

Abstract
Purpose
To determine the effect of mycophenolate mofetil (MMF) on retinal degeneration on two mouse models of retinitis pigmentosa.
Methods
Intraperitoneal injections of MMF were administered daily in rd10 and c57 mice starting at postoperative day 12 (P12) and rd1 mice starting at P8. The effect of MMF was assessed with optical coherence tomography, immunohistochemistry, electroretinography, and OptoMotry. Whole retinal cyclic guanosine monophosphate (cGMP) and mycophenolic acid levels were quantified with mass spectrometry. Photoreceptor cGMP cytotoxicity was evaluated with cell counts of cGMP immunostaining.
Results
MMF treatment significantly delays the onset of retinal degeneration and cGMP-dependent photoreceptor cytotoxicity in rd10 and rd1 mice, albeit a more modest effect in the latter. In rd10 mice, treatment with MMF showed robust preservation of the photoreceptors up to P22 with associated suppression of cGMP immunostaining and microglial activation; The neuroprotective effect diminished after P22, but outer retinal thickness was still significantly thicker by P35 and OptoMotry response was significantly better up to P60. Whereas cGMP immunostaining of the photoreceptors were present in rd10 and rd1 mice, hyperphysiological whole retinal cGMP levels were observed only in rd1 mice.
Conclusions
Early treatment with MMF confers potent neuroprotection in two animal models of RP by suppressing the cGMP-dependent common pathway for photoreceptor cell death. The neuroprotective effect of MMF on cGMP-dependent cytotoxicity occurs independently of the presence of hyperphysiological whole retinal cGMP levels. Thus our data suggest that MMF may be an important new class of neuroprotective agent that could be useful in the treatment of patients with RP.
Keywords: cGMP, retinitis pigmentosa, mycophenolate, rd10, rd1
Retinitis pigmentosa (RP), a genetically heterogeneous group of inherited retinal dystrophies, is one of the leading causes of visual disability in the developed world with an incidence of one in 4000.1 Mutations in numerous genes have been associated with RP, which typically leads to progressive degeneration of rod photoreceptors, followed by gradual loss of cone photoreceptors and subsequent atrophy of the retina and retinal pigment epithelium (RPE). Early symptoms include night-blindness and peripheral vision loss, which progress to severe tunnel vision and profound loss of visual acuity in advanced disease.
There is currently only one treatment approved by the Food and Drug Administration (FDA) for RP in the form of gene augmentation therapy for patients with biallelic mutations in the RPE65 gene; however, there are no treatments for RP caused by mutations in any of the other genes. The other two main strategies for developing treatments for RP include cell-based therapy and small molecule neuroprotection, both of which have the potential advantage of being broadly applicable among patients with RP regardless of the genetic mutation. However, whereas cell-based therapies aim to replace photoreceptors or RPE in advanced disease, neuroprotection would also benefit patients early in the disease course by slowing the rate of degeneration. Thus neuroprotection remains an important strategy that is complementary and synergistic to gene and cell-based approaches.
The development of novel neuroprotective compounds requires a better understanding of the pathways for photoreceptor degeneration. Although both apoptotic and nonapoptotic cell death pathways have been implicated in RP,2–7 neuroprotective strategies are more likely to be effective targeting upstream events and triggers of photoreceptor dysfunction. It has long been known that whole retinal cyclic guanosine monophosphate (cGMP) concentrations are abnormally elevated in the retina of rd1 mice and correlates with a decline in expression of phosphodiesterase 6 (PDE6) because of a nonsense mutation in the Pde6b gene.8,9 The rd10 mouse is an even more popular model of RP, which possesses a missense mutation in the same gene and has a less-severe attenuation of PDE6 expression.10,11 However, to our knowledge, there have been no publications on the whole retinal levels of cGMP in rd10 mice.
In the rod photoreceptor, PDE6 is the primary enzyme that hydrolyzes cGMP and regulates the activity of cGMP-gated Na+ and Ca+2 channels. Indeed, the abnormal presence of cGMP immunolabeling was shown to be specific to the photoreceptors on immunohistochemistry (IHC) in both rd1 and rd10 mice.4,10,12 Paquet-Durand and colleagues13 have shown that this anti-cGMP antibody signal is an intracellular biomarker for cGMP-dependent cytotoxicity and an alternative pathway for photoreceptor cell death.4,14–18 Interestingly, cGMP immunolabeling on IHC is also seen in other rodent models with mutations in other genes not directly related to cGMP hydrolysis, namely those associated with RP (Prph2, Cngb1, Rho), Leber congenital amaurosis (Aipl1), and achromatopsia (Cnga3).4,19 This indicates that cGMP-dependent photoreceptor toxicity may be a common phenomenon that occurs independently of mutations affecting cGMP metabolism and whole retinal cGMP levels. Together, these studies show that cGMP dysregulation may be a common mechanism of photoreceptor cell death in up to 30% of RP and Leber congenital amaurosis.15
There have been myriad promising neuroprotection studies in rd1 and rd10 mice11,17,18,20–69; however, only one directly targets cGMP cytotoxicity.63 Here, we present another promising drug candidate that could potentially mitigate cGMP-dependent photoreceptor cell death. Mycophenolate mofetil (MMF) is a prodrug of mycophenolic acid (MPA), which potently suppresses de novo guanine nucleotide production by reversibly inhibiting inosine monophosphate dehydrogenase (IMPDH).70 Isoforms of IMPDH are strongly expressed by photoreceptors,71–73 which makes MMF a potentially ideal neuroprotective agent that specifically targets the degenerating cells in RP. Moreover, MMF has been shown to be neuroprotective after excitatory injury, inhibit microglia activation, inhibit leukocytes, and act as an antioxidant.74–78 In addition, MMF is an FDA-approved medication for organ transplantation and is commonly used off-label as an immunomodulatory medication for the treatment of ocular inflammation.79 Here, we present evidence that MMF is also a neuroprotective agent that potently inhibits retinal degeneration in rd10 and rd1 mice.
Methods
Animals
All animal care and tissue collection were performed with the approval of Institutional Animal Care and Use Committee at Oregon Health & Science University and conformed to United States Department of Agriculture standards and the Association for Research in Vision and Ophthalmology statement for the use of animals in ophthalmic and vision research. Breeding mice of the following genotypes were obtained from The Jackson Laboratory (Bar Harbor, ME, USA); wild-type C57BL/6J (c57), Pde6brd10/rd10(rd10), and Pde6brd1/rd1(C3H/HeOuJ or rd1). A DNeasy Blood & Tissue Kit from Qiagen (Hilden, Germany) was used to isolate gDNA from rd10 mice and PCR (Applied Biosystems, Coster City, CA, USA) was used to confirm the coding sequence containing the missense mutation in Pde6b according to manufacturer's instructions. Mice were housed in standard conditions under a 12-hour light-dark cycle. Mycophenolate Mofetil (CellCept; Genentech, San Francisco, CA, USA) was dissolved in 5% dextrose (Baxter, Deerfield, IL, USA). Mice were given daily intraperitoneal injections of MMF 25 to 100 mg/kg or vehicle (5% dextrose) up to postnatal day (P) 60. Pilot studies using MMF up to 200 mg/kg was not tolerated by the animals because of diarrhea, so 100 mg/kg was chosen as the maximum dose. To limit exposure to the breeding pair to the drug, rd10 mice receiving the first injection at P12 were weaned at P13, and mice receiving the first injection at P13 to P14 were weaned the same day. Retinal tissue and plasma were harvested 24 hours after the last dose in animals receiving injections.
Optical Coherence Tomography Imaging
In vivo retinal imaging was performed throughout the experiment using spectral domain optical coherence tomography (SD-OCT) (Envisu R2200-HR SD-OCT; Bioptigen, Durham, NC, USA) as previously reported.80 Mice were sedated using 1.5% isoflurane, and sedation was maintained through a nose cone. Corneas were anesthetized with proparacaine, and pupils were dilated with 1% tropicamide and 2.5% phenylephrine. Horizontal (nasal and temporal) and vertical (superior and inferior) linear scans were obtained. SD-OCT scans were exported from InVivoVue software as AVI files then loaded into ImageJ (version 2.0; National Institutes of Health, Bethesda, MD, USA) and averaged as a z-stack using a custom plug-in. A custom segmentation program in IGOR Pro (version 6.37; WaveMetrics Inc., Lake Oswego, OR, USA) was used to quantify the nasal and temporal outer retinal thickness. Outer retinal thickness segmentation included both the photoreceptor and retinal pigment epithelial layer (receptor plus, REC+) and is measured from Bruch's membrane to the inner nuclear layer/outer plexiform layer junction (Fig. 1B).
Figure 1.
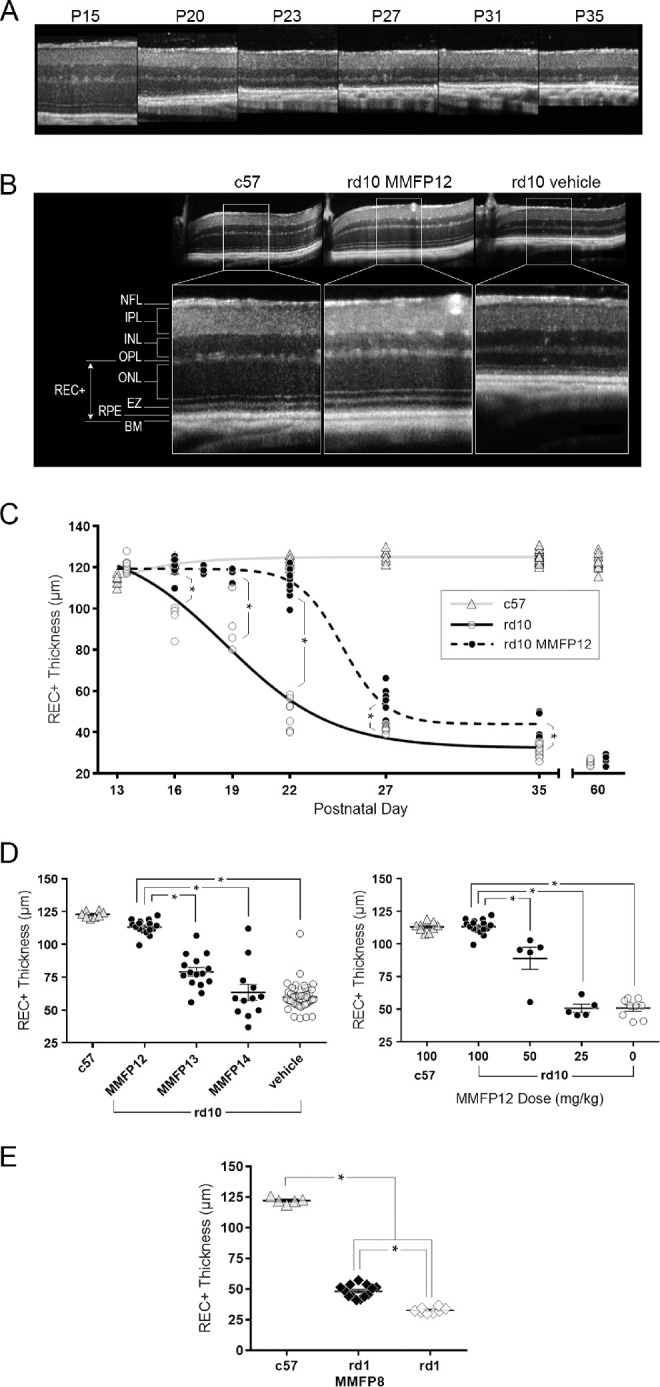
Effect of MMF on the outer retina in rd10 and rd1 mice. (A) OCT images of naïve rd10 mice degeneration over time from P15 to P35, which shows considerable loss in the outer retina by P23. (B) Representative OCT images of the retina at P22 showing that rd10 mice treated with MMF 100 mg/kg starting at P12 (MMFP12) had similar outer retinal structure to c57 mice, as compared with the degeneration seen in rd10 mice treated with vehicle. (C) REC+ thickness from rd10 mice treated with MMFP12 were significantly greater than naïve rd10 mice up to P35 even though the benefit declined after P22: P16 (n = 8 and 4, P < 0.0001), P19 (n = 6 and 4, P <0.0001), P22 (n = 16 and 9, P < 0.0001), P27 (n = 8 and 7, P = 0.0021), P35 (n = 4 and 12, P = 0.0029), and P60 (n = 8 and 8, P > 0.99). The REC+ thickness in c57 mice was stable across time. (D) MMF treatment of rd10 mice starting at P12 (n = 16) produced greater REC+ protection by P22 as compared with initiating treatment at P13 (MMFP13, n = 15, P < 0.0001) or P14 (MMFP14, n = 12, P < 0.0001) as well as vehicle-treated rd10 mice (n = 49, P < 0.0001). The dose response of MMFP12 treatment in rd10 mice showed that 100 mg/kg (n = 16) produced significantly better efficacy compared with 50 mg/kg (n = 5, P < 0.0001), 25 mg/kg (n = 5, P < 0.0001), and no treatment (n = 9, P < 0.0001). (E) The rd1 mice treated with 100 mg/kg MMF initiated at P8 (MMFP8) had significantly thicker REC+ than naïve rd1 mice at P15 (n = 13 and 8, P < 0.0001), albeit the effect was more modest than in rd10 mice. INL, inner nuclear layer; EZ, ellipsoid zone; BM, Bruch's membrane; *P ≤ 0.05.
Electroretinograms
Electroretinograms (ERGs) were recorded the day after SD-OCT imaging. Before ERG recording, mice were dark adapted for 40 minutes. The mice were then anesthetized under dim red light with an intraperitoneal injection of ketamine (100 mg/kg) and xylazine (10 mg/kg). The cornea was anesthetized with administration of proparacaine, and then pupils were dialed with application of 1% tropicamide and 2.5% phenylephrine. Anesthetized mice were placed on a heated platform (37°C) inside of a Ganzfeld dome coated with highly reflective white paint. Flash ERGs were recorded from platinum electrodes placed on the cornea, and eyes were lubricated with 2.5% hypromellose ophthalmic lubricant (Goniovisc; Dynamic Diagnostics, Westland, MI, USA). Platinum references and ground electrodes were placed subcutaneously in the forehead and tail. The signal was amplified with a preamplifier (Model CP511; Grass Instruments, West Warwick, RI, USA). ERG traces were averaged and then analyzed using custom software to export a- and b-wave amplitudes. Mice were than light-adapted for 10 minutes before eliciting photopic cone-dependent responses to white light, UV (365 nm), or green (500 nm) stimuli.
Immunohistochemistry
Mice were dark adapted for 40 minutes, and tissue collection was performed under dim red light. Mice were euthanized through intraperitoneal injections of xylazine/ketamine overdose. Fine-tipped permanent marker was used to mark the superior 12 o'clock position, eyes were enucleated and fixed in 4% paraformaldehyde (PFA) on ice for 5 minutes, then transferred to phosphate-buffered saline solution (PBS). Eyes were dissected under normal light conditions in PBS to remove the cornea and lens. For cross-sectional IHC, the eye-cups were fixed in 4% PFA for one hour, cryopreserved with sucrose, and frozen in OCT Compound (Tissue-Tek; Sakura Finetek, Torrance, CA, USA). Cups were cryosectioned horizontally at 12 µm thickness and stored at −20°C on glass slides. Tissue sections were rehydrated and permeabilized using primary incubation buffer (5% horse serum, 0.3% Triton X, 0.0025% sodium azide) for one hour and then incubated with the primary antibody overnight at 4°C. Sections were washed with PBS three times for five minutes and then incubated with the secondary antibody (1:800) in secondary incubation buffer (5% horse serum and 0.0025% sodium azide) for one hour at room temperature. Slides were washed with PBS three times, stained with DAPI (1:12,000) for 30 seconds, and then washed with PBS twice. For whole retinal flat mount IHC, the eye-cups were fixed in 4% PFA overnight at 4°C, sclera was removed, and the whole retinal cup was transferred to a 96-well plate. Whole retinas were incubated in primary antibodies for one week, washed with PBS four times for 30 minutes each, and incubated in secondary antibodies for two days. After four 30-minute washes, whole retinas were stained with DAPI overnight at 4°C and then washed two times for 10 minutes each. Whole retinas were cut into a cloverleaf pattern and mounted flat on a slide. The following primary antibodies were used: cGMP 1:500 (Prof. Harry Steinbush, Maastricht University, Maastricht, the Netherlands),4,81 visual arrestin 1:50 (E-3; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), cone arrestin 1:30,000 (AB15282, Millipore, Temecula, CA, USA), CD11b 1:500-1000 (Serotec/Bio-Rad, Hercules, CA, USA), GFAP 1:50,000 (G9269; Sigma, St. Louis, MO, USA), and IBA1 1:500 (Novus Biologics, Centennial, CO, USA). Cone arrestin dilution was adjusted to 1:1000 for whole retinal flat mounts.
Microscopy and Cell Counting
For cross-sectional IHC, microscopy was performed on an Olympus FV1000 Laser Scanning Confocal Microscope (Olympus, Tokyo, Japan). Depth scan images were captured in Fluoview software using a ×10 or ×40 objective. Images were obtained only from optic nerve-containing tissue sections to ensure the horizontal location was consistent across the study. Three tissue sections were imaged nasally and temporally for one eye from each mouse. Images were either z-stacked in Fluoview or in ImageJ (version 2.0; National Institutes of Health). ImageJ was used to count the number of DAPI-stained cell rows of the outer nuclear layer (ONL) and cGMP-stained ONL cell bodies. The number of ONL rows was counted within a ×40 field at 10°, 30°, 50°, and 70° from the optic disc. The number of cGMP-positive cells in the ONL were counted within a ×10 field and normalized to the ONL area. The ONL row count and cGMP count for each mouse eye was an average of three tissue sections.
For whole retinal flat mount IHC, microscopy was performed on a Leica TCS SP8 X laser confocal microscope (Leica Microsystems, Buffalo Grove, IL, USA) using a ×20 objective. Imaris (Zurich, Switzerland) 3-D cell counting software was optimized to automatically quantify the number of cones by detecting the apical cell bodies. ImageJ z-stack was used to visualize microglia within the photoreceptor layer, outer plexiform layer (OPL), inner plexiform layer (IPL), and the layers composed of the vitreoretinal interface (VRI), nerve fiber layer (NFL), and ganglion cell layer (GCL). Microglia counts from each image were an average of two masked graders. The total count for each eye was an average of the four retinal quadrants.
OptoMotry
Visuospatial sensitivity of mice was assessed as cycles per degree through observing the optokinetic head-tracking (OKT) response to vertical sine wave grating (OptoMotry, Cerebral Mechanics)82,83 with 100% contrast and the brightest light condition (62 cd m−2). The observer was masked to the grating frequency. The minimum observation time was five minutes before determining that a mouse had no measureable OKT response. Both eyes were assessed separately and then averaged for analysis.
Mass Spectrometry
Mycophenolate is a prodrug of mycophenolic acid (MPA), which is the active metabolite. Thus the concentration of MPA was measured through liquid chromatography-tandem mass spectrometry (LC-MS). Whole blood was collected from rd10 and c57 mice at the ocular or saphenous vein using ethylenediamine tetra-acetic acid–treated microvette collection tubes (Starstedt Inc., Nümbrecht, Germany). Plasma was isolated through 10 minutes’ centrifugation at 2000 g. An MPA (Sigma-Aldrich, St. Louis, MO, USA) standard curve was prepared using c57 mouse plasma (Innovative, Novi, MI, USA) in methanol solution. A 3-µm particle size, 100 mm × 2.1 mm Hypersil GOLD LC column (Thermo, Waltham, MA, USA) was used. Samples were analyzed using a 5500 QTRAP hybrid/triple quadrupole linear ion trap mass spectrometer (AB SCIEX, Framingham, MA, USA) with electrospray ionization (ESI) in positive mode. The mass spectrometer was interfaced to a SIL-20AC XR auto-sampler followed by 2 LC-20AD XR LC pumps (Shimadzu, Columbia, MD, USA). The instrument was operated with the following source settings: 3500 V, source gas 1 (GS1) 30 psi, source gas 2 (GS2) 35 psi, curtain gas (CUR) 10, ion source temperature (TEM) 450°C, and collision activated dissociation (CAD) gas on high.
For retinal cGMP concentration levels, mice were dark-adapted as previously discussed. After euthanization, retinas were collected under dim red light and immediately frozen in liquid nitrogen. Whole retina samples were homogenized in 0.1 mol/L HCl then spun in a centrifuge at 500 g for 10 minutes, and supernatant was preserved. A cGMP standard curve was prepared with cGMP stock (Cat. 80-0153; Enzo, Farmingdale, NY, USA) in 0.1 mol/L HCl. A 2.6 µm 150 mm × 3 mm Kinetex F5 Core-shell LC Column (Phenomenex, Torrance, CA, USA) was used. Samples were analyzed using a 4000 QTRAP hybrid/triple quadrupole linear ion trap mass spectrometer (AB Sciex) with ESI in positive mode. The mass spectrometer was interfaced as described above. The instrument was operated with the following source settings: 4500 V, GS1 40 psi, GS2 50 psi, CUR 15, TEM 550°C, and CAD gas on medium.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 7.03 (San Diego, CA, USA). A t test or one-way analysis of variance was used. For nonparametric comparisons, Mann-Whitney or Kruskal-Wallis test was used. Dunn's or Šidák correction was performed for multiple comparisons. A P value ≤ 0.05 was considered significant. To determine intergrader agreement for masked microglia counts, intraclass correlation coefficient with 95% confidence intervals (CI) was calculated.
Results
MMF Delays Outer Retinal Thinning in rd10 and rd1 Mice
In vivo imaging using OCT showed that naïve rd10 mice exhibit rapid retinal degeneration starting at P15 with the majority of outer retinal thinning occurring by P20-23 (Figs. 1A–1C). Representative OCT images of age-matched animals at P22 show severe thinning of the photoreceptor layer as measured by REC+ in rd10 mice compared to c57 mice, whereas the inner retinal thickness appears stable (Fig. 1B). The rd10 mice treated with MMF 100 mg/kg starting at P12 (MMFP12) had profound preservation of REC+ that was significant thicker than in naïve rd10 mice by 26% at P16 (119.8 ± 1.69 µm vs. 95.0 ± 3.71 µm, P < 0.0001), 28% at P19 (117.4 ± 1.08 µm vs. 91.9 ± 6.58 µm, P < 0.0001), 123% at P22 (113.2 ± 1.38 µm vs. 50.7 ± 2.35 µm, P < 0.0001), 28% at P27 (52.80 ± 3.03 µm vs. 41.39 ± 0.63 µm, P = 0.0021) and 40% at P35 (43.99 ± 3.27 µm vs. 31.55 ± 0.86 µm, P = 0.0029; Fig. 1C). However, at P60, there was no significant effect of MMFP12 in rd10 mice (27.0 ± 0.77 µm vs. 25.3 ± 0.46 µm, P > 0.99). The neuroprotective effect of MMF was the greatest at P22, and representative OCT images show normal-appearing ONL, ellipsoid zone, and RPE in MMFP12-treated rd10 mice (Fig. 1B). Furthermore, at P22, the REC+ in MMFP12-treated rd10 mice and was significantly greater than vehicle-treated rd10 mice (113.2 ± 1.38 µm vs. 59.7 ± 1.42 µm, P < 0.0001), but not significantly different than c57 mice that were naïve (122.8 ± 0.88 µm, P = 0.17) or treated with MMFP12 (113.0 ± 1.26 µm, P > 0.99; Fig. 1D). There was also no significant difference in c57 mice that were naïve versus treated with MMFP12 (P = 0.27), which suggests that MMF had no effect on normal outer retinal structure. Early weaning alone at P13 had no effect on outer retinal degeneration by P22 (P > 0.99; Supplemental Fig. S1). Postnatal day 22 was also the earliest time point at which the majority of REC+ thinning was observed, so additional studies were performed at P22 to elucidate the therapeutic window and dose-response (Fig. 1D). The effect of MMF 100 mg/kg on REC+ preservation in rd10 mice was significantly lower when treatment was initiated just 24 to 48 hours later at P13 (MMFP13, 79.0 ± 3.30 µm, P < 0.0001) or P14 (MMFP14, 63.4 ± 6.09 µm, P < 0.0001). There was a clear dose response to MMFP12 treatment, wherein lower concentrations of MMF (50 and 25 mg/kg) had progressively reduced efficacy on REC+ preservation at P22 (88.9 ± 8.52 µm, P < 0.0001, and 50.6 ±3.03 µm, P <0.0001, respectively). As further proof of concept, MMF was also used to treat rd1 mice, which undergo earlier and more rapid retinal degeneration than rd10 mice (Fig. 1E). In rd1 mice, retinal degeneration is known to begin as early as P8 with significant retinal degeneration occurring by P15. Thus treatment with MMF 100 mg/kg was initiated at P8 (MMFP8) and assessed with OCT at P15. The treated rd1 mice had significantly thicker outer retina than naïve rd1 mice at P15 (+48%, 48.24 ± 1.45 vs. 32.65 ± 0.78 µm, P < 0.0001), albeit the effect was more modest than in rd10 mice.
MMF Preserves Visual Function in rd10 Mice
In our lab the minimum age for a mouse to reliably tolerate a sedated full-field ERG was P22, thus ERG was not performed in rd1 P15 mice, but only rd10 mice at P22 and later time points. Full-field scotopic ERGs of MMFP12-treated rd10 mice showed significant preservation compared to naïve rd10 mice, but only at P22 for the a-wave of the mixed rod and cone response to maximal 3.39 log cd ∙ s/m2 stimulus (+254 µV: 354.5 ± 26.9 µV vs. 100.3 ±7.1 µV, P < 0.0001; Figs. 2A, 2B). The a-wave amplitude from MMFP12-treated rd10 tended to be lower than naïve c57 mice (432 ±26.3 µV), but it did not reach statistical significance (P = 0.07). There was no treatment effect at P35 or P60. Treatment with MMFP12 did not confer any benefit to the scotopic b-wave amplitude of rd10 mice at any age for either the rod-dependent or mixed rod and cone response to −1.68 log cd ∙ s/m2 or 3.39 log cd ∙ s/m2 stimulus, respectively. To confirm the effect of MMFP12 at P22, an expanded spectrum (−0.1 to 3.39 log cd ∙ s/m2) of scotopic stimuli was also performed, which showed progressively greater a-wave amplitudes in treated rd10 mice as the intensity of the stimuli increased (Fig. 2C). In addition to a significant increase of +254 µV at 3.39 log cd ∙ s/m2 as stated above, rd10 MMFP12 mice also had larger a-wave amplitudes than naïve rd10 mice for the following stimuli: −0.14 log cd ∙ s/m2 (+80 µV: 102.0 ± 11.1 µV vs. 22.1 ± 3.0 µV, P = 0.008), 0.41 (+130 µV: 142.0 ±12.4 µV vs. 36.8 ± 3.5 µV, P = 0.013), 1.52 (+144 µV: 221.2 ±15.1 µV vs. 76.9 ± 8.8 µV, P = 0.0001). There was no effect of MMFP12 on the b-wave amplitude for any scotopic stimuli. Additionally, there was no effect of MMFP12 on the ERG of c57 mice, which suggests that MMF is not detrimental to the development of normal retinal function.
Figure 2.

Effect of MMF on scotopic retinal function in rd10 mice. (A) Naïve c57 mice have significantly greater scotopic b-wave amplitudes than either naïve rd10 or rd10 mice treated with MMF starting at P12 (MMFP12) for each stimuli at all ages (P22: n = 14, 8, 11; P35: n = 19, 18, 6; P60: n = 16, 7, and 6, respectively). The same was true for the a-wave response to 3.39 log cd*s/m2, except at P22 where MMFP12-treated rd10 mice (n = 11) had amplitudes that were not significantly different than c57 mice (n = 14, P = 0.07), and both were significantly greater than that of naïve rd10 mice (n = 8, P < 0.0001). There was no significant effect of MMFP12 treatment on the scotopic responses to 3.39 log cd ∙ s/m2 from rd10 mice at either P35 or P60. (B) Representative scotopic waveforms at P22 from naïve or MMFP12-treated rd10 and c57 mice in response to −1.68 or 3.39 log cd ∙ s/m2 stimuli. For the 3.39 log cd ∙ s/m2 stimuli, the rd10 mice treated with MMFP12 showed larger amplitude responses that were similar in appearance to c57 mice. (C) The expanded scotopic ERG protocol (stimulus: −4.3 to 3.39 log cd ∙ s/m2) showed that rd10 mice treated with MMFP12 (n = 11) had significant preservation of the a-wave amplitude as compared with naïve rd10 mice (n = 8): −0.14 (P = 0.008), 0.41 (P = 0.013), 1.52 (P = 0.0001), and 3.39 (P < 0.0001) log cd ∙ s/m2 stimulus. The b-wave amplitude only showed a mild trend toward preservation that was not significant. There was no effect of MMFP12 treatment on the retinal function of c57 mice, which shows that MMF is not detrimental to the development of normal retinal function. cd ∙ s/m2, candela-seconds per square meter; *P ≤ 0.05.
Full-field photopic ERGs did not show any effect of MMFP12 treatment in rd10 mice at P35 and P60. Photopic ERGs were not planned at P22 because the majority of cone degeneration occurs between P35 to P60 in rd10 mice, and we felt the time under anesthesia was better spent performing an expanded scotopic protocol at P22. The b-wave amplitude in c57 mice was significantly greater than rd10 mice that were either naïve or MMFP12-treated at P35 and P60 for all photopic stimuli. The c57 a-wave amplitude was significantly greater only for the 0.82 log cd ∙ s/m2 stimulus.
To correlate the findings of ERG with visual behavior, OKT was also performed. In our hands, mice that were P22 or younger were not sufficiently cooperative to produce reliable OKT responses. Thus, OKT could not be done on rd1 P15 mice, and rd10 mice were tested only at P35 and P60. Although ERG responses showed no benefit of MMFP12 at P35 and P60, OKT responses showed significantly better thresholds in MMFP12-treated rd10 mice, as compared with naïve rd10 mice at P35 (0.25 ± 0.01 vs. 0.18 ± 0.02 cycles/degree, P = 0.001; Fig. 4A). Moreover, MMFP12-treated rd10 mice continued to show significantly better performance on OKT up to P60 when naïve rd10 mice no longer had any evidence of a response (0.07 ± 0.02 vs. 0.0 ± 0.0 cycles/degree, P < 0.0001; Fig. 4B).
Figure 4.
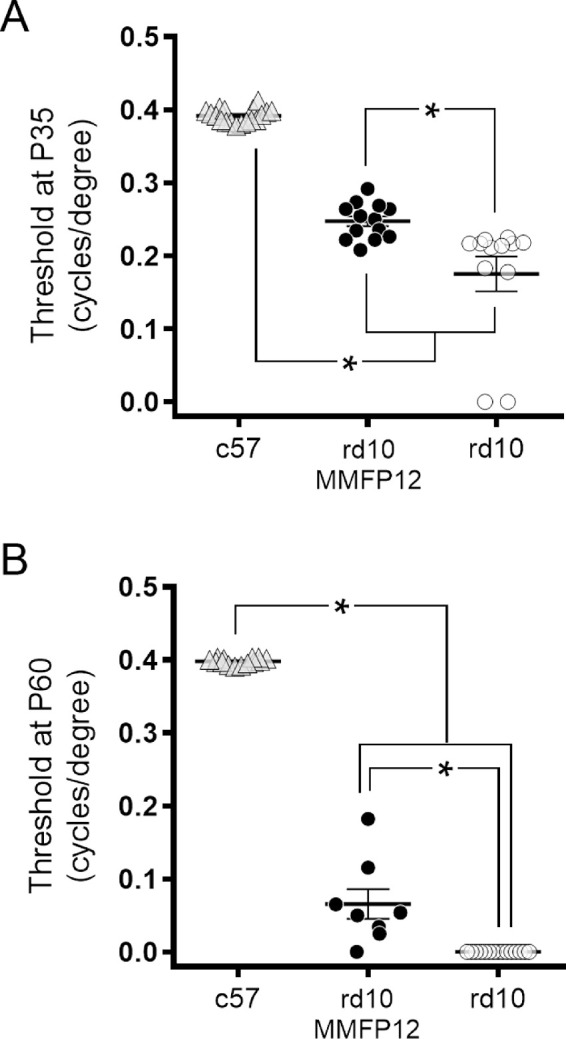
Effect of MMF on OKT Responses in rd10 Mice. (A) At P35, OKT responses showed significantly better thresholds in rd10 mice treated with MMF beginning at P12 (MMFP12; n = 13), as compared with naïve rd10 mice (n = 12, P = 0.001). (B) At P60, OKT responses in MMFP12-treated rd10 mice (n = 8) were still significantly better than naïve rd10 mice (n = 15) which no longer exhibited OKT behavior (P < 0.0001). *P ≤ 0.05.
MMF Preserves Photoreceptor Structure in rd10 Mice and rd1 Mice
Naive rd10 mice experienced a persistent decline in the number of ONL cell body rows from P13 to P19 (13.3 ± 0.4 to 8.0 ± 1.7) with a temporary trough to P20 (8.3 ± 0.4) and a subsequent rapid and severe loss by P22 (4.0 ± 0.5; Fig. 5A). In comparison, rd1 mice had an initial slower decline of ONL cell body rows from P8 to P11/12 (10.2 ± 0.2 to 9.3 ± 0.5), followed by a rapid loss by P15 (3.4 ± 0.4). Similar to the findings on OCT imaging, IHC show a robust neuroprotective effect of MMFP12 and MMFP8 treatment on photoreceptor structure in rd10 and rd1 mice, respectively. Compared with naïve rd10 mice at P22, MMFP12-treated rd10 mice had significantly greater ONL cell body rows (11.3 ± 0.5, P <0.0001) that was in the normal range and not significantly different from c57 mice (11.7 ± 0.1, P = 0.85; Figs. 5A, 5B). However, at P27 and P35, MMFP12 did not have a significant effect on rd10 mice. Compared with naïve rd1 mice at P15, MMFP8-treated rd1 mice had significantly more than double the number of ONL cell body rows (7.6 ± 0.4, P < 0.0001).
Figure 5.

Effect of MMF on photoreceptor structure at P22 in rd10 and rd1 mice. (A) Quantification of the average number of ONL cell body rows in rd10 and rd1 mice show progressive decline over P8-19 and P13-P35, respectively. Wild-type c57 mice had stable ONL cell body counts at P22 and P35. Compared with naïve animals, treatment with MMF beginning at P8 (MMFP8) in rd1 mice or P12 (MMFP12) in rd10 mice showed significant preservation of ONL cell body rows at P15 (n = 8 and 3, P < 0.0001) and P22 (n = 11 and 7, P < 0.0001), respectively. However, there was no difference between MMFP12-treated and naïve rd10 mice at P27 (n = 6 and 4, P = 0.44) or P35 (n = 3 and 3, P = 0.97). (B) IHC at P22 of MMFP12-treated rd10 mice showed normal appearance of photoreceptor structure similar to c57 naïve mice as visualized with DAPI stain and immunolabeling for visual arrestin 1:50 (red) and cone arrestin 1:30,000 (white). Naïve rd10 mice had severely thinned ONL layer and disorganized cone and rod outer segments. A spider plot of the average number of ONL cell body rows, quantified by DAPI staining, per degrees of horizontal eccentricity from the optic nerve showed that MMFP12-treated rd10 mice (n = 10) had significantly greater ONL rows than naïve rd10 mice (n = 11) at −70° (P = 0.02), −50° (P = 0.02), −30° (P = 0.05), −10° (P = 0.02), +10° (P = 0.005), +30° (P = 0.002), +50° (P = 0.02), and +70 degrees (P = 0.01). There was no difference in ONL cell body rows between MMFP12-treated rd10 mice, MMFP12-treated c57 mice (n = 5), or naïve c57 mice (n = 4). (C) Cone arrestin 1:1000 whole retinal flat mount IHC also showed that MMFP12-treated rd10 mice had normal-appearing cone tips as compared with degenerated shortened cone tips in naïve rd10 mice. The rd10 mice treated with MMFP12 (n = 4) had significantly greater cone tip counts than naïve rd10 mice (n = 4), P < 0.0001), but still less than that of c57 mice (n = 4, P = 0.03). INL, inner nuclear layer; ns, non-significant; *P ≤ 0.05.
To further evaluate the level of neuroprotection, additional structural IHC studies were performed on rd10 mice at p22. The rod and cone photoreceptor layer in MMFP12-treated rd10 mice appears to have cross-sectional morphology similar to c57 mice compared with severe photoreceptor degeneration in naïve rd10 mice, as visualized with immunolabeling for visual arrestin and cone arrestin, respectively (Fig. 5B). A spider plot of the ONL cell body rows, by degrees of horizontal eccentricity from the optic nerve, showed that the number of ONL cell body rows of in MMFP12-treated rd10 mice was significantly greater than naïve rd10 mice at all degrees measured: −70° (10.02 ± 0.9 vs. 3.72 ± 0.6, P = 0.02), −50° (10.0 ± 1.0 vs. 3.8 ± 0.5, P = 0.02), −30° (10.3 ± 0.9 vs. 3.4 ± 0.4, P = 0.05), −10° (10.7 ± 1.1 vs. 3.7 ± 0.4, P = 0.02), +10° (10.8 ± 1.3 vs. 3.9 ± 0.5, P = 0.005), +30° (11.1 ± 0.9 vs. 4.5 ± 0.6, P = 0.002), +50° (10.5 ± 0.8 vs. 5.0 ± 0.6, P = 0.02), and +70° (10.2 ± 0.8 vs. 4.5 ± 0.6, P = 0.01). There was no difference in ONL cell body rows among MMFP12-treated rd10 mice, MMFP12-treated c57 mice, and naïve c57 mice. Whole retinal flat mount IHC also showed that MMFP12-treated rd10 mice had normal appearance of cone arrestin-positive cone tip structure as compared to abnormally shortened morphology in naïve rd10 mice (Fig. 5C). The rd10 mice treated with MMFP12 also had significantly greater cone tip counts than naïve rd10 mice (3438 ± 27 vs. 2839 ± 56, P < 0.0001), but still less than that of c57 mice (3670 ± 65, P = 0.03).
Immunohistochemistry showed that CD11b-positive microglia in MMFP12-treated rd10 mice at P22 appeared to be similar to that of c57 mice, in an inactive highly-ramified state limited to the nerve fiber layer/ganglion cell layer, inner plexiform layer, and outer plexiform layer, as opposed to naïve rd10 mice at p22 where activated microglia/macrophages develop an amoeboid morphology and migrate into the outer retina and subretinal space (Fig. 6A).84 However, by P35, the microglia/macrophages in the retina of MMFP12-treated rd10 mice were similar to naïve rd10 mice, showing an activated phenotype and migrating into the ONL and subretinal space (Supplementary Fig. S2). To confirm the findings at P22, microglia IHC was also quantified en-face using whole retinal flat mounts with CD11b and Iba1 antibodies (Fig. 6B). The microglia counts between the two masked graders were highly correlated with an intraclass correlation coefficient of 0.847 (95% CI: 0.747-0.903). Within the VRI/NFL+GCL, rd10 mice treated with MMFP12 tended to have less microglia than naïve rd10 mice but was not statistically significant (45 ± 2 vs. 53 ± 3, P = 0.12). For all the other layers, MMFP12-treated rd10 mice had significantly less microglia than naïve rd10 mice: IPL (59 ± 4 vs. 73 ± 2, P = 0.02), OPL (68 ± 4 vs. 81 ± 3, P = 0.046), and photoreceptor layer (16 ± 4 vs. 91 ± 5, P < 0.0001). Representative IHC images show that the microglia within the IPL and OPL were more ramified and less densely packed in MMFP12-treated rd10 than naïve rd10 mice. The microglia appeared amoeboid within the photoreceptor layer in naïve and MMFP12-treated rd10 mice; however, they were dramatically less numerous in the latter.
Figure 6.

Effect of MMF on microglia in rd10 mice. (A) Immunostaining of CD11b 1:1000 (green) showed that microglia at P22 in rd10 mice treated with MMF beginning at P12 (MMFP12) appeared to be similar to that of c57 mice; they were in an inactive highly-ramified state within the inner retinal layers, as opposed to naïve rd10 mice where infiltrating microglia/macrophages develop an amoeboid morphology and migrate into the outer retina and subretinal space (white arrows). (B) En-face microglia IHC using whole retinal flat mounts and CD11b and Iba1 antibodies (1:500) show that microglia within the IPL and OPL were more ramified and less densely packed in MMFP12-treated rd10 than naïve rd10 mice. The microglia had amoeboid morphology within the photoreceptor layer in both naïve and MMFP12-treated rd10 mice, but dramatically less numerous in the latter. Microglia counts were significantly less numerous in the IPL (P = 0.02), OPL (P = 0.046), and photoreceptor layer (P < 0.0001) for MMFP12-treated rd10 mice (n = 4) than naïve rd10 mice (n = 4). There was no significant difference within the VRI/NFL+GCL (p =0.12).*P ≤ 0.05.
MMF Delays Activation of Mueller Cells in rd10 Mice
Consistent with the microglial/macrophage results, immunolabeling for glial fibrillary acidic protein (GFAP) showed predominant staining limited to the footplates, indicating the Mueller cells were in the resting state in c57 mice and not overtly activated in MMFP12-treated rd10 mice, compared with extensive GFAP upregulation of activated Mueller cells in naïve rd10 at P22 (Supplementary Fig. S3). At P35, GFAP was upregulated in the retina of both MMFP12-treated and naïve rd10 mice.
MMF Inhibits cGMP-Dependent Photoreceptor Cytotoxicity in rd10 and rd1 Mice
The anti-cGMP primary antibodies used in this study were validated in the brain and retina in prior studies,81,85,86 and have been used extensively by Paquet-Durand and colleagues to demonstrate cGMP-dependent photoreceptor cell death in rodent models of RP.4 Similar to these prior studies, we also observed that scattered cGMP staining was evident on the ONL cell bodies and residual outer segments of degenerating photoreceptors in naïve rd10 and rd1 mice, whereas the retina from c57 mice exhibited no evidence of staining (Fig. 7A). Quantification of cGMP immunolabeling in the ONL of rd1 and rd10 mice showed no appreciable staining at P8 and P15, respectively (0.23 ± 0.06 and 0.19 ± 0.05 counts/µm2; Fig. 7B). Subsequently, photoreceptor cGMP cytotoxicity became evident and increased to peak levels in approximately one week in both rd1 mice (P15: 74.6 ±8.6 counts/µm2) and rd10 mice (P24: 15.0 ± 3.5 counts/µm2). Interestingly, before reaching peak levels, rd1 mice had an inflection at P11-13 (range 17.6–20.5 counts/µm2) and rd10 mice had an initial plateau at P16-19 (range 4.4–5.4 counts/µm2). Wild type c57 mice had no appreciable cGMP positive cell counts at all time points analyzed: P15, P22, and P35. Treatment with MMF showed dramatic suppression of cGMP immunoreactivity in both rd1 and rd10 mice. Compared with the peak levels of cGMP staining at P15 in naïve rd1 mice, MMFP8-treated rd1 mice had a 59% lower level of staining (30.9 ± 2.2 counts/µm2, P = 0.0001).
Figure 7.

Effect of MMF on cGMP IHC in rd10 and rd1 mice. (A) Nuclear stain with DAPI (blue) and immunostaining of cGMP 1:500 (red) show that rd10 mice treated with MMF starting at P12 (MMFP12) and c57 mice had no cGMP staining of the ONL as compared with naïve rd10 mice, which had scattered staining of the ONL cell bodies and residual photoreceptor outer segments. Compared with naïve rd1 mice, treatment with MMF starting at P8 (MMFP8) inhibited cGMP immunostaining when normalized to the ONL area. (B) cGMP IHC quantified as positive cell counts per ONL area showed that naïve rd10 mice exhibited an abrupt onset of cGMP staining beginning at P16 and spiked even higher at P22-24. Naïve rd1 mice had an even more dramatic rise in cGMP immunostaining from P8 to P11/12 with an inflection at P13 and an even greater peak at P15. Wild type c57 mice had no appreciable cGMP positive cell counts at all time points analyzed: P15, P22, and P35. Compared with naïve rd10 mice, treatment with MMFP12 significantly suppressed cGMP staining at P16 (n = 4 and 4, P = 0.0019), P17 (n = 4 and 4; P = 0.0003), P19 (n = 3 and 4; P = 0.0071), and P22 (n = 11 and 9; P < 0.0001). At P27, MMFP12-treated rd10 mice still had significantly lower cGMP staining than naïve rd10 mice (n = 6 and 4; P = 0.0013), but at P35 it was not significantly different (n = 3 and 3; P = 0.40). Compared with naive rd1 mice, MMFP8 treatment significantly suppressed cGMP immunoreactivity at P15 (n = 8 and 7; P < 0.0001). (C) The inhibitory effect of MMF on cGMP staining in rd10 mice at P22 was significantly diminished when treatment initiation was delayed from P12 to P14 (MMFP14, n = 6; P = 0.05). INL, inner nuclear layer; ns, nonsignificant; *P ≤ 0.05.
Compared with naïve rd10 mice, MMFP12-treated rd10 mice had completely suppressed cGMP staining at P16 (5.02 ± 0.96 vs. 0.003 ± 0.003 counts/µm2, P = 0.0019), P17 (4.36 ± 0.57 vs. 0.0022 ± 0.0010 counts/µm2, P = 0.0003), P19 (5.44 ± 1.47 vs. 0.025 ± 0.023 counts/µm2, P = 0.0071), and P22 (14.14 ± 2.46 vs. 0.076 ± 0.040 counts/µm2, P < 0.0001). At the height of cGMP cytotoxicity at P22 in naïve rd10 mice, MMFP12-treated rd10 mice appeared normal and were not significantly different from c57 mice (P > 0.9999). However, MMFP12-treated rd10 mice eventually exhibited cGMP immunoreactivity after P22, which was similar in magnitude to the smaller initial cGMP spike in naïve rd10 mice at P16, but never approached the highest levels in naïve rd10 observed at P24. At P27, MMFP12-treated rd10 mice still had significantly lower cGMP staining than naïve rd10 mice (5.13 ± 1.04 vs. 12.35 ± 0.91 counts/µm2, P = 0.0013), but at P35 the results were not significantly different (5.48 ± 0.38 vs. 6.14 ± 0.59 counts/µm2, P = 0.40). Given that the greatest effect of MMF treatment was at P22, additional studies were performed at this age to further characterize the window of treatment initiation (Fig. 7C). When the initiation of MMF treatment in rd10 mice was delayed by one or two days, there was a progressive increase in cGMP staining. Compared with rd10 MMFP12 mice, the rd10 mice where treatment began at P14 (MMFP14) had significantly greater cGMP immunostaining of the ONL by P22 (5.66 ± 0.86 counts/µm2, P = 0.05).
The Effect of MMF on Whole Retinal cGMP Levels in rd10 and rd1 Mice
The first evidence that cGMP was associated with retinal degeneration was in the form of elevated whole retinal cGMP in rd1 mice.8 However, to our knowledge, whole retina levels of cGMP in rd10 mice have not previously been published. In this study, we validated our LC-MS method with each batch of samples by using masked positive (purified cGMP) controls of different concentrations and masked negative controls. Comparable to prior studies, we show that whole retinal levels of cGMP in rd1 mice are significantly greater than c57 mice at P13 (29.0 ± 5.9 vs. 4.8 ± 0.3 pmol/retina, P = 0.0024), P15 (30.0 ± 4.3 vs. 7.4 ± 0.9 pmol/retina, P < 0.0001), and P17 (23.5 ± 8.2 vs. 5.5 ± 0.5 pmol/retina, P = 0.0136; Fig. 8). In addition, c57 mice exhibited a gradual rise in whole retinal cGMP levels that plateaued at P27. The cGMP levels in rd1 mice were significantly greater than rd10 mice at P12 (3.3 ±0.5 pmol/retina, P < 0.0001), P15 (3.1 ± 0.4 pmol/retina, P < 0.0001), and P17 (3.3 ± 0.2 pmol/retina, P = 0.008).
Figure 8.
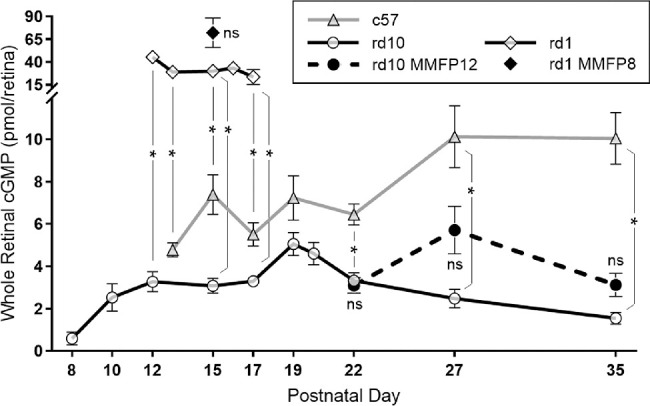
Effect of MMF on whole retinal cGMP levels in rd10 and rd1 mice. Whole retinal levels of cGMP quantified by LC-MS show that in c57 mice, there was a gradual rise that plateaued by P27. Naïve rd1 mice had cGMP levels significantly greater than c57 mice at P13 (n = 4 and 5, P = 0.0024), P15 (n = 17 and 11, P < 0.0001), and P17 (n = 3 and 4, P = 0.0136), whereas rd10 mice had cGMP levels that trended lower than c57 from P15 to P19 and were significantly lower than c57 mice at P22 (n = 14 and 13, P < 0.0001), P27 (n = 8 and 9, P = 0.0002), and P35 (n = 9 and 10, P < 0.0001). The cGMP levels in rd1 mice were significantly greater than rd10 mice at P12 (n = 3 and 6, P < 0.0001), P15 (n = 17 and 13, P < 0.0001), and P17 (n = 3 and 5, P = 0.008). Naïve rd10 mice exhibited a relative spike in cGMP levels at P19, which appeared to be delayed until P27 in rd10 mice treated with MMF beginning at P12 (MMFP12). However, for each time point, cGMP levels were not significantly different between naïve and MMFP12-treated rd10 mice at P22 (n = 13, P = 0.70), P27 (n = 6, P = 0.08), and P35 (n = 6, P = 0.26). Compared with naïve rd1 mice, the cGMP levels were also not significantly different in MMFP8-treated rd1 mice at P15 (n = 7, P = 0.32). ns, nonsignificant; *P ≤ 0.05.
However, naïve rd10 mice had cGMP levels that trended lower than c57 from P15 to P19 and were significantly lower than c57 mice at P22 (3.3 ± 0.4 vs. 6.5 ± 0.5 pmol/retina, P < 0.0001), P27 (2.5 ± 0.4 vs. 10.1 ± 1.5 pmol/retina, P = 0.0002), and P35 (1.5 ± 0.3 vs. 10.0 ± 1.2 pmol/retina, P < 0.0001). Interestingly, rd10 mice had a relative peak in cGMP levels at P19 (5.06 ± 0.54 pmol/retina), which then declined thereafter. Compared with naïve animals, treatment with MMF had no significant effect on cGMP levels at P15 in rd1 MMFP8 mice (72.0 ± 16.3 pmol/retina, P = 0.32) and P22 in rd10 MMFP12 mice (3.1 ± 0.4 pmol/retina, P = 0.70), respectively. The cGMP levels in MMFP12-treated rd10 mice were also not significantly different than naïve rd10 mice at P27 (5.7 ± 1.1 pmol/retina, P = 0.08) and P35 (3.1 ± 0.5 pmol/retina, P = 0.26). However, MMFP12-treated rd10 mice tended to have higher cGMP levels at P27, creating a relative peak, which suggests that MMF delays the peak of cGMP in naïve rd10 mice by eight days.
MPA Levels in the Plasma and Retina
Whole retinal levels of MPA quantified by LC-MS showed that the retinal concentration was about 3000 to 4000 times lower than the concentration in plasma (Supplementary Fig. S4). Moreover, there was a decline in plasma MPA levels from P16 (25.4 ± 7.0 µmol/L, n = 6) to P18 (4.23 ± 0.12 µmol/L, n = 4) along with a corresponding decline in whole retinal levels that also occurred after P16 (8.41 ± 0.80 nmol/L, n = 4) reaching a trough concentration after P22 (1.80 ± 0.44 nmol/L, n = 21).
Discussion
We show that early treatment with MMF potently inhibits cGMP-dependent photoreceptor cytotoxicity and slows the initial rate of retinal degeneration in two mouse models of RP with mutations in the gene Pde6b, albeit with a greater effect in rd10 than rd1 mice (Figs. 1, 5, 7). The neuroprotective effect of MMF in rd10 mice was specific to the rescue of cone tip morphology/density and scotopic cone function (Figs. 234, 5). Indeed, it has recently been shown that inhibitory cGMP analogs also rescue photoreceptor degeneration in the rd1 and rd10 mice.63 Similar to our results, Vighi et al.63 showed that early treatment with these cGMP analogs produced greater neuroprotection in rd10 mice by P24 than in rd1 mice by P14. The beneficial effect of cGMP analogs in rd10 mice also waned thereafter with a smaller effect by P30. In contrast, the cGMP analogs produced no significant effect at P18 and partial preservation at P24 in rd10 mice, whereas our data showed that MMF produced complete structural preservation of the photoreceptor layer in rd10 mice between P16 and P22. Thus there may be additional benefit of a drug like MPA that may mitigate cGMP dysregulation further upstream (Fig. 9). It is well-established that MPA inhibits IMPDH and reduces the intracellular pool of guanine nucleotides (GMP, guanosine diphosphate [GDP], guanosine trisphosphate [GTP]).70 This suggests that MPA can effectively modulate the precursors of cGMP at the cellular level and thereby inhibit cGMP-dependent photoreceptor cell death. Alternatively, a relative reduction in guanine nucleotides may inhibit other potential pathways of cell death that may be dependent on high levels of GDP and GTP. A recent study indicates that these rhodopsin-mediated transducin-independent pathways may very well exist in rd10 mice.87 The mechanisms of photoreceptor pathophysiology upstream of cGMP cytotoxicity require further elucidation. Nevertheless, these studies demonstrate that manipulation of cGMP cytotoxicity via the use of cGMP analogues or IMPDH inhibitors is a viable strategy that may represent a new class of neuroprotective agents.
Figure 3.

Effect of MMF on photopic retinal function in rd10 mice. Treatment with MMF beginning at P12 (MMFP12) had no significant effect on photopic responses in rd10 mice at P35 or P60. At P35, c57 mice (n = 19) had significantly greater photopic b-wave amplitudes than either naïve rd10 (n = 18) or MMFP12-treated rd10 mice (n = 6). Similarly, at P60, c57 mice (n = 16) had significantly greater b-wave amplitudes than naïve rd10 (n = 7) and MMFP12-treated rd10 mice (n = 6). For a-wave amplitudes, c57 mice were significantly greater only for the 0.82 log cd ∙ s/m2 photopic stimulus. cd ∙ s/m2, candela-seconds per square meter; *P ≤ 0.05.
Figure 9.

Mechanism of action of MPA. This schematic shows the guanine nucleotide synthesis via the de novo and salvage pathway, and the mechanism of action of MPA inhibition of the de novo pathway. The phototransduction cascade is also shown, highlighting the mutant PDE6b subunit and PDE6 enzyme. In rd10 mice, the mutation leads to intracellular elevation of cGMP and calcium, triggering cell death via an alternative pathway or calpain, respectively. The alternative pathway is associated with PKG, HDAC, and PARP.4 PRPP, phoribosyl-1-pyrophosphate; HGPRTase, hypoxanthine-guanine phosphoribosyltransferase; IMP, inosine 5′-monophosphate; XMP, xanthosine monophosphate; GMPS, GMP synthetase; RDR, ribonucleotide diphosphate reductase; GK, guanylate kinase; NDPK, nucleoside diphosphate kinase; GC, guanylate cyclase; GCAP, GC-activating protein; PKG, protein kinase G; HDAC, histone deacetylase; PARP, poly-ADP-ribose-polymerase; CNG, cyclic nucleotide-gated ion channel; NAD, nicotinamide adenine dinucleotide; RHO, rhodopsin; T, transducin; ATP, adenosine triphosphate; *activate form of molecule.
The difference in efficacy of neuroprotection between the two mouse models is most likely associated with the severity of the genetic mutation in Pde6b. Although rd1 mice have an intronic MVL virus insertion and a second nonsense point mutation in exon 7 of Pde6b, the rd10 mice harbor a missense mutation, Arg560Cys in exon 13. These mutations result in a retinal degeneration in rd1 mice that is more severe and rapid than rd10 mice (Fig. 5A). The level of PDE6B expression and the level of intracellular cGMP immunolabeling are inversely correlated in rd1 and rd10 mice, with rd1 mice having lower levels of PDE6B and higher levels of cGMP staining.4,10 Our results not only confirm the findings of Arango-Gonzalez et al.,4 but we also show that the longitudinal dynamics of cGMP cytotoxicity in rd10 and rd1 mice were distinct (Fig. 7B). We observed a peak in cGMP cytotoxicity at P15 in rd1 mice that was 5 times greater than the peak in rd10 mice at P24. Our results also show that the rise in cGMP staining had an inflection point in rd1 mice at P11-13, and a temporary plateau in rd10 mice at P16-19. These time points correlate with the onset of rapid degeneration of ONL cell bodies in rd1 mice versus a trough in ONL degeneration in rd10 mice. In rd10 mice, cGMP staining rise rapidly again after the plateau to reach a second peak at P22 to P24 that is correlated with an even more rapid rate of ONL loss. Interestingly, studies of cell death using TUNEL assay show two peaks of cell death at P18 and P22-24 in rd10 mice,4 which correlate with our data. Together, these findings illustrate a complex interplay of cGMP cytotoxicity and subsequent photoreceptor cell death, wherein there may be multiple waves of cGMP-dependent photoreceptor degeneration according to photoreceptor type and/or retinal geography.
Our study confirms that cGMP immunostaining is a specific biomarker of cGMP-dependent cell death, whereas whole retinal cGMP levels were divergent between the rd1 and rd10 mice and do not correlate well with the dynamics of retinal degeneration or neuroprotection. In 1974, Farber et al.8 showed that retinal degeneration in rd1 mice was associated with highly elevated whole retinal levels of cGMP, and it was subsequently thought to be explained by the mutation in Pde6b. However, this reasoning is incompatible with our whole retinal cGMP data. Using LC-MS to quantify whole retinal cGMP levels, we confirm the findings of Farber et al.8 in rd1 mice (Fig. 8), but show that whole retinal cGMP levels in rd10 mice are surprisingly lower than normal. One obvious hypothesis is that the mutation in rd10 mice may cause constitutive cGMP hydrolysis, however PDE enzyme activity has been shown to be severely attenuated in rd10 mice.11 Our results are supported by studies in another mouse model with an alternative missense mutation in Pde6b (H620Q), which showed that whole retinal levels of cGMP were not significantly different than normal.88 Thus, even though the various Pde6b mutations in the rd1, rd10, and Pde6bH620Q mice have all been shown to result in decreased Pde6b expression and/or enzyme activity,10,88 the subsequent effect on the whole retinal levels of cGMP is unpredictably variable with concentrations that are high, low, or normal, respectively. These observations indicate that different mutations in the same gene, Pde6b, can result in distinct consequences in pathophysiology, which is a concept well known for other genes such as Rho.89 A complete multimeric structure for the PDE6 heterodimer is not yet available, making it difficult to predict specific changes in overall enzyme stability or function because of mutations in Pde6b.90 Moreover, PDE6 interactions with other molecules is complex and its enzymatic activity is itself influenced by the cGMP microenvironment via its noncatalytic cGMP binding sites.91 As such, the pathophysiology of PDE6-mediated cGMP dysregulation in the context of retinal degeneration is likely much more complex than our current understanding of cGMP homeostasis in photoreceptors.16 Taken together with the literature, our data show that the effect of different mutations in Pde6b on whole retinal cGMP dysregulation is difficult to predict and do not correlate with cGMP-dependent photoreceptor cell death.
This seeming contradiction in the results of cGMP IHC versus whole retinal cGMP is due to the inherent differences in the two assays as follows: (1) Whole retinal cGMP is quantitative but is an average measurement of all retinal cell types, while cGMP IHC is semiquantitative but is a biomarker specific to the photoreceptors. Although the main contributor of whole retinal cGMP is from the photoreceptors in rd1 mice,8 it is unknown if this assumption holds for all mouse models of RP. (2) Whole retinal cGMP measurement using LC-MS detects free cGMP with high sensitivity and no floor effect in c57 mice, whereas the signal in cGMP IHC is mediated by an antibody that binds to an unknown photoreceptor protein that is cross-linked with cGMP. Thus it is unknown whether c57 mice have no cGMP staining because the intracellular cGMP levels are below the threshold of detection by IHC or that the sequestration of cGMP with an unknown protein only occurs in photoreceptors undergoing cell death. This latter explanation may be the reason why cGMP immunostaining is also found in mouse models of retinal degeneration with mutations in genes not directly associated with cGMP metabolism.
The neuroprotective effect of MMF was also correlated with a reduction in microglia activation and infiltration, as well as Mueller cell activation in rd10 mice (Fig. 6789, Supplementary Fig. S3). Indeed, microglial-dependent photoreceptor cell death has been implicated in RP,84 and modulation of microglia has been shown to be neuroprotective.22,30,31,49,55,84 Although MMF is known to inhibit microglial activation in vitro,76 the main neuroprotective mechanism of MMF in rd10 mice is most likely due to mitigation of upstream cGMP-associated cell death pathways. Our data shows very clearly that the therapeutic window of MMF treatment in rd10 mice is very early at P12, which is nearly one week before microglia activate and migrate to the ONL.84
Early treatment with MMF appeared to be safe in wildtype mouse eyes, having no detrimental effect on retinal structure or function (Fig. 1D, 2C, 5B). The most common dose-dependent side effects include stomach upset, diarrhea, and susceptibility to infections. There is also risk of reversible liver, kidney, or bone marrow dysfunction, which is routinely monitored with blood work in patients taking MMF. Our murine dose of MMF at 100 mg/kg/day, given an estimated ∼7 fold metabolic difference between humans and rodents, is equivalent to a human dose of 14.3 mg/kg/day, which is two to three times lower than the dose typically used in the treatment of uveitis (∼33.3 to 50 mg/kg/day). Given that adverse events from MMF are typically dose-related, the relatively low therapeutic dose that is anticipated suggests that the neuroprotective dose for MMF should have a better safety profile. Although MMF treatment in rd10 mice was limited by metabolism and bioavailability (Supplementary Fig. S4), durable neuroprotection of visual behavior was observed up to P60 (Figs. 1, 3), which support potential benefit of long-term therapy. Clinically, it has already been shown that MMF can be well tolerated for long periods in patients with organ transplant, rheumatologic disease, and uveitis.79,92,93 Future studies are aimed at developing an alternative MPA prodrug with better retinal bioavailability, and intraocular delivery strategies. Either way, the use of MMF or MPA is not without risk, and we hope these studies will eventually lead to a clinical trial to establish the benefit-risk ratio in patients with RP.
Even with the recent success of gene therapy for RPE65, there remains an unmet need to develop neuroprotective agents that can be applied more broadly to mitigate common pathways of retinal degeneration. We show that MMF inhibits cGMP-dependent photoreceptor cytotoxicity in two models of RP even though the different mutations in Pde6b cause divergent consequences in whole retinal cGMP pathophysiology. Although additional studies in other RP genes associated with cGMP cytotoxicity will be required to establish generalizability of the effect of MMF in RP, we present proof of principle that MMF may be an important new class of neuroprotective agent that could be useful in the treatment of patients with RP.
Supplementary Material
Acknowledgments
Analytical support was provided by the Bioanalytical Shared Resource/Pharmacokinetics Core Facility which is part of the University Shared Resource Program at Oregon Health and Sciences University. Yuquan Wen is credited with writing the custom OCT segmentation software used in this study.
Supported by the Foundation Fighting Blindness (Columbia, MD): CD-NMT-0714-0648-OHSU (PY), CD-NMT-0914-0659-OHSU (MEP), C-CL-0711-0534-OHSU01 (Unrestricted, CEI); the Research to Prevent Blindness (New York, NY): Career Development Award (MEP); Unrestricted Grant from RPB (CEI), P30EY010572 (CEI), the Medical Research Foundation (Portland, OR) (PY); and the National Institutes of Health (Bethesda, MD): 1K08 EY0231186 (MEP), 1K08 EY026650 (PY), and 1R01 EY029985 (RMD, CWM).
Disclosure: P. Yang, AGTC (C), Nanoscope Therapeutics (C); R. Lockard, None; H. Titus, None; J. Hiblar, None; K. Weller, None; D. Wafai, None; R.G. Weleber, AGTC (F, S), Sanofi (F), FFB (F, S); these relationships have been reviewed and managed by OHSU; R.M. Duvoisin: None; C.W. Morgans, None; M.E. Pennesi, AGTC (F, C), Sanofi (F), Spark (C), Editas (C), ProQR (C), Ionis Pharm (C)
References
- 1. Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006; 368: 1795–1809. [DOI] [PubMed] [Google Scholar]
- 2. Nakazawa M. Effects of calcium ion, calpains, and calcium channel blockers on retinitis pigmentosa. J Ophthalmol. 2011; 2011: 292040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Doonan F, Donovan M, Cotter TG. Activation of multiple pathways during photoreceptor apoptosis in the rd mouse. Invest Ophthalmol Vis Sci. 2005; 46: 3530–3538. [DOI] [PubMed] [Google Scholar]
- 4. Arango-Gonzalez B, Trifunovic D, Sahaboglu A, et al.. Identification of a common non-apoptotic cell death mechanism in hereditary retinal degeneration. PloS One. 2014; 9: e112142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lohr HR, Kuntchithapautham K, Sharma AK, Rohrer B. Multiple, parallel cellular suicide mechanisms participate in photoreceptor cell death. Exp Eye Res. 2006; 83: 380–389. [DOI] [PubMed] [Google Scholar]
- 6. Paquet-Durand F, Silva J, Talukdar T, et al.. Excessive activation of poly(ADP-ribose) polymerase contributes to inherited photoreceptor degeneration in the retinal degeneration 1 mouse. J Neurosci. 2007; 27: 10311–10319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang J, Richmond AM, Ogilvie JM. Inhibition of dopamine signaling suppresses cGMP accumulation in rd1 retinal organ cultures. Neuroreport. 2014; 25: 601–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Farber DB, Lolley RN. Cyclic guanosine monophosphate: elevation in degenerating photoreceptor cells of the C3H mouse retina. Science. 1974; 186: 449–451. [DOI] [PubMed] [Google Scholar]
- 9. Farber DB, Park S, Yamashita C. Cyclic GMP-phosphodiesterase of rd retina: biosynthesis and content. Exp Eye Res. 1988; 46: 363–374. [DOI] [PubMed] [Google Scholar]
- 10. Chang B, Hawes NL, Pardue MT, et al.. Two mouse retinal degenerations caused by missense mutations in the beta-subunit of rod cGMP phosphodiesterase gene. Vis Res. 2007; 47: 624–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang T, Reingruber J, Woodruff ML, et al.. The PDE6 mutation in the rd10 retinal degeneration mouse model causes protein mislocalization and instability and promotes cell death through increased ion influx. J Biol Chem. 2018; 293: 15332–15346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stasheff SF, Shankar M, Andrews MP. Developmental time course distinguishes changes in spontaneous and light-evoked retinal ganglion cell activity in rd1 and rd10 mice. J Neurophysiol. 2011; 105: 3002–3009. [DOI] [PubMed] [Google Scholar]
- 13. Paquet-Durand F, Beck S, Michalakis S, et al.. A key role for cyclic nucleotide gated (CNG) channels in cGMP-related retinitis pigmentosa. Hum Mol Genet. 2011; 20: 941–947. [DOI] [PubMed] [Google Scholar]
- 14. Paquet-Durand F, Marigo V, Ekstrom P. RD Genes Associated with High Photoreceptor cGMP-Levels (Mini-Review). Adv Exp Med Biol. 2019; 1185: 245–249. [DOI] [PubMed] [Google Scholar]
- 15. Power M, Das S, Schutze K, Marigo V, Ekstrom P, Paquet-Durand F. Cellular mechanisms of hereditary photoreceptor degeneration—Focus on cGMP. Prog Retin Eye Res. 2019; 74: 100772. [DOI] [PubMed] [Google Scholar]
- 16. Tolone A, Belhadj S, Rentsch A, Schwede F, Paquet-Durand F. The cGMP pathway and inherited photoreceptor degeneration: targets, compounds, and biomarkers. Genes (Basel). 2019; 10: 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Trifunovic D, Dengler K, Michalakis S, Zrenner E, Wissinger B, Paquet-Durand F. cGMP-dependent cone photoreceptor degeneration in the cpfl1 mouse retina. J Comp Neurol. 2010; 518: 3604–3617. [DOI] [PubMed] [Google Scholar]
- 18. Sahaboglu A, Tanimoto N, Bolz S, et al.. Knockout of PARG110 confers resistance to cGMP-induced toxicity in mammalian photoreceptors. Cell Death Dis 2014; 5: e1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ramamurthy V, Niemi GA, Reh TA, Hurley JB. Leber congenital amaurosis linked to AIPL1: a mouse model reveals destabilization of cGMP phosphodiesterase. Proc Natl Acad Sci USA. 2004; 101: 13897–13902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang J, Saul A, Roon P, Smith SB. Activation of the molecular chaperone, sigma 1 receptor, preserves cone function in a murine model of inherited retinal degeneration. Proc Natl Acad Sci USA. 2016; 113: E3764–E3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang J, Saul A, Smith SB. Activation of sigma 1 receptor extends survival of cones and improves visual acuity in a murine model of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2019; 60: 4397–4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Arroba AI, Alvarez-Lindo N, van Rooijen N, de la Rosa EJ. Microglia-mediated IGF-I neuroprotection in the rd10 mouse model of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2011; 52: 9124–9130. [DOI] [PubMed] [Google Scholar]
- 23. Boatright JH, Moring AG, McElroy C, et al.. Tool from ancient pharmacopoeia prevents vision loss. Mol Vis. 2006; 12: 1706–1714. [PubMed] [Google Scholar]
- 24. Corrochano S, Barhoum R, Boya P, et al.. Attenuation of vision loss and delay in apoptosis of photoreceptors induced by proinsulin in a mouse model of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2008; 49: 4188–4194. [DOI] [PubMed] [Google Scholar]
- 25. Delyfer MN, Simonutti M, Neveux N, Leveillard T, Sahel JA. Does GDNF exert its neuroprotective effects on photoreceptors in the rd1 retina through the glial glutamate transporter GLAST? Mol Vis. 2005; 11: 677–687. [PubMed] [Google Scholar]
- 26. Drack AV, Dumitrescu AV, Bhattarai S, et al.. TUDCA slows retinal degeneration in two different mouse models of retinitis pigmentosa and prevents obesity in Bardet-Biedl syndrome type 1 mice. Invest Ophthalmol Vis Sci. 2012; 53: 100–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Frasson M, Picaud S, Leveillard T, et al.. Glial cell line-derived neurotrophic factor induces histologic and functional protection of rod photoreceptors in the rd/rd mouse. Invest Ophthalmol Vis Sci. 1999; 40: 2724–2734. [PubMed] [Google Scholar]
- 28. Frasson M, Sahel JA, Fabre M, Simonutti M, Dreyfus H, Picaud S. Retinitis pigmentosa: rod photoreceptor rescue by a calcium-channel blocker in the rd mouse. Nature Med. 1999; 5: 1183–1187. [DOI] [PubMed] [Google Scholar]
- 29. Garcia-Delgado AB, Valdes-Sanchez L, Calado SM, Diaz-Corrales FJ, Bhattacharya SS. Rasagiline delays retinal degeneration in a mouse model of retinitis pigmentosa via modulation of Bax/Bcl-2 expression. CNS Neurosci Ther. 2018; 24: 448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Glybina IV, Kennedy A, Ashton P, Abrams GW, Iezzi R. Photoreceptor neuroprotection in RCS rats via low-dose intravitreal sustained-delivery of fluocinolone acetonide. Invest Ophthalmol Vis Sci. 2009; 50: 4847–4857. [DOI] [PubMed] [Google Scholar]
- 31. Glybina IV, Kennedy A, Ashton P, Abrams GW, Iezzi R. Intravitreous delivery of the corticosteroid fluocinolone acetonide attenuates retinal degeneration in S334ter-4 rats. Invest Ophthalmol Vis Sci. 2010; 51: 4243–4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Valdes-Sanchez L, Garcia-Delgado AB, Montero-Sanchez A, et al.. The resveratrol prodrug JC19 delays retinal degeneration in rd10 mice. Adv Exp Med Biol. 2019; 1185: 457–462. [DOI] [PubMed] [Google Scholar]
- 33. Hasegawa T, Ikeda HO, Iwai S, et al.. Branched chain amino acids attenuate major pathologies in mouse models of retinal degeneration and glaucoma. Heliyon. 2018; 4: e00544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hu SL, Zheng CP. (3R)-5,6,7-trihydroxy-3-isopropyl-3-methylisochroman-1-one ameliorates retinal degeneration in Pde6b(rd10) mice. Cutan Ocul Toxicol. 2018; 37: 245–251. [DOI] [PubMed] [Google Scholar]
- 35. Ikeda HO, Sasaoka N, Koike M, et al.. Novel VCP modulators mitigate major pathologies of rd10, a mouse model of retinitis pigmentosa. Sci Rep. 2014; 4: 5970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Isiegas C, Marinich-Madzarevich JA, Marchena M, et al.. Intravitreal Injection of Proinsulin-Loaded Microspheres Delays Photoreceptor Cell Death and Vision Loss in the rd10 Mouse Model of Retinitis Pigmentosa. Invest Ophthalmol Vis Sci. 2016; 57: 3610–3618. [DOI] [PubMed] [Google Scholar]
- 37. Jackson AC, Roche SL, Byrne AM, Ruiz-Lopez AM, Cotter TG. Progesterone receptor signalling in retinal photoreceptor neuroprotection. J Neurochem. 2016; 136: 63–77. [DOI] [PubMed] [Google Scholar]
- 38. Kakizuka A. VCP, a major ATPase in the cells, as a novel drug target for currently incurable disorders. In: Nakao K, Minato N, Uemoto S, eds. Innovative Medicine: Basic Research and Development. Tokyo: Springer Nature; 2015: 61–69. [PubMed] [Google Scholar]
- 39. Kang K, Tarchick MJ, Yu X, Beight C, Bu P, Yu M. Carnosic acid slows photoreceptor degeneration in the Pde6b(rd10) mouse model of retinitis pigmentosa. Sci Rep. 2016; 6: 22632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kang K, Yu M. Protective effect of sulforaphane against retinal degeneration in the Pde6(rd10) mouse model of retinitis pigmentosa. Curr Eye Res. 2017; 42: 1684–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Komeima K, Rogers BS, Campochiaro PA. Antioxidants slow photoreceptor cell death in mouse models of retinitis pigmentosa. J Cell Physiol. 2007; 213: 809–815. [DOI] [PubMed] [Google Scholar]
- 42. Kucharska J, Del Rio P, Arango-Gonzalez B, et al.. Cyr61 activates retinal cells and prolongs photoreceptor survival in rd1 mouse model of retinitis pigmentosa. J Neurochem. 2014; 130: 227–240. [DOI] [PubMed] [Google Scholar]
- 43. Lee SY, Usui S, Zafar AB, et al.. N-Acetylcysteine promotes long-term survival of cones in a model of retinitis pigmentosa. J Cell Physiol. 2011; 226: 1843–1849. [DOI] [PubMed] [Google Scholar]
- 44. Li Y, Li T, Li JZ, Wu QS. (2R, 3S)-Pinobanksin-3-cinnamate ameliorates photoreceptor degeneration in Pde6(rd)(10) mice. Cutan Ocul Toxicol. 2017; 36: 273–277. [DOI] [PubMed] [Google Scholar]
- 45. Lu J, Luo L, Huang D, et al.. Photoreceptor protection by mesencephalic astrocyte-derived neurotrophic factor (MANF). eNeuro. 2018; 5(2): e0109–e0118, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Obolensky A, Berenshtein E, Lederman M, et al.. Zinc-desferrioxamine attenuates retinal degeneration in the rd10 mouse model of retinitis pigmentosa. Free Radic Biol Med. 2011; 51: 1482–1491. [DOI] [PubMed] [Google Scholar]
- 47. Olivares-Gonzalez L, Martinez-Fernandez de la Camara C, Hervas D, Millan JM, Rodrigo R. HIF-1alpha stabilization reduces retinal degeneration in a mouse model of retinitis pigmentosa. FASEB J 2018; 32: 2438–2451. [DOI] [PubMed] [Google Scholar]
- 48. Oveson BC, Iwase T, Hackett SF, et al.. Constituents of bile, bilirubin and TUDCA, protect against oxidative stress-induced retinal degeneration. J Neurochem. 2011; 116: 144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Peng B, Xiao J, Wang K, So KF, Tipoe GL, Lin B. Suppression of microglial activation is neuroprotective in a mouse model of human retinitis pigmentosa. J Neurosci. 2014; 34: 8139–8150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Phillips MJ, Walker TA, Choi HY, et al.. Tauroursodeoxycholic acid preservation of photoreceptor structure and function in the rd10 mouse through postnatal day 30. Invest Ophthalmol Vis Sci. 2008; 49: 2148–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Picard E, Jonet L, Sergeant C, et al.. Overexpressed or intraperitoneally injected human transferrin prevents photoreceptor degeneration in rd10 mice. Mol Vis. 2010; 16: 2612–2625. [PMC free article] [PubMed] [Google Scholar]
- 52. Platon-Corchado M, Barcelona PF, Jmaeff S, et al.. p75(NTR) antagonists attenuate photoreceptor cell loss in murine models of retinitis pigmentosa. Cell Death Dis. 2017; 8: e2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Punzo C, Kornacker K, Cepko CL. Stimulation of the insulin/mTOR pathway delays cone death in a mouse model of retinitis pigmentosa. Nat Neurosci. 2009; 12: 44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Patel AK, Surapaneni K, Yi H, et al.. Activation of Wnt/beta-catenin signaling in Muller glia protects photoreceptors in a mouse model of inherited retinal degeneration. Neuropharmacology. 2015; 91: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Roche SL, Ruiz-Lopez AM, Moloney JN, Byrne AM, Cotter TG. Microglial-induced Muller cell gliosis is attenuated by progesterone in a mouse model of retinitis pigmentosa. Glia. 2018; 66: 295–310. [DOI] [PubMed] [Google Scholar]
- 56. Sanchez-Cruz A, Villarejo-Zori B, Marchena M, et al.. Modulation of GSK-3 provides cellular and functional neuroprotection in the rd10 mouse model of retinitis pigmentosa. Mol Neurodegener 2018; 13: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sanz MM, Johnson LE, Ahuja S, Ekstrom PA, Romero J, van Veen T. Significant photoreceptor rescue by treatment with a combination of antioxidants in an animal model for retinal degeneration. Neuroscience. 2007; 145: 1120–1129. [DOI] [PubMed] [Google Scholar]
- 58. Sasahara M, Otani A, Oishi A, et al.. Activation of bone marrow-derived microglia promotes photoreceptor survival in inherited retinal degeneration. Am J Pathol. 2008; 172: 1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Strettoi E, Gargini C, Novelli E, et al.. Inhibition of ceramide biosynthesis preserves photoreceptor structure and function in a mouse model of retinitis pigmentosa. Proc Natl Acad Sci USA. 2010; 107: 18706–18711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Takano Y, Ohguro H, Dezawa M, et al.. Study of drug effects of calcium channel blockers on retinal degeneration of rd mouse. Biochem Biophys Res Comm. 2004; 313: 1015–1022. [DOI] [PubMed] [Google Scholar]
- 61. Venkatesh A, Ma S, Le YZ, Hall MN, Ruegg MA, Punzo C. Activated mTORC1 promotes long-term cone survival in retinitis pigmentosa mice. J Clin Invest. 2015; 125: 1446–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Vidal-Gil L, Sancho-Pelluz J, Zrenner E, Oltra M, Sahaboglu A. Poly ADP ribosylation and extracellular vesicle activity in rod photoreceptor degeneration. Sci Rep. 2019; 9: 3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Vighi E, Trifunovic D, Veiga-Crespo P, et al.. Combination of cGMP analogue and drug delivery system provides functional protection in hereditary retinal degeneration. Proc Natl Acad Sci USA. 2018; 115: E2997–E3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang K, Peng B, Xiao J, Weinreb O, Youdim MBH, Lin B. Iron-chelating drugs enhance cone photoreceptor survival in a mouse model of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2017; 58: 5287–5297. [DOI] [PubMed] [Google Scholar]
- 65. Wang K, Xiao J, Peng B, et al.. Retinal structure and function preservation by polysaccharides of wolfberry in a mouse model of retinal degeneration. Sci Rep. 2014; 4: 7601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang X, Zhao L, Zhang Y, et al.. Tamoxifen provides structural and functional rescue in murine models of photoreceptor degeneration. J Neurosci. 2017; 37: 3294–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang YY, Wang CG, Qi SN, Liu ZX, Su GF, Zheng YJ. 3,5-Dimethoxy-4-hydroxy myricanol ameliorates photoreceptor cell degeneration in Pde6b(rd)(10) mouse model. Cutan Ocul Toxicol. 2019; 38: 36–43. [DOI] [PubMed] [Google Scholar]
- 68. Xiang Z, Bao Y, Zhang J, et al.. Inhibition of non-NMDA ionotropic glutamate receptors delays the retinal degeneration in rd10 mouse. Neuropharmacology. 2018; 139: 137–49. [DOI] [PubMed] [Google Scholar]
- 69. Xu XJ, Wang SM, Jin Y, Hu YT, Feng K, Ma ZZ. Melatonin delays photoreceptor degeneration in a mouse model of autosomal recessive retinitis pigmentosa. J Pineal Res. 2017; 63. [DOI] [PubMed] [Google Scholar]
- 70. Allison AC, Kowalski WJ, Muller CD, Eugui EM. Mechanisms of action of mycophenolic acid. Ann NY Acad Sci. 1993; 696: 63–87. [DOI] [PubMed] [Google Scholar]
- 71. Gunter JH, Thomas EC, Lengefeld N, et al.. Characterisation of inosine monophosphate dehydrogenase expression during retinal development: differences between variants and isoforms. Int J Biochem Cell Biol. 2008; 40: 1716–1728. [DOI] [PubMed] [Google Scholar]
- 72. Kennan A, Aherne A, Bowne SJ, et al.. On the role of IMPDH1 in retinal degeneration. Adv Exp Med Biol. 2003; 533: 13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Spellicy CJ, Daiger SP, Sullivan LS, et al.. Characterization of retinal inosine monophosphate dehydrogenase 1 in several mammalian species. Mol Vis. 2007; 13: 1866–1872. [PubMed] [Google Scholar]
- 74. Dalmarco EM, Budni P, Parisotto EB, Wilhelm Filho D, Frode TS. Antioxidant effects of mycophenolate mofetil in a murine pleurisy model. Transpl Immunol. 2009; 22: 12–17. [DOI] [PubMed] [Google Scholar]
- 75. Dehghani F, Hischebeth GT, Wirjatijasa F, Kohl A, Korf HW, Hailer NP. The immunosuppressant mycophenolate mofetil attenuates neuronal damage after excitotoxic injury in hippocampal slice cultures. Eur J Neurosci. 2003; 18: 1061–1072. [DOI] [PubMed] [Google Scholar]
- 76. Dehghani F, Sayan M, Conrad A, et al.. Inhibition of microglial and astrocytic inflammatory responses by the immunosuppressant mycophenolate mofetil. Neuropathol Appl Neurobiol. 2010; 36: 598–611. [DOI] [PubMed] [Google Scholar]
- 77. Papadimitriou JC, Cangro CB, Lustberg A, et al.. Histologic features of mycophenolate mofetil-related colitis: a graft-versus-host disease-like pattern. Int J Surg Pathol. 2003; 11: 295–302. [DOI] [PubMed] [Google Scholar]
- 78. Zandman-Goddard G, Shoenfeld Y. Mycophenolate mofetil in animal models of autoimmune disease. Lupus. 2005; 14(Suppl 1): s12–s16. [DOI] [PubMed] [Google Scholar]
- 79. Daniel E, Thorne JE, Newcomb CW, et al.. Mycophenolate mofetil for ocular inflammation. Am J Ophthalmol. 2010; 149: 423–432 e1–e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Pennesi ME, Michaels KV, Magee SS, et al.. Long-term characterization of retinal degeneration in rd1 and rd10 mice using spectral domain optical coherence tomography. Invest Ophthalmol Vis Sci. 2012; 53: 4644–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. de Vente J, Steinbusch HW, Schipper J. A new approach to immunocytochemistry of 3′,5′-cyclic guanosine monophosphate: preparation, specificity, and initial application of a new antiserum against formaldehyde-fixed 3′,5′-cyclic guanosine monophosphate. Neuroscience. 1987; 22: 361–373. [DOI] [PubMed] [Google Scholar]
- 82. Maekawa S, Sato K, Fujita K, et al.. The neuroprotective effect of hesperidin in NMDA-induced retinal injury acts by suppressing oxidative stress and excessive calpain activation. Sci Rep. 2017; 7: 6885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Nishiguchi KM, Carvalho LS, Rizzi M, et al.. Gene therapy restores vision in rd1 mice after removal of a confounding mutation in Gpr179. Nat Commun. 2015; 6: 6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zhao L, Zabel MK, Wang X, et al.. Microglial phagocytosis of living photoreceptors contributes to inherited retinal degeneration. EMBO Mol Med. 2015; 7: 1179–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Gamm DM, Barthel LK, Raymond PA, Uhler MD. Localization of cGMP-dependent protein kinase isoforms in mouse eye. Invest Ophthalmol Vis Sci. 2000; 41: 2766–2773. [PubMed] [Google Scholar]
- 86. Tanaka J, Markerink-van Ittersum M, Steinbusch HW, De Vente J. Nitric oxide-mediated cGMP synthesis in oligodendrocytes in the developing rat brain. Glia 1997; 19: 286–2897. [PubMed] [Google Scholar]
- 87. Sundar JC, Munezero D, Bryan-Haring C, Saravanan T, Jacques A, Ramamurthy V. Rhodopsin signaling mediates light-induced photoreceptor cell death in rd10 mice through a transducin-independent mechanism. Hum Mol Genet. 2020; 29: 394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Davis RJ, Tosi J, Janisch KM, et al.. Functional rescue of degenerating photoreceptors in mice homozygous for a hypomorphic cGMP phosphodiesterase 6 b allele (Pde6bH620Q). Invest Ophthalmol Vis Sci. 2008; 49: 5067–5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Athanasiou D, Aguila M, Bellingham J, et al.. The molecular and cellular basis of rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Progr Retin Eye Res. 2018; 62: 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Maryam A, Vedithi SC, Khalid RR, et al.. The molecular organization of human cGMP specific phosphodiesterase 6 (PDE6): structural implications of somatic mutations in cancer and retinitis pigmentosa. Comput Struct Biotechnol J. 2019; 17: 378–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Cote RH, Bownds MD, Arshavsky VY. cGMP binding sites on photoreceptor phosphodiesterase: role in feedback regulation of visual transduction. Proc Natl Acad Sci USA. 1994; 91: 4845–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Becker BN. Mycophenolate mofetil. Transplant Proc. 1999; 31: 2777–2778. [DOI] [PubMed] [Google Scholar]
- 93. Sahin A. Mycophenolate mofetil in the treatment of systemic lupus erythematosus. Eurasian J Med. 2009; 41: 180–5. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.