

Targeting the main protease (Mpro) of SARS-CoV-2, we identified two indole-chloropyridinyl-ester derivatives, GRL-0820 and GRL-0920, active against SARS-CoV-2, employing RNA-qPCR and immunocytochemistry and show that the two compounds exerted potent activity against SARS-CoV-2. While GRL-0820 and remdesivir blocked SARS-CoV-2 infection, viral breakthrough occurred as examined with immunocytochemistry. In contrast, GRL-0920 completely blocked the infectivity and cytopathic effect of SARS-CoV-2 without significant toxicity. Structural modeling showed that indole and chloropyridinyl of the derivatives interacted with two catalytic dyad residues of Mpro, Cys145 and His41, resulting in covalent bonding, which was verified using HPLC/MS. The present data should shed light on the development of therapeutics for COVID-19, and optimization of GRL-0920 based on the present data is essential to develop more-potent anti-SARS-CoV-2 compounds for treating COVID-19.
KEYWORDS: COVID-19, SARS-CoV-2, main protease, antiviral agents
ABSTRACT
We assessed various newly generated compounds that target the main protease (Mpro) of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and various previously known compounds reportedly active against SARS-CoV-2, employing RNA quantitative PCR (RNA-qPCR), cytopathicity assays, and immunocytochemistry. Here, we show that two indole-chloropyridinyl-ester derivatives, GRL-0820 and GRL-0920, exerted potent activity against SARS-CoV-2 in cell-based assays performed using VeroE6 cells and TMPRSS2-overexpressing VeroE6 cells. While GRL-0820 and the nucleotide analog remdesivir blocked SARS-CoV-2 infection, viral breakthrough occurred. No significant anti-SARS-CoV-2 activity was found for several compounds reportedly active against SARS-CoV-2 such as lopinavir, nelfinavir, nitazoxanide, favipiravir, and hydroxychroloquine. In contrast, GRL-0920 exerted potent activity against SARS-CoV-2 (50% effective concentration [EC50] = 2.8 μM) and dramatically reduced the infectivity, replication, and cytopathic effect of SARS-CoV-2 without significant toxicity as examined with immunocytochemistry. Structural modeling shows that indole and chloropyridinyl of the derivatives interact with two catalytic dyad residues of Mpro, Cys145 and His41, resulting in covalent bonding, which was verified using high-performance liquid chromatography–mass spectrometry (HPLC/MS), suggesting that the indole moiety is critical for the anti-SARS-CoV-2 activity of the derivatives. GRL-0920 might serve as a potential therapeutic for coronavirus disease 2019 (COVID-19) and might be optimized to generate more-potent anti-SARS-CoV-2 compounds.
INTRODUCTION
The pandemic of novel coronavirus disease 2019 (COVID-19) caused by a positive-strand RNA virus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (formerly 2019-nCoV), started in Wuhan, Hubei province, China (1–4). As of 1 July 2020, more than 10 million COVID-19 cases, with nearly 500,000 deaths, had been reported in over 210 countries (5). The common symptoms of COVID-19 include fever, cough, fatigue, and dyspnea. Individuals with severe COVID-19 develop viral pneumonia and hypoxia requiring intubation and mechanical ventilation (6). Currently, no specific therapeutics or vaccines are available and rapid identification of efficacious drugs is urgently needed, while some drugs are still under investigation, including remdesivir (7). An efficient approach to drug discovery for treating a new pathogenic agent includes testing of existing compounds that are known to be active against related pathogens and the ensuing optimization of lead compounds. At the same time, structure-based design, synthesis, and identification of novel compounds potent against SARS-CoV-2 are needed.
Since the outbreaks of SARS and Middle East respiratory syndrome (MERS) in 2003 and in 2012, respectively, a large part of medicinal chemistry efforts for the control of SARS-CoV and MERS-CoV has been dedicated to design, synthesis, and identification of small compounds such as inhibitors of the main protease (Mpro) and papain-like protease (PLpro) of SARS-CoV. Structurally, the 5′ two-thirds of the viral genome encodes two overlapping polyproteins, pp1a and pp1ab, which are processed to generate the viral replication complex replicase polyprotein, which undergoes processing by two viral proteases, Mpro and papain-like protease (PLpro) (8–12). The two proteases are essential for replication of SARS-CoV, and both proteases have been recognized as attractive targets for developing antiviral agents (13–23). In addition, PLpro reportedly modifies host proteins posttranslationally to evade antiviral immune responses (24). We previously (between 2005 and 2009) designed, synthesized, and identified a series of SARS-CoV Mpro (also known as 3CLpro) inhibitors that bound to Mpro, enzymatically blocked Mpro activity, and exerted significant activity against SARS-CoV (20–23, 25). The present study shows that two compounds, GRL-0920 and GRL-0820, exert potent activity against SARS-CoV-2, while a number of previously known compounds such as favipiravir, nelfinavir, chloroquine, and hydroxychloroquine (except the nucleotide analogue remdesivir) failed to show significant activity in examinations of their antiviral effect using RNA-quantitative PCR (RNA-qPCR), cytopathicity, and immunocytochemistry.
RESULTS
Mpro is highly conserved between SARS-CoV and SARS-CoV-2.
SARS-CoV-2, which causes COVID-19, belongs to the family of betacoronaviruses that includes SARS-CoV and MERS-CoV. The genome of SARS-CoV-2 has ∼80% overall nucleotide identity with that of SARS-CoV (26), and the main proteases (Mpros) of these two viruses have 96% amino acid sequence identity (Fig. 1A). Superimposition of the structures of the main proteases of both SARS-CoV-2 and SARS-CoV exhibit nearly identical tertiary structures (Fig. 1B).
FIG 1.

Sequence conservation and structural comparison of four of coronaviruses’ main proteases. (A) The amino acid sequences of SARS-CoV-2 Mpro and SARS-CoV Mpro have 96% identity (12 amino acids [highlighted in blue] of 306 amino acids differ between the two). Conserved amino acids among the four Mpro sequences are shown in red. Distant coronavirus variants MERS-CoV and CoV-NL63 exhibit much less sequence conservation. Sequences were aligned using ENDscript server. (B) Structures of Mpro of SARS-CoV-2 (PDB entry 6LU7; in green) superimposed on Mpro of SARS-CoV (2AMQ; in blue), MERS-CoV (5C3N; in salmon), and CoV-NL63 (5GWY; in yellow) are shown. Catalytic dyad residues Cys145 and His41 are indicated in stick mode. For clarity, only a monomer structure is shown and the covalent protease inhibitor N3 was omitted. The picture was generated using UCSF Chimera.
In this regard, we previously designed, synthesized, and identified a series of Mpro inhibitors between 2005 and 2009 that bound to Mpro and exerted significant activity against SARS-CoV (19–23, 25). On the basis of the high structure homology between SARS-CoV Mpro and SARS-CoV-2 Mpro mentioned above, we examined the potential activity of such experimental SARS-CoV Mpro inhibitors against SARS-CoV-2. We also newly synthesized a series of potential SARS-CoV-2 Mpro inhibitors. In the present study, we determined their activity against SARS-CoV-2 using VeroE6 cells or TMPRSS2-overexpressing VeroE6 cells (VeroE6TMPRSS2) as target cells and a SARS-CoV-2 strain, JPN/TY/WK-521 (SARS-CoV-2WK-521), employing a quantitative RNA-qPCR assay with cell culture supernatants, cytotoxicity assays, and immunocytochemistry.
GRL-0820 and GRL-0920 exert potent activity against SARS-CoV-2.
We first tested the antiviral activity of the compounds against SARS-CoV-2WK-521 by employing a series of compounds that had previously been reported to be active against SARS-CoV (17–19) and identified GRL-0920 and a newly synthesized compound, GRL-0820, as potent inhibitors of SARS-CoV-2 (Table 1). When VeroE6 cells were exposed to SARS-CoV-2WK-521 at a multiplicity of infection (MOI) of 0.05 and cultured in the presence of various concentrations of the two indole chloropyridinyl esters GRL-0820 and GRL-0920, the compounds were found to be highly potent against SARS-CoV-2WK-521 with 50% effective concentration (EC50) values of 15 ± 18 and 2.8 ± 0.3 μM, respectively, using RNA-qPCR (Table 1). We further examined the antiviral activity of these two compounds together with other selected compounds that had previously been reported to be active against SARS-CoV and/or SARS-CoV-2, including remdesivir, lopinavir, nelfinavir, favipiravir, hydroxychloroquine, nitazoxanide, and nafamostat (7, 27–29). To compare the antiviral activities of the 9 compounds as accurately as possible, the compounds were examined at the same time in one assay. The data shown in Fig. 2 (see also Table S1 in the supplemental material) confirmed that GRL-0820 and GRL-0920 were active against SARS-CoV-2WK-521 with EC50 values of 23 ± 5 and 8.6 ± 2.8 μM, respectively. Remdesivir was also found to be active against SARS-CoV-2WK-521 (EC50 = 1.2 ± 0.06 μM). The EC50 values for lopinavir, nelfinavir, and nitazoxanide were 19 ± 8, 3.1 + 0.06, and 31 ± 6 μM, respectively; however, their specificity index values were only 2.7, 17, and 2.8, respectively (Table S1). Thus, such EC50 values were thought possibly to be affected by their cytotoxicity (see below). While favipiravir and nafamostat were apparently nontoxic at all concentrations tested, the EC50 values of both were >100 μM, suggesting that neither of the compounds was effective against the virus (Fig. 2; see also Table S1). Hydroxychloroquine appeared to be nontoxic at up to 100 μM, and its EC50 turned out to be 5.1 ± 3.4 μM; however, its cytotoxicity was suspected to have contributed to its apparent antiviral activity (see below).
TABLE 1.
Activity of GRL-0820 and GRL-0920 against the infectivity and replication of SARS-CoV-2 in VeroE6 cellsa
| Compound | Structure | Anti-SARS-CoV (μM) | Anti-SARS-CoV-2 (μM) |
|---|---|---|---|
| GRL-0820 | 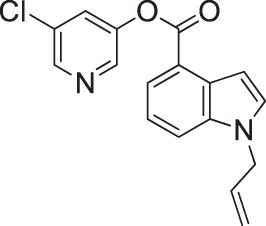 |
ND | 15 ± 18 |
| GRL-0920 | 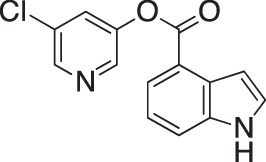 |
6.9 ± 0.9 | 2.8 ± 0.3 |
The data shown represent mean EC50 values ± standard deviations (SD). The assays were conducted on at least 3 different occasions, and the data show representative results. All assays were performed in duplicate. ND, not done.
FIG 2.

Antiviral activity of selected compounds against SARS-CoV-2. VeroE6 cells were exposed to SARS-CoV-2WK-521. The viral copy numbers in the culture supernatants (left axis, open bars) and percent inhibition (right axis, red lines) of each compound in VeroE6 cells were determined using RT-qPCR. Each compound was tested on at least 3 different occasions. For details of the EC50 and CC50 values, see Table S1. The data illustrate representative ones and are shown as means ± standard deviations (SD). All compounds were tested and compared in one assay performed in duplicate.
Antiviral activity and cytotoxicity of compounds against SARS-CoV-2. A lowercase superscript “a” indicates that each selectivity index value denotes a ratio of CC50 to EC50. The data shown represent mean EC50 and CC50 values ± standard deviations (SD). The assays were conducted on at least 3 different occasions, and the data shown here are representative of the results. All assays were performed in duplicate. Download Table S1, DOCX file, 0.01 MB (14.3KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Immunocytochemistry features of VeroE6 and VeroE6TMPRSS2 cells exposed to SARS-CoV-2.
We first examined the immunocytochemistry features of VeroE6 cells under conditions of exposure of the cells to SARS-CoV-2WK-521. When VeroE6 cells were cultured alone and stained with Texas Red-X dye conjugated phalloidin, the cytoskeleton filamentous actin (F-actin) was well visualized as a mesh-like structure (see top left inset in Fig. S1 in the supplemental material), suggesting that the cells were healthy and replicating. In contrast, when the cells were exposed to SARS-CoV-2WK-521, the F-actin was rearranged, disrupted, and destroyed upon infection by SARS-CoV-2WK-521. Superimposition of 3-color images (red, blue, and green, indicating F-actin, nuclei, and SARS-CoV-2 antigens, respectively) showed that no nonspecific staining with the antibody against SARS-CoV-2 (an IgG fraction from the serum of a COVID-19-convalescent patient [convIgG] was used as the primary antibody for immunofluorescence staining) had occurred in SARS-CoV-2WK-521-unexposed VeroE6 cells (top right inset in Fig. S1). However, most of the SARS-CoV-2WK-521-exposed VeroE6 cells stained in green, indicating most of those cells were infected and producing viral components (bottom right inset in Fig. S1).
Visualization of SARS-CoV-2 infection using immunocytochemistry. VeroE6 cells were exposed to SARS-CoV-2WK-521 at MOI of 0.05 and cultured in the absence of test compound. After 3 days, the cells were fixed with 4% paraformaldehyde and immunofluorescent staining using an IgG fraction from serum of a COVID-19 individual was performed. SARS-CoV-2 antigens, F-actin, and nuclei are shown in green, red, and blue, respectively. The upper three insets show images of VeroE6 cells cultured alone, while the lower three insets show images of VeroE6 cells exposed to SARS-CoV-2WK-521 and cultured in the absence of test compound. The upper and lower extreme right insets represent merged images, which are composed of images obtained from 3-color fluorescence. The lower left panel shows that the structure of one of the cytoskeletons, the actin filament, was destroyed by SARS-CoV-2WK-521 infection. Almost all cells are stained in green (SARS-CoV-2 positive) in the lower right panel, and no nonspecific staining (in green) is seen in the upper right panel. Download FIG S1, PDF file, 0.3 MB (291.8KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
We also examined the cell destruction effects of the virus by the use of immunocytochemistry performed with two cell lines, VeroE6 and VeroE6TMPRSS2 cells. The images in Fig. S2 show that robust actin filaments were observed when the VeroE6 and VeroE6TMPRSS2 cells were cultured alone (insets a and d), while actin filaments were all gone (inset b) and most of those cells were stained in green when convIgG was used as the primary antibody in VeroE6 cells exposed to SARS-CoV-2WK-521 and cultured in the absence of test compounds. When the highly SARS-CoV-2-susceptible VeroE6TMPRSS2 cells (30) were exposed to SARS-CoV-2WK-521, most of the cells were infected, their actin filaments were totally destroyed, and the cells were killed by the virus detached from the bottom of microculture titer plate wells, resulting in the loss of the cells (inset e). We also employed a murine anti-spike monoclonal antibody as the primary antibody in our immunostaining (insets c and f in Fig. S2). convIgG detected much higher levels of SARS-CoV-2-infected cells than were seen when the murine anti-spike monoclonal antibody was used as the primary antibody (compare insets b and c). Thus, we chose the IgG fraction from the convalescent COVID-19 individual for use as the primary antibody for further study.
Different multiplicities of SARS-CoV-2 infection. VeroE6 and VeroE6TMPRSS2 cells were exposed to SARS-CoV-2WK-521, and immunocytochemistry using the IgG fraction from a convalescent individual with COVID-19 (convIgG) (insets b and e) or murine (insets c and f) anti-spike monoclonal antibody (MAb) as the primary antibody was performed. SARS-CoV-2 antigens (insets b and and e) and SARS-CoV-2 spike antigen (insets c and f), F-actin, and nuclei are shown in green, red, and blue, respectively. Insets a and d show images of VeroE6 and VeroE6TMPRSS2 cells cultured alone and stained, respectively, while insets b and c show those of VeroE6 cells and insets e and f show those of VeroE6TMPRSS2 cells exposed to SARS-CoV-2WK-521. Download FIG S2, PDF file, 0.2 MB (235.4KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
It is known that the infectivity and replication of SARS-CoV-2 significantly vary depending on the types of target cells used. Therefore, we quantitatively examined the infectivity and replication of SARS-CoV-2WK-521 in two cell lines, VeroE6 cells and VeroE6TMPRSS2 cells plus SARS-CoV-2WK-521. Upon exposure of VeroE6 cells to SARS-CoV-2WK-521, the viability of the VeroE6 cells was reduced to close to 55% by 48 h following the viral exposure and the viability was found to have further reduced to ∼50% by 96 h in culture (Fig. S3A). In contrast, the viability of VeroE6TMPRSS2 cells exposed to SARS-CoV-2WK-521 went down quickly to ∼20% by 48 h in culture and further down to ∼0% to ∼5% by 72 h in culture. We also determined the numbers of SARS-CoV-2WK-521 RNA copies in the supernatants in the same experiment. In the 24 and 48 h following the viral exposure, VeroE6TMPRSS2 cells produced greater numbers of SARS-CoV-2WK-521 RNA copies than VeroE6 cells, while at 72 h and after, the VeroE6 cells and VeroE6TMPRSS2 cells produced comparable numbers of RNA copies (Fig. S3B). Images of the cultures captured under light microscopy and immunocytochemistry results confirmed that the VeroE6TMPRSS2 cells were more susceptible to the infectivity and cytopathicity of SARS-CoV-2WK-521 (Fig. S3C). Thus, the assay performed using VeroE6 cells and SARS-CoV-2WK-521 is referred to as a “low-stringency assay” in the present report, while that performed using VeroE6TMPRSS2 cells and SARS-CoV-2WK-521 is referred to as a “high-stringency assay.”
VeroE6TMPRSS2 cells have greater susceptibility to SARS-CoV-2WK-521 than VeroE6 cells. (A and B) Cell viability (A) and viral RNA copy numbers (B) of SARS-CoV-2WK-521 in VeroE6 (○) and VeroE6TMPRSS2 (●) cells. VeroE6 and VeroE6TMPRSS2 cells were exposed to SARS-CoV-2WK-521 (300 TCID50) for 1 h, and the virus was then washed off. The viability of VeroE6TMPRSS2 cells was lower than that of VeroE6 cells from 48 h postinfection (hpi), and virus copy numbers were higher in VeroE6TMPRSS2 cells than in VeroE6 cells at 24 and 48 hpi. Cell viability (the level of viability of virus-unexposed cells is assigned a value of 100%) and viral copy numbers were determined using WST-8 assay and RNA-qPCR, respectively. All assays were performed in quadruplicate, and each plot denotes each value. (C) Visualization of SARS-CoV-2 infection using immunocytochemistry. Images of VeroE6 and VeroE6TMPRSS2 cells exposed to SARS-CoV-2WK-521 were captured using light microscopy (upper lines) and immunocytochemistry (lower lines). SARS-CoV-2 antigens, F-actin, and nuclei are shown in green, red, and blue, respectively. A higher number of SARS-CoV-2-positive cells was seen in VeroE6TMPRSS2 cells than in VeroE6 cells at 24 hpi. At 48 hpi and beyond, most of VeroE6TMPRSS2 cells were destroyed by the cytopathicity of the virus, but VeroE6 cells remained on the bottom of the microtiter culture plate. Download FIG S3, PDF file, 0.5 MB (529.3KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Association of apparent antiviral activity with the cytostatic/cytotoxic nature of test compounds.
We carefully investigated whether the two compounds GRL-0920 and remdesivir exerted antiviral activity at 1, 10, and 100 μM (Fig. 3A) without significant cytostatic or cytotoxic effects. We first examined light microscopic images of VeroE6 cells exposed to SARS-CoV-2WK-521 and cultured in the presence of GRL-0920 or remdesivir (Fig. 3A). VeroE6 cells were exposed to SARS-CoV-2WK-521 for 1 h, the virus was washed off, and the cells were cultured in the presence of various concentrations of test compound for 3 days. VeroE6 cells cultured alone (left top inset, Fig. 3A) appear to be robust; however, many of the cells exposed to SARS-CoV-2WK-521 and cultured in the absence of test compounds showed granular patterns (bottom inset at left in Fig. 3A). All of the VeroE6 cells exposed to the virus and cultured with GRL-0920 or remdesivir (at 100 μM) appeared to be protected (top row of insets at right in Fig. 3A).
FIG 3.

GRL-0920 and remdesivir virtually completely block the infectivity and cytopathic effect of SARS-CoV-2WK-521 in VeroE6 and VeroE6TMPRSS2 cells. (A) GRL-0920 potently blocks the cytopathic effect of SARS-CoV-2 in VeroE6 cells. VeroE6 cells were exposed to SARS-CoV-2WK-521, cultured in the presence of GRL-0920 or remdesivir, and examined under a microscope (magnification, ×200). (B) GRL-0920, GRL-0820, and remdesivir potently blocked the infectivity and cytopathic effect of SARS-CoV-2WK-521 in VeroE6TMPRSS2 cells, while lopinavir, nelfinavir, and favipiravir failed to block the infection. (C) GRL-0920 at 100 μM completely blocked SARS-CoV-2WK-521 infection in VeroE6 cells, while nafamostat and hydroxychloroquine did not exert significant antiviral activity. Nafamostat did not exert significant toxicity to the cells, but it totally failed to block the infectivity of the virus. For immunocytochemistry, the IgG fraction of serum from a convalescent COVID-19 individual was employed as the primary antibody. The SARS-CoV-2 antigens, F-actin, and nuclei are shown in green, red, and blue, respectively. The image in each inset in panels B and C represents the merged image.
In order to corroborate the data representing the antiviral activity of GRL-0920 and remdesivir described above, we further examined the inhibitory activity of those compounds using detailed immunocytochemistry. When VeroE6TMPRSS2 cells, which are known to be highly susceptible to the infectivity of SARS-CoV-2 (30), were cultured alone, robust cellular cytoskeleton filamentous actin (F-actin) was seen in the form of mesh-like structures in red and a number of nuclei (in blue) were identified, signifying that those cells were healthy and replicating (top panel at extreme left in Fig. 3B). However, when VeroE6TMPRSS2 cells were exposed to SARS-CoV-2WK-521 and cultured in the absence of test compounds, the results showed that most of the cells had been infected and destroyed by the virus, were detached from the bottom of the microtiter culture plates, and had been lost during the staining procedure as seen with fluorescence microscopy (bottom, extreme left in Fig. 3B). Most of the residual cells stained in green, indicating that those cells were infected and producing viral proteins and that the cellular F-actin was almost totally lost due to rearrangements and destruction caused by the virus (31). When VeroE6TMPRSS2 cells were exposed to SARS-CoV-2WK-521 and cultured in the presence of various concentrations of lopinavir and nelfinavir, many virus-infected cells were seen at 1 and 10 μM and stained in green, indicating that these two compounds had no detectable antiviral activity in the assay. At 100 μM, most of the cells were lost (Fig. 3B; see also Fig. S4A), presumably due to both the infection by the virus and the toxicity of the agents. These data strongly suggest that the apparent antiviral effect of nelfinavir and lopinavir stemmed from their cytotoxicity, producing “false” antiviral effects as assessed using RNA-qPCR (Fig. 2). In contrast, when SARS-CoV-2WK-521-exposed VeroE6TMPRSS2 cells were cultured in the presence of 100 μM GRL-0920 or remdesivir, there were essentially no infected cells seen. The cellular F-actin was almost completely preserved, indicating that the SARS-CoV-2WK-521-exposed VeroE6TMPRSS2 cells remained healthy and replicating (Fig. 3B). Of note, the level of antiviral activity of GRL-0820 was moderate and a substantial number of cells stained in green, indicating that viral breakthrough had occurred even with 100 μM GRL-0820 (Fig. 3B).
GRL-0920 and remdesivir block SARS-CoV-2 infection. (A) Lopinavir (d to g), nelfinavir (h to k), and nafamostat (l to o) failed to block SARS-CoV-2 infection in VeroE6 cells. Images of VeroE6 cells exposed to the virus and cultured alone are shown in insets a to c. (B) Remdesivir (d to g) significantly blocked SARS-CoV-2 infection but allowed viral breakthrough in VeroE6 cells. Images of virus-exposed cells cultured alone are shown in insets a to c. Chloroquine (h to k) and hydroxychloroquine (i to o) failed to block the infectivity of SARS-CoV-2WK-521, and almost all the cells were shed from the wells at 100 μM. Favipiravir (p to s) also failed to block SARS-CoV-2 infection. (C) GRL-0920 completely blocked the infectivity of SARS-CoV-2WK-521, but GRL-0820 permitted viral breakthrough in VeroE6 cells. Images of virus-exposed VeroE6 cells cultured alone are shown in insets a to c. (d to n) Images of VeroE6 cells exposed to SARS-CoV-2WK-521 and cultured in the absence (d to f) or presence (g to n) of each compound. The cells were stained using the IgG fraction from serum of a convalescent COVID-19 individual as the primary antibody. SARS-CoV-2 antigens, F-actin, and nuclei are shown in green, red, and blue, respectively. Download FIG S4, PDF file, 0.9 MB (965.6KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
GRL-0920 completely blocks the infectivity of SARS-CoV-2, while various compounds reportedly active against SARS-CoV-2 fail to block infectivity.
VeroE6TMPRSS2 cells are highly susceptible to the infectivity of SARS-CoV-2WK-521 (insets b and e in Fig. S2). In order to ensure detection of even moderate antiviral activity of test compounds against SARS-CoV-2, we subsequently used the low-stringency combination of VeroE6 cells and SARS-CoV-2WK-521 to further examine selected compounds, including nitazoxanide, nafamostat, chloroquine, and hydroxychloroquine. As shown in Fig. 3C (see also Fig. S4A), nitazoxanide did not exert activity against the virus at 1 and 10 μM and only a few nuclei remained at 100 μM, strongly suggesting that nitazoxanide had no antiviral activity and that its cytotoxicity resulted in a false antiviral effect as assessed using RNA-qPCR and shown in Fig. 2. Nafamostat was not significantly cytotoxic but was inactive against the virus at all the concentrations tested (Fig. 3C; see also Fig. S4A). Two compounds, chloroquine and hydroxychloroquine, appeared to slightly block infectivity at 10 μM, allowing actin filaments to remain, although a number of the cells had been infected with the virus (Fig. 3C) (Fig. S4B; see also Fig. S5). In this regard, chloroquine reportedly blocks the catabolism and/or degradation of cellular proteins through increasing endosomal pH, possibly resulting in the maintenance of actin filaments despite of SARS-CoV-2WK-521 infection (32). Chloroquine also reportedly interferes with glycosylation of the ACE2 receptor, one of the cellular entry points of SARS-CoV, possibly resulting in moderating the infection by SARS-CoV-2 (7, 33). However, almost all VeroE6 cells were lost when the cells were cultured in the presence of 100 μM chloroquine because of its toxicity (Fig. 3C) (Fig. S4B; see also Fig. S5). The effects of hydroxychloroquine were almost the same as those of chloroquine. Of note, when VeroE6 cells were exposed to SARS-CoV-2WK-521 and cultured in the presence of 10 μM chloroquine or hydroxychloroquine, a number of the cells were infected by the virus; nevertheless, the actin filaments were moderately conserved (Fig. S5B). In this regard, the WST-8 assay used in the present study (34) evaluated the mitochondrial NAD(P)H-dependent cellular oxidoreductase enzyme activity in living cells that results in reduction of WST-8 to formazan dyes. Therefore, the data from WST-8 assays, obtained under defined conditions, reflect the number of viable cells. Chloroquine reportedly does not target mitochondria but damages other cellular organelles involved in protein synthesis and metabolism such as lysosome, endoplasmic reticulum, and Golgi apparatus, resulting in compromised cellular growth and functions (34). Thus, the results of WST-8 assays did not reflect cellular robustness or functionality, especially with regard to chloroquine and hydroxychloroquine. As shown in Fig. S4B, in the VeroE6 cells exposed to relatively low concentrations (1 and 10 μM) of chloroquine and hydroxychloroquine, following infection, the virus did not replicate well because the protein synthesis in the VeroE6 cells was compromised by the compounds, moderately maintaining actin filament structures. However, the damaging effects of chloroquine and hydroxychloroquine at 100 μM were significant and virtually no cells were left in the wells of microtiter culture plates (Fig. 3C) (Fig. S4B; see also Fig. S4).
Chloroquine and hydroxychloroquine fail to block the infectivity of SARS-CoV-2WK-521 in VeroE6 cells. VeroE6 cells were exposed to SARS-CoV-2WK-521 and cultured in the presence of chloroquine or hydroxychloroquine. Immunocytochemistry using an IgG fraction from serum of a convalescent COVID-19 individual was performed. SARS-CoV-2 antigens, F-actin, and nuclei are shown in green, red, and blue, respectively. The images in the upper and lower left insets show those acquired under a light microscope (magnification, ×200), showing that whereas the VeroE6 cells cultured alone were all alive (upper inset), the VeroE6 cells exposed to SARS-CoV-2WK-521 were killed and detached from the culture wells (lower inset). VeroE6 cells exposed to the virus and cultured in the presence of chloroquine or hydroxychloroquine at 1 and 10 μM were infected with the virus, and virtually all of those cultured at 100 μM were seen to have been shed from the wells of culture plates due to the damage to the cells imposed by the compounds. Download FIG S5, PDF file, 0.5 MB (556.4KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
GRL-0920 reproducibly and completely blocked the infectivity and cytopathic effect of SARS-CoV-2WK-521 as examined with the low-stringency assay using VeroE6 cells (Fig. 3C; see also Fig. S4C). When VeroE6 cells were exposed to SARS-CoV-2WK-521 and cultured in the presence of remdesivir or GRL-0920 at 0.01, 0.1, 1, 10, and 100 μM, there was further suppression of viral replication at 10 μM and either nearly complete (remdesivir) or complete (GRL-0920) suppression at 100 μM as assessed using either the convIgG or the murine anti-spike monoclonal antibody as the primary antibody (Fig. S6). Examining the activity of 100 μM remdesivir and GRL-0920 against SARS-CoV-2WK-521 with VeroE6 cells, the appearance of actin filaments proved to be virtually the same as seen in the high-stringency assay performed using VeroE6TMPRSS2 cells (Fig. 3B). These data strongly suggest that the VeroE6 cells protected by remdesivir and GRL-0920 were viable and that the integrity of the cells was likely maintained.
GRL-0920 completely blocks the infectivity and cytopathic effect of SARS-CoV-2. VeroE6 cells were exposed to SARS-CoV-2WK-521 at an MOI of 0.05 and cultured in the presence of 0.01, 0.1, 1, 10, and 100 μM remdesivir or GRL-0920. After 3 days, the cells were fixed with 4% paraformaldehyde and immunofluorescent staining using the IgG fraction from serum of a convalescent individual with COVID-19 or a murine anti-SARS-CoV-2 spike monoclonal antibody was performed. SARS-CoV-2 antigens, F-actin, and nuclei are shown in green, red, and blue, respectively. The inset at the extreme upper left shows an image of VeroE6 cells cultured alone, and the inset at the extreme lower left shows that of VeroE6 cells exposed to SARS-CoV-2WK-521 and cultured in the absence of test compound. All the images except the two insets described above show images captured with 3-color fluorescence. Download FIG S6, PDF file, 0.3 MB (319.8KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
GRL-0920 and GRL-0820 covalently bind to Mpro of SARS-CoV-2.
Finally, we built molecular models to understand the structural interactions of GRL-09-20 and GRL-0820 with Mpro of SARS-CoV-2. We started with the recently published crystal structure of SARS-CoV-2 Mpro (RCSB PDB identifier [ID] 6Y2F) (35). GRL-0920 represents an indole chloropyridinyl ester. GRL-0820 has the same moiety as GRL-0920 but with an additional propene substituent on the indole nitrogen. When Mpro Cys145 attacked GRL-0920 and formed an initial complex in the active site (Fig. 4a), the hydroxyl oxygen (or the oxyanion) formed hydrogen bond interactions with the backbone amide nitrogens of Gly143, Ser144, and Cys145. The indole moiety is appropriately positioned to form pi-pi interactions with His41 and His163. The pyridinyl nitrogen is positioned to form polar interactions with Gly143, and the chlorine atom forms halogen bond interactions with the backbone amide nitrogen of Thr26. These interactions of different moieties of GRL-0920, including the chloropyridinyl group, may help stabilize the initial reaction intermediate. Following acylation, the chloropyridinyl group departs, and the carbonyl indole is bound to Cys145 with a covalent bond. The presence of covalently bound carbonyl indole was verified by high-performance liquid chromatography–mass spectrometry (HPLC/MS). Newly generated recombinant SARS-CoV-2-Mpro was incubated with dimethyl sulfoxide (DMSO), GRL-0820, or GRL-0920 for 30 min. DMSO-treated Mpro gave a molecular weight of 33,853.36 consistent with full-length Mpro (expected molecular weight, 33,853.53) (Fig. S7A). GRL-0920 treatment and GRL-0820 treatment increased the mass by 143.96 and 183.85 amu (Fig. S7B and C), respectively. The increase in the observed mass of Mpro is consistent with acylation of Mpro by two compounds (expected increases of 143.51 and 183.2). The modeled complex of Mpro with GRL-0920 was further minimized, and the interactions are shown in Fig. 4b. The carbonyl carbon has hydrogen bond interactions with the backbone amine nitrogens of Gly143, Ser144, and Cys145. Slight rotations of the rings make favorable pi-pi interactions of both His41 and His163 with the indole moiety. We also examined the interactions of GRL-0820 with Mpro. The initial and final complexes are shown in Fig. 4c and d, respectively. The presence of the propene substituent seems to have drastically reduced the binding interactions of GRL-0820 in comparison to GRL-0920. While the chloropyridyl group has polar interactions, there was only one polar interaction of the hydroxyl (or oxyanion) moiety with the backbone of Cys145. The polar interactions with Gly143 and Ser144 seen for GRL-0920 were lost. These reductions in interactions are most likely due to the change in conformation with the additional substituent and to the shallow binding site of Mpro. The GRL-0820 complex with SARS-CoV-2 Mpro seen following completion of the acylation reaction is shown in Fig. 4d. This complex also had a lower number of polar interactions with Mpro than the GRL-0920 complex. Overall, the difference in these interactions may at least in part explain the greater potency of GRL-0920 than GRL-0820.
FIG 4.

Molecular models of interactions of GRL-0920 and GRL-0820 with SARS-CoV-2 Mpro. The proposed mechanism is the formation of a covalent bond of GRL-0920 and GRL-0820 with Cys145 of SARS-CoV-2 Mpro (RCSB PDB ID 6Y2F) through acylation, followed by the departure of the chloropyridine moiety. The location and interactions of the active site residues with GRL-0920 prior to cleavage are shown in panel a and after the loss of the chloropyridine moiety are shown in panel b. Similar interactions of SARS-CoV-2 Mpro with GRL-0820 are shown in panels c and d. The reaction may be reversible with the existence of both forms. The carbons of GRL-0920 and GRL-0820 are shown in magenta, whereas the carbons of Mpro in gray. Nitrogen, oxygen, sulfur, chlorine, and polar hydrogens are shown in blue, red, yellow, dark green, and white, respectively. The covalent bond is shown with a green arrow. Hydrogen and halogen bonds are shown with yellow and magenta dashed lines, respectively.
Mass analysis of the complex of Mpro of SARS-CoV-2 with GRL-0920 or GRL-0820. (A) Mass detection of Mpro of SARS-CoV-2 alone was done with an Agilent 6230 time of flight (TOF) mass spectrometer, and data are shown with the total ion current (TIC). The major protease species had an average mass of 33,853.36 (expected mass, 33,853.53). The inset shows the molecular ion profile for the main eluting Mpro peak, and the deconvolution result for that ion profile is shown at the bottom. (B and C) Mass detection of the complex of Mpro with GRL-0920 (B) or GRL-0820 (C) was done, and data are shown with the total ion current (TIC). The major Mpro species with GRL-0920 and GRL-0820 had average masses of 33,997.27 (expected mass, 33,996.68 for acylation of the Mpro with GRL-0920) and 34,037.21 (expected mass, 34,036.73 for acylation of the protease), respectively. The upper inset shows the molecular ion profile for the main eluting protease peak, and the lower inset shows the deconvolution results for that ion profile. Download FIG S7, PDF file, 1.3 MB (1.3MB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Thermal stability of Mpro in the absence or presence of GRL-0920.
We also examined the thermal stability of Mpro in the presence of GRL-0920 using differential scanning fluorimetry (DSF) and CYPRO orange (36, 37). As illustrated in Fig. 5, the melting temperature (Tm) of Mpro (5 μM) alone in experiment 1 was 53.63°C, while the Tm value decreased to 51.04, 49.13, and 46.98°C in the presence of 5, 50, and 100 μM GRL-0920, respectively. The shift of Tm to lower temperatures is reportedly due to the destabilization of the protein by covalent binding compounds (37). Thus, the present data corroborate that GRL-0920 forms a covalent bond with Mpro. As a control, we determined the Tm value for HIV-1 protease in the presence of an HIV-1 protease inhibitor, GRL-142 (36). The Tm value determined for HIV-1 protease (5 μM) alone in experiment 2 was 54.78°C, while when the same HIV-1 protease was made to interact with GRL-142, the Tm value significantly shifted to the right, giving a Tm value of 87.13°C and signifying that GRL-142 had very effective noncovalent interactions. Therefore, these thermal stability data corroborate our nanoLC-MS results indicating that GRL-0920 forms covalent interactions with Mpro and that GRL-0920 exerts its antiviral activity against SARS-CoV-2 by performing covalent binding with Mpro.
FIG 5.

Thermal stability of Mpro in the presence or absence of GRL-0920. Thermal stability of Mpro in the presence of GRL-0920 was analyzed using differential scanning fluorimetry (DSF) as described for experiment 1. Note that the Tm value of Mpro (5 μM) alone was 53.63°C, while the Tm values decreased to 51.04, 49.13, and 46.98°C in the presence of 5, 50, and 100 μM GRL-0920, respectively, suggesting that the Mpro get destabilized when GRL-0920 covalently bound to Mpro. Note that, as expected, the presence of HIV-1 protease inhibitor GRL-142 (5 μM) increased the thermal stability of HIV-1 protease (5 μM) as shown for experiment 2.
DISCUSSION
While GRL-0820 and remdesivir significantly blocked the infectivity and replication of SARS-CoV-2, they still permitted viral breakthrough (Fig. 3B; see also Fig. S6 in the supplemental material). However, GRL-0920 completely blocked the infectivity, replication, and cytopathic effect of SARS-CoV-2 in both the high-stringency and low-stringency assays. In our study, the EC50 value of nitazoxanide was 31 ± 6 μM; however, its specificity index was only 2.8 (see Table S1 in the supplemental material). Thus, such an EC50 value was thought to be affected by its inherent cytotoxicity. In fact, nitazoxanide did not block the infectivity of SARS-CoV-2WK-521 as examined with immunocytochemistry (Fig. 3C). With regard to another compound, Hoffman et al. recently reported that nafamostat, a serine protease inhibitor, blocked SARS-CoV-2 infection in Calu-3 cells exposed to SARS-CoV-2 with an EC50 value of as low as 5 nM (38). Note, however, that Calu-3 cells are not very susceptible to SARS-CoV-2 infection and do not support viral replication. Thus, in the assays performed using Calu-3 cells and in an alternative assay that was performed without using live infectious SARS-CoV-2, instead using “pseudotype entry” as an endpoint, it was found that the EC50 values can be excessively low (i.e., 5 nM) (28, 38) compared to the EC50 values obtained in the assays using other cell lines such as VeroE6 cells. Thus, we refer to the cell-based assay systems using Calu-3 cells as a “very-low-stringency” assays, where moderately or only slightly active compounds tend to get very low EC50 values. Using both VeroE6 cells and VeroE6TMPRSS2 cells with SARS-CoV-2WK-521 in the present study, nafamostat failed to show significant antiviral activity (EC50 values of >100 μM). Thus, we conclude that neither nitazoxanide nor nafamostat exerted significant activity against SARS-CoV-2WK-521 in the present study.
As described above, cytostatic and cytotoxic effects of test compounds in cell-based assays are often mistakenly interpreted as representative of apparent antiviral activity since the production of the relevant virus is reduced by the inherent cytostatic and cytotoxic effects of the test compounds (39, 40). Indeed, none of such toxic agents (i.e., daunorubicin and adriamycin) have proven to be of clinical utility as antiviral agents. Of note, we seriously attempted to sever the data representing reduction of virus copy numbers due to cytostatic/cytotoxic effects from the data representing the virus-specific inhibitory activity of the test compounds. We believe that our detailed immunocytochemistry results clearly segregated the effects of virus-specific antiviral activity from the cytostatic/cytotoxic effects of the test compounds. We conclude that in the present study, no detectable anti-SARS-CoV-2 activity was present in compounds such as HIV-1 protease inhibitors (nelfinavir and lopinavir), favipriavir, hydroxychloquine, and others reportedly active against SARS-CoV-2 and that GRL-0920 potently blocked the infectivity and cytopathicity of SARS-CoV-2. Indeed, the results from the present study show that the previously reported activity of nelfinavir, lopinavir, nitazoxanide, chroloquine, and hydroxychroloquine against SARS-CoV-2 (7, 27–29) was incorrectly judged to represent specific activity against SARS-CoV-2 and that the reduction of viral production observed was due to inherent cytostatic and/or cytotoxic effects of those compounds. Favipiravir and nafamostat were not very toxic but did not show detectable antiviral activity in the present study.
Our modeling studies strongly suggest that GRL-0820 and GRL-0920 interact with Mpro and exert their activity against SARS-CoV-2. The results of our Mpro molecular weight analysis performed with HPLC/MS (Fig. S7) show that both compounds covalently bond with Mpro. Furthermore, the data representing thermal stability of Mpro in the presence of GRL-0920 showed a relatively odd feature, i.e., a shift of the stability curve to a lower temperature, corroborating the idea that GRL-0920 forms covalent bonds with Mpro. In regard with the covalent bonding, it is concerning that compounds forming irreversible covalent interactions may cause permanent injuries to critical cellular components by binding covalently to produce serious adverse effects. However, the duration of administration of a therapeutic(s) to patients with COVID-19 could be as short as 5 days or weeks at the longest. Thus, certain adverse effects due to such remedies might be acceptable if lives are expected to be saved. Also, it is possible that GRL-0920-Mpro covalently linking ketal may convert to a carbonyl with noncovalent binding with Mpro in a reversible manner. Therefore, the covalent interactions of GRL-0920 and its analogs may not pose serious toxicity. GC-376, reportedly one of the most potent SARS-CoV-2 Mpro inhibitors, has an EC50 value of 3.37 μM as examined with regard to the inhibition of cytopathic effect of SARS-CoV-2 using VeroE6 cells (41). The EC50 value representing the potency of GC-376 is comparable to the EC50 value (8.6 ± 2.8 μM) determined for GRL-0920 (Fig. 2; see also Table S1); however, for an accurate comparison, both compounds have to be tested within the same assay using the same cell type and the same virus strain. GRL-0920 might thus serve as a potential therapeutic agent against COVID-19, and optimization of GRL-0920 based on the present data is essential to develop more-potent anti-SARS-CoV-2 compounds for treating COVID-19. However, the potency of GRL-0920 is moderate, and optimization is essential. It is noteworthy that a series of SARS-CoV PLpro inhibitors have been reported (17–19, 25). Combination therapy consisting of a potent SARS-CoV-2 Mpro inhibitor and a potent SARS-CoV-2 PLpro inhibitor could benefit infected individuals significantly more than monotherapy performed with a SARS-CoV-2 protease inhibitor. Moreover, if the combination of a potent novel Mpro inhibitor and a potent SARS-CoV-2 RNA polymerase inhibitor proves to be significantly more effective against the virus than each class alone, such combined therapy could be more effective in controlling SARS-CoV-2 infection in a manner comparable to that witnessed in the area of the therapy against HIV-1 infection and AIDS (42, 43). If the combination of GRL-0920 and remdesivir proves to be significantly more potent than administration of GRL-0920 or remdesivir alone, combined therapy might be more effective in controlling SARS-CoV-2 infection.
MATERIALS AND METHODS
Cells, viruses, and antiviral compounds.
VeroE6 cells and TMPRSS2-overexpressing VeroE6 (VeroE6TMPRSS2) cells were obtained from the Japanese Collection of Research Bioresources (JCRB) Cell Bank (Osaka, Japan). VeroE6 cells were maintained in Dulbecco’s modified Eagle’s medium (d-MEM) supplemented with 10% fetal bovine serum (FCS), 100 μg/ml of penicillin, and 100 μg/ml of streptomycin. VeroE6TMPRSS2 cells were maintained in d-MEM as mentioned above in the presence of 1 mg/ml of G418. SARS-CoV-2 strain JPN/TY/WK-521 (SARS-CoV-2WK-521) was obtained from the National Institute of Infectious Diseases (Tokyo, Japan).
The antiviral agents lopinavir (Sigma-Aldrich, St. Louis, MO); nelfinavir, nafamostat, hydroxychloroquine, and nitazoxanide (Tokyo Chemical Industry, Tokyo, Japan); favipiravir (MedChemExpress, Monmouth Junction, NJ); and chloroquine (Selleck, Sylvanfield Drive, Houston, TX) were purchased. Remdesivir was obtained from Clifford Lane, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD. GRL-0820 and GRL-0920 were synthesized by A. K. Ghosh. Each compound except remdesivir was dissolved in DMSO at 20 mM, and remdesivir was prepared with saline solution at 5 mM concentrations as stock solutions.
Antiviral activity, cytotoxicity, cytopathicity, and virus replication assays.
For antiviral assay, cells were seeded in a 96-well plate (2 × 104 cells/well) and incubated. After 1 day, virus was inoculated into cells at multiplicity of infection (MOI) of 0.05. After an additional 3 days, cell culture supernatants were harvested and viral RNA was extracted using a QIAamp viral RNA minikit (Qiagen, Hilden, Germany), and quantitative RT-PCR (RT-qPCR) was then performed using One Step PrimeScript III RT-qPCR mix (TaKaRa Bio, Shiga, Japan) following the instructions of the manufacturers. The primers and probe used for detecting SARS-CoV-2 envelope (6) were 5′-ACT TCT TTT TCT TGC TTT CGT GGT-3′ (forward), 5′-GCA GCA GTA CGC ACA CAA TC-3′ (reverse), and 5′-FAM-CTA GTT ACA CTA GCC ATC CTT ACT GC-black hole quencher 1 (BHQ1)-3′ (probe). To determine the cytotoxicity of each compound, cells were seeded in a 96-well plate (2 × 104 cells/well). One day later, various concentrations of each compound were added, and cells were incubated for additional 3 days. The 50% cytotoxic concentrations (CC50) values were determined using the WST-8 assay and Cell Counting Kit-8 (Dojindo, Kumamoto, Japan).
For cytopathicity and virus replication assay, cells (104 cells/well in a 96-well plate) were exposed to SARS-CoV-2WK-521 (300 50% tissue culture infective doses [TCID50]) for 1 h, washed, and cultured in fresh culture medium. At 1, 24, 48, 72, or 96 h postinfection (hpi), viral RNA copy numbers were determined using RNA-qPCR as mentioned above, and the cytopathicity of SARS-CoV-2WK-521 was determined using the WST-8 assay. The percentage of cell viability was calculated using the following formula: percent cell viability = [OD (450 nm) value of cells − mean OD value of VeroE6TMPRSS2 cells at 96 hpi]/[mean OD value of uninfected cells − mean OD value of VeroE6TMPRSS2 cells at 96 hpi] × 100 (where “OD” represents optical density). In this formula, the mean OD value of VeroE6TMPRSS2 cells at 96 hpi was the same as in the wells without cells, indicating that the virus-exposed VeroE6TMPRSS2 cells had been totally destroyed by the virus as examined at 96 hpi.
Immunocytochemistry.
Cells in a 96-well microtiter culture plate were fixed with 4% paraformaldehyde–phosphate-buffered saline (PBS) for 15 min, washed with PBS (300 μl/well) three times for 5 min each time, and then blocked with a blocking buffer (10% goat serum, 1% bovine serum albumin [BSA], 0.3% Triton X-100, PBS 1×) for 1 h. After removal of the blocking buffer, the cells were immediately stained with the primary antibody mouse monoclonal anti-SARS-CoV/SARS-CoV-2 (COVID-19) spike antibody (1A9) (GeneTex, Alton Pkwy Irvine, CA, USA) or a convalescent IgG fraction, which was isolated from serum of a convalescent COVID-19 individual using a spin column-based antibody purification kit (Cosmo Bio, Tokyo, Japan) overnight at 4°C. The stained cells were washed with PBS (300 μl/well) three times for 5 min each time, and the cells were incubated with secondary antibody goat polyclonal anti-mouse IgG-Alexa Fluor 488 antibody (Thermo Fisher Scientific, Waltham, MA, USA) or goat polyclonal anti-human IgG-Alexa Fluor 488 Fab fragment antibody (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA), together with Texas Red-X dye-conjugated phalloidin (Thermo Fisher Scientific) for F-actin visualization for 2 h. After washing of the cells with PBS (300 μl/well) three times for 5 min each time, DAPI (4′,6-diamidino-2-phenylindole) solution (Thermo Fisher Scientific)–PBS (50 μl/well) was added to stain nuclei. Signals were acquired with a Cytation 5 cell imaging multimode reader (BioTek, Winooski, VT, USA).
Molecular modeling of the interaction of GRL-0920 and GRL-0820 with SARS-CoV-2 protease.
We started with a crystal structure of SARS-CoV-2 main protease (Mpro) with RCSB PDB ID 6Y2F. We deleted the dimethyl sulfoxide and added hydrogens to the protein atoms, water oxygens, and the crystalized inhibitor. The protonation states of asparagines, glutamines, and histidines were determined, and the orientations of all hydrogen atoms, including those attached to crystal waters, were optimized to improve hydrogen bonding interactions. Using the OPLS3 force field, restrained minimization was performed (with a cutoff value of 0.30 Å for the root mean square differences of heavy atoms from the crystal structure coordinates). The assay steps described above were performed with the Protein Preparation wizard present in Maestro. The structure thus obtained was used for molecular docking. The inhibitor molecules GRL-0920 and GRL-0820 were built in Maestro, and minimized conformations were generated using the LigPrep module. The covalent docking submodule of Glide was used, and a docking grid encompassing the volume occupied by the inhibitor from the crystal structure was generated. A nucleophilic attack by Cys-145 of SARS-CoV2 Mpro on the ligand ester carbonyl was chosen as the mode of reaction. This choice was made because the literature on SARS-CoV demonstrates that the active site cysteine residue undergoes acylation reaction with ester groups, with formation of a covalent bond with the carbonyl carbon followed by the departure of a part of the ligand. Recently determined crystal structures also demonstrate that Cys-145 of SARS-CoV-2 protease forms covalent bond with inhibitors. An initial mode of interaction that involves the whole ligand molecule was thus generated. Subsequently, GRL-0920 and GRL-0820 were manually cleaved from these docked complexes to generate the appropriate thiocarbonyl complexes. These complexes were further minimized using the OPLS3 force field. All simulations were done using software versions/modules present in Maestro Version 10.7.015 (Schrödinger LLC, New York, NY).
Expression and purification of Mpro.
The SARS-CoV2 Mpro-encoding sequence was cloned into pGEX-4T1 vector (Genscript, Piscataway, NJ). The plasmid construct was transformed into BL21 Star (DE3) cells (Thermo Fisher Scientific). The culture was grown in Terrific Broth media supplemented with ampicillin. Protein expression was induced by adding 1 mM isopropyl β-d-thiogalactopyranoside at an optical density at 600 nm of 0.5. Protein expression continued at 20°C overnight. SARS-CoV-2 Mpro was purified first by affinity chromatography using glutathione S-transferase (GST) Sepharose 4B (GE Healthcare, Piscataway, NJ). The GST tag was cleaved off by the use of thrombin and separated from Mpro via GST affinity chromatography, providing the intact Mpro with an additional N-terminal glycine residue. The cleaved Mpro was further purified by size exclusion chromatography using a HiLoad Superdex 200-pg column (GE Healthcare) in a reaction mixture containing 20 mM Tris (pH 7.5), 150 mM NaCl, and 2 mM dithiothreitol. The protease was confirmed to be >99% pure based on SDS-gel electrophoresis and HPLC/MS chromatography as shown in Fig. S7 in the supplemental material.
Mpro molecular weight analysis with HPLC/MS.
The newly generated SARS-CoV-2 Mpro (see above) was diluted to approximately 10 μM (300 μg/ml) in 20 mM Tris buffer (pH 7.4) with 150 mM sodium chloride and 2 mM dithiothreitol, and 9.5 μl of Mpro was treated either with DMSO (0.5 μl)–2 mM GRL-0920 (0.5 μl) or with DMSO (0.5 μl)–2 mM GRL-0820 (0.5 μl) at a final concentration of 100 μM for each compound. The preparation was incubated at 37°C for 30 min and then diluted 10-fold (90 μl) with high-performance liquid chromatography–mass spectrometry (HPLC/MS) running buffer A (water with 0.1% formic acid and 0.02% trifluoroacetic acid). To detect the molecular weight of the protease, analysis was done using a quadrupole time of flight (QTOF) mass spectrometer (Agilent 6230) in positive mode with liquid chromatography (Agilent 1260) (Agilent, Santa Clara, CA). A sample (25 μl) was separated on a Zorbax Extend C18 column (2.1 by 50 mm, 1.8-μm pore size) (Agilent) over 35 min using a 2% acetonitrile gradient. Separations started with 100% buffer A and 0% buffer B (acetonitrile with 0.1% formic acid and 0.02% trifluoroacetic acid) to 70% buffer B and then ramped to 100% in the following 5 min and then returned to starting conditions 2 min later. The intact and modified forms of the protease eluted at approximately 30 min. The TOF settings were as follows: gas temperature, 350°C; drying gas rate, 13 liters/min; nebulizer, 55 pounds per square inch gauge (psig); sheath gas temperature, 350°C; fragmenter, 350 V; skimmer, 65 V. Molecular weights were determined by protein deconvolution using Agilent Mass Hunter software (Agilent).
Thermal stability analysis of Mpro complexed with GRL-0920 using differential scanning fluorimetry.
Thermal stability was examined using differential scanning fluorimetry. An Mpro preparation (5 μM dissolved in 10 mM Tris [pH 7.5]) that included 1 mM EDTA was mixed with various amounts of a test compound and incubated at 37°C for 3 h. Subsequently, 30 μl of the solution was gradually heated from 15°C to 95°C, and the changes of fluorescence intensity were documented using a real-time PCR system (Applied Biosystems). The 50% Tm (50% melting temperature) values were determined as the temperature at which the relative fluorescent intensity level reached 50%.
ACKNOWLEDGMENTS
We thank Clifford Lane (National Institute of Allergy and Infectious Diseases, National Institutes of Health) for kindly providing remdesivir, Thomas Misteli (National Cancer Institute) for his critical reading and editing of the manuscript, and Asuka Fujiwara (National Center for Global Health and Medicine) for technical help. This study utilized the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health, Bethesda, MD (https://hpc.nih.gov).
The present work was supported in part by a grant for Development of Novel Drugs for Treating COVID-19 (H. Mitsuya, 19A3001) from the Intramural Research Program of National Center for Global Health and Medicine, in part by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health (H. Mitsuya and R. Yarchoan), and in part by the National Institutes of Health (AI150466, A. K. Ghosh). The work was also supported in part by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under contract HHSN27220170006C (A. K. Ghosh).
Our contributions were as follows: conceptualization, S.-i. Hattori, A. K. Ghosh, and H. Mitsuya; investigation, S.-i. Hattori, N. Higshi-Kuwata, J. Raghavaiah, D. Das, H. Bulut, D. Davis, Y. Takamatsu, K. Matsuda, N. Takamune, N. Kishimoto, T. Okamura, and S. Misumi; data curation, S.-i. Hattori, N. Higshi-Kuwata, D. Das, H. Bulut, D. Davis, N. Takamune, N. Kishimoto, S. Misumi, and H. Mitsuya; methodology, S.-i. Hattori, N. Higshi-Kuwata, D. Das, H. Bulut, K. Maeda, A. K. Ghosh, and H. Mitsuya; writing—original draft, S.-i. Hattori, N. Higshi-Kuwata, and H. Mitsuya; writing—review and editing, all of us; funding acquisition and supervision, R. Yarchoan, A. K. Ghosh, and H. Mitsuya.
All of us declare no competing interests.
Footnotes
This article is a direct contribution from Robert Yarchoan, a Fellow of the American Academy of Microbiology, who arranged for and secured reviews by Douglas Richman, Departments of Pathology and Medicine, UCSD, and Robert Shafer, Stanford University School of Medicine.
Citation Hattori S-i, Higshi-Kuwata N, Raghavaiah J, Das D, Bulut H, Davis DA, Takamatsu Y, Matsuda K, Takamune N, Kishimoto N, Okamura T, Misumi S, Yarchoan R, Maeda K, Ghosh AK, Mitsuya H. 2020. GRL-0920, an indole chloropyridinyl ester, completely blocks SARS-CoV-2 infection. mBio 11:e01833-20. https://doi.org/10.1128/mBio.01833-20.
REFERENCES
- 1.Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, Niu P, Zhan F, Ma X, Wang D, Xu W, Wu G, Gao GF, Tan W, China Novel Coronavirus Investigating Research Team. 2020. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen HD, Chen J, Luo Y, Guo H, Jiang RD, Liu MQ, Chen Y, Shen XR, Wang X, Zheng XS, Zhao K, Chen QJ, Deng F, Liu LL, Yan B, Zhan FX, Wang YY, Xiao GF, Shi ZL. 2020. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Q, Guan X, Wu P, Wang X, Zhou L, Tong Y, Ren R, Leung KSM, Lau EHY, Wong JY, Xing X, Xiang N, Wu Y, Li C, Chen Q, Li D, Liu T, Zhao J, Liu M, Tu W, Chen C, Jin L, Yang R, Wang Q, Zhou S, Wang R, Liu H, Luo Y, Liu Y, Shao G, Li H, Tao Z, Yang Y, Deng Z, Liu B, Ma Z, Zhang Y, Shi G, Lam TTY, Wu JT, Gao GF, Cowling BJ, Yang B, Leung GM, Feng Z. 2020. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N Engl J Med 382:1199–1207. doi: 10.1056/NEJMoa2001316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mitsuya H, Kokudo N. 2020. Sustaining containment of COVID-19: global sharing for pandemic response. Glob Health Med 2:53–55. doi: 10.35772/ghm.2020.01040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.World Health Organization. 2020. Coronavirus disease (COVID-19) situation report. https://www.who.int/emergencies/diseases/novel-coronavirus-2019. Accessed 30 June 2020.
- 6.Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z, Yu T, Xia J, Wei Y, Wu W, Xie X, Yin W, Li H, Liu M, Xiao Y, Gao H, Guo L, Xie J, Wang G, Jiang R, Gao Z, Jin Q, Wang J, Cao B. 2020. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, Shi Z, Hu Z, Zhong W, Xiao G. 2020. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res 30:269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hilgenfeld R. 2014. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J 281:4085–4096. doi: 10.1111/febs.12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muramatsu T, Kim YT, Nishii W, Terada T, Shirouzu M, Yokoyama S. 2013. Autoprocessing mechanism of severe acute respiratory syndrome coronavirus 3C-like protease (SARS-CoV 3CLpro) from its polyproteins. FEBS J 280:2002–2013. doi: 10.1111/febs.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li C, Qi Y, Teng X, Yang Z, Wei P, Zhang C, Tan L, Zhou L, Liu Y, Lai L. 2010. Maturation mechanism of severe acute respiratory syndrome (SARS) coronavirus 3C-like proteinase. J Biol Chem 285:28134–28140. doi: 10.1074/jbc.M109.095851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lindner HA, Fotouhi-Ardakani N, Lytvyn V, Lachance P, Sulea T, Ménard R. 2005. The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J Virol 79:15199–15208. doi: 10.1128/JVI.79.24.15199-15208.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barretto N, Jukneliene D, Ratia K, Chen Z, Mesecar AD, Baker SC. 2005. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J Virol 79:15189–15198. doi: 10.1128/JVI.79.24.15189-15198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jacobs J, Grum-Tokars V, Zhou Y, Turlington M, Saldanha SA, Chase P, Eggler A, Dawson ES, Baez-Santos YM, Tomar S, Mielech AM, Baker SC, Lindsley CW, Hodder P, Mesecar A, Stauffer SR. 2013. Discovery, synthesis, and structure-based optimization of a series of N-(tert-butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. J Med Chem 56:534–546. doi: 10.1021/jm301580n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xue X, Yu H, Yang H, Xue F, Wu Z, Shen W, Li J, Zhou Z, Ding Y, Zhao Q, Zhang XC, Liao M, Bartlam M, Rao Z. 2008. Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J Virol 82:2515–2527. doi: 10.1128/JVI.02114-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang J, Pettersson HI, Huitema C, Niu C, Yin J, James MN, Eltis LD, Vederas JC. 2007. Design, synthesis, and evaluation of inhibitors for severe acute respiratory syndrome 3C-like protease based on phthalhydrazide ketones or heteroaromatic esters. J Med Chem 50:1850–1864. doi: 10.1021/jm061425k. [DOI] [PubMed] [Google Scholar]
- 16.Jain RP, Pettersson HI, Zhang J, Aull KD, Fortin PD, Huitema C, Eltis LD, Parrish JC, James MN, Wishart DS, Vederas JC. 2004. Synthesis and evaluation of keto-glutamine analogues as potent inhibitors of severe acute respiratory syndrome 3CLpro. J Med Chem 47:6113–6116. doi: 10.1021/jm0494873. [DOI] [PubMed] [Google Scholar]
- 17.Lee H, Lei H, Santarsiero BD, Gatuz JL, Cao S, Rice AJ, Patel K, Szypulinski MZ, Ojeda I, Ghosh AK, Johnson ME. 2015. Inhibitor recognition specificity of MERS-CoV papain-like protease may differ from that of SARS-CoV. ACS Chem Biol 10:1456–1465. doi: 10.1021/cb500917m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee H, Cao S, Hevener KE, Truong L, Gatuz JL, Patel K, Ghosh AK, Johnson ME. 2013. Synergistic inhibitor binding to the papain-like protease of human SARS coronavirus: mechanistic and inhibitor design implications. ChemMedChem 8:1361–1372. doi: 10.1002/cmdc.201300134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghosh AK, Takayama J, Aubin Y, Ratia K, Chaudhuri R, Baez Y, Sleeman K, Coughlin M, Nichols DB, Mulhearn DC, Prabhakar BS, Baker SC, Johnson ME, Mesecar AD. 2009. Structure-based design, synthesis, and biological evaluation of a series of novel and reversible inhibitors for the severe acute respiratory syndrome-coronavirus papain-like protease. J Med Chem 52:5228–5240. doi: 10.1021/jm900611t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghosh AK, Gong G, Grum-Tokars V, Mulhearn DC, Baker SC, Coughlin M, Prabhakar BS, Sleeman K, Johnson ME, Mesecar AD. 2008. Design, synthesis and antiviral efficacy of a series of potent chloropyridyl ester-derived SARS-CoV 3CLpro inhibitors. Bioorg Med Chem Lett 18:5684–5688. doi: 10.1016/j.bmcl.2008.08.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghosh AK, Xi K, Johnson ME, Baker SC, Mesecar AD. 2007. Progress in anti-SARS coronavirus chemistry, biology and chemotherapy. Annu Rep Med Chem 41:183–196. doi: 10.1016/S0065-7743(06)41011-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghosh AK, Xi K, Grum-Tokars V, Xu X, Ratia K, Fu W, Houser KV, Baker SC, Johnson ME, Mesecar AD. 2007. Structure-based design, synthesis, and biological evaluation of peptidomimetic SARS-CoV 3CLpro inhibitors. Bioorg Med Chem Lett 17:5876–5880. doi: 10.1016/j.bmcl.2007.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghosh AK, Xi K, Ratia K, Santarsiero BD, Fu W, Harcourt BH, Rota PA, Baker SC, Johnson ME, Mesecar AD. 2005. Design and synthesis of peptidomimetic severe acute respiratory syndrome chymotrypsin-like protease inhibitors. J Med Chem 48:6767–6771. doi: 10.1021/jm050548m. [DOI] [PubMed] [Google Scholar]
- 24.Shin D, Mukherjee R, Grewe D, Bojkova D, Baek K, Bhattacharya A, Schulz L, Widera M, Mehdipour AR, Tascher G, Geurink PP, van der Heden van Noort GJ, Ovaa H, Knobeloch K-P, Rajalingam K, Schulman BA, Cinatl J, Hummer G, Ciesek S, Dikic I. Inhibition of papain-like protease PLpro blocks SARS-CoV-2 spread and promotes anti-viral immunity. Nature Res doi: 10.21203/rs.3.rs-27134/v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ratia K, Pegan S, Takayama J, Sleeman K, Coughlin M, Baliji S, Chaudhuri R, Fu W, Prabhakar BS, Johnson ME, Baker SC, Ghosh AK, Mesecar AD. 2008. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc Natl Acad Sci U S A 105:16119–16124. doi: 10.1073/pnas.0805240105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. 2020. The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol 5:536–544. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu J, Cao R, Xu M, Wang X, Zhang H, Hu H, Li Y, Hu Z, Zhong W, Wang M. 2020. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov 6:16. doi: 10.1038/s41421-020-0156-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu N-H, Nitsche A, Müller MA, Drosten C, Pöhlmann S. 2020. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamamoto N, Yang R, Yoshinaka Y, Amari S, Nakano T, Cinatl J, Rabenau H, Doerr HW, Hunsmann G, Otaka A, Tamamura H, Fujii N, Yamamoto N. 2004. HIV protease inhibitor nelfinavir inhibits replication of SARS-associated coronavirus. Biochem Biophys Res Commun 318:719–725. doi: 10.1016/j.bbrc.2004.04.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsuyama S, Nao N, Shirato K, Kawase M, Saito S, Takayama I, Nagata N, Sekizuka T, Katoh H, Kato F, Sakata M, Tahara M, Kutsuna S, Ohmagari N, Kuroda M, Suzuki T, Kageyama T, Takeda M. 2020. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc Natl Acad Sci U S A 117:7001–7003. doi: 10.1073/pnas.2002589117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Owczarek K, Szczepanski A, Milewska A, Baster Z, Rajfur Z, Sarna M, Pyrc K. 2018. Early events during human coronavirus OC43 entry to the cell. Sci Rep 8:7124. doi: 10.1038/s41598-018-25640-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohkuma S, Chudzik J, Poole B. 1986. The effects of basic substances and acidic ionophores on the digestion of exogenous and endogenous proteins in mouse peritoneal macrophages. J Cell Biol 102:959–966. doi: 10.1083/jcb.102.3.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vincent MJ, Bergeron E, Benjannet S, Erickson BR, Rollin PE, Ksiazek TG, Seidah NG, Nichol ST. 2005. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol J 2:69. doi: 10.1186/1743-422X-2-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oda K, Koriyama Y, Yamada E, Ikehara Y. 1986. Effects of weakly basic amines on proteolytic processing and terminal glycosylation of secretory proteins in cultured rat hepatocytes. Biochem J 240:739–745. doi: 10.1042/bj2400739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, Becker S, Rox K, Hilgenfeld R. 2020. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aoki M, Hayashi H, Rao KV, Das D, Higashi-Kuwata N, Bulut H, Aoki-Ogata H, Takamatsu Y, Yedidi RS, Davis DA, Hattori SI, Nishida N, Hasegawa K, Takamune N, Nyalapatla PR, Osswald HL, Jono H, Saito H, Yarchoan R, Misumi S, Ghosh AK, Mitsuya H. 2017. A novel central nervous system-penetrating protease inhibitor overcomes human immunodeficiency virus 1 resistance with unprecedented aM to pM potency. Elife 6:e28020. doi: 10.7554/eLife.28020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simeonov A. 2013. Recent developments in the use of differential scanning fluorometry in protein and small molecule discovery and characterization. Expert Opin Drug Discov 8:1071–1082. doi: 10.1517/17460441.2013.806479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoffmann M, Schroeder S, Kleine-Weber H, Müller MA, Drosten C, Pöhlmann S. 2020. Nafamostat mesylate blocks activation of SARS-CoV-2: new treatment option for COVID-19. Antimicrob Agents Chemother 64:e00754-20. doi: 10.1128/AAC.00754-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Filion LG, Logan D, Gaudreault R, Izaguirre CA. 1993. Inhibition of HIV-1 replication by daunorubicin. Clin Invest Med 16:339–347. [PubMed] [Google Scholar]
- 40.Bergamini A, Perno CF, Balzarini J, Capozzi M, Marinelli L, Milanese G, Pesce CD, Calio R, Rocchi G. 1992. Selective inhibition of HIV replication by adriamycin in macrophages but not in lymphocytes. AIDS Res Hum Retroviruses 8:1239–1247. doi: 10.1089/aid.1992.8.1239. [DOI] [PubMed] [Google Scholar]
- 41.Ma C, Sacco MD, Hurst B, Townsend JA, Hu Y, Szeto T, Zhang X, Tarbet B, Marty MT, Chen Y, Wang J. 2020. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res 2020:1–15. doi: 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fontela C, Aguinaga A, Moreno-Iribas C, Reparaz J, Rivero M, Gracia M, Floristan Y, Fresan U, Miguel RS, Ezpeleta C, Castilla J. 2020. Trends and causes of mortality in a population-based cohort of HIV-infected adults in Spain: comparison with the general population. Sci Rep 10:8922. doi: 10.1038/s41598-020-65841-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anglemyer A, Sturt A, Maldonado Y. 2018. The effect of combination antiretroviral therapy use among HIV positive children on the hazard of AIDS using calendar year as an instrumental variable. Curr HIV Res 16:151–157. doi: 10.2174/1570162X16666180409150826. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Antiviral activity and cytotoxicity of compounds against SARS-CoV-2. A lowercase superscript “a” indicates that each selectivity index value denotes a ratio of CC50 to EC50. The data shown represent mean EC50 and CC50 values ± standard deviations (SD). The assays were conducted on at least 3 different occasions, and the data shown here are representative of the results. All assays were performed in duplicate. Download Table S1, DOCX file, 0.01 MB (14.3KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Visualization of SARS-CoV-2 infection using immunocytochemistry. VeroE6 cells were exposed to SARS-CoV-2WK-521 at MOI of 0.05 and cultured in the absence of test compound. After 3 days, the cells were fixed with 4% paraformaldehyde and immunofluorescent staining using an IgG fraction from serum of a COVID-19 individual was performed. SARS-CoV-2 antigens, F-actin, and nuclei are shown in green, red, and blue, respectively. The upper three insets show images of VeroE6 cells cultured alone, while the lower three insets show images of VeroE6 cells exposed to SARS-CoV-2WK-521 and cultured in the absence of test compound. The upper and lower extreme right insets represent merged images, which are composed of images obtained from 3-color fluorescence. The lower left panel shows that the structure of one of the cytoskeletons, the actin filament, was destroyed by SARS-CoV-2WK-521 infection. Almost all cells are stained in green (SARS-CoV-2 positive) in the lower right panel, and no nonspecific staining (in green) is seen in the upper right panel. Download FIG S1, PDF file, 0.3 MB (291.8KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Different multiplicities of SARS-CoV-2 infection. VeroE6 and VeroE6TMPRSS2 cells were exposed to SARS-CoV-2WK-521, and immunocytochemistry using the IgG fraction from a convalescent individual with COVID-19 (convIgG) (insets b and e) or murine (insets c and f) anti-spike monoclonal antibody (MAb) as the primary antibody was performed. SARS-CoV-2 antigens (insets b and and e) and SARS-CoV-2 spike antigen (insets c and f), F-actin, and nuclei are shown in green, red, and blue, respectively. Insets a and d show images of VeroE6 and VeroE6TMPRSS2 cells cultured alone and stained, respectively, while insets b and c show those of VeroE6 cells and insets e and f show those of VeroE6TMPRSS2 cells exposed to SARS-CoV-2WK-521. Download FIG S2, PDF file, 0.2 MB (235.4KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
VeroE6TMPRSS2 cells have greater susceptibility to SARS-CoV-2WK-521 than VeroE6 cells. (A and B) Cell viability (A) and viral RNA copy numbers (B) of SARS-CoV-2WK-521 in VeroE6 (○) and VeroE6TMPRSS2 (●) cells. VeroE6 and VeroE6TMPRSS2 cells were exposed to SARS-CoV-2WK-521 (300 TCID50) for 1 h, and the virus was then washed off. The viability of VeroE6TMPRSS2 cells was lower than that of VeroE6 cells from 48 h postinfection (hpi), and virus copy numbers were higher in VeroE6TMPRSS2 cells than in VeroE6 cells at 24 and 48 hpi. Cell viability (the level of viability of virus-unexposed cells is assigned a value of 100%) and viral copy numbers were determined using WST-8 assay and RNA-qPCR, respectively. All assays were performed in quadruplicate, and each plot denotes each value. (C) Visualization of SARS-CoV-2 infection using immunocytochemistry. Images of VeroE6 and VeroE6TMPRSS2 cells exposed to SARS-CoV-2WK-521 were captured using light microscopy (upper lines) and immunocytochemistry (lower lines). SARS-CoV-2 antigens, F-actin, and nuclei are shown in green, red, and blue, respectively. A higher number of SARS-CoV-2-positive cells was seen in VeroE6TMPRSS2 cells than in VeroE6 cells at 24 hpi. At 48 hpi and beyond, most of VeroE6TMPRSS2 cells were destroyed by the cytopathicity of the virus, but VeroE6 cells remained on the bottom of the microtiter culture plate. Download FIG S3, PDF file, 0.5 MB (529.3KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
GRL-0920 and remdesivir block SARS-CoV-2 infection. (A) Lopinavir (d to g), nelfinavir (h to k), and nafamostat (l to o) failed to block SARS-CoV-2 infection in VeroE6 cells. Images of VeroE6 cells exposed to the virus and cultured alone are shown in insets a to c. (B) Remdesivir (d to g) significantly blocked SARS-CoV-2 infection but allowed viral breakthrough in VeroE6 cells. Images of virus-exposed cells cultured alone are shown in insets a to c. Chloroquine (h to k) and hydroxychloroquine (i to o) failed to block the infectivity of SARS-CoV-2WK-521, and almost all the cells were shed from the wells at 100 μM. Favipiravir (p to s) also failed to block SARS-CoV-2 infection. (C) GRL-0920 completely blocked the infectivity of SARS-CoV-2WK-521, but GRL-0820 permitted viral breakthrough in VeroE6 cells. Images of virus-exposed VeroE6 cells cultured alone are shown in insets a to c. (d to n) Images of VeroE6 cells exposed to SARS-CoV-2WK-521 and cultured in the absence (d to f) or presence (g to n) of each compound. The cells were stained using the IgG fraction from serum of a convalescent COVID-19 individual as the primary antibody. SARS-CoV-2 antigens, F-actin, and nuclei are shown in green, red, and blue, respectively. Download FIG S4, PDF file, 0.9 MB (965.6KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Chloroquine and hydroxychloroquine fail to block the infectivity of SARS-CoV-2WK-521 in VeroE6 cells. VeroE6 cells were exposed to SARS-CoV-2WK-521 and cultured in the presence of chloroquine or hydroxychloroquine. Immunocytochemistry using an IgG fraction from serum of a convalescent COVID-19 individual was performed. SARS-CoV-2 antigens, F-actin, and nuclei are shown in green, red, and blue, respectively. The images in the upper and lower left insets show those acquired under a light microscope (magnification, ×200), showing that whereas the VeroE6 cells cultured alone were all alive (upper inset), the VeroE6 cells exposed to SARS-CoV-2WK-521 were killed and detached from the culture wells (lower inset). VeroE6 cells exposed to the virus and cultured in the presence of chloroquine or hydroxychloroquine at 1 and 10 μM were infected with the virus, and virtually all of those cultured at 100 μM were seen to have been shed from the wells of culture plates due to the damage to the cells imposed by the compounds. Download FIG S5, PDF file, 0.5 MB (556.4KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
GRL-0920 completely blocks the infectivity and cytopathic effect of SARS-CoV-2. VeroE6 cells were exposed to SARS-CoV-2WK-521 at an MOI of 0.05 and cultured in the presence of 0.01, 0.1, 1, 10, and 100 μM remdesivir or GRL-0920. After 3 days, the cells were fixed with 4% paraformaldehyde and immunofluorescent staining using the IgG fraction from serum of a convalescent individual with COVID-19 or a murine anti-SARS-CoV-2 spike monoclonal antibody was performed. SARS-CoV-2 antigens, F-actin, and nuclei are shown in green, red, and blue, respectively. The inset at the extreme upper left shows an image of VeroE6 cells cultured alone, and the inset at the extreme lower left shows that of VeroE6 cells exposed to SARS-CoV-2WK-521 and cultured in the absence of test compound. All the images except the two insets described above show images captured with 3-color fluorescence. Download FIG S6, PDF file, 0.3 MB (319.8KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Mass analysis of the complex of Mpro of SARS-CoV-2 with GRL-0920 or GRL-0820. (A) Mass detection of Mpro of SARS-CoV-2 alone was done with an Agilent 6230 time of flight (TOF) mass spectrometer, and data are shown with the total ion current (TIC). The major protease species had an average mass of 33,853.36 (expected mass, 33,853.53). The inset shows the molecular ion profile for the main eluting Mpro peak, and the deconvolution result for that ion profile is shown at the bottom. (B and C) Mass detection of the complex of Mpro with GRL-0920 (B) or GRL-0820 (C) was done, and data are shown with the total ion current (TIC). The major Mpro species with GRL-0920 and GRL-0820 had average masses of 33,997.27 (expected mass, 33,996.68 for acylation of the Mpro with GRL-0920) and 34,037.21 (expected mass, 34,036.73 for acylation of the protease), respectively. The upper inset shows the molecular ion profile for the main eluting protease peak, and the lower inset shows the deconvolution results for that ion profile. Download FIG S7, PDF file, 1.3 MB (1.3MB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.