

Summary
Alzheimer's disease (AD) is the most common cause of dementia with around 50 million people suffering from this disease worldwide. Mutations in the ATP-binding cassette sub-family A member 7 (ABCA7) have been reported to cause susceptibility to AD 9 (OMIM #608907). In this study, we report a novel variant in ABCA7 in a Saudi patient with susceptibility to AD 9 and a strong family history of neurodegenerative disorders, which may be explained by the same variant. We studied a single 57-year-old female patient with typical symptoms of AD supported by MRI findings from a Saudi family with a positive history of a similar disease in multiple individuals. The case study was conducted in King Abdulaziz Medical City in Jeddah, Saudi Arabia. Whole-exome sequencing identified the novel heterozygous variant c.3706C>T p.(Avg 1236Cys) in the ABCA7 gene, which leads to an amino acid exchange. Furthermore, bioinformatics in silico programs predict a pathogenic effect for this variant. To the best of our knowledge, the variant has not been described in the literature so far as evidenced by a thorough literature review using multiple databases such as Ovid, Medline, EMBASE, ProQuest, Science Direct, Google Scholar, and PubMed. In this article, we reported a middle-aged Saudi woman with a novel variant in ABCA7 who had clinical features of both AD and Parkinson's disease. Given the reported function of this gene, it is most likely that it is etiological and pathological because of the presenting complex neurological disease due to decreased clearance of β-amyloid and α-Synuclein. We illustrate the importance of this interesting gene that could be implicated in several neurodegenerative disorders.
Keywords: ABCA7, Alzheimer disease, dementia, mutation, Saudi Arabia
1. Introduction
Alzheimer's disease (AD) is a major public health issue because of the rising cost of caring for the increasing number of people suffering from this disorder (1). It is a neurodegenerative protein-conformational disease in which soluble neuronal proteins attain altered conformations leading to abnormal neuronal function and cell death. AD is the most common cause of dementia with around 50 million people suffering from this disease worldwide. It usually affects people above the age of 65 years (2). The early cardinal cognitive symptoms of AD are impairment of memory, executive function, and problem-solving ability. Behavioral and psychological manifestations such as apathy, social disengagement, and psychosis constitute the middle-late manifestations of the disease. Atypical presentation in which memory loss is not the initial manifestation could occur, especially in those with an early-onset disease in the fourth and fifth decade of life (3). Despite advances in medical therapy and intensive research efforts, currently available treatments have only marginal benefits with no cure or preventive intervention (4).
The sporadic form of AD is the most frequent type of AD with an age of onset usually after 60 years. The familial form represents less than 1% of all cases with early onset of the disease (the fourth and fifth decades of life) (5). A Mendelian autosomal dominant pattern of inheritance is seen in early-onset AD. The identified causative mutations in 60 to 70% of early-onset AD are in the following three genes: APP, PSEN1, and PSEN2 located at ch 21q, ch14q, and ch 1q, respectively (6). In contrast to early-onset AD, the genetic basis of late-onset AD is more complex with various and more common genetic factors but with a lesser degree of penetrance. However, the most penetrating genetic factor acknowledged is the presence of the Apolipoprotein E (APOE) epsilon 4 (ε4) allele on chromosome 19. The presence of two ε4 copies increases odds of AD by 8-12 fold, while one ε4 allele increases odds of AD by 2-3 fold compared with non-carriers (7). More than 20 other genes were identified by genome-wide association studies, yet the increased AD odds ratio is modest, ranging from 1-1.5 fold. These genes include CLU, PICALM, BIN1, EPHA1, and ABCA7 (8-10). Mutations in the ATP-binding cassette sub-family A member 7 (ABCA7) have been reported to cause susceptibility to AD 9 (OMIM #608907). In this study, we report a novel variant in ABCA7 for the first time in a Saudi patient with susceptibility to AD 9 and a strong family history of neurodegenerative disorders, which could be explained by the same variant.
2. Materials and Methods
2.1. Genetic analysis
We included a female patient with typical symptoms of AD supported by MRI findings from a Saudi family with a positive history of a similar disease in other family members. The diagnosis of AD was made according to the DSM-5 (Diagnostic and Statistical Manual of Mental Disorders, fifth edition) criteria. The case study was conducted in King Abdulaziz Medical City in Jeddah, Saudi Arabia. The genetic analysis was done by DNA fragmentation, and the exons of the known genes in the human genome, as well as the corresponding exon-intron boundaries, were enriched using Roche NimbleGen (SeqCap MedExome) capture technology, amplified and sequenced simultaneously by Illumina technology (next-generation sequencing, NGS) using an Illumina system. The target regions were sequenced with an average coverage of 106 fold. For about 99% of the regions of interest, a 15 fold coverage and for about 98%, a 20 fold coverage was obtained.
NGS data were aligned to the hg19 genome assembly. Variant calling and annotation was performed using an in-house developed bioinformatics pipeline. Identified single nucleotide variants and indels were filtered against external and internal databases focusing on rare variants with a minor allele-frequency (MAF) in genomAD of 1% or less and removing known artifacts and variants in regions with highly homologous regions. Classification of variants was conducted based on the American College of Medical Genetics guidelines considering database entries (including the Human Gene Mutation Database), bioinformatics prediction tools, and literature status. Variants annotated as common polymorphisms in databases or the literature or that were classified as (likely) benign were neglected.
Putatively pathogenic differences between the wildtype sequence (human reference genome according to the University of California Santa Cruz genome browser, hg19 GRCh37) and the patient's sequence mentioned and interpreted in this report were assessed using the in-house established quality score. Variants not passing the quality threshold were verified using polymerase chain reaction amplification followed by conventional Sanger sequencing. The sample identity was ensured by internal quality management procedures. This study was approved by the Institutional Review Board (IRB) of the King Abdullah International Medical Research Center (KAIMRC).
2.2. Case report
A 57-year-old lady presented to the neurology clinic with memory impairment, difficulty in walking, and abnormal muscle twitches of both upper and lower extremities. The onset of her complaints started four years ago after the death of her brother (her age at onset was 53 years). In addition, her family has noticed low mood, sleep deprivation, and episodic irritability. The course of the disease has been gradually progressive, and she struggles to remember her work-related tasks as she is a school administrator. She is a university graduate with a bachelor's degree in education. Her memory loss has worsened over a period of two years, which eventually has caused social impairment since she cannot remember the names of her relatives. In addition, she has complained of urinary incontinence for more than two years. In terms of her medical background, she is a known case of hypertension, diabetes, and hypothyroidism for more than ten years as well as atrial fibrillation for more than six years. She has been on aspirin, levothyroxine, simvastatin, omeprazole, gliclazide, insulin, propranolol, and mirtazapine. She is wheelchair-bound and dependent on others in most of her daily living activities. She is divorced and lives with her nephew. There is a strong family history of neurodegenerative diseases in both the central and peripheral nervous system affecting her mother, sisters, and brothers (Figure 1). Two brothers died at the age of 1 and 4 years, respectively, with no obvious medical illness. Two other brothers died at the ages of 38 and 45 years, respectively, with a neurological disorder manifesting as gait difficulty, ataxia, dysarthria, and nystagmus with normal cognitive functions. Two sisters died at the ages of 4 years and three days, respectively, with severe hypotonia and weakness. Her mother and one older sister who died at the age of 60 years had similar clinical features to our index case. On physical examination, she was afebrile and alert but disoriented to time, place, and person. She recalled 0/3 objects upon short memory testing with a mini-mental status examination score of 19/30. General knowledge questions showed a partial impairment of her long term memory as well. However, her language was intact, and her speech tone, volume, and fluency were normal. Her cranial nerve examination from 2 to 12 was normal. She has pyramidal signs with increased tone in both upper and lower extremities and brisk symmetrical reflexes, but her plantar responses were downgoing. She also has extrapyramidal signs in the form of mild axial rigidity. She was not able to walk, and her legs were stiff and weak with a power of 2/5 in both proximal and distal muscles. MRI of the brain showed severe diffuse atrophy for her age with confluent periventricular white matter and bilateral centrum semiovale abnormal signal intensity in keeping with moderate to severe microangiopathic leukoencephalopathic changes (Figure 2). To exclude other differential diagnoses that may explain our patient's clinical and radiological findings, spinal cord MRI and electroencephalogram studies were done, which were unremarkable. The final diagnosis was familial AD, and her management was modified to include warfarin, memantine, rivastigmine, and aggressive physiotherapy.
Figure 1.
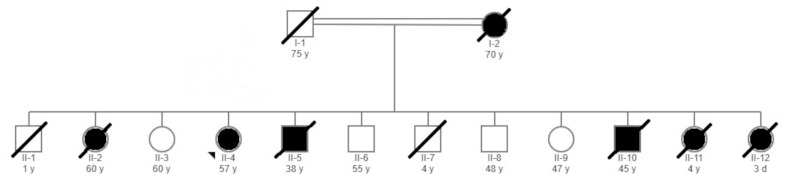
Family pedigree showing the details of the members of the family. Arrowhead indicates the proband included in this study.
Figure 2.
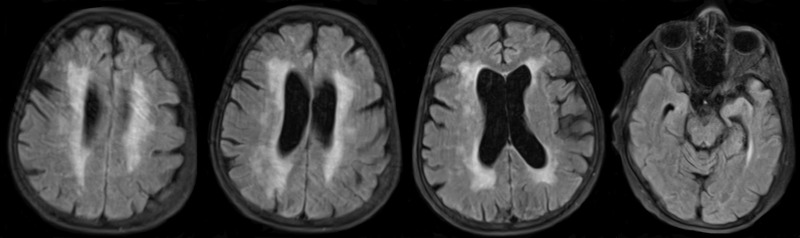
MRI of the brain showing severe diffuse atrophy for her age with confluent periventricular white matter and bilateral centrum semiovale abnormal signal intensity in keeping with moderate to severe microangiopathic leukoencephalopathic changes.
3. Results and Discussion
Whole exome sequencing identified the heterozygous variant c.3706C>T p.(Avg 1236Cys) in the ABCA7 gene, which leads to an amino acid exchange. Bioinformatics in silico studies predicted a pathogenic effect for this variant. To the best of our knowledge, the variant has not been described in the literature as far as is evident from a thorough literature review using multiple databases including Ovid, Medline, EMBASE, ProQuest, Science Direct, Google Scholar, and PubMed. The variant is found in 0.003% of the overall population (4 heterozygous (ages range from 40 - 45 to > 80), 0 homozygous; genomAD). No variants in other genes related to AD or genes related to leukoencephalopathy were identified. Considering the heterozygous variant in ABCA7 and the supportive phenotype of the patient, a genetic diagnosis of susceptibility to AD 9 (OMIM #608907) was made. Genetic testing for the living family members was suggested, but the idea was rejected by the family.
The prevalence of AD in Arab countries is not accurately evaluated in comprehensive epidemiological studies (11). In Saudi Arabia, there are approximately 130,000 persons suffering from AD representing 0.4% of the Saudi population. Due to the shifting of the age of the population towards the elderly, this number is expected to increase in the coming two decades (12). In a recent study done by El Bitar et al. (13), a comprehensive screening for point mutations in 117 AD cases from Saudi Arabia was carried out by direct sequencing of coding regions in four AD-related genes (PSEN1, PSEN2, APP, and SORL1). They identified a total of eight potential pathogenic missense variants in all studied genes. The ABCA7 gene was not tested in that cohort of patients. This indicates the importance of genetic testing of cases of AD, especially those with early-onset or strong family history of dementia. In addition, performing whole-exome sequencing yields more than targeted gene sequencing.
Familial AD (affecting three or more members in a family) represents 25% of all cases of AD (14). Both familial and non-familial AD have the same clinical and pathologic phenotype, and the distinction can be made only by family history and/or molecular genetic testing. Familial AD can be either early-onset (before age 65 years) or late-onset (age 65 years and above) (14). Susceptibility genes in late-onset familial AD (more than 20 genes) have a role in brain development, immune functions, and cytoskeletal organization. In early-onset familial AD, more than four genes have been identified. Although the heritability of AD is about 70%, only 30% of those cases can be explained from the known genes. This can be explained by the phenomenon of complex genetic disease/traits (15).
ATP-binding cassette sub-family A member 7, abbreviated as (ABCA7) gene, is located on chromosome 19p13.3. This gene encodes a protein member of the superfamily ATP-binding cassette (ABC) transporters (16). ABCA7 expression is widely distributed in the human body since it is found in the brain, lung, adrenal gland, kidney, spleen, thymus, lymph node, testis, keratinocytes, and pancreatic islets. In the brain, the mRNA of ABCA7 was most abundant in microglial cells. The exact function of these (ABC) transporters are not fully understood, yet it is proposed that they interfere with lipid metabolism as well as phagocytosing apoptotic cells (17). In mouse models, ABCA7 knock-out results in an interruption of the uptake and the proteolytic functions of microglial cells to degrade amyloid-beta peptides (Aβ). Therefore, brain aggregations of Aβ plaques were enhanced in those mice. Additionally, it is established that even the production of (Aβ) is increased and accelerated in primary neurons of mice in the absence of ABCA7 by further activation of the amyloid precursor protein (APP) cleaving enzymes (18). These findings provide us with a better insight into the ABCA7 role in the pathogenesis of AD. However, human model-based research studies are needed for better understanding.
To date, the most common ABCA7 mutation variants, with minor allele frequency (MAF) of more than 5%, are rs3764650, rs3764647, rs115550680, rs142076058, rs4147929, and rs3752246. These variants' genetic penetrating capability and the predisposition of developing AD are largely dependent on the ethnicity of the studied population. ABCA7 variants rs3764650, rs3752246, and rs4147929 are significantly linked and reproduced among Caucasians (19-21). The African American population shows a large susceptibility for the following ABCA7 variants: rs3764647, rs115550680, and rs142076058 (22-24). In addition, a rare (MAF < 1%) ABCA7 missense variant, rs3752239, was detected within African Americans as well (25). A low frequency (MAF 1-5%) ABCA7 variant, rs78117248, with high penetrating capability was reported by a Belgian cohort study (26). It is hypothesized that the characteristics of ABCA7 are different, and only loss of function variants can be causal variants. Missense mutations of this gene are considered risk variants and not causal variants. More time and further research are needed to clarify such a theory.
In our patient, the presence of an ABCA7 variant in this patient with early-onset AD with the presence of clinical features of both AD and Parkinson's disease suggests that this gene might be a risk factor for neurodegeneration. This gene is hypothesized to be involved in transport of phospholipid and cholesterol across membranes to APOE. In addition, it has been implicated in the activation of phagocytosis to clear amyloid plaques and apoptotic cells. It is functional throughout the brain and has been reported to be involved in several disorders including Parkinson's disease, cystic fibrosis, and tangier disease (27).
The definitive diagnosis of AD requires a histopathological confirmation through biopsy. However, this is rarely done in clinical practice due to its inconvenience (28). Therefore, the diagnosis is made upon the insidious progression of cognitive symptoms and the implementation of the National Institute on Aging and the Alzheimer's Association (NIA-AA) diagnosing criteria (29). Radiological evaluation is of great value in all suspected AD cases. MRI is the modality of choice in which a characteristic focal reduced hippocampal volume or medial temporal lobe atrophy is demonstrated. In addition, functional brain imaging with 18-F fluorodeoxyglucose positron emission tomography (FDG-PET) or single-photon emission computed tomography (SPECT) could be utilized as well (30). In our patient, we think that the MRI findings of severe white matter hyperintensity with subcortical white matter changes are not related to her genetic abnormality. This was confirmed by a thorough literature review that yielded no articles discussing the relationship between ABCA7 variants and such radiological abnormality.
The limitation of this study involves the lack of confirmation of the presence of amyloid in the brain, which can be detected through amyloid PET imaging or cerebrospinal fluid analysis. In addition, the pathogenicity was not supported by a segregation study since the other family members either have died or refused genetic study. Furthermore, functional studies of the variant in animal models were not performed due to the unavailability of such technology in our center.
In this article, we reported a middle-aged Saudi woman with a novel variant in ABCA7 who had clinical features of both AD and Parkinson's disease. Given the reported function of this gene, it is most likely that it is etiological and pathological of the presenting complex neurological disease due to decreased clearance of Aβ and α-Synuclein. We illustrate the importance of this interesting gene that could be implicated in several neurodegenerative disorders. Further studies using whole-exome sequencing should be utilized to screen familial cases of AD in the Saudi population.
References
- 1. Scheltens P, Blennow K, Breteler MM, de Strooper B, Frisoni GB, Salloway S, Van der Flier WM. Alzheimer's disease. Lancet. 2016; 388:505-517. [DOI] [PubMed] [Google Scholar]
- 2. Prince M, Ali GC, Guerchet M, Prina AM, Albanese E, Wu YT. Recent global trends in the prevalence and incidence of dementia, and survival with dementia. Alzheimers Res Ther. 2016; 8:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. Alzheimer's disease. Lancet. 2011; 377:1019-1031. [DOI] [PubMed] [Google Scholar]
- 4. Cummings J, Lee G, Ritter A, Zhong K. Alzheimer's disease drug development pipeline: 2018. Alzheimers Dement (N Y). 2018; 4:195-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cacace R, Sleegers K, Van broeckhoven C. Molecular genetics of early-onset Alzheimer's disease revisited. Alzheimers Dement. 2016; 12:733-748. [DOI] [PubMed] [Google Scholar]
- 6. Reitz C. Genetic diagnosis and prognosis of Alzheimer's disease: challenges and opportunities. Expert Rev Mol Diagn. 2015; 15:339-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Balasa M, Gelpi E, Antonell A, Rey MJ, Sánchez-Valle R, Molinuevo JL, Lladó A. Clinical features and APOE genotype of pathologically proven early-onset Alzheimer disease. Neurology. 2011; 76:1720-1725. [DOI] [PubMed] [Google Scholar]
- 8. Chouraki V, Seshadri S. Genetics of Alzheimer's disease. Adv Genet. 2014; 87:245-294. [DOI] [PubMed] [Google Scholar]
- 9. Seshadri S, Fitzpatrick AL, Ikram MA, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA. 2010; 303:1832-1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Le guennec K, Nicolas G, Quenez O, et al. ABCA7 rare variants and Alzheimer disease risk. Neurology. 2016; 86:2134-2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. El-metwally A, Toivola P, Al-rashidi M, Nooruddin S, Jawed M, AlKanhal R, Abdul Razzak H, Albawardi N. Epidemiology of Alzheimer's disease and dementia in Arab Countries: a systematic review. Behav Neurol. 2019; 2019:3935943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Algahtani H, Shirah B, Alhazmi A, Alshareef A, Bajunaid M, Samman A. Perception and attitude of the general population towards Alzheimer's disease in Jeddah, Saudi Arabia. Acta Neurol Belg. 2020; 120:313-320. [DOI] [PubMed] [Google Scholar]
- 13. El bitar F, Qadi N, Al rajeh S, Majrashi A, Abdulaziz S, Majrashi N, Al Inizi M, Taher A, Al Tassan N. Genetic study of Alzheimer's disease in Saudi population. J Alzheimers Dis. 2019; 67:231-242. [DOI] [PubMed] [Google Scholar]
- 14. Dorszewska J, Prendecki M, Oczkowska A, Dezor M, Kozubski W. Molecular basis of familial and sporadic Alzheimer's disease. Curr Alzheimer Res. 2016; 13:952-963. [DOI] [PubMed] [Google Scholar]
- 15. Kim JH. Genetics of Alzheimer's Disease. Dement Neurocogn Disord. 2018; 17:131-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Allen M, Lincoln SJ, Corda M, Watzlawik JO, Carrasquillo MM, Reddy JS, Burgess JD, Nguyen T, Malphrus K, Petersen RC, Graff-Radford NR, Dickson DW, Ertekin-Taner N. ABCA7 loss-of-function variants, expression, and neurologic disease risk. Neurol Genet. 2017; 3:e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aikawa T, Holm ML, Kanekiyo T. ABCA7 and Pathogenic Pathways of Alzheimer's Disease. Brain Sci. 2018; 8:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim WS, Li H, Ruberu K, Chan S, Elliott DA, Low JK, Cheng D, Karl T, Garner B. Deletion of Abca7 increases cerebral amyloid-β accumulation in the J20 mouse model of Alzheimer's disease. J Neurosci. 2013; 33:4387-4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet. 2011; 43:429-435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat Genet. 2011; 43:436-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lambert JC, Ibrahim-verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013; 45:1452-1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Logue MW, Schu M, Vardarajan BN, et al. A comprehensive genetic association study of Alzheimer disease in African Americans. Arch Neurol. 2011; 68:1569-1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Reitz C, Jun G, Naj A, et al. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E ϵ4,and the risk of late-onset Alzheimer disease in African Americans. JAMA. 2013; 309:1483-1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cukier HN, Kunkle BW, Vardarajan BN, et al. ABCA7 frameshift deletion associated with Alzheimer disease in African Americans. Neurol Genet. 2016; 2:e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. N'Songo A, Carrasquillo MM, Wang X, Burgess JD, Nguyen T, Asmann YW, Serie DJ, Younkin SG, Allen M, Pedraza O, Duara R, Custo MTG, Graff-Radford NR, Ertekin-Taner N. African American exome sequencing identifies potential risk variants at Alzheimer disease loci. Neurol Genet. 2017; 3:e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cuyvers E, De roeck A, Van den bossche T, Van Cauwenberghe C, Bettens K, Vermeulen S, Mattheijssens M, Peeters K, Engelborghs S, Vandenbulcke M, Vandenberghe R, De Deyn PP, Van Broeckhoven C, Sleegers K. Mutations in ABCA7 in a Belgian cohort of Alzheimer's disease patients: a targeted resequencing study. Lancet Neurol. 2015; 14:814-822. [DOI] [PubMed] [Google Scholar]
- 27. Nuytemans K, Maldonado L, Ali A, John-Williams K, Beecham GW, Martin E, Scott WK, Vance JM. Overlap between Parkinson disease and Alzheimer disease in ABCA7 functional variants. Neurol Genet. 2016; 2:e44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Atri A. The Alzheimer's disease clinical spectrum: diagnosis and management. Med Clin North Am. 2019; 103:263-293. [DOI] [PubMed] [Google Scholar]
- 29. Mckhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging- Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011; 7:263-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Risacher SL, Saykin AJ. Neuroimaging in aging and neurologic diseases. Handb Clin Neurol. 2019; 167:191-227. [DOI] [PMC free article] [PubMed] [Google Scholar]