

Abstract
Purpose
To identify the role of the BBSome protein Bardet–Biedl syndrome 5 (BBS5) in photoreceptor function, protein trafficking, and structure using a congenital mutant mouse model.
Methods
Bbs5–/– mice (2 and 9 months old) were used to assess retinal function and morphology. Hematoxylin and eosin staining of retinal sections was performed to visualize histology. Electroretinography was used to analyze rod and cone photoreceptor function. Retinal protein localization was visualized using immunofluorescence (IF) within retinal cryosections. TUNEL staining was used to quantify cell death. Transmission electron microscopy (TEM) was used to examine retinal ultrastructure.
Results
In the Bbs5–/– retina, there was a significant loss of nuclei in the outer nuclear layer accompanied by an increase in cell death. Through electroretinography, Bbs5–/– mice showed complete loss of cone photoreceptor function. IF revealed mislocalization of the cone-specific proteins M- and S-opsins, arrestin-4, CNGA3, and GNAT2, as well as a light-dependent arrestin-1 mislocalization, although perpherin-2 was properly localized. TEM revealed abnormal outer segment disk orientation in Bbs5–/–.
Conclusions
Collectively, these data suggest that, although BBS5 is a core BBSome component expressed in all ciliated cells, its role within the retina mediates specific photoreceptor protein cargo transport. In the absence of BBS5, cone-specific protein mislocalization and a loss of cone photoreceptor function occur.
Keywords: BBS5, BBSome, ciliopathy, cone–rod dystrophy, retinal degeneration
Primary cilia are evolutionarily conserved sensory structures that function as critical signaling nodes of numerous biological processes, including sensory perception. Photoreceptors are light-sensitive neurons of the retina that contain highly modified primary cilia. Within these structures, protein movement is governed by multifaceted complexes at the base of the cilium, one of which is made up of Bardet–Biedl Syndrome (BBS) proteins. Eight BBS proteins (BBS1, BBS2, BBS4, BBS5, BBS7, BBS8, BBS9, and BBS18) participate in the formation of a stable protein complex named the BBSome. The overall function of the BBSome complex is understood to be a coat complex required for sorting specific membrane proteins to and from the primary cilia, specifically with regard to retrograde trafficking.1,2
Mutations in Bbs genes that result in deficiencies in primary cilia function and receptor trafficking are associated with pathologies in a group of disorders with overlapping phenotypes, termed ciliopathies.3–5 BBS is a rare autosomal recessive ciliopathy in which phenotypes such as renal anomalies, polydactyly, hypogonadism, cognitive impairment, and obesity evolve throughout the first decade of life.6–8 Although there is phenotypic variability within patients, the most highly penetrant feature is retinal dystrophy, which occurs in over 90% of BBS patients.9,10 Cases have been reported as cone–rod dystrophy and rod–cone dystrophy with secondary findings such as strabismus, cataracts, and astigmatism.5,9–12 Several studies of BBS knockout animal models have been shown to emulate some phenotypes found in patients, specifically pertaining to retinal degeneration.13–21
A component of the BBSome, Bardet–Biedl Syndrome 5 (BBS5), is one of the most widely conserved components across eukaryotes and was discovered using a comparative genomics approach.22,23 BBS5 contains two pleckstrin homology domains and a three-helix bundle.24 One of the observed molecular functions of BBS5 is its interaction with arrestin-1 in a light-dependent manner.25 Currently, few animal models of Bbs5 absence have been generated to study its role in retinal development, function, and homeostasis. Containing a novel frameshift mutation found in a cohort of patients, a zebrafish animal model of this BBS5 mutation was found to have retinal development issues and extensive protein mislocalization.26 Recently, it has been reported that the lack of Bbs5 in mice results in a cone–rod dystrophy conveyed through the loss of photopic function by electroretinography (ERG) analyses.27 To further analyze the role of BBS5 in the development and maintenance of photoreceptors and its potential role in photoreceptor protein trafficking and structure, we generated a congenital knockout mouse model, Bbs5−/−. We have found cone phototransduction component mislocalization, accompanied by light-dependent arrestin-1 mislocalization. Additionally, we have found abnormal outer segment disks through transmission electron microscopy (TEM). These data bring to the surface some of the differences in BBSome function between rod and cone photoreceptors with respect to protein trafficking and photoreceptor function.
Methods
Animals
BBS5 knockout (Bbs5tm1a; Bbs5–/–) embryonic stem cells were obtained from the European Conditional Mouse Mutagenesis (EUCOMM) program and were used to generate Bbs5–/– mice on a mixed genetic background (B6J to B6N albino). Primers used for genotyping included 5′ ttcagttggtcagttttgtatcgt (common forward in 5′ homology arm of Bbs5), 5′ tcagcaccggataacagagc (reverse WT), and 5′ catagttggcagtgtttgggg (reverse in the tm1a cassette). Sequencing of the Bbs5–/– genomic and cDNA was performed via fluorescence-based Sanger sequencing. There were non-Mendelian ratios of surviving animals. All animal studies were conducted in compliance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research using protocols approved by the Institutional Animal Care and Use Committee at the University of Alabama at Birmingham.
Quantitative Western Blotting
Retinas from Bbs5–/– and wild-type (WT) mice (n = 5 per group) were homogenized in sample application buffer as described previously.28 They were then separated by standard polyacrylamide gel electrophoresis, transferred to a supported nitrocellulose membrane (1212590; GVS North America, Sanford, ME, USA), and blocked for 1 hour at room temperature with 4% non-fat dry milk in TBST (20-mM Tris-Cl, pH 7.6; 0.1% [v/v] Tween 20), supplemented with 0.02% sodium azide. They were incubated overnight at 4°C in the same solution containing primary antibody (anti-BBS525 and β-actin antibody, 4967; Cell Signaling Technology, Danvers, MA, USA). Binding of secondary antibodies conjugated to horseradish peroxidase (62-6520, 31460; Thermo Fisher Scientific, Waltham, MA, USA) was detected using chemiluminescent reagents by exposure to film. For quantification, individual band intensity was measured using ImageJ (National Institutes of Health, Bethesda, MD, USA). Bands were normalized to the β-actin loading control.
Tissue Preparation and Immunofluorescence Microscopy
Animals were housed with 12-hour light/dark cycles. For dark-adapted studies, animals were dark-adapted for at least 4 hours. After euthanasia, retinal tissues were fixed in 4% paraformaldehyde and were then cryoprotected in 30% sucrose. Tissues were embedded and frozen in optimal cutting temperature compound and sliced into 12-µm-thick sections. Retinal sections were then permeabilized with 0.1% Triton X-100 in PBS. Blocking and primary antibody incubations were in 10% normal goat serum in PBS with 0.01% sodium azide and 0.3% Triton X-100, and sections were washed with PBS. Primary antibody incubations were performed for 16 to 24 hours at 4°C. Primary antibodies used included rhodopsin (1D4, 1:2000; courtesy of Robert Molday29–31); arrestin-1 (sc-67130, 1:250; Santa Cruz Biotechnology, Santa Cruz, CA, USA); transducin (K-20, 1:250; Santa Cruz Biotechnology); M-opsin, S-opsin, and arrestin-4 (1:1000; courtesy of Cheryl Craft32,33); GNAT2 (PA5-22340, 1:100; Thermo Fisher Scientific); peripherin-2 (MABN293, 1:100; MilliporeSigma, Burlington, MA, USA); and cyclic nucleotide-gated channel alpha 3 (CNGA3, 1:100; courtesy of Xin-Qin Ding34). All secondary antibodies and mounting media were purchased from Thermo Fisher Scientific, and incubations were performed for 1 hour at room temperature using a 1:500 dilution. Secondary antibodies used included Alexa Fluor 488-conjugated Goat anti-Mouse IgG (A32723) and Alexa Fluor 488-conjugated Goat anti-Rabbit IgG (A-11034), and tissue nuclei were visualized with nuclear stain 4′,6-diamidino-2-phenylindole (DAPI, 62248; Thermo Fisher Scientific). Coverslips were mounted using Immu-Mount (9990402; Thermo Fisher Scientific).
Retinal tissue images were taken either on a PerkinElmer Ultra ERS-6 spinning disk confocal microscope (Waltham, MA, USA) with a 60× Apo TIRF oil immersion objective (1.49 NA) and a Hamamatsu C9100 EM-CCD (Hamamatsu Photonics, Hamamatsu, Japan) or on a Zeiss LSM-800 Airyscan confocal microscope (Carl Zeiss Microscopy, White Plains, NY, USA) with a 60× Apo TIRF oil immersion objective (1.49 NA) and a Zeiss Axiocam-506 CCD camera. Retinal tissue hematoxylin (72704; Thermo Fisher Scientific) and eosin (17372-87-1; Acros Organics, Morris Plains, NJ, USA) staining was performed and imaged on a Nikon Eclipse TE2000 with a 40× objective and a MicroPublisher 3.3 RTV camera (Teledyne QImaging, Surrey, BC, Canada).
Retinal spidergrams were constructed by plotting the number of outer nuclear layer (ONL) nuclei as a function of position in the retina relative to the optic nerve (µm). To detect cell death, TUNEL labeling was performed using the ApopTag Red kit (S7165; EMD Millipore, Darmstadt, Germany) and counting the number of positively labeled apoptotic nuclei from 15 retinal sections from each animal. All images were compiled using ImageJ software.
Electroretinography
Mice were dark adapted for at least 4 hours and anesthetized with 2.5% isoflurane. Following general anesthesia, animals were placed on a heating pad, corneas were anesthetized with proparacaine (0.5%), and pupils were dilated with topical phenylephrine HCl (2.5%) and tropicamide (1%). Electroretinogram recordings were collected using a HMsERG unit (OcuScience, Henderson, NV, USA) and loop electrodes of 37-gauge platinum wire as previously described.35 Protocols Scotopic 2 and Photopic (OcuScience) were used over a range of flash intensities (0.1–25 cd·s/m2). Prior to testing, a background light adaptation of 10 minutes at 30 cd·s/m2 was used for photopic responses. ERGView software (OcuScience) was used to analyze the ERG recordings and to determine a- and b-wave amplitudes. Averaged recordings were exported to text files and graphed in KalediaGraph (Synergy Software, Inc., Reading, PA, USA).
Transmission Electron Microscopy
Mouse eyecups were fixed by immersion in 2.5% paraformaldehyde and 2.5% glutaraldehyde in 100-mM sodium cacodylate buffer (pH 7.4) at room temperature for 30 minutes, then at 4°C for 2 hours with gentle rotation. The eyecups were then washed with 60-mM sodium phosphate (pH 7.4) supplemented with 3% sucrose and 150-mM CaCl2 two times, 15 minutes each. They were then secondarily fixed with 1% OsO4 in 60-mM sodium phosphate (pH 7.4) supplemented with 3% sucrose and 150-mM CaCl2. The eyecups were dehydrated through an ethanol series and transitioned to the embedding medium with propylene oxide. The eyecups were embedded for sectioning in 29% (w/w) Araldite 502, 17.8% (w/w) LX-112, and 53.2% (w/w) dodecenyl succinic anhydride supplemented with 0.2-mL benzyldimethylamine/10 g resin at 70°C for 8 hours. All reagents were obtained from Electron Microscopy Sciences (Hatfield, PA, USA).
Statistical Analyses
Groups were compared by the Student's t-test or, in the case of multiple comparisons, by one-way ANOVA followed by appropriate post hoc tests. Kruskal–Wallis nonparametric ANOVA with post hoc Mann–Whitney U test was used for TUNEL quantifications. Greenhouse–Geisser correction was used for ERG analyses. A value of P < 0.05 was considered statistically significant.
Results
Generation of Bbs5 Congenital Knockout First Allele Line
The BBSome is a stable protein complex whose role has been evaluated in numerous ciliated cell types.1,2 Whether there are differential roles for the BBSome components among ciliated cell types is currently unknown. To assess the role of BBS5 in photoreceptors, we generated a congenital knockout first allele mouse line (Bbs5–/–) (Fig. 1A). Sequencing of cDNA obtained from retinal extract of Bbs5–/– mice indicates that the allele contains a cryptic splice site in the engineered exon, which skips the LacZ cassette and resplices back into the fourth exon without maintaining the reading frame. This prevents the use of LacZ to evaluate Bbs5 expression. The novel splicing results in a frameshift and premature stop codon. Embryos maintained on a mixed background were found to be viable and were aged to 2, 3, or 9 months old. BBS5 protein expression levels were examined through quantitative western blot analyses using retinal extract and revealed significant decreased expression in Bbs5–/– samples compared to controls (Figs. 1B–1D).
Figure 1.
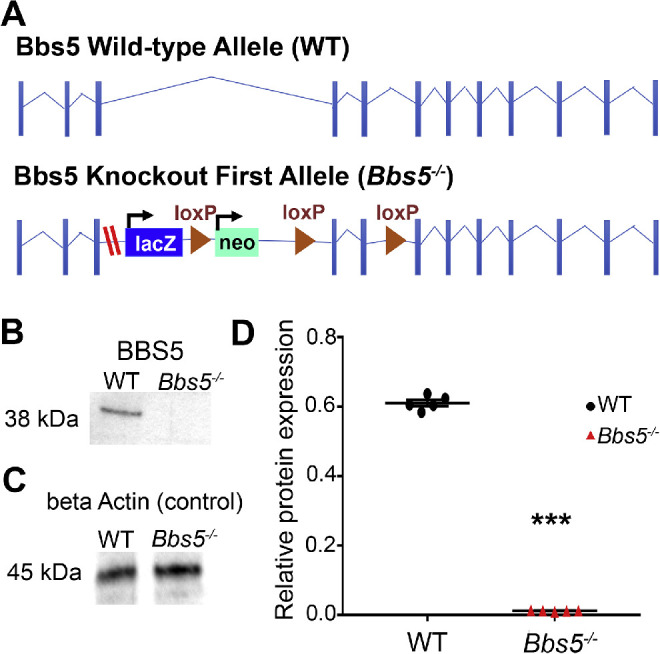
Generation of Bbs5 congenital knockout first allele line. (A) Allele schematics: WT and Bbs5 knockout first allele (Bbs5–/–; red dashes indicate cryptic splice site). (B, C) Quantitative western blot analyses show significantly reduced expression of BBS5 from Bbs5–/– retinal extract compared to WT. Loading control probing for β-actin was used to normalize band intensity. (D) Statistical analyses of protein level: BBS5 was significantly reduced (P < 0.05) based on Student's t-test (n = 5 per group; mean ± SEM; ***P < 0.05).
Congenital Absence of BBS5 Results in Retinal Degeneration
To investigate the role of BBS5 in photoreceptors, Bbs5–/– mice were analyzed at 2- and 9-months-old time points. At 2 months, Bbs5–/– retinas underwent a slight reduction in the number of nuclei in the ONL with minimal cell death (Figs. 2A, 2C–2E). In contrast, 9-month-old Bbs5–/– retinas had a significant reduction in nuclei in the ONL accompanied with a significant increase of cell death (Figs. 2D–2F).
Figure 2.
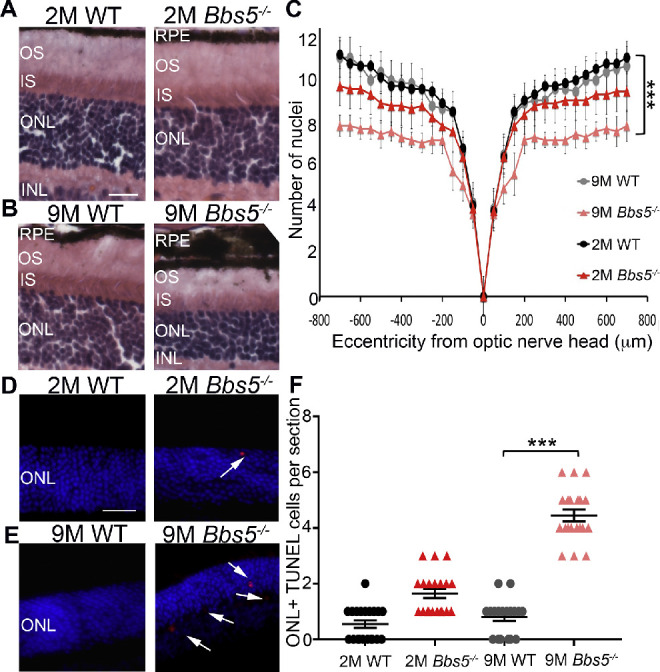
Congenital absence of BBS5 results in retinal degeneration. (A, B) H&E staining of retina sections from WT and Bbs5–/– 2-month-old mice (2M) and 9-month-old mice (9M). Retina layers are indicated as follows: retinal pigment epithelium (RPE), outer segment (OS), inner segment (IS), outer nuclear layer (ONL), and inner nuclear layer (INL). Scale bar: 50 µm. (C) Morphometric analysis of nuclei counts at different distances from the optic nerve head using Student's t-test revealed only slight retinal degeneration in 2M Bbs5–/– compared to statistically significant retinal degeneration in 9M Bbs5–/–. (D, E) TUNEL staining images for apoptosis (red) in 2M and 9M retinas. DAPI stained nuclei are blue. Scale bar: 50 µm. (F) Graph of TUNEL quantification in 2M and 9M retinas. Kruskal–Wallis nonparametric ANOVA with post hoc Mann–Whitney comparisons yielded ***P < 0.05 (n = 6 per group; mean ± SEM).
BBS5 Plays a Role in the Proper Trafficking of Specific Retinal Proteins
One of the proposed functions of the BBSome is mediating vesicular trafficking to and from the ciliary membrane. To evaluate whether the absence of BBS5 disrupted normal regulation of phototransduction protein transport, we analyzed the localization of several rod- and cone-specific phototransduction components in 2-month-old (2M) WT and Bbs5–/–. In both light- and dark-adapted mice, rhodopsin and rod transducin localize normally to the outer segment (OS) and inner segment (IS), respectively, in 2M Bbs5–/– mice (Figs. 3A, 3B). In dark conditions, arrestin-1 is restricted from the OS and properly localizes to the IS and ONL of both WT and Bbs5−/− mice. However, the absence of BBS5 did have a noticeable effect on arrestin-1 localization in the response to light (Fig. 3C). In WT retinas, light induces the near complete transfer of arrestin-1 into the OS. Although arrestin-1 does accumulate in the OS of Bbs5–/– mice following light adaptation, there are significant remnants of arrestin-1 in the IS and ONL and at the synapse that is not observed in WT retinas.
Figure 3.
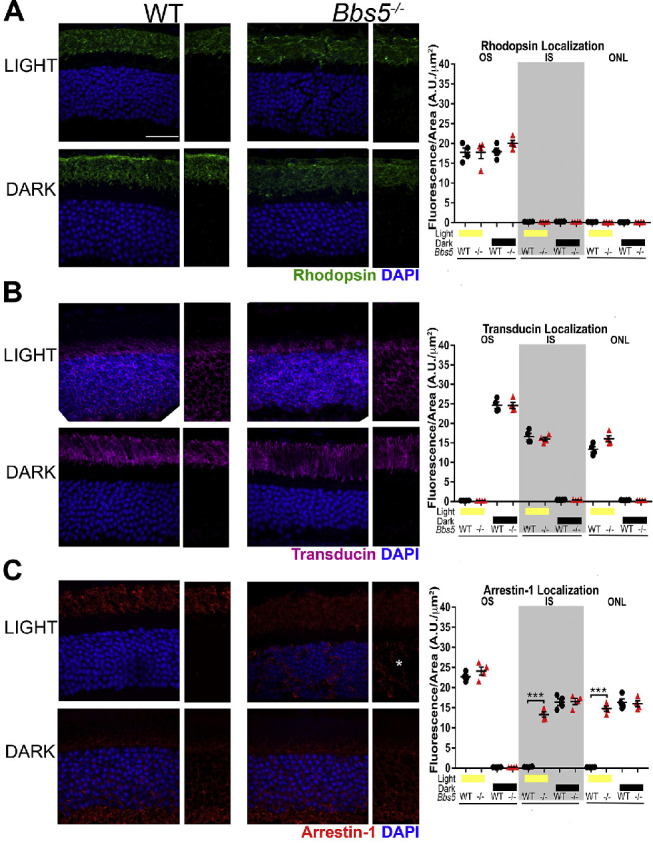
Analysis of rod cell phototransduction protein localization in Bbs5–/– retinas. (A) Rhodopsin staining (green) in both light- and dark-adapted conditions reveals normal localization to the outer segment in 2M Bbs5–/– and WT mice. (B) Transducin staining (magenta) is similar between Bbs5–/– and WT controls in both the light and dark conditions. (C) Arrestin-1 staining (red) is present in the outer segment and is mislocalized to the inner segment and outer nuclear layer in the light-adapted Bbs5–/– retinas (asterisk) but has normal localization in dark adapted conditions. One-way ANOVA was used for quantification of the average fluorescence intensity distribution, as indicated for each protein in the graphs (***P < 0.05). DAPI-stained nuclei are blue. *Areas of mislocalization. Scale bar: 25 µm.
In light-adapted animals, we found abnormal localization of cone phototransduction components, such as arrestin-4, M- and S-opsin, cone transducin (GNAT2), and the cone photoreceptor CNGA3 in Bbs5–/– mice (Figs. 4A–4E). We found that cone M- and S-opsin expression was not confined to the cone outer segments but was also localized to the ONL and synapse (Figs. 4B, 4C). However, we did observe proper localization of peripherin-2, which is expressed in both rods and cones, which suggests that its trafficking is not mediated by BBS5 (Fig. 4F).
Figure 4.
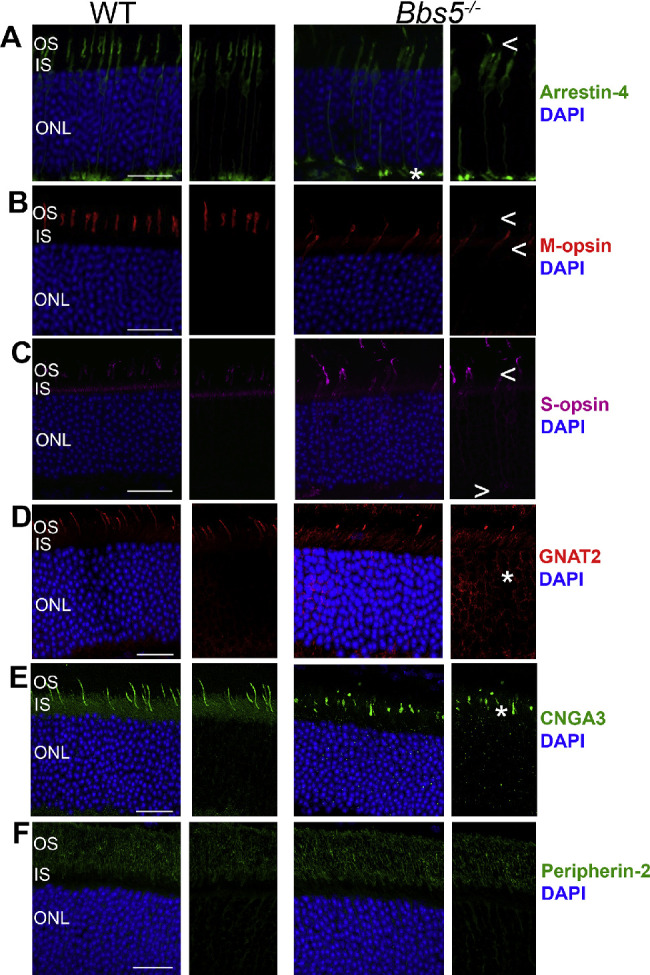
Bbs5–/– retinas have aberrant cone phototransduction component localization but have proper peripherin-2 trafficking. (A) Arrestin-4 staining (green) revealed abnormal localization in 2M Bbs5–/–, with minimal present in the outer segments and increased localization at the synapse compared to WT mice. (B, C) M-opsin (red) and S-opsin staining (magenta) is present in the outer segment and is mislocalized to the inner segment and outer nuclear layer in Bbs5–/– retinas. (D) Cone transducin (GNAT2) staining (magenta) is dissimilar between Bbs5–/– and WT. (E) CNGA3 staining (green) appears to have abnormal localization. (F) Peripherin-2 (green) labeling shows normal localization in Bbs5–/– as in WT. All animals were light adapted. DAPI-stained nuclei are blue. *Mislocalization. Arrows (< and >) indicate greater absence of protein staining. Scale bar: 25 µm.
BBS5 Is Needed for Optimal Rod Cell Response But Also Proves Vital for Cone Photoreceptor Function
To assess rod and cone photoreceptor function in the absence of BBS5, ERG was conducted in 2M WT and Bbs5–/– mice. Scotopic ERG responses revealed a decrease in a- and b-wave amplitudes, although latencies were unaffected (Figs. 5A, 5B, 5D). However, photopic ERG responses were flatlined, resulting in a complete loss of signal compared to age-matched WT animals (Figs. 5C, 5D). This indicates that BBS5 is needed for optimal rod photoreceptor function and is vital for cone photoreceptor function.
Figure 5.
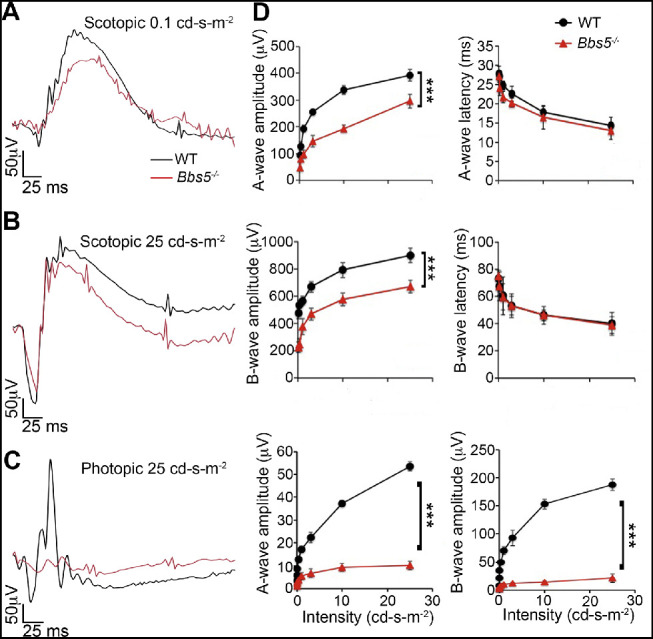
BBS5 is needed for optimal rod cell response and is vital for cone photoreceptor function. (A, B, C) Overlaid representative scotopic and photopic ERG traces at the lowest and highest flash intensities (0.1 and 25 cd·s/m2) from dark-adapted 2-month-old WT (black) and Bbs5–/– (red) animals. (D) Summary data averaged across WT (black circles) and Bbs5–/– (red triangles). The a-wave amplitude was measured from baseline to trough, and the b-wave amplitude was measured from baseline to peak at each flash intensity. Latencies represent time to peak/trough. Bbs5–/– scotopic a- and b-wave amplitudes were significantly decreased compared to WT, but latencies were comparable to those of WT. Bbs5–/– photopic responses were flatlined compared to WT controls (n = 6 per group; mean ± SD; ***P < 0.05, repeated-measures ANOVA).
Absence of BBS5 Results in Abnormal Outer Segment Orientation
Transmission electron microscopy revealed altered disk orientation in 3-month-old Bbs5–/– retinas (Figs. 6A–6D). It was found that some photoreceptor OS appeared completely disorganized, with perpendicularly oriented disk membranes running along the sides of the OS and parallel disks on the inside (Fig. 6B). Aberrant OS disk membranes were also observed apically localized in OS disk membranes in Bbs5–/– mice (Fig. 6D), which was not found in WT mice (Fig. 6C).
Figure 6.
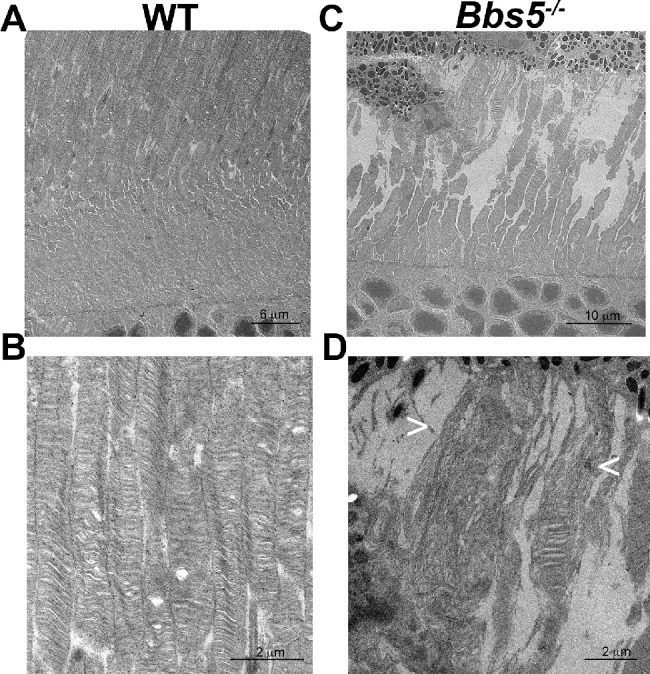
Bbs5–/– mice have aberrant outer segment disk membranes. Sagittal sectioned retinas from 3-month-old WT (A, B) and Bbs5–/– (C, D) mice show aberrant outer segment membranes. In WT mice, disks are oriented parallel, whereas in Bbs5–/– mice not only are the disks oriented parallel but there is the appearance of perpendicularly oriented disks and membranous churns (indicated by arrows, < and >).
Discussion
Bardet–Biedl syndrome is a devastating ciliopathic syndrome, with a majority of affected patients displaying a retinal dystrophy. The cause and underlying molecular mechanisms still remain unknown. Through the use of a congenital BBS5 knockout mouse model, Bbs5–/–, we have been able to provide some insight into the role BBS5 plays within photoreceptors. We found that Bbs5–/– mice developed retinal degeneration, with minimal loss of nuclei in the ONL at 2 months, but hematoxylin and eosin (H&E) staining showed overall normal retinal morphology. Aging mice out to 9 months showed a significant loss of nuclei in the ONL that was accompanied by an increased number of apoptotic nuclei as revealed by TUNEL staining. These data concur with the previously reported studies in other BBS mutant animal models that photoreceptor cilia formation was undisrupted; however, photoreceptor maintenance was affected.15,21,36
One of the proposed mechanisms of the BBSome is governing vesicular protein trafficking to and from the cilium, thus playing a major role in retrograde trafficking. To evaluate whether the retinal phenotypes are related with distorted regulation of phototransduction protein transport, we analyzed the localization of rod- and cone-specific proteins. We found a light-dependent mislocalization of arrestin-1 and abnormal staining of cone phototransduction components: arrestin-4 and M- and S-opsin, as well as GNAT2 and CNGA3. We also observed normal localization of peripherin-2 in Bbs5–/– mice. ERG revealed complete loss of cone photoreceptor response with decreased scotopic a- and b-wave amplitudes.27 In arrestin-1:arrestin-4 double knockout mice, ERGs showed a complete loss of photopic response, revealing that both arrestin-1 and arrestin-4 are necessary for cone survival.37–39 Absence of arrestin-1, arrestin-4, and CNG channel subunits have been shown to elicit cone opsin mistrafficking, resulting in opsin degradation.40–42 Likewise, several retinal dystrophies are elicited by mistrafficking of cone phototransduction components.43–45 Adding to this list, our data show that BBS5 plays an important role in specific cone protein trafficking or sorting.
Our data and those of others support the suggestion that the BBSome and its components are involved in outer segment maintenance.17,46 In order to analyze OS ultra-structure, we performed TEM and observed that Bbs5–/– photoreceptors had abnormal disk orientation. This has also been observed in other BBS knockout mice and has been portrayed as a trait of abnormal OS morphogenesis.46,47 Due to the role of the BBSome in other ciliated cells in both recruitment and cargo trafficking toward the ciliary base, it is possible that BBS5 plays a role in the trafficking process of photoreceptor disk organization and orientation.1,17,48 In a study assessing photoreceptor disk membrane formation, it was found that retrograde intraciliary trafficking of opsin during the maintenance of cone-shaped photoreceptor OSs occurred, but peripherin-2 was not retrogradely trafficked.49 It has been proposed in other ciliated cell types that the BBSome is involved in retrograde trafficking of proteins from the cilia.50–52 BBS5 could potentially play a pivotal role in the retrograde trafficking of specific cone phototransduction components that comprise cone disks, and the absence of BBS5 could explain cone-specific protein mislocalization.
Interestingly, this work supports the concept that not only do these connecting cilium compartments have specific protein trafficking roles but there are also molecular differences between rod and cone photoreceptor ciliary compartments. Our data show that cone photoreceptors are primarily affected by the absence of BBS5 with complete loss of function, as well as cone-specific protein mislocalization; rod photoreceptors were only minimally affected, with the exception of light-dependent arrestin-1 mislocalization. Perpherin-2, which is found in both rod and cone photoreceptors, has proper localization, suggesting a BBS5-independent mode of peripherin-2 trafficking. In a recent study investigating ciliary diversity, it was shown that differential regulation of transition zone and centriole proteins contributes to ciliary base diversity.53 Arguably, this potentially could be the case for rod and cone connecting cilium diversity, especially due to their distinct functional and morphological differences. Sheffield and colleagues54,55 have shown that other BBSome subunits can still integrate into the BBSome in the absence of specific BBSome components, supporting the notion that components are incorporated independently into the BBSome. Although not directly tested in these studies, further experiments using this knockout model could help uncover the BBSome assembly.
This work provides insight into the distinct role that the BBSome component BBS5 has in photoreceptors. We have shown that BBS5 is required for photoreceptor outer segment maintenance and, in particular, cone-specific photoreceptor function and protein trafficking. Our data support the conclusion that the absence of BBS5 has differential effects between rod and cone photoreceptors, revealing distinct requirements with regard to function and protein trafficking. Overall, these data address some of the functional and molecular mechanisms involved in the absence of function in the BBSome that are imperative for understanding the retinal degeneration associated with these devastating ciliopathic disorders.
Acknowledgments
The authors thank Cheryl M. Craft, PhD, and the Mary D. Allen Laboratory for Vision Research/USC ROSKI Eye Institute; Robert Molday and Xin-Qin Ding for antibodies; and Courtney J. Haycraft and Meredith G. Hubbard for helpful comments and discussions.
Supported by grants from the National Institutes of Health (R01EY019311 to AKG; R01DK065655 and R01DK115752 to BKY). Center support for the project was provided by P30DK074038, P30DK079337, P30EY003039, and P30DK079626 grants, as well as by an instrumentation grant (S10 RR026887). Services utilized in this study were provided by the Civitan International Research Center Cellular Imaging Facility and UAB Transgenic & Genetically Engineered Models (TGEMS) facility. Additional support includes NIH P30 CA13148, P30 AR048311, P30 DK074038, P30 DK05336, and P60 DK079626 awards (RAK).
Disclosure: K.L. Bales, None; M.R. Bentley, None; M.J. Croyle, None; R.A. Kesterson, None; B.K. Yoder, None; A.K. Gross, None
References
- 1. Nachury MV, Loktev AV, Zhang Q, et al.. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007; 129: 1201–1213. [DOI] [PubMed] [Google Scholar]
- 2. Klink BU, Zent E, Juneja P, Kuhlee A, Raunser S, Wittinghofer A. A recombinant BBSome core complex and how it interacts with ciliary cargo. Elife. 2017; 6: e27434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med. 2011; 364: 1533–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hurd TW, Hildebrandt F. Mechanisms of nephronophthisis and related ciliopathies. Nephron Exp Nephrol. 2011; 118: e9–e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet. 1999; 36: 437–446. [PMC free article] [PubMed] [Google Scholar]
- 6. Forsythe E, Beales PL. Bardet-Biedl syndrome. Eur J Hum Genet. 2013; 21: 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Daniels AB, Sandberg MA, Chen J, Weigel-DiFranco C, Fielding Hejtmancic J, Berson EL. Genotype-phenotype correlations in Bardet-Biedl syndrome. Arch Ophthalmol. 2012; 130: 901–907. [DOI] [PubMed] [Google Scholar]
- 8. Sheffield VC, Carmi R, Kwitek-Black A, et al.. Identification of a Bardet-Biedl syndrome locus on chromosome 3 and evaluation of an efficient approach to homozygosity mapping. Hum Mol Genet. 1994; 3: 1331–1335. [DOI] [PubMed] [Google Scholar]
- 9. Myers MH, Iannaccone A, Bidelman GM. A pilot investigation of audiovisual processing and multisensory integration in patients with inherited retinal dystrophies. BMC Ophthalmol. 2017; 17: 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Adams NA, Awadein A, Toma HS. The retinal ciliopathies. Ophthalmic Genet. 2007; 28: 113–125. [DOI] [PubMed] [Google Scholar]
- 11. Scheidecker S, Hull S, Perdomo Y, et al.. Predominantly cone-system dysfunction as rare form of retinal degeneration in patients with molecularly confirmed Bardet-Biedl syndrome. Am J Ophthalmol. 2015; 160: 364–372.e361. [DOI] [PubMed] [Google Scholar]
- 12. Tattoli F, Falconi D, Bottaro C, et al.. [Bardet-Biedl syndrome and kidney failure: a case report]. G Ital Nefrol. 2018; 35: 2018–vol1. [PubMed] [Google Scholar]
- 13. Abd-El-Barr MM, Sykoudis K, Andrabi S, et al.. Impaired photoreceptor protein transport and synaptic transmission in a mouse model of Bardet-Biedl syndrome. Vision Res. 2007; 47: 3394–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eichers ER, Abd-El-Barr MM, Paylor R, et al.. Phenotypic characterization of Bbs4 null mice reveals age-dependent penetrance and variable expressivity. Hum Genet. 2006; 120: 211–226. [DOI] [PubMed] [Google Scholar]
- 15. Nishimura DY, Fath M, Mullins RF, et al.. Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc Natl Acad Sci U S A. 2004; 101: 16588–16593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ross AJ, May-Simera H, Eichers ER, et al.. Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat Genet. 2005; 37: 1135–1140. [DOI] [PubMed] [Google Scholar]
- 17. Hsu Y, Garrison JE, Kim G, et al.. BBSome function is required for both the morphogenesis and maintenance of the photoreceptor outer segment. PLoS Genet. 2017; 13: e1007057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Datta P, Allamargot C, Hudson JS, et al.. Accumulation of non-outer segment proteins in the outer segment underlies photoreceptor degeneration in Bardet-Biedl syndrome. Proc Natl Acad Sci U S A. 2015; 112: E4400–E4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Murphy D, Singh R, Kolandaivelu S, Ramamurthy V, Stoilov P. Alternative splicing shapes the phenotype of a mutation in BBS8 to cause nonsyndromic retinitis pigmentosa. Mol Cell Biol. 2015; 35: 1860–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang Q, Nishimura D, Seo S, et al.. Bardet-Biedl syndrome 3 (Bbs3) knockout mouse model reveals common BBS-associated phenotypes and Bbs3 unique phenotypes. Proc Natl Acad Sci U S A. 2011; 108: 20678–20683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mykytyn K, Mullins RF, Andrews M, et al.. Bardet-Biedl syndrome type 4 (BBS4)-null mice implicate Bbs4 in flagella formation but not global cilia assembly. Proc Natl Acad Sci U S A. 2004; 101: 8664–8669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hodges ME, Scheumann N, Wickstead B, Langdale JA, Gull K. Reconstructing the evolutionary history of the centriole from protein components. J Cell Sci. 2010; 123: 1407–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li JB, Gerdes JM, Haycraft CJ, et al.. Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell. 2004; 117: 541–552. [DOI] [PubMed] [Google Scholar]
- 24. Jin H, White SR, Shida T, et al.. The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell. 2010; 141: 1208–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Smith TS, Spitzbarth B, Li J, et al.. Light-dependent phosphorylation of Bardet-Biedl syndrome 5 in photoreceptor cells modulates its interaction with arrestin1. Cell Mol Life Sci. 2013; 70: 4603–4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Al-Hamed MH, van Lennep C, Hynes AM, et al.. Functional modelling of a novel mutation in BBS5. Cilia. 2014; 3: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kretschmer V, Patnaik SR, Kretschmer F, Chawda MM, Hernandez-Hernandez V, May-Simera HL. Progressive characterization of visual phenotype in Bardet-Biedl syndrome mutant mice. Invest Ophthalmol Vis Sci. 2019; 60: 1132–1143. [DOI] [PubMed] [Google Scholar]
- 28. Sandoval IM, Price BA, Gross AK, et al.. Abrupt onset of mutations in a developmentally regulated gene during terminal differentiation of post-mitotic photoreceptor neurons in mice. PLoS One. 2014; 9: e108135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Molday RS, MacKenzie D. Monoclonal antibodies to rhodopsin: characterization, cross-reactivity, and application as structural probes. Biochemistry. 1983; 22: 653–660. [DOI] [PubMed] [Google Scholar]
- 30. MacKenzie D, Arendt A, Hargrave P, McDowell JH, Molday RS. Localization of binding sites for carboxyl terminal specific anti-rhodopsin monoclonal antibodies using synthetic peptides. Biochemistry. 1984; 23: 6544–6549. [DOI] [PubMed] [Google Scholar]
- 31. Gross AK, Rao VR, Oprian DD. Characterization of rhodopsin congenital night blindness mutant T94I. Biochemistry. 2003; 42: 2009–2015. [DOI] [PubMed] [Google Scholar]
- 32. Zhu X, Brown B, Li A, Mears AJ, Swaroop A, Craft CM. GRK1-dependent phosphorylation of S and M opsins and their binding to cone arrestin during cone phototransduction in the mouse retina. J Neurosci. 2003; 23: 6152–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhu X, Li A, Brown B, Weiss ER, Osawa S, Craft CM. Mouse cone arrestin expression pattern: light induced translocation in cone photoreceptors. Mol Vis. 2002; 8: 462–471. [PubMed] [Google Scholar]
- 34. Matveev AV, Quiambao AB, Browning Fitzgerald J, Ding XQ. Native cone photoreceptor cyclic nucleotide-gated channel is a heterotetrameric complex comprising both CNGA3 and CNGB3: a study using the cone-dominant retina of Nrl–/– mice. J Neurochem. 2008; 106: 2042–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Reish NJ, Maltare A, McKeown AS, et al.. The age-regulating protein klotho is vital to sustain retinal function. Invest Ophthalmol Vis Sci. 2013; 54: 6675–6685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fath MA, Mullins RF, Searby C, et al.. Mkks-null mice have a phenotype resembling Bardet-Biedl syndrome. Hum Mol Genet. 2005; 14: 1109–1118. [DOI] [PubMed] [Google Scholar]
- 37. Craft CM, Deming JD. Cone arrestin: deciphering the structure and functions of arrestin 4 in vision. Handb Exp Pharmacol. 2014; 219: 117–131. [DOI] [PubMed] [Google Scholar]
- 38. Brown BM, Ramirez T, Rife L, Craft CM. Visual Arrestin 1 contributes to cone photoreceptor survival and light adaptation. Invest Ophthalmol Vis Sci. 2010; 51: 2372–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Deming JD, Pak JS, Brown BM, et al.. Visual cone Arrestin 4 contributes to visual function and cone health. Invest Ophthalmol Vis Sci. 2015; 56: 5407–5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Deming JD, Pak JS, Shin JA, et al.. Arrestin 1 and cone Arrestin 4 have unique roles in visual function in an all-cone mouse retina. Invest Ophthalmol Vis Sci. 2015; 56: 7618–7628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xu J, Morris L, Fliesler SJ, Sherry DM, Ding XQ. Early-onset, slow progression of cone photoreceptor dysfunction and degeneration in CNG channel subunit CNGB3 deficiency. Invest Ophthalmol Vis Sci. 2011; 52: 3557–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xu J, Morris LM, Michalakis S, et al.. CNGA3 deficiency affects cone synaptic terminal structure and function and leads to secondary rod dysfunction and degeneration. Invest Ophthalmol Vis Sci. 2012; 53: 1117–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li L, Khan N, Hurd T, et al.. Ablation of the X-linked retinitis pigmentosa 2 (Rp2) gene in mice results in opsin mislocalization and photoreceptor degeneration. Invest Ophthalmol Vis Sci. 2013; 54: 4503–4511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang T, Zhang N, Baehr W, Fu Y. Cone opsin determines the time course of cone photoreceptor degeneration in Leber congenital amaurosis. Proc Natl Acad Sci USA. 2011; 108: 8879–8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McClements M, Davies WI, Michaelides M, et al.. X-linked cone dystrophy and colour vision deficiency arising from a missense mutation in a hybrid L/M cone opsin gene. Vision Res. 2013; 80: 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Swiderski RE, Nishimura DY, Mullins RF, et al.. Gene expression analysis of photoreceptor cell loss in Bbs4-knockout mice reveals an early stress gene response and photoreceptor cell damage. Invest Ophthalmol Vis Sci. 2007; 48: 3329–3340. [DOI] [PubMed] [Google Scholar]
- 47. Gilliam JC, Chang JT, Sandoval IM, et al.. Three-dimensional architecture of the rod sensory cilium and its disruption in retinal neurodegeneration. Cell. 2012; 151: 1029–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kim JC, Badano JL, Sibold S, et al.. The Bardet-Biedl protein BBS4 targets cargo to the pericentriolar region and is required for microtubule anchoring and cell cycle progression. Nat Genet. 2004; 36: 462–470. [DOI] [PubMed] [Google Scholar]
- 49. Tian G, Lodowski KH, Lee R, Imanishi Y. Retrograde intraciliary trafficking of opsin during the maintenance of cone-shaped photoreceptor outer segments of Xenopus laevis. J Comp Neurol. 2014; 522: 3577–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nozaki S, Katoh Y, Kobayashi T, Nakayama K. BBS1 is involved in retrograde trafficking of ciliary GPCRs in the context of the BBSome complex. PLoS One. 2018; 13: e0195005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nachury MV. The molecular machines that traffic signaling receptors into and out of cilia. Curr Opin Cell Biol. 2018; 51: 124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wei Q, Zhang Y, Li Y, Zhang Q, Ling K, Hu J. The BBSome controls IFT assembly and turnaround in cilia. Nat Cell Biol. 2012; 14: 950–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jana SC, Mendonca S, Machado P, et al.. Differential regulation of transition zone and centriole proteins contributes to ciliary base diversity. Nat Cell Biol. 2018; 20: 928–941. [DOI] [PubMed] [Google Scholar]
- 54. Seo S, Zhang Q, Bugge K, et al.. A novel protein LZTFL1 regulates ciliary trafficking of the BBSome and Smoothened. PLoS Genet. 2011; 7: e1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhang Q, Yu D, Seo S, Stone EM, Sheffield VC. Intrinsic protein-protein interaction-mediated and chaperonin-assisted sequential assembly of stable Bardet-Biedl syndrome protein complex, the BBSome. J Biol Chem. 2012; 287: 20625–20635. [DOI] [PMC free article] [PubMed] [Google Scholar]