

This cross-sectional study examines whether sex-related factors could alter the clinical presentation of Stargardt disease among individuals with a low-penetrant ABCA4 allele.
Key Points
Question
Are mild ABCA4 alleles associated with the patient’s sex in Stargardt disease?
Findings
In this cross-sectional study of 550 patients with genetically confirmed Stargardt disease, a female predilection was observed among patients who carried a mild ABCA4 genotype compared with patients who harbored (moderately) severe ABCA4 genotypes.
Meaning
This study found that, in the 25% of patients with Stargardt disease who carried a combination of a mild and a severe allele, sex may play a modifying role in the disease.
Abstract
Importance
The mechanisms behind the phenotypic variability and reduced penetrance in autosomal recessive Stargardt disease (STGD1), often a blinding disease, are poorly understood. Identification of the unknown disease modifiers can improve patient and family counseling and provide valuable information for disease management.
Objective
To assess the association of incompletely penetrant ABCA4 alleles with sex in STGD1.
Design, Setting, and Participants
Genetic data for this cross-sectional study were obtained from 2 multicenter genetic studies of 1162 patients with clinically suspected STGD1. Unrelated patients with genetically confirmed STGD1 were selected. The data were collected from June 2016 to June 2019, and post hoc analysis was performed between July 2019 and January 2020.
Main Outcomes and Measures
Penetrance of reported mild ABCA4 variants was calculated by comparing the allele frequencies in the general population (obtained from the Genome Aggregation Database) with the genotyping data in the patient population (obtained from the ABCA4 Leiden Open Variation Database). The sex ratio among patients with and patients without an ABCA4 allele with incomplete penetrance was assessed.
Results
A total of 550 patients were included in the study, among which the mean (SD) age was 45.7 (18.0) years and most patients were women (311 [57%]). Five of the 5 mild ABCA4 alleles, including c.5603A>T and c.5882G>A, were calculated to have incomplete penetrance. The women to men ratio in the subgroup carrying c.5603A>T was 1.7 to 1; the proportion of women in this group was higher compared with the subgroup not carrying a mild allele (difference, 13%; 95% CI, 3%-23%; P = .02). The women to men ratio in the c.5882G>A subgroup was 2.1 to 1, and the women were overrepresented compared with the group carrying no mild allele (difference, 18%; 95% CI, 6%-30%; P = .005).
Conclusions and Relevance
This study found an imbalance in observed sex ratio among patients harboring a mild ABCA4 allele, which concerns approximately 25% of all patients with STGD1, suggesting that STGD1 should be considered a polygenic or multifactorial disease rather than a disease caused by ABCA4 gene mutations alone. The findings suggest that sex should be considered as a potential disease-modifying variable in both basic research and clinical trials on STGD1.
Introduction
Autosomal recessive Stargardt disease (STGD1) is the most frequent inherited macular dystrophy.1 It is caused by mutations in both copies of the gene encoding the transmembrane ATP-binding cassette transporter type A4 (ABCA4 gene [OMIM 601691]).2 Patients generally report progressive central loss of vision in the first or second decade of life,3,4,5 but cases with a much later disease onset, even past the age of 80 years, have been described as well.6 This large clinical variability has also been observed in siblings sharing the same ABCA4 gene mutations and has remained largely unexplained to date.7,8,9 The lack of knowledge about the mechanisms underlying this clinical variability impairs not only patient and family counseling but also informed decision-making regarding study design and patient inclusion. Moreover, deciphering these mechanisms might lead to new targets for treating this incurable disease.
Late-onset STGD1 and clinical variability are associated with mild variants, which generally are clinically relevant only when in trans with a severe ABCA4 variant.8,10,11,12 To date, 8 mild ABCA4 variants have been reported.8,10,11,12,13 Four coding variants—c.2588G>C, p.[Gly863Ala,Gly863del]; c.3113C>T, p.(Ala1038Val); c.5882G>A, p.(Gly1961Glu); and c.6089G>A, p.(Arg2030Gln)—and 1 noncanonical splice site variant—c.5714+5G>A, p.[=,Glu1863Leufs*33]—were deemed mild on the basis of their considerably lower than expected homozygous occurrence in patients, considering the respective allele frequencies in the general population.13 An additional mild coding variant—ie, c.5603A>T, p.(Asn1868Ile)—and 2 mild deep-intronic variants—c.769-784C>T, p.[=,Leu257Aspfs*3] and c.4253+43G>A, p.[=,Ile1377Hisfs*3]—were identified.10,12,14 Variant c.5603A>T was calculated to cause disease only in a small proportion of individuals (approximately 5%) who carried this allele in combination with a severe one.11,15 Variant c.2588G>C is likely benign and only has complete penetrance when present in the same gene copy as c.5603A>T (c.[2588G>C;5603A>T]).10 Similarly, in most cases of STGD1, with the deep-intronic variant c.769-784C>T, and in some cases with c.4253+43G>A, c.5603A>T was found in the same gene copy, and arguably only the complex alleles (c.[769-784C>T;5603A>T] and c.[4253+43G>A;5603A>T]) have complete penetrance.8 The mechanisms underlying incomplete penetrance, found for some ABCA4 genotypes, are still unknown; both cis- or trans-acting genetic modifiers and environmental factors might alter ABCA4 gene expression and the clinical presentation of STGD1.16
In a small cohort of patients carrying the c.5603A>T allele, 22 of 34 were female patients (65%) and all 3 siblings unaffected by compound heterozygous were male patients.11 In another study, which compared siblings with STGD1, a sex difference appeared to be present; in 5 of 5 families in which a wide variation in age at onset was observed (13-39 years), the patient with the latest onset was male.9 A sex ratio imbalance in STGD1 has never been established despite abundant research into this disease. We hypothesized that only a subgroup of patients (ie, those carrying a low penetrant ABCA4 allele) might harbor sex-related risk factors for STGD1. Because sex-related differences are associated with the causes and severity of many common diseases as well as the treatment outcomes, we investigated the sex distribution in a large STGD1 patient cohort. To assess the penetrance of mild ABCA4 alleles and the sex distribution among patients with biallelic mutations, we used data from 2 recent genetic studies in which the ABCA4 gene of 1162 patients with a potential ABCA4-associated retinal dystrophy was fully sequenced.17,18 The ABCA4 Disease Consortium Study Group allowed us to validate the sex ratio imbalance for c.5603A>T and to identify the incomplete penetrance and sex ratio imbalance for c.5882G>A, another ABCA4 allele.
Methods
This cross-sectional study adhered to the tenets of the Declaration of Helsinki.19 Approval from institutional research ethics committees were obtained from all hospitals and medical facilities that participated in the 2 studies we analyzed (specified at the end of the article), and patients provided written informed consent before participation. No compensation or incentive was offered to patients for participation in the study. Data were collected from June 2016 to June 2019.
Patient Selection
Patients with genetically confirmed STGD1 were selected from 2 studies that involved 21 international and 4 national centers.17,18 In these studies, unrelated patients with a potential ABCA4-associated retinal dystrophy (eg, STGD; cone-rod dystrophy; unspecified macular dystrophies; and pattern dystrophy, a well-known phenocopy) underwent clinical examination by a local ophthalmologist specializing in inherited retinal diseases. The coding and noncoding regions of the ABCA4 gene were sequenced using 3866 single-molecule molecular inversion probes. In 555 of 1162 cases, STGD1 was genetically confirmed by the presence of either 2 or more (likely) pathogenic ABCA4 variants or c.5603A>T combined with a (likely) pathogenic ABCA4 variant. In addition, we collected data on sex, age, and age at onset. Age at onset was defined as the onset of initial symptoms reported by the patient. Patients with missing data on sex were excluded.
In this study, we used the term sex, defined by biological characteristics, rather than gender, which refers to a person’s identity and the sociocultural expectations of behavior associated with a given sex. However, the 2 terms could potentially be used interchangeably in the given context.
Penetrance Calculation
As previously described for variants c.5603A>T, p.(Asn1868Ile); c.769-784C>T, p.[=,Leu257Aspfs*3]; and c.4253+43G>A, p.[=,Ile1377Hisfs*3],8,11,15 we aimed to first assess whether additional mild variants could have reduced penetrance. For this purpose, we performed penetrance calculations for 5 other alleles, which were deemed mild because of their absence or low occurrence in a homozygous state in patients; these alleles were c.5882G>A, p.(Gly1961Glu); c.5714+5G>A, p.[=,Glu1863Leufs*33]; c.6089G>A, p.(Arg2030Gln); c.2588G>C, p.[Gly863Ala,Gly863del]; and c.3113C>T, p.(Ala1038Val).13
Penetrance was calculated by comparing the ABCA4 allele frequency data in the general population that were obtained from gnomAD (Genome Aggregation Database, version 2.1.1.),20 with the genotyping data in the patient population that were obtained from the ABCA4 LOVD (Leiden Open Variation Database, version 3.0). Given that most patients in the LOVD have a non-Finnish European ethnic background, only the non-Finnish European population in gnomAD was considered, which included data on 7718 genomes and 56 885 exomes. Patients in the LOVD who were reported to have an ethnic origin other than non-Finnish European were excluded, as were patients in whom fewer than 2 ABCA4 variants had been identified. Penetrance was calculated according to the same methods used in previous studies,8,11,15 which could be summarized as the ratio of the observed frequency of patients (cases) with the genotype of interest to the theoretically expected frequency of patients with this genotype. Expected frequency was calculated according to the respective allele frequency (AF) in the general population (controls). Thus, the formula, given an estimated STGD1 prevalence of 1:10 000,1 was as follows:
 |
The genotype of interest consisted of the combination mild variant of interest and severe variant. Variants in the gnomAD and LOVD were annotated as severe if they constituted stop mutations, frameshift mutations, or canonical splice site variants (eTable 1 in the Supplement). We also added noncanonical splice site variants21 and deep-intronic variants shown to result in less than 25% normal messenger RNA, which we deemed to be a conservative cutoff for allocating variants to the status of severe.21
Statistical Analysis
To examine whether the proportion of men and women in the patient group harboring an allele with reduced penetrance was different from the patient group without any of these alleles, we conducted a 2-tailed Fisher exact test. For the statistical analysis, the 2 most frequent mild variants c.5603A>T and c.5882G>A were selected from the total of 8 mild alleles. We applied Bonferroni correction for multiple testing in the main analysis, which consisted of 2 tests (α = .025). For patient groups who showed a sex ratio imbalance, we tested whether the age at onset differed between men and women using the Mann-Whitney test. A 2-tailed α = .05 indicated statistical significance.
Analyses were performed with SPSS for Windows, version 25 (SPSS IBM), between July 2019 and January 2020.
Results
Description of the Cohort
After exclusion of 5 patients for missing data on sex, we included a total of 550 patients in this study. In this cohort, the mean (SD) age was 45.7 (18.0) years, 456 patients (83%) were of non-Finnish European descent (eTable 2 in the Supplement), and 311 patients were women (57%) (Table 1). Mild alleles were frequently identified in 266 (48%) participants, often in women (169 of 266 [64%]). Because c.5603A>T was formerly not considered to be a causal variant, it was (not unexpectedly) found to be the most frequent variant in this cohort, present as a noncomplex allele (ie, no additional potentially pathogenic variant on the same gene copy) in 125 patients (23%). The other most frequent variant was c.5882G>A, which was found in a noncomplex configuration in 79 patients (14%). The 6 other mild variants were identified in a total of 64 patients (12%). These 6 variants were c.4253+43G>A, c.5714+5G>A, c.6089G>A, c.3113C>T, c.769-784C>T, and c.2588G>C.
Table 1. Study Participants.
| ABCA4 allele | No. (%)a | |
|---|---|---|
| Women | Men | |
| Mild allele | ||
| c.5603A>T | 79 (63) | 46 (37) |
| c.5882G>A | 54 (68) | 25 (32) |
| c.4253+43G>A | 18 (58) | 13 (42)b |
| c.5714+5G>A | 10 (56) | 8 (44) |
| c.6089G>A | 5 | 1 |
| c.3113C>T | 1b | 2 |
| c.769-784C>T | 1 | 2 |
| c.2588G>C | 2 | 1 |
| Total | 169 | 97 |
| No mild allele | 142 (50) | 142 (50) |
Percentages were presented if the total was 10 or more.
Two of 125 cases who carried c.5603A>T also harbored the mild allele c.3113C>T or c.4253+43G>A.
Reduced Penetrance of 5 Additional Mild Variants
A total of 2031 cases in the LOVD met the criteria of having a non-Finnish European ethnic background and at least 2 ABCA4 variants. For variants c.5882G>A, c.5714+5G>A, and c.6089G>A, dividing the frequency of the genotype of interest in cases by the frequency of this genotype in controls and then multiplying the numbers by the STGD1 prevalence of 1:10 000 yielded reduced penetrance of 50.4% for c.5882G>A, 58.6% for c.5714+5G>A, and 36.4% for c.6089G>A (Table 2). For variants c.3113C>T and c.2588G>C, which can be part of frequent complex alleles c.[1622T>C;3113C>T] and c.[2588G>C;5603A>T], penetrance was calculated in the same manner, not taking potential cis-variants into consideration. The relative proportion of these complex alleles is generally higher in patients than in controls. Consequently, both the observed and the expected numbers of patients carrying the noncomplex alleles were overestimated, but the observed number was overestimated to a greater extent. Therefore, the penetrance 16.8% for c.3113C>T and 11.0% for c.2588G>C should be interpreted as an overestimation of the actual penetrance.
Table 2. Penetrance of Mild ABCA4 Alleles When Accompanied by a Severe ABCA4 Allele.
| Variable in calculation | c.5882G>A | c.5714+5G>A | c.6089G>A | c.3113C>T | c.2588G>C |
|---|---|---|---|---|---|
| LOVD (cases) | |||||
| Observed number of patients with mild/severe genotype | 177 | 30 | 21 | 36 | 80 |
| Frequency of mild/severe genotypea | 0.087149 | 0.014771 | 0.010340 | 0.017725 | 0.039389 |
| gnomAD (controls) | |||||
| AF mild variant | 0.003778 | 0.000550 | 0.000620 | 0.002302 | 0.007840 |
| Sum AF severe variants | 0.002290 | 0.002290 | 0.002290 | 0.002290 | 0.002290 |
| Frequency of mild/severe genotypeb | 0.000017 | 0.000003 | 0.000003 | 0.000011 | 0.000036 |
| Penetrance | 50.4% | 58.6% | 36.4% | 16.8% | 11.0% |
Abbreviations: AF, allele frequency; gnomAD, Genome Aggregation Database; LOVD, Leiden Open Variation Database.
The observed number of patients in LOVD with a mild variant in combination with a severe variant was divided by 2031, the total number of non-Finnish European patients with at least 2 ABCA4 variants in LOVD.
The expected frequency of the mild variant in combination with a severe variant in the general population was obtained by 2 × mild AF × sum severe AF (Hardy-Weinberg principle).
Sex Ratio in STGD1 Associated With c.5603A>T and c.5882G>A
The sex ratio in the subgroup of patients with none of the mild alleles was exactly 1 to 1. The women to men ratio in the subgroup carrying c.5603A>T was 1.7 to 1; the proportion of women in this group was higher compared with the subgroup not harboring a mild allele (difference, 13%; 95% CI, 3%-23%; P = .02) (Figure 1A and B). Furthermore, the sex ratio in the c.5882G>A group was 2.1 to 1 and the women were overrepresented compared with the group carrying no mild allele (difference, 18%; 95% CI, 6%-30%; P = .005) (Figure 1A and B). Among the patients who harbored any of the 6 other mild alleles (c.4253+43G>A, c.5714+5G>A, c.6089G>A, c.3113C>T, c.769-784C>T, and c.2588G>C), the overall women to men ratio was 1.4 to 1.
Figure 1. Comparison of Sex Distribution Among Patients With and Without a Mild ABCA4 Allele.
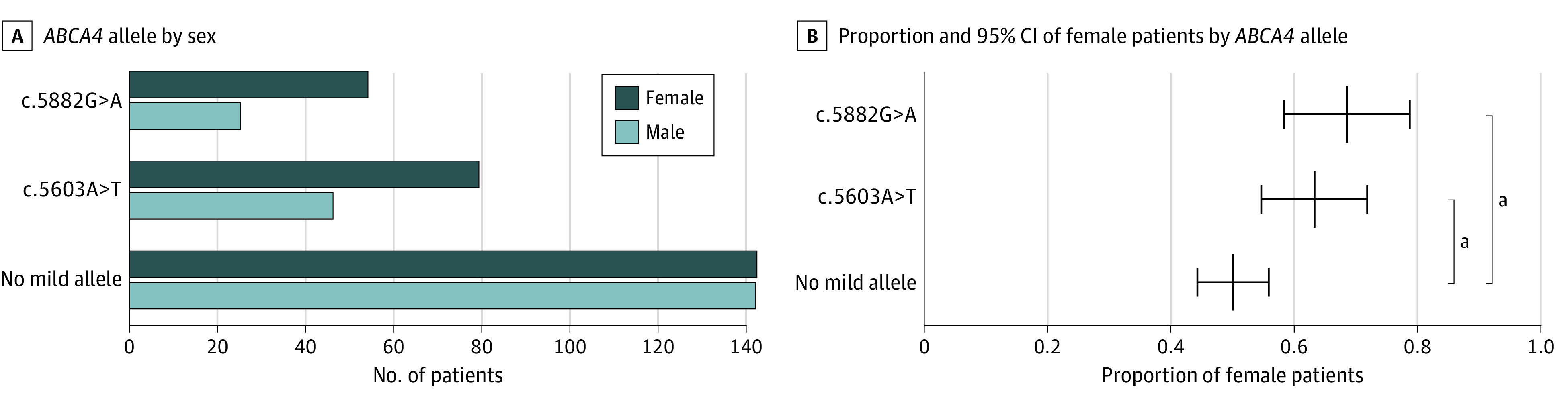
A, More women than men carried the mild allele c.5882G>A (54:25) or c.5603A>T (79:46), whereas the female to male ratio among patients not carrying a mild allele was exactly 1 (142:142). B, In Stargardt disease (STGD1) caused by the mild allele c.5882G>A or c.5603A>T, the proportion of women (68% and 63%, respectively) was significantly higher than in STGD1 not caused by a mild ABCA4 allele (13% [95% CI, 3%-23%; P = .02] vs 18% [95% CI, 6%-30%; P = .005]).
aP < .03.
To assess whether a survival bias from longer life expectancy of women could have a statistically significant implication for this study, we compared the current age of the women and men in the subgroups. The median age was not statistically significantly different between the sexes in any of the subgroups (eFigure 1 in the Supplement). Among patients harboring c.5603A>T or c.5882G>A, the sex ratio imbalance was observed regardless of age and was pronounced before the age of 60 years (eFigure 2 in the Supplement).
Based on the observed sex ratio imbalance and the calculated penetrance, the sex-specific penetrance of c.5603A>T and c.5882G>A, when in trans with a severe allele, was approximately 6% for women and 4% for men with c.5603A>T, and 68% for women and 32% for men with c.5882G>A (eFigure 3 in the Supplement).
Age at Disease Onset Among Patients
No difference was observed in the age of disease onset between the sexes in any of the genotypic subgroups (Figure 2). Among patients who were carrying c.5603A>T, women reported initial symptoms at a median (range) age of 40.0 (9.0-86.0) years, whereas men reported symptoms at a median (range) age of 43.0 (5.0-65.0) years (difference, 1.0 year; 95% CI, –7.0 to 10.0 years; P = .78). In the c.5882G>A subgroup, age at onset was slightly right-skewed, with a median (range) of 21.0 (1.0-51.0) years in women and 19.5 (10.0-40.0) years in men (difference, 1.0 year; 95% CI, –7.0 to 9.0 years; P = .82) (Figure 2; eFigure 2B in the Supplement). As expected, the age at onset among patients who were carrying none of the mild alleles was the lowest, with a median (range) of 13.0 (5.0-82.0) years in women and 12.0 (1.0-52.0) years in men (difference, 2.0 years; 95% CI, 0.0-4.0 years; P = .27) (Figure 2). In this latter subgroup, the distribution of age at onset was clearly right-skewed, likely because most of these individuals carried 2 (moderately) severe alleles and only some variants could behave like mild variants (eFigure 2C in the Supplement).
Figure 2. Age at Onset by Sex and Carriers of ABCA4 Allele.

Horizontal lines and error bars represent the median with the interquartile range. No difference in age at onset was observed between men and women in any of the 3 subgroups (ie, biallelic patients not carrying a known mild allele, patients carrying the c.5882G>A allele, and patients carrying the c.5603A>T allele).
Discussion
To our knowledge, no sex ratio imbalance has been established before in STGD1, nor has it been observed in other non-sex-related inherited retinal diseases, except for the maternally inherited Leber hereditary optic neuropathy, in which incomplete penetrance was observed and more men than women were affected.22,23 This international collaboration, complete ABCA4 sequencing data, and advanced understanding of mildly pathogenic ABCA4 alleles allowed us to establish that, in a genetically predefined patient group, those who harbored ABCA4 alleles with potential incomplete penetrance were predominantly women. This observation corroborated the evidence of reduced penetrance in STGD1 and pointed to the existence of sex-related modifiers of the expression of reduced penetrant ABCA4 alleles (Figure 3).24,25,26
Figure 3. Sex and Other Modifying Factors Associated With Risk for Stargardt Disease (STGD1).

Shown is a modification of the previously proposed ABCA4 disease model that associated genotypes with phenotypes on the basis of the residual activity of the ABCA4 protein.24,25,26 In individuals heterozygous for 1 severe variant, ABCA4 activity was reduced to 50%, which does not lead to STGD1. In a previous in vitro study, individually tested variants were considered mild if the percentage of wild-type messenger RNA, which we extrapolated to the residual protein activity, was 60% to 80%.21 Therefore, a combination of a severe and a mild allele leads to a total residual activity of approximately 30% to 40% and results in classic STGD1 (mild/severe bar). Because the expression of a mild, low-penetrant allele was variable, its combination with a severe variant could result either in generally late-onset STGD1 or in a normal phenotype (Mild, low penetrant/severe bar). Only in this latter category of individuals might sex and other genetic, epigenetic, and nongenetic modifiers alter the risk for developing STGD1.
Women were overrepresented among patients who were carrying 1 of the 2 most frequent alleles, c.5603A>T or c.5882G>A, compared with patients who were not carrying a known mild allele. Approximately 10%10,11 of all patients with STGD1 harbored c.5603A>T and 15%13,27 had c.5882G>A as 1 of the alleles. A search for the mechanisms underlying incomplete penetrance and female preponderance in approximately 25% of all patients with STGD1 may yield new hints for the pathogenesis and management of this blinding disease. Although many future studies are likely needed to elucidate the cause of sex differences in STGD1 and other diseases, these findings warrant careful consideration of sex or gender as a variable in future preclinical and clinical trials. Currently, medical practice in general (diagnosis and treatment) is less evidence based for women than for men because of the underrepresentation of women in biomedical research.28 However, considering sex as a study variable is indispensable in this era of personalized medicine, given that sex differences in the prevalence, manifestation, progression, and outcome of multifactorial disorders are increasingly identified. For instance, studies have found that ischemic heart disease has a younger onset, a higher incidence, and a different manifestation in men vs women, and dilated cardiomyopathy has a higher prevalence, younger onset, and worse outcome in men, whereas left ventricular hypertrophy is more prevalent and less modifiable in women.29,30 Female predilection has been shown in many autoimmune diseases, most strikingly (7-10:1) in Sjögren syndrome, systemic lupus erythematosus, and thyroiditis.31
The biological mechanisms underlying the sex-based differences in multifactorial diseases are not completely understood, but among the likely factors are sex chromosomes, sex hormones, and mitochondria.29,32,33 As estrogen, progesterone, and androgen receptors’ messenger RNAs have been identified in the human retina and specifically the retinal pigment epithelium,34,35 each of these sex hormones may play a causal role in sexual differences in retinal health and pathology. Research to date has mainly focused on estrogen, which had neuroprotective properties for retinal ganglion cells in a glaucoma rat model.36,37 Estrogen receptor β protected mice against retinal pigment epithelium injury.38 Moreover, sex hormone receptors, which are nuclear transcription factors that bind to DNA, are known to regulate gene expression. Sex-based differences in retinal gene expression have been observed in male and female mice.39
Mitochondria are involved in steroid hormone synthesis, production of reactive oxygen species, and cell death.32 Sex hormones, in turn, regulate mitochondrial function. Two retinal disease models, PDE6B-associated retinitis pigmentosa40 and neuronal ceroid lipofuscinoses,41 showed an increased susceptibility to photoreceptor degeneration of female mice compared with age-matched male mice.
In addition to biological processes, socioeconomic and behavioral factors seem obvious variables to consider in future studies of modifiers for STGD1. Environmental factors that could directly be associated with the pathophysiological processes (eg, accumulation of vitamin A derivatives and phototoxic reaction) are diet and sunlight exposure.
Genetic modifiers independent of sex exist that could also explain why some individuals harboring a mild and a severe allele develop STGD1 whereas others do not. First, despite sequencing of the complete 128-kb ABCA4 gene using a targeted, short-read sequencing method, the presence of deep-intronic variants, inversions or insertions in cis with a small proportion of the mild variants that make them fully penetrant, could not be ruled out.18 Second, sequence variants within or outside the ABCA4 locus could change disease expression, for instance, through effects on the transcription machinery (eg, owing to variants in enhancer or silencer regions) or through protein-protein interaction (eg, variants in PROM1 [OMIM *604365], ROM1 [OMIM *180721], and PRPH2 [OMIM *179605]42,43). However, these genetic sequence variants are not likely to be factors in the observed sex ratio imbalance.
Although we hypothesized that the factors associated with a female predilection in STGD1 could also be associated with a more severe or earlier onset phenotype in women, the data on age at onset did not support this hypothesis. Instead, specific disease modifiers might play a role at specific stages in life and could be different for men and women. Therefore, female patients are not necessarily expected to have more severe cases.
Limitations
This study has limitations that are mostly associated with its cross-sectional design. Cause and effect could not be determined. Selection bias (eg, potential sex differences in health seeking behavior or willingness to participate in research) could not be eliminated but was unlikely a major factor given that a reference group of patients from the same cohort was used. In addition, the finding that age at onset was not associated with sex argues against a substantial difference in health-seeking behavior that explains the sex ratio imbalance. This study corroborated a finding of a small, independent cohort study that a female predilection existed in the c.5603A>T genotypic subgroup.11 The female predilection in c.5882G>A cases was a novel finding and should be corroborated in an independent cohort. Self-reported age at onset might not reflect the actual disease onset, although this method is often used because of lack of a superior, accessible measure. The limitations of penetrance calculations have been previously discussed.11,15 These calculations rely on assumptions regarding disease prevalence, ethnic diversity, and absence of unidentified causal variants in cis with the mild variants. If disease prevalence was higher, the actual penetrance would be higher. If patients carried unidentified causal variants in cis with the mild variant, the actual penetrance of the mild variant would be lower. Thus, these calculations provided robust indications of incomplete penetrance rather than the exact penetrance rates.
Conclusions
This cross-sectional study demonstrated a female predilection in STGD1 among all patients who were carrying either of the 2 most frequent and mild ABCA4 alleles compared with patients who were not carrying a putative mild ABCA4 allele. This finding adds to the evidence of reduced penetrance for some frequent ABCA4 genotypes and highlights the potentially crucial role of sex in human health and disease that needs to be taken into consideration in future studies of STGD1.
eTable 1. The Allele Frequencies of Mild and Severe ABCA4 Variants in the General Non-Finnish European Population
eTable 2. Origin of Participants
eFigure 1. Age Distribution by Sex
eFigure 2. Age of Onset Distribution by Sex
eFigure 3. Sex-Specific Penetrance Estimates for ABCA4 Alleles c.5603A>T and c.5882G>A
eReferences
References
- 1.Blacharski P. Fundus flavimaculatus. In: Newsome DA, ed. Retinal Dystrophies and Degenerations. Vol 135-159 Raven Press; 1988. [Google Scholar]
- 2.Allikmets R, Singh N, Sun H, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;15(3):236-246. doi: 10.1038/ng0397-236 [DOI] [PubMed] [Google Scholar]
- 3.Rotenstreich Y, Fishman GA, Anderson RJ. Visual acuity loss and clinical observations in a large series of patients with Stargardt disease. Ophthalmology. 2003;110(6):1151-1158. doi: 10.1016/S0161-6420(03)00333-6 [DOI] [PubMed] [Google Scholar]
- 4.Fujinami K, Zernant J, Chana RK, et al. Clinical and molecular characteristics of childhood-onset Stargardt disease. Ophthalmology. 2015;122(2):326-334. doi: 10.1016/j.ophtha.2014.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lambertus S, van Huet RA, Bax NM, et al. Early-onset Stargardt disease: phenotypic and genotypic characteristics. Ophthalmology. 2015;122(2):335-344. doi: 10.1016/j.ophtha.2014.08.032 [DOI] [PubMed] [Google Scholar]
- 6.Westeneng-van Haaften SC, Boon CJ, Cremers FP, Hoefsloot LH, den Hollander AI, Hoyng CB. Clinical and genetic characteristics of late-onset Stargardt’s disease. Ophthalmology. 2012;119(6):1199-1210. doi: 10.1016/j.ophtha.2012.01.005 [DOI] [PubMed] [Google Scholar]
- 7.Burke TR, Tsang SH, Zernant J, Smith RT, Allikmets R. Familial discordance in Stargardt disease. Mol Vis. 2012;18:227-233. [PMC free article] [PubMed] [Google Scholar]
- 8.Runhart EH, Valkenburg D, Cornelis SS, et al. Late-onset Stargardt disease due to mild, deep-intronic ABCA4 alleles. Invest Ophthalmol Vis Sci. 2019;60(13):4249-4256. doi: 10.1167/iovs.19-27524 [DOI] [PubMed] [Google Scholar]
- 9.Valkenburg D, Runhart EH, Bax NM, et al. Highly variable disease courses in siblings with Stargardt disease. Ophthalmology. 2019;126(12):1712-1721. doi: 10.1016/j.ophtha.2019.07.010 [DOI] [PubMed] [Google Scholar]
- 10.Zernant J, Lee W, Collison FT, et al. Frequent hypomorphic alleles account for a significant fraction of ABCA4 disease and distinguish it from age-related macular degeneration. J Med Genet. 2017;54(6):404-412. doi: 10.1136/jmedgenet-2017-104540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Runhart EH, Sangermano R, Cornelis SS, et al. The common ABCA4 Variant p.Asn1868Ile shows nonpenetrance and variable expression of Stargardt disease when present in trans with severe variants. Invest Ophthalmol Vis Sci. 2018;59(8):3220-3231. doi: 10.1167/iovs.18-23881 [DOI] [PubMed] [Google Scholar]
- 12.Zernant J, Lee W, Nagasaki T, et al. Extremely hypomorphic and severe deep intronic variants in the ABCA4 locus result in varying Stargardt disease phenotypes. Cold Spring Harb Mol Case Stud. 2018;4(4):a002733. doi: 10.1101/mcs.a002733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cornelis SS, Bax NM, Zernant J, et al. In silico functional meta-analysis of 5,962 ABCA4 variants in 3,928 retinal dystrophy cases. Hum Mutat. 2017;38(4):400-408. doi: 10.1002/humu.23165 [DOI] [PubMed] [Google Scholar]
- 14.Sangermano R, Garanto A, Khan M, et al. Deep-intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genet Med. 2019;21(8):1751-1760. doi: 10.1038/s41436-018-0414-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cremers FPM, Cornelis SS, Runhart EH, Astuti GDN. Author response: penetrance of the ABCA4 p.Asn1868Ile allele in Stargardt disease. Invest Ophthalmol Vis Sci. 2018;59(13):5566-5568. doi: 10.1167/iovs.18-25944 [DOI] [PubMed] [Google Scholar]
- 16.Cremers FPM, Lee W, Collin RWJ, Allikmets R. Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Prog Retin Eye Res. 2020;100861. doi: 10.1016/j.preteyeres.2020.100861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khan M, Cornelis SS, Khan MI, et al. Cost-effective molecular inversion probe-based ABCA4 sequencing reveals deep-intronic variants in Stargardt disease. Hum Mutat. 2019;40(10):1749-1759. doi: 10.1002/humu.23787 [DOI] [PubMed] [Google Scholar]
- 18.Khan M, Cornelis SS, Pozo-Valero MD, et al. Resolving the dark matter of ABCA4 for 1054 Stargardt disease probands through integrated genomics and transcriptomics. Genet Med. 2020;22(7):1235-1246. doi: 10.1038/s41436-020-0787-4 [DOI] [PubMed] [Google Scholar]
- 19.World Medical Association World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310(20):2191-2194. doi: 10.1001/jama.2013.281053 [DOI] [PubMed] [Google Scholar]
- 20.Karczewski KJ, Francioli LC, Tiao G, et al. ; Genome Aggregation Database Consortium . The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434-443. doi: 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sangermano R, Khan M, Cornelis SS, et al. ABCA4 midigenes reveal the full splice spectrum of all reported noncanonical splice site variants in Stargardt disease. Genome Res. 2018;28(1):100-110. doi: 10.1101/gr.226621.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pisano A, Preziuso C, Iommarini L, et al. Targeting estrogen receptor β as preventive therapeutic strategy for Leber’s hereditary optic neuropathy. Hum Mol Genet. 2015;24(24):6921-6931. doi: 10.1093/hmg/ddv396 [DOI] [PubMed] [Google Scholar]
- 23.Oostra RJ, Kemp S, Bolhuis PA, Bleeker-Wagemakers EM. No evidence for ‘skewed’ inactivation of the X-chromosome as cause of Leber’s hereditary optic neuropathy in female carriers. Hum Genet. 1996;97(4):500-505. doi: 10.1007/BF02267075 [DOI] [PubMed] [Google Scholar]
- 24.Cremers FP, van de Pol DJ, van Driel M, et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum Mol Genet. 1998;7(3):355-362. doi: 10.1093/hmg/7.3.355 [DOI] [PubMed] [Google Scholar]
- 25.van Driel MA, Maugeri A, Klevering BJ, Hoyng CB, Cremers FP. ABCR unites what ophthalmologists divide(s). Ophthalmic Genet. 1998;19(3):117-122. doi: 10.1076/opge.19.3.117.2187 [DOI] [PubMed] [Google Scholar]
- 26.Maugeri A, van Driel MA, van de Pol DJ, et al. The 2588G→C mutation in the ABCR gene is a mild frequent founder mutation in the Western European population and allows the classification of ABCR mutations in patients with Stargardt disease. Am J Hum Genet. 1999;64(4):1024-1035. doi: 10.1086/302323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schulz HL, Grassmann F, Kellner U, et al. Mutation spectrum of the ABCA4 gene in 335 Stargardt disease patients from a multicenter German cohort - impact of selected deep intronic variants and common SNPs. Invest Ophthalmol Vis Sci. 2017;58(1):394-403. doi: 10.1167/iovs.16-19936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Franconi F, Campesi I, Colombo D, Antonini P. Sex-gender variable: methodological recommendations for increasing scientific value of clinical studies. Cells. 2019;8(5):E476. doi: 10.3390/cells8050476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerdts E, Regitz-Zagrosek V. Sex differences in cardiometabolic disorders. Nat Med. 2019;25(11):1657-1666. doi: 10.1038/s41591-019-0643-8 [DOI] [PubMed] [Google Scholar]
- 30.De Bellis A, De Angelis G, Fabris E, Cannatà A, Merlo M, Sinagra G. Gender-related differences in heart failure: beyond the “one-size-fits-all” paradigm. Heart Fail Rev. 2020;25(2):245-255. doi: 10.1007/s10741-019-09824-y [DOI] [PubMed] [Google Scholar]
- 31.Billi AC, Kahlenberg JM, Gudjonsson JE. Sex bias in autoimmunity. Curr Opin Rheumatol. 2019;31(1):53-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ventura-Clapier R, Moulin M, Piquereau J, et al. Mitochondria: a central target for sex differences in pathologies. Clin Sci (Lond). 2017;131(9):803-822. doi: 10.1042/CS20160485 [DOI] [PubMed] [Google Scholar]
- 33.Sampathkumar NK, Bravo JI, Chen Y, et al. Widespread sex dimorphism in aging and age-related diseases. Hum Genet. 2020;139(3):333-356. doi: 10.1007/s00439-019-02082-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wickham LA, Gao J, Toda I, Rocha EM, Ono M, Sullivan DA. Identification of androgen, estrogen and progesterone receptor mRNAs in the eye. Acta Ophthalmol Scand. 2000;78(2):146-153. doi: 10.1034/j.1600-0420.2000.078002146.x [DOI] [PubMed] [Google Scholar]
- 35.Marin-Castaño ME, Elliot SJ, Potier M, et al. Regulation of estrogen receptors and MMP-2 expression by estrogens in human retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2003;44(1):50-59. doi: 10.1167/iovs.01-1276 [DOI] [PubMed] [Google Scholar]
- 36.Hulsman CA, Westendorp IC, Ramrattan RS, et al. Is open-angle glaucoma associated with early menopause? The Rotterdam Study. Am J Epidemiol. 2001;154(2):138-144. doi: 10.1093/aje/154.2.138 [DOI] [PubMed] [Google Scholar]
- 37.Prokai-Tatrai K, Xin H, Nguyen V, et al. 17β-estradiol eye drops protect the retinal ganglion cell layer and preserve visual function in an in vivo model of glaucoma. Mol Pharm. 2013;10(8):3253-3261. doi: 10.1021/mp400313u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elliot SJ, Catanuto P, Espinosa-Heidmann DG, et al. Estrogen receptor beta protects against in vivo injury in RPE cells. Exp Eye Res. 2010;90(1):10-16. doi: 10.1016/j.exer.2009.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Du M, Mangold CA, Bixler GV, et al. Retinal gene expression responses to aging are sexually divergent. Mol Vis. 2017;23:707-717. [PMC free article] [PubMed] [Google Scholar]
- 40.Li B, Gografe S, Munchow A, Lopez-Toledano M, Pan ZH, Shen W. Sex-related differences in the progressive retinal degeneration of the rd10 mouse. Exp Eye Res. 2019;187:107773. doi: 10.1016/j.exer.2019.107773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guarneri R, Russo D, Cascio C, et al. Retinal oxidation, apoptosis and age- and sex-differences in the mnd mutant mouse, a model of neuronal ceroid lipofuscinosis. Brain Res. 2004;1014(1-2):209-220. doi: 10.1016/j.brainres.2004.04.040 [DOI] [PubMed] [Google Scholar]
- 42.Poloschek CM, Bach M, Lagrèze WA, et al. ABCA4 and ROM1: implications for modification of the PRPH2-associated macular dystrophy phenotype. Invest Ophthalmol Vis Sci. 2010;51(8):4253-4265. doi: 10.1167/iovs.09-4655 [DOI] [PubMed] [Google Scholar]
- 43.Lee W, Paavo M, Zernant J, et al. Modification of the PROM1 disease phenotype by a mutation in ABCA4. Ophthalmic Genet. 2019;40(4):369-375. doi: 10.1080/13816810.2019.1660382 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eTable 1. The Allele Frequencies of Mild and Severe ABCA4 Variants in the General Non-Finnish European Population
eTable 2. Origin of Participants
eFigure 1. Age Distribution by Sex
eFigure 2. Age of Onset Distribution by Sex
eFigure 3. Sex-Specific Penetrance Estimates for ABCA4 Alleles c.5603A>T and c.5882G>A
eReferences