

Publisher's Note: There is a Blood Commentary on this article in this issue.
Key Points
RNA-binding proteins with roles in regulating alternative splicing, DAZAP1, EWSR1, HNRNPH1, are frequently mutated in MCL.
Most somatic HNRNPH1 mutations are intronic and disrupt regulation of HNRNPH1 through alternative splicing.
Abstract
Mantle cell lymphoma (MCL) is an uncommon B-cell non-Hodgkin lymphoma (NHL) that is incurable with standard therapies. The genetic drivers of this cancer have not been firmly established, and the features that contribute to differences in clinical course remain limited. To extend our understanding of the biological pathways involved in this malignancy, we performed a large-scale genomic analysis of MCL using data from 51 exomes and 34 genomes alongside previously published exome cohorts. To confirm our findings, we resequenced the genes identified in the exome cohort in 191 MCL tumors, each having clinical follow-up data. We confirmed the prognostic association of TP53 and NOTCH1 mutations. Our sequencing revealed novel recurrent noncoding mutations surrounding a single exon of the HNRNPH1gene. In RNA-seq data from 103 of these cases, MCL tumors with these mutations had a distinct imbalance of HNRNPH1 isoforms. This altered splicing of HNRNPH1 was associated with inferior outcomes in MCL and showed a significant increase in protein expression by immunohistochemistry. We describe a functional role for these recurrent noncoding mutations in disrupting an autoregulatory feedback mechanism, thereby deregulating HNRNPH1 protein expression. Taken together, these data strongly imply a role for aberrant regulation of messenger RNA processing in MCL pathobiology.
Visual Abstract

Introduction
Mantle cell lymphoma (MCL) is an uncommon B-cell lymphoma representing 4% to 9% of non-Hodgkin lymphoma (NHL) diagnoses worldwide.1 It can be broadly divided into 2 clinical subtypes, nodal and leukemic nonnodal disease, with each displaying distinct natural history and clinical and genetic features.2 MCL commonly follows an aggressive clinical course in patients, including nonsustained responses to frontline chemoimmunotherapy and frequent relapses, although some patients, including the majority of those with the leukemic nonnodal variant, exhibit significantly longer survival.3 Clinical prognostic metrics such as the MCL International Prognostic Index have enabled patient stratification and improvements in frontline therapy, such as the inclusion of active agents (rituximab, bendamustine, and cytarabine) as well as consolidative strategies (autologous stem cell transplantation), have significantly improved outcomes over the past 2 decades.1,4,5
The unifying genetic feature of MCL is a chromosomal translocation that places cyclin D1 (CCND1) proximal to the immunoglobulin heavy chain enhancer, causing constitutive CCND1 expression.2,6 The translocated CCND1 allele can also accrue secondary mutations including noncoding mutations in the 3′ untranslated region (UTR), thereby enhancing CCND1 messenger RNA (mRNA) stability and further elevating CCND1 protein abundance.7,8 Through exome and targeted sequencing efforts, largely focused on nonnodal leukemic subtype, several genes have been identified as commonly mutated in MCL, including those involved in DNA damage response (ATM, TP53), epigenetic regulation (KMT2D, WHSC1), Notch signaling (NOTCH1, NOTCH2), NFκB signaling (CARD11, BIRC3, SYK), and ubiquitin-mediated proteolysis (UBR5).9-11 The genomic features of MCL have proven to be heterogeneous and diverse, and therefore larger comprehensive explorations are necessary to further understand its biological spectrum.
A limited number of recurrent mutations have been associated with prognosis in MCL treated with standard therapy. The most firmly established of these include nonsilent mutations affecting TP53, NOTCH1, and CCND1,12-15 as well as amplifications of 3q or deletions in 17p.13,16 With the ongoing evaluation of new therapeutics for MCL, mutations associated with acquired treatment resistance are beginning to be identified.17 Despite a broad collection of MCL-related genes and mutations, stratification of patients by proliferation, whereby patients are separated into low-, intermediate-, and high-risk categories, remains more robust than any individual driver mutation.14,18
The present study describes driver mutations in MCL and nominates the perturbation of mRNA processing as an important feature of MCL biology. Specifically, we report novel recurrent mutations affecting genes that encode 3 RNA-binding proteins HNRNPH1, DAZAP1, and EWSR1, including intronic mutations affecting exon 4 splicing in HNRNPH1. We demonstrate that select mutations in HNRNPH1 alter this splicing and are associated with higher HNRNPH protein expression in patient tissues. Our functional characterization in MCL patient samples and cell lines indicate that HNRNPH1 splicing is regulated by the HNRNPH1 protein via a negative feedback loop leading to exclusion of exon 4 in the alternative transcript.
Methods
Study design and sequencing
We assembled a discovery cohort of paired fresh-frozen (FF) tumor-normal exome sequencing from 51 novel Canadian cases and 33 previously published cases.9,11 Our validation cohort consisted of targeted sequencing performed on formalin-fixed, paraffin-embedded tissue representing 191 diagnostic tumor samples from British Columbia (BC) (170 unique cases). Sixteen validation samples and 18 additional FF biopsies from BC underwent whole-genome sequencing (WGS). We performed RNA sequencing (RNA-seq) on a subset of the BC cases (103 total). Patient characteristics are available in Table 1. See supplemental Data and supplemental Figure 1 (available on the Blood Web site) for more details. This study was approved by the BC Cancer Research Ethics Board. All participants recruited provided informed consent.
Table 1.
Characteristics of patient samples
| Total (n = 213) | |
|---|---|
| Median age (range; n = 189) | 64 (31-84) |
| Male (%) | 160/211 (76) |
| Performance status >1 (%) | 35/176 (20) |
| Blastoid (%) | 20/212 (9) |
| MIPI (%) | |
| Low | 59/138 (43) |
| Intermediate | 34/138 (25) |
| High | 45/138 (33) |
| Treatment | |
| R-CHOP | 147 |
| Observation | 39 |
| Other | 5 |
| Unknown | 22 |
| HSCT/AUTO | 14 |
MCL tumor samples from 213 patients were obtained from patients living in British Columbia.
MIPI, MCL International Prognostic Index.
Data analysis
Sequencing reads were aligned to GRCh38 using Burroughs-Wheeler alignment19 (exomes, WGS), Geneious (targeted), or STAR20 (RNA-seq; individual-nucleotide resolution crosslinking and immunoprecipitation [iCLIP]). Simple somatic mutations (SSMs) in exomes and WGS were called, by using Strelka (v1.0.14)21 and Strelka2 (v2.9.6),22 respectively. Variants were annotated with Variant Effect Predictor23 using Ensembl release 83 (exomes) and Ensembl release 95 (WGS, targeted). Significantly mutated genes in the discovery cohort were identified by using a voting strategy with results from MutSigCV,24 OncodriveFM,25 OncodriveFML,26 and OncodriveCLUST27 (false discovery rate [FDR] < 0.1). Targeted sequencing was performed on significantly mutated genes identified by 2 or more methods, as well as NOTCH1, CARD11, and NFKBIE.11,12,28 Variants from all sequencing approaches were consolidated on a per patient basis. We compared the mutation patterns observed in MCL to diffuse large B-cell lymphoma (DLBCL) by using targeted and exome sequencing data from 1616 unique patients.29-31
Experimental approaches
All cell lines were grown as previously described.12,29 None of the cell lines REC-1, JVM2, Z-138 (in-house sequencing), or HEK cells32 have HNRNPH1 mutations at the hot spots identified herein. The HNRNPH1_ex2-6 “minigene” was assembled from human genomic DNA. pEGFP-HNRNPH1 was generated by inserting cDNA encoding the entire open reading frame into the pEGFP-C1 plasmid. Droplet digital polymerase chain reaction (ddPCR), western blot analysis, and tissue microarray immunohistochemistry were performed as described in the supplemental Methods. All ddPCR samples were normalized to the geometric mean of 3 reference genes.
Statistical analysis
Associations between gene mutation status and binary clinical characteristics were assessed, using Fisher’s exact test. Overall survival (OS) correlates were separately tested in all patients, regardless of treatment (n = 213), and in the subset of R-CHOP (rituximab, cyclophosphamide, doxorubicin hydrochloride, vincristine, prednisone)–treated, hematopoietic stem cell transplantation–untreated cases (n = 133), using univariate Cox proportional hazard modeling, and P values were corrected for multiple-hypothesis testing by the Bonferroni-Hochberg method. Corrected P < .1 was considered significant. The HNRNPH1 exon 4–skipping ratio was derived from RNA-seq data, and survival analysis using this ratio was limited to these cases (n = 102), using the median skipping ratio for all HNRNPH1-mutated cases as a cutoff for “mutant-like” splicing. Multivariate survival associations were examined with the Cox proportional hazard model on RNA-seq cases so that the HNRNPH1 exon 4–skipping ratio could be included in the model (n = 102). The final tested multivariate model included the TP53 and NOTCH1 mutations, HNRNPH1 mutant-like splicing, and morphology.
Results
Resolving the frequency of SSMs and recurrently mutated genes in MCL
Several genes have previously been implicated as recurrent targets of SSMs in MCL,9-11,33 although the relevant genes and their mutation incidence has varied considerably among these studies.34 This variability can be attributed to both genetic heterogeneity in this malignancy and the limited cohort sizes included in each study. To address this problem, we sequenced paired tumor/normal exomes from 51 MCLs diagnosed in Canada and analyzed the data alongside available paired exome data. Three of the 87 available samples exhibited significantly higher mutation burdens (median 5 112; range 1 621-14 959) and were excluded because of the effect of hypermutation on the detection of drivers. In the remaining “discovery cohort,” comprising 84 cases, tumor exomes harbored an average of nonsilent SSMs affecting 76 genes (range, 30-219).
Through our analysis of this cohort, 16 genes were deemed recurrently mutated by 2 or more algorithms used to identify driver genes. Three of the algorithms found each of ATM, BIRC3, TP53, S1PR1, and B2M to be significantly mutated, and each of MEF2B and WHSC1 were identified by 2 methods (supplemental Table 1). Notably, CCND1, often affected by somatic hypermutation, was identified by OncodriveCLUST,27 which relies on spatial clustering of mutations. Of the candidate MCL genes, those frequently mutated were ATM, CCND1, TP53, WHSC1, and KMT2D, each gene having been previously nominated by other studies. Three genes not previously associated with MCL (HNRNPH1, DAZAP1, and EWSR1) were also identified by at least 2 methods. Each of these 3 genes encode RNA-binding proteins that play a role in regulating RNA maturation, including alternative splicing.35,36
Novel mutation patterns in MCL
Based on these results and those of prior studies, we performed targeted sequencing of the coding exons of 18 genes in 191 additional MCLs and separately performed WGS on 34 cases to broadly resolve the exonic and intronic mutation patterns (supplemental Figure 1). We consolidated variants across all samples sequenced by more than one approach and used the resulting nonredundant variants from 272 cases for subsequent analyses. Mutation patterns and prevalence in established MCL genes were largely consistent with prior reports (Figure 1A; supplemental Table 2). Each of NOTCH1, MEF2B, and CCND1 have been shown to have mutation hot spots in MCL and other cancers, but the pattern of MEF2B mutations in MCL was distinct from that in other cancers (supplemental Figure 2).7,12,37,38
Figure 1.

Recurrent mutations are identified in MCL. (A) Mutations observed across 273 MCL samples in 18 candidate MCL genes. Mutations shown here are limited to nonsilent mutations of all genes with the exception of HNRNPH1. For this gene, intronic and silent mutations affecting or immediately surrounding exon 4 are included. Spatial distribution of mutations observed in (B) EWSR1, and (C) DAZAP1 in MCL compared with DLBCL.
Unsurprisingly, the incidence of nonsilent mutations in newly identified genes was generally lower than that in established MCL genes. EWSR1 was mutated in 8 cases (3%) and DAZAP1, in 13 cases (5%; Figure 1A). EWSR1 predominantly harbored frameshift or nonsense mutations in MCL and exhibited a similar pattern at a lower prevalence in a larger compendium of DLBCLs (0.3%; Figure 1B), suggesting that EWSR1 has an unappreciated tumor-suppressor function in MCL and possibly in DLBCL. DAZAP1 had a distinctive pattern, with mutations clustered near the C terminus in a region containing a nuclear localization signal (p.G383-R407)39 and a proline-rich, protein-binding domain (Figure 1C).40,41 Nine cases harbored putative truncating mutations, with each predicted to remove or disrupt the nuclear localization signal while leaving most of the open reading frame intact. Nonsynonymous mutations in this region mainly affected highly conserved residues (ie, p.F402, p.R406, and p.R407). Previous work indicates that substitution of these residues causes cytoplasmic accumulation of DAZAP1 in human kidney epithelial (293T) and simian (COS7) cells.39
HNRNPH1 intronic mutations disrupt HNRNPH1 binding motifs
HNRNPH1 was mutated in 26 cases (10%) when we consider both coding and noncoding mutations, placing it as the eighth most commonly mutated gene overall (Figure 1A; supplemental Figure 3). Despite limited coverage of introns by our sequencing assay, intronic variants were the most common type of SSM detected in this gene, particularly in the regions surrounding exon 4 (Figure 2A). Paired tumor/normal sequencing confirmed that all recurrent variants were somatic, and the WGS data confirmed that the pattern was restricted to this exon and the immediate flanking regions (Figure 2B). HNRNP proteins are widely involved in regulating splicing by binding to pre-mRNA at specific motifs and either promoting or inhibiting usage of nearby splice sites. Distinct from other HNRNPs, HNRNPH1 (and its paralog HNRNPH2) preferentially binds RNA at poly-G motifs.42 Strikingly, 73% (19 of 26) of patients with HNRNPH1 mutations had mutations affecting a poly-G motif within or near this exon. Each of the affected bases are deeply conserved in the homologous region of all available vertebrate genomes, supporting their functional importance.
Figure 2.
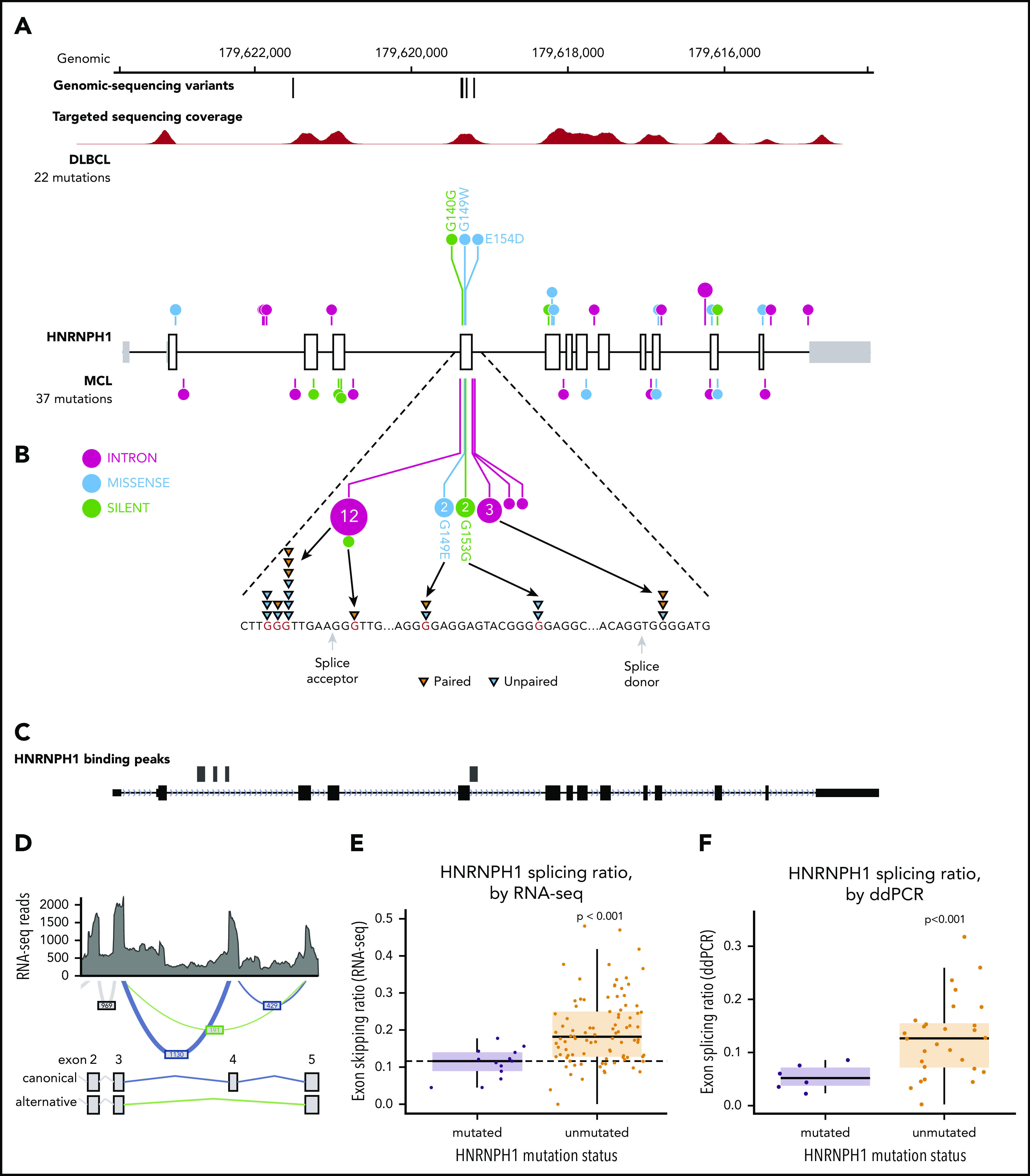
HNRNPH1 mutations in MCL cluster near exon 4 in poly-G motifs. (A) Somatic mutations found in genomic sequencing cases and targeted sequencing coverage of a representative sample. The prevalence and pattern of mutations in HNRNPH1 is compared between DLBCL and MCL. (B) Splice site and intronic mutations affecting poly-G motifs were observed both upstream and downstream of exon 4. Paired mutations (orange triangles) are those found to be somatic by sequencing matched constitutional DNA (n = 7). Unpaired mutations (blue triangles) are mutations found in tumor-only DNA sequencing (n = 12). (C) HNRNPH1 iCLIP binding peaks show that HNRNPH1 binds near exon 4 of the transcript (shown is Ref-seq isoform NM_001257293). (D) A representative Sashimi plot of splicing events in HNRNPH1 are indicated. The canonical splicing events are shown in blue, and the alternative (exon 4 skipping) splice event is shown in green. RNA-seq splicing ratios were calculated by the sum of reads supporting the alternative (green) event, divided by the number of reads supporting the canonical (blue) splicing event. (E) Mutated HNRNPH1 cases showed significantly lower exon-skipping ratios compared with unmutated cases, as measured by RNA-seq. Cases below the dotted horizontal line (skipping ratio ≤ median mutant skipping ratio) are referred to as mutantlike in further analyses. (F) Digital PCR was used to separately quantify alternative and canonical HNRNPH1 transcripts in mutant (n = 6) and wild-type (n = 30) cases. Mutant cases exhibit lower rate of exon skipping and higher overall abundance of HNRNPH1 mRNA.
Further confirming the recurrence of this event, we found mutations in this region of HNRNPH1 by reanalyzing published and unpublished exomes from 2 recent studies. We observed mutations consistent with the same pattern in 3 of 16 (18.8%) relapsed/refractory MCL exomes sequenced from a recent clinical trial17 and 4 of 24 (16.7%) exomes from another recent study43 (supplemental Table 3). We also sequenced this gene in diagnostic tumor tissue from 145 patients treated with either ibrutinib or temsirolimus on a recent clinical trial (clinicaltrials.gov #NCT01646021)44 and found mutations in 11 (7.5%) of these cases (supplemental Table 3). Among the available WGS data from Burkitt lymphoma45 (n = 106), DLBCL (n = 153), chronic lymphocytic leukemia (n = 144), and follicular lymphoma (n = 110), we only identified 2 DLBCL patients with HNRNPH1 mutations in this region (1.3%), suggesting the potential for these to be driver mutations with a highly specific function in MCL biology.29 This highly reproducible mutation pattern provides strong evidence that these mutations have a regulatory function, most likely affecting the expression and/or splicing of HNRNPH1 mRNA.
There is a growing list of splicing regulators, including multiple HNRNP family members, that modulate splicing of their mRNA to tightly regulate expression.46,47 Taking this into consideration along with the prevalence of HNRNPH1 mutations in sequence contexts resembling HNRNP motifs led us to speculate that HNRNPH1 protein regulates its own expression by modulating the splicing of the HNRNPH1 transcript. We reanalyzed HNRNPH1 iCLIP-seq data from Uren et al.42 and confirmed multiple sites of interaction between HNRNPH1 and its pre-mRNA including exon 4 (Figure 2C), supporting a model of direct association at the region affected by mutation.
HNRNPH1 has multiple alternative isoforms including several transcripts that result from skipping of exon 4, which are predicted to be targets of nonsense-mediated decay (NMD) (Figure 2D). Although they do not directly affect canonical splice signals, we hypothesized that the mutations in these poly-G motifs impact the splicing or skipping of exon 4. We analyzed RNA-seq data from 103 cases with known HNRNPH1 mutation status to evaluate splicing differences between mutated (n = 15) and unmutated (n = 88) tumors. By comparing the number of reads supporting the exon-skipping event to reads supporting inclusion of exon 4, we found that mutated cases exhibited a ratio of isoforms that favors inclusion of exon 4 (P = 1.13 × , Wilcox rank sum; Figure 2E). We implemented a custom ddPCR assay to separately quantify canonical and alternative HNRNPH1 transcripts. Using this assay, we corroborated these findings in selected cases (P < .001; Figure 2F), which showed a strong correlation (R = 0.66; P < .01) with splicing ratios determined from RNA-seq data from the corresponding cases (supplemental Figure 4A-B). These results support the notion that HNRNPH1 mutations favor the inclusion of exon 4, or suppress the skipping of this exon, promoting the formation of the full-length transcript. Based on our model, these mutations disrupt the binding of HNRNPH1 to poly-G motifs surrounding exon 4 and dampen the normal feedback inhibition (supplemental Figure 4C).
HNRNPH1 splicing is associated with inferior outcomes in MCL
We examined whether any mutations identified in this study were associated with patient outcome. This entailed 2 separate analyses: first using all cases with available survival data and then separately, within the subset of cases with R-CHOP treatment. In univariate comparisons, mutations in NOTCH1 (hazard ratio [HR] = 2.05; Q = 8.6 × 10−2) or TP53 (HR = 3.38; Q = 2.8 × 10−7) were associated with shorter OS in the complete cohort. Consistent with previous reports, there was also a significant prognostic association of NOTCH1 (HR = 2.38; Q = 3.6 × 10−2) and TP53 (HR = 3.53; Q = 8.4 × 10−6) mutations in patients treated with R-CHOP. Notably, although KMT2D mutations have been recently implicated as a prognostic feature in MCL,48 our analysis did not reproduce this result (supplemental Figure 5). In addition, in R-CHOP–treated cases EWSR1 was associated with shorter OS (HR = 8.71; Q = 3.5 × 10−3), although the number of mutated cases was small. In contrast, a significant association was not observed when these patients were stratified by mutation status of other genes, including HNRNPH1 (supplemental Data). We separately evaluated the effect of HNRNPH1 mutation status in the relapsed/refractory MCL patients treated with either ibrutinib or temsirolimus. Because of the limited number of mutated cases, patients on both arms of the trial were considered together in this analysis. In contrast to the R-CHOP cohort, patients with HNRNPH1 mutations had significantly shorter progression-free survival (Figure 3A), providing further support for contribution of these mutations to the biology of MCL.
Figure 3.
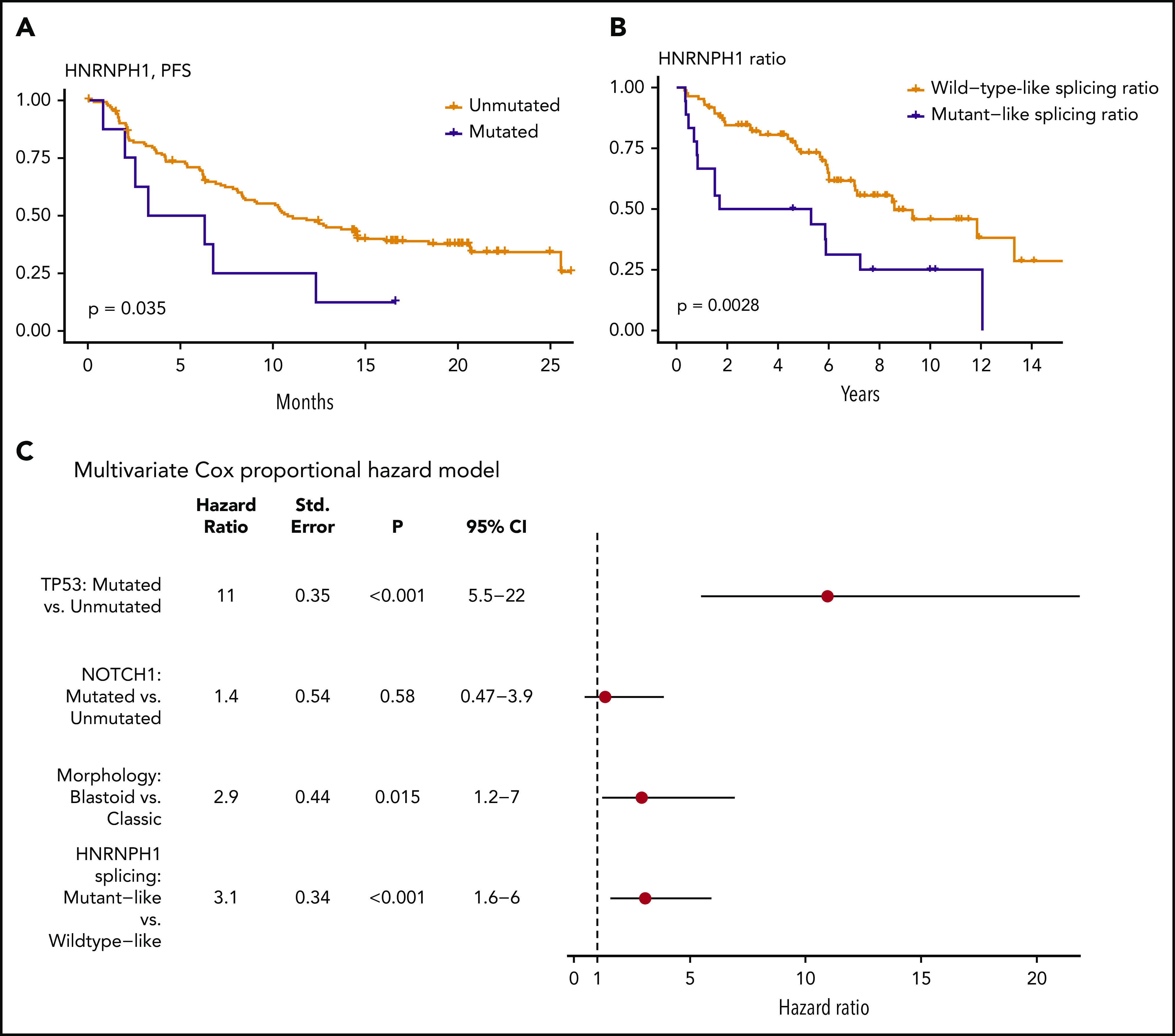
HNRNPH1 splicing is independently associated with survival in MCL. (A) Survival data and HNRNPH1 mutation status was obtained for an independent clinical trial cohort (n = 145); cases with HNRPNH1 mutations exhibit significantly worse progression-free survival (n = 8). (B) All MCL cases with available RNA-seq data (n = 103) were classified as having a mutantlike (n = 18) or wild-type (n = 85) HNRNPH1 splicing ratio as in Figure 2E, and OS was plotted. Mutantlike HNRNPH1 splicing was significantly associated with poorer OS. (C) Multivariate Cox proportional modeling was applied to MCL cases with available RNA-seq data (n = 103). TP53 nonsynonymous mutations, HNRNPH1 mutant-like splicing ratio, and blastoid morphology were independently associated with increased hazard.
Given the strong association between HNRNPH1 alternative splicing and mutation status, we rationalized that the proportion of HNRNPH1 mRNAs containing exon 4 could be used as a proxy for HNRNPH1 protein expression. We selected a conservative threshold to assign cases with mutantlike exon skipping based on the median value in all cases with HNRNPH1 mutations (Figure 2E). In patients with RNA-seq data available (n = 102), this stratification revealed significantly shorter OS in patients with mutantlike splicing of HNRNPH1 (Figure 3B; HR = 2.50; P = .00388). In support of the utility of this information, the splicing ratio was also significantly associated with OS when treated as a continuous variable. In a multivariate analysis, TP53 mutations, HNRNPH1 mutant-like splicing, and blastoid morphology were each independently associated with shorter OS (Figure 3C). We conclude that the mutant-like splicing pattern of HNRNPH1, which favors the productive isoform, is a novel biomarker of inferior outcome in MCL and is independent of established prognostic genetic features and morphology.
Mutations and alternative splicing influence HNRNPH1 protein expression in MCL
We hypothesized that HNRNPH1 mutations in poly-G tracts disrupt an autoregulatory negative feedback loop, which predicts a higher HNRNPH protein expression in HNRNPH1-mutant tumors. To address this directly, we evaluated HNRNPH expression in 170 MCL tumors by immunohistochemistry (Figure 4A). Of these cases, all had at least 1 type of sequencing data available and 79 had RNA-seq performed. Consistent with our hypothesis, tissues with strong HNRNPH staining intensity were significantly enriched for HNRNPH1 mutations (P = .0007214; Fisher’s exact test). Tissues with strong staining were also enriched for cases with mutant-like splicing (P = .001251; Fisher’s exact test) and the distribution of splicing ratios was significantly different between tissues with moderate and strong staining (Figure 4B). Based on the RNA-seq data, the total mRNA level of HNRNPH1 was not significantly higher in samples with strong staining (Figure 4C), which suggests that the relative proportion of canonical transcripts, rather than the total mRNA abundance, is more directly related to HNRNPH1 protein expression. Our initial finding showing that an association between productive splicing and survival (Figure 3A) is consistent with the trend observed here: namely, the association between strong HNRNPH staining and shorter survival (Figure 4D).
Figure 4.

Evaluating HNRNPH expression by tissue microarray. (A) Reactive tonsil tissue stained for HNRNPH expression was used as the reference. Representative images of tumor cores scored as weak (0), moderate (1), or strong (2) HNRNPH staining. (B) Based on the tissues on this tissue microarray with available RNA-seq data (n = 79), higher intensity of HNRNPH staining was associated with a lower splice ratio or a relatively lower proportion of alternative (unproductive transcripts; P = .01376, Wilcoxon rank sum). (C) For the same cases as in panel B, cases with higher intensity staining did not have higher levels of HNRNPH1 mRNA based on normalized read counts. In contrast, cases with high-intensity staining were associated with lower HNRNPH1 expression (P = .02004; Wilcoxon rank sum). (D) Stratification of patients by moderate or strong HNRNPH staining did not show a significant association with OS, but there was a trend toward inferior outcomes.
Common HNRNPH1 mutations disrupt productive splicing and translation
The correlation between HNRNPH1 isoform usage and increased protein levels only indirectly implicates NMD in this process. To substantiate the role of NMD in vitro, we inhibited this process by using the eukaryotic translation inhibitor cycloheximide. Cycloheximide is a widely used indirect inhibitor of NMD, owing to the essential role of translation in the NMD process.49-52 In 3 MCL cell lines (JVM2, REC-1, and Z-138) and in HEK cells, cycloheximide treatment caused a significant and dose-dependent increase in the alternative nonproductive HNRNPH1 transcript, compared with total HNRNPH1 transcript (alternative plus canonical; Figure 5A). This was consistent with the change in splicing pattern observed in SRSF3, which has alternative isoforms that are targeted to NMD due to inclusion of a poison exon (Figure 5B).49 These results suggest that the HNRNPH1 isoforms lacking exon 4 are degraded in an NMD-dependent manner in MCL cells.
Figure 5.

Mutations in HNRNPH1 prevent negative regulation via nonsense-mediated decay. We separately quantified canonical and alternative HNRNPH1 (A) and SRSF3 (B) transcripts by digital PCR in MCL cells (REC1, JVM2, and Z138) and HEK cells cultured with cycloheximide (an indirect inhibitor of NMD). This revealed an increasing proportion of the alternative transcript (skipped exon 4 in HNRNPH1) with increasing concentrations of cycloheximide. (C) Output from the minigene reporter is represented schematically. Differential splicing of the transcribed pre-mRNA results in inclusion or exclusion of HNRNPH1 exon 4. The out-of-frame transcript (right) results in the introduction of a premature termination codon (PTC) in exon 5 and translation of a truncated peptide. Translated peptides are represented in cartoon form. (D) HA tag abundance from the wild-type HNRNPH1 minigene is detected by western blot analysis. HA expression, representing productive splicing, is lost in the presence of HNRNPH1 overexpression. (E) The 3 independent G>T mutations introduced into the HNRNPH1 minigene are schematically represented in relation to the patient-identified mutations. (F) Expression of the HA tag from wild-type and mutant HNRNPH1 minigenes are detected by western blot analysis. HA expression, again representing productive splicing, is substantially higher when mutated minigenes are transfected into HEK cells as compared with the wild-type minigene.
To functionally demonstrate that mutations found in MCL disrupt regulation of alternative splicing, we constructed a minigene containing the genomic sequence for HNRNPH1 from exons 2 through 6 (HNRNPH1_ex2_6), including all intronic sequences. Productive splicing of the minigene created a full-length, in-frame peptide containing the hemagglutinin (HA) tag at the C terminus. Unproductive splicing, resulting from skipping of exon 4, forced the C terminus out of frame and caused translated peptides to lack the terminal HA tag. We first transiently transfected the minigene in HEK cells, along with a vector bearing the cDNA for HNRNPH1 tagged with EGFP. Ectopic expression of HNRNPH1 notably impaired expression of the HA tag, suggesting an HNRNPH1-dependent switch from productive to unproductive splicing of the minigene (Figure 5C). Subsequently, we separately generated 3 distinct mutants of this minigene by site-directed mutagenesis (Figure 5D; Table 2) and transiently transfected each mutant into HEK cells. The presence of any 1 of the 3 mutations tested markedly increased abundance of the HA-tagged peptide, confirming a shift toward productive splicing (Figure 5E). This suggests that all 3 poly-G tracts are individually essential for proper regulation of HNRNPH1 splicing and adds further support to a model in which these mutations disrupt the autoregulatory feedback cycle and favor HNRNPH1 protein expression (supplemental Figure 4).
Table 2.
Specific patient-derived mutations introduced into the HNRNPH1 minigene
| Minigene mutant | Mutation introduced | Corresponding patient mutation | |
|---|---|---|---|
| ΔA | G>T | chr5:179619414 C>A | Intronic |
| ΔB | G>T | chr5:179619406 C>A | G133G |
| ΔC | G>A | chr5:179619359 C>T | G149E |
Minigene mutations corresponding to patient-identified HNRNPH1 mutations were introduced.
Discussion
Using 272 MCL cases, we validated the incidence and pattern of mutations in genes with known relevance to MCL, including ATM, KMT2D, TP53, CCND1, and NOTCH1. Using clinical data available for the bulk of these cases, we confirmed the prognostic association of mutations in both TP53 and NOTCH1. NOTCH1 was not independently prognostic in a multivariate model that included TP53 mutations. We note that in the current data, WHSC1 mutations were also not associated with OS, in contrast to another study,53 which may be attributable to the limited sample size of that study. Motivated by the putative role of mutations in HNRNPH1, we also identified a strong association between HNRNPH1 splicing and outcome. Although the likely consequence of an imbalance of HNRNPH1 isoforms is an increase in HNRNPH1 protein abundance, we found a stronger association between the splicing ratio and patient outcome. This can be attributed to a limited dynamic range available for scoring HNRNPH expression by immunohistochemistry. In the absence of higher resolution methods for quantifying this protein in tissues, our results indicate that direct measurement of splicing may be a robust biomarker for HNRNPH activity.
Although the incidence was low, we consider the mutation pattern of EWSR1 to be a notable finding. EWSR1 is an established cancer gene that is typically discussed in the context of the EWSR1-FLI1 fusion oncoprotein that drives Ewing sarcoma.54 The pattern of mutations observed here implies a separate tumor-suppressor role of this gene in MCL. Notably, EWSR1 has been implicated in regulating CCND1 by promoting formation of the less oncogenic CCND1a isoform relative to the shorter CCND1b isoform.55 Although the targets of EWSR1 have not been established in MCL, our data are consistent with the notion that loss of EWSR1 activity alters RNA metabolism and splicing of genes relevant to MCL.
The DAZAP1 mutations described here are similar to previous reports in a subset of DLBCLs.29,31 The existence of recurrent EWSR1 and DAZAP1 mutations in both malignancies add to the limited genetic features shared between DLBCL and MCL, along with inactivating mutations in KMT2D and TP53. Based on previous mutagenesis experiments,39 we hypothesize that the more common DAZAP1 mutations cause reduced nuclear occupancy and affect interactions with other proteins, which could disrupt several processes, including transcription, alternative splicing, mRNA transport, and translation.35,41,56
Although HNRNPH1 has been identified as overexpressed in other cancer types,57-59 our description of regulatory mutations in HNRNPH1 is novel and suggests an unappreciated role for HNRNPH1 in B-cell development and/or lymphomagenesis. HNRNPH1 is a member of the HNRNPH/F family of heterogeneous nuclear ribonucleoproteins60 and binds to various cis-regulatory elements that, depending on the sequence context and interacting proteins, can promote or suppress the use of nearby splice sites.61 Our data support a model wherein HNRNPH1 protein normally limits its own accumulation by favoring the skipping of exon 4, thus directing its mRNA to NMD. Self-regulation by modulating unproductive splicing, and translation is an emerging theme among other RNA binding proteins, including HNRNPA2B1, HNRNPL, and SRSF3.46,47,62-64 We have shown, using RNA-seq and ddPCR, that tissues with mutations near exon 4 have a biased representation of the productive isoform containing this exon. The effect of this on protein expression was confirmed through immunohistochemical analysis of tumor tissue. Similar to the predicted effects of other RNA binding proteins with a multiplicity of targets,65-67 increased expression of HNRNPH1 is expected to have widespread effects on the splicing landscape in MCL.42,61 The relative paucity of mutations in this region in other B-cell NHL is consistent with a more important role of HNRNPH1 in MCL biology. This warrants further exploration of the suite of genes and splicing events regulated by HNRNPH1 in MCL.
The concurrent identification of 3 novel MCL-related genes (EWSR1, DAZAP1, and HNRNPH1) with related function is compelling, as it may implicate mRNA maturation, splicing, and/or trafficking as a general feature of lymphomagenesis in MCL. Accordingly, there is growing evidence relating alterations in RNA-binding proteins and splice factors in numerous cancers, including other B-cell lymphomas, to various aspects of cancer cell biology.68-70 Specifically, small changes in RNA-binding proteins can have large downstream effects on gene expression and can thus affect multiple hallmarks of cancer.71 For example, the splicing factor SF3B1 was identified as recurrently mutated in chronic lymphocytic leukemia,72-74 and further detailed investigations have identified widespread alternative splicing affecting multiple cellular pathways.75,76 The identification of pleiotropic effects downstream of SF3B1, including DNA damage response, apoptosis, and Notch signaling, indicate that widespread disruptions to RNA processing can enhance cancer cell survival by multiple pathways.75,77 Further work will identify these downstream effects in MCL.
In summary, through genomic analysis of 273 MCL tumors, we identified novel recurrently mutated genes with a range of mutation incidences. We implicate an important role for RNA-binding proteins and RNA processing in MCL as compared with other B-cell lymphomas, suggesting that RNA metabolism and splicing have a specific role in MCL pathology. We specifically attribute mutations in HNRNPH1 to disruptions in HNRNPH1 autoregulation, leading to increased HNRNPH1 protein expression in MCL. Further work that links these mutations to dysregulation of specific RNA molecules will highlight the relevance of RNA processing in MCL.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Canada’s Michael Smith Genome Sciences Centre for library construction, sequencing, and bioinformatics support; Arezoo Mohajeri for the construction of exome libraries; Qiang Pan-Hammarström for allowing us to use the exome sequencing data from Wu et al11; and all patients who provided tissue samples for this study.
This work was supported by the Canadian Institutes of Health Research (CIHR; 300738) and the Terry Fox Research Institute (1021, 1043, and 1061). Some of the biological materials were provided by the Ontario Tumour Bank, which is supported by the Ontario Institute for Cancer Research through funding provided by the Government of Ontario. K.M.C. is supported by a CIHR postdoctoral fellowship.
Footnotes
All genome and exome data will be submitted to the European Genome-Phenome Archive (EGA) for controlled-access data sharing (accession number EGAS00001004289).
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: P.P. analyzed and interpreted the data; S.E.A. and M.A. performed library preparation; K.M.C. and N.T. analyzed CLIP-seq and RNA-sequencing data; K.M.C. performed cell-based and formalin-fixed, paraffin-embedded tissue–based assays; M.A. performed sequencing and targeted sequencing analysis; B.M.G. provided bioinformatic support and interpreted the data; C.R. analyzed DLBCL mutations; B.M., M.B., G.W.S., A.J.M., and Q.Q. performed nucleic acid extractions and sample quality control; G.W.S. performed tissue microarray scoring; D.V. and D.W.S. provided clinical data and reviewed the cases; D.W.S., T.E.A., M.A.M., N.A.J., R.D.G., C.S., J.C., S.B., S.-J.D., G.L., C.S.T., R.A., and R.D.M. interpreted data, designed the study, and, with P.P. and K.M.C., wrote the manuscript.
Conflict-of-interest disclosure: R.D.G., J.M.C., D.V., and D.W.S. are inventors of the Nanostring nCounter–based MCL35 assay. The remaining authors declare no competing financial interests.
Correspondence: Ryan D. Morin, Molecular Biology and Biochemistry, Simon Fraser University, 8888 University Dr, Burnaby, BC V5A 1S6, Canada; e-mail: rdmorin@sfu.ca.
REFERENCES
- 1.Vose JM. Mantle cell lymphoma: 2017 update on diagnosis, risk-stratification, and clinical management. Am J Hematol. 2017;92(8):806-813. [DOI] [PubMed] [Google Scholar]
- 2.Chen D, Viswanatha DS, Zent CS, et al. Indolent Mantle Cell Lymphoma: A Distinct Subgroup Characterized by Leukemic Phase Disease without Lymphadenopathy [abstract]. Blood. 2009;114(22). Abstract 3937. [Google Scholar]
- 3.Abrisqueta P, Scott DW, Slack GW, et al. Observation as the initial management strategy in patients with mantle cell lymphoma. Ann Oncol. 2017;28(10):2489-2495. [DOI] [PubMed] [Google Scholar]
- 4.Lim SY, Horsman JM, Hancock BW. The Mantle Cell Lymphoma International Prognostic Index: Does it work in routine practice? Oncol Lett. 2010;1(1):187-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maddocks K. Update on mantle cell lymphoma. Blood. 2018;132(16):1647-1656. [DOI] [PubMed] [Google Scholar]
- 6.Cheah CY, Seymour JF, Wang ML. Mantle Cell Lymphoma. J Clin Oncol. 2016;34(11):1256-1269. [DOI] [PubMed] [Google Scholar]
- 7.Mohanty A, Sandoval N, Das M, et al. CCND1 mutations increase protein stability and promote ibrutinib resistance in mantle cell lymphoma. Oncotarget. 2016;7(45):73558-73572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wiestner A, Tehrani M, Chiorazzi M, et al. Point mutations and genomic deletions in CCND1 create stable truncated cyclin D1 mRNAs that are associated with increased proliferation rate and shorter survival. Blood. 2007;109(11):4599-4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beà S, Valdés-Mas R, Navarro A, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci USA. 2013;110(45):18250-18255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J, Jima D, Moffitt AB, et al. The genomic landscape of mantle cell lymphoma is related to the epigenetically determined chromatin state of normal B cells. Blood. 2014;123(19):2988-2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu C, de Miranda NF, Chen L, et al. Genetic heterogeneity in primary and relapsed mantle cell lymphomas: Impact of recurrent CARD11 mutations. Oncotarget. 2016;7(25):38180-38190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kridel R, Meissner B, Rogic S, et al. Whole transcriptome sequencing reveals recurrent NOTCH1 mutations in mantle cell lymphoma. Blood. 2012;119(9):1963-1971. [DOI] [PubMed] [Google Scholar]
- 13.Halldórsdóttir AM, Lundin A, Murray F, et al. Impact of TP53 mutation and 17p deletion in mantle cell lymphoma. Leukemia. 2011;25(12):1904-1908. [DOI] [PubMed] [Google Scholar]
- 14.Rosenwald A, Wright G, Wiestner A, et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell. 2003;3(2):185-197. [DOI] [PubMed] [Google Scholar]
- 15.Tam CS, Anderson MA, Pott C, et al. Ibrutinib plus Venetoclax for the Treatment of Mantle-Cell Lymphoma. N Engl J Med. 2018;378(13):1211-1223. [DOI] [PubMed] [Google Scholar]
- 16.Espinet B, Salaverria I, Bea S, et al. Incidence and prognostic impact of secondary cytogenetic aberrations in a series of 145 patients with mantle cell lymphoma. Genes Chromosome Canc. 49(5):439-451. [DOI] [PubMed] [Google Scholar]
- 17.Agarwal R, Chan Y-C, Tam CS, et al. Dynamic molecular monitoring reveals that SWI-SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma. Nat Med. 2019;25(1):119-129. [DOI] [PubMed] [Google Scholar]
- 18.Scott DW, Abrisqueta P, Wright GW, et al. ; Lymphoma/Leukemia Molecular Profiling Project . New Molecular Assay for the Proliferation Signature in Mantle Cell Lymphoma Applicable to Formalin-Fixed Paraffin-Embedded Biopsies. J Clin Oncol. 2017;35(15):1668-1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 2013;1303.3997. [Google Scholar]
- 20.Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15-21. doi:10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28(14):1811-1817. [DOI] [PubMed] [Google Scholar]
- 22.Kim S, Scheffler K, Halpern AL, et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods. 2018;15(8):591-594. [DOI] [PubMed] [Google Scholar]
- 23.McLaren W, Gil L, Hunt SE, et al. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17(1):122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gonzalez-Perez A, Lopez-Bigas N. Functional impact bias reveals cancer drivers. Nucleic Acids Res. 2012;40(21):e169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mularoni L, Sabarinathan R, Deu-Pons J, Gonzalez-Perez A, López-Bigas N. OncodriveFML: a general framework to identify coding and non-coding regions with cancer driver mutations. Genome Biol. 2016;17(1):128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tamborero D, Gonzalez-Perez A, Lopez-Bigas N. OncodriveCLUST: exploiting the positional clustering of somatic mutations to identify cancer genes. Bioinformatics. 2013;29(18):2238-2244. [DOI] [PubMed] [Google Scholar]
- 28.Mansouri L, Noerenberg D, Young E, et al. Frequent NFKBIE deletions are associated with poor outcome in primary mediastinal B-cell lymphoma. Blood. 2016;128(23):2666-2670. [DOI] [PubMed] [Google Scholar]
- 29.Arthur SE, Jiang A, Grande BM, et al. Genome-wide discovery of somatic regulatory variants in diffuse large B-cell lymphoma. Nat Commun. 2018;9(1):4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reddy A, Zhang J, Davis NS, et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell. 2017;171(2):481-494.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmitz R, Wright GW, Huang DW, et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med. 2018;378(15):1396-1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin Y-C, Boone M, Meuris L, et al. Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations. Nat Commun. 2014;5(1):4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meissner B, Kridel R, Lim RS, et al. The E3 ubiquitin ligase UBR5 is recurrently mutated in mantle cell lymphoma. Blood. 2013;121(16):3161-3164. [DOI] [PubMed] [Google Scholar]
- 34.Ahmed M, Zhang L, Nomie K, Lam L, Wang M. Gene mutations and actionable genetic lesions in mantle cell lymphoma. Oncotarget. 2016;7(36):58638-58648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith RWP, Anderson RC, Smith JWS, Brook M, Richardson WA, Gray NK. DAZAP1, an RNA-binding protein required for development and spermatogenesis, can regulate mRNA translation. RNA. 2011;17(7):1282-1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paronetto MP, Miñana B, Valcárcel J. The Ewing sarcoma protein regulates DNA damage-induced alternative splicing. Mol Cell. 2011;43(3):353-368. [DOI] [PubMed] [Google Scholar]
- 37.Fabbri G, Rasi S, Rossi D, et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med. 2011;208(7):1389-1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morin RD, Mendez-Lago M, Mungall AJ, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476(7360):298-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin Y-T, Yen PH. A novel nucleocytoplasmic shuttling sequence of DAZAP1, a testis-abundant RNA-binding protein. RNA. 2006;12(8):1486-1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dai T, Vera Y, Salido EC, Yen PH. Characterization of the mouse Dazap1 gene encoding an RNA-binding protein that interacts with infertility factors DAZ and DAZL. BMC Genomics. 2001;2(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choudhury R, Roy SG, Tsai YS, Tripathy A, Graves LM, Wang Z. The splicing activator DAZAP1 integrates splicing control into MEK/Erk-regulated cell proliferation and migration. Nat Commun. 2014;5(1):3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uren PJ, Bahrami-Samani E, de Araujo PR, et al. High-throughput analyses of hnRNP H1 dissects its multi-functional aspect. RNA Biol. 2016;13(4):400-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khodadoust MS, Olsson N, Wagar LE, et al. Antigen presentation profiling reveals recognition of lymphoma immunoglobulin neoantigens. Nature. 2017;543(7647):723-727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rule S, Jurczak W, Jerkeman M, et al. Ibrutinib versus temsirolimus: 3-year follow-up of patients with previously treated mantle cell lymphoma from the phase 3, international, randomized, open-label RAY study. Leukemia. 2018;32(8):1799-1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grande BM, Gerhard DS, Jiang A, et al. Genome-wide discovery of somatic coding and noncoding mutations in pediatric endemic and sporadic Burkitt lymphoma. Blood. 2019;133(12):1313-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rossbach O, Hung L-H, Schreiner S, et al. Auto- and cross-regulation of the hnRNP L proteins by alternative splicing. Mol Cell Biol. 2009;29(6):1442-1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martinez FJ, Pratt GA, Van Nostrand EL, et al. Protein-RNA Networks Regulated by Normal and ALS-Associated Mutant HNRNPA2B1 in the Nervous System. Neuron. 2016;92(4):780-795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ferrero S, Rossi D, Rinaldi A, et al. KMT2D mutations and TP53 disruptions are poor prognostic biomarkers in mantle cell lymphoma receiving high-dose therapy: a FIL study [published online ahead of print 19 September 2019]. Haematologica. doi:haematol.2018.214056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Änkö M-L, Müller-McNicoll M, Brandl H, et al. The RNA-binding landscapes of 2 SR proteins reveal unique functions and binding to diverse RNA classes. Genome Biol. 2012;13(3):R17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saltzman AL, Pan Q, Blencowe BJ. Regulation of alternative splicing by the core spliceosomal machinery. Genes Dev. 2011;25(4):373-384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pereverzev AP, Gurskaya NG, Ermakova GV, et al. Method for quantitative analysis of nonsense-mediated mRNA decay at the single cell level. Sci Rep. 2015;5(1):7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Noensie EN, Dietz HC. A strategy for disease gene identification through nonsense-mediated mRNA decay inhibition. Nat Biotechnol. 2001;19(5):434-439. [DOI] [PubMed] [Google Scholar]
- 53.Yang P, Zhang W, Wang J, Liu Y, An R, Jing H. Genomic landscape and prognostic analysis of mantle cell lymphoma. Cancer Gene Ther. 2018;25(5-6):129-140. [DOI] [PubMed] [Google Scholar]
- 54.Grünewald TGP, Cidre-Aranaz F, Surdez D, et al. Ewing sarcoma. Nat Rev Dis Primers. 2018;4(1):5. [DOI] [PubMed] [Google Scholar]
- 55.Sanchez G, Delattre O, Auboeuf D, Dutertre M. Coupled alteration of transcription and splicing by a single oncogene: boosting the effect on cyclin D1 activity. Cell Cycle. 2008;7(15):2299-2305. [DOI] [PubMed] [Google Scholar]
- 56.Chen H-Y, Yu Y-H, Yen PH. DAZAP1 regulates the splicing of Crem, Crisp2 and Pot1a transcripts. Nucleic Acids Res. 2013;41(21):9858-9869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Honoré B, Baandrup U, Vorum H. Heterogeneous nuclear ribonucleoproteins F and H/H′ show differential expression in normal and selected cancer tissues. Exp Cell Res. 2004;294(1):199-209. [DOI] [PubMed] [Google Scholar]
- 58.Rauch J, O’Neill E, Mack B, et al. Heterogeneous nuclear ribonucleoprotein H blocks MST2-mediated apoptosis in cancer cells by regulating A-Raf transcription. Cancer Res. 2010;70(4):1679-1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sun Y-L, Liu F, Liu F, Zhao X-H. Protein and gene expression characteristics of heterogeneous nuclear ribonucleoprotein H1 in esophageal squamous cell carcinoma. World J Gastroenterol. 2016;22(32):7322-7331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Geuens T, Bouhy D, Timmerman V. The hnRNP family: insights into their role in health and disease. Hum Genet. 2016;135(8):851-867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang E, Aslanzadeh V, Papa F, Zhu H, de la Grange P, Cambi F. Global profiling of alternative splicing events and gene expression regulated by hnRNPH/F. PLoS One. 2012;7(12):e51266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Änkö M-L, Morales L, Henry I, Beyer A, Neugebauer KM. Global analysis reveals SRp20- and SRp75-specific mRNPs in cycling and neural cells. Nat Struct Mol Biol. 2010;17(8):962-970. [DOI] [PubMed] [Google Scholar]
- 63.Jumaa H, Nielsen PJ. The splicing factor SRp20 modifies splicing of its own mRNA and ASF/SF2 antagonizes this regulation. EMBO J. 1997;16(16):5077-5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pervouchine D, Popov Y, Berry A, Borsari B, Frankish A, Guigó R. Integrative transcriptomic analysis suggests new autoregulatory splicing events coupled with nonsense-mediated mRNA decay. Nucleic Acids Res. 2019;47(10):5293-5306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cannell IG, Merrick KA, Morandell S, et al. A Pleiotropic RNA-Binding Protein Controls Distinct Cell Cycle Checkpoints to Drive Resistance of p53-Defective Tumors to Chemotherapy. Cancer Cell. 2015;28(5):623-637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Díaz-Muñoz MD, Turner M. Uncovering the Role of RNA-Binding Proteins in Gene Expression in the Immune System. Front Immunol. 2018;9:1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Saha S, Murmu KC, Biswas M, et al. Transcriptomic Analysis Identifies RNA Binding Proteins as Putative Regulators of Myelopoiesis and Leukemia. Front Oncol. 2019;9:692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.LeFave CV, Squatrito M, Vorlova S, et al. Splicing factor hnRNPH drives an oncogenic splicing switch in gliomas. EMBO J. 2011;30(19):4084-4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Golan-Gerstl R, Cohen M, Shilo A, et al. Splicing factor hnRNP A2/B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer Res. 2011;71(13):4464-4472. [DOI] [PubMed] [Google Scholar]
- 70.Xu C, Xie N, Su Y, et al. HnRNP F/H associate with hTERC and telomerase holoenzyme to modulate telomerase function and promote cell proliferation [published online ahead of print 20 December 2019]. Cell Death Differ. 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hong S. RNA Binding Protein as an Emerging Therapeutic Target for Cancer Prevention and Treatment. J Cancer Prev. 2017;22(4):203-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Quesada V, Conde L, Villamor N, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2011;44(1):47-52. [DOI] [PubMed] [Google Scholar]
- 73.Rossi D, Bruscaggin A, Spina V, et al. Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: association with progression and fludarabine-refractoriness. Blood. 2011;118(26):6904-6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang L, Lawrence MS, Wan Y, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365(26):2497-2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang L, Brooks AN, Fan J, et al. Transcriptomic Characterization of SF3B1 Mutation Reveals Its Pleiotropic Effects in Chronic Lymphocytic Leukemia. Cancer Cell. 2016;30(5):750-763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ten Hacken E, Valentin R, Regis FFD, et al. Splicing modulation sensitizes chronic lymphocytic leukemia cells to venetoclax by remodeling mitochondrial apoptotic dependencies. JCI Insight. 2018;3(19):e121438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Te Raa GD, Derks IA, Navrkalova V, et al. The impact of SF3B1 mutations in CLL on the DNA-damage response. Leukemia. 2015;29(5):1133-1142. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.