

Abstract
In β-thalassemia, accumulated free α-globin forms intracellular precipitates that impair erythroid cell maturation and viability. Protein quality control systems mitigate β-thalassemia pathophysiology by degrading toxic free α-globin, although the associated mechanisms are poorly understood. We show that loss of the autophagy-activating Unc-51–like kinase 1 (Ulk1) gene in β-thalassemic mice reduces autophagic clearance of α-globin in red blood cell precursors and exacerbates disease phenotypes, whereas inactivation of the canonical autophagy-related 5 (Atg5) gene has relatively minor effects. Systemic treatment with the mTORC1 inhibitor rapamycin reduces α-globin precipitates and lessens pathologies in β-thalassemic mice via an ULK1-dependent pathway. Similarly, rapamycin reduces free α-globin accumulation in erythroblasts derived from CD34+ cells of β-thalassemic individuals. Our findings define a drug-regulatable pathway for ameliorating β-thalassemia.
INTRODUCTION
β-Thalassemia is a common, frequently debilitating, inherited anemia caused by HBB gene mutations that reduce or eliminate expression of the β-globin subunit of adult hemoglobin (HbA, α2β2) (1). The pathophysiology arises from two major consequences of β-globin deficiency. First, insufficient HbA production reduces red blood cell (RBC) oxygen-carrying capacity, causing tissue hypoxia. Second, unstable free α-globin protein generates cytotoxic reactive oxidant species and cellular precipitates that impair the maturation and viability of RBC precursors, resulting in ineffective erythropoiesis and premature lysis of circulating RBCs (2–7). Heterozygosity for β-globin null mutations (β-thalassemia trait) is usually clinically silent but becomes symptomatic with coinheritance of extra α-globin genes (HBA1 or HBA2) due to the increased amount of free α-globin protein (8–11). Hence, globin chain imbalance with accumulation of free α-globin is a major determinant of β-thalassemia pathophysiology.
Normal and β-thalassemic RBC precursors can detoxify excess α-globin [reviewed in (9)]. Specifically, free α-globin is stabilized by the molecular chaperone α-hemoglobin stabilizing protein (AHSP) (12, 13) and is eliminated by the ubiquitin-proteasome system and autophagy (7). Together, these observations suggest mechanisms through which individuals with β-thalassemia mutations can tolerate a modest pool of free α-globin, with pathologies arising when the amount of free α-globin exceeds the capacity of endogenous protective mechanisms. In this respect, β-thalassemia resembles other “protein aggregation disorders” that are characterized by the accumulation of various toxic protein precipitates in disease-associated target tissues [reviewed in (7, 14–16)]. Efforts are underway to treat protein aggregation disorders via the pharmacologic induction of protein quality control pathways to eliminate relevant unstable proteins (17–19). In the current study, we sought to better define the mechanisms of free α-globin clearance by autophagy and to explore related therapies for β-thalassemia.
Autophagy is a process that delivers proteins or organelles to lysosomes for degradation (20). Autophagy pathways are essential for eukaryotic tissue development, cellular homeostasis, and protection against metabolic and proteotoxic stresses (21–24). The major pathway, termed “macroautophagy” (hereafter referred to as “autophagy”), is a multistep process that envelops cargo to be degraded within a double-membrane vesicle (an autophagosome) that eventually fuses with lysosomes [reviewed in (20, 25, 26)]. Research by several groups, including ours, indicates that free α-globin is degraded by autophagy in β-thalassemia (7, 17,27). Here, we examined the contributions of two core autophagy proteins, Unc-51-like kinase 1 (ULK1) and autophagy-related 5 (ATG5), to disease progression in a mouse model of β-thalassemia. ULK1 initiates autophagosome formation by phosphorylating several proteins in response to cellular stresses and has additional roles in vesicular transport (28, 29). Although ULK2, a related protein with overlapping functions, is expressed in most tissues, ULK1 is the predominant member of this family during erythropoiesis (30). ATG5 is part of a protein complex that conjugates microtubule-associated protein 1 light chain 3β (Map1LC3b; hereafter referred to as LC3) and related proteins to membrane-associated phosphatidylethanolamine. This process, termed “lipidation,” creates a scaffold that facilitates cargo recognition and autophagosome maturation (18, 20). ULK1 and ATG5 are essential for most, but not all, autophagy processes. During RBC formation, ULK1 has a nonredundant role in eliminating mitochondria (mitophagy) and ribosomes from reticulocytes, whereas ATG5 is dispensable for these processes (30, 31).
We used an Hbb gene–disrupted β-thalassemic mouse model to show that the accumulation of toxic insoluble α-globin is inhibited by autophagy that is ULK1 mediated and largely ATG5 independent. In β-thalassemic mice, inhibition of the ULK1 inhibitor mammalian target of rapamycin complex 1 (mTORC1) by rapamycin reduced the accumulation of insoluble free α-globin and alleviated the phenotype in an ULK1-dependent fashion. Moreover, rapamycin treatment reduced free α-globin in cultured human RBC precursors from β-thalassemic individuals. Our findings indicate that autophagy relieves the effects of β-thalassemia mutations by degrading excess α-globin, and they identify an associated mechanism that can be targeted to develop therapies.
RESULTS
Hematopoietic ablation of Ulk1 exacerbates pathologic hallmarks of β-thalassemia
To determine whether ULK1 mediates free α-globin degradation in β-thalassemia, we introduced a null allele into β-thalassemic mice (strain HbbTh3/+) (32,33). Double-mutant HbbTh3/+Ulk1−/− mice were born at normal Mendelian ratios, but most died perinatally, with only 15% surviving to 30 days, which contrasted with the 80% survival for HbbTh3/+Ulk1+/+ mice (P < 0.01) (Fig. 1A). Before birth at embryonic day 18.5 (E18.5), HbbTh3/+Ulk1−/− and HbbTh3/+Ulk1+/+ embryos were similar in appearance, with no differences in blood hematocrit, reticulocyte count, or weight (fig. S1, A and B). Therefore, the higher death rate for double-mutant newborn mice, as compared to mice with either mutation alone, is not due to exacerbated anemia. ULK1 regulates energy metabolism and redox homeostasis by modulating the activity of autophagy-related proteins and glycolytic enzymes in response to metabolic stress (28,34–36). Thus, we speculate that ULK1 activity in non-erythroid tissues is required to meet the increased metabolic demands resulting from ineffective erythropoiesis in β-thalassemia, which become manifest postnatally upon the loss of maternal circulation.
Fig. 1. Ablation of Ulk1 in hematopoietic stem cells exacerbates β-thalassemia.
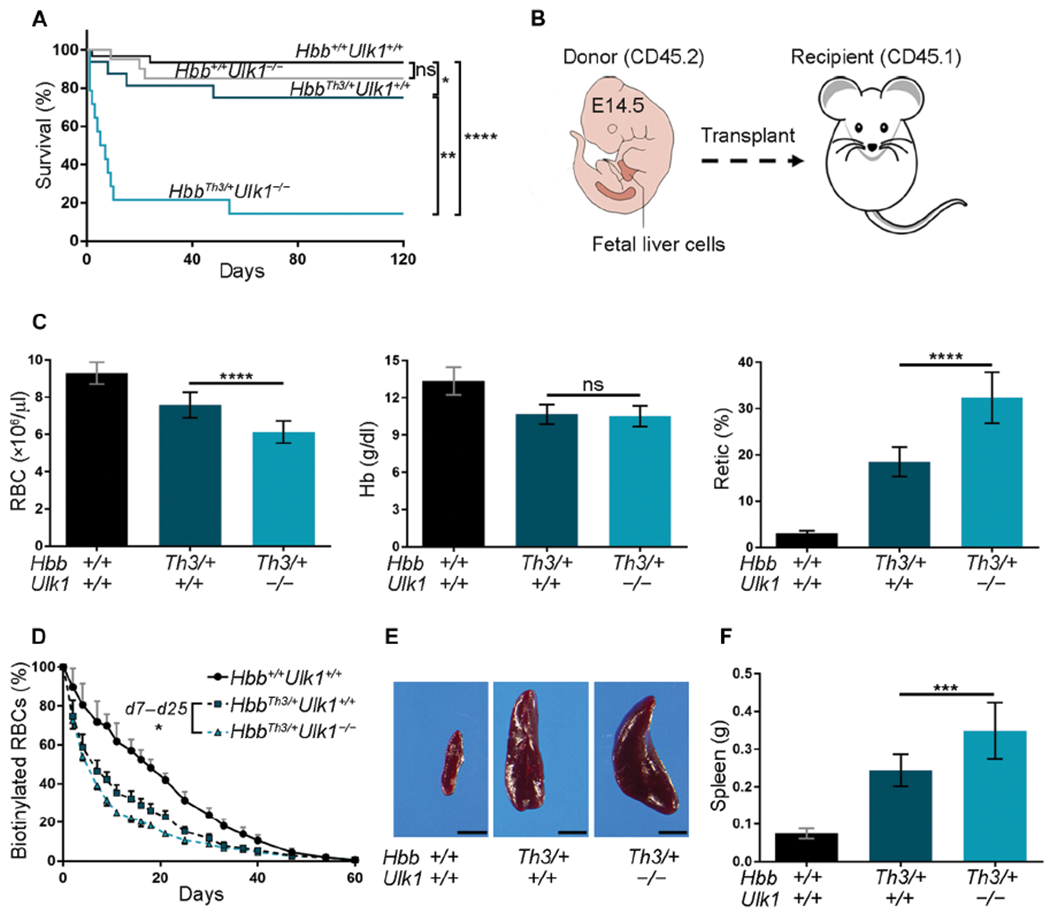
(A) Kaplan-Meier survival plot of mice with germline genotypes Hbb+/+Ulk1+/+ (n = 30), Hbb+/+Ulk1−/− (n = 20), HbbTh3/+Ulk1+/+ (n = 20), and HbbTh3/+Ulk1−/− (n = 15). (B) Embryonic E14.5 fetal liver cells with the genotypes in (A) (C57BL/6, CD45.2) were injected intravenously into lethally irradiated WT (Hbb+/+Ulk1+/+) mice (C57BL/6, CD45.1). (C) Erythroid indices (y axis) according to donor HSC genotype (x axis) at 90 days after HSC transplantation. Hbb+/+Ulk1+/+, n = 22; HbbTh3/+Ulk1+/+, n = 29; HbbTh3/+Ulk1−/−, n = 25. RBC, red blood cell number; Hb, hemoglobin; Retic, reticulocyte. (D) RBC survival at time 0 (about 90 days after HSC transplantation), sulfo-NHS-biotin was injected into the tail vein of mice with HSCs of the indicated genotypes. The fraction of biotinylated RBCs over time was quantified by streptavidin labeling followed by flow cytometry: Hbb+/+Ulk1+/+, n = 8; HbbTh3/+Ulk1+/+, n = 7; HbbTh3/+Ulk1−/−, n = 4. Differences between HbbTh3/+Ulk1+/+ and HbbTh3/+Ulk1−/− mice were statistically significant at all times measured between days 7 and 25 (false discovery rate-adjusted P = 0.002 to 0.02 by the Benjamini and Hochberg method). (E) Representative spleens from mice transplanted with HSC donors of the indicated genotypes. Scale bars, 0.5 cm. (F) Summary of spleen weights: Hbb+/+Ulk1+/+, n = 19; HbbTh3/+Ulk1+/+, n = 15; HbbTh3/+Ulk1−/−, n = 12. Data are mean values ± SDs; ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05; ns, not significant.
To examine the hematopoietic cell-intrinsic consequences of ULK1 loss in β-thalassemia, we transplanted E14.5 fetal liver cells from HbbTh3/+Ulk1−/− or control strains (CD45.2) into lethally irradiated wild-type (WT) CD45.1 hosts (Fig. 1B). Donor myelo-lymphoid cell engraftment exceeded 82 and 94% after 30 and 90 days, respectively (fig. S1C). Loss of Ulk1 exacerbated β-thalassemia, with a 20% reduction in the RBC count (P < 0.0001) and a twofold increase in the reticulocyte count (P < 0.0001) to maintain the same concentration of blood Hb (Fig. 1C and table S1). Loss of Ulk1 reduced the half-life of circulating β-thalassemia RBCs from 7.5 to 5.4 days (P < 0.0001) versus 17.5 days for WT (Hbb+/+Ulk1+/+) RBCs (Fig. 1D). Loss of ULK1 in HbbTh3/+ mice also caused increased splenomegaly, bone marrow erythroid precursor hyperplasia, and extramedullary erythropoiesis in spleen and liver (Fig. 1, E and F; fig. S2; and table S2). The distribution of β-thalassemic erythroid precursors was shifted toward more immature forms by the loss of ULK1 (fig. S3), consistent with worsened ineffective erythropoiesis. Thus, loss of ULK1 reduces RBC life span and worsens ineffective erythropoiesis in β-thalassemia.
ULK1 loss promotes accumulation of free α-globin in β-thalassemia
To investigate whether loss of ULK1 influenced the steady-state concentrations of insoluble α-globin, we lysed circulating RBCs and fractionated the contents by centrifugation followed by triton–acetic acid–urea (TAU) gel electrophoresis to resolve the α- and β-globin proteins (7). As expected, HbbTh3/+ RBCs contained insoluble α-globin, which was increased approximately twofold by the loss of ULK1 (P < 0.001) (Fig. 2, A and B). Transmission electron microscopy (TEM) analysis of flow cytometry–purified reticulocytes from β-thalassemic mice revealed electron-dense material that was shown previously to be precipitated α-globin, some of which was within double-membrane structures (Fig. 2C and fig. S4A) (37, 38). Automated image analysis of 100 to 200 cells from multiple mice demonstrated an approximately twofold increase in the area of electron-dense material in HbbTh3/+Ulk1−/− reticulocytes, as compared to HbbTh3/+Ulk1+/+ reticulocytes (P < 0.05; Fig. 2D).
Fig. 2. ULK1 loss in β-thalassemia increases accumulation of insoluble α-globin.
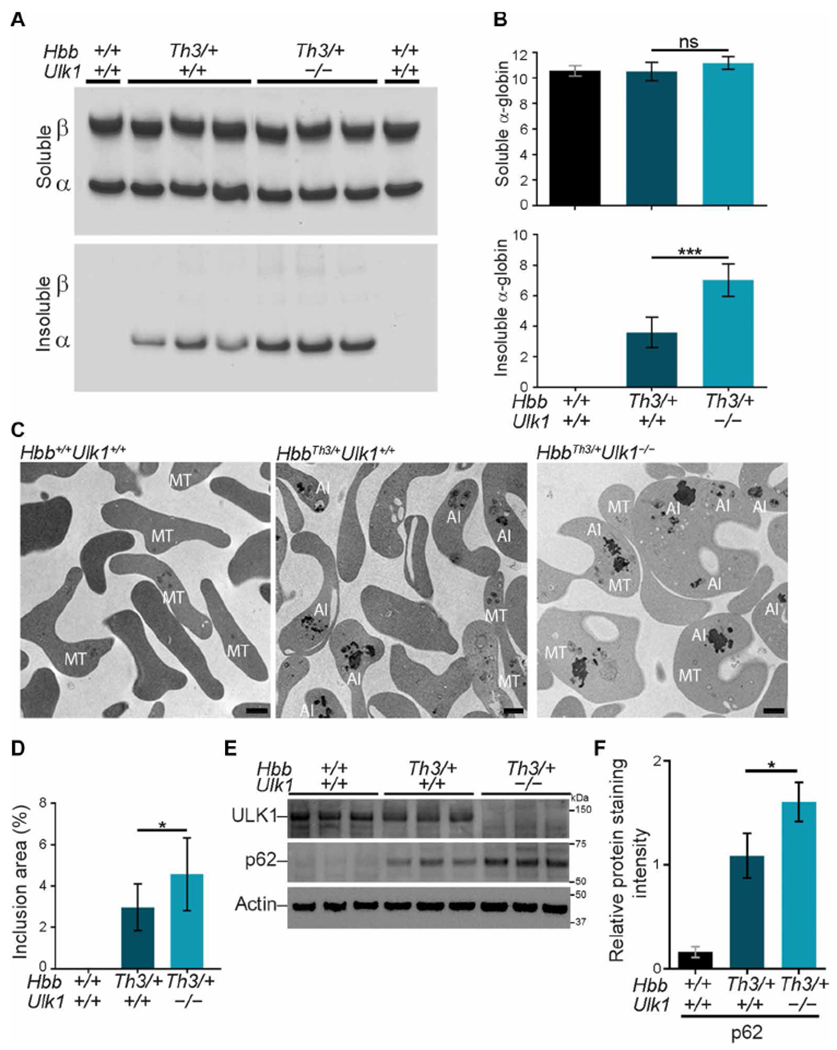
WT mice were transplanted with fetal liver HSCs as in Fig. 1B and analyzed 90 to 120 days later. (A) Soluble and insoluble globins in RBCs. Equal volumes of RBCs (normalized by hematocrit) were lysed, centrifuged to separate insoluble and soluble proteins, fractionated by TAU gel electrophoresis to resolve α- and β-globin proteins, and stained with Coomassie brilliant blue. (B) Results of multiple experiments performed as in (A). The y axes represent the relative soluble and insoluble α-globin staining intensities on TAU gels measured by automated image analysis and expressed in arbitrary units. Hbb+/+Ulk1+/+, n = 4; HbbTh3/+Ulk1+/+, n = 11; HbbTh3/+Ulk1−/−, n = 8. (C) Thiazole orange–stained reticulocytes were isolated by fluorescence-activated cell sorting (FACS) to a purity of about 95% and then analyzed by TEM. AI, α-globin inclusions; MT, mitochondria. Scale bars, 1 μm. (D) Areas of electron-dense α-globin inclusions in reticulocytes (y axis) determined by automated image analysis of electron micrographs. Hbb+/+Ulk1+/+, n = 7; HbbTh3/+Ulk1+/+, n = 11; HbbTh3/+Ulk1−/−, n = 12. About 100 to 200 cells from each mouse were analyzed. (E) Western blot analysis of autophagy proteins ULK1 and p62 in FACS-purified reticulocytes from mice with the indicated genotypes. Migration of protein molecular weight standards is shown on the right. (F) Results of multiple experiments performed as in (E). The y axis represents the relative protein staining intensity on Western blots, measured by automated image analysis, normalized to β-actin, and shown in arbitrary units: n = 3 mice for each genotype. Data are mean values ± SDs; ***P < 0.001; *P < 0.05; ns, not significant.
The amount of autophagy adaptor protein SQSTM1/p62 was increased in HbbTh3/+Ulk1+/+ reticulocytes and, to a greater extent, in HbbTh3/++Ulk1−/− reticulocytes, as compared to WT reticulocytes (Fig. 2, E and F), although the amounts of the corresponding mRNA (Sqstm1) were similar for all three genotypes (fig. S4B). Together, these findings indicate that ULK1 is required for the autophagic clearance of free α-globin and of some autophagy proteins in β-thalassemia.
Autophagy of free α-globin in β-thalassemic mice is largely ATG5-independent and ULK1-dependent
Although ATG5 is required for most autophagy processes, mitochondria are eliminated from reticulocytes via an ATG5-independent, ULK1-dependent form of autophagy (30, 31). We investigated the role of ATG5 in eliminating free α-globin by generating HbbTh3/+ β-thalassemic mice in which Atg5 alleles were disrupted specifically in erythroid precursors (Atg5fl/fl × EpoR-Cre, hereafter referred to as Atg5fl/fl) (fig. S5A). Double-mutant (HbbTh3/+Atg5fl/fl) mice were born at normal Mendelian ratios, and at age 4 to 6 months, Atg5 transcripts and protein content were reduced by about 90% in Ter119+ bone marrow erythroid precursors (fig. S5B). The erythroid indices of HbbTh3/+Atg5fl/fl × EpoR-Cre mice were mildly worsened compared to those of HbbTh3/+ mice, with an approximately 10% reduction in RBC count (P < 0.001) and a 20% increase in reticulocyte count (P < 0.05), but there was no significant difference in spleen weight (Fig. 3, A and B, and table S3). However, in contrast to ULK1 deficiency, ATG5 deficiency did not increase the accumulation of insoluble RBC α-globin in β-thalassemic mice (Fig. 3, C and D). Consistent with impaired autophagy [reviewed in (39)], SQSTM1/p62 was increased in HbbTh3/+Atg5fl/f reticulocytes, as compared to β-thalassemic HbbTh3/+ reticulocytes (fig. S5, C and D). As expected, phosphoethanolamine-conjugated LC3 (referred to as LC3-II or lipidated LC3) was not detected in HbbTh3/+Atg5fl/fl reticulocytes (Fig. 3, E and F). In contrast, LC3-II was increased in Hbbth3/+ and Hbbth3/+Ulk1−/− reticulocytes after chloroquine treatment to inhibit lysosomal proteases, indicating that LC3 is lipidated and delivered to lysosomes (40). We also noted accumulation of nonlipidated LC3 (LC3-I) in HbbTh3/+Atg5fl/fl and HbbTh3/+Ulk1−/− reticulocytes, as compared to HbbTh3/+ reticulocytes. Together, these data indicate that ATG5 mediates some important autophagy processes during β-thalassemic erythropoiesis but that most insoluble free α-globin is eliminated by an ATG5-independent, ULK1-dependent pathway (compare Fig. 2, A and B, to Fig. 3, C and D). The relatively minor worsening of RBC indices conferred by ATG5 deficiency could be due to small increases in insoluble α-globin that were not detected in our assays or failure to eliminate other proteins.
Fig. 3. Clearance of free α-globin in β-thalassemia is largely ATG5 independent.
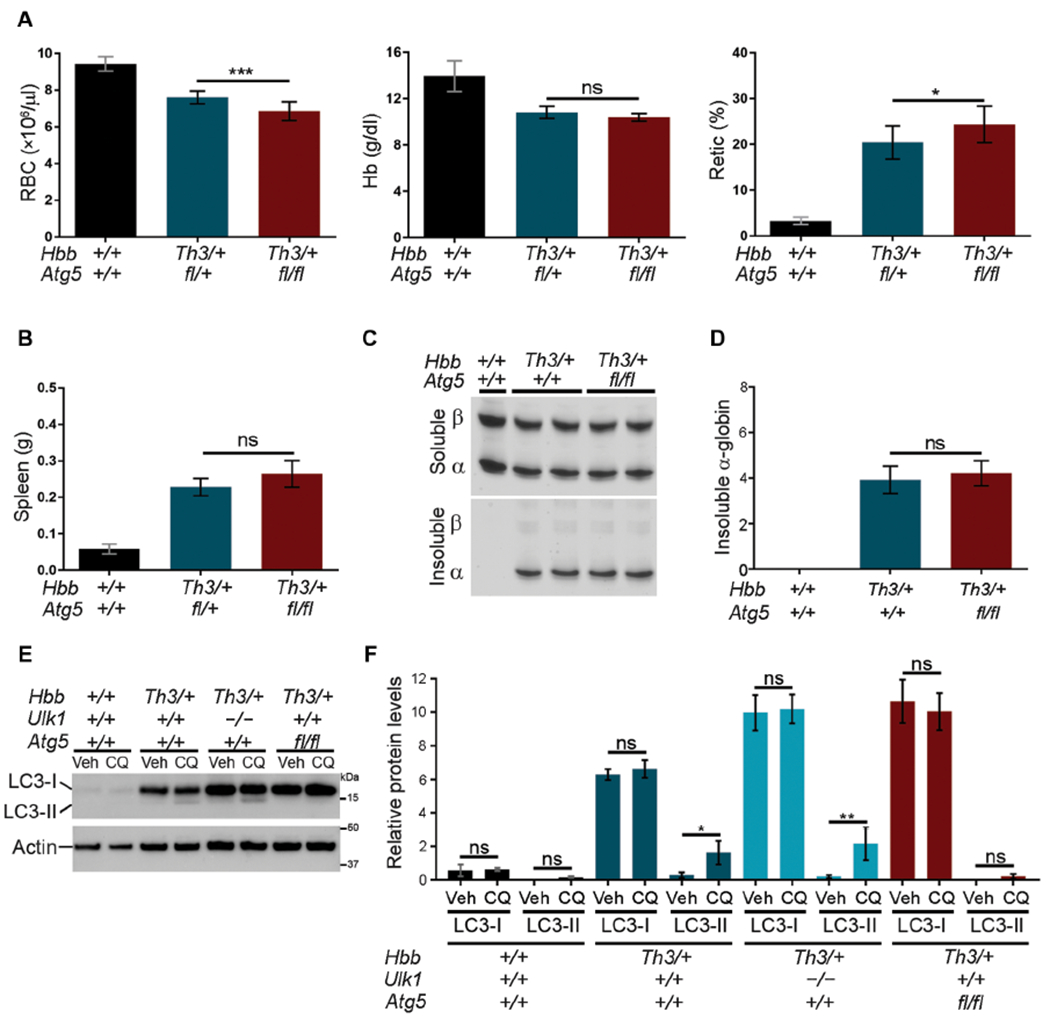
(A) Erythroid indices (y axis) of mice with the indicated germline genotypes (x axis) at 5 months of age: Hbb+/+Atg5+/+, n = 10; HbbTh3/+Atg5+/+, n = 10; HbbTh3/+Atg5fl/fl, n = 12. The Atg5 fl allele refers to an erythroid-specific targeted disruption of Atg5 (fig. S5). (B) Spleen weight according to genotype. Hbb+/+Atg5+/+, n = 10; HbbTh3/+Atg5+/+, n = 5; HbbTh3/+Atg5fl/fl, n = 4. (C) Soluble and insoluble α-globin in RBCs determined as described for Fig. 2A. (D) Results of multiple experiments performed as in (C) and quantified as described for Fig. 2B. Hbb+/+Atg5+/+, n = 5; HbbTh3/+Atg5+/+, n = 8; HbbTh3/+Atg5fl/fl, n = 8. (E) Western blot analysis of LC3 in FACS-purified reticulocytes cultured for 3 hours with chloroquine (CQ) to inhibit lysosomal proteases or with vehicle (Veh). Migration of protein molecular weight standards is shown on the right. (F) Results of multiple experiments performed as in (E), measured by automated image analysis, normalized to β-actin, and shown in arbitrary units: n = 4 mice for each genotype. Data are mean values ± SDs; ***P < 0.001; **P < 0.01; *P < 0.05; ns, not significant.
Previous work by our group (7) and others [reviewed in (9)] demonstrated that the ubiquitin-proteasome system and autophagy degrade excess α-globin in β-thalassemic erythroid cells. Systemic administration of the proteasome inhibitor bortezomib to HbbTh3/+ mice caused up-regulation of autophagy with no change in the amount of insoluble α-globin (7). To further investigate how loss of ULK1 or ATG5 affect the turnover of insoluble α-globin in β-thalassemia, we pulse-labeled mutant reticulocytes with 35S-labeled amino acids, chased with unlabeled amino acids and various inhibitors or vehicle, and quantified soluble and insoluble nascent radiolabeled α-globin in cell lysates. Immediately after pulse labeling, the amounts ofnascent insoluble α-globin were similar in HbbTh3/+ Ulk1−/−, HbbTh3/+Ulk1+/+, and HbbTh3/+Atg5fl/fl reticulocytes (fig. S6A). After a 6-hour chase, Hbbth3/+ reticulocytes had cleared about 60% of the labeled insoluble α-globin through mechanisms that were sensitive to proteasome inhibition by MG132 or lysosomal inhibition by chloroquine or bafilomycin A1 (Fig. 4 and fig. S6, B and C). Soluble α-globin present within HbA tetramers was stable throughout the entire chase period (fig. S6, B, D, and E). Insoluble α-globin was partially cleared by the proteasome in HbbTh3/+Ulk1−/− reticulocytes; however, lysosome-mediated clearance was fully eliminated (Fig. 4). In contrast, lysosomal degradation of insoluble α-globin remained intact in HbbTh3/+ reticulocytes lacking ATG5 (Fig. 4). Thus, insoluble free α-globin in β-thalassemic erythroid cells is cleared by autophagy that is largely ULK1 dependent and ATG5 independent.
Fig. 4. Lysosome-dependent flux of insoluble free α-globin in reticulocytes is ATG5 independent and ULK1 dependent.
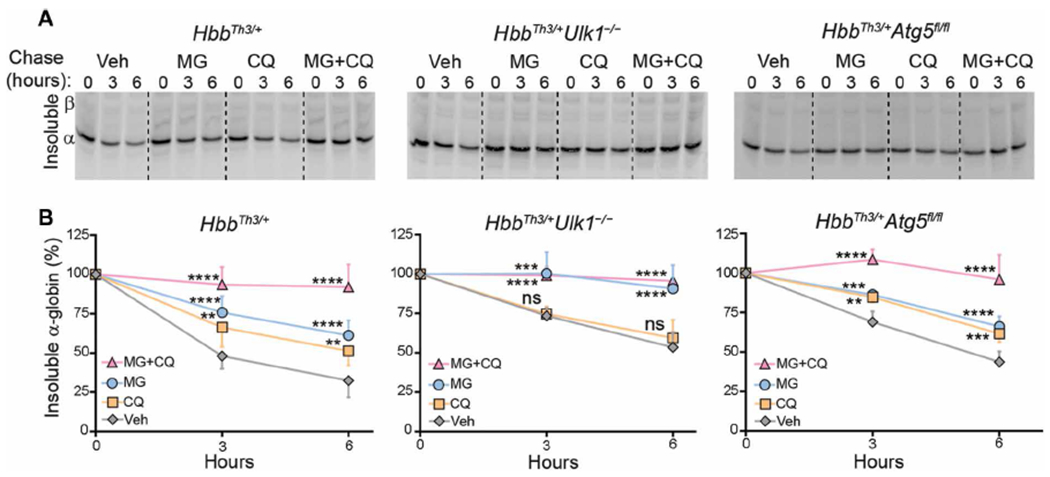
(A) Reticulocytes In whole blood from mice with the indicated genotypes were pulse-labeled with 35S-amino acids and then chased with unlabeled amino acids ± a proteasome inhibitor [MG132 (MG), 10 μM] and/or lysosome inhibitor (chloroquine, 100 μM). Insoluble α-globin in cell lysates was resolved by TAU gel electrophoresis and then visualized by autoradiography. (B) Results of multiple experiments performed as in (A), quantified as described for Fig. 2B: HbbTh3/+, n = 8; HbbTh3/+Ulk1−/−, n = 5; HbbTh3/+Atg5fl/fl, n = 4. Data are mean values ± SDs; ****P < 0.0001; ***P < 0.001; **P < 0.01; ns, not significant versus vehicle for each genotype, by two-way analysis of variance (ANOVA).
Rapamycin increases free α-globin clearance and reduces pathologies in β-thalassemia via ULK1 activation
mTORC1 inhibits autophagy by phosphorylating ULK1 (36, 41–43). Therefore, we tested whether mTORC1 inhibition could ameliorate β-thalassemia by stimulating autophagic clearance of free α-globin. Rapamycin (4 mg/kg once daily) was administered intraperitoneally to β-thalassemic HbbTh3/+ or HbbTh3/+Ulk1−/− mice for 30 days, followed by necropsy. In vivo drug activity in reticulocytes was indicated by reduced phosphorylation of the mTORC1 target ribosomal protein S6 (P < 0.001) (fig. S7, A and B). Rapamycin treatment caused significantly reduced protein synthesis rates in immature erythroblasts (Ery.A) from bone marrow (P < 0.05) but not spleen, as measured by in vivo labeling with O-propargyl-puromycin (Fig. 5A). Rapamycin treatment did not significantly alter protein synthesis in mature erythroblast populations Ery.B and Ery.C in bone marrow or spleen.
Fig. 5. Rapamycin acts through ULK1 to alleviate clinical phenotypes of β-thalassemia in mice.

WT mice were transplanted with fetal liver HSCs of the indicated genotypes, as in Fig. 1B. Twelve weeks later, rapamycin (Rap; 4 mg/kg) or vehicle was administered intraperitoneally, once daily for 30 days. (A) Protein synthesis rates in splenic and bone marrow erythroblasts, determined by O-propargyl-puromycin incorporation. Ery.A, Ery.B, and Ery.C represent increasingly mature Ter119+ erythroblast populations designated according to forward scatter and CD71 expression (see fig. S3): vehicle, n = 3; rapamycin, n = 4. (B) Erythroid indices (y axis) according to HSC genotype (x axis) after treatment with rapamycin or vehicle. HbbTh3/+Ulk1+/+, n = 15; HbbTh3/+Ulk1−/−, n = 12. (C) Sulfo-NHS-biotin was injected intravenously on day 13 of rapamycin treatment, and the fraction of biotinylated RBCs was quantified serially by streptavidin labeling and flow cytometry. (D) Spleens of β-thalassemic mice (HbbTh3/+Ulk1+/+) after rapamycin or vehicle treatment. Scale bars, 0.5 cm. (E) Summary of spleen weights in multiple HbbTh3/+Ulk1+/+ mice after treatment with rapamycin (n = 12) or vehicle (n = 12). Data are mean values ± SDs; ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05; ns, not significant.
β-Thalassemic mice with intact Ulk1 alleles treated with rapamycin exhibited numerous indications of reduced RBC pathology, including a 27% increase in RBC count (P < 0.0001), a 9% increase in Hb (P < 0.01), a 47% decrease in reticulocyte count (P < 0.0001), and an increase in the half-life of circulating RBCs from 6.9 to 12.2 days (P < 0.0001; half-life of WT RBCs was 18 days) (Fig. 5, B and C, and table S4). Rapamycin-treated HbbTh3/+Ulk1+/+ mice also showed reductions in spleen weight (P < 0.01), spleen cell number (P < 0.01), total splenic Ter119+ erythroblasts (P < 0.05), bone marrow erythroid hyperplasia, and extramedullary erythropoiesis in the spleen and liver (Fig. 5, D and E; fig. S7, C and D; and table S5). Rapamycin treatment of HbbTh3/+Ulk1+/+ mice caused a shift of spleen and bone marrow erythroid precursors toward more mature forms, indicating reduced ineffective erythropoiesis (fig. S7, E and F). Rapamycin had no effect on RBC indices or RBC half-life in WT or HbbTh3/+Ulk1−/− mice (Fig. 5, B and C).
Rapamycin treatment reduced insoluble α-globin by about 50% in RBCs of Hbbth3/+ mice (P < 0.001) (Fig. 6, A and B) but not in those of HbbTh3/+Ulk1−/− mice. Similarly, TEM showed an approximately 60% reduction in electron-dense aggregates in reticulocytes from rapamycin-treated Hbbth3/+ mice (P < 0.0001) but not in those from HbbTh3/+Ulk1−/− mice (Fig. 6, C and D). The amounts of LC3-I, LC3-II, and p62 declined by more than 50% in reticulocytes of HbbTh3/+Ulk1+/+ mice treated with rapamycin (Fig. 6, E and F), with no significant change being observed in the corresponding mRNAs (fig. S8). In contrast, rapamycin had no effect on LC3 or p62 protein expression in HbbTh3/+Ulk1−/− mice. Together, these studies indicate that rapamycin stimulates ULK1-dependent autophagy in β-thalassemic mice to reduce free α-globin precipitates, alleviate ineffective erythropoiesis, and extend RBC life span.
Fig. 6. Rapamycin induces ULK1-dependent elimination of free α-globin.
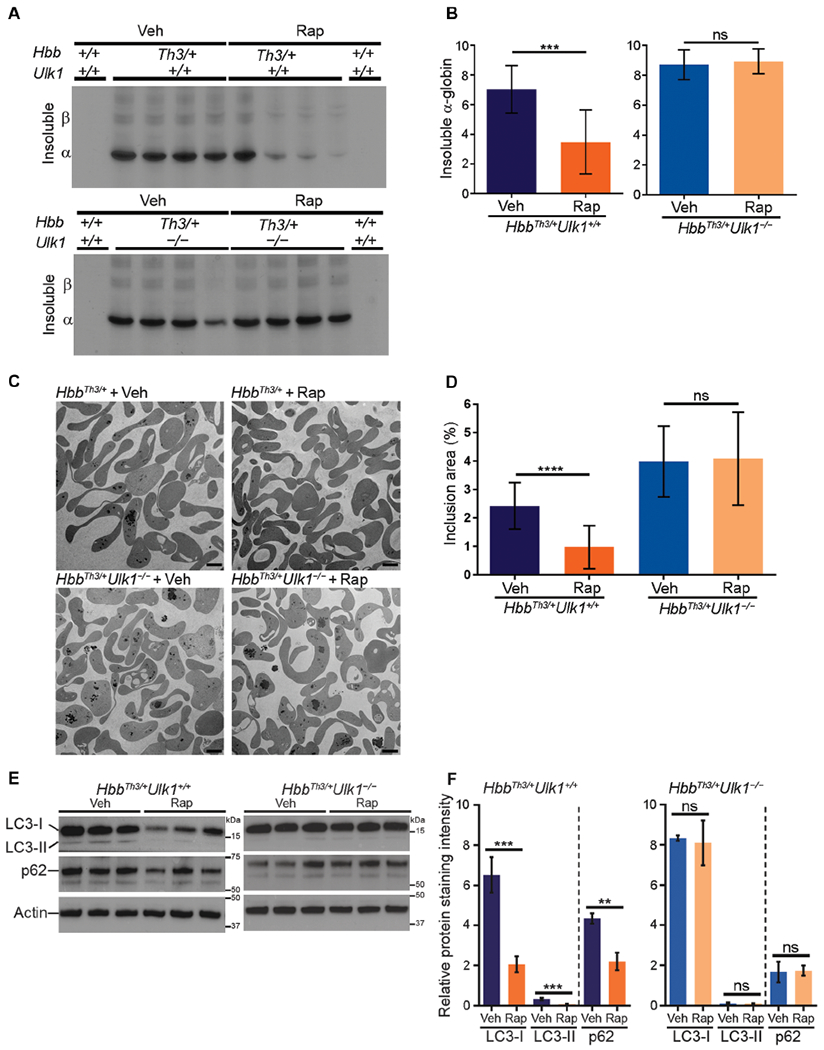
(A) Relative concentrations of insoluble α-globin in RBCs after 30-day treatment with rapamycin or vehicle in mice of the indicated genotypes. RBCs were analyzed as described in Fig. 2A. (B) Results of multiple experiments performed as in (A) and quantified as described for Fig. 2B. n = 7 mice for each condition shown. (C) Transmission electron micrographs of purified reticulocytes in mice with HSCs of the indicated genotypes treated with rapamycin or vehicle. Scale bars, 2 μm. (D) Areas of electron-dense α-globin inclusions in reticulocytes (y axis) determined by analysis of electron micrographs, as described for Fig. 2D. HbbTh3/+Ulk1+/+ + Rap, n = 7; HbbTh3/+Ulk1+/+ + Veh, n = 8; HbbTh3/+Ulk1−/− + Rap, n = 7; HbbTh3/+Ulk1−/− + Veh, n = 7. (E) Western blot analysis of LC3 and p62 in reticulocytes from mice with HSCs of the indicated genotypes treated with rapamycin or vehicle. Migration of protein molecular weight standards is shown on the right. (F) Results of multiple experiments performed as in (E). The y axis represents the relative protein staining intensity on Western blots, measured by automated image analysis, normalized to β-actin, and shown in arbitrary units: n = 4 mice for each genotype. Data are mean values ± SDs; ****P < 0.0001; ***P < 0.001; **P < 0.01; ns, not significant.
Rapamycin reduces free α-globin in human β-thalassemic erythroblasts
To investigate the therapeutic potential of rapamycin for human β-thalassemia, we treated erythroid precursors generated by in vitro differentiation of CD34+ hematopoietic stem and progenitor cells from affected individuals who require regular RBC transfusions [transfusion-dependent (TD)] (n = 5) and from those with variable degrees of anemia who require transfusions intermittently or not at all [non-transfusion-dependent (NTD)] (n = 5) (tables S6 and S7). β-Thalassemic erythroblasts appeared to mature more rapidly than control cells (fig. S9, A to C), as reported previously (44), although the current studies were not designed to test this conclusion. Reversed-phase high-performance liquid chromatography (RP-HPLC) analysis of β-thalassemic erythroblast lysates revealed an α-globin excess (fig. S9, D and E). Nondenaturing ion exchange (IE)–HPLC identified a free α-globin peak in β-thalassemic erythroblasts, but not in control cells (Fig. 7 and fig. S9, F and G). Rapamycin (10 or 20 μM) or the proteasome inhibitor MG132 was added to day 13 cultures that contained mainly mid- to late-stage erythroblasts, and free α-globin was quantified by IE- and RP-HPLC 2 days later (Fig. 7 and fig. S10A). As expected, proteasome inhibition by MG132 increased free α-globin and induced cell death specifically in β-thalassemic erythroblasts but not control cells (Fig. 7 and fig. S10B). In contrast, rapamycin treatment reduced free α-globin by about 40 and 85% in TD β-thalassemia (P < 0.0001) and NTD β-thalassemic erythroblasts (P < 0.001), respectively (Fig. 7), with no alterations in cell survival (fig. S10B). Rapamycin did not alter the maturation stage or protein synthesis rate of control or β-thalassemic erythroblasts over the 2-day treatment period (fig. S10, C and D). Previous studies showed that rapamycin could increase fetal Hb in human CD34+ cell-derived erythroblasts (45, 46). We did not observe this effect in our studies (fig. S10E), perhaps because rapamycin treatment occurred at a relatively late maturation stage and over only 2 days. The current findings demonstrate that rapamycin reduces the accumulation of free α-globin in erythroid cells derived from individuals with β-thalassemia.
Fig. 7. Rapamycin reduces free α-globin in CD34+ cell-derived erythroblasts from individuals with β-thalassemia.
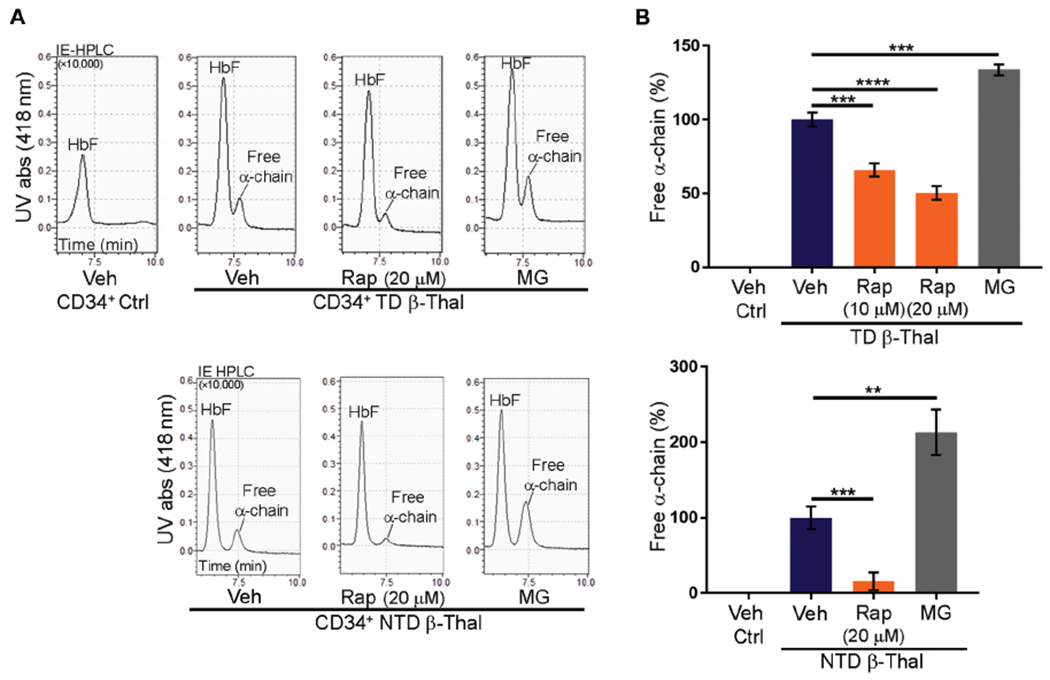
CD34+ cells from normal donors (n = 7) or individuals with transfusion-dependent (TD; n = 5) or non-transfusion-dependent (NTD; n = 5) β-thalassemia were grown in culture under conditions that promote erythroid differentiation, treated with rapamycin (10 or 20 μM), MG132 (2.5 μM), or vehicle beginning on day 13 for 2 days, and then analyzed for free α-globin by ion exchange (IE)-HPLC. (A) Representative chromatograms. UV abs, ultraviolet absorbance. (B) Quantification of free α-globin fractions in multiple experiments performed as in (A). Data are mean values ± SDs; ****P < 0.0001; ***P < 0.001; **P < 0.01.
DISCUSSION
We have shown here that ULK1-dependent autophagy of free α-globin reduces ineffective erythropoiesis and improves RBC life span in β-thalassemia and that this process can be stimulated in a mouse model by the mTORC1 inhibitor rapamycin (fig. S11). Our findings are consistent with the long-standing concept that globin subunit imbalance with excessive buildup of toxic free α-chain is a major determinant of β-thalassemia severity (7,47,48) and indicate that disease severity is modulated by the ubiquitin-proteasome system and autophagy (9). The identification of ULK1 as a central regulator of free α-globin autophagy elucidates a poorly understood facet of β-thalassemia biology and highlights a potentially druggable therapeutic pathway.
According to our current model for protein quality control in β-thalassemic erythropoiesis, unpaired α-chains are polyubiquitinated by ubiquitin ligases including UBE2O, followed by proteasomal degradation (49, 50). Alternatively, free α-globin is eliminated by ULK1-dependent autophagy (fig. S11). We showed previously that proteasome inhibition in β-thalassemic mice did not cause an increase in free α-globin because compensatory up-regulation of autophagy occurred (7, 51). Conversely, we have shown here that loss of ULK1-mediated autophagy increases free α-globin, consistent with findings that autophagy inhibition can impair the degradation of proteasome substrates (52).
Autophagy can occur via multiple pathways depending on the physiologic context, cell type, and cargo, with substantial variation in the requirements for specific effector proteins [reviewed in (20)]. Here, we have shown that autophagy of most free α-globin in β-thalassemia requires ULK1 but not the canonical autophagy protein ATG5, which mediates the conjugation of LC3 and related proteins to autophagosome membrane–associated phosphatidylethanolamine (53). ULK1-dependent, ATG5-independent autophagy occurs with DNA damage (54), lysosomal targeting of ferritin (55), mitophagy during reticulocyte maturation (31, 56), reprogramming of fibroblasts to induced pluripotent stem cells (57), and cardiac ischemia (58). In several of these examples, autophagy was shown to occur independently of LC3 lipidation and specifically required the guanosine triphosphatase RAB9, most likely to facilitate trafficking of cargo-associated Golgi vesicles to lysosomes. Our observation that nonlipidated LC3 (LC3-I) accumulated in ULK1- and ATG5-deficient β-thalassemic erythroblasts may reflect inhibition of the canonical (ULK1- and ATG5-dependent) autophagy pathway by aggregated α-globin (59) and/or nonspecific binding of LC3-I to aggregated free α-globin, independent of the autophagy pathway (60).
A previous study showed that rapamycin improved the RBC count in β-thalassemic mice (61). Here, we have extended these findings by showing that rapamycin alleviates multiple pathological abnormalities of β-thalassemia. We have elucidated the underlying mechanism by showing that rapamycin inhibition of mTORC1 activates ULK1 to stimulate autophagy of free α-globin. Through genetic studies, we have shown that the beneficial effects of rapamycin on β-thalassemia in mice are ULK1 dependent. Moreover, rapamycin treatment reduces the accumulation of free α-globin in CD34+ cell–derived erythroblasts from individuals with β-thalassemia. Rapamycin treatment of human erythroblasts in culture has been shown to induce the synthesis of γ-globin, a fetal-expressed β-like globin that can bind, stabilize, and detoxify free α-globin (45). This effect did not occur in our studies, probably because of differences in the experimental design. Moreover, the reduction of free α-globin by rapamycin did not occur via a decrease in the protein synthesis rate. Overall, these data support our model that rapamycin stimulates autophagy of free α-globin via an ULK1-dependent pathway. However, one limitation of our study is the failure to show directly that rapamycin stimulates the delivery of free α-globin to lysosomes in β-thalassemic cells, in part due to technical difficulties in distinguishing intracellular free α-globin from that in stable HbA (α2β2) or HbF (α2γ2) tetramers, which is present in much higher concentrations. Further studies are required to better define the specific proteins that cooperate with ULK1 to degrade free α-globin during β-thalassemic erythropoiesis.
Although our findings in β-thalassemic mice predict similar beneficial effects for human patients, distinct clinical responses may result from interspecies differences in erythropoiesis. For example, the spleen is a more dominant organ for β-thalassemic erythropoiesis in mice, contrasting with the bone marrow in humans. Thus, reduced ineffective erythropoiesis should manifest mainly as decreased splenomegaly in mice and as decreased bone marrow erythroid hyperplasia in humans. In contrast, damaged RBCs are eliminated by splenic macrophages in both species (62). Therefore, enhanced RBC survival should reduce spleen size in both mice and humans with β-thalassemia.
Considering that rapamycin was approved by the U.S. Food and Drug Administration almost 20 years ago, can be administered safely, and is relatively inexpensive, our findings indicate the utility of a proof-of-principle clinical study to determine its safety and efficacy for treating NTD β-thalassemia. Inhibition of mTORC1 to stimulate autophagy is under investigation for treating neurodegenerative disorders associated with accumulation of aggregated proteins (63–65). However, variable effects have been reported in preclinical studies, depending on the specific neurologic disease, the drug used, and the dosing schedule. Compared to neurodegenerative diseases, β-thalassemia may be more amenable to therapeutic benefits from mTORC1 inhibitor-induced autophagy because erythroid cells undergo rapid turnover, whereas neurons are static and accumulate aggregated protein more slowly. Hence, clinical responses to mTORC1 inhibition in β-thalassemia could be achieved within a relatively short time frame. This study detected beneficial effects of rapamycin in β-thalassemic mice after only 1 month.
Several caveats exist regarding the potential use of rapamycin for treating β-thalassemia patients. First, the doses used in the current studies are standard for preclinical models but are difficult to translate to therapeutic dosing in humans. Second, chronic rapamycin administration can have deleterious effects on immunity and metabolism (63–65), although low doses are reported to extend life span in animal models (66). Thus, future studies should seek to optimize rapamycin dosing regimens based on the therapeutic index for β-thalassemia. It may also be possible to increase the therapeutic index by using newer mTORC1 inhibitors (alone or in combination) and/or drugs that activate ULK1 directly (67). Moreover, the identification of ULK1 as a regulatable effector of free α-globin autophagy suggests the potential for additional therapeutic targets. For example, adenosine monophosphate (AMP)–activated protein kinase (AMPK) activates ULK1 via direct phosphorylation (43). The development of drugs that activate AMPK and ULK1 is underway (68, 69), and their utility as therapies for β-thalassemia should be assessed.
MATERIALS AND METHODS
Study design
Our objective was to study the role of autophagy in eliminating unstable free α-globin in β-thalassemia, focusing on the canonical autophagy proteins ULK1 and ATG5. We introduced null alleles for Ulk1 and Atg5 into a mouse model for β-thalassemia and examined the effects on accumulation of insoluble free α-globin, its clearance by autophagy and the ubiquitin-proteasome pathway, and β-thalassemic phenotypes. Accumulation of free α-globin was measured in soluble and insoluble cell fractions by TAU gel electrophoresis with Coomassie brilliant blue staining or by TEM of purified reticulocytes followed by automated image analysis. Turnover of nascent free α-globin was quantified by pulse-labeling reticulocytes with 35S-labeled amino acids followed by unlabeled amino acid chase with a proteasome inhibitor (MG132) and/or a lysosome inhibitor (chloroquine or bafilomycin A1), fractionation on TAU gels, and autoradiography. Hematologic phenotypes of various β-thalassemia strains were examined by standard analyses, including automated blood counts, tissue histology, flow cytometry of erythroblasts for maturation stage markers, and biotin labeling of RBCs followed by serial flow cytometry to determine half-life. To assess the effects of rapamycin on clearance of free α-globin, 3-month-old β-thalassemic mice with intact or disrupted Ulk1 alleles were treated with rapamycin (sirolimus; Pfizer) intraperitoneally (4 mg/kg daily) for 30 days before phenotypic analysis was performed. The interpretation of tissue histology in all cohorts was carried out in a blinded fashion by a board-certified veterinary pathologist. Quantification of Western blot data and α-globin aggregates in electron micrographs was performed by automated image analysis. To examine the effects of rapamycin on free α-globin concentrations in human erythropoiesis, we cultured CD34+ cell–derived erythroblasts from individuals with β-thalassemia under conditions that favor erythroid differentiation and then measured the free α-globin by HPLC. No data from our experiments were discarded for this study. All studies included at least three biological replicates.
Mice
Mouse experimental protocols were approved by the Institutional Animal Care and Use Committee at St. Jude Children’s Research Hospital. The breeding and analysis of HbbTh3+/− and Ulk1−/− mice have been described previously (7, 30). EpoR-Cre mice, with Cre recombinase being expressed from the endogenous erythropoietin receptor locus (70), were crossed with Atg5flox mice (Atg5fl/fl) with loxP sites flanking exon 3 of Atg5 (22). All Atg5fl/fl mice analyzed in this study also carried the EpoR-Cre allele. All mice were backcrossed onto the C57BL/6J background (The Jackson Laboratory) for five to seven generations. Experiments were conducted with mice aged 6 to 24 weeks, with WT littermates being used as controls. Fetal liver transplants were performed as described previously (71). Briefly, 8-week-old C57BL/6 CD45.1 recipient mice were lethally irradiated (with 11.25 Gy); then, after a 24-hour interval, they received a transplant of 2 × 106 fetal liver cells from CD45.2 E14.5 embryo donors via tail vein injection. Engraftment was determined by flow cytometry for CD45 alleles at 4 and 12 weeks after transplant.
Hematologic analysis
Mice were analyzed at 1 to 6 months of age. Blood was collected by submandibular bleeding, anticoagulated with EDTA, and analyzed on a FORCYTE Veterinary Hematology Analyzer. Reticulocytes were quantified with thiazole orange (Retic-COUNT, BD Biosciences) in accordance with the manufacturer’s protocol, using an LSRFortessa cell analyzer (BD Biosciences), and cell counts were analyzed using FlowJo 10.4.1 software (FlowJo, LLC).
Drug treatment
Beginning at 90 days after hematopoietic stem cell (HSC) transplantation, mice received daily injections intraperitoneally of rapamycin (sirolimus; Pfizer) (4 mg/kg) or vehicle (sirolimus vehicle; 0.5% ethanol, 5% Tween 80, and 5% polyethylene glycol 400) for 30 days, at which point the animals were euthanized. Mice were bled from the tail vein once or twice per week to determine erythrocyte life span and the percentage of reticulocytes by flow cytometry. Blood samples obtained by cardiac puncture were submitted for hematologic analysis and detection of globin precipitates and for reticulocyte sorting for TEM. Tissue samples were submitted for pathology analysis.
In vivo measurement of protein synthesis
Mice received an intraperitoneal injection of O-propargyl-puromycin solution (OP-Puro; Medchem Source LLP) (50 mg/kg) or vehicle [phosphate-buffered saline (PBS)]. Bone marrow and spleen were harvested after 1 hour and then kept on ice. Cells were stained with antibodies against cell surface markers (Ter119 and CD71), fixed in 100% methanol (≥99.8%; Aldrich-Sigma), and permeabilized in tris-buffered saline (TBS) with 0.2% Tween 20 (Sigma-Aldrich). The azide-alkyne cyclo addition was performed using the Click-iT Plus OP-Puro Alexa Fluor 488 Protein Synthesis Assay Kit (Thermo Fisher Scientific). Cells were washed twice and then analyzed by flow cytometry.
Erythroid differentiation and drug treatment of human CD34+ cells
Human CD34+ cells were obtained under human subject research protocols that were approved by local ethical committees: St. Jude Children’s Research Hospital protocol “Bone marrow for hemoglobinopathy research” (NCT00669305) and Foundation IRCCS Ca’ Granda Ospedale Maggiore Policlinico of Milan “In vitro study to evaluate the role of ULK1 in the clearance of free α-globin in β-thalassemia” (312_2018bis). The CD34+ cells were isolated from the bone marrow of individuals with TD β-thalassemia and from the peripheral blood of those with NTD β-thalassemia or healthy donors (table S7). The CD34+ cells were cultured according to a three-phase erythroid differentiation protocol and monitored by flow cytometry with an LSRFortessa cell analyzer (BD Biosciences), using the following antibodies: anti-CD235a (clone GA-R2, BD Biosciences), anti-Band3 (from X. An at the New York Blood Center), and anti–α4-integrin (clone MZ18-24A9; Miltenyi). At the beginning of phase 3 (day 13), cells were treated with rapamycin (LC Laboratories) (10 or 20 μM) or MG132 (Enzo Life Sciences) (1 or 2.5 μM) for 48 hours.
Statistics
Statistical analyses were performed using GraphPad Prism 6.0 software and R-project (R-3.2.5). Data are shown as means ± SD. A two-sample Student’s t test or an exact Mann-Whitney-Wilcoxon test was used to compare continuous variables in two groups, depending on the normality of the data. The Benjamini and Hochberg method was used to control the false discovery rate at a level of 0.05 in the analyses of RBC survival at different time points. Otherwise, P values of less than 0.05 were considered statistically significant. All tests were two tailed.
Supplementary Material
Acknowledgments:
We are grateful to S. Frase, C. Robinson, L. Horner, and R. Gursky (St. Jude Cellular Imaging Shared Research Core); S. Savage, M. Anderson, P. Varner, B. Gallini, and M. Straign (St. Jude Veterinary and Pathology Core); D. Ashmun and all the staff (Flow Cytometry and Cell Sorting Shared Resource); C. Savage, M. Inoue, and other staff at the St. Jude Animal Resource Center; T. Pestina (St. Jude Department of Hematology); G. Kang (St. Jude Department of Biostatistics); and V. Brancaleoni (University of Milan, Genotyping Resource Center). We thank U. Klingmüller (German Cancer Research Center, Heidelberg, Germany) and N. Mizushima (Tokyo Medical and Dental University, Tokyo, Japan) for sharing EpoR-Cre and Atg5flox mouse models, respectively. We thank X. An for sharing anti-Band3 antibody (New York Blood Center). We thank D. Green for helpful discussions, A. Impagliazzo (impag1@lumos.net) for graphic design, and K. A. Laycock (St. Jude Department of Scientific Editing) for scientific editing of the manuscript.
Funding: This work was supported by grant R01 DK61692 (to M.J.W.) from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), grant R01 HL114697 (to M.K.) from the National Heart, Lung, and Blood Institute (NHLBI), EMBO Long-Term Fellowship ALTF 1526-2016 (to E.B.R.), and the American Lebanese Syrian Associated Charities (ALSAC).
Footnotes
Competing interests: An international patent application on the work described here was published on 2 May 2019: PCT/US2018/057709; “Method for treating thalassemia”; authors C.L., M.K., and M.J.W.; St. Jude Children’s Research Hospital. M.J.W. is a consultant for GlaxoSmithKline, Rubius Inc., Cellarity Inc., Beam Therapeutics, and Esperion; none of the consulting work is relevant to the current project. M.D.C. is a consultant for Celgene, Sanofi Genzyme, Novartis, CRISP, VIFOR, and IONIS; none of the consulting work is relevant to the current project. All other authors declare that they have no competing interests.
Data and materials availability: All data are available in the main text or the Supplementary Materials.
stm.sciencemag.org/cgi/content/full/11/506/eaav4881/DC1
Fig. S1. Ablation of Ulk1 in hematopoietic cells.
Fig. S2. Phenotyping of ablation of Ulk1 in hematopoietic cells.
Fig. S3. Developmental staging of erythroblasts in spleen and bone marrow of HbbTh3/+Ulk1+/+ and HbbTh3/+Ulk1−/− mice.
Fig. S4. Characteristics of autophagy in β-thalassemic RBC precursors.
Fig. S5. Ablation of Atg5 in erythroid cells.
Fig. S6. Elimination of insoluble α-globin by the proteasome and autophagy in β-thalassemic reticulocytes.
Fig. S7. Effects of systemic rapamycin on β-thalassemic erythropoiesis.
Fig. S8. Expression of Map1lc3bm (LC3) and Sqstm1 (p62) mRNAs in reticulocytes of HbbTh3/+Ulk1+/+ mice treated with rapamycin.
Fig. S9. Presence of free α-globin in CD34+ cell-derived erythroblasts from individuals with β-thalassemia.
Fig. S10. Rapamycin and MG132 treatment of CD34+ cells from normal donors or from individuals with β-thalassemia.
Fig. S11. Model for ULK1 kinase–dependent clearance of free α-globin in β-thalassemia.
Table S1. Effects of Ulk1 gene disruption on erythroid indices of β-thalassemic mice.
Table S2. Effects of Ulk1 loss on erythroid hyperplasia in β-thalassemic mice.
Table S3. Effects of Atg5 gene disruption on erythroid indices of β-thalassemic mice.
Table S4. Effects of systemic rapamycin on erythroid indices of β-thalassemic mice ± Ulk1 gene disruption.
Table S5. ULK1-dependent reduction of erythroid hyperplasia in β-thalassemic mice treated with rapamycin.
Table S6. Erythroid indices of individuals with β-thalassemia who provided CD34+ cells for this study.
Table S7. Genotypes of individuals with β-thalassemia who provided CD34+ cells for this study.
SUPPLEMENTARY MATERIALS http://stm.sciencemag.org/content/suppl/2019/08/19/11.506.eaav4881.DC1
REFERENCES AND NOTES
- 1.Higgs DR, Engel JD, Stamatoyannopoulos G, Thalassaemia. Lancet 379, 373–383 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Steinberg MH, Forget BG, Higgs DR, Weatherall DJ, Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management (Cambridge Univ. Press, ed. 2, 2009). [Google Scholar]
- 3.Shinar E, Shalev O, Rachmilewitz EA, Schrier SL, Erythrocyte membrane skeleton abnormalities in severe beta-thalassemia. Blood 70, 158–164 (1987). [PubMed] [Google Scholar]
- 4.Advani R, Rubin E, Mohandas N, Schrier SL, Oxidative red blood cell membrane injury in the pathophysiology of severe mouse beta-thalassemia. Blood 79, 1064–1067 (1992). [PubMed] [Google Scholar]
- 5.Rachmilewitz E, Schrier S, in Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management, Steinberg MH, Forget BG, Higgs R, Nagel RL, Eds. (Cambridge Univ. Press, ed. 1, 2001). [Google Scholar]
- 6.Orkin SH, Nathan DG, Ginsburg D, Fisher DE, Look AT, Lux SE, Nathan and Oski’s Hematology of Infancy and Childhood (Saunders, ed. 7, 2008). [Google Scholar]
- 7.Khandros E, Thom CS, D’Souza J, Weiss MJ, Integrated protein quality-control pathways regulate free α-globin in murine β-thalassemia. Blood 119, 5265–5275 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Premawardhena A, Fisher CA, Olivieri NF, de Silva S, Sloane-Stanley J, Wood WG, Weatherall DJ, A novel molecular basis for thalassemia intermedia poses new questions about its pathophysiology. Blood 106, 3251–3255 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Khandros E, Weiss MJ, Protein quality control during erythropoiesis and haemoglobin synthesis. Hematol. Oncol. Clin. North Am 24, 1071–1088 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Higgs DR, The thalassaemia syndromes. Q. J. Med 86, 559–564 (1993). [PubMed] [Google Scholar]
- 11.Birgens H, Ljung R, The thalassaemia syndromes. Scand. J. Clin. Lab. Invest 67, 11–25 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Kong Y, Zhou S, Kihm AJ, Katein AM, Yu X, Gell DA, Mackay JP, Adachi K, Foster-Brown L, Louden CS, Gow AJ, Weiss MJ, Loss of α-hemoglobin–stabilizing protein impairs erythropoiesis and exacerbates β-thalassemia. J. Clin. Invest 114, 1457–1466 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kihm AJ, Kong Y, Hong W, Russell JE, Rouda S, Adachi K, Simon MC, Blobel GA, Weiss MJ, An abundant erythroid protein that stabilizes free α-haemoglobin. Nature 417, 758–763 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Aigelsreiter A, Janig E, Stumptner C, Fuchsbichler A, Zatloukal K, Denk H, How a cell deals with abnormal proteins. Pathogenetic mechanisms in protein aggregation diseases. Pathobiology 74, 145–158 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Ciechanover A, Brundin P, The ubiquitin proteasome system in neurodegenerative diseases: Sometimes the chicken, sometimes the egg. Neuron 40, 427–446 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Cox D, Raeburn C, Sui X, Hatters DM, Protein aggregation in cell biology: An aggregomics perspective of health and disease. Semin. Cell Dev. Biol (2018). [DOI] [PubMed] [Google Scholar]
- 17.Lithanatudom P, Wannatung T, Leecharoenkiat A, Svasti S, Fucharoen S, Smith DR, Enhanced activation of autophagy in β-thalassemia/Hb E erythroblasts during erythropoiesis. Ann. Hematol 90, 747–758 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Mizushima N, Yoshimori T, Ohsumi Y, The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol 27, 107–132 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Ciechanover A, Kwon YT, Degradation of misfolded proteins in neurodegenerative diseases: Therapeutic targets and strategies. Exp. Mol. Med 47, e147 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, Choi AM, Chu CT, Codogno P, Colombo MI, Cuervo AM, Debnath J, Deretic V, Dikic I, Eskelinen EL, Fimia GM, Fulda S, Gewirtz DA, Green DR, Hansen M, Harper JW, Jäättelä M, Johansen T, Juhasz G, Kimmelman AC, Kraft C, Ktistakis NT, Kumar S, Levine B, Lopez-Otin C, Madeo F, Martens S, Martinez J, Melendez A, Mizushima N, Münz C, Murphy LO, Penninger JM, Piacentini M, Reggiori F, Rubinsztein DC, Ryan KM, Santambrogio L, Scorrano L, Simon AK, Simon HU, Simonsen A, Tavernarakis N, Tooze SA, Yoshimori T, Yuan J, Yue Z, Zhong Q, Kroemer G, Molecular definitions of autophagy and related processes. EMBO J. 36, 1811–1836 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, Metzger D, Reggiani C, Schiaffino S, Sandri M, Autophagy is required to maintain muscle mass. Cell Metab. 10, 507–515 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N, Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K, The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med 13, 619–624 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Komatsu M, Ichimura Y, Selective autophagy regulates various cellular functions. Genes Cells 15, 923–933 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Mizushima N, A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol 20, 521–527 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Codogno P, Mehrpour M, Proikas-Cezanne T, Canonical and non-canonical autophagy: Variations on a common theme of self-eating? Nat. Rev. Mol. Cell Biol 13, 7–12 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Travassos LH, Vasconcellos LR, Bozza MT, Carneiro LA, Heme and iron induce protein aggregation. Autophagy 13, 625–626 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang B, Kundu M, Canonical and noncanonical functions of ULK/Atg1. Curr. Opin. Cell Biol 45, 47–54 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Noda NN, Fujioka Y, Atg1 family kinases in autophagy initiation. Cell. Mol. Life Sci 72, 3083–3096 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kundu M, Lindsten T, Yang C-Y, Wu J, Zhao F, Zhang J, Selak MA, Ney PA, Thompson CB, Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 112, 1493–1502 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Honda S, Arakawa S, Nishida Y, Yamaguchi H, Ishii E, Shimizu S, Ulk1-mediated Atg5-independent macroautophagy mediates elimination of mitochondria from embryonic reticulocytes. Nat. Commun 5, 4004 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Kundu M, ULK1, mammalian target of rapamycin, and mitochondria: Linking nutrient availability and autophagy. Antioxid. Redox Signal 14, 1953–1958 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang B, Kirby S, Lewis J, Detloff PJ, Maeda N, Smithies O, A mouse model for β0-thalassemia. Proc. Natl. Acad. Sci. U.S.A 92, 11608–11612 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li TY, Sun Y, Liang Y, Liu Q, Shi Y, Zhang CS, Zhang C, Song L, Zhang P, Zhang X, Li X, Chen T, Huang HY, He X, Wang Y, Wu YQ, Chen S, Jiang M, Chen C, Xie C, Yang JY, Lin Y, Zhao S, Ye Z, Lin SY, Chiu DT, Lin SC, ULK1/2 constitute a bifurcate node controlling glucose metabolic fluxes in addition to autophagy. Mol. Cell 62, 359–370 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Jung CH, Seo M, Otto NM, Kim D-H, ULK1 inhibits the kinase activity of mTORC1 and cell proliferation. Autophagy 7, 1212–1221 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S.-i, Natsume T, Takehana K, Yamada N, Guan J-L, Oshiro N, Mizushima N, Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 20, 1981–1991 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ho PJ, Wickramasinghe SN, Rees DC, Lee MJ, Eden A, Thein SL, Erythroblastic inclusions in dominantly inherited β thalassemias. Blood 89, 322–328 (1997). [PubMed] [Google Scholar]
- 38.Wickramasinghe SN, Hughes M, Ultrastructural studies of erythropoiesis in β-thalassaemia trait. Br. J. Haematol 46, 401–407 (1980). [DOI] [PubMed] [Google Scholar]
- 39.Liu WJ, Ye L, Huang WF, Guo LJ, Xu ZG, Wu HL, Yang C, Liu HF, p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett 21, 29 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mizushima N, Yoshimori T, How to interpret LC3 immunoblotting. Autophagy 3, 542–545 (2007). [DOI] [PubMed] [Google Scholar]
- 41.Laplante M, Sabatini DM, mTOR signaling in growth control and disease. Cell 149, 274–293 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X, ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem 284, 12297–12305 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim J, Kundu M, Viollet B, Guan K-L, AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol 13, 132–141 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ribeil J-A, Arlet J-B, Dussiot M, Moura IC, Courtois G, Hermine O, Ineffective erythropoiesis in β-thalassemia. Sci. World J. 2013, 394295 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fibach E, Bianchi N, Borgatti M, Zuccato C, Finotti A, Lampronti I, Prus E, Mischiati C, Gambari R, Effects of rapamycin on accumulation of α-, β- and γ-globin mRNAs in erythroid precursor cells from β-thalassaemia patients. Eur. J. Haematol 77, 437–441 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Pecoraro A, Troia A, Calzolari R, Scazzone C, Rigano P, Martorana A, Sacco M, Maggio A, Di Marzo R, Efficacy of rapamycin as inducer of Hb F in primary erythroid cultures from sickle cell disease and β-thalassemia patients. Hemoglobin 39, 225–229 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Mettananda S, Gibbons RJ, Higgs DR, α-Globin as a molecular target in the treatment of β-thalassemia. Blood 125, 3694–3701 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Olivieri NF, The β-thalassemias. N. Engl. J. Med 341, 99–109 (1999). [DOI] [PubMed] [Google Scholar]
- 49.Yanagitani K, Juszkiewicz S, Hegde RS, UBE2O is a quality control factor for orphans of multiprotein complexes. Science 357, 472–475 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nguyen AT, Prado MA, Schmidt PJ, Sendamarai AK, Wilson-Grady JT, Min M, Campagna DR, Tian G, Shi Y, Dederer V, Kawan M, Kuehnle N, Paulo JA, Yao Y, Weiss MJ, Justice MJ, Gygi SP, Fleming MD, Finley D, UBE2O remodels the proteome during terminal erythroid differentiation. Science 357, eaan0218 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lilienbaum A, Relationship between the proteasomal system and autophagy. Int. J. Biochem. Mol. Biol 4, 1–26 (2013). [PMC free article] [PubMed] [Google Scholar]
- 52.Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC, Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol. Cell 33, 517–527 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Geng J, Klionsky DJ, The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy.’Protein modifications: Beyond the usual suspects’ review series. EMBO Rep. 9, 859–864 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maskey D, Yousefi S, Schmid I, Zlobec I, Perren A, Friis R, Simon H-U, ATG5 is induced by DNA-damaging agents and promotes mitotic catastrophe independent of autophagy. Nat. Commun 4, 2130 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goodwin JM, Dowdle WE, DeJesus R, Wang Z, Bergman P, Kobylarz M, Lindeman A, Xavier RJ, McAllister G, Nyfeler B, Hoffman G, Murphy LO, Autophagy-independent lysosomal targeting regulated by ULK1/2-FIP200 and ATG9. Cell Rep. 20, 2341–2356 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S, Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 461, 654–658 (2009). [DOI] [PubMed] [Google Scholar]
- 57.Ma T, Li J, Xu Y, Yu C, Xu T, Wang H, Liu K, Cao N, Nie B-M, Zhu S-Y, Xu S, Li K, Wei W-G, Wu Y, Guan K-L, Ding S, Atg5-independent autophagy regulates mitochondrial clearance and is essential for iPSC reprogramming. Nat. Cell Biol 17, 1379–1387 (2015). [DOI] [PubMed] [Google Scholar]
- 58.Saito T, Nah J, Oka SI, Mukai R, Monden Y, Maejima Y, Ikeda Y, Sciarretta S, Liu T, Li H, Baljinnyam E, Fraidenraich D, Fritzky L, Zhai P, Ichinose S, Isobe M, Hsu C-P, Kundu M, Sadoshima J, An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. J. Clin. Invest 129, 802–819 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu Q, Chen L, Atkinson JM, Claxton DF, Wang HG, Atg5-dependent autophagy contributes to the development of acute myeloid leukemia in an MLL-AF9-driven mouse model. Cell Death Dis. 7, e2361 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kuma A, Matsui M, Mizushima N, LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: Caution in the interpretation of LC3 localization. Autophagy 3, 323–328 (2007). [DOI] [PubMed] [Google Scholar]
- 61.Bayeva M, Khechaduri A, Puig S, Chang HC, Patial S, Blackshear PJ, Ardehali H, mTOR regulates cellular iron homeostasis through tristetraprolin. Cell Metab. 16, 645–657 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Klei TR, Meinderts SM, van den Berg TK, van Bruggen R, From the cradle to the grave: The role of macrophages in erythropoiesis and erythrophagocytosis. Front. Immunol 8, 73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jahrling JB, Laberge RM, Age-related neurodegeneration prevention through mTOR inhibition: Potential mechanisms and remaining questions. Curr. Top. Med. Chem 15, 2139–2151 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bové J, Martínez-Vicente M, Vila M, Fighting neurodegeneration with rapamycin: Mechanistic insights. Nat. Rev. Neurosci. 12, 437–452 (2011). [DOI] [PubMed] [Google Scholar]
- 65.Li J, Kim SG, Blenis J, Rapamycin: One drug, many effects. Cell Metab. 19, 373–379 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kondratov RV, Kondratova AA, Rapamycin in preventive (very low) doses. Aging 6, 158–159 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mannick JB, Morris M, Hockey H-UP, Roma G, Beibel M, Kulmatycki K, Watkins M, Shavlakadze T, Zhou W, Quinn D, Glass DJ, Klickstein LB, TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci. Transl. Med 10, eaaq1564 (2018). [DOI] [PubMed] [Google Scholar]
- 68.Zhang L, Fu L, Zhang S, Zhang J, Zhao Y, Zheng Y, He G, Yang S, Ouyang L, Liu B, Discovery of a small molecule targeting ULK1-modulated cell death of triple negative breast cancer in vitro and in vivo. Chem. Sci 8, 2687–2701 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li J, Zhong L, Wang F, Zhu H, Dissecting the role of AMP-activated protein kinase in human diseases. Acta Pharm. Sin. B 7, 249–259 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Heinrich AC, Pelanda R, Klingmüller U, A mouse model for visualization and conditional mutations in the erythroid lineage. Blood 104, 659–666 (2004). [DOI] [PubMed] [Google Scholar]
- 71.Gudmundsson KO, Stull SW, Keller JR, Transplantation of mouse fetal liver cells for analyzing the function of hematopoietic stem and progenitor cells. Methods Mol. Biol 879, 123–133 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Alter BP, Gel electrophoretic separation of globin chains. Prog. Clin. Biol. Res 60, 157–175 (1981). [PubMed] [Google Scholar]
- 73.Beauchemin H, Blouin MJ, Trudel M, Differential regulatory and compensatory responses in hematopoiesis/erythropoiesis in α- and β-globin hemizygous mice. J. Biol. Chem 279, 19471–19480 (2004). [DOI] [PubMed] [Google Scholar]
- 74.Liu Y, Pop R, Sadegh C, Brugnara C, Haase VH, Socolovsky M, Suppression of Fas-FasL coexpression by erythropoietin mediates erythroblast expansion during the erythropoietic stress response in vivo. Blood 108, 123–133 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A, Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Arganda-Carreras I, Kaynig V, Rueden C, Eliceiri KW, Schindelin J, Cardona A, Sebastian Seung H, Trainable Weka Segmentation: A machine learning tool for microscopy pixel classification. Bioinformatics 33, 2424–2426 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.