

Abstract
Fibroblast growth factor receptors (FGFRs) comprise a subfamily of receptor tyrosine kinases (RTKs) that are master regulators of a broad spectrum of cellular and developmental processes, including apoptosis, proliferation, migration and angiogenesis. Due to their broad impact, FGFRs and other RTKs are highly regulated and normally only basally active. Deregulation of FGFR signaling by activating mutations or ligand/receptor overexpression could allow these receptors to become constitutively active, leading to cancer development, including both hematopoietic and solid tumors, such as breast, bladder and prostate carcinomas. In this review, we focus on potential modes of FGFR-mediated tumorigenesis, in particular, the role of FGFR1 during prostate cancer progression.
Keywords: prostate cancer, fibroblast growth factor receptor (FGFR), chemical inducer of dimerization (CID), epithelial-mesenchymal transition (EMT), angiogenesis, androgen independence
Role of FGFR1 in Neoplastic Prostate Growth
Aberrant expression of multiple FGF family members and their cognate receptors are found in multiple cancers, including PCa, providing a strong indication of their role in neoplastic progression.1-7 While FGFR2 signaling is key in regulating both prostate morphogenesis and homeostasis,8-10 FGFR1 signaling has been more tightly correlated with PCa progression, evidenced by elevated expression of FGFR1 and some of its ligands in both human PCa11,12 and mouse prostate tumor models, such as SV40 T/t antigen (T/tag)-based TRAMP.13-15 There have been a number of studies to date of the expression of FGFR1 in human PCa.12,16-19 Both our studies12 and those of Hamaguchi et al.20 demonstrate that in benign prostate glands, staining is seen primarily in cells in a basal location within the gland (encompassing basal cells, transit amplifying cells and prostatic progenitor cells) although the exact cell type expressing FGFR1 is unclear. All studies to date using immunohistochemistry (IHC) have shown increased expression of FGFR1 in PCa.12,16-19 Our group has confirmed this IHC finding by western blotting (WB).12 More recently Sahadevan et al. have shown that FGFR1 transcripts are significantly upregulated in PCa relative to normal prostate epithelium by quantitative RT-PCR of laser captured benign and cancer epithelium.19 Thus, it is unequivocal that FGFR1 is increased in PCa. The linkage of FGFR1 to outcome is less clear. Our group has shown that FGFR1 expression is correlated with poor differentiation in cancer from radical prostatectomy (RP) specimens,12 and other groups have shown association with extracapsular spread18 and high pre-operative PSA17 in RP tissues. The study of Sahadevan et al.19 did not find a correlation with outcome but their study used both RP and TURP (advanced cancer) tissues and is derived from a population that is not PSA-screened (as is the rule in the U.S.). In all cases, the cohorts studied were small, limiting the power of the analysis. In addition, the cohort of Gravdal et al.17 had a very high percentage of aggressive cancers (33% with seminal vesicle invasion, which is much higher than is standard in the U.S.), again limiting the applicability of their findings to U.S. cohorts. Thus, for RP tissues, the current data suggests, but does not categorically prove, that FGFR1 expression is associated with aggressive disease.
Correspondingly, FGFR1 binding partners, FGF1 and FGF2, FGF8b as well as FGF17, are also upregulated.5,6,21,22 In addition, transgenic mice expressing constitutively active FGFR1 (caFGFR1) in prostate epithelium develop progressive hyperplasia and prostatic intra-epithelial neoplasia (PIN).23 The activation of FGFR1 signaling in the prostates of JOCK1 (Juxtaposition of CID and Kinase-1) mice that overexpress a genetically modified CID (Chemical Inducer of Dimerization)-activated FGFR1 in prostate epithelium, induces epithelial hyperplasia (after one month of CID treatment), high-grade PIN (after six months) and prostate adenocarcinoma with coincident focal epithelial to mesenchymal transition (EMT) (>30 weeks).24-26 Together, these studies strongly suggest a tumor-promoting role of FGFR1 signaling in PCa.
FGFR Structure and Signaling
In total, the FGF signaling axis includes about 18 different functionally defined ligands (excluding FGF homologous factors FHF1/FGF11-FHF4/FGF14 that cannot bind FGFRs27) along with four membrane-localized FGFRs, many of which have multiple isoforms that are made up mostly of alternative splicing in the third extracellular immunoglobulin-like domains.28,29 Changes in these extracellular domains directly influence ligand binding through different affinities of both these ligands and various associated, negatively charged heparin sulfate moieties.27,30,31 In addition to extracellular (“ecto”) domains, FGFRs are also comprised of transmembrane and intracellular “split-kinase” domains, FRS2-binding juxta-membrane domains, and PLC©-1-binding C-terminal “tails”.27 Ligand-induced dimerization of these molecules leads to successful activation through FGFR sequential trans-phos- phorylation of several tyrosine residues within their intracellular domains. This increases the FGFR kinase activities and creates docking sites for downstream signaling molecules, such as PLC©-1 and Crk.32-34 PLC© molecules contain a Src-homology domain 2 (SH2) that, similar to the phospho-tyrosine binding (PTB) domain found within adaptor Shc2 (and other related signaling molecules), bind to phosphorylated tyrosine residues.33 Similarly, the adapter Crk may bind directly to FGFR1 (at Y463), via its SH2 domain, and this may help propagate mitogenic signals.34
Unlike other RTKs, FGF (as well as NGF) receptors depend on the adaptor molecule FRS2/SNT1 to execute their mitogenic effects, representing an additional signaling molecule that is essential for the mitogenicity of FGFR signaling.35 The myristoylated protein FRS2 (FRS2〈 and FRS2®/SNT2/FRS3) localizes to the plasma membrane where it associates with the juxtamembrane domain of FGFRs via a PTB domain, leading to FRS2 tyrosine phosphorylation at multiple sites by active FGFRs.36 In turn, tyrosine phosphorylation of FRS2 recruits MAPK adapters, like Grb2, and the p85 subunit of PI3K, linking FGFR activation with the MAPK and PI3K/Akt pathways, respectively. Although direct binding between FGFR1 and the non-receptor tyrosine kinase Src has been more difficult to demonstrate, recent evidence has shown that FRS2 also bridges this interaction.37,38 FGFR activation of Src molecules may link FGF signaling to cortactin and the cytoskeleton, contributing to cell motility (Fig. 1).39 FRS2 can also form an FGFR inhibitory complex through the recruitment of the E3 ubiquitin ligase, Cbl, leading to the ubiquitination and proteosomal degradation of FGFRs.40 Loss of FRS2 leads to defects in prostate branching morphogenesis during development and also a reduction in tumor formation, thus linking FRS2 to FGFR1-mediated cancer progression.41
Figure 1.
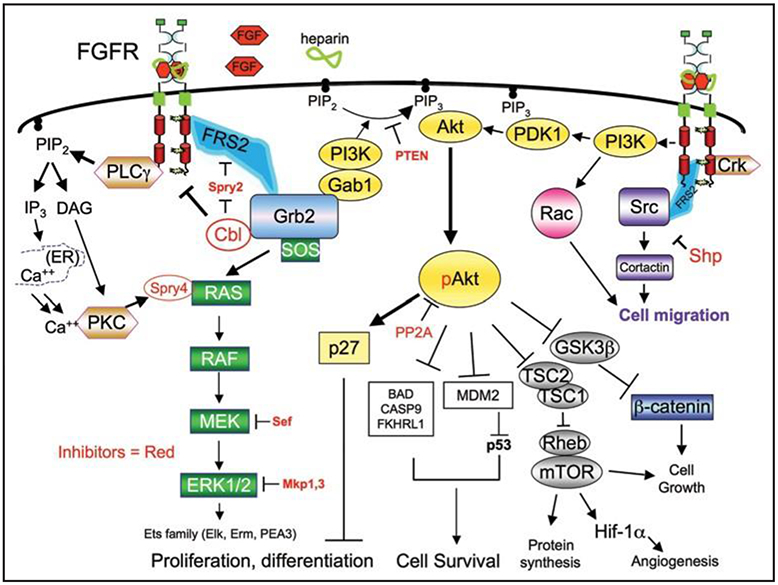
FGFR signaling pathways. FGFRs regulate distinct biological process via several downstream signaling pathways. Activation of these signaling cascades is subject to the recruitment and activation of specific adaptor or downstream signaling molecules. A number of inhibitory molecules (red) play a role at distinct steps of FGFR downstream signaling, and serve to keep signaling activity under control.
Several other negative feedback mechanisms exist to control the dangers of FGFR over-activation. Signaling by FGFRs and other RTKs (e.g., EGF) can induce the activity or transcription of MAPK feedback inhibitors, such as Sprouty,42 Sprouty-related proteins, Spred-1, 2 (43), Mkp/Dusp family members,44 as well as “similar expression to FGF” (Sef ).45 For example, Sprouty2 becomes phosphorylated at Y55 by Src family kinases, creating a binding site for adapter Grb2 and blocking its interaction with FRS2 and MAPK signaling, while Sprouty4 association with Ras blocks PKC©-mediated ERK activation (Fig. 1).46,47 Together, these inhibitory molecules play an important role in mitigating FGFR activity and hence, these molecules may also serve as tumor suppressors. Consistent with this concept, loss of expression of Sprouty and Sef proteins have been demonstrated in prostate cancer by several groups.45,48-51
Disruption of Epithelial-Stroma Communication
The mitogenic role of FGFR1 can contribute to prostate tumorigenesis by disrupting normal prostate epithelial/stromal communication that ensures that the epithelium never outgrows its dependence on a supportive underlying stroma. In fact, normal prostate development and maintenance can be regulated by stromal cells producing FGF7 and FGF10, which act in a paracrine fashion with their cognate receptor, FGFR2IIIb, on neighboring epithelial cells.52 The importance of this paracrine network is highlighted by experiments showing that the loss of FGFR2IIIb expression by prostate epithelial cells results in defects of prostate ductal morphogenesis (similar to loss of FRS2a) and its responsiveness to androgens.53 In addition, prostate epithelial cells can produce factors that stimulate stromal growth, including angiogenesis. Although such studies are challenging in physiological models, cellular studies based on the Dunning rat tumor model, demonstrate that paracrine-acting FGF9 from transformed prostate epithelial cells can stimulate stromal-derived cells, which express high levels of its cognate receptor FGFR3.54 This type of paracrine epithelial-stroma communication is believed to be altered by alternative splicing of FGFR2. During PCa progression, epithelial FGFR2 can undergo alternative splicing from FGFR2IIIb to FGFR2IIIc, a more ligand-promiscuous conformation.55 Moreover, ectopic expression of normally stroma-localized FGFR1IIIc by epithelial cells, along with the FGFR2 isoform change and/or downregulation can destroy the communication and homeostasis between the stromal and epithelial compartments.56-58 These changes of the FGF axis within the prostate and tumor microenvironment likely lead to widespread autocrine signaling and stromal-independence, ultimately leading to uncontrolled epithelial cell proliferation.6 In particular, the FGFR1 and FGFR2IIIc ligand, FGF2, is highly upregulated in advanced and metastatic PCa, suggesting that the establishment of an autocrine signal promotes a more aggressive phenotype.5,59 Another recent study based on mesenchymal overexpression of FGF10 showed that paracrine FGF10 can induce neoplasia in an apparently basal epithelial-targeted, FGFR1-dependent manner.60 Thus, expression changes in FGFs and FGFRs can render epithelial cells independent of normally tight stromal-mediated regulation disrupting prostate homeostasis.
One of the challenges of most in vivo signaling studies is the lack of strict spatio-temporal control of target pathways, complicating distinguishing causal relationships from epiphenomena. Using a unique, synthetic ligand-inducible model (mentioned above) called JOCK1 (Juxtaposition of CID and Kinase), we examined the direct effects of ectopic FGFR1 signaling in the mouse prostate. JOCK1 mice express a prostate-specific, inducible chimeric version of FGFR1 (iFGFR1), comprised of a membrane-bound myristoylated cytoplasmic signaling domain of FGFR1 and two tandem dimerizer drug-binding domains.26 Activation of iFGFR1 can be controlled by the use of a synthetic chemical inducer of dimerization (CID). To study the direct effects of FGFR1 signaling in vivo, JOCK1 mice were treated with CID for 0 (untreated), 6 and 24 hours and compared to 0 hours (untreated) wild type mice to obtain an iFGFR1 gene expression signature. The iFGFR1 expression profile was initially analyzed by hierarchical gene clustering as previously described.25 This type of analysis showed minimal differences between wild type and JOCK1 at 0 hours, indicating the low basal signaling by untreated JOCK1 mice. In contrast, a total of 6 different clusters were identified after 6 and 24 hours of iFGFR1 activity. One cluster represented genes that are expressed after 6 hours but regress to basal levels within 24 hours, while another represented genes being expressed at 6 hours and peaking after 24 hours (Fig. 2).
Figure 2.
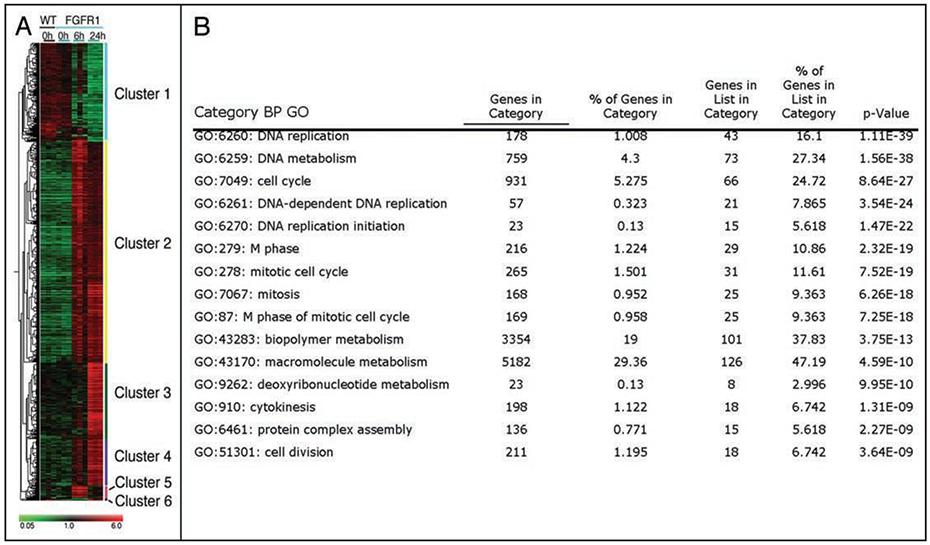
iFGFR1 gene signature in prostate-epithelia. In vivo activation of iFGFR1 led to the identification of immediate FGFR1-target genes within prostate epithelium, which was grouped into 6 clusters (A). Gene ontology classification of genes present in clusters 3 and 4 showed an enrichment for genes involved in cellular proliferation (B).
To help determine if a particular type(s) of cellular change(s) is associated with these clusters of genes, gene annotation by gene ontology (GO) was performed. GO results for genes present in clusters 3 and 4 showed an increase in genes involved in the process of DNA replication and mitosis (Fig. 2). Correspondingly, chronic biweekly CID stimulation led to highly synchronized progression from hyperplasia to adenocarcinoma within 2 and 30 weeks of CID stimulation, respectively. Together, these results suggest that FGFR1 can drive many distinct processes, while its first effect within prostate cells is growth and proliferation.
Tumor Angiogenesis
The formation of new blood vessels or neovascularization is critical for normal tissue development and homeostasis, and has been described during embryonic development, inflammation and wound healing. Similarly, in order for tumors to evolve, they must escape hypoxic and low nutrient conditions associated with an increase in cellularity. Therefore, only the subset of tumors that can induce new blood vessels from established vasculature can progress. This angiogenic process can increase oxygen availability and nutrients, significantly promoting tumor growth. Expansion and migration of endothelial cells, the foundation of blood vessels, is necessary for angiogenesis to occur. The tyrosine kinase receptors, FGFR1, vascular endothelial grow factor receptors (VEGFR) and Tie2, are the main receptors that promote angiogenesis, with Tie2 and both VEGFR1 and 2 being selectively expressed by endothelial cells.61 In fact, FGFs were the first identified angiogenic factors and their role in endothelial cell proliferation, migration, cell adhesion and other angiogenic promoting process has been extensively studied.61,62 Although FGF1 and FGF2 were among the first molecules found that could promote angiogenesis, the six members of the VEGF ligand family and the three VEGF receptors are now known to be the main factors that drive embryonic vascularization, angiogenesis and lymphangiogenesis.63 However, there is ample evidence that both FGFs and VEGF cooperate to promote angiogenesis. For example, FGF2 is known to induce the expression of VEGF in vascular endothelial cells, while blocking antibodies against VEGF reduce the expression of endogenous FGF2, suggestive of a positive feedback mechanism.64,65 Moreover, inhibition of FGFR1 and FGFR2 activity can reduce tumor vascularization as well as VEGF expression.66 Thus, promotion of angiogenesis by FGFs may be dependent on crosstalk between FGF-VEGF signaling axes.
More specifically, we asked how activation of FGFR1 in prostate epithelial cells could promote tumor vascularization? In JOCK1 studies focused on the in vivo effect of FGFR1 signaling, two weeks of iFGFR1 induction was sufficient to elicit an increase in the pro-angiogenic factor Hif-1〈 and the concomitant upregulation of VEGF and VEGFR-1 and -2.24 Thus, even in normal prostate epithelial cells, activation of FGFR1 promotes the production and secretion of VEGF that acts as a paracrine factor on endothelial cell to promote angiogenesis. Moreover, termination of iFGFR1 signaling by CID withdrawal stops further neovascularization. Similar results were seen when mice deficient for FGF2 expression were crossed to an autochthonous model of prostate cancer (TRAMP). In this study, FGF2−/−/TRAMP mice had highly reduced PCa progression, metastasis, as well as a (likely) compensatory increase in the angiogenic factors, FGF-binding protein (FGF-BP), and VEGF expression when compared to normal TRAMP mice.67,68 However, the FGF-mediated angiogenic effect is not strictly dependent on the VEGF axis, as FGF2 overexpression can further increase vascularization even in the presence of high VEGF levels.69,70 Furthermore, the expression of a secreted, dominant negative version of FGFR1 by distinct cancer cell lines affected a reduction in angiogenesis, even in cells that failed to respond to anti-VEGF treatment, highlighting the importance of FGFR1 in tumor vascularization.71 Together, these results suggest that FGFs have a role in prostate cancer progression and tumor vascularization, and that despite working synergistically with VEGF, FGFs could also promote tumor angiogenesis independent of VEGF.
In addition to controlling VEGF expression, FGFR1 signaling upregulates Angiopoietin 2 (Ang2) expression, while reducing the levels of Ang1,24 leading to complex microenvironment changes. Angiopoietins are known angiogenic factors that interact with their cognate RTK, Tie2, on endothelial cells.72 Despite the high homology between Ang1 and Ang2, the former is known to stabilize blood vessels and is essential for normal embryonic vascular development, while Ang2 acts as an Ang1 antagonist, disrupting mouse embryonic blood vessels.72,73 However, in an adult organisms, Ang2 and VEGF are found at sites of neovascular development, suggesting that destabilization of endothelial cells by Ang2 facilitates or sensitizes endothelial cells to VEGF-mediated angiogenesis.74 Other studies suggest that Ang1 and Ang2 play opposing roles in inflammation-induced angiogenesis, whereas Ang1 blocks angiogenesis by interacting with Tie2 and stabilizing blood vessels.75 In contrast, Ang2 destabilization can also sensitize cells to TNF〈, which increases the expression of cell adhesion molecules and subsequent recruitment of pro-inflammatory cells, like macrophages and neutrophils.76,77 Recruitment of these leukocytes can create a proinflammatory environment, further contributing to both angiogenesis and tumorigenesis. Moreover, constant activation of the MAPK/Ras pathway by tumor cells can also promote the recruitment of proinflammatory cells through increased expression and secretion of the chemokine IL-8, capable of supporting both angiogenesis and tumor growth.78,79
Although some leukocytes, like cytotoxic T lymphocytes (CTLs) can target and eliminate tumor cells, others, such as a subset of tumor associated macrophages, called Type II TAMs (or M2), can promote tumor development.80 Type II TAMs can not only contribute to angiogenesis by releasing VEGF, TNF〈, IL-8 and FGF-2/[basic (b)FGF], but can also secrete a series of proteases, such as matrix metalloproteinase (MMP)-2 and MMP-9. These MMPs are able to disrupt basement membranes, consequently promoting cell migration and the release of VEGF molecules trapped within the extracellular matrix (Fig. 3).81 These results suggest that activation of FGFR1 in prostate epithelial cells can enhance angiogenesis by increasing VEGF and Ang2 availability. Ang2 production and Ang1 downregulation could sensitize endothelial cells, creating an angiogenic switch by allowing these cells to become responsive to the microenvironment or FGFR-driven, pro-angiogenic factors (Fig. 3). Alternatively, release of cytokines and recruitment of inflammatory cells that produce matrix-degrading proteases, TNF〈, and other pro-angiogenic factors involved in wound healing could be an independent cause of tumor angiogenesis.
Figure 3.
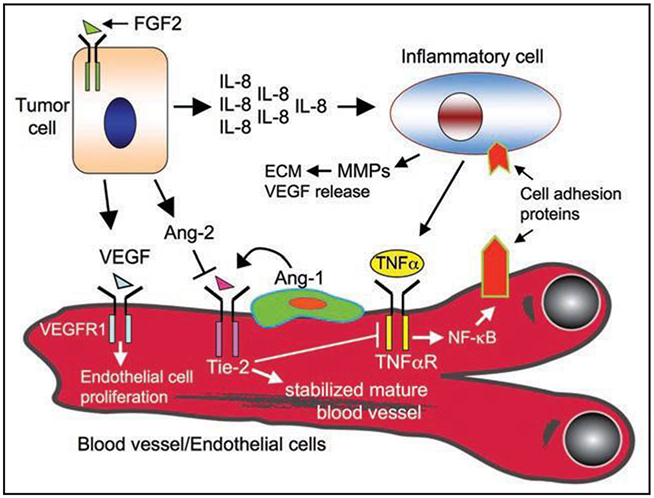
Activation of FGFR1 in tumor cells promotes angiogenesis. FGFR1 signaling leads to an increase in VEGF expression. Paracrine VEGF signaling by tumor cells exerts a mitogenic effect on endothelial cells and promotes migration. Moreover, FGFR1-Ras signaling within tumor cells allows for the production and secretion of IL-8, which serves to recruit inflammatory cells that can further promote angiogenesis. Another FGFR1 target, Ang2, is known to play an antagonist role distinct from Ang1 through the destabilization of the vasculature. Destabilized endothelial cells can then become sensitive to VEGF and/or TNF〈 produced by inflammatory cells.
Androgen-Independence Achieved through Tyrosine Kinase Receptor Regulation of the Androgen Receptor
The androgen receptor (AR) is a member of the steroid-binding, nuclear receptor family of transcription factors. AR normally becomes active after binding to dihydroxytestosterone (DHT) or, to lesser extent, testosterone. This hormone-receptor interaction leads to phosphorylation, homodimerization, nuclear translocation and transcription or suppression of AR target genes involved in cellular growth and survival. Interactions between androgens and AR play an important role during all stages of prostate development, as well as in normal glandular function.82 Moreover, androgens can also contribute to aberrant prostate growth. Work performed by Huggins in the 1940’s revealed that androgen ablation reduced prostate cancer, suggesting an immediate link. To date, this type of hormonal therapy, either by (surgical) orchiectomy or modern (chemical) anti-androgen treatments is commonly used in advanced, treatment-recalcitrant prostate cancer. Nonetheless, after initial positive outcome, 80–90% of patients have a relapse after 12–33 months of a more aggressive, androgen-independent (AI) disease.83 An increasing amount of evidence points to a necessary role for AR persistence in the development of AI.84,85 Several different mechanisms have been proposed for the development of AI through the AR, including: (1) an increase in the levels of AR or coactivators, (2) ligand-binding domain mutations or ligand promiscuity, (3) high local production of androgen and (4) phosphorylation of AR by kinases downstream of RTKs.85 Thus, the AR is a hub of prostate homeostasis and disregulation.
Growth factors, such as insulin-like growth factor (IGF), EGF and FGFs have been shown to activate AR and AR-targeted genes in the absence of androgens.86 While the exact mechanism by which growth factors promote AI is not completely understood, many of the commonly regulated growth factor signaling pathways have been shown to regulate AI. The use of Genetically Engineered Mouse (GEM) models has shed light into the molecular factors associated with AI. In mice that are null for the tumor suppressor gene Pten, or heterozygous for both Pten and the Pten-targeted gene Nkx3.1 (Pten+/−/Nkx3.1+/−), an increase in AI disease after androgen ablation has been observed.87,88 This tumor suppressor plays a key role in regulating the activity of the growth factor induced PI3K/Akt pathway. Pten dephosphorylates the C-3 phosphate group of the PI3K-product, phosphatidyl inositol, 3,4,5-triphosphate, PIP3, thereby, blocking Akt recruitment via its pleckstrin homology domain and activation via membrane-associated PDK1 and Rictor. These findings suggest that activation of the PI3K/Akt pathway by RTKs allows prostate cells to become androgen-independent, similar to Pten deficiency. Analysis of Pten and Nkx3.1 GEM prostate cancer models suggests that the mechanism by which Pten leads to AI may be Nkx3.1-dependent. For example, Pten has been shown to maintain Nkx3.1 levels,88 while reduced Nkx3.1 complements Pten in promoting AI, in contrast to relatively normal Pten+/− mice.89 When Nkx3.1 is reintroduced into Pten/Nkx3.1 null cells, proliferation is reduced concomitant with an increase in p53-dependent apoptosis.90 Lei and coworkers further showed that Nkx3.1 was able to bind to the AR promoter and repress AR-mediated target gene transcription. Cell culture experiments have confirmed that Pten-mediated AI is still AR-dependent, as knocking down AR in Pten-deficient cells leads to a reduction in tumorigenesis.91 These results suggest that activation of the PI3K/Akt pathway may allow for AI by blocking Nkx3.1 transcriptional inhibition of AR expression.
Other proposed mechanisms for PI3K/Akt-mediated AI include increased ®-catenin transcriptional activity after inactivation of Akt target, GSK3®, and inhibition of pro-apoptotic molecules by Akt.85,92 Akt is also capable of inhibiting a number of pro-apoptotic molecules, like Bcl-2 family member, BAD, forkhead family transcription factor, FOXO3a and pro-caspase-9. Since androgen blockade leads to induction of both prostate and tumor cell apoptosis, Akt-mediated anti-apoptotic activity could potentially compensate for reduced AR-mediated pro-survival signaling. In addition, Akt may directly target AR for phosphorylation.93 Other studies have shown that Her2/Neu activation of PI3K/Akt leads to AR phosphorylation and activation, which can be blocked by dominant negative Akt.94 However, contradictory studies have argued that Akt phosphorylation may have an inhibitory effect on AR.95 As always, these discrepancies may be dependent on cell context or on the specific residues that are post-translationally modified.
In addition to PI3K/Akt, other FGFR-activated signaling pathways can contribute to AI. Recent studies have shown that the Sex-Determining Region (SRY) transcription factor family member, Sox9, directly increases AR mRNA levels in prostate cells.96 Gene expression studies in JOCK1 mice showed that Sox9 is upregulated by iFGFR1 within 6 hours of activation.25 While studies performed on chondrocytes have shown that FGF2 increases Sox9 expression in a MAPK-dependent manner.97 Taken together, these results suggest that FGFR1 activation of the MAPK pathway can increase Sox9, which in turn, increases AR levels. Further evidence linking FGF signaling with AI comes from reconstitution studies using FGF10-overexpressing stromal cells, which lead to survival and proliferation of prostate epithelial cell in the absence of androgens.98 Furthermore, androgen ablation of JOCK1 mice after the development of prostate cancer leads to castration-resistant proliferation.25 Consistent with these results, stimulation of the RTK, Her2/Neu, in androgen-dependent LAPC-4 cells, triggers AI, accelerating tumor progression in castrated mice.99 This study also provided a mechanism by which Her2/Neu relies on AR sensitization to promote AI. Related studies show that Her2/Neu-mediated AR nuclear translocation depends on the phosphorylation of AR’s N-terminus by Erk, consequently promoting AR transactivation and possible interaction with co-activators.100 Thus, RTK signaling via the MAPK pathway could lead to an increase in AR levels through Sox9 transcription, and further post-translational modifications may activate AR-transcription in the absence of androgens (Fig. 3).
Finally, RTK-activated Src kinase can also phosphorylate AR (at Y514), leading to nuclear translocation and augmentation of transcriptional activity (Fig. 4).101 Interestingly, addition of androgens to AR-expressing cells can increase IGF-1R expression, previously implicated in Src-dependent AI. However, this effect may be independent of AR transcriptional activity since a DNA-binding domain mutant had similar efficacy.102 These results support a second, extra-nuclear function for AR. For example, Kousteni et al. have shown that cytosolic AR-Src interaction can help regulate Erk activity and inhibit apoptosis.103 Altogether, these studies show how different FGFR- and other RTK-regulated pathways and cellular processes emerge in the regulation of AR- and androgen-independent prostate cancer progression.
Figure 4.
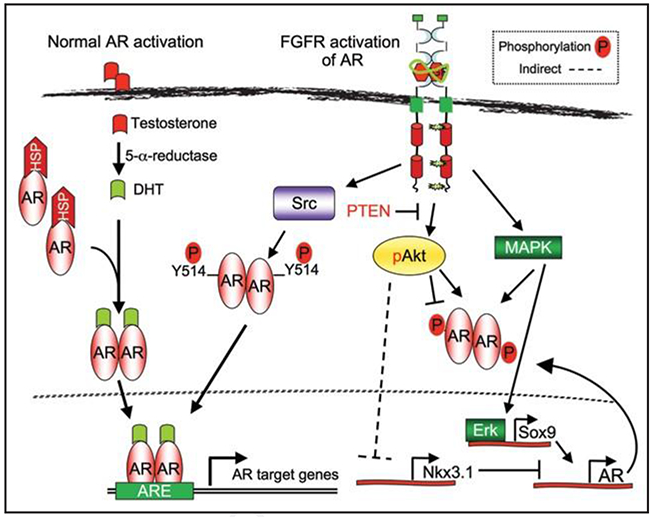
FGFR1 can promote androgen-independence. Androgens, such as dihydroxytestosterone (DHT), regulate the activity of the androgen receptor (AR) in normal physiological conditions. During androgen-independence, FGFR1 may regulate AR via a combination of different pathways.
Epithelial-to-Mesenchymal Transition and Epithelial Cell Plasticity
An increase in the migratory capacity of epithelial cells is associated with epithelial cell plasticity, described as an EMT. During an EMT, epithelial cells lose their cell polarity, E-cadherin expression and cell-cell interactions, increase their motility, and begin expressing mesenchymal markers, such as vimentin and fibronectin, becoming “mesenchymal-like”. This process is now well characterized during embryonic gastrulation and neuroectoderm development. The FGF signaling axis has been shown to be necessary during these developmental stages. For example, null FGFR1−/− mutations are embryonic lethal, due to a lack of cell migration of the primitive streak, forming an aberrant mesoderm during gastrulation.104 Additionally, FGFR1 null embryos lack the expression of the EMT-associated zinc finger transcription factor, Snail, a transcriptional repressor of the epithelial phenotype caretaker, E-cadherin.105,106 E-cadherin is a membrane-localized epithelial molecule that plays a key role in cell-cell adhesion, while its downregulation permits cells to detach and migrate. In fact, mouse embryos that lack p38〈 expression, which can target E-cadherin for proteosomal degradation, fail to downregulate E-cadherin protein levels. As a result, there is continuous cell-cell adhesion and lack of cell migration, resulting in a failed EMT and gastrulation.107
EMT has also been linked to cancer progression, characterized by the loss of epithelial cell differentiation-markers and an acquired increase in migratory capacity that serves to mediate tumor-cell invasion and metastasis.106 As with development, multiple changes in the FGFR axis are coincident with EMT in adult tissues. Along with ectopic FGFR1 expression, changes in FGFR2 isoforms from epithelial-type FGFR2IIIb to mesenchymal-type FGFR2IIIc have been postulated to be a manifestation of an EMT.55,108,109 In addition to sharing FGFR changes, common downstream molecular mechanisms may drive an EMT during cancer progression. The FGFR1-targeted Snail family genes seem to be at the center of FGF-mediated tumorigenic-EMT, as bladder carcinoma cells lacking Slug (Snail2) expression are resistant to FGF1-mediated EMT.110 Moreover, Snail has been linked to recurrence in a spontaneous breast cancer model.111 These results support a role for the FGF axis in driving a tumorigenic-EMT. This type of epithelial cell plasticity may also contribute to tumor heterogeneity with features of both sarcomas and carcinomas.
Interestingly, unlike the FGFR1’s effects on Snail/Slug expression during gastrulation, JOCK1 studies showed that iFGFR1 signaling does not lead directly to increases in Snail expression within 24 hours, but Snail/Slug upregulation was eventually found in advanced and metastatic lesions.25 Instead, the EMT-associated gene, Sox9, a member of the SRY family of transcription factors, is increased in vivo after 6 hours of CID-mediated iFGFR1 activation. Sox9 may promote an EMT by ultimately inducing the expression of both Snail and Slug genes, as mice deficient for Sox9 lack Snail expression, while co-expression of Sox9 and Snail/Slug promotes neural crest cell migration.112,113 Moreover, a protein complex formed by the physical interaction between Sox9 and Slug, works synergistically to promote an EMT by targeting the Slug-promoter region and increasing Slug’s transcription levels.114 These results suggest that Sox9 enhances Snail gene expression. Other signals, such as TGF®, may also regulate the initial upregulation of Snail molecules and “jump start” FGFR1-Sox9-mediated expression of Snail genes. In fact, TGF® induction of the MAPK and Akt pathways can lead to an increase in Snail levels and repression of E-cadherin, while addition of FGF-2 synergistically cooperates with TGF® to promote Snail transcription and EMT.115,116 TGF®R signaling can also lead to an increase in another E-cadherin repressor, Sip1, as well as interactions with Par proteins, resulting in tight-junction disas- sembly. However, this TGF®R-mediated EMT induction may vary according to cell type and TGF® receptor/ligand usage.117,118 While the MAPK pathway contributes to increased Snail mRNA levels, the PI3K/Akt pathway promotes Snail protein stability by inhibiting the repressor protein, GSK-3®. Modifications of Snail by GSK-3® in two consensus motifs lead to changes in cellular localization and eventu- ally proteosomal degradation.119 Taken together, these results suggest that FGFR1 can promote both the expression and protein stability of Snail genes via the MAPK and Akt pathways respectively, hence promoting an EMT event.
Other transcription factors, like the basic helix-loop-helix family member, Twist, also play a crucial role in promoting EMT and metastasis,120 but it is not completely understood whether FGFRs also regulate Twist to drive EMT. However, at minimum, FGFR1-stimulated EMT seems to be dependent on Snail/Slug and the downregulation of E-cadherin. It is this loss of E-cadherin and resulting cell-cell contact and polarity that permits tumor cells to break free and invade surrounding and distant sites.107 Twist-mediated EMT has also been shown to allow cells to escape the safeguards of senescence, hence, providing a potential mechanism for malignant cells to escape this barrier and dormancy.121
Lastly, the EMT effectors TGF®, Snail and Twist have recently been shown to confer cells with cancer stem cell (CSC) charac- teristics, as cells that had undergone an EMT have an increased colony forming ability.122 CSCs are cells with stem cell properties, including self-renewal and differentiation into different progenies, contributing to tumor heterogeneity.123 Moreover, these cells may circumvent traditional therapies targeting highly proliferative cells, leading to tumor relapse. One of the best-characterized CSC popu- lations in solid tumors is CD44+/CD24low from breast cancers, which can recapitulate the heterogeneity of breast tumors.124 These CD44+/CD24low cells have been shown to express FGFR1 and TGF® with the latter helping to maintain a more mesenchymal phenotype.125 Interestingly, CD44 was also upregulated in JOCK1 mice following iFGFR1 activation (as well as members of the Wnt signaling axis), suggesting a possible role in driving “reversion” to a more stem-like phenotype.25 It will be interesting to test whether recently identified Lin(CD31, CD45, Ter-119)−Sca-1+CD49fhigh cells126 or Lin−Sca-1+CD44+CD133+CD127+ cells127 with stem cell activity express FGFR1. Taken together, FGFR and other EMT-associated molecules may promote the formation of metastatic epithelial cells with CSC properties, while at the same time generating a more chemotherapy-resistant group of cells with the potential for tumor relapse.
Conclusions
Control of tumor cell growth, angiogenesis, and a likely role in tumor metastasis and recurrence make the FGF/FGFR signaling axis an attractive therapeutic target. Here, we provide an overview of potential modes of FGF-mediated tumorigenesis, in particular, its role in prostate cancer progression. Novel therapies may include the development and use of dominant-negative, isoform-specific FGFR genes, as well as crossreacting antibodies and inhibitors. As opposed to typical xenograft and cell culture models, the use of GEM models is a potentially more physiologically relevant resource to test novel therapies. These models are able to recapitulate the tumor microenvironment with distinct stages of tumor progression in an immunocompetent environment, similar to human disease. Our studies using the JOCK1 prostate cancer model have allowed us to validate the importance of this signaling axis during tumori- genesis along with helping elucidate the molecular and phenotypical changes associated with FGFR1 during cancer progression. This conditional and reversible model led to the identification of temporal differences in responsiveness to iFGFR1 inhibition, indicative of a “susceptibility window” for targeting FGFR1. Rapidly inducible and reversible models, like JOCK1, can advance the goal of developing novel and valid therapies with a higher probability of success in treating human disease.
Acknowledgements
We are grateful to David Rowley for a careful reading of the manuscript. This work was supported by NIH grant U01-CA84296 (V.D.A., M.I., D.M.S.) and F31-GM069044 (V.D.A.).
Abbreviations:
- AI
androgen independence
- AR
androgen receptor
- CID
chemical inducer of dimerization
- EMT
epithelial to mesenchymal transition
- FGF
fibroblast growth factor
- FGFR
fibroblast growth factor receptor
- iFGFR1
inducible fibroblast growth factor receptor 1
- GEM
genetically engineered mouse
- PCa
prostate cancer
- RTK
receptor tyrosine kinase
References
- 1.Grose R, Dickson C. Fibroblast growth factor signaling in tumorigenesis. Cytokine Growth Factor Rev 2005; 16:179–86. [DOI] [PubMed] [Google Scholar]
- 2.Penault-Llorca F, Bertucci F, Adelaide J, Parc P, Coulier F, Jacquemier J, et al. Expression of FGF and FGF receptor genes in human breast cancer. Int J Cancer 1995; 61:170–6. [DOI] [PubMed] [Google Scholar]
- 3.Roumiantsev S, Krause DS, Neumann CA, Dimitri CA, Asiedu F, Cross NCP, et al. Distinct stem cell myeloproliferative/T lymphoma syndromes induced by ZNF198-FGFR1 and BCR-FGFR1 fusion genes from 8p11 translocations. Cancer Cell 2004; 5:287–98. [DOI] [PubMed] [Google Scholar]
- 4.Cappellen D, De Oliveira C, Ricol D, de Medina S, Bourdin J, Sastre-Garau X, et al. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet 1999; 23:18–20. [DOI] [PubMed] [Google Scholar]
- 5.Giri D, Ropiquet F, Ittmann M. Alterations in expression of basic fibroblast growth factor (FGF) 2 and its receptor FGFR-1 in human prostate cancer. Clin Cancer Res 1999; 5:1063–71. [PubMed] [Google Scholar]
- 6.Kwabi-Addo B, Ozen M, Ittmann M. The role of fibroblast growth factors and their recep- tors in prostate cancer. Endocr Relat Cancer 2004; 11:709–24. [DOI] [PubMed] [Google Scholar]
- 7.Toll A, Real FX. Somatic oncogenic mutations, benign skin lesions and cancer progression: where to look next? Cell Cycle 2008; 7:2674–81. [DOI] [PubMed] [Google Scholar]
- 8.Lin Y, Liu G, Zhang Y, Hu YP, Yu K, Lin C, et al. Fibroblast growth factor receptor 2 tyrosine kinase is required for prostatic morphogenesis and the acquisition of strict androgen dependency for adult tissue homeostasis. Development 2007; 134:723–34. [DOI] [PubMed] [Google Scholar]
- 9.Yan G, Fukabori Y, McBride G, Nikolaropolous S, McKeehan WL. Exon switching and activation of stromal and embryonic fibroblast growth factor (FGF)-FGF receptor genes in prostate epithelial cells accompany stromal independence and malignancy. Mol Cell Biol 1993; 13:4513–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu W, Luo Y, Kan M, McKeehan WL. Fibroblast growth factor-10. A second candidate stromal to epithelial cell andromedin in prostate. J Biol Chem 1999; 274:12827–34. [DOI] [PubMed] [Google Scholar]
- 11.Dorkin TJ, Robinson MC, Marsh C, Bjartell A, Neal DE, Leung HY. FGF8 overexpression in prostate cancer is associated with decreased patient survival and persists in androgen independent disease. Oncogene 1999; 18:2755–61. [DOI] [PubMed] [Google Scholar]
- 12.Giri D, Ropiquet F, Ittmann M. Alterations in expression of basic fibroblast growth fac- tor (FGF) 2 and its receptor FGFR-1 in human prostate cancer. Clin Cancer Res 1999; 5:1063–71. [PubMed] [Google Scholar]
- 13.Foster BA, Kaplan PJ, Greenberg NM. Characterization of the FGF axis and identification of a novel FGFR1iiic isoform during prostate cancer progression in the TRAMP model. Prostate Cancer and Prostatic Diseases 1999; 76–82. [DOI] [PubMed] [Google Scholar]
- 14.Greenberg NM, DeMayo F, Finegold MJ, Medina D, Tilley WD, Aspinall JO, et al. Prostate cancer in a transgenic mouse. ProcNatlAcadSciUSA 1995; 92:3439–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huss WJ, Barrios RJ, Foster BA, Greenberg NM. Differential expression of specific FGF ligand and receptor isoforms during angiogenesis associated with prostate cancer progres- sion. Prostate 2003; 54:8–16. [DOI] [PubMed] [Google Scholar]
- 16.Valve EM, Nevalainen MT, Nurmi MJ, Laato MK, Martikainen PM, Harkonen PL. Increased expression of FGF-8 isoforms and FGF receptors in human premalignant prostatic intraepithelial neoplasia lesions and prostate cancer. Lab Invest 2001; 81:815–26. [DOI] [PubMed] [Google Scholar]
- 17.Gravdal K, Halvorsen OJ, Haukaas SA, Akslen LA. Expression of bFGF/FGFR-1 and vascular proliferation related to clinicopathologic features and tumor progress in localized prostate cancer. Virchows Arch 2006; 448:68–74. [DOI] [PubMed] [Google Scholar]
- 18.Devilard E, Bladou F, Ramuz O, Karsenty G, Dales JP, Gravis G, et al. FGFR1 and WT1 are markers of human prostate cancer progression. BMC Cancer 2006; 6:272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sahadevan K, Darby S, Leung HY, Mathers ME, Robson CN, Gnanapragasam VJ. Selective overexpression of fibroblast growth factor receptors 1 and 4 in clinical prostate cancer. J Pathol 2007; 213:82–90. [DOI] [PubMed] [Google Scholar]
- 20.Hamaguchi A, Tooyama I, Yoshiki T, Kimura H. Demonstration of fibroblast growth factor receptor-I in human prostate by polymerase chain reaction and immunohistochemistry. Prostate 1995; 27:141–7. [DOI] [PubMed] [Google Scholar]
- 21.Trevor J Dorkin MCRCMDENHYL. aFGF immunoreactivity in prostate cancer and its co-localization with bFGF and FGF8. The Journal of Pathology 1999; 189:564–9. [DOI] [PubMed] [Google Scholar]
- 22.Zhang X, Ibrahimi OA, Olsen SK, Umemori H, Mohammadi M, Ornitz DM. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J Biol Chem 2006; 281:15694–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang F, McKeehan K, Yu C, Ittmann M, McKeehan WL. Chronic activity of ectopic type 1 fibroblast growth factor receptor tyrosine kinase in prostate epithelium results in hyperplasia accompanied by intraepithelial neoplasia. Prostate 2004; 58:1–12. [DOI] [PubMed] [Google Scholar]
- 24.Winter SF, Acevedo VD, Gangula RD, Freeman KW, Spencer DM, Greenberg NM. Conditional activation of FGFR1 in the prostate epithelium induces angiogenesis with concomitant differential regulation of Ang1 and Ang2. Oncogene 2007; 26:4897–907. [DOI] [PubMed] [Google Scholar]
- 25.Acevedo VD, Gangula RD, Freeman KW, Li R, Zhang Y, Wang F, et al. Inducible FGFR-1 activation leads to irreversible prostate adenocarcinoma and an epithelial-to-mesenchymal transition. Cancer Cell 2007; 12:559–71. [DOI] [PubMed] [Google Scholar]
- 26.Freeman KW, Welm BE, Gangula RD, Rosen JM, Ittmann M, Greenberg NM, et al. Inducible prostate intraepithelial neoplasia with reversible hyperplasia in conditional FGFR1-expressing mice. Cancer Research 2003; 63:8256–63. [PubMed] [Google Scholar]
- 27.Mohammadi M, Olsen SK, Ibrahimi OA. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev 2005; 16:107–37. [DOI] [PubMed] [Google Scholar]
- 28.Ornitz DM, Itoh N. Fibroblast growth factors. Genome Biol 2001; 2:3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Powers CJ, McLeskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocr Relat Cancer 2000; 7:165–97. [DOI] [PubMed] [Google Scholar]
- 30.McKeehan WL, Wang F, Kan M. The heparan sulfate-fibroblast growth factor family: diversity of structure and function. Prog Nucleic Acid Res Mol Biol 1998; 59:135–76. [DOI] [PubMed] [Google Scholar]
- 31.Ye S, Luo Y, Lu W, Jones RB, Linhardt RJ, Capila I, et al. Structural basis for interaction of FGF-1, FGF-2 and FGF-7 with different heparan sulfate motifs. Biochemistry 2001; 40:14429–39. [DOI] [PubMed] [Google Scholar]
- 32.Furdui CM, Lew ED, Schlessinger J, Anderson KS. Autophosphorylation of FGFR1 kinase is mediated by a sequential and precisely ordered reaction. Mol Cell 2006; 21:711–7. [DOI] [PubMed] [Google Scholar]
- 33.Schlessinger J Cell signaling by receptor tyrosine kinases. Cell 2000; 103:211–25. [DOI] [PubMed] [Google Scholar]
- 34.Larsson H, Klint P, Landgren E, Claesson-Welsh L. Fibroblast growth factor receptor-1-mediated endothelial cell proliferation is dependent on the Src homology (SH) 2/SH3 domain-containing adaptor protein Crk. J Biol Chem 1999; 274:25726–34. [DOI] [PubMed] [Google Scholar]
- 35.Kouhara H, Hadari YR, T S-K, Schilling J, D B-S, Lax I, et al. A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell 1997; 89:693–702. [DOI] [PubMed] [Google Scholar]
- 36.Ong SH, Guy GR, Hadari YR, Laks S, Gotoh N, Schlessinger J, et al. FRS2 proteins recruit intracellular signaling pathways by binding to diverse targets on fibroblast growth factor and nerve growth factor receptors. Mol Cell Biol 2000; 20:979–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sandilands E, Akbarzadeh S, Vecchione A, McEwan DG, Frame MC, Heath JK. Src kinase modulates the activation, transport and signalling dynamics of fibroblast growth factor receptors. EMBO Rep 2007; 8:1162–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li X, Brunton VG, Burgar HR, Wheldon LM, Heath JK. FRS2-dependent SRC activation is required for fibroblast growth factor receptor-induced phosphorylation of Sprouty and suppression of ERK activity. J Cell Sci 2004; 117:6007–17. [DOI] [PubMed] [Google Scholar]
- 39.Zhan X, Plourde C, Hu X, Friesel R, Maciag T. Association of fibroblast growth factor receptor-1 with c-Src correlates with association between c-Src and cortactin. J Biol Chem 1994; 269:20221–4. [PubMed] [Google Scholar]
- 40.Wong A, Lamothe B, Lee A, Schlessinger J, Lax I. FRS2 alpha attenuates FGF receptor signaling by Grb2-mediated recruitment of the ubiquitin ligase Cbl. Proc Natl Acad Sci USA 2002; 99:6684–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Zhang J, Lin Y, Lan Y, Lin C, Xuan JW, et al. Role of epithelial cell fibroblast growth factor receptor substrate 2alpha in prostate development, regeneration and tumorigenesis. Development 2008; 135:775–84. [DOI] [PubMed] [Google Scholar]
- 42.Kim HJ, Bar-Sagi D. Modulation of signalling by Sprouty: a developing story. Nat Rev Mol Cell Biol 2004; 5:441–50. [DOI] [PubMed] [Google Scholar]
- 43.Wakioka T, Sasaki A, Kato R, Shouda T, Matsumoto A, Miyoshi K, et al. Spred is a Sprouty-related suppressor of Ras signalling. Nature 2001; 412:647–51. [DOI] [PubMed] [Google Scholar]
- 44.Eblaghie MC, Lunn JS, Dickinson RJ, Munsterberg AE, Sanz-Ezquerro JJ, Farrell ER, et al. Negative feedback regulation of FGF signaling levels by Pyst1/MKP3 in chick embryos. Curr Biol 2003; 13:1009–18. [DOI] [PubMed] [Google Scholar]
- 45.Darby S, Sahadevan K, Khan MM, Robson CN, Leung HY, Gnanapragasam VJ. Loss of Sef (similar expression to FGF) expression is associated with high grade and metastatic prostate cancer. Oncogene 2006; 25:4122–7. [DOI] [PubMed] [Google Scholar]
- 46.Sasaki A, Taketomi T, Kato R, Saeki K, Nonami A, Sasaki M, et al. Mammalian Sprouty4 suppresses Ras-independent ERK activation by binding to Raf1. Nat Cell Biol 2003; 5:427–32. [DOI] [PubMed] [Google Scholar]
- 47.Fong CW, Leong HF, Wong ES, Lim J, Yusoff P, Guy GR. Tyrosine phosphorylation of Sprouty2 enhances its interaction with c-Cbl and is crucial for its function. J Biol Chem 2003; 278:33456–64. [DOI] [PubMed] [Google Scholar]
- 48.Kwabi-Addo B, Wang J, Erdem H, Vaid A, Castro P, Ayala G, et al. The expression of Sprouty1, an inhibitor of fibroblast growth factor signal transduction, is decreased in human prostate cancer. Cancer Res 2004; 64:4728–35. [DOI] [PubMed] [Google Scholar]
- 49.McKie AB, Douglas DA, Olijslagers S, Graham J, Omar MM, Heer R, et al. Epigenetic inactivation of the human sprouty2 (hSPRY2) homologue in prostate cancer. Oncogene 2005; 24:2166–74. [DOI] [PubMed] [Google Scholar]
- 50.Zisman-Rozen S, Fink D, Ben-Izhak O, Fuchs Y, Brodski A, Kraus MH, et al. Downregulation of Sef, an inhibitor of receptor tyrosine kinase signaling, is common to a variety of human carcinomas. Oncogene 2007; 26:6093–8. [DOI] [PubMed] [Google Scholar]
- 51.Wang J, Thompson B, Ren C, Ittmann M, Kwabi-Addo B. Sprouty4, a suppressor of tumor cell motility, is downregulated by DNA methylation in human prostate cancer. Prostate 2006; 66:613–24. [DOI] [PubMed] [Google Scholar]
- 52.Thomson AA. Role of androgens and fibroblast growth factors in prostatic development. Reproduction 2001; 121:187–95. [DOI] [PubMed] [Google Scholar]
- 53.Lin Y, Liu G, Zhang Y, Hu Y-P, Yu K, Lin C, et al. Fibroblast growth factor receptor 2 tyrosine kinase is required for prostatic morphogenesis and the acquisition of strict androgen dependency for adult tissue homeostasis. Development 2007; 134:723–34. [DOI] [PubMed] [Google Scholar]
- 54.Jin C, Wang F, Wu X, Yu C, Luo Y, McKeehan WL. Directionally specific paracrine communication mediated by epithelial FGF9 to stromal FGFR3 in two-compartment premalignant prostate tumors. Cancer Res 2004; 64:4555–62. [DOI] [PubMed] [Google Scholar]
- 55.Yan G, Fukabori Y, McBride G, Nikolaropolous S, McKeehan WL. Exon switching and activation of stromal and embryonic fibroblast growth factor (FGF)-FGF receptor genes in prostate epithelial cells accompany stromal independence and malignancy. Mol Cell Biol 1993; 13:4513–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gnanapragasam VJ, Robinson MC, Marsh C, Robson CN, Hamdy FC, Leung HY. FGF8 isoform b expression in human prostate cancer. Br J Cancer 2003; 88:1432–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carstens RP, Eaton JV, Krigman HR, Walther PJ, Garcia-Blanco MA. Alternative splicing of fibroblast growth factor receptor 2 (FGF-R2) in human prostate cancer. Oncogene 1997; 15:3059–65. [DOI] [PubMed] [Google Scholar]
- 58.Feng S, Wang F, Matsubara A, Kan M, McKeehan WL. Fibroblast growth factor receptor 2 limits and receptor 1 accelerates tumorigenicity of prostate epithelial cells. Cancer Res 1997; 57:5369–78. [PubMed] [Google Scholar]
- 59.Cronauer MV, Hittmair A, Eder IE, Hobisch A, Culig Z, Ramoner R, et al. Basic fibroblast growth factor levels in cancer cells and in sera of patients suffering from proliferative disorders of the prostate. Prostate 1997; 31:223–33. [DOI] [PubMed] [Google Scholar]
- 60.Memarzadeh S, Xin L, Mulholland DJ, Mansukhani A, Wu H, Teitell MA, et al. Enhanced paracrine FGF10 expression promotes formation of multifocal prostate adenocarcinoma and an increase in epithelial androgen receptor. Cancer Cell 2007; 12:572–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer 2003; 3:401–10. [DOI] [PubMed] [Google Scholar]
- 62.Presta M, Dell’Era P, Mitola S, Moroni E, Ronca R, Rusnati M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev 2005; 16:159–78. [DOI] [PubMed] [Google Scholar]
- 63.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med 2003; 9:669–76. [DOI] [PubMed] [Google Scholar]
- 64.Seghezzi G, Patel S, Ren CJ, Gualandris A, Pintucci G, Robbins ES, et al. Fibroblast growth factor-2 (FGF-2) induces vascular endothelial growth factor (VEGF) expression in the endothelial cells of forming capillaries: an autocrine mechanism contributing to angiogenesis. J Cell Biol 1998; 141:1659–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tsunoda S, Nakamura T, Sakurai H, Saiki I. Fibroblast growth factor-2-induced host stroma reaction during initial tumor growth promotes progression of mouse melanoma via vascular endothelial growth factor A-dependent neovascularization. Cancer Sci 2007; 98:541–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Auguste P, Gursel DB, Lemiere S, Reimers D, Cuevas P, Carceller F, et al. Inhibition of fibroblast growth factor/fibroblast growth factor receptor activity in glioma cells impedes tumor growth by both angiogenesis-dependent and -independent mechanisms. Cancer Res 2001; 61:1717–26. [PubMed] [Google Scholar]
- 67.Polnaszek N, Kwabi-Addo B, Peterson LE, Ozen M, Greenberg NM, Ortega S, et al. Fibroblast growth factor 2 promotes tumor progression in an autochthonous mouse model of prostate cancer. Cancer Res 2003; 63:5754–60. [PubMed] [Google Scholar]
- 68.Czubayko F, Liaudet-Coopman ED, Aigner A, Tuveson AT, Berchem GJ, Wellstein A. A secreted FGF-binding protein can serve as the angiogenic switch in human cancer. Nat Med 1997; 3:1137–40. [DOI] [PubMed] [Google Scholar]
- 69.Giavazzi R, Giuliani R, Coltrini D, Bani MR, Ferri C, Sennino B, et al. Modulation of tumor angiogenesis by conditional expression of fibroblast growth factor-2 affects early but not established tumors. Cancer Res 2001; 61:309–17. [PubMed] [Google Scholar]
- 70.Hori A, Sasada R, Matsutani E, Naito K, Sakura Y, Fujita T, et al. Suppression of solid tumor growth by immunoneutralizing monoclonal antibody against human basic fibroblast growth factor. Cancer Res 1991; 51:6180–4. [PubMed] [Google Scholar]
- 71.Ogawa T, Takayama K, Takakura N, Kitano S, Ueno H. Anti-tumor angiogenesis therapy using soluble receptors: enhanced inhibition of tumor growth when soluble fibroblast growth factor receptor-1 is used with soluble vascular endothelial growth factor receptor. Cancer Gene Ther 2002; 9:633–40. [DOI] [PubMed] [Google Scholar]
- 72.Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 1997; 277:55–60. [DOI] [PubMed] [Google Scholar]
- 73.Gale NW, Thurston G, Davis S, Wiegand SJ, Holash J, Rudge JS, et al. Complementary and coordinated roles of the VEGFs and angiopoietins during normal and pathologic vascular formation. Cold Spring Harb Symp Quant Biol 2002; 67:267–73. [DOI] [PubMed] [Google Scholar]
- 74.Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, Zagzag D, et al. Vessel cooption, regression and growth in tumors mediated by angiopoietins and VEGF. Science 1999; 284:1994–8. [DOI] [PubMed] [Google Scholar]
- 75.Thurston G, Suri C, Smith K, McClain J, Sato TN, Yancopoulos GD, et al. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science 1999; 286:2511–4. [DOI] [PubMed] [Google Scholar]
- 76.Fiedler U, Reiss Y, Scharpfenecker M, Grunow V, Koidl S, Thurston G, et al. Angiopoietin-2 sensitizes endothelial cells to TNFalpha and has a crucial role in the induction of inflammation. Nat Med 2006; 12:235–9. [DOI] [PubMed] [Google Scholar]
- 77.Lemieux C, Maliba R, Favier J, Theoret JF, Merhi Y, Sirois MG. Angiopoietins can directly activate endothelial cells and neutrophils to promote proinflammatory responses. Blood 2005; 105:1523–30. [DOI] [PubMed] [Google Scholar]
- 78.Schwertfeger KL, Xian W, Kaplan AM, Burnett SH, Cohen DA, Rosen JM. A critical role for the inflammatory response in a mouse model of preneoplastic progression. Cancer Res 2006; 66:5676–85. [DOI] [PubMed] [Google Scholar]
- 79.Sparmann A, Bar-Sagi D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell 2004; 6:447–58. [DOI] [PubMed] [Google Scholar]
- 80.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer 2004; 4:71–8. [DOI] [PubMed] [Google Scholar]
- 81.Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res 2006; 66:605–12. [DOI] [PubMed] [Google Scholar]
- 82.Cunha GR, Alarid ET, Turner T, Donjacour AA, Boutin EL, Foster BA. Normal and abnormal development of the male urogenital tract. Role of androgens, mesenchymal-epithelial interactions, and growth factors. J Androl 1992; 13:465–75. [PubMed] [Google Scholar]
- 83.Hellerstedt BA, Pienta KJ. The current state of hormonal therapy for prostate cancer. CA Cancer J Clin 2002; 52:154–79. [DOI] [PubMed] [Google Scholar]
- 84.Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, Ogata GK, et al. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med 1995; 332:1393–8. [DOI] [PubMed] [Google Scholar]
- 85.Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer 2001; 1:34–45. [DOI] [PubMed] [Google Scholar]
- 86.Culig Z, Hobisch A, Cronauer MV, Radmayr C, Trapman J, Hittmair A, et al. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res 1994; 54:5474–8. [PubMed] [Google Scholar]
- 87.Abate-Shen C, Banach-Petrosky WA, Sun X, Economides KD, Desai N, Gregg JP, et al. Nkx3.1; Pten mutant mice develop invasive prostate adenocarcinoma and lymph node metastases. Cancer Res 2003; 63:3886–90. [PubMed] [Google Scholar]
- 88.Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 2003; 4:209–21. [DOI] [PubMed] [Google Scholar]
- 89.Shen MM, Abate-Shen C. Pten inactivation and the emergence of androgen-independent prostate cancer. Cancer Res 2007; 67:6535–8. [DOI] [PubMed] [Google Scholar]
- 90.Lei Q, Jiao J, Xin L, Chang CJ, Wang S, Gao J, et al. NKX3.1 stabilizes p53, inhibits AKT activation, and blocks prostate cancer initiation caused by PTEN loss. Cancer Cell 2006; 9:367–78. [DOI] [PubMed] [Google Scholar]
- 91.Jiao J, Wang S, Qiao R, Vivanco I, Watson PA, Sawyers CL, et al. Murine cell lines derived from Pten null prostate cancer show the critical role of PTEN in hormone refractory prostate cancer development. Cancer Res 2007; 67:6083–91. [DOI] [PubMed] [Google Scholar]
- 92.Mulholland DJ, Dedhar S, Wu H, Nelson CC. PTEN and GSK3beta: key regulators of progression to androgen-independent prostate cancer. Oncogene 2006; 25:329–37. [DOI] [PubMed] [Google Scholar]
- 93.Wang Y, Kreisberg JI, Ghosh PM. Cross-talk between the androgen receptor and the phosphatidylinositol 3-kinase/Akt pathway in prostate cancer. Curr Cancer Drug Targets 2007; 7:591–604. [DOI] [PubMed] [Google Scholar]
- 94.Wen Y, Hu MC, Makino K, Spohn B, Bartholomeusz G, Yan DH, et al. HER-2/neu promotes androgen-independent survival and growth of prostate cancer cells through the Akt pathway. Cancer Res 2000; 60:6841–5. [PubMed] [Google Scholar]
- 95.Lin HK, Yeh S, Kang HY, Chang C. Akt suppresses androgen-induced apoptosis by phosphorylating and inhibiting androgen receptor. Proc Natl Acad Sci USA 2001; 98:7200–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang H, McKnight NC, Zhang T, Lu ML, Balk SP, Yuan X. SOX9 is expressed in normal prostate basal cells and regulates androgen receptor expression in prostate cancer cells. Cancer Res 2007; 67:528–36. [DOI] [PubMed] [Google Scholar]
- 97.Murakami S, Kan M, McKeehan WL, de Crombrugghe B. Upregulation of the chondrogenic Sox9 gene by fibroblast growth factors is mediated by the mitogen-activated protein kinase pathway. Proc Natl Acad Sci USA 2000; 97:1113–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Memarzadeh S, Xin L, Mulholland DJ, Mansukhani A, Wu H, Teitell MA, et al. Enhanced paracrine FGF10 expression promotes formation of multifocal prostate adenocarcinoma and an increase in epithelial androgen receptor. Cancer Cell 2007; 12:572–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Craft N, Shostak Y, Carey M, Sawyers CL. A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat Med 1999; 5:280–5. [DOI] [PubMed] [Google Scholar]
- 100.Yeh S, Lin HK, Kang HY, Thin TH, Lin MF, Chang C. From HER2/Neu signal cascade to androgen receptor and its coactivators: a novel pathway by induction of androgen target genes through MAP kinase in prostate cancer cells. Proc Natl Acad Sci USA 1999; 96:5458–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Guo Z, Dai B, Jiang T, Xu K, Xie Y, Kim O, et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell 2006; 10:309–19. [DOI] [PubMed] [Google Scholar]
- 102.Pandini G, Mineo R, Frasca F, Roberts CT Jr, Marcelli M, Vigneri R, et al. Androgens upregulate the insulin-like growth factor-I receptor in prostate cancer cells. Cancer Res 2005; 65:1849–57. [DOI] [PubMed] [Google Scholar]
- 103.Kousteni S, Bellido T, Plotkin LI, O’Brien CA, Bodenner DL, Han L, et al. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell 2001; 104:719–30. [PubMed] [Google Scholar]
- 104.Yamaguchi TP, Harpal K, Henkemeyer M, Rossant J. fgfr-1 is required for embryonic growth and mesodermal patterning during mouse gastrulation. Genes Dev 1994; 8:3032–44. [DOI] [PubMed] [Google Scholar]
- 105.Ciruna B, Rossant J. FGF signaling regulates mesoderm cell fate specification and morphogenetic movement at the primitive streak. Dev Cell 2001; 1:37–49. [DOI] [PubMed] [Google Scholar]
- 106.Thiery JP. Epithelial-mesenchymal transition in tumor progression. Nat Rev Cancer 2002; 2:442–54. [DOI] [PubMed] [Google Scholar]
- 107.Zohn IE, Li Y, Skolnik EY, Anderson KV, Han J, Niswander L. p38 and a p38-interacting protein are critical for downregulation of E-cadherin during mouse gastrulation. Cell 2006; 125:957–69. [DOI] [PubMed] [Google Scholar]
- 108.Oltean S, Sorg BS, Albrecht T, Bonano VI, Brazas RM, Dewhirst MW, et al. Alternative inclusion of fibroblast growth factor receptor 2 exon IIIc in Dunning prostate tumors reveals unexpected epithelial mesenchymal plasticity. Proc Natl Acad Sci USA 2006; 103:14116–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Savagner P, Valles AM, Jouanneau J, Yamada KM, Thiery JP. Alternative splicing in fibroblast growth factor receptor 2 is associated with induced epithelial-mesenchymal transition in rat bladder carcinoma cells. Mol Biol Cell 1994; 5:851–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Savagner P, Yamada KM, Thiery JP. The zinc-finger protein slug causes desmosome dissociation, an initial and necessary step for growth factor-induced epithelial-mesenchymal transition. J Cell Biol 1997; 137:1403–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Moody SE, Perez D, Pan TC, Sarkisian CJ, Portocarrero CP, Sterner CJ, et al. The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell 2005; 8:197–209. [DOI] [PubMed] [Google Scholar]
- 112.Bolos V, Peinado H, Perez-Moreno MA, Fraga MF, Esteller M, Cano A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. J Cell Sci 2003; 116:499–511. [DOI] [PubMed] [Google Scholar]
- 113.Cheung M, Chaboissier MC, Mynett A, Hirst E, Schedl A, Briscoe J. The transcriptional control of trunk neural crest induction, survival and delamination. Dev Cell 2005; 8:179–92. [DOI] [PubMed] [Google Scholar]
- 114.Sakai D, Suzuki T, Osumi N, Wakamatsu Y. Cooperative action of Sox9, Snail2 and PKA signaling in early neural crest development. Development 2006; 133:1323–33. [DOI] [PubMed] [Google Scholar]
- 115.Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol 2005; 17:548–58. [DOI] [PubMed] [Google Scholar]
- 116.Peinado H, Quintanilla M, Cano A. Transforming growth factor {beta}-1 induces snail transcription factor in epithelial cell lines: Mechanisms for epithelial mesenchymal transitions. J Biol Chem 2003; 278:21113–23. [DOI] [PubMed] [Google Scholar]
- 117.Rees JRE, Onwuegbusi BA, Save VE, Alderson D, Fitzgerald RC. In vivo and in vitro evidence for transforming growth factor-{beta}1-mediated epithelial to mesenchymal transition in esophageal adenocarcinoma. Cancer Res 2006; 66:9583–90. [DOI] [PubMed] [Google Scholar]
- 118.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions 2006; 7:131–42. [DOI] [PubMed] [Google Scholar]
- 119.Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, et al. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol 2004; 6:931–40. [DOI] [PubMed] [Google Scholar]
- 120.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004; 117:927–39. [DOI] [PubMed] [Google Scholar]
- 121.Ansieau S, Bastid J, Doreau A, Morel A-P, Bouchet BP, Thomas C, et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell 2008; 14:79–89. [DOI] [PubMed] [Google Scholar]
- 122.Mani SA, Guo W, Liao M-J, Eaton EN, Ayyanan A, Zhou AY, et al. The epitheli- almesenchymal transition generates cells with properties of stem cells. Cell 2008; 133:704–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Clarke MF, Fuller M. Stem cells and cancer: Two faces of eve. Cell 2006; 124:1111–5. [DOI] [PubMed] [Google Scholar]
- 124.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. From the cover: Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 2003; 100:3983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell 2007; 11:259–73. [DOI] [PubMed] [Google Scholar]
- 126.Lawson DA, Xin L, Lukacs RU, Cheng D, Witte ON. Isolation and functional characterization of murine prostate stem cells. Proc Natl Acad Sci USA 2007; 104:181–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Leong KG, Wang BE, Johnson L, Gao WQ. Generation of a prostate from a single adult stem cell. Nature 2008. [DOI] [PubMed] [Google Scholar]