage was calculated as the number of mapped reads divided by the total number of reads in the sample. Next, we used a Spearman rank-sum test to estimate the association between crAssphage abundance and the phenotypes or microbial taxa of interest. Adjustment for multiple testing was conducted using the Benjamini–Hochberg procedure80. The results are listed in Supplementary Files 3 and 4.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Sequence data that support the findings of this study have been deposited in GenBank under BioProject accession PRJNA510571 and at https://github.com/linsalrob/crAssphage. Each of the samples has a unique BioSample accession number (SAMN10656826–SAMN10658627, SAMN10658653 and SAMN10659294). The SRA runs used in this analysis are included in Supplementary File 5. The data that support the findings of this study are also available from the corresponding authors on reasonable request.
Code availability
The code used to generate the data can be accessed at https://github.com/linsalrob/crAssphage. The current release81 is v.2.0.
Supplementary Material
Acknowledgements
We thank R. Matthews, M. Wright, J. Alexander, S. Arredondo, N. Branch, D. Campbell, R. Chea, D. McDougle, J. Parks and V. Vipatapat for providing access to wastewater treatment samples; the members of the Mountain Gorilla Veterinary Project and the staff of Maryland Zoo for collecting the gorilla faecal samples in Rwanda; G. Britton for collecting the baboon faecal samples in Ethiopia; staff of the CSWCT, the UWA and the UNCST for collecting the chimpanzee faecal samples in Uganda; J. Manor at Central Virology Laboratory, Chaim Sheba Medical Center, Tel-Hashomer Hospital and G. Steward, Department of Oceanography, University of Hawai’i at Manoa for help with sample collection; the COMPARE and LifeLines-DEEP projects for sharing data; O.D.N. thanks G. Steward, University of Hawai’i, Manoa for support. P.C.F. thanks C. Taylor for support with the PCR. Primate samples were provided by the PMC at the University of Illinois Urbana-Champaign; D.T.M. thanks the Australian Research Council’s Linkage Project LP160100408, Melbourne Water and EPA Victoria for funding the collection of samples in Melbourne. Gorilla samples were originally obtained by M.K. and the Mountain Gorilla Veterinary Project in Rwanda. G.R. and N.J.D. provided the wild baboon samples from Ethiopia. Howler samples were provided by M.K. and lemur samples were provided by R.E.J. and M.T.I., R.M.S. and L.M. provided the chimpanzee samples with permission from the CSWCT, the UWA and the UNCST. The primate microbiome project was supported by NSF BCS 0935347 to S.L., R. Stumpf, B.W. and K. Nelson. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. This work used the XSEDE Jetstream resources at Indiana University and Texas Advanced Computing Center through allocation MCB170036 to R.A.E., which is supported by National Science Foundation grant number ACI-1548562. Some of this work was supported by San Diego State University Grants Programs to R.A.E., including the Summer Undergraduate Research Program. This work was supported by National Science Foundation grant numbers MCB-1441985 to R.A.E. and DUE-1323809 to E.A.D; the Department of Energy Lawrence Livermore National Laboratory grant B618146 to R.A.E., P.A.d.J. and B.E.D. were supported by the NWO Vidi grant 864.14.004; F.L.N. by the NWO Veni grant 016. Veni.181.092; S.J.J.B. by the European Research Council Stg grant (638707) and the Vidi grant 864.11.005; O.C. and K. Mazankova by the Ministry of Health of the Czech Republic grant numbers 15-31426A and 15-29078A; P.C.F. by a Rutherford Discovery Fellowship from the Royal Society of New Zealand. J.J.B. by the ARC Discovery Early Career Researcher Award (DE170100525); S.L.D.M. by an NIH Pathway to Independence Fellowship (1K99AI119401-01A1); K.B. by award number 1510925 from the United States National Science Foundation; M.T.I. by National Geographic Society (CRE) and NSERC; and C.D. by the Agence Nationale de la Recherche JCJC grant ANR-13-JSV6-0004 and Investissements d’Avenir Méditerranée Infection 10-IAHU-03. The LifeLines-DEEP sample collection and analysis was funded by the Netherlands Heart Foundation (IN-CONTROL CVON grant 2012-03) to A.Z. and J.F., by the Top Institute Food and Nutrition, Wageningen, the Netherlands (TiFN GH001) to C.W., by NWO Vidi grants 864.13.013 to J.F. and 016.178.056 to A.Z., NWO Spinoza Prize SPI 92-266 to C.W., and by the ERC FP7/2007-2013/ERC Advanced Grant agreement 2012-322698 to C.W., ERC Starting Grant 715772 to A.Z. A.Z. also holds a Rosalind Franklin Fellowship from the University of Groningen. The COMPARE data collection was funded by The Novo Nordisk Foundation (NNF16OC0021856).
Footnotes
Competing interests
The authors declare no competing interests.
Supplementary information is available for this paper at https://doi.org/10.1038/s41564-019-0494-6.
References
- 1.Sender R, Fuchs S & Milo R Are we really vastly outnumbered? Revisiting the ratio of bacterial to host cells in humans. Cell 164, 337–340 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Nguyen S et al. Bacteriophage transcytosis provides a mechanism to cross epithelial cell layers. mBio 8, e01874–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reyes A et al. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 466, 334–338 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Minot S et al. The human gut virome: inter-individual variation and dynamic response to diet. Genome Res. 21, 1616–1625 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reyes A, Semenkovich NP, Whiteson K, Rohwer F & Gordon JI Going viral: next-generation sequencing applied to phage populations in the human gut. Nat. Rev. Microbiol 10, 607–617 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paterson S et al. Antagonistic coevolution accelerates molecular evolution. Nature 464, 275–278 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pedulla ML et al. Origins of highly mosaic mycobacteriophage genomes. Cell 113, 171–182 (2003). [DOI] [PubMed] [Google Scholar]
- 8.Heldal M & Bratbak G Production and decay of viruses in aquatic environments. Mar. Ecol. Prog. Ser 72, 205–212 (1991). [Google Scholar]
- 9.Breitbart M, Wegley L, Leeds S, Schoenfeld T & Rohwer F Phage community dynamics in hot springs. Appl. Environ. Microbiol 70, 1633–1640 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Steward GF, Smith DC & Azam F Abundance and production of bacteria and viruses in the Bering and Chukchi Seas. Mar. Ecol. Prog. Ser 131, 287–300 (1996). [Google Scholar]
- 11.Minot S et al. Rapid evolution of the human gut virome. Proc. Natl Acad. Sci. USA 110, 12450–12455 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dutilh BE et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun 5, 4498 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yutin N et al. Discovery of an expansive bacteriophage family that includes the most abundant viruses from the human gut. Nat. Microbiol 3, 38–46 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shkoporov A et al. ΦCrAss001, a member of the most abundant bacteriophage family in the human gut, infects Bacteroides. Preprint at 10.1101/354837 (2018). [DOI] [PMC free article] [PubMed]
- 15.Barylski J et al. Analysis of spounaviruses as a case study for the overdue reclassification of tailed bacteriophages. Preprint at 10.1101/220434 (2018). [DOI] [PMC free article] [PubMed]
- 16.Adriaenssens E & Brister JR How to name and classify your phage: an informal guide. Viruses 9, 70 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Callahan BJ, McMurdie PJ & Holmes SP Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 11, 2639–2643 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.NCBI Resource Coordinators Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 44, D7–D19 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nicholls SM et al. Probabilistic recovery of cryptic haplotypes from metagenomic data. Preprint at 10.1101/117838 (2017). [DOI]
- 20.Lim ES et al. Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat. Med 21, 1228–1234 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang YY, Zhang W, Tong YG & Chen SP crAssphage is not associated with diarrhoea and has high genetic diversity. Epidemiol. Infect 144, 3549–3553 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Piper HG et al. Severe gut microbiota dysbiosis is associated with poor growth in patients with short bowel syndrome. JPEN J. Parenter. Enter. Nutr 41, 1202–1212 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Vatanen T et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell 165, 842–853 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huerta-Cepas J, Serra F & Bork P ETE 3: reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol 33, 1635–1638 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stachler E et al. Quantitative crAssphage PCR assays for human fecal pollution measurement. Environ. Sci. Technol 51, 9146–9154 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stachler E & Bibby K Metagenomic evaluation of the highly abundant human gut bacteriophage crAssphage for source tracking of human fecal pollution. Environ. Sci. Technol. Lett 1, 405–409 (2014). [Google Scholar]
- 27.García-Aljaro C, Ballesté E, Muniesa M & Jofre J Determination of crAssphage in water samples and applicability for tracking human faecal pollution. Microb. Biotechnol 10, 1775–1780 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahmed W et al. Evaluation of the novel crAssphage marker for sewage pollution tracking in storm drain outfalls in Tampa, Florida. Water Res. 131, 142–150 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Yatsunenko T et al. Human gut microbiome viewed across age and geography. Nature 486, 222–227 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Santiago-Rodriguez TM et al. Natural mummification of the human gut preserves bacteriophage DNA. FEMS Microbiol. Lett 363, fnv219 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maixner F et al. The 5300-year-old Helicobacter pylori genome of the Iceman. Science 351, 162–165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Altschul SF, Gish W, Miller W, Myers EW & Lipman DJ Basic local alignment search tool. J. Mol. Biol 215, 403–410 (1990). [DOI] [PubMed] [Google Scholar]
- 33.Guerin E et al. Biology and taxonomy of crAss-like bacteriophages, the most abundant virus in the human gut. Cell Host Microbe 24, 653–664 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Raymond F et al. The initial state of the human gut microbiome determines its reshaping by antibiotics. ISME J. 10, 707–720 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moeller AH et al. Rapid changes in the gut microbiome during human evolution. Proc. Natl Acad. Sci. USA 111, 16431–16435 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moeller AH et al. Cospeciation of gut microbiota with hominids. Science 353, 380–382 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhernakova A et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 352, 565–569 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tigchelaar EF et al. Cohort profile: LifeLines DEEP, a prospective, general population cohort study in the northern Netherlands: study design and baseline characteristics. BMJ Open 5, e006772 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.David LA et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Turnbaugh PJ et al. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med 1, 6ra14 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singh RK et al. Influence of diet on the gut microbiome and implications for human health. J. Transl. Med 15, 73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Filippo C et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl Acad. Sci. USA 107, 14691–14696 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kovatcheva-Datchary P et al. Dietary fiber-induced improvement in glucose metabolism is associated with increased abundance of Prevotella. Cell Metab. 22, 971–982 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Edwards RA, McNair K, Faust K, Raes J & Dutilh BE Computational approaches to predict bacteriophage-host relationships. FEMS Microbiol. Rev 40, 258–272 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Manrique P et al. Healthy human gut phageome. Proc. Natl Acad. Sci. USA 113, 10400–10405 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kupczok A et al. Rates of mutation and recombination in Siphoviridae phage genome evolution over three decades. Mol. Biol. Evol 35, 1147–1159 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schrago CG & Russo CAM Timing the origin of New World monkeys. Mol. Biol. Evol 20, 1620–1625 (2003). [DOI] [PubMed] [Google Scholar]
- 48.Bankevich A et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol 19, 455–477 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hyatt D et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11, 119 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sievers F et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol 7, 539 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nguyen L-T, Schmidt HA, von Haeseler A & Minh BQ IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol 32, 268–274 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A & Jermiin LS ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou X, Shen X, Hittinger CT & Rokas A Evaluating fast maximum likelihood-based phylogenetic programs using empirical phylogenomic data sets. Mol. Biol. Evol 35, 486–503 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dutilh BE et al. Assessment of phylogenomic and orthology approaches for phylogenetic inference. Bioinformatics 23, 815–824 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Cinek O et al. Quantitative crAssphage real-time PCR assay derived from data of multiple geographically distant populations. J. Med. Virol 90, 767–771 (2018). [DOI] [PubMed] [Google Scholar]
- 56.Liang Y, Jin X, Huang Y & Chen S Development and application of a real-time polymerase chain reaction assay for detection of a novel gut bacteriophage (crAssphage). J. Med. Virol 90, 464–468 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ewing B & Green P Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8, 186–194 1998). [PubMed] [Google Scholar]
- 60.Ewing B, Hillier L, Wendl MC & Green P Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8, 175–185 (1998). [DOI] [PubMed] [Google Scholar]
- 61.Rice P, Longden I & Bleasby A EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16, 276–277 (2000). [DOI] [PubMed] [Google Scholar]
- 62.Knudsen BE, Bergmark L & Pamp SJ SOP—DNA isolation QIAamp Fast DNA Stool modified. Figshare 10.6084/m9.figshare.3475406.v4 (2016). [DOI] [Google Scholar]
- 63.National Center for Biotechnology Information SRA Handbook (National Center for Biotechnology Information, 2009). [Google Scholar]
- 64.Stewart CA et al. Jetstream: a self-provisioned, scalable science and engineering cloud environment. In Proc. 2015 XSEDE Conference Scientific Advancements Enabled by Enhanced Cyberinfrastructure 29 (ACM, 2015). [Google Scholar]
- 65.Towns J et al. XSEDE: accelerating scientific discovery. Comput. Sci. Eng 16, 62–74 (2014). [Google Scholar]
- 66.Edwards R SearchSRA (2017); 10.5281/zenodo.1043562 [DOI]
- 67.Torres PJ, Edwards RA & McNair K PARTIE: a partition engine to separate metagenomics and amplicon projects in the Sequence Read Archive. Bioinformatics 33, 2389–2391 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schmieder R & Edwards R Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cantu VA, Sadural J & Edwards R PRINSEQ++, a multi-threaded tool for fast and efficient quality control and preprocessing of sequencing datasets. Preprint at 10.7287/peerj.preprints.27553v1 (2019). [DOI]
- 70.Levi K, Rynge M, Eroma A & Edwards RA Searching the Sequence Read Archive using Jetstream and Wrangler. In Proc. Practice and Experience on Advanced Research Computing (2018). [Google Scholar]
- 71.Stallman RM, McGrath R & Smith PD GNU Make: A Program for Directing Recompilation, for version 3.81 (Free Software Foundation, 2004). [Google Scholar]
- 72.Edgar RC MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Letunic I & Bork P Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Berke L & Snel B The histone modification H3K27me3 is retained after gene duplication and correlates with conserved noncoding sequences in Arabidopsis. Genome Biol. Evol 6, 572–579 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Meyer F et al. The metagenomics RAST server—a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 9, 386 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhao Y, Tang H & Ye Y RAPSearch2: a fast and memory-efficient protein similarity search tool for next-generation sequencing data. Bioinformatics 28, 125–126 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vlčková K et al. Impact of stress on the gut microbiome of free-ranging western lowland gorillas. Microbiology 164, 40–44 (2018). [DOI] [PubMed] [Google Scholar]
- 78.McNair K, Zhou C, Dinsdale EA, Souza B & Edwards RA PHANOTATE: a novel approach to gene identification in phage genomes. Bioinformatics 10.1093/bioinformatics/btz265 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Katoh K & Standley DM MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol 30, 772–780 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Benjamini Y & Hochberg Y Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol 57, 289–300 (1995). [Google Scholar]
- 81.Dutilh Bas E., and Edwards Robert A. crAssphage Data Repository on GitHub (Github, 2018); 10.5281/zenodo.1230436 [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence data that support the findings of this study have been deposited in GenBank under BioProject accession PRJNA510571 and at https://github.com/linsalrob/crAssphage. Each of the samples has a unique BioSample accession number (SAMN10656826–SAMN10658627, SAMN10658653 and SAMN10659294). The SRA runs used in this analysis are included in Supplementary File 5. The data that support the findings of this study are also available from the corresponding authors on reasonable request.