

Abstract
Plasmacytoid dendritic cells are the major producers of IFN-α, an antiviral cytokine involved in immunomodulation as well as control of HIV-1 replication, while Toxoplasma gondii is a life-threatening opportunistic infection in AIDS patients. During infection with HIV-1, human pDC decrease in circulation and remaining pDC produce lower amounts of IFN-a in response to viral stimulation. In this study, we investigated the impact of coinfection with T. gondii on the innate virus-directed responses of human pDCs. Using intracellular flow cytometry and fluorescence microscopy, we determined that T. gondii invaded but did not induce IFN-α or TNF-α in human pDC. However, T. gondii inhibited IFN-α and TNF-α produced in response to HSV and HIV, thus functionally inactivating pDC. IFN-α production was inhibited only in cells infected by T. gondii. T. gondii inhibited neither, uptake of GFP-HSV nor localization of TLR9 in CD71+ endosomes, directing us to investigate downstream events. Using imaging flow cytometry, we found that both T. gondii and IL-10 inhibited virus-induced nuclear translocation, but not phosphorylation, of IRF7. Blockade of IRF7 nuclear translocation and inhibition of the IFN-α response was partially reversed by a deficiency in the T. gondii-derived ROP16 kinase, known to directly phosphorylate STAT3, a critical mediator of IL-10’s anti-inflammatory effects. Taken together, our results indicate that T. gondii suppresses pDC activation by mimicking IL-10’s regulatory effects through a ROP16 kinase-dependent mechanism. Our findings further imply a convergent mechanism of inhibition of TLR signaling by T. gondii and IL-10 and suggest potential negative consequences of HIV/T. gondii co-infection.
INTRODUCTION
Human plasmacytoid dendritic cells (pDC) are part of the innate immune system with the intrinsic ability to produce large quantities of Type I IFN in response to viral nucleic acids. pDC constitutively express endosomal toll-like receptors −7 and −9 (TLR7 and −9), which recognize viral RNA and DNA molecules, respectively (reviewed in (1)). Following recognition of nucleic acid ligands by TLR7/9, an intracellular signaling pathway is triggered in pDC that leads to phosphorylation and nuclear translocation of interferon response factor (IRF)-7 (2). Human pDC can mount this effective response through their constitutively high expression of IRF-7 (3, 4). As a result of activation, phosphorylated IRF-7 binds to the promoter of Type I IFN genes and initiates production of Type I IFN (mostly IFN-α). Secreted IFN-α then controls viral replication and spread by inducing upregulation of various antiviral molecules and by enhancing immune responses. The status of pDC and IFN-α production in pDC from individuals with HIV infection is complex: In individuals with HIV infection, both numbers of pDC and their production of IFN-α in response to virus is diminished (reviewed in (5, 6)), thus resulting in suppression of normal antiviral immune responses. Decreased pDC and decreased IFN-α production is strongly associated with progression to opportunistic infections (OI) in HIV-infected individuals. Other evidence, however suggests that chronic stimulation of remaining pDC contributes to chronic immune activation in HIV infection (reviewed in in (5, 6)).
Although it is clear that HIV infection leads to enhanced susceptibility to opportunistic pathogens, whether OI reciprocally and adversely impact the innate immune response to HIV is not known. A common OI in AIDS patients is caused by Toxoplasma gondii (T. gondii), an intracellular parasite that causes fatal encephalitis upon reactivation of the latent cyst stages of the parasite in the CNS (7, 8). Worldwide distribution of T. gondii has been estimated at 14% in the US and up to 75% in Western Europe and sub-Saharan Africa, making it the leading CNS infection in regions with the highest frequencies of HIV infection (9). Upon active entry of the host cell, T. gondii, through its apical end, injects multiple protein factors from a specialized secretory organelle, known as rhoptries, to modify host cell signaling and immune responses (10). Prominent examples of such parasite-derived secreted factors include ROP18 and ROP16 kinases, which have been implicated in inhibition of IFNg-dependent antimicrobial effector responses and TLR-dependent IL-12 and TNF-α production, respectively (11), but. In mouse cells, the ROP-16 mediated negative modulation of TLR-responses was reported to be mediated by inhibition of nuclear translocation of NFκB and by inducing phosphorylation of STAT3 and STAT6 (11–15). Given the global prevalence of T. gondii in the HIV-infected population and the known prowess for immunomodulation by the parasite, we used T. gondii coinfection of human pDCs to investigate (or model) the impact of opportunistic pathogens on the innate immune response by these cells to HIV and herpes simplex virus (HSV), which stimulate pDC through TLR7 and TLR9, respectively. Herein, we report that T. gondii infection inhibits anti-viral responses of human pDCs by functional mimicry of IL-10.
MATERIALS AND METHODS
Isolation and culture of PBMC and pDC
Heparinized peripheral blood from healthy donors was collected with informed consent and PBMC were separated using Lymphocyte Separation Media (Mediatech, Inc). PBMC were cultured in RPMI-1640 media containing 10% FCS, 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM L-glutamine and 25 mM HEPES (RPMI-10%) at a final concentration of 2×106 cells/mL at 37°C in 5% CO2. For experiments requiring highly-enriched pDC, a negative selection kit (Miltenyi Biotec) was used according to the manufacturer’s protocol. Average purity of enriched pDC using this kit was 93% with average yield of 0.16% of total PBMC. For ImageStream experiments, partial enrichment of pDC was conducted using the Miltenyi negative enrichment kit using half the recommended antibody and bead mixture, and purity of 60–80 percent was obtained.
T. gondii
Parasite strains RH, GFP-RH were obtained from the ATCC; mCherry-RH was kindly provided by Marc-Jan Gubbels (Boston College, Boston, MA). RHΔku80 (RH) and RHΔku80Δrop16 (RHΔrop16) knockout strains were kindly provided by David Bzik (Dartmouth Medical School, Hanover, NH) (13). All T. gondii strains were grown in a monolayer of human foreskin fibroblasts (HFF) for 48 hours prior to harvest. One hour before infection of pDC, parasites were collected from HFF by passage through a 25-gauge needle. Parasites were then washed at 2,000g and resuspended in RPMI-10%. Infection of 1 million PBMC was conducted at multiplicities of infection (MOI) as described in the figures for 40,000 purified pDC. PBMC with added T. gondii were then spun down at 100g for 3 min to enhance contact between parasites and cells. Parasite cultures were tested for Mycoplasma contamination using a PCR primer set (Agilent Technologies). Parasite/PBMC cultures were given a 1-hour head start at 37°C in 5% CO2 prior to stimulation of cells with viruses.
HSV-1 and HIV-1
Vero monkey kidney cells (ATCC) were infected with HSV-1 strain 2931 (originally obtained from Dr. Carlos Lopez, then of Sloan-Kettering Inst., New York, NY) until development of cytopathic effect. Cells were then lysed using a sonicator, debris was centrifuged down, and virus aliquots were stored at −80°C. HSV-1 titer was assessed using plaque formation assay in Vero cells, and virus was used at MO1 of 1. HSV K26-GFP was originally provided by P. Desai (Johns Hopkins University, Baltimore, MD), grown in Vero cells and concentrated using a sucrose density gradient. AT-2 inactivated HIVMN was kindly provided by Dr. Jeff Lifson (SAIC, Frederick, MD).
Pre-treatment of pDC with IL-10
PBMC were pretreated with recombinant human IL-10 (Peprotech), 50 ng/ml, for 1 hr. prior to addition of virus.
Flow cytometry
For detection of pDC in PBMC populations, Fc receptors were blocked using 2% heat inactivated pooled human serum (PHS) for 5min, and cells were stained using CD123-APC (clone 6H6, BioLegend) and CD303-PE (BDCA-2, clone AC144, Miltenyi Biotec), at 4°C in 0.1% BSA in PBS buffer for 15min. Following washes, cells were fixed overnight in 2% paraformaldehyde (PFA) in PBS. Intracellular staining for IFN-a as conducted by permeabilizing cells using 0.5% Saponin in 2% FCS PBS followed by incubation with biotinylated anti-IFN-a Ab (PBL) which was then detected with streptavidin-PeCy7 (BioLegend); at the same time cells were stained with anti-TNF-a-Pacific Blue (clone MAb-11, BioLegend). A BD LSR II was used to collect 300,000 events and analysis was performed using FlowJo software (TreeStar, Inc.).
Determination of effect of T. gondii on Annexin V/7AAD staining
Annexin V/7AAD (BD) staining was carried out in purified pDC after 18 hr exposure to GFP-T. gondii. Surface staining was carried out as described above and Annexin V-biotin followed by streptavidin-Alexa680 on cells gated for T. gondii-GFP.
Immunofluorescence microscopy
Highly-enriched pDC were surface stained and fixed in 4% PFA for 10 min at room temperature and stained and permeabilized using the same protocol as for flow cytometry. Following the last wash and fixation, cells were cytospun using a Shandon Cytospin 2 at 1000 rpm for 6 min. ProLong Antifade Gold (Invitrogen) was used to mount coverslips.
Phospho-flow analyses
Freshly isolated PBMC were allowed to rest for 30 min at 37°C in 5% CO2. Cells were then stimulated as described in the figure legends and fixed with an equal volume of 4% PFA at RT for 15 min. Next, cells were washed, surface stained with HLA-DR-PacificBlue (clone G46-2.6, BD Biosciences) and CD123-APC (clone 6H6, Biolegend), followed by washing and another 10 min 4% PFA fixation. Permeabilization was carried out with 70% ethanol in H2O at −20°C, which was added dropwise to tubes incubated on ice; cells were then stored at −20°C overnight. Permeabilized cells were washed twice with PBS containing 2% FCS and stained using anti-Phospho IRF7-PE (pS477/pS479, clone K47-671, BD Biosciences) or anti-Phospho STAT3-PerCpCy5.5 (pY705, clone 4/P-STAT3, BD Biosciences). T. gondii infection was measured using anti-SAG1 mAb (Clone p30, IgG2a, GenWay Biotech Inc.) that was labeled with Zenon Kit-488 (Invitrogen). Matched isotypes at the same concentration as the phospho-flow antibodies were used in each experiment.
Virus Internalization
Enriched pDC were pre-infected with T. gondii mCherry-RH for one hour followed by addition of HSV-GFP at an MOI of 10 and cells were co-cultured for 20, 40, or 60 min. Cells were then washed with cold PBS buffer containing 5 mM EDTA to remove surface attached virus and were fixed with 2% PFA overnight. Samples were acquired using an ImageStream IS100 or ImageStreamX Mark II (Amnis, Inc.) with an extended depth of field element. Uptake of virus was analyzed using a combination of algorithms to quantify internalization of GFP particles. First, single cells were gated using a singlet gate based on Aspect Ratio to the Area of Brightfield, followed by creation of an eroded cell morphology mask to gate on GFP positive events within cell area. Percentage of pDC positive for HSV-GFP was calculated by dividing the number of internalized GFP events by the total number of collected pDC.
Determination of cytokine levels
40,000 highly-enriched pDC were incubated in wells of a 96-well round-bottom plate with a combination of T. gondii and HSV or HIV as described above, in a final volume of 200μL. Culture supernatants were collected 18 hrs post addition of virus and analyzed using the BenderMed Systems human IFN-a module set and the OptEIA® Human TNF-a Set. Absorbance was recorded at 450nm using TECAN GENios reader and concentration of cytokines was calculated using Microsoft Excel from a linear curve fit. For determination of IL-10 levels, samples were sent to Myriad RBM and analyzed for expression of inflammatory cytokines using Human InflammationMAP v.1.0.
Nuclear Translocation of IRF7
100,000 enriched pDC were pre-infected with T. gondii RH as described above. Surface staining was conducted at 4°C in 0.1%BSA in PBS using a combination of BDCA2-PE and BDCA4-PE (Miltenyi) antibodies for detection of pDC. Cells were then fixed overnight using 2%PFA in PBS, and permeabilized using 0.1% TritonX in 2% FCS PBS. Intracellular IRF7 staining was done at room temperature using a monoclonal Ab (clone G-8, Santa Cruz) labeled with Zenon- Alexa Fluor 488 (Invitrogen), and the parasite’s parasitophorous vacuole membrane was visualized using rabbit anti-GRA7 antiserum followed by anti-rabbit Alexa610-R-PE (Invitrogen); the rabbit anti-GRA7 antiserum was produced by the Yap laboratory by immunization of a rabbit with full-length GRA7 protein. Following washing, cells were resuspended in 50uL 1% PFA in PBS. All staining was performed in siliconized Eppendorf tubes (Sigma) and tubes were spun in a swinging rotor centrifuge to minimize cell loss. DRAQ5® (1:500 final dilution, Cell Signaling Technology) was added immediately before acquiring cell images to visualize nuclei. Cells were acquired with the ImageStream IS100 or ImageStreamX Mark II (Amnis, Inc.) T. gondii infection and IRF7 translocation was analyzed using Amnis IDEAS application 3.0. First, single cells were gated based on aspect ratio intensity of DRAQ5 (Ch6) versus the area of (Ch6). Dot plot of IRF7 Intensity (Ch3) versus BDCA2 intensity (Ch4) was used to gate pDC positive for IRF7 and BDCA2 signal. Further refinement of single cells was conducted by exclusion of non-circular pDC events using circularity feature of BDCA2 events. Similarity object function between default nuclear mask (Ch6) and IRF-7 channel was used to calculate the scores for IRF7 nuclear translocation. Translocation gate was then defined based on mock stimulated sample and confirmed by visual inspection of corresponding images. All images shown were collected using the 40X camera.
Statistical Analyses
Experiments were analyzed using one-way ANOVA with Tukey’s or Dunnet’s post-test with GraphPad PRISM. In cases where only two experimental groups were compared, a two-tailed student’s t-test was used. All bar graphs represent mean ± SEM of at least three independent experiments from different donors. *p<0.05; ** p<0.01; ***p<0.001; NS, not significant.
RESULTS
Infection of human pDC by Toxoplasma gondii
To determine whether human pDC are permissive to infection by T. gondii, we utilized intracellular flow cytometry. pDC were infected with T. gondii expressing GFP and were stained for intracellular GRA7, to mark the formation of the parasitophorous vacuole membrane (PVM) inside the host’s cytoplasm (Figure 1A). The majority of T. gondii GFP positive pDC were also positive for GRA7 (61%, upper right quadrant) by 6 hrs of infection, indicating active parasitization of the human pDC cytoplasm. Next, we used purified pDC to visualize establishment of PVM by immunofluorescence microscopy (Figure 1B). pDC can be seen either in the process of being infected by T. gondii (GFP, green) or infected by T. gondii with established PVM (GRA7-positive, pink).
Figure 1. T. gondii infects human pDC and establishes a parasitophorous vacuole membrane (PVM).
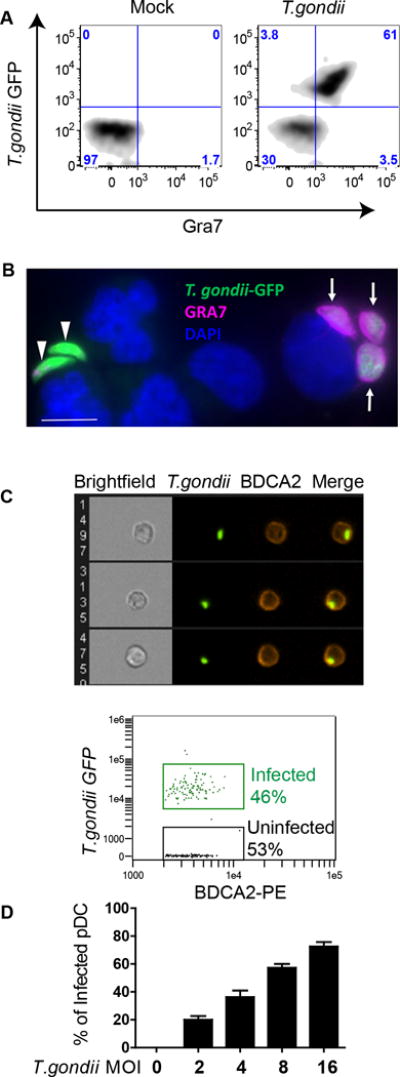
A, Establishment of PVM was quantified using intracellular flow cytometric staining against GRA7 in BDCA2 and CD123 positive pDC infected with T. gondii RH- GFP at MOI 4. A representative flow figure showing Gra7+/T. gondii-GFP+ cells is shown. B, Fluorescent microscopy images of purified pDC infected with T. gondii-GFP and stained with anti-GRA7 (pink) to visualize parasitophorous vacuole membrane (PVM) and with DAPI (blue) to visualize nuclei. Arrows indicate GRA7+/GFP+ parasitophorous vesicles in an infected cell, while green T. gondii indicate extracellular parasites. Scale bar representing 5μ is shown on figure, lower left, in white. C, Purified pDC were infected with T. gondii RH- GFP at MOI 8 for 1hr and stained with anti-BDCA2-PE. Infection of pDC was visualized and quantified using AMNIS ImageStream. Upper panel: representative images of single pDC (BDCA2+ cells) that have internalized T. gondii. Lower panel: Percent pDC that internalized T. gondii from a representative ImageStream experiment. Data shown are from a single experiment representative of three independent experiments. D, Flow cytometry was used to determine optimal MOI of T. gondii 1 hr post infection from three independent experiments (Mean ± SEM).
Results from these experiments were confirmed using the Amnis ImageStream, which allowed us to visualize and quantify the infection of BDCA2 positive pDC by GFP-tagged T. gondii. Using the Ideas software, we quantified the infection of pDC while excluding extracellular parasites through application of an internalization algorithm. In the representative experiment shown, 46% of the pDC were infected with T. gondii (Figure 1C lower panel), with representative images of T. gondii-infected cells shown in 1C, upper panel. To identify the optimal MOI of T. gondii for infection of pDC that would allow us to analyze both infected and uninfected pDC, we used traditional flow cytometry to gate on T. gondii-GFP positive pDC within the PBMC population. An MOI of 4–8 yielded infection rates of 40–60% (Figure 1D). In spite of low abundance of pDC in the PBMC population, T. gondii efficiently infected these cells of the innate immune system that are crucial for recognition and clearance of viral infections. Taken together, these data identify that an active infection of human pDC and establishment of PVM can be quantified and visualized using ImageStream and flow cytometry. Moreover, we determined that subsets of T. gondii-infected and -uninfected pDC can be identified and investigated independently.
T. gondii infection inhibits pDC responses to HSV-1 and HIV-1
Having found that T. gondii infects human pDC, we next investigated the effects of T. gondii infection on virus-induced production of IFN-a in pDC. Pepper et al. (16) reported that murine pDC produced IFN-α in response to T. gondii; in our hands, T. gondii by itself did not induce production of IFN -a in human pDC (Figure 2A), nor did stimulation of these cells with profilin (data not shown), which is a T. gondii-encoded TLR11 ligand that has been reported to induce IL-12 production in murine pDC (16). These data indicate that human pDC are either unable to recognize T. gondii pathogen associated molecular patterns (PAMPS) resulting in an IFN-a response or that T. gondii actively inhibits IFN-a production. To address the latter hypothesis, PBMC were pre-infected with T. gondii at an increasing MOI, followed by stimulation of the cells with HSV-1. With an increasing ratio of T. gondii to PBMC, there was an increasing inhibition of IFN -a production in response to HSV (Figure 2A). This loss of IFN-α production could not be explained by additional cell toxicity after infection with T. gondii as determined by Annexin V/7AAD staining (Figure 2B upper right for T. gondii vs. mock in upper right panel); as expected given the culture-labile nature of pDC, there was loss of viability in the mock cultured pDC, and a similar level of cell viability was seen in the T. gondii-infected pDC. For the combination treatment of T. gondii and HSV, there was actually improved survival of the pDC at 18 hours post-infection compared to the other three groups (Figure 2B, lower right).
Figure 2. T. gondii inhibits HSV-1 and HIVMN triggered production of IFN-a and TNF-a in parasite infected pDC.
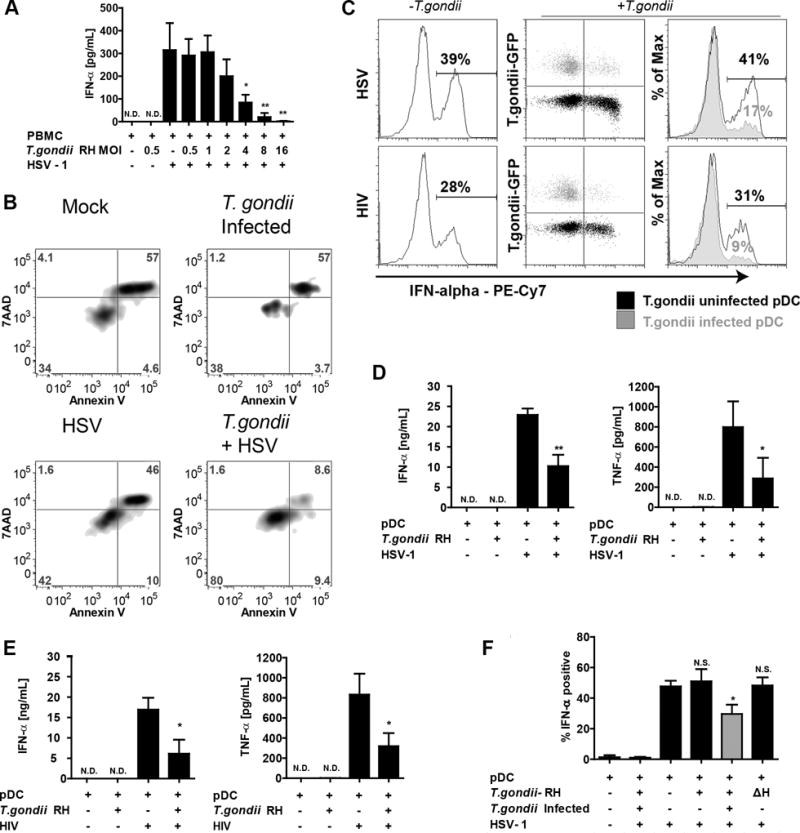
A, PBMC were pre-infected with increasing MOI of T. gondii RH for 1 hr followed by 18 hrs stimulation with HSV-1. IFN-a levels in supernatants were quantified using sandwich ELISA. Data are from mean ± SEM for three independent experiments. Inhibition of HSV-1 induced IFN-α by T. gondii was determined using 1-way ANOVA with Dunnett’s Multiple Comparison Test. B, The effect of T. gondii on pDC was determined by Annexin V/7AAD 18 hours after infection with T. gondii. C, Purified pDC were pre-infected with T. gondii-GFP at MOI 4 and exposed to HSV-1 at MOI 1 or 500ng/mL p24 equivalents of AT-2 inactivated HIVMN. Cells were collected at 6 hrs and analyzed by intracellular flow cytometry for expression of IFN-a. On the right, overlay of T. gondii infected population (gray) and T. gondii uninfected population (black) from the same sample, numbers represent % of IFN-a positive cells in T. gondii uninfected or infected populations. Data are representative of three independent experiments. D,E, Purified pDC were pre-infected with T. gondii-GFP followed by HSV-1 (D) or AT-2 inactivated HIVMN (E) treatments for 18 hrs. Supernatants were collected and analyzed for production of IFN-a and TNF-α production using ELISA. Data are mean ± SEM for N=3 experiments. F, Inhibition of IFN-a by T. gondii RH in pDC from PBMC population. BDCA2 and CD123 positive pDC were gated and analyzed for intracellular expression of IFN-a in T. gondii uninfected (GFP-, black) or infected (GFP+, gray) cells as described in panel B. Heat inactivated parasite (ΔH) was used to determine whether parasite needs to be alive to inhibit IFN-a. Data are mean ± SEM of three independent experiments. Data analyzed using one-way ANOVA with Tukey’s post-test using GraphPad Prism. *p<0.05, **p<0.01, **p<0.001, ns-not significant vs. virus treatment.
To determine whether pDC function is directly or indirectly down-modulated by T. gondii, we examined IFN-a production under conditions where only a subset of the pDC was infected. Suppression of IFN-a was observed only in T. gondii infected cells (green) while uninfected cells from the same sample responded normally to HSV stimulation (Figure 2C, upper panels, black). Similar results were found for IFN-α production in response to AT2-inactivated HIV, which induces IFN-α in pDC through TLR7 signaling (Figure 2C, lower panels).
To further investigate the inhibitory effects of T. gondii infection on production of cytokines, highly-enriched pDC were stimulated with HSV. pDC produced neither IFN-a nor TNF-a in response to T. gondii; however strong inhibition of IFN-a production (55% inhibition), and of TNF-a (64% inhibition) was observed in T. gondii-infected pDC stimulated with HSV (Figure 2D), suggesting that the effect of T. gondii on pDC was not dependent on the presence of other cell types. Since recognition of HSV-1 by pDC occurs through TLR9 (17), we next assessed whether T. gondii also inhibits HIV-1 induced IFN-a production in pDC, which is mediated through TLR7 (18). Similar to what we observed with HSV-1, T. gondii strongly inhibited production of both IFN-a and TNF-a by purified pDC in response to AT2-inactivated HIVMN (Figure 2E). To further evaluate whether the effect of T. gondii on pDC requires infection of the pDC by the parasite, we carried out experiments in purified pDC infected with a sub-optimal MOI of T. gondii to yield populations that contained both infected and uninfected pDC (Figure 2F). Similar to what we observed in pDC within the whole PBMC population (Figure 2C), significant inhibition of IFN-a occurred only in actively parasitized pDC which were positive for PVM marker GRA7 (Figure 2F); pDC from the population exposed to T. gondii but not actively infected by the parasite produced IFN-a at the same level as those stimulated with HSV in the absence of T. gondii. Heat-inactivated T. gondii, which is unable to actively invade cells, failed to inhibit virus-induced IFN-a production by pDC (Figure 2F).
T. gondii infection of pDC affects neither uptake of GFP-tagged HSV nor the endosomal localization of TLR9
To determine whether failure to take-up virus accounted for the deficiency in IFN-a and TNF-a production in T. gondii-infected pDC, we evaluated the uptake of GFP-tagged HSV by these cells using imaging flow cytometry. T. gondii infection did not reduce the capacity or alter the kinetics of pDC for virus uptake (Figure 3A, left panel) and cells were clearly visible that had taken up both HSV and T. gondii (Figure 3A right panel). To further characterize the effect of T. gondii infection on the TLR9 pathway, we quantified the co-localization of TLR9 with CD71, a marker of early and recycling endosomes, in pDC either infected or uninfected with T. gondii and in the presence or absence of HSV-1 stimulation. Unstimulated pDC expressed high levels of TLR9 in CD71-positive endosomes as quantified by high constitutive co-localization of these two molecules. Similarity scores quantify the co-localization of the two markers, with scores higher than 1 indicating co-localization of TLR9 and CD71 as confirmed by visual inspection of representative events on the right in panel B. Neither HSV stimulation nor T. gondii infection affected the median similarity score of 1.9 across all experimental conditions (Figure 3B). Moreover, T. gondii infection did not decrease cytoplasmic levels of TLR9 (as measured by TLR9 mean fluorescent intensity), indicating that TLR9 in T. gondii infected pDC remains accessible for engagement by HSV (data not shown). Together, these data strongly suggest that T. gondii infection neither prevents uptake of virus nor affects the endosomal localization of TLR9, indicating that the target of inhibition is located downstream of TLR9 engagement.
Figure 3. T. gondii infection neither affects the uptake of HSV-1 nor the subcellular localization of TLR9.
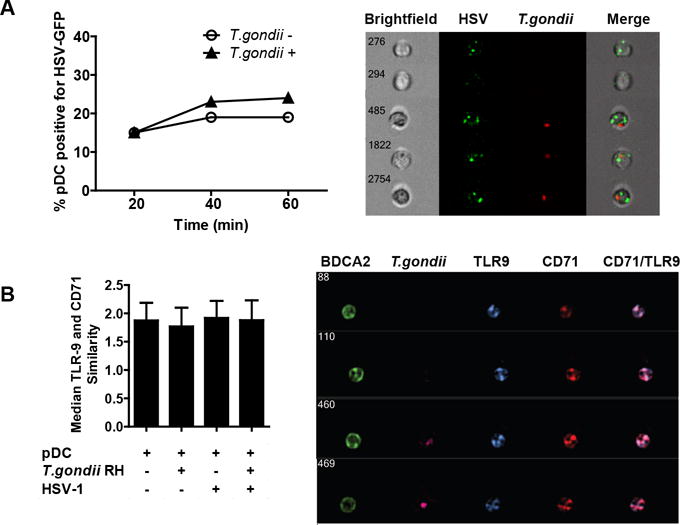
A, Purified pDC were infected with T. gondii-mCherry-RH at an MOI of 4 for 1 hr, then incubated with HSV expressing GFP capsid at MOI of 10. At indicated times, cells were extensively washed with ice cold PBS EDTA to remove surface bound virus and fixed with PFA. Samples were collected with the Amnis ImageStream and percent of pDC positive for HSV-GFP was determined using IDEAS analysis software and compared between T. gondii uninfected and infected pDC. On the right are the representative images of T. gondii uninfected and infected HSV-GFP positive pDC and on the left is a time-course analysis of HSV-GFP uptake in the presence or absence of T. gondii. B, Amnis ImageStream was used to determine the similarity score between TLR9 and recycling endosome marker CD71 following 30 min exposure to HSV. Mean similarity scores ± SEM from three independent experiments are shown. On the right, representative images of four cells showing TLR9 distribution in CD71-positive compartments are shown.
T. gondii infection does not inhibit phosphorylation of IRF7 but abrogates IRF7 nuclear translocation in virus-stimulated pDC
Having ruled out inhibition of initial steps in viral uptake and TLR9 cellular localization in CD71 positive endosomes as being responsible for the inhibitory effects of T. gondii infection on pDC, we next investigated downstream events of TLR9-mediated recognition of HSV-1. We have previously shown that pDC express high constitutive levels of IRF7 (3) the major transcription factor required for IFN-a production by pDC (2). First, we measured the phosphorylation of IRF7 in response to virus, which is crucial for production of IFN-a in pDC. Phospho-flow cytometry results indicate that by 3 hrs post exposure of pDC to HSV, IRF7 is phosphorylated on serine 477/479. T. gondii infection of pDC did not inhibit the HSV-induced phosphorylation of IRF7. Interestingly, T. gondii infected pDC (in the absence of virus stimulation) showed slightly higher pIRF7 mean fluorescent intensities than did the mock stimulated cells (Figure 4A). Even though phosphorylation of IRF7 was not inhibited by T. gondii, infection by the parasite resulted in slightly decreased cytoplasmic levels of IRF7 and T. gondii prevented HSV-1 induced upregulation of this transcription factor (Figure 4B).
Figure 4. T. gondii inhibits nuclear translocation of IRF-7 in response to HSV-1 without affecting phosphorylation of IRF-7.
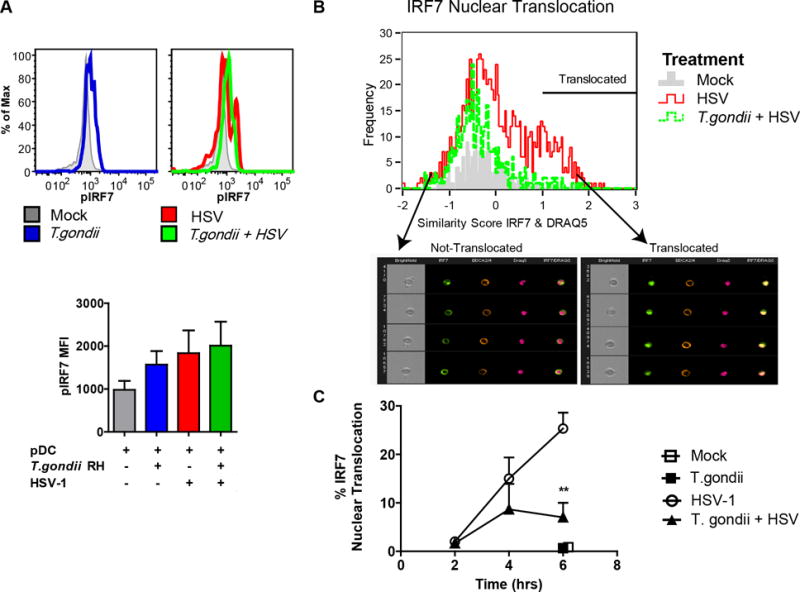
A, PBMC were pre-infected with T. gondii at an MOI of 4, followed by stimulation with HSV, MOI 1. 3hrs after stimulation with virus, fixed pDC were stained using anti-HLA-DR and CD123; following ethanol permeabilization intracellular phosphorylation of IRF7 was assessed using pIRF7-PE (pS477/pS479) and measured using flow cytometry. Representative flow plots from a single experiment are shown in the top panel, and summary data are shown in the lower panel for 3 independent experiments. B, Enriched pDC were pre-infected with T. gondii as described above and exposed to HSV-1 for 2, 4 and 6 hrs; BDCA2 and BDCA4 positive pDC were then analyzed for cytoplasmic and nuclear distribution of IRF7 using ImageStream and Amnis IDEAS software. Representative data are shown from the 4-hr timepoint. IRF7 nuclear translocation in pDC was determined by first creating an object mask for nuclear stain (DRAQ5), and then analyzed using built in function of similarity between IRF7 and nuclear mask. A histogram overlay was generated for comparison of IRF7 nuclear translocation in Mock (gray), HSV-1 (red), and in T. gondii pre-infected and HSV-1 treated pDC (green) pDC. Arrows indicate bin with representative images of not-translocated (left) and translocated (right) events. C, Kinetics of IRF7 nuclear translocation and the effect of T. gondii infection on IRF7 translocation in B were analyzed for statistical significance (n=3).
Next, we assessed whether HSV-triggered nuclear translocation of IRF7 is affected by T. gondii infection. In agreement with our previous results (4, 19), HSV treatment of pDC resulted in strong nuclear translocation of IRF7. T. gondii alone did not result in IRF7 translocation (black square, at 6 hr). However, T. gondii infection of pDC blocked HSV-induced IRF7 nuclear translocation, reaching statistical significance at 6 hr (Figure 4C). Taken together these data convincingly show that T. gondii infection of pDC inhibited nuclear translocation of IRF7 in response to stimulation with HSV-1 without attenuating phosphorylation of IRF7.
T. gondii mimics IL-10 anti-inflammatory signaling
To understand the molecular basis of T. gondii-mediated inhibition of IRF7 nuclear translocation, we investigated the hypothesis that the parasite mimics IL-10 anti-inflammatory signaling in pDC without signaling through IL-10, as previously suggested by Butcher et al. in a murine macrophage system (11). Using purified pDC, we observed that pDC did not produce IL-10 in response to HSV-1, HIV-1 or T. gondii following overnight stimulation with these pathogens (data not shown). Common to IL-10 signaling and T. gondii infection is the phosphorylation of STAT3 (11, 20). Similar to previous findings in other cell types, T. gondii infection of pDC resulted in strong phosphorylation of STAT3 on tyrosine 705 by 5 hr (Figure 5A, upper right quadrant), whereas T. gondii uninfected pDC (lower left quadrant) did not exhibit STAT3 phosphorylation. These findings indicate that STAT3 phosphorylation is mediated in cis, only in T. gondii-infected cells, ruling out the effect of soluble extracellular factors in STAT3 phosphorylation, such as IL-10 or IL-6. As expected, treatment of pDC with recombinant human IL-10 resulted in rapid phosphorylation of STAT3 in the entire population of pDC (Figure 5B).
Figure 5. T. gondii, like IL-10 inhibits STAT3 phosphorylation and IFN-α production through blockade of IRF-7 translocation but not phosphorylation.
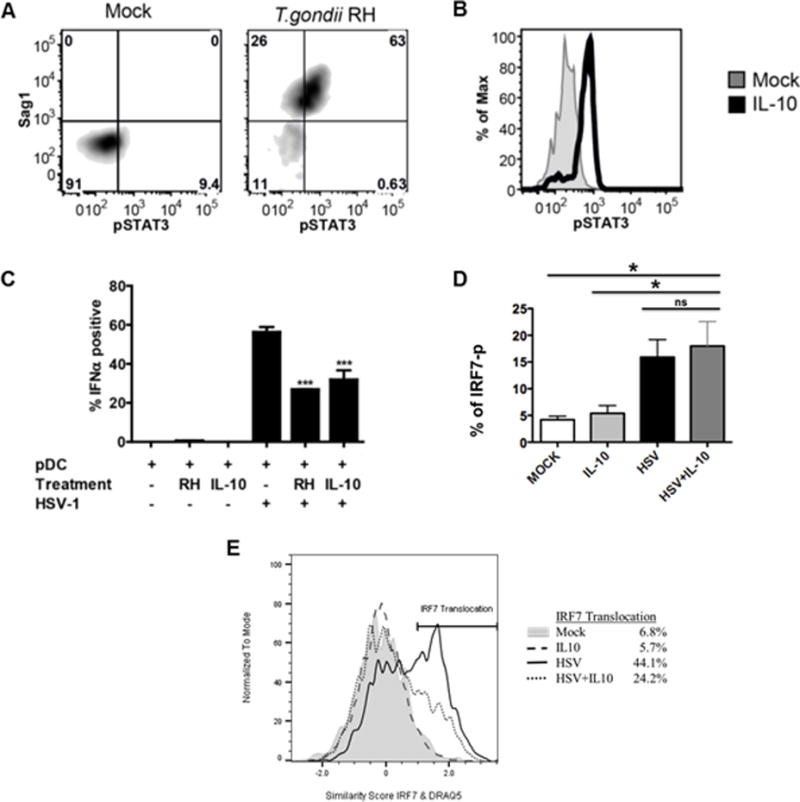
A, Phosphorylation of STAT3 (pY705) was measured in T. gondii-infected pDC 5-hr post- infection using intracellular phospho-flow cytometry. T. gondii infection of pDC (identified by HLA-DR-Pac-blue and CD123-APC) was assessed using intracellular staining against Sag1. Data are representative of three independent experiments. B, PBMC were treated with IL-10 (50 ng/ml) for 15 minutes. Data shown are histogram overlays of IL-10-induced phosphorylation of STAT3 in pDC from PBMC population. Data are representative of three independent experiments. C, Summary figure for IL-10 and T. gondii inhibition of IFN-α production as compared to HSV stimulation of pDC using intracellular flow cytometry as described in A, B (n=3, *** p<0.01 vs. HSV-stimulated). D, PBMC were pretreated with or without IL-10 (50 ng/ml) for 1hr, then the cells were stimulated with HSV-1 (MOI=1) for 3hrs. Phospho-flow analysis was performed for IRF-7 activation in pDC as described in Figure 4A. Data represent the mean ± SEM of three independent experiments; E, IL-10 mediated inhibition of HSV-induced IRF7 nuclear translocation. Enriched pDC were pretreated with or without IL-10 (50 ng/ml) for 1hr, then the cells were stimulated with HSV-1 for 5hrs. IL-10 mediated inhibition of HSV-induced IRF7 nuclear translocation was quantified using Amnis ImageStream as described for Figure 4. Data are representative of three independent experiments.
We next directly compared the inhibitory effect of IL-10 treatment to the effect of T. gondii infection on HSV-induced IFN-a production. T. gondii infection and IL-10 treatment inhibited IFN-a to a similar extent (Figure 5C). This experimental system confirms our previous published findings that IL-10 inhibits HSV-induced IFN-a production (21). Furthermore, to address the question of how IL-10 mediates inhibition of IFN-a, we focused on phosphorylation and nuclear translocation of IRF7. As was seen with T. gondii co-infection, IL-10 stimulation of pDC along with HSV stimulation did not inhibit phosphorylation of IRF-7 (Figure 5D) but did inhibit subsequent HSV-1-induced nuclear translocation of IRF7 (Figure 5E). These data thus reveal a previously unreported mechanism of inhibition of TLR signaling that is shared by IL-10 and T. gondii.
ROP16 is responsible for inhibition of IRF7 nuclear translocation and inhibition of IFN-α production
To further test the hypothesis that parasite-mediated phosphorylation of STAT3 is responsible for the inhibitory effect of T. gondii, we used rop16 knockout parasite (13). ROP16 is a T. gondii virulence factor responsible for direct phosphorylation and activation of STAT3 and STAT6 (14, 22). STAT3 is a signal transducer activated by IL-10 that mediates anti-inflammatory effect of this cytokine (20); therefore, by directly phosphorylating STAT3 in infected cells, T. gondii mimics IL-10 anti-inflammatory signaling. As expected, there was significantly less phosphorylation of STAT3 on tyrosine 705 in the pDC infected with rop16 knockout parasite than in the cells infected with the RH strain (Figure 6A, 6B). To corroborate the involvement of ROP16 kinase in abrogation of TLR9 signaling, the effects of loss of ROP16 kinase on IRF7 nuclear translocation and induction of IFN-a were investigated. rop16 knockout RH parasites failed to inhibit IRF-7 nuclear translocation as compared to the RH strain (Figure 6C). This release of the IRF7 blockade resulted in significantly higher production of IFN-α in response to HSV stimulation with the rop16 knockout vs. RH strain (Figure 6D). To normalize for the effect of parasite infection rate between the wild type parasites and the knockout parasites, the intracellular expression of IFN-a in pDC infected with T. gondii was quantified using flow cytometry. No difference in infection rate between RH and RHΔrop16 was observed (data not shown). As shown in Figure 6E, with gating on T. gondii infected pDC (SAG1 positive, gray bars), rop16 knockout parasites were significantly less efficient than the RH strain in inhibiting IFN -α production. Interestingly, we were not able to fully reverse T. gondii mediated inhibition of IFN-α, and we did not observe rescue in inhibition of TNF-α production in response to HSV-1 (data not shown). Therefore, these data suggest that ROP16 mediated phosphorylation of STAT3 an important mechanism for T. gondii inhibition of FN-α production, but that the parasite uses additional mechanisms for inhibition of TNF-α.
Figure 6. ROP16 kinase is involved in inhibition of IFN-α production and nuclear translocation of IRF7.
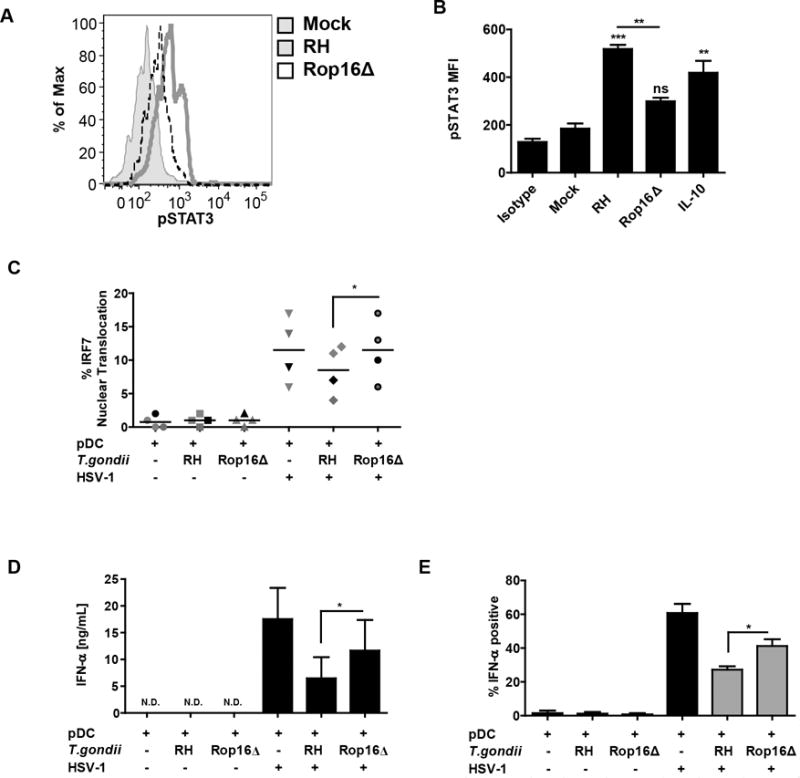
A, Histogram overlay compares pSTAT3 signal between the parental RH strain and the rop16 knockout parasite (RHΔrop16). Loss of ROP16 mediated phosphorylation of STAT3 was measured as described in Figure 5B. B, Quantification of result from panel A from four independent experiments with 15 min IL-10 stimulation used as a control for STAT3 phosphoryation. *’s and “ns” are for statistical analysis of each treatment vs Mock, while the bar represents comparison of wild type vs. Rop16 knockout parasite, as carried out by Anova with Tukey’s post-hoc test. C, Ability of T. gondii to inhibit IRF7 nuclear translocation was lost in RHΔrop16, experiments conducted as described in Figure 4B 5 hrs after stimulation with HSV. D, T. gondii mediated inhibition of IFN-α was compared between the parental RH strain and the rop16 knockout parasite (RHΔrop16). Purified pDC were pre-infected with indicated strain of T. gondii followed by 18 hrs stimulation of HSV, secreted IFN-α was measured using ELISA. E, Flow cytometric analysis of IFN-α production in pDC infected with RH or RHΔrop16. BDCA2+/CD123+ pDC were stained for intracellular expression of IFN-α and SAG1. Gray bars indicate T. gondii infected cells (SAG1 positive). For C, D and E, data are from four independent experiments and were analyzed by Anova with Tukey’s post hoc test comparing each of the groups exposed to HSV.
DISCUSSION
Immunocompromised HIV infected patients suffer from opportunistic infections including toxoplasmosis (8). In this investigation, we have shown that infection of human pDC by the opportunistic pathogen, T. gondii, results in the inability of pDC to respond to viral nucleic acids with the production of IFN-α, linking our study to T. gondii/HIV-1 co-infection in AIDS patients. Although T. gondii/HIV co-infections are widely known, to date no investigation has focused on understanding of the molecular interaction of these two pathogens in the context of host antiviral responses. Based on our findings that T. gondii infection blocks IFN-α production by pDC without inducing death of pDC, we hypothesize that this mechanism of immune-modulation by reactivated parasites could further result in exacerbation of immunocompromised state in HIV-infected patients.
T. gondii infects multiple cell types of the innate immune system including monocyte/macrophages, dendritic cells and pDC (16, 23, 24). Pepper et al. previously reported that mouse pDC became activated in response to T. gondii and responded with production of both IFN-α and IL-12 (16). These intriguing findings led us to investigate the effect of T. gondii infection on human pDC in the context of viral co-infection and innate recognition. In our investigation, an IFN-α response was not detected in human PBMC population infected with T. gondii or in purified human pDC infected by T. gondii. This difference in host response vs. the report of Pepper et al. most likely reflects interspecific variability in the immune responsiveness of mouse and human pDCs. A major difference between murine and human pDC relevant to this study is that human pDC, unlike mouse pDC, generally do not produce IL-12 (25). Tosh et al. described human monocytes and CD1c+ DC as capable of producing IL-12 and TNF-α in response to T. gondii, but did not assess the pDC subset of DC (24). Moreover, Pepper et al. (16) reported that murine pDC recognized parasite ligand profilin through TLR11, a TLR that is not present in humans (26). Moreover, human pDC, unlike their mouse counterparts, express CD4, CCR5, and CXCR4 and are themselves susceptible to HIV-1 infection (27).
Toxoplasma gondii, a protozoan eukaryotic pathogen, has been reported to use multiple mechanisms for evasion and modulation of immune responses. There are numerous reports of inhibition of NFκB and MAPK signaling that result in their down-modulation of TNF-α and IL-12 production by macrophages (28, 29). Additional reports implicate induction of SOCS-1 and modulation of chromatin rearrangement as a mechanism for inhibiting TLR4 signaling in response to LPS (30–32). Because TLR4 signaling can utilize both MyD88 dependent or independent pathways, it was not clear whether the T. gondii inhibition of LPS effects were restricted to a specific pathway. The human pDC system we used in this study allowed us to investigate the inhibition of purely MyD88-dependent, TLR7 and TRL-9-mediated signaling response to HIV-1 and HSV-1, respectively, with IFN-α as our major readout. With this system, we were able to identify that T. gondii targets inhibition of MyD88-dependent signaling through blocking the nuclear translocation of IRF7, an inhibitory mechanism that has not been previously reported for T. gondii. This inhibition was not due to T. gondii toxicity on the pDC, as determined by Annexin V/7AAD binding. Further evidence of a lack of general toxicity of the T. gondii on the pDC was found in that there was normal phosphorylation of IRF7 and STAT1, as discussed below, in pDC infected with T. gondii. Although it is established that IL-10 acts as anti-inflammatory cytokine through STAT3 signaling and induction of the suppressor of cytokine signaling (33), precisely how it mediates immunomodulation of pDC innate responses remains unclear. Our novel finding in Fig. 5 showing that IL-10 inhibited IRF-7 nuclear translocation but not phosphorylation provides a mechanism for our previously reported observation that IL-10 potently inhibits IFN-α production by human pDCs (21). In agreement with our previous observations (21), purified pDC failed to produce IL-10 in response to viral stimulation, nor did they produce IL-10 in response to T. gondii, thus indicating that the inhibition by T. gondii functionally mimicked the IL-10 pathway without being dependent on IL-10. Lack of a soluble mediator responsible for the effects of T. gondii on pDC IFN-α production was further indicated by experiments that showed within mixed populations of pDC that were infected or uninfected with T. gondii, that only the actively parasitized pDC were deficient in IFN-α production. Future investigations should address the presumed STAT3 requirement for IL-10’s effect on IRF7 translocation and whether interference with IRF7 signaling requires STAT3-driven de novo synthesis of new factors or STAT3/protein interactions in the cytosol. Moreover, the ability of T. gondii to induce STAT3 phosphorylation in pDC did not extend to STAT1 phosphorylation: T. gondii did not induce STAT1 phosphorylation, nor did it inhibit STAT1 phosphorylation in pDC in response to IFN-α (data not shown).
Upon infection of the host cell, T. gondii rapidly injects ROP16 into the cytoplasm and tranlocated into the host cell nucleus (34). ROP16 is a tyrosine kinase, which has been shown to mimic IL-10 and IL-4/IL-13 signaling through phosphorylation of STAT3 and STAT6 (14, 22). These reports of immune evasion through ROP16-mediated phosphorylation of STAT3 prompted us to investigate the effect of T. gondii infection on this purely MyD88-dependent signaling of TLR7 and TLR9 in pDC. Both of these TLRs are crucial for viral recognition and clearance, and MyD88 is crucial for host survival to T. gondii infection in mice. Our investigation of TLR7/9 signaling in the context of viral and T. gondii co-infection resulted in identification of inhibition of IRF7 nuclear translocation in response to HIV and HSV that was similar to that observed with IL-10 inhibition of HSV-induced IRF7 translocation. Knockout of rop16 from the T. gondii resulted in the release of IRF7 blockade and partially restored the IFN-α response; although statistically significant, this release of the IRF7 blockade was not complete, suggesting that an additional mechanism responsible for the residual inhibition of IFN-α remains to be identified. It is possible that the remaining inhibition is also mediated through STAT3 since rop16 knockout parasites retain the ability to phosphorylate STAT3 on serine 727 (13).
Inhibition of Type I IFN in pDC by T.gondii may have consequences for viral infections. IFN-α is crucial for control of viral replication through induction of multiple antiviral host response genes. These IFN-stimulated genes (ISG) include, among many others: IRF7, Trim5, Mx protein, PKR, and 2′ 5′ OAS, which inhibit translation, induce apoptosis, sequester viral nucleoproteins and induce RNA degradation to control viral replication as well as stimulate a variety of anti-viral host immune responses (35–38). Therefore, the loss in function in any of these genes can lead to decreased immune responses making the host more susceptible to viral infections like HIV-1. This is the first report of inhibition of innate immune responses to HIV-1 and HSV-1 by T. gondii through inhibition of IFN-α production. Evasion of immune responses through blockade of IRF7 nuclear translocation by T. gondii further adds to the complexity of immune-evasive maneuvers employed by this highly adaptable opportunistic pathogen.
Acknowledgments
The authors thank Marc-Jan Gubbels (Boston College, Boston, MA) for the provision of the mCherry-RH. RHΔku80 (RH) and RHΔku80Δrop16 (RHΔrop16) knockout strains were kindly provided by David Bzik (Dartmouth Medical School, Hanover, NH). The HSV K26-GFP virus was kindly provided by P. Desai (Johns Hopkins University, Baltimore, MD).
PLP, GSY and PFB designed the experiments and interpreted the data. PLP, YZ, and JD carried out the experiments. SS assisted with flow cytometry and imaging flow cytometry design and analysis. PLP, GSY and PFB prepared the manuscript with the assistance of JD, SS and YZ.
Abbreviations
- DC
dendritic cell
- IRF
interferon response factor
- IRG
immunity-related GTPase
- MOI
multiplicity of infection
- ND
none detected
- pDC
plasmacytoid dendritic cell
- T. gondii
Toxoplasma gondii
Footnotes
This work was supported by grants from the National Institute of Allergy and Infectious Diseases, National Institutes of Health (R01AI062806 and R01AI106125 to PFB and R56AI124691 and RO1AI083405 to GSY) and the National Institute for Research Resources grant S10ODO18103 to PFB. PLP was supported by a pre-doctoral fellowship from the New Jersey Commission on Cancer Research
References
- 1.Fitzgerald-Bocarsly P, Dai J, Singh S. Plasmacytoid dendritic cells and type I IFN: 50 years of convergent history. Cytokine Growth Factor Rev. 2008;19:3–19. doi: 10.1016/j.cytogfr.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 3.Izaguirre A, Barnes B, Amrute S, Yeow YZ, Megjugorac N, Dai J, Feng D, Chung E, Pitha P, Fitzgerald-Bocarsly P. Comparative analysis of IRF and IFN-alpha expression in human plasmacytoid and monocyte derived dendritic cells. J Leuk Biol. 2003;74:1125–1138. doi: 10.1189/jlb.0603255. [DOI] [PubMed] [Google Scholar]
- 4.Dai J, Megjugorac NJ, Amrute SB, Fitzgerald-Bocarsly P. Regulation of IFN regulatory factor-7 and IFN-alpha production by enveloped virus and lipopolysaccharide in human plasmacytoid dendritic cells. J Immunol. 2004;173:1535–1548. doi: 10.4049/jimmunol.173.3.1535. [DOI] [PubMed] [Google Scholar]
- 5.Fitzgerald-Bocarsly P, Jacobs ES. Plasmacytoid dendritic cells in HIV infection: striking a delicate balance. J Leukoc Biol. 2010;87:609–620. doi: 10.1189/jlb.0909635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feldman S, Stein D, Amrute S, Denny T, Garcia Z, Kloser P, Sun Y, Megjugorac N, Fitzgerald-Bocarsly P. Decreased interferon-alpha production in HIV-infected patients correlates with numerical and functional deficiencies in circulating type 2 dendritic cell precursors. Clin Immunol. 2001;101:201–210. doi: 10.1006/clim.2001.5111. [DOI] [PubMed] [Google Scholar]
- 7.Luft BJ, Conley F, Remington JS, Laverdiere M, Wagner KF, Levine JF, Craven PC, Strandberg DA, File TM, Rice N, Meunier-Carpentier F. Outbreak of central-nervous-system toxoplasmosis in western Europe and North America. Lancet. 1983;1:781–784. doi: 10.1016/s0140-6736(83)91847-0. [DOI] [PubMed] [Google Scholar]
- 8.Richards FO, Jr, Kovacs JA, Luft BJ. Preventing toxoplasmic encephalitis in persons infected with human immunodeficiency virus. Clin Infect Dis. 1995;21(Suppl 1):S49–56. doi: 10.1093/clinids/21.supplement_1.s49. [DOI] [PubMed] [Google Scholar]
- 9.Jones JL, K-M D, Wilson M. Toxoplasma gondii prevalence, United States [letter] Emerg Infect Dis. 2007;13(4):2. [Google Scholar]
- 10.Dubremetz JF. Rhoptries are major players in Toxoplasma gondii invasion and host cell interaction. Cell Microbiol. 2007;9:841–848. doi: 10.1111/j.1462-5822.2007.00909.x. [DOI] [PubMed] [Google Scholar]
- 11.Butcher BA, Kim L, Panopoulos AD, Watowich SS, Murray PJ, Denkers EY. IL-10-independent STAT3 activation by Toxoplasma gondii mediates suppression of IL-12 and TNF-alpha in host macrophages. J Immunol. 2005;174:3148–3152. doi: 10.4049/jimmunol.174.6.3148. [DOI] [PubMed] [Google Scholar]
- 12.Saeij JP, Boyle JP, Coller S, Taylor S, Sibley LD, Brooke-Powell ET, Ajioka JW, Boothroyd JC. Polymorphic secreted kinases are key virulence factors in toxoplasmosis. Science. 2006;314:1780–1783. doi: 10.1126/science.1133690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Butcher BA, Fox BA, Rommereim LM, Kim SG, Maurer KJ, Yarovinsky F, Herbert DR, Bzik DJ, Denkers EY. Toxoplasma gondii Rhoptry Kinase ROP16 Activates STAT3 and STAT6 Resulting in Cytokine Inhibition and Arginase-1-Dependent Growth Control. PLoS Pathog. 2011;7:e1002236. doi: 10.1371/journal.ppat.1002236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamamoto M, Standley DM, Takashima S, Saiga H, Okuyama M, Kayama H, Kubo E, Ito H, Takaura M, Matsuda T, Soldati-Favre D, Takeda K. A single polymorphic amino acid on Toxoplasma gondii kinase ROP16 determines the direct and strain-specific activation of Stat3. J Exp Med. 2009;206:2747–2760. doi: 10.1084/jem.20091703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taylor GA, Feng CG, Sher A. p47 GTPases: regulators of immunity to intracellular pathogens. Nat Rev Immunol. 2004;4:100–109. doi: 10.1038/nri1270. [DOI] [PubMed] [Google Scholar]
- 16.Pepper M, Dzierszinski F, Wilson E, Tait E, Fang Q, Yarovinsky F, Laufer TM, Roos D, Hunter CA. Plasmacytoid dendritic cells are activated by Toxoplasma gondii to present antigen and produce cytokines. J Immunol. 2008;180:6229–6236. doi: 10.4049/jimmunol.180.9.6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peng WM, Yu CF, Allam JP, Oldenburg J, Bieber T, Hoch J, Eis-Hubinger AM, Novak N. Inhibitory oligodeoxynucleotides downregulate herpes simplex virus-induced plasmacytoid dendritic cell type I interferon production and modulate cell function. Hum Immunol. 2007;68:879–887. doi: 10.1016/j.humimm.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lepelley A, Louis S, Sourisseau M, Law HK, Pothlichet J, Schilte C, Chaperot L, Plumas J, Randall RE, Si-Tahar M, Mammano F, Albert ML, Schwartz O. Innate sensing of HIV-infected cells. PLoS Pathog. 2011;7:e1001284. doi: 10.1371/journal.ppat.1001284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fanning SL, George TC, Feng D, Feldman SB, Megjugorac NJ, Izaguirre AG, Fitzgerald-Bocarsly P. Receptor cross-linking on human plasmacytoid dendritic cells leads to the regulation of IFN-alpha production. J Immunol. 2006;177:5829–5839. doi: 10.4049/jimmunol.177.9.5829. [DOI] [PubMed] [Google Scholar]
- 20.Williams L, Bradley L, Smith A, Foxwell B. Signal transducer and activator of transcription 3 is the dominant mediator of the anti-inflammatory effects of IL-10 in human macrophages. J Immunol. 2004;172:567–576. doi: 10.4049/jimmunol.172.1.567. [DOI] [PubMed] [Google Scholar]
- 21.Payvandi F, Amrute S, Fitzgerald-Bocarsly P. Exogenous and endogenous IL-10 regulate interferon-alpha production by PBMC in response to viral stimulatoin. J Immunol. 1998;160:5861–5868. [PubMed] [Google Scholar]
- 22.Ong YC, Reese ML, Boothroyd JC. Toxoplasma rhoptry protein 16 (ROP16) subverts host function by direct tyrosine phosphorylation of STAT6. J Biol Chem. 2010;285:28731–28740. doi: 10.1074/jbc.M110.112359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leng J, Butcher BA, Denkers EY. Dysregulation of macrophage signal transduction by Toxoplasma gondii: past progress and recent advances. Parasite Immunol. 2009;31:717–728. doi: 10.1111/j.1365-3024.2009.01122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tosh KW, Mittereder L, Bonne-Annee S, Hieny S, Nutman TB, Singer SM, Sher A, Jankovic D. The IL-12 Response of Primary Human Dendritic Cells and Monocytes to Toxoplasma gondii Is Stimulated by Phagocytosis of Live Parasites Rather Than Host Cell Invasion. J Immunol. 2016;196:345–356. doi: 10.4049/jimmunol.1501558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ito T, Kanzler H, Duramad O, Cao W, Liu YJ. Specialization, kinetics, and repertoire of type 1 interferon responses by human plasmacytoid predendritic cells. Blood. 2006;107:2423–2431. doi: 10.1182/blood-2005-07-2709. [DOI] [PubMed] [Google Scholar]
- 26.Beutler B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature. 2004;430:257–263. doi: 10.1038/nature02761. [DOI] [PubMed] [Google Scholar]
- 27.Schmidt B, Scott I, Whitmore RG, Foster H, Fujimura S, Schmitz J, Levy JA. Low-level HIV infection of plasmacytoid dendritic cells: onset of cytopathic effects and cell death after PDC maturation. Virology. 2004;329:280–288. doi: 10.1016/j.virol.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 28.Shapira S, Harb OS, Margarit J, Matrajt M, Han J, Hoffmann A, Freedman B, May MJ, Roos DS, Hunter CA. Initiation and termination of NF-kappaB signaling by the intracellular protozoan parasite Toxoplasma gondii. J Cell Sci. 2005;118:3501–3508. doi: 10.1242/jcs.02428. [DOI] [PubMed] [Google Scholar]
- 29.Denkers EY, Butcher BA, Del Rio L, Kim L. Manipulation of mitogen-activated protein kinase/nuclear factor-kappaB-signaling cascades during intracellular Toxoplasma gondii infection. Immunol Rev. 2004;201:191–205. doi: 10.1111/j.0105-2896.2004.00180.x. [DOI] [PubMed] [Google Scholar]
- 30.Zimmermann S, Murray PJ, Heeg K, Dalpke AH. Induction of suppressor of cytokine signaling-1 by Toxoplasma gondii contributes to immune evasion in macrophages by blocking IFN-gamma signaling. J Immunol. 2006;176:1840–1847. doi: 10.4049/jimmunol.176.3.1840. [DOI] [PubMed] [Google Scholar]
- 31.Leng J, Butcher BA, Egan CE, Abdallah DS, Denkers EY. Toxoplasma gondii prevents chromatin remodeling initiated by TLR-triggered macrophage activation. J Immunol. 2009;182:489–497. doi: 10.4049/jimmunol.182.1.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim L, Butcher BA, Denkers EY. Toxoplasma gondii interferes with lipopolysaccharide-induced mitogen-activated protein kinase activation by mechanisms distinct from endotoxin tolerance. J Immunol. 2004;172:3003–3010. doi: 10.4049/jimmunol.172.5.3003. [DOI] [PubMed] [Google Scholar]
- 33.El Kasmi KC, Holst J, Coffre M, Mielke L, de Pauw A, Lhocine N, Smith AM, Rutschman R, Kaushal D, Shen Y, Suda T, Donnelly RP, Myers MG, Jr, Alexander W, Vignali DA, Watowich SS, Ernst M, Hilton DJ, Murray PJ. General nature of the STAT3-activated anti-inflammatory response. J Immunol. 2006;177:7880–7888. doi: 10.4049/jimmunol.177.11.7880. [DOI] [PubMed] [Google Scholar]
- 34.Saeij JP, Coller S, Boyle JP, Jerome ME, White MW, Boothroyd JC. Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature. 2007;445:324–327. doi: 10.1038/nature05395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Imaizumi T, Hatakeyama M, Yamashita K, Ishikawa A, Yoshida H, Satoh K, Taima K, Mori F, Wakabayashi K. Double-stranded RNA induces the synthesis of retinoic acid-inducible gene-I in vascular endothelial cells. Endothelium. 2005;12:133–137. doi: 10.1080/10623320500191885. [DOI] [PubMed] [Google Scholar]
- 36.Levy DE, Marie I, Smith E, Prakash A. Enhancement and diversification of IFN induction by IRF-7-mediated positive feedback. J Interferon Cytokine Res. 2002;22:87–93. doi: 10.1089/107999002753452692. [DOI] [PubMed] [Google Scholar]
- 37.Turan K, Mibayashi M, Sugiyama K, Saito S, Numajiri A, Nagata K. Nuclear MxA proteins form a complex with influenza virus NP and inhibit the transcription of the engineered influenza virus genome. Nucleic Acids Res. 2004;32:643–652. doi: 10.1093/nar/gkh192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rebouillat D, Hovanessian AG. The human 2′,5′-oligoadenylate synthetase family: interferon-induced proteins with unique enzymatic properties. J Interferon Cytokine Res. 1999;19:295–308. doi: 10.1089/107999099313992. [DOI] [PubMed] [Google Scholar]