

Abstract
The organization of genomic DNA into nucleosomes profoundly affects all DNA-related processes in eukaryotes. The histone chaperone ‘FAcilitates Chromatin Transcription’ (FACT, consisting of subunits SPT16 and SSRP11) promotes both disassembly and reassembly of nucleosomes during gene transcription, DNA replication, and repair2. The mechanism by which FACT causes these opposing outcomes is unknown. Here we report two cryo-EM structures of human FACT in complex with partially assembled ‘sub-nucleosomes’, with supporting biochemical and hydrogen-deuterium exchange (HDX) data. FACT is engaged in extensive interactions with nucleosomal DNA and all histones. The large DNA-binding surface on FACT appears to be protected by the C-terminal domains of both subunits, and this inhibition is released by interaction with H2A-H2B, allowing FACT-H2A-H2B to dock onto a (H3-H4)2-DNA complex3. SPT16 binds nucleosomal DNA and tethers H2A-H2B through its C-terminal domain by acting as a placeholder for DNA. SSRP1 also contributes to DNA binding, and can assume two conformations, depending on whether a second H2A-H2B dimer is present. Our data suggest a compelling mechanism for how FACT maintains chromatin integrity during polymerase passage, by facilitating H2A-H2B dimer removal, stabilizing intermediate ‘subnucleosomal’ states, and promoting nucleosome reassembly. Our findings reconcile discrepancies regarding the many roles of FACT and underscore the dynamic interactions between histone chaperones and nucleosomes.
Nucleosomes are the basic repeating structural units of eukaryotic chromatin, consisting of two H2A-H2B dimers and one (H3-H4)2 tetramer and wrapped by 147 bp of DNA4 They represent formidable barriers to the transcription and replication machinery, and therefore must be at least partially dismantled ahead of polymerases and re-assembled in their wake, to maintain the structural and regulatory functions of chromatin5. Histone chaperones, a diverse group of structurally unrelated proteins, facilitate these processes6. FACT (Fig. 1a) is an essential histone chaperone1,7,8 (reviewed in reference2) that in vitro promotes nucleosome assembly by tethering histone and DNA components, and also facilitates nucleosome disassembly by displacing H2A-H2B and promoting the partial unraveling of DNA3,8-13. Although the structure of each individual domain in both FACT subunits Spt16 and SSRP1 is known2, the structure of the intact FACT heterodimer alone or with its relevant substrate(s) has been elusive, and insight into its mechanism is therefore largely indirect.
Figure 1: FACT forms two complexes with a partially assembled nucleosome.

a. Domain structure of the two FACT subunits SPT16 and SSRP1. NTD – N-terminal domain; DD – dimerization domain; MD – middle domain; CTD – C-terminal domain; IDD – intrinsically disordered domain; HMG – HMG box domain. Domains for which no density is visible shown in white.
b. Biochemical characterization of FACT-sub-nucleosome complexes by native- and SDS-PAGE, stained by SYBR Gold and Blazin Blue. This experiment was repeated more than 10 times with similar results.
c. Ribbon diagram of DNA, histones and FACT domains fit into class 1 (top) and class 2 (bottom) electron density maps (80% transparency).
d. Schematic representation of sub-nucleosomal particles and FACT (top row), and of the two complexes in the same orientation as in c).
FACT-nucleosome complex: a ‘unicycle’
We previously identified an intermediate state of FACT-mediated nucleosome assembly3. This complex is biochemically reconstituted by combining 79 base pairs (bp) of the ‘601’ DNA pre-bound to a (H3-H4)2 tetramer (‘tetrasome’), with FACT pre-bound to H2A-H2B (Fig. 1b). During grid preparation for cryo-EM, very few particles escaped being damaged by the water-air interface in conventional blotting and plunge-freezing procedures. Addition of the zwitterionic detergent CHAPS, followed by sample aspiration and rapid freezing using ‘Spotiton’14 resulted in well-dispersed particles (Extended Data Fig. 1a).
Through extensive 3D classification (Extended Data Fig. 1-3, and Extended Data Table 1), we obtained two maps at 4.9 Å (complex 1) and 7.4 Å (complex 2) resolution, which on first inspection differ in the number of H2A-H2B dimers bound to the (H3-H4)2-DNA-FACT complex (Fig. 1c, d). In both structures, histones bind DNA and each other in a manner that is identical to their interactions in a complete nucleosome4, 15. Although full length FACT was used, no density was observed for the N-terminal domain (NTD) of SPT16 and several of the C-terminal domains of SSRP1 (Fig. 1a).
The overall architecture of both structures resembles a unicycle, consisting of a saddle and fork formed by the FACT heterodimer, a wheel (the tetrasome), and one or two pedals (H2A-H2B dimers; Fig. 1c, d, Supplementary video 1). The SPT16 dimerization domain (DD) straddles nucleosomal DNA at the dyad, while the SSRP1 DD stacks on top of the SPT16 DD, together forming the saddle. SPT16 and SSRP1 middle domains (MD) project down on either side of the tetrasome wheel and interact with DNA and histones, forming the fork. On the SPT16 side, one H2A-H2B dimer pedal is docked onto the tetrasome through a nucleosomal four-helix bundle arrangement between H2B and H4, while the SPT16 C-terminal domain (CTD) wraps around the exposed H2A-H2B DNA-binding surface. The SSRP1 MD is located on the other side of the tetrasome ‘wheel’. While no H2A-H2B is present on the SSRP1 side in complex 1, clear electron density for a second H2A-H2B dimer is observed in complex 2, comprising 20-30 % of the intact particles (Fig. 1c, lower panel). This second H2A-H2B dimer also is engaged with the tetrasome in the same manner as in a complete nucleosome, despite the absence of DNA, and is in close proximity with the rearranged SSRP1 MD. The presence of two complexes is consistent with biochemical data3.
FACT has an extensive DNA binding surface
The extensive interaction of both FACT subunits with nucleosomal DNA is surprising, as in absence of histones FACT (in its phosphorylated form) does not bind DNA (see below). Approximately 19 bp of DNA are contacted by FACT (Fig. 2a), burying a combined surface area of ~2300 Å2. Elements from SPT16 DD and both MDs contribute to DNA binding. The inner surface of the FACT saddle and fork is positively charged, while the tips of the fork are negatively charged, promoting FACT interactions with DNA and histones in the configuration of the tetrasome (Fig. 2a).
Figure 2: FACT makes extensive contacts with tetrasome DNA.

a. Surface potential of the FACT-sub-nucleosome complex. Coloring: −5 kT/e (red) to +5 kT/e (blue), generated with APBS Tools 2.1 (Pymol 2.3.2). Histones are omitted from middle and right panels.
b. Regions of significant Δ average HDX (from parts c and d) mapped onto complex 1. Significant regions are colored according to domain location (Fig. 1a). Regions with no detectable change are white, regions where no peptides were recovered are grey. Grey arrows: protected regions at the inter-subunit interface. Sub-nucleosome is in wheat. Representative uptake plots are listed in Extended Data Fig. 5
c. Volcano plot comparing average HDX of SPT16 peptides in FACT and FACT bound to sub-nucleosomes at all timepoints. Each time point was collected in triplicate (n=3); the Welch's t-test was one-sided.
d. Volcano plot comparing average HDX of SSRP1 peptides in FACT and FACT bound to sub-nucleosomes at all timepoints. Each time point was collected in triplicate (n=3) and the Welch's t-test was one-sided. For c and d, dotted lines show significance cut-offs of Δ average HDX>0.3 Da and p-value<0.01 from a Welch’s t-test. Insignificant peptides are light grey, while significant peptides are colored according to their domain location and listed.
e. DNA binding of FACT C-terminal domain deletions. 400 nM FACT or deletion constructs were incubated with 100 nM Atto-647 labeled 30 bp DNA and analyzed by native PAGE, visualized by DNA fluorescence. This was repeated two times with similar results, and multiple times with DNA of different lengths.
f. Fluorescence polarization assay of FACT (0 to 2500 nM) with 20 nM Alexa-488 labeled tetrasome. Kd (in nM): full length FACT (magenta) 204.2 ± 19 (R2=0.9723), SPT16ΔC-SSRP1 (blue) 184.8 ± 14 (R2=0.9807), SPT16-SSRP1ΔC (orange) 201.0 ± 50.6 (R2=0.8266) and double ΔC (black) 28.15 ± 1.9 (R2=0.9843). Kd and standard deviations are derived from 4 biological replicates.
The current map does not allow assignment of amino acids in FACT that directly participate in sub-nucleosome binding. To validate our structures, we employed HDX coupled to mass spectrometry, comparing deuterium uptake in FACT and FACT bound to sub-nucleosomes (Extended Data Fig. 4 and Extended Data Fig. 5). Based on volcano plot analysis16, a significant decrease in deuterium uptake (ΔHX>0.3 Da and p-value <0.01) occurs in regions of the DDs and MDs of SPT16 and SSRP1. These primarily map to the inner face of the fork that interfaces directly with DNA and histones (Fig. 2b-d). A decrease in deuterium is also observed at the interface between the SSRP1 MD and both DDs (Fig. 2b, ‘hinge region’), suggesting decreased domain flexibility upon sub-nucleosome binding. These solution-based results are entirely consistent with the structures, and almost no changes in deuterium uptake are observed in regions outside those present in the structure.
Despite the extensive DNA interface, phosphorylated FACT purified from insect cells is unable to bind free DNA (Fig. 2e). We hypothesized that the disordered CTDs of one or both FACT subunits block DNA binding by preventing access to the concave side of the saddle, and that this auto-inhibition could be relieved by H2A-H2B interaction which is known to occur through either CTD12. To test this, we performed DNA-binding assays with FACT C-terminal deletion constructs (Fig. 2e and Extended Data Fig. 6a). Neither SPT16 CTD nor SSRP1 CTD deletion alone had an effect, but deletion of both CTDs simultaneously enabled FACT to shift free DNA on a native gel. DNA interaction was also facilitated (to a lesser degree) when full length FACT was pre-bound to H2A-H2B dimer (Extended Data Fig. 6b). We previously found that FACT does not produce a gel shift with tetrasome3. To test whether this is also due to auto-inhibition by FACT CTDs, we used fluorescence polarization (FP) binding assays. As was the case for DNA binding, deletion of both CTDs resulted in enhanced affinity of FACT to the tetrasome (Fig. 2f).
The SPT16 C-terminal tail shields H2A-H2B
The acidic SPT16 CTD (pI of 4.3) wraps around the DNA binding surface of H2A-H2B (Fig. 1c; Fig. 3a). The precise SPT16 residues involved cannot be identified from the density or the HDX due to low coverage in that region (Extended Data Fig. 4a), but we observe slight yet significant protection of residues 1016-1030 (Extended Data Fig. 4c). H2A-H2B interfaces can be identified by comparing HDX of H2A-H2B that is bound by FACT, to H2A-H2B in the FACT-bound sub-nucleosome (Fig. 3b and Extended Data Fig. 7). Direct comparison with free H2A-H2B is convoluted by the global stabilization of histone-folds upon protein interaction17. Volcano plot analysis shows a significant decrease in deuterium uptake (ΔHDX>0.3 Da and p-value<0.01) in H2A-H2B regions involved in nucleosome formation. In H2B the decrease is localized to its DNA-binding L2 loop and interface with H4, while in H2A it is localized to the docking domain that binds H3 αN and the SPT16 MD (see later) and to the L2 loop (Fig. 3c). Notably, H2A L2 binds the SPT16 CTD rather than DNA, supporting our interpretation that the SPT16 CTD acts the part of DNA12 and might counteract the ‘foreign DNA invasion’ observed in transcription through nucleosomes18,19.
Figure 3: SPT16 interactions with the sub-nucleosome.

a. All visible domains of SPT16 interact extensively with the sub-nucleosome. The SPT16 CTD occupies the H2A-H2B DNA-binding surface.
b. Volcano plot comparing average HDX of H2A-H2B with FACT and H2A-H2B with FACT and tetrasomes at 100 s exchange. Data was collected in triplicate (n=3) and the Welch's t-test was one-sided. Dotted lines show significance cut-offs of Δ average HDX>0.3 Da and p-value<0.01 from a Welch’s t-test. Peptides from H2A are black and peptides from H2B are grey. Significant peptides are listed.
c. Regions of significant Δ average HDX (from part b) mapped onto H2A-H2B from complex 1. Parts of the docking domain are shown in space filling mode. For H2A-H2B, regions with change are yellow (H2A) and red (H2B); regions with no peptide coverage are grey. FACT and tetrasome are shown in wheat.
d. Comparison of SPT16 MD in complex 1, and bound to (H3-H4)2 (4Z2M). Only DNA and SPT16 MD from complex 1 are shown; the two structures were aligned based on the (H3-H4)2 tetramer (RMSD <0.1 Å).
SPT16 has two binding modes on (H3-H4)2
A crystal structure of the SPT16 MD with the (H3-H4)2 tetramer reveals two regions of contact, neither of which is possible in the context of the sub-nucleosome10 Our structures confirm the prediction10 that one region of interaction is occluded by H3 αN and the H2A docking domain, while the other is occupied by nucleosomal DNA. Consequently, the SPT16 MD (and with it the entire FACT complex) moves and rotates substantially by ~ 30 Å and 23° when the substrate transitions from free (H3-H4)2 to tetrasome (Fig. 3d). This frees up space for the H2A docking domain, while the SPT16 MD binds to the nearby DNA (interface 1, Extended Data Fig. 8). It also displaces a segment of FACT that in absence of DNA covers the DNA-binding L1 loop of histone H4. Instead, this FACT region is now near the H4 N-terminal tail and H3 α1 (interface 2, Extended Data Fig. 8). Overall, this massive rearrangement allows SPT16 MD to make interactions with nucleosomal DNA while also forging new interactions with histones, thereby stabilizing a sub-nucleosomal particle. Of note, the previously demonstrated interaction between SPT16 MD and a fused H2A-H2B is not observed 20 (see ref. 12).
SSRP1 can exclude or hold a second H2A-H2B
The most significant difference between our two structures are on the SSRP1 side of the complex: they differ in the absence (complex 1) or presence (complex 2) of a second H2A-H2B dimer, likely representing two stages of the assembly in equilibrium (Fig. 1b, c, d; Fig. 4, and Extended Data Fig. 9). In both structures, the SSRP1 MD (consisting of two Pleckstrin Homology (PH) domains21) interacts with both DDs, and the PH1 domain makes multiple contacts with the DNA (interface 1 in Fig. 4b). Although the relative orientation of PH1 with respect to the DNA is slightly different in the two structures, the same structural elements are likely engaged with DNA (Fig. 4a, b, interface 1 and 1’). In complex 1, the SSRP1 PH2 domain is positioned across the (H3-H4)2 four helix bundle and on top of H3 α2, thereby precluding the placement of the second H2A-H2B and H3 αN in its rightful position in the nucleosome (interface 2 in Fig. 4b). In complex 2, a second H2A-H2B has successfully docked onto the histone core, facilitated by interactions with the PH2 domain of SSRP1 MD (interface 2’ in Fig. 4b). H3 αN and an additional 5 bp DNA (which binds the L1L2 interface of the second H2A-H2B dimer) are clearly visible in the electron density map for complex 2 but absent in complex 1. The relocation of the SSRP1 PH2 domain on the tetrasome surface by 10.9 Å and 9.5° (upon addition of the second H2A-H2B dimer; Fig. 4c), is partially transmitted to the dimerization domains, and also results in a subtle movement of the SPT16 MD (Supplementary video 2).
Figure 4: SSRP1 can assume two different positions on the sub-nucleosome, depending on presence of a second H2A-H2B dimer.

a. Schematic of SSRP1 domain structure
b. Comparison of binding interfaces between SSRP1 and the sub-nucleosome in complex 1 (top panel) and complex 2 (bottom panel). Interfaces are enlarged in insets. The molecular surface of SSRP1 is shown with the main chain trace.
c. SSRP1 MD and DD in complex 1 and 2 undergo substantial rearrangement on the sub-nucleosome surface when superimposing the two complexes via the (H3-H4)2 tetramer. SSRP1 MD in complex 1 would clash with the H2A docking domain.
Implications for FACT mechanism
The structures reported here and previously10, together with HDX, highlight a plasticity of FACT interactions (Supplementary videos 2 and 3) that is likely required for its diverse functions. In the absence of DNA, FACT maintains the tetrameric conformation of (H3-H4)2 during early steps of nucleosome (re)assembly. In this mode, FACT might also bind H2A-H2B dimer through the SPT16 CTD. DNA then binds the tetramer and displaces FACT which assumes its position on top of nucleosomal DNA while forging new contacts with H3-H4. This promotes incorporation of the tethered H2A-H2B by freeing up H3-H4 to engage its H2A docking domain. FACT maintains interactions with the H2A-H2B DNA-binding surface through the SPT16 CTD, thereby stabilizing the sub-nucleosome. A similar interaction between the SPT16 CTD and the H2A-H2B has been observed recently, although no other FACT domains were visible in that structure 22.
Our structures also suggest a potential mechanism for FACT-mediated nucleosome disassembly11,13 through a reversal of these steps. When the DNA at superhelix location −1 is peeled off from H3-H4 by DNA or RNA polymerase18, SPT16 MD protects the newly exposed sites on the histones on the proximal side of the nucleosome, allowing FACT to contact the H3-H4 four helix bundle resulting in the displacement of the H2A docking domain (Supplementary video 3) to facilitate passage of polymerase. Similarly, the SSRP1 MD can also switch between two binding modes to accommodate either addition or destabilization of H2A-H2B on the distal side of the nucleosome. This mechanism is consistent with observations of histone occupancy in an Spt16 or Ssrp1 knockdown in Drosophila cells23. Both processes might be facilitated by the HMG domain of SSRP124 and other domains not visible in our structure, and this is the topic of ongoing investigations.
Methods
FACT/FACT mutant expression and purification
Sf9 cells were infected with AcMNPV to express either the wildtype (WT) human FACT or FACT C-terminal deletions (SPT16ΔC(Δ934-1047)-SSRP1, SPT16-SSRP1ΔC(Δ625-709) or SPT16ΔC(Δ934-1047)-SSRP1ΔC(Δ625-709)). Proteins were purified as previously described3, with minor modifications. A 6xHis tag was fused to the N-terminus of SPT16, and FACT was purified over a 5 ml prepacked HisTrap HP column, followed by a 5 ml prepacked HiTrap Q HP column. The last step was a Superdex 200 10/300 size exclusion column run in 200 mM NaCl, 20 mM Tris-Cl (pH 8.0), 0.01% CHAPS, 0.01% octylglucoside (OG), 5% glycerol, and 1 mM TCEP. Columns were purchased from GE Healthcare.
Complex formation
Recombinant histones (Homo sapiens) were expressed and purified as described27. 1 μM FACT was pre-mixed with equimolar amounts of refolded H2A-H2B for 10 min at room temperature, followed by addition of an equimolar of (H3-H4)2 tetrasome reconstituted onto 79 bp 601 DNA28 in 20 mM Tris-Cl (pH 8.0), 150 mM NaCl, 1 mM EDTA and 1 mM TCEP27. After a 30 min incubation, the complex was visualized by 5% native PAGE.
Binding assays
400 nM FACT (wild type or deletion constructs) were mixed with 100 nM of Atto-647 labeled 30 bp DNA in 20 mM Tris-Cl (pH 8.0), 100 mM NaCl, 1 mM EDTA and 1 mM TCEP. After incubation for 10 min in room temperature, DNA shifting was analyzed by 5% native PAGE.
To measure FACT - tetrasome interactions, varying amounts of FACT (up to 2500 nM) were mixed with 20 nM Alexa-488 labeled H3-H4 tetrasome in 20 mM Tris-Cl (pH 8.0), 0.01% CHAPS, 0.01% NP40, 100 mM NaCl, 1 mM EDTA and 1 mM TCEP. The reaction was incubated in a 384-well microplate for 10 min at room temperature and fluorescence polarization (FP) data (obtained on a CLARIOstar microplate reader from BMG LABTECH) were analyzed by Graphpad Prism. Four replicates were done. We point out that absolute Kd values obtained with labeled tetrasome have to be taken with caution because of known tetrasome heterogeneity between batches. Nevertheless, comparative values obtained with the same batch are reproducible. Original data are found in two Supplementary Information Tables S1a, b.
Single-Particle Cryo-EM sample vitrification
Reconstituted complexes were concentrated to 7-10 μM by Amicon Ultra −4 centrifugal filter (Ultracel 50K, Millipore). Fresh detergent (CHAPS, 0.25%-0.5%) was added before grid preparation. In order to minimize sample interaction with the air-water interface and to keep the complex intact29, the complex was vitrified using a commercial prototype (from TTP Labtech) of the Spotiton robot14,30,31. The time between sample being spotted onto the nanowire grid32 and vitrification into liquid ethane was 133 ms. Grids were plasma-cleaned (Gatan Solarus) for 5 s using an H2/O2 mixture prior to vitrification. The robot chamber was operated at room temperature and ~80% humidity in the sample and plunging area.
Data acquisition
Images were recorded on a Titan Krios electron microscope (FEI) equipped with a Cs corrector and a K2 summit direct detector with a Quantum energy filter (Gatan) at 1.0961 A per pixel in counting mode using the Leginon software package33. Pixel size was calibrated in-house using a proteasome test sample. Energy filter slit width of 30 eV was used during the collection and aligned automatically every hour using Leginon. The first data collection was performed using a dose of ~66.37 e-/Å2 across 50 frames (200 msec per frame) at a dose rate of ~8.0 e–/pix/sec, using a set defocus range of −1.9 μm to −2.0 μm. The second data collection was performed using a dose of ~ 63.61 e-/Å2 across 50 frames (200 msec per frame) at a dose rate of ~7.6 e–/pix/sec, using a set defocus range of −1.5 μm to −2.5 μm. 100 μm objective aperture was used. A total of 8,318 micrographs were recorded over two separate data collection sessions using an image beam shift data collection strategy34.
Data processing
Data from the two sessions were processed separately and combined towards the end of the processing pipeline. For the first dataset, movie frames were aligned using MotionCor235 with 5 by 5 patches and B-factor of 100 using the Appion software package36. Micrograph CTF estimation was performed using CTFFind437. DoG picker38, a template free particle picking algorithm, was used to pick 744,552 particles (240 box size binned by 2) that were extracted in Relion 3.039-41 and transferred into CryoSPARC 242 for 2D classification. 2D class averages that were clearly contaminated or showed no features were discarded. CryoSPARC ab initio using 2 classes was then run iteratively for 5 rounds, keeping particles in the better class for the next iteration, producing a final stack of 17,369 particles. These particles were processed using non-uniform refinement to produce a map of around 6 Å resolution. The Euler angles and shifts were used to re-extract the particles in Relion 3.0, followed by CTF refinement and Bayesian polishing43. The particles were brought back into CryoSPARC 2 for non-uniform refinement. Improved Euler angles and shifts were used to re-extract the particles in Relion 3.0 for two more rounds of Bayesian polishing. The final non-uniform refinement of 17,369 particles (extracted using 256 box size binned by 1.28 to 200 box size) resulted in a map at 4.4 Å resolution based on the FSC 0.143 criterion44,45.
For the second dataset, movie frames were aligned using patch motion in CryoSPARC 2. CTF was estimated in a patch manner and particles were picked with the CryoSPARC 2 blob picker. A total of 522,050 picked particles were then extracted (256 box size binned by 2) and 2D classification was performed. 4 rounds of iterative CryoSPARC ab initio using 3 classes was used to clean up the dataset – the particles in the best class after each round was carried forward to the next iteration. The remaining 8,399 particles were refined to around 8 Å resolution using non-uniform refinement. The Euler angles and shifts were then used to re-extract the particles in Relion 3.0, followed by CTF refinement and Bayesian polishing43. The particles were brought back into CryoSPARC 2 for non-uniform refinement. Improved Euler angles and shifts were then used to re-extract the particles in Relion 3.0 for two more rounds of Bayesian polishing. The final non-uniform refinement of 8,399 particles (extracted using 256 box size binned by 1.28 to 200 box size) resulted in a map at 7.4 Å resolution based on the FSC 0.143 criterion44,45.
Heterogeneous refinement was performed using 4 classes for these particles. Two distinct classes were classified – differing by the presence of absence of one of the histone dimers. Class 1, with 10,317 particles and absent the second histone H2A-H2B dimer, resulted in a map at 6.6 Å resolution using non-uniform refinement; Class 2, with 4,212 particles and the second H2A-H2B dimer resulted in a map at 7.3 Å resolution using non-uniform refinement.
Both datasets were then combined in CryoSPARC 2.0 and a 3-class ab initio run was performed to sort out heterogeneity in the dataset. Two distinct classes were classified out – differing by the presence of absence of one of the histone dimers. Class 1, with 16,317 particles and absent the second histone H2A-H2B dimer, resulted in a map at 4.9 Å resolution using non-uniform refinement; Class 2, with 6,990 particles and the second H2A-H2B dimer resulted in a map at 7.4 Å resolution using non-uniform refinement. The locally sharpened maps from CryoSPARC 2.0 were used for subsequent model building and analysis. 3DFSC and sphericity of the maps were calculated using the 3DFSC server46.
All conversions between Relion and CryoSPARC were performed using Daniel Asarnow’s pyem script (unpublished, https://github.com/asarnow/pyem).
Model building and refinement
The initial models were built by fitting published high resolution crystal structures, or homology models into the final 3D electron maps in UCSF Chimera26. The crystal structures (Protein data bank ID) used for initial modeling are: 3LZ0 (‘601 Widom DNA), 3AFA (human histone core), 4Z2M (SPT16 MD) and 4IFS (SSRP1 MD). The initial model for DDs was a combination of a crystal structure (5UMR, SSRP1; amino acid 1-100) and a homology model based on the crystal structure of the Chaetomium thermophilum Spt16 Pob3N heterodimer (4KHB) using SWISS-MODEL47. The initial models were then manually edited by COOT48. Poly-UNK was used to fit the visible density without assignment of amino acids. The models were refined by real-space refinement in PHENIX49.
HDX-MS
7 μM H2A-H2B, FACT, FACT-H2A-H2B and FACT-sub-nucleosome were prepared in 20 mM Tris-HCl, 150 mM NaCl, 1 mM TCEP, pH 7.5 at 25°C. Samples were diluted 1:6 with the same buffer containing H2O for controls (pHread=7.5 at 25°C) or D2O (final D2O of 83% (v/v) with pHread=7.1 at 25°C). After 10 s, 102 s, 103 s, and 104 s, samples were mixed 1:1 with 1% Formic Acid, 3.84 M Guanidinium Chloride, pH 1.75 to give a final pH of 2.3 at 0 °C, and flash frozen in liquid nitrogen for storage at −80°C. Samples were thawed and LC-MS performed using a Waters HDX manger and SYNAPT G2-Si Q-Tof. Three technical replicates of each sample were analyzed in a random order. Samples were digested on-line by Sus scrofa Pepsin A (Waters Enzymate BEH) at 15°C and peptides trapped on a C4 pre-column (Waters Acquity UPLC Protein BEH C4) for 3 min at 100 μL/min and 1°C. The LC buffer was 0.1% Formic Acid. Peptides were separated over a C18 column (Waters Acquity UPLC BEH) and eluted with a linear 3-40% (v/v) Acetonitrile gradient for 7 min at 40 uL/min and 1 °C. Raw data of control samples were processed by PLGS (Waters Protein Lynx Global Server 3.0.2) using a database containing Sus scrofa Pepsin A and Homo sapiens SSRP1, SPT16, H2A and H2B. In PLGS, the minimum fragment ion matches per peptide was 3, and methionine oxidation and serine/threonine/tyrosine phosphorylation were allowed. The low and elevated energy thresholds were 250 and 50 counts respectively, and overall intensity threshold was 750 counts. Labelled data were analyzed in DynamX 3.0 with 0.3 products per amino acid and 1 consecutive product.
All mass spectrometry data were acquired using positive ion mode in either HDMS or HDMSE modes. HDMSE mode was used to collect both low (6V) and high (ramping 22-44 V) energy data-independent peptide fragmentation data for peptide identification. HDMS mode was used to collect low energy ion data for all deuterated samples. All samples were acquired in resolution mode. Capillary voltage was set to 2.8 kV for the sample sprayer. Desolvation gas was set to 650 L/hour at 175 °C. The source temperature was set to 80°C. Cone and nebulizer gas was flowed at 90 L/hour and 6.5 bar, respectively. The sampling cone and source offset were both set to 30 V. Data were acquired at a scan time of 0.4 s with a range of 100-2000 m/z. Mass correction was done using [Glu1]-fibrinopeptide B as a reference mass. To allow access to the HDX data of this study, the HDX data summary table (Supplementary Information Table S2) and the HDX data table (Supplementary Information Table S3) are included. This is per consensus guidelines50.
Extended Data
Extended Data Figure 1: EM data analysis.

a. Representative micrograph. Inset is a lower magnification view showing the holy substrate. The particles in the final picking were highlighted by the green circles. This experiment was repeated with 6 independent grids in two Krios sessions, with similar results.
b. 2D class averages generated from the dataset obtained from CryoSPARC 2.0.
c. Data processing strategy flow chart.
Extended Data Figure 2: EM data validation.
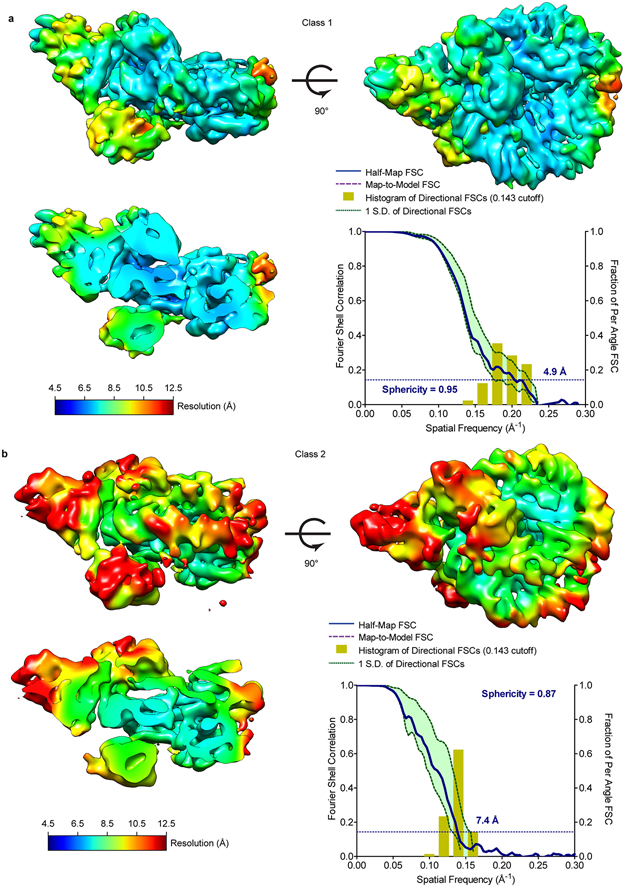
a. Local resolution map and FSC curve of class 1 (complex 1). Local resolution was calculated in CryoSPARC 2.0 and 3DFSC was calculated using 3DFSC server.
b. Local resolution map and FSC curve of class 2 (complex 2). Local resolution was calculated as in a.
Extended Data Figure 3: Electron density and models.

a. Density maps of class 1 (top panel) and class 2 (bottom panel) in three orthogonal orientations. Color coding of subunits and domains as indicated.
b. Representative views of model fit into electron density maps for FACT-sub-nucleosome complex 1, DNA, H2A-H2B, (H3-H4)2, SPT16 MD, SSRP1 MD and SPT16-SSRP1 DDs.
Extended Data Figure 4: Coverage maps of FACT HDX experiments.

a. and b. SPT16 and SSRP1 coverage maps from HDX experiments. Each rectangle is one peptide repeatably monitored through all time points and samples. Significant peptides (see Fig. 2b) are colored and listed. Secondary structure elements from the structures of the published domains that were used for fitting are shown.
c. Deuterium uptake plot for SPT16 residues 1016-1030 showing FACT (black) and FACT with sub-nucleosomes (red). Each data point is the average of three replicates with error bars of ±2 standard deviations.
Extended Data Figure 5: Changes in deuterium uptake mapped onto complex 1.

Deuterium uptake plots for example peptides that show a significant change (see Fig. 2c-d) when comparing FACT (black) and FACT with sub-nucleosomes (red). Each data point is the average of three replicates with error bars of ±2 standard deviations. The sub-nucleosome is shown in wheat. For FACT, regions with no detectable change are white, regions with change are colored according to their domain location (see Fig. 1a), and regions with no peptide coverage are grey.
Extended Data Figure 6: FACT deletion mutants in DNA binding.
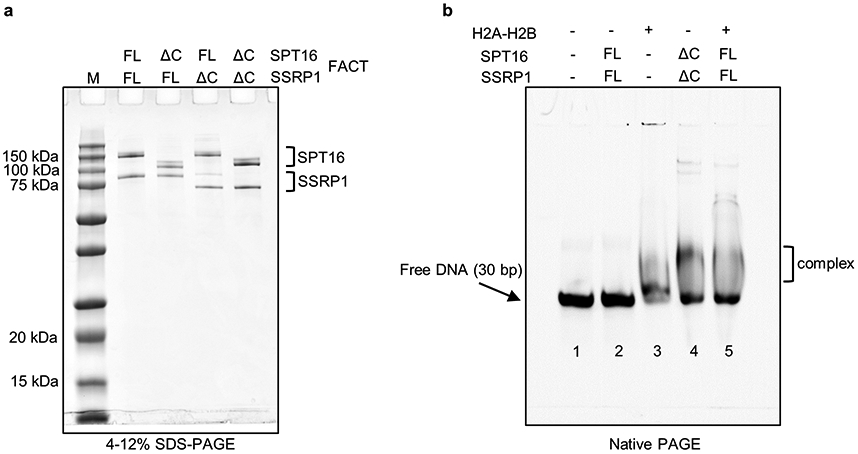
a. FACT and FACT C-terminal domain deletions SPT16ΔC-SSRP1, SPT16-SSRP1ΔC or SPT16ΔC-SSRP1ΔC were purified from Sf9 cells and visualized on 4-12% SDS-PAGE and stained by Blazin Blue. This particular gel was only run once, although proteins were purified and analyzed by SDS-PAGE four times.
b. As in Fig. 2e, but full-length FACT was pre-incubated with an equimolar amount of H2A-H2B dimer at 400 nM. The pre-incubation of wild type FACT with H2A-H2B dimer has a similar effect on DNA binding as FACT C-terminal deletions. Visualized by Atto N (647). The experiment was done twice with similar results.
Extended Data Figure 7: Coverage Maps of H2A-H2B HDX experiments.

a. and b. H2A and H2B coverage maps from HDX experiments. Each rectangle is one peptide repeatably monitored through all time points and samples. Significant peptides (see Fig. 3b) are colored and listed. Secondary structure elements are shown.
c. Deuterium uptake plots for example peptides that show a significant change (see Fig. 3c) when comparing H2A-H2B with FACT (black) and H2A-H2B with FACT and tetrasomes (red). Each data point is the average of three replicates with error bars of ±2 standard deviations. FACT, H3-H4 and DNA are wheat. For H2A-H2B, regions with no detectable change are white, regions with change are yellow for H2A and red for H2B, and regions with no peptide coverage are grey.
Extended Data Figure 8: Interactions between H3-H4 and SPT16 MD (4Z2M) are incompatible with FACT interactions made with the sub-nucleosome.

a. Schematic of SPT16 domain structure, and cartoon of overall complex architecture
b. Interaction between H3-H4 and SPT16 MD (4Z2M) are incompatible with FACT interactions made with the sub-nucleosome. Complex 1 showing only the SPT16 MD. Two sites of interaction with the sub-nucleosome (interface 1: near the H2A docking domain and nearby DNA; interface 2: near H4 N-tail and DNA) are indicated by boxes.
c. The presence of DNA and the H2A-H2B dimer completely occludes interactions between the SPT16 MD and the (H3-H4)2 tetramer in 4Z2M (clash 1 and clash 2, indicated by red arrows).
Extended Data Figure 9: Model-to-map fitting for class 2 map of SSRP1 MD.

a. Class 1 model of SSRP1 MD does not fit into class 2 map. The contour level of the map is 0.153. Left panel shows entire map. The orange oval highlights how class 1 model overlaps with the density assigned to H2A-H2B. Right panel: close-up of the fit between model 1 and class 2 density for the SSRP1 MD. Arrows highlight problematic fits; a significant part of PH2 is outside the density, while other parts of the density are empty. The correlation between model 1 (from class 1) and map 2 for the entire SSRP1 MD is 0.84 (calculated by UCSF Chimera).
b. Class 2 model of SSRP1 MD fits into class 2 density map. The contour level of the map is 0.153 as indicated. Left panel shows entire map; no clashes are identified. Right panel: close-up of the fit between model 2 and density for SSRP1 MD (map 2). Arrows indicate the same parts of the model or map as in a. The correlation between model 2 (from class 2) and the map 2 for the entire SSRP1 MD is 0.93 (calculated by UCSF Chimera).
c. Local resolution heat map for class 2 map. The resolution of the highlighted region (PH2 of SSRP1 MD at interface 2) is between 9-10 Å. The contour level is 0.177
d. The relative movement of highlighted region (PH2 of SSRP1 MD at interface 2) between models of class 1 and class 2. The distances/movements of the identical amino acids in the two models indicated in the highlighted region are indicated.
Extended Table 1: Summary of cryoEM data collection and refinement.
To obtain the best model-map fit, ADP real space refinement was applied in PHENIX. The B factors increase significantly during ADP real-space refinement. These numbers do not represent the physical meaning of B factors as them in crystallography25.
| Class 1 (EMDB-20840 (PDB 6UPK) |
Class 2 (EMDB-20841) (PDB 6UPL) |
||
|---|---|---|---|
| Data collection and processing | |||
| Magnification | 105,000 | ||
| Voltage (kV) | 300 | ||
| Electron exposure (e-/Å2) | 70 | ||
| Defocus range (μm) | 1.5-2.5 | ||
| Pixel size (Å) | 1.0961 | ||
| Symmetry imposed | C1 | C1 | |
| Initial particle images (no.) | 162,798 | ||
| Final particle images (no.) | 16,784 | 4212 | |
| Map resolution (Å) | 4.9 | 7.4 | |
| FSC threshold | 0.143 | ||
| Map resolution range (Å) | 4.9-12.3 | 6.5-20.0 | |
| Directional resolution range (Å) | 4.4-6.7 | 6.2-9.4 | |
| Sphericity of 3DFSC | 0.95 | 0.87 | |
| Refinement | |||
| Initial model used (PDB code) | 3LZ0,3AFA,4Z2M,4IFS | ||
| Model resolution (Å) | 5.25 | 7.3 | |
| FSC threshold | 0.143 | 0.143 | |
| Model resolution range (Å) | 4.8-7.1 | 6.6-7.9 | |
| Map sharpening B factor (Å2) | 151.0 | 382.7 | |
| Model composition | |||
| Non-hydrogen atoms | 13861 | 15882 | |
| Protein residues | 1368 | 1589 | |
| Nucleotide | 144 | 158 | |
| B factors (Å2)* | |||
| Protein | 524 | 408 | |
| Nucleotide | 470 | 411 | |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.006 | 0.005 | |
| Bond angles (°) | 1.101 | 1.090 | |
| Validation | |||
| MolProbity score | 2.26 | 2.25 | |
| Clashscore | 20.32 | 21.65 | |
| Poor rotamers (%) | 0.00 | 0.23 | |
| Ramachandran plot | |||
| Favored (%) | 92.85 | 93.55 | |
| Allowed (%) | 7.15 | 6.32 | |
| Disallowed (%) | 0.00 | 0.13 | |
Supplementary Material
Acknowledgements
Supported by the Howard Hughes Medical Institute (YL, KZ, KL), and by startup funds to S.D. Screening of initial cryo-EM condition was performed at CU Boulder Electron Microscopy Service, with help from C. Page and G.P. Morgan. Some of this work was performed at the Simons Electron Microscopy Center and National Resource for Automated Molecular Microscopy located at the New York Structural Biology Center, supported by grants from the Simons Foundation (SF349247) and the NIH National Institute of General Medical Sciences (GM103310) with additional support from Agouron Institute (F00316) and NIH (OD019994). We thank D. Reinberg for assistance in initial stages of the project.
Footnotes
Competing interest statement:
B.C and S.C.P. have an IP licensing arrangement with TTP Labtech Ltd. for the Chameleon sample vitrification instrument.
Data availability statement
HDX-MS raw data is available in SI Tables 2 and 3. Atomic coordinates and cryo-EM maps have been deposited in the Protein Data Bank (PDB) and Electron Microscopy Data Bank (EMDB) under accession codes EMDB-20840; PDB 6UPK and EMDB-20841; PDB 6UPL, according to policies.
References
- 1.Orphanides G, LeRoy G, Chang CH, Luse DS & Reinberg D FACT, a factor that facilitates transcript elongation through nucleosomes. Cell 92, 105–116 (1998). [DOI] [PubMed] [Google Scholar]
- 2.Gurova K, Chang HW, Valieva ME, Sandlesh P & Studitsky VM Structure and function of the histone chaperone FACT - Resolving FACTual issues. Biochim Biophys Acta Gene Regul Mech, doi: 10.1016/j.bbagrm.2018.07.008 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang T et al. The histone chaperone FACT modulates nucleosome structure by tethering its components. Life Sci Alliance 1, e201800107, doi: 10.26508/lsa.201800107 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luger K, Mader AW, Richmond RK, Sargent DF & Richmond TJ Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389, 251–260, doi: 10.1038/38444 (1997). [DOI] [PubMed] [Google Scholar]
- 5.Lai WKM & Pugh BF Understanding nucleosome dynamics and their links to gene expression and DNA replication. Nat Rev Mol Cell Biol 18, 548–562, doi: 10.1038/nrm.2017.47 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Das C, Tyler JK & Churchill ME The histone shuffle: histone chaperones in an energetic dance. Trends Biochem Sci 35, 476–489, doi: 10.1016/j.tibs.2010.04.001 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belotserkovskaya R et al. FACT facilitates transcription-dependent nucleosome alteration. Science 301, 1090–1093 (2003). [DOI] [PubMed] [Google Scholar]
- 8.Hsieh FK et al. Histone chaperone FACT action during transcription through chromatin by RNA polymerase II. Proc Natl Acad Sci U S A 110, 7654–7659, doi: 10.1073/pnas.1222198110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Winkler DD, Muthurajan UM, Hieb AR & Luger K Histone chaperone FACT coordinates nucleosome interaction through multiple synergistic binding events. J Biol Chem 286, 41883–41892, doi: 10.1074/jbc.M111.301465 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsunaka Y, Fujiwara Y, Oyama T, Hirose S & Morikawa K Integrated molecular mechanism directing nucleosome reorganization by human FACT. Genes Dev 30, 673–686, doi: 10.1101/gad.274183.115 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Valieva ME et al. Large-scale ATP-independent nucleosome unfolding by a histone chaperone. Nat Struct Mol Biol, doi: 10.1038/nsmb.3321 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kemble DJ, McCullough LL, Whitby FG, Formosa T & Hill CP FACT Disrupts Nucleosome Structure by Binding H2A-H2B with Conserved Peptide Motifs. Mol Cell 60, 294–306, doi: 10.1016/j.molcel.2015.09.008 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen P et al. Functions of FACT in Breaking the Nucleosome and Maintaining Its Integrity at the Single-Nucleosome Level. Mol Cell 71, 284–293 e284, doi: 10.1016/j.molcel.2018.06.020 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Dandey VP et al. Spotiton: New features and applications. J Struct Biol 202, 161–169, doi: 10.1016/j.jsb.2018.01.002 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tachiwana H et al. Structural basis of instability of the nucleosome containing a testis-specific histone variant, human H3T. Proc Natl Acad Sci U S A 107, 10454–10459, doi: 10.1073/pnas.1003064107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hageman TS & Weis DD Reliable Identification of Significant Differences in Differential Hydrogen Exchange-Mass Spectrometry Measurements Using a Hybrid Significance Testing Approach. Anal Chem 91, 8008–8016, doi: 10.1021/acs.analchem.9b01325 (2019). [DOI] [PubMed] [Google Scholar]
- 17.D'Arcy S et al. Chaperone Nap1 Shields Histone Surfaces Used in a Nucleosome and Can Put H2A-H2B in an Unconventional Tetrameric Form. Mol Cell 51, 662–677, doi: 10.1016/j.molcel.2013.07.015 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kujirai T et al. Structural basis of the nucleosome transition during RNA polymerase II passage. Science 362, 595–598, doi: 10.1126/science.aau9904 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Ehara H et al. Structural insight into nucleosome transcription by RNA polymerase II with elongation factors. Science 363, 744–747, doi: 10.1126/science.aav8912 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Hondele M et al. Structural basis of histone H2A-H2B recognition by the essential chaperone FACT. Nature 499, 111–114, doi: 10.1038/nature12242 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Zhang W et al. Crystal Structure of Human SSRP1 Middle Domain Reveals a Role in DNA Binding. Sci Rep 5, 18688, doi: 10.1038/srep18688 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mayanagi K et al. Structural visualization of key steps in nucleosome reorganization by human FACT. Sci Rep 9, 10183, doi: 10.1038/s41598-019-46617-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramachandran S, Ahmad K & Henikoff S Transcription and Remodeling Produce Asymmetrically Unwrapped Nucleosomal Intermediates. Mol Cell 68, 1038–1053 e1034, doi: 10.1016/j.molcel.2017.11.015 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCullough LL et al. Functional roles of the DNA-binding HMGB domain in the histone chaperone FACT in nucleosome reorganization. J Biol Chem, doi: 10.1074/jbc.RA117.000199 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wlodawer A, Li M & Dauter Z High-Resolution Cryo-EM Maps and Models: A Crystallographer's Perspective. Structure 25, 1589–1597 e1581, doi: 10.1016/j.str.2017.07.012 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pettersen EF et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612, doi: 10.1002/jcc.20084 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Dyer PN et al. Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol 375, 23–44 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Thastrom A et al. Sequence motifs and free energies of selected natural and non-natural nucleosome positioning DNA sequences. J Mol Biol 288, 213–229. (1999). [DOI] [PubMed] [Google Scholar]
- 29.Noble AJ et al. Routine single particle CryoEM sample and grid characterization by tomography. Elife 7, doi: 10.7554/eLife.34257 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Razinkov I et al. A new method for vitrifying samples for cryoEM. J Struct Biol 195, 190–198, doi: 10.1016/j.jsb.2016.06.001 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jain T, Sheehan P, Crum J, Carragher B & Potter CS Spotiton: a prototype for an integrated inkjet dispense and vitrification system for cryo-TEM. J Struct Biol 179, 68–75, doi: 10.1016/j.jsb.2012.04.020 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wei H et al. Optimizing "self-wicking" nanowire grids. J Struct Biol 202, 170–174, doi: 10.1016/j.jsb.2018.01.001 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suloway C et al. Automated molecular microscopy: the new Leginon system. J Struct Biol 151, 41–60, doi: 10.1016/j.jsb.2005.03.010 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Cheng A et al. High resolution single particle cryo-electron microscopy using beam-image shift. J Struct Biol 204, 270–275, doi: 10.1016/j.jsb.2018.07.015 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng SQ et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14, 331–332, doi: 10.1038/nmeth.4193 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lander GC et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J Struct Biol 166, 95–102 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rohou A & Grigorieff N CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol 192, 216–221, doi: 10.1016/j.jsb.2015.08.008 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Voss NR, Yoshioka CK, Radermacher M, Potter CS & Carragher B DoG Picker and TiltPicker: software tools to facilitate particle selection in single particle electron microscopy. J Struct Biol 166, 205–213 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scheres SH RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530, doi: 10.1016/j.jsb.2012.09.006 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kimanius D, Forsberg BO, Scheres SH & Lindahl E Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. Elife 5, doi: 10.7554/eLife.18722 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zivanov J et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, doi: 10.7554/eLife.42166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Punjani A, Rubinstein JL, Fleet DJ & Brubaker MA cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290–296, doi: 10.1038/nmeth.4169 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Zivanov J, Nakane T & Scheres SHW A Bayesian approach to beam-induced motion correction in cryo-EM single-particle analysis. IUCrJ 6, 5–17, doi: 10.1107/S205225251801463X (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rosenthal PB & Henderson R Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol 333, 721–745 (2003). [DOI] [PubMed] [Google Scholar]
- 45.Scheres SH & Chen S Prevention of overfitting in cryo-EM structure determination. Nat Methods 9, 853–854, doi: 10.1038/nmeth.2115 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tan YZ et al. Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nat Methods 14, 793–796, doi: 10.1038/nmeth.4347 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Waterhouse A et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46, W296–W303, doi: 10.1093/nar/gky427 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501, doi: 10.1107/S0907444910007493 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221, doi: 10.1107/S0907444909052925 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Masson GR et al. Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments. Nat Methods 16, 595–602, doi: 10.1038/s41592-019-0459-y (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.