

Abstract
Background
Insulin shares a limited physiological concentration range with other endocrine hormones. Not only too low, but also too high systemic insulin levels are detrimental for body functions.
Main body
The physiological function and clinical relevance of insulin are usually seen in association with its role in maintaining glucose homeostasis. However, insulin is an anabolic hormone which stimulates a large number of cellular responses. Not only too low, but also excess insulin concentrations are detrimental to the physiological balance. Although the glucoregulatory activity of insulin is mitigated during hyperinsulinemia by dampening the efficiency of insulin signaling (“insulin resistance”), this is not the case for most other hormonal actions of insulin, including the promotion of protein synthesis, de novo lipogenesis, and cell proliferation; the inhibition of lipolysis, of autophagy-dependent cellular turnover, and of nuclear factor E2-related factor-2 (Nrf2)-dependent antioxidative; and other defense mechanisms. Hence, there is no general insulin resistance but selective impairment of insulin signaling which causes less glucose uptake from the blood and reduced activation of endothelial NO synthase (eNOS). Because of the largely unrestricted insulin signaling, hyperinsulinemia increases the risk of obesity, type 2 diabetes, and cardiovascular disease and decreases health span and life expectancy. In epidemiological studies, high-dose insulin therapy is associated with an increased risk of cardiovascular disease. Randomized controlled trials of insulin treatment did not observe any effect on disease risk, but these trials only studied low insulin doses up to 40 IU/day. Proof for a causal link between elevated insulin levels and cardiovascular disease risk comes from Mendelian randomization studies comparing individuals with genetically controlled low or high insulin production.
Conclusions
The detrimental actions of prolonged high insulin concentrations, seen also in cell culture, argue in favor of a lifestyle that limits circadian insulin levels. The health risks associated with hyperinsulinemia may have implications for treatment regimens used in type 2 diabetes.
Keywords: Hyperinsulinemia, Insulin resistance, Nrf2, Autophagy, eNOS, Obesity, Type 2 diabetes mellitus, Inflammation, Oxidative stress, Cardiovascular morbidity and mortality
Background
Most endocrine hormones exhibit a window of optimal physiological concentrations, with compromised function of the organism at levels below or above that range. For instance, subnormal levels of thyroid hormone define the clinical condition of hypothyroidism, above normal levels represent hyperthyroidism which usually requires therapy. Addison’s disease is characterized by insufficient cortisol production, while excess synthesis is seen in Cushing syndrome.
For insulin, we argue here that not only hypoinsulinemia but also hyperinsulinemia is detrimental to body functions. Hypoinsulinemia causes insulin-deficient diabetes, and the hormonal actions of insulin are necessary for the life of complex organisms [1]. On the other hand, permanently elevated levels of insulin may cause disturbance of normal cellular physiology and organ function. We describe the molecular basis of these undesired insulin actions and consequences of hyperinsulinemia for health-relevant endpoints, such as obesity or cardiovascular diseases.
Main text
Insulin signaling pathways
Binding of insulin to its cognate cell surface-bound receptor causes a conformational change which initiates a cascade of signaling events. Autophosphorylation by the insulin receptor tyrosine kinase is accompanied by tyrosine phosphorylation of receptor substrates, such as insulin receptor substrate (IRS) and Src homology 2 domain-containing transforming proteins (SHC) proteins. Phosphorylation of IRS allows binding of phosphatidylinositol-3-kinase (PI3K) and synthesis of phosphatidylinositol (3,4,5)-trisphosphate (PIP3), which eventually leads to the phosphorylation and activation of the serine/threonine-specific protein kinase B (AKT). Upon activation, AKT interacts with several substrates which mediate anabolic effects of insulin; these include glucose uptake, glycogen synthesis, de novo lipogenesis, and protein synthesis [2]. Additional pathways triggered by the activated insulin receptor comprise phosphorylation of SHC, followed by activation of the Rat sarcoma (Ras)–rapidly accelerated fibrosarcoma (Raf)–mitogen-activated protein kinase kinase (MEK)–extracellular signal-regulated kinase (ERK) pathway. The terminal kinase ERK is a mitogen-activated kinase promoting cell proliferation and further cellular activities including protein synthesis [3]. Another pathway triggered by the engaged insulin receptor involves activation of NADPH oxidase 4 and subsequent hydrogen peroxide-mediated inhibition of phosphatase and tensin homolog (PTEN), which is an important negative regulator of PI3K signaling [4] (Fig. 1).
Fig. 1.
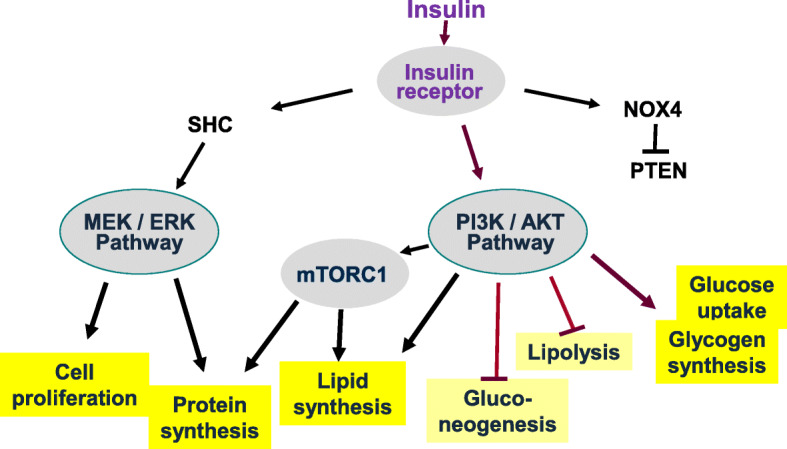
Metabolic signaling of insulin is anabolic. Insulin signaling through the insulin receptor engages several pathways and results in an anabolic state of metabolism. The canonical pathway via phosphokinases PI3K and AKT/PKB promotes glucose uptake and glycogen and lipid syntheses, whereas lipolysis is inhibited in adipocytes, as well as hepatic gluconeogenesis. In addition, AKT kinases activate mTORC1 which supports de novo lipogenesis and protein synthesis. The insulin signaling pathway via SHC and the MAP kinases MEK and ERK promotes cell proliferation and protein synthesis. Another insulin signaling pathway involves NOX4 and the inhibition of PTEN, an inhibitor of the PI3K-AKT pathway
Insulin secretion
Insulin secretion by pancreatic islet β cells responds to the level of circulating nutrients such as glucose, amino acids, and free fatty acids. Sweeteners may further increase carbohydrate-induced insulin secretion. A large number of endogenous factors contribute to the regulation of β cell activity, either stimulatory, inhibitory, or both context-dependent. These include hormones, neurotransmitters, and immune mediators [5–12]. Insulin is essential for maintaining glucose homeostasis, primarily by facilitating the post-meal uptake of glucose into muscle and fat cells via translocation of the glucose transporter 4 [13]. In the absence of dietary glucose supply and after depletion of glycogen stores, glucose in circulation primarily comes from gluconeogenesis in the liver. If circulating insulin levels are below the concentrations required for stimulating glucose uptake from the blood, endogenous stores of fat and protein must be used for energy production. For the maintenance of life in the fasting state, circulating insulin levels range between approx. 25 and 70 pmol/l (25–75% percentile), as determined for healthy adult persons in the National Health and Nutrition Examination Survey (NHANES) [14]. In response to meals with varying carbohydrate content, insulin levels may rise to the range of approx. 300–800 pmol/l [15].
Insulin promotes obesity
Almost 100 years ago, insulin injections were one of the options of therapy in nondiabetic persons suffering from undernutrition in the context of various diseases. Insulin doses were in the range of those applied in type 1 diabetes and led to increased appetite and weight gain [16]. Indeed, one major function of insulin as an anabolic hormone is to favor energy storage over usage. This is reflected by the finding that insulin infusion (1 mU/kg/min) significantly inhibits lipolysis in the skeletal muscle (about 43%) and even more effective in adipose tissue (about 75%) [17]. Doubling fasting insulin levels suffices to inhibit lipolysis by approx. 50% and to promote lipogenesis (for both, mean insulin concentration for 50% effect (EC50) of approx. 80 pmol/l) [18]. At this insulin level, gluconeogenesis is still ongoing. For half-maximal inhibition of gluconeogenesis, insulin concentrations must rise to approx. 160 pmol/l in arterial circulation. In order to stimulate glucose uptake to half maximum, insulin levels must rise to even higher levels, approx. ten times the fasting insulin concentrations (25–75% percentiles for stimulating glucose uptake approx. 350–480 pmol/l) [18]. Thus, a modest rise (doubling) of fasting insulin levels will already substantially inhibit lipolysis and promote lipogenesis while gluconeogenesis is not yet inhibited. Since such small increases of systemic insulin concentrations are enough for favoring adipogenesis, fasting and diurnal insulin levels are a determinant of obesity risk. Indeed, several data support the obesity-promoting role of insulin (for a detailed review see [18]) (Fig. 2).
Fig. 2.
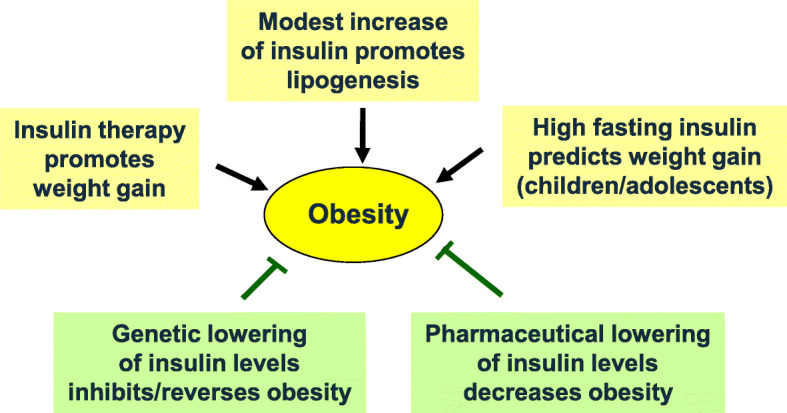
Insulin promotes obesity. Several independent types of observations support the conclusion that insulin promotes adipogenesis and obesity. For details, see description in the general text
These include epidemiological studies, which found high fasting insulin levels (and concomitant insulin resistance) in children and adolescents to be associated with higher weight gain in later years [19]. Studies in adults are less consistent [20]. Pharmaceutical interventions that lower insulin secretion, such as treatment with diazoxide or octreotide, led to significant body weight loss [21–23]. This fits with the observation that insulin therapy promotes weight gain [24]. One probable reason is that insulin levels in the high normal range are close to EC50 concentrations for inhibition of lipolysis [18].
In mice, modest lowering of circulating insulin concentrations by genetic manipulation of insulin genes caused resistance to weight gain despite a high-fat diet [25]. Decreasing insulin gene expression in adult mice via partial gene ablation reversed diet-induced obesity [26]. In men, the Hph1 “T” polymorphism in the insulin gene region was found to be associated with higher fasting insulin levels and a more rapid weight gain in obese persons [27]. A Mendelian randomization analysis showed that persons with genetically determined higher insulin secretion to oral glucose exhibited a higher body mass index (BMI) [28], supporting a causal relationship between insulin and obesity risk.
Taken together, moderate to high normal levels of insulin in metabolic healthy persons appear to be a risk factor for the development of obesity.
Elevated insulin concentrations impair cellular functions—insulin “toxicity”
There is ample evidence that transient increases of metabolic or immune mediator levels are benign physiological responses to biochemical challenges, such as the rise of systemic glucose or cytokines following meals. However, chronic elevations of such mediators, even when modest in amplitude, are usually detrimental to cellular functions [29]. In the case of glucose, the term glucose toxicity was coined to describe this phenomenon [30]. Prolonged conditions of elevated glucose concentrations cause dysfunction of numerous cell types in the body, including beta cells, neurons, and the endothelium, via several pathways, including increased oxidative stress and activation of the sorbitol pathway [31–33]. As described below, there seems to be a similar detrimental outcome of long-term elevated insulin concentrations on cellular functions, a corresponding term would be insulin toxicity.
When cells are exposed to continuously elevated insulin levels, there is a partial downregulation of insulin signaling. The resulting “insulin resistance” is not primarily due to less insulin receptor expression on the cell surface but due to impaired insulin signal transduction as a result of receptor dysfunction. In response to prolonged hyperinsulinemia, there is diminished autophosphorylation of the insulin receptor, compared to that observed after short-term exposure to insulin, and subsequent steps of the PI3K–AKT signaling pathway are affected [34, 35]. Consequently, in muscle and fat cells, there is less AKT-stimulated translocation of GLUT 4 to the cell surface (Fig. 3). Thus, insulin resistance can be seen as a protective mechanism for preventing excess activation of glucose transport from the blood despite chronically elevated insulin levels, for maintaining glucose homeostasis in vivo and for mitigating metabolic and oxidative stress due to excess glucose influx [36–39]. Limiting glucose export from the blood does not necessarily require dampening of insulin signaling. During the early weeks of feeding with a high caloric diet, mice show decreased insulin-dependent glucose uptake despite unperturbed insulin-stimulated AKT phosphorylation [40, 41] (Fig. 3). An interesting aspect is that the partitioning of insulin receptor isoforms A and B and of hybrid insulin/insulin-like growth factor-1 receptors among cell types may contribute to insulin resistance in some tissues, but the pathophysiological relevance is unknown [42].
Fig. 3.
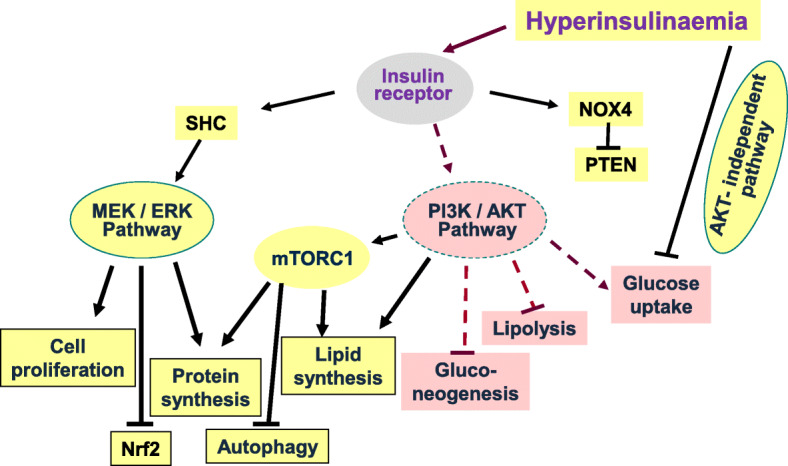
Signaling of insulin during insulin resistance. During insulin resistance, signaling through AKT kinases is partially impaired. Not all AKT-dependent pathways are affected, as well as other signaling pathways, indicating that insulin resistance is selective. Therefore, hyperinsulinemia, in the presence of insulin resistance, promotes anabolic cell activities via the MEK–ERK pathway and via mTORC1. Although the PI3K/AKT pathway is impaired during insulin resistance, and provides only insufficient translocation of GLUT4 for glucose uptake and deficient activation of eNOS, there appears to be a normal activation of mTORC1. In addition to the anabolic consequences of signaling via the MEK/ERK pathway depicted in the figure, there is enhanced expression of ET-1 and PAI-1 (not shown), as well as inhibition of autophagy and of the nuclear factor Nrf2, which compromises cell constituent turnover and cell defense mechanisms to radical stress, respectively. Hyperinsulinemia downregulates glucose uptake not only via dampening the PI3K/AKT pathway (“insulin resistance”) but also via as yet unknown other pathways
The phenomenon of insulin toxicity partly arises from the fact that there are additional cellular responses to elevated insulin levels which are not toned down during insulin resistance (Fig. 3). These comprise the upregulation of protein synthesis and the accumulation of ubiquitinated or otherwise modified proteins, probably due to insufficient degradation of these polypeptides [43]. A major role of insulin signaling via the canonical mitogen-activated protein (MAP) kinase pathway Ras–MEK–ERK, as well as via activation of NADPH oxidase 4, has been observed [4]. Even some AKT-dependent pathways do not appear to be suppressed by insulin resistance, such as de novo lipogenesis in hepatocytes or the upregulation of mechanistic target of rapamycin complex 1 (mTORC1) [44–47]. Enhanced activity of mTORC1 leads to increased protein synthesis and to deteriorated cell functions largely because of suppressed autophagy [48].
Hence, chronic exposure of cells to high ambient insulin concentrations causes an imbalance of cellular responses because of the downregulation of some insulin signaling pathways (“insulin resistance”) but not of others. The resulting functional state of cells is characterized by an unbalanced anabolic activity of insulin favoring protein synthesis while suppressing autophagy. The latter inhibits autophagic removal and turnover of proteins and lipids, which favors cell senescence [49]. In short-term experiments of exposure to high insulin levels, a protective cellular stress response is observed, the unfolded protein response, probably due to the accumulation of derivatized proteins in the absence of enough disposal. In experimentally induced or diabetes-associated chronic insulin resistance (and hyperinsulinemia), such a protective stress response of the endoplasmic reticulum to high insulin levels is diminished or absent [50].
Another activity of insulin is the suppression of transcription of the nuclear factor Nrf2 via induction of heterogeneous ribonucleoproteins F and K [51]. Nrf2 is the central regulator of the protective response of cells against oxidative and other types of electrophile stress [52]. Suppression of Nrf2 expression is expected to impair the antioxidant and cytoprotective defense capacity of cells. Insulin signaling required for Nrf2 inhibition occurs via the MAP kinase pathway and thus is not mitigated by insulin resistance [53] (Fig. 3). It therefore can be assumed that hyperinsulinemia increases the susceptibility of cells against oxidative or other electrophile stress caused by environmental insults. Prolonged exposure of cells to high insulin concentrations can therefore be regarded as toxic. Indeed, exposure to 0.5 nmol/l insulin has been found to cause DNA damage in a number of cell types, including human lymphocytes [42, 54]. At the only concentration tested (100 nmol/l), insulin impairs oxygen radical defense and sensitizes apoptosis pathways in human islets [55]. In the brain of mice, hyperinsulinemia impairs electrophysiological functions of neurons and protein turnover, causing a transition to a senescent cell state and an accompanying cognitive decline [56]. The direct toxic property of insulin deserves further study.
Chronically elevated insulin concentrations impair body functions
Longevity
The above list of detrimental cellular responses to high ambient insulin concentrations suggests concomitant functional impairments at the level of the organism. This fits with the observed impact of insulin on longevity. Studies in nonvertebrate model systems such as the nematode Caenorhabditis elegans or the fruit fly Drosophila melanogaster find that moderate to high insulin activity shortens lifespan [57, 58]. A consistent finding from mouse model studies is that decreased signaling of anabolic hormones like insulin, insulin-like growth factor, or growth hormone results in a prolonged lifespan [59]. Disruption of the insulin-receptor substrate 1 gene caused insulin-resistance with defects in insulin signaling [60] and led to an extension of lifespan by 14–16% [61]. A knockout of the insulin receptor in adipose tissue of mice resulted in an 18% increase of lifespan [62]. Disruption of the Ins1 gene and one of the two mouse Ins2 alleles lowered insulin levels by 25–34% (Ins2+/− mice versus Ins2+/+ controls) in aged female mice without altering circulating insulin-like growth factor (IGF)-1 levels. These aged experimental mice exhibited lower fasting glucose, improved insulin sensitivity, and 3–11% lifespan extension across two different diets [63]. Concomitantly, the proteome and transcriptome indicated a profile associated with healthy aging. An important aspect is that this study selectively addressed insulin. Other interventions for promoting longevity or extending healthspan, such as caloric restriction, not only lower circadian insulin levels; but several additional hormones, including IGF-1, are also affected [64].
Insulin, IGF-1, and hybrid insulin/IGF-1 receptors share signaling via PI3K and AKT. The subsequent activation of the protein kinase mTORC1 is a major pathway for supporting somatic growth, protein synthesis, and fertility, while impeding autophagy and lifespan. Suppression of mTOR signaling by treatment with rapamycin prolongs life in model organisms and mice [65]. In humans, hyperinsulinemia in (pre) type 2 diabetes is associated with increased mTORC1 activity which may have a negative impact on beta cell survival, healthspan, and longevity [66]. In the Leiden Longevity Study, follow-up of nonagenarians for 10 years showed a strong association of low insulin and glucose levels with healthy aging [67].
Since both IGF-1 and insulin employ PI3K and AKT for signal transduction, it is difficult to disentangle the contribution of insulin versus IGF-1 to the modulation of longevity. In animal models, selective downregulation of circulating insulin levels improved the lifespan of mice, and in elderly persons of the Leiden Longevity Study, only insulin and glucose, but not IGF-1, consistently met all four pre-defined criteria of healthy aging [63, 67]. Therefore, it may be concluded that low circulating insulin concentrations are not only a marker of longevity but are causally involved in promoting healthspan or lifespan extension.
Detrimental combination of hyperinsulinemia with insulin resistance
Insulin resistance is defined as an attenuated effect of insulin on blood glucose homeostasis, primarily by less efficient export of glucose from the blood into skeletal muscle, adipose, and liver tissue. Permanently elevated insulin concentrations in the blood are often considered as an attempt to overcome insulin resistance. Indeed, induction of insulin resistance by genetic disruption of insulin signaling, as well as by increased growth hormone levels or an inflammatory milieu, causes hyperinsulinemia [68–70]. The opposite causality is of more relevance. Hyperinsulinemia during insulin infusion in humans leads to systemic insulin resistance [71], while in vitro, high ambient insulin concentrations cause an increase in insulin resistance in isolated adipocytes [72]. A summary analysis of nine studies in rodents and seven trials in humans confirmed that the first detectable change in the fasting state, after feeding a high caloric diet for several days, is an increase of basal insulin concentrations, but not of blood glucose concentrations or insulin resistance [73]. Both increased secretion of insulin by ß cells and decreased insulin clearance in the liver contribute to elevated insulin levels post-meal, the latter being of primary importance in the case of carbohydrate-rich food [74].
The combination of hyperinsulinemia and insulin resistance appears to promote hypertension and atherogenesis (Fig. 4). One important molecule for maintaining vessel function, including relaxation of the arterial smooth muscle layer, is nitric oxide (NO) which is generated by endothelial NO synthase (eNOS). Insulin increases NO production via posttranslational modification of eNOS via PI3K/AKT activity; however, this mechanism is suppressed during insulin resistance [75, 76]. Decreased local NO production impairs arterial smooth muscle relaxation and concomitant vasodilatation. An important factor in this context is the calcium ion homeostasis of vascular smooth muscle cells. Under physiological conditions, insulin promotes both calcium influx into the cytoplasm of smooth muscle cells via several ion channels, including L-type and store-operated Ca2+ channels, and counterregulatory NO-mediated efflux of Ca2+ and K+ ions which prevents calcium ion-induced myosin light chain phosphorylation and concomitant vascular contractility. During insulin resistance, NO production is impaired while the supportive effect of insulin on calcium ion influx (via PI3K delta and possibly the MEK–ERK pathway) and vasoconstriction is still present (Fig. 4) [77, 78].
Fig. 4.
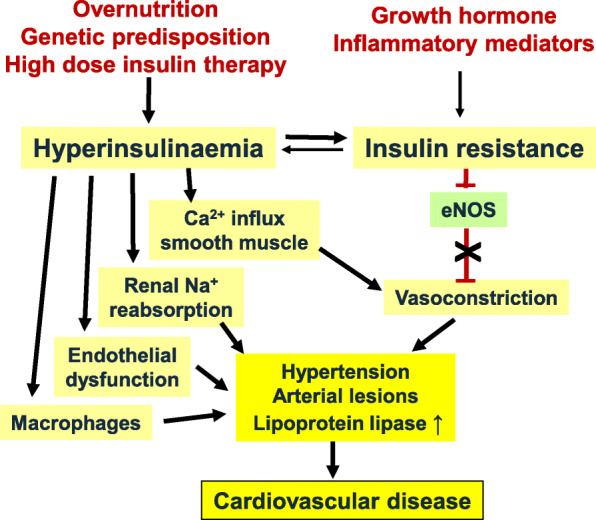
Hyperinsulinemia, insulin resistance, and cardiovascular disease. High insulin concentrations in the blood may occur due to genetic predisposition, overnutrition, or high-dose insulin treatment of type 2 diabetes. Hyperinsulinemia induces “insulin resistance” as a defense response to maintain glucose homeostasis. Conversely, insulin resistance may be directly induced such as by growth hormone or pro-inflammatory cytokines. Hyperinsulinemia and insulin resistance enhance the risk of cardiovascular disease, by inducing endothelial dysfunction, suppression of endothelial nitric oxide synthase (eNOS), and activation and promotion of calcium ion influx into smooth muscle cells, resulting in increased vascular tone, enhanced reabsorption of sodium ions in renal tubules, adhesion of macrophages to the vessel wall, and development of arterial lesions with increased lipoprotein lipase activity and cardiovascular disease
At the same time, insulin signals through the mitogen-activated protein (MAP) kinase pathway to upregulate the expression of endothelin-1 (ET-1), plasminogen activator inhibitor-1 (PAI-1), adhesion molecules, and pro-inflammatory cytokines [79, 80]. The renin-angiotensin system is activated in the context of endothelial dysfunction and contributes together with decreased NO production and increased ET-1 secretion to vascular stiffening and upregulation of vascular tone [81–83]. In the absence of hyperinsulinemia/insulin resistance, the lower insulin levels exert less potential proatherogenic activities which are counteracted by insulin-stimulated local NO production [83, 84].
Elevated insulin levels also increase the risk of hypertension by enhancing renal reabsorption of sodium ions by several transport systems in different segments of the nephron (Fig. 4). Signaling of insulin occurs via insulin receptor substrate 2 (IRS2) and is not suppressed during insulin resistance, while signaling via IRS1 for counterregulatory mechanisms, including local NO production, is impaired [85, 86]. These detrimental actions may be mitigated during chronic hyperinsulinemia/insulin resistance [87]. However, a meta-analysis of 11 prospective epidemiological studies showed that the pooled relative risk of hypertension was 1.54 when comparing the highest to the lowest category of fasting insulin levels, and 1.43 for comparing highest to lowest (selective) insulin resistance categories, calculated as homeostasis model assessment of insulin resistance (HOMA-IR) [88].
As a consequence of endothelial dysfunction during prolonged treatment with insulin, arterial lesions rich in lipids are formed [89]. The progression of early fatty streak lesions to plaques is accompanied by the adhesion and pro-inflammatory activity of macrophages, which eventually develop into foam cells. This process is driven by endothelial and macrophage lipoprotein lipase activity, as demonstrated by the observation of less atherosclerosis in mice with inactivated lipoprotein lipase gene [90–92]. Lipoprotein lipase activity in macrophages is enhanced with higher insulin levels in vivo, but there is no direct stimulatory effect of insulin on isolated macrophages [93].
The concern, that hyperinsulinemia might promote arterial disease in diabetic persons, developed in the late 1960s, due to the steady increase of incidences of atherosclerosis in diabetic persons, despite improved glycemia and decreased risk of ketosis due to insulin therapy [94]. Since then, a wealth of data supports the observation that insulin resistance (and hyperinsulinemia) is a marker of increased risk of cardiovascular disease in the general population and in patients with diabetes [95]. Although observational studies suggested an approximately linear relation between the severity of hyperglycemia and vascular damage, several large randomized controlled trials have shown that intense glycemic control per se does not decrease the risk of macrovascular/cardiovascular events [96]; indeed, insulin therapy may even increase the risk [95, 97, 98]. However, these trials were not randomized for insulin treatment, and treatment of CVD risk factors was not kept similar between patient subgroups. In the United Kingdom Prospective Diabetes Study (UKPDS), hyperinsulinemia and insulin resistance were not mitigated by insulin treatment, and fasting plasma insulin levels even rose [97]. By contrast, in UKPDS and other trials [97, 99–101], oral treatment with the biguanide metformin reduced the risk of cardiovascular events and in parallel decreased insulin resistance and hyperinsulinemia.
In epidemiological studies of type 2 diabetes, it has been consistently observed that the addition of insulin to the treatment regimen or the intensification of insulin treatment result in a higher rate of cardiovascular events [102–121] (Fig. 5). Indeed, it has been shown that the risk increases with increasing insulin dosage [111, 116]. These epidemiological studies may suffer from residual confounding, since it is difficult to account for the possibly more advanced disease stage of patients receiving insulin. A higher rate of hypoglycemic events may be an additional confounder. However, covariates considered in the statistical analyses cover a broad range of potential risk factors from 18 different categories (Supplement Table 1). Large randomized controlled trials such as UKPDS [122] or the Outcome Reduction With Initial Glargine Intervention (ORIGIN) Trial [123] did not observe an increased incidence of cardiovascular disease with insulin therapy, but these trials focused on low-dose insulin therapy of up to a median of 40 IU/day (or 0.4 IU/kg/day), respectively. Similar randomized trials of higher-dose insulin therapy, as typical for real-world conditions, have not been conducted. Recent studies of real-world clinical settings report mean daily basal insulin doses of close to 0.60 IU/kg in the Canadian REALITY Study for insulin-experienced patients with type 2 diabetes [124] and of 0.73 IU/kg in a physician survey in New York [125]. In the European multi-centre EU-TREAT Study, mean baseline insulin doses were between 32 and 54 U per day, depending on the type of insulin therapy regimen applied [126]. It can be concluded that under real-world conditions, the majority of insulin-experienced patients with type 2 diabetes receive higher insulin doses per day than those tried in UKPDS or ORIGIN.
Fig. 5.

Hazard ratio of insulin medication versus different reference medications. Shown are adjusted hazard ratios (HR) for each study with 95% confidence interval. #Moderate insulin exposure; +high insulin exposure; *moderate insulin dose (75 to < 100 units per day); §high insulin dose (> 100 units per day)
In the absence of randomized controlled trials, a Mendelian randomization is an appropriate approach of testing for a causal relationship in humans. Mendelian randomization studies made use of the finding that some genotypes are associated with high or low fasting insulin levels. When comparing individuals carrying ≥ 17 alleles that raise fasting insulin levels with those exhibiting genetically determined low fasting insulin levels, an increased risk of elevated blood pressure, cardiovascular disease, and type 2 diabetes was observed [127]. In two large recent Mendelian randomization studies, a genetic profile predicting high insulin levels in the blood, after adjustment for BMI, was also associated with increased systolic blood pressure and risk of myocardial infarction [128].
Conclusions
As discussed above, insulin signaling engages at least three different pathways and modifies a large number of cellular responses (Table 1). Transient elevations of systemic insulin concentrations are physiological responses to dietary stimuli or other challenges such as environmental toxins [129]. In case of prolonged upregulation of insulin levels, such as in response to overnutrition, glucose homeostasis is maintained by mitigating insulin signaling via PI3K/AKT for glucose export from the blood into tissues. Consequently, insulin resistance has been considered as a defense response in order to avoid hypoglycemia [38]. However, other hormonal actions of insulin via the MAP kinase MEK/ERK pathway and in part via PI3K/AKT are not or poorly inhibited by “insulin resistance.” These pathways promote a host of anabolic responses including protein synthesis. Concomitantly, there is an accumulation of ubiquinated and otherwise modified proteins. Activation of mTORC1 results in the suppression of autophagy, i.e., the removal and turnover of proteins and lipids. Signaling via MEK/ERK causes inhibition of Nrf2 activation, with the consequence of a compromised cytoprotective response to oxidative and other chemical stress. This may be the reason for increased DNA damage in the presence of high insulin concentrations. Insulin resistance suppresses the activation of eNOS by AKT, and the resulting endothelial dysfunction is enhanced by MEK/ERK-dependent expression of ET-1 and PAI-1. Further detrimental actions of insulin during insulin resistance are the promotion of calcium ion influx into smooth vascular cells favoring contractility/vascular stiffening and the enhanced sodium reabsorption in renal tubules increasing the risk of hypertension.
Table 1.
Key messages
| • Insulin employs at least three different pathways of signal transduction. One pathway involves phosphorylation steps via IRS–PI3K–AKT, another is the MAP kinases Ras–MEP–ERK, and third leads to the activation of NOX4. | |
| • Insulin resistance is selective because it partially mitigates the PI3K/AKT pathway for limiting glucose uptake and eNOS activation despite hyperinsulinemia, but many other hormonal actions of insulin are not suppressed. | |
| • Signaling via mTOR and the MEP/ERK pathway causes suppression of autophagy and NRF2, leading to deficient turnover and impaired cell defense. | |
| • Moderate to high normal insulin levels inhibit lipolysis and promote lipogenesis/obesity. | |
| • Insulin resistance and hyperinsulinemia are interdependent. Diet-induced hyperinsulinemia precedes insulin resistance. | |
| • In epidemiological studies, insulin therapy of type 2 diabetes is associated with a higher risk of cardiovascular events or death. | |
| • Randomized trials of insulin therapy and associated risks only studied dosages up to 40 IU/day. | |
| • Mendelian randomization studies found that genetically determined high insulin levels lead to cardiovascular disease. | |
| • Suppression of hyperinsulinemia and concomitant “insulin resistance” provides substantial health benefits. |
These mechanistic insights lend support to the view that the association of hyperinsulinemia with several detrimental health outcomes is of causal nature. Outcomes include obesity, endothelial dysfunction, hypertension, myocardial infarction, and decreased lifespan. We did not discuss the possible contribution of hyperinsulinemia to cancer development or to the deterioration of cognitive functions. Final proof of a causal relationship between hyperinsulinemia and disease risk cannot be obtained by randomized controlled trials, due to problems with masking the type of intervention, long-term compliance, and because of ethical concerns. Alternatively, Mendelian randomization studies are suitable tools to test for causality in humans, and such studies have found hyperinsulinemia to increase the risk of obesity [27, 28] and cardiovascular disease [127, 128].
A straightforward approach for lowering circulating insulin levels is restricting the exposure of islet ß cells to insulin secretagogues. One option is limiting calorie uptake, either continuously or during defined periods of the day or week [130–132]. Another effective way of lowering insulin levels in the blood is the stimulation of insulin clearance via exercise [133]. A different approach is a bariatric surgery [134–136]. Gastric bypass leads to rapid regression of hyperinsulinemia and later of insulin resistance; additionally, there are substantial benefits with regard to health outcomes and mortality. It seems improbable that such marked clinical improvement could have happened in the presence of persistent hyperinsulinemia and insulin resistance.
We conclude that low fasting or circadian insulin levels should be a primary aim of healthy lifestyle guidelines. Insulin treatment of type 2 diabetes seems only warranted if hyperinsulinemia and concomitant (selective) insulin resistance can be avoided. This favors insulin treatment only in the late phases of type 2 diabetes as has been suggested in recent guidelines [137].
Supplementary information
Additional file 1: Supplementary Table 1. List of all regression confounders for the creation of adjusted hazard ratios.
Acknowledgements
We thank Yasemin Kempf for proofreading the manuscript.
Abbreviations
- AKT
Serine/threonine-specific protein kinase B
- BMI
Body mass index
- DNA
Deoxyribonucleic acid
- EC50
Mean insulin concentration for 50% effect
- eNOS
Endothelial nitric oxide synthase
- ERK
Extracellular signal-regulated kinase
- ET-1
Endothelin-1
- GLUT4
Glucose transporter 4
- HOMA-IR
Homeostasis model assessment of insulin resistance
- HR
Hazard ratio
- IGF
Insulin-like growth factor
- IRS
Insulin receptor substrate
- MAP
Mitogen-activated protein
- MEK
Mitogen-activated protein kinase kinase
- mTORC1
Mechanistic target of rapamycin complex 1
- NHANES
National Health and Nutrition Examination Survey
- NO
Nitric oxide
- NOX4
NADPH oxidase 4
- Nrf2
Nuclear factor E2-related factor-2
- ORIGIN
Outcome Reduction With Initial Glargine Intervention
- PAI-1
Plasminogen activator inhibitor-1
- PI3K
Phosphatidylinositol 3-kinase
- PIP3
Phosphatidylinositol (3,4,5)-trisphosphate
- PTEN
Phosphatase and tensin homolog
- Raf
Rapidly accelerated fibrosarcoma
- Ras
Rat sarcoma
- SHC
Src homology 2 domain-containing transforming proteins
- UKPDS
United Kingdom Prospective Diabetes Study
Authors’ contributions
H. Kolb and S. Martin were responsible for the concept and conducted the bibliographic research. H. Kolb was responsible for the initial drafting and final writing. S. Martin, K. Kempf, and M. Röhling participated in the initial drafting, manuscript review, and final writing. The last author, S. Martin, is the guarantor of the review; he had full access to all the data and had final responsibility for the decision to submit for publication. All the authors agreed to publish the review; they reviewed and approved the final version.
Funding
Not applicable.
Availability of data and materials
Data for this review were identified by searches from MEDLINE, PubMed, and references from relevant articles using the search terms “hyperinsulinaemia,” “insulin and longevity or Nrf2 or autophagy,” “insulin treatment and type 2 diabetes,” and “insulin signaling pathways,” In order to limit the number of references, more recently published papers referring to several previously published articles were cited, if possible. Only articles published in English were selected.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
S. Martin has received non-restricted support for the conduct of trials of lifestyle change in persons at risk or with type 2 diabetes by Novartis Pharma GmbH, Boehringer Ingelheim Pharma GmbH & Co. KG, Almased Wellness GmbH, Nintendo of Europe GmbH, HMM Holding AG, and Gesellschaft von Freunden und Förderern der Heinrich-Heine-Universität Düsseldorf e.V.. H. Kolb, K. Kempf, and M. Röhling have nothing to disclose.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Hubert Kolb, Email: hubert.kolb@hhu.de.
Kerstin Kempf, Email: kerstin.kempf@wdgz.de.
Martin Röhling, Email: martin.roehling@wdgz.de.
Stephan Martin, Email: stephan.martin@uni-duesseldorf.de.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12916-020-01688-6.
References
- 1.Accili D, Drago J, Lee EJ, Johnson MD, Cool MH, Salvatore P, et al. Early neonatal death in mice homozygous for a null allele of the insulin receptor gene. Nat Genet. 1996;12:106–109. doi: 10.1038/ng0196-106. [DOI] [PubMed] [Google Scholar]
- 2.Haeusler RA, McGraw TE, Accili D. Biochemical and cellular properties of insulin receptor signalling. Nat Rev Mol Cell Biol. 2018;19:31–44. doi: 10.1038/nrm.2017.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Unal EB, Uhlitz F, Bluthgen N. A compendium of ERK targets. FEBS Lett. 2017;591:2607–2615. doi: 10.1002/1873-3468.12740. [DOI] [PubMed] [Google Scholar]
- 4.Williams KJ, Wu X. Imbalanced insulin action in chronic over nutrition: clinical harm, molecular mechanisms, and a way forward. Atherosclerosis. 2016;247:225–282. doi: 10.1016/j.atherosclerosis.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 5.Rorsman P, Braun M. Regulation of insulin secretion in human pancreatic islets. Annu Rev Physiol. 2013;75:155–179. doi: 10.1146/annurev-physiol-030212-183754. [DOI] [PubMed] [Google Scholar]
- 6.Prentki M, Matschinsky FM, Madiraju SR. Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 2013;18:162–185. doi: 10.1016/j.cmet.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 7.Newsholme P, Cruzat V, Arfuso F, Keane K. Nutrient regulation of insulin secretion and action. J Endocrinol. 2014;221:R105–R120. doi: 10.1530/JOE-13-0616. [DOI] [PubMed] [Google Scholar]
- 8.Jones B, Bloom SR, Buenaventura T, Tomas A, Rutter GA. Control of insulin secretion by GLP-1. Peptides. 2018;100:75–84. doi: 10.1016/j.peptides.2017.12.013. [DOI] [PubMed] [Google Scholar]
- 9.Boden G. Free fatty acids and insulin secretion in humans. Curr Diab Rep. 2005;5:167–170. doi: 10.1007/s11892-005-0004-5. [DOI] [PubMed] [Google Scholar]
- 10.Pingitore A, Gonzalez-Abuin N, Ruz-Maldonado I, Huang GC, Frost G, Persaud SJ. Short chain fatty acids stimulate insulin secretion and reduce apoptosis in mouse and human islets in vitro: role of free fatty acid receptor 2. Diabetes Obes Metab. 2019;21:330–339. doi: 10.1111/dom.13529. [DOI] [PubMed] [Google Scholar]
- 11.Dror E, Dalmas E, Meier DT, Wueest S, Thevenet J, Thienel C, et al. Postprandial macrophage-derived IL-1β stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat Immunol. 2017;18:283–292. doi: 10.1038/ni.3659. [DOI] [PubMed] [Google Scholar]
- 12.Dalenberg JR, Patel BP, Denis R, Veldhuizen MG, Nakamura Y, Vinke PC, et al. Short-term consumption of sucralose with, but not without, carbohydrate impairs neural and metabolic sensitivity to sugar in humans. Cell Metab. 2020;31:493–502. doi: 10.1016/j.cmet.2020.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klip A, McGraw TE, James DE. Thirty sweet years of GLUT4. J Biol Chem. 2019;294:11369–11381. doi: 10.1074/jbc.REV119.008351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li C, Ford ES, McGuire LC, Mokdad AH, Little RR, Reaven GM. Trends in hyperinsulinemia among nondiabetic adults in the U.S. Diabetes Care. 2006;29:2396–2402. doi: 10.2337/dc06-0289. [DOI] [PubMed] [Google Scholar]
- 15.Rijkelijkhuizen JM, McQuarrie K, Girman CJ, Stein PP, Mari A, Holst JJ, et al. Effects of meal size and composition on incretin, α-cell, and β-cell responses. Metabolism. 2010;59:502–511. doi: 10.1016/j.metabol.2009.07.039. [DOI] [PubMed] [Google Scholar]
- 16.Appel KE, Farr CB. Marshall HK (Eds): insulin in undernutrition in the psychoses. In Archives of Neurology and Psychiatry. 1928;90:149–164. [Google Scholar]
- 17.Jacob S, Hauer B, Becker R, Artzner S, Grauer P, Loblein K, et al. Lipolysis in skeletal muscle is rapidly regulated by low physiological doses of insulin. Diabetologia. 1999;42:1171–1174. doi: 10.1007/s001250051288. [DOI] [PubMed] [Google Scholar]
- 18.Kolb H, Stumvoll M, Kramer W, Kempf K, Martin S. Insulin translates unfavourable lifestyle into obesity. BMC Med. 2018;16:232. doi: 10.1186/s12916-018-1225-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen YY, Wang JP, Jiang YY, Li H, Hu YH, Lee KO, et al. Fasting plasma insulin at 5 years of age predicted subsequent weight increase in early childhood over a 5-year period-the Da Qing Children Cohort Study. PLoS One. 2015;10:e0127389. doi: 10.1371/journal.pone.0127389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Howard BV, Adams-Campbell L, Allen C, Black H, Passaro M, Rodabough RJ, et al. Insulin resistance and weight gain in postmenopausal women of diverse ethnic groups. Int J Obes Relat Metab Disord. 2004;28:1039–1047. doi: 10.1038/sj.ijo.0802645. [DOI] [PubMed] [Google Scholar]
- 21.Loves S, van Groningen L, Filius M, Mekking M, Brandon T, Tack CJ, et al. High-dose, diazoxide-mediated insulin suppression boosts weight loss induced by lifestyle intervention. J Clin Endocrinol Metab. 2018;103:4014–4022. doi: 10.1210/jc.2018-01147. [DOI] [PubMed] [Google Scholar]
- 22.Alemzadeh R, Langley G, Upchurch L, Smith P, Slonim AE. Beneficial effect of diazoxide in obese hyperinsulinemic adults. J Clin Endocrinol Metab. 1998;83:1911–1915. doi: 10.1210/jcem.83.6.4852. [DOI] [PubMed] [Google Scholar]
- 23.Lustig RH, Greenway F, Velasquez-Mieyer P, Heimburger D, Schumacher D, Smith D, et al. A multicenter, randomized, double-blind, placebo-controlled, dose-finding trial of a long-acting formulation of octreotide in promoting weight loss in obese adults with insulin hypersecretion. Int J Obes. 2006;30:331–341. doi: 10.1038/sj.ijo.0803074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koivisto VA. Insulin therapy in type II diabetes. Diabetes Care. 1993;16(Suppl 3):29–39. doi: 10.2337/diacare.16.3.29. [DOI] [PubMed] [Google Scholar]
- 25.Mehran AE, Templeman NM, Brigidi GS, Lim GE, Chu KY, Hu X, et al. Hyperinsulinemia drives diet-induced obesity independently of brain insulin production. Cell Metab. 2012;16:723–737. doi: 10.1016/j.cmet.2012.10.019. [DOI] [PubMed] [Google Scholar]
- 26.Page MM, Skovso S, Cen H, Chiu AP, Dionne DA, Hutchinson DF, et al. Reducing insulin via conditional partial gene ablation in adults reverses diet-induced weight gain. FASEB J. 2018;32:1196–1206. doi: 10.1096/fj.201700518R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Le SC, Fallin D, Schork NJ, Bougneres P. The insulin gene VNTR is associated with fasting insulin levels and development of juvenile obesity. Nat Genet. 2000;26:444–446. doi: 10.1038/82579. [DOI] [PubMed] [Google Scholar]
- 28.Astley CM, Todd JN, Salem RM, Vedantam S, Ebbeling CB, Huang PL, et al. Genetic evidence that carbohydrate-stimulated insulin secretion leads to obesity. Clin Chem. 2018;64:192–200. doi: 10.1373/clinchem.2017.280727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kolb H, Eizirik DL. Resistance to type 2 diabetes mellitus: a matter of hormesis? Nat Rev Endocrinol. 2011;8:183–192. doi: 10.1038/nrendo.2011.158. [DOI] [PubMed] [Google Scholar]
- 30.Rossetti L, Giaccari A, DeFronzo RA. Glucose toxicity. Diabetes Care. 1990;13:610–630. doi: 10.2337/diacare.13.6.610. [DOI] [PubMed] [Google Scholar]
- 31.Roma LP, Jonas JC. Nutrient metabolism, subcellular redox state, and oxidative stress in pancreatic islets and beta-cells. J Mol Biol. 2020;432:1461–1493. doi: 10.1016/j.jmb.2019.10.012. [DOI] [PubMed] [Google Scholar]
- 32.Yagihashi S. Glucotoxic mechanisms and related therapeutic approaches. Int Rev Neurobiol. 2016;127:121–149. doi: 10.1016/bs.irn.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 33.Dou L, Jourde-Chinche N. Endothelial toxicity of high glucose and its by-products in diabetic kidney disease. In Toxins. 2019;11. [DOI] [PMC free article] [PubMed]
- 34.Bertacca A, Ciccarone A, Cecchetti P, Vianello B, Laurenza I, Maffei M, et al. Continually high insulin levels impair Akt phosphorylation and glucose transport in human myoblasts. Metabolism. 2005;54:1687–1693. doi: 10.1016/j.metabol.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 35.Catalano KJ, Maddux BA, Szary J, Youngren JF, Goldfine ID, Schaufele F. Insulin resistance induced by hyperinsulinemia coincides with a persistent alteration at the insulin receptor tyrosine kinase domain. PLoS One. 2014;9:e108693. doi: 10.1371/journal.pone.0108693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoehn KL, Salmon AB, Hohnen-Behrens C, Turner N, Hoy AJ, Maghzal GJ, et al. Insulin resistance is a cellular antioxidant defense mechanism. Proc Natl Acad Sci U S A. 2009;106:17787–17792. doi: 10.1073/pnas.0902380106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taegtmeyer H, Beauloye C, Harmancey R, Hue L. Insulin resistance protects the heart from fuel overload in dysregulated metabolic states. Am J Physiol Heart Circ Physiol. 2013;305:H1693–H1697. doi: 10.1152/ajpheart.00854.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nolan CJ, Ruderman NB, Kahn SE, Pedersen O, Prentki M. Insulin resistance as a physiological defense against metabolic stress: implications for the management of subsets of type 2 diabetes. Diabetes. 2015;64:673–686. doi: 10.2337/db14-0694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boden G, Homko C, Barrero CA, Stein TP, Chen X, Cheung P, et al. Excessive caloric intake acutely causes oxidative stress, GLUT4 carbonylation, and insulin resistance in healthy men. Sci Transl Med. 2015;7:304re7. doi: 10.1126/scitranslmed.aac4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoehn KL, Hohnen-Behrens C, Cederberg A, Wu LE, Turner N, Yuasa T, et al. IRS1-independent defects define major nodes of insulin resistance. Cell Metab. 2008;7:421–433. doi: 10.1016/j.cmet.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Turner N, Kowalski GM, Leslie SJ, Risis S, Yang C, Lee-Young RS, et al. Distinct patterns of tissue-specific lipid accumulation during the induction of insulin resistance in mice by high-fat feeding. Diabetologia. 2013;56:1638–1648. doi: 10.1007/s00125-013-2913-1. [DOI] [PubMed] [Google Scholar]
- 42.Belfiore A, Malaguarnera R, Vella V, Lawrence MC, Sciacca L, Frasca F, et al. Insulin receptor isoforms in physiology and disease: an updated view. Endocr Rev. 2017;38:379–431. doi: 10.1210/er.2017-00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boden G, Cheung P, Salehi S, Homko C, Loveland-Jones C, Jayarajan S, et al. Insulin regulates the unfolded protein response in human adipose tissue. Diabetes. 2014;63:912–922. doi: 10.2337/db13-0906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu X, Chen K, Williams KJ. The role of pathway-selective insulin resistance and responsiveness in diabetic dyslipoproteinemia. Curr Opin Lipidol. 2012;23:334–344. doi: 10.1097/MOL.0b013e3283544424. [DOI] [PubMed] [Google Scholar]
- 45.Semple RK, Sleigh A, Murgatroyd PR, Adams CA, Bluck L, Jackson S, et al. Postreceptor insulin resistance contributes to human dyslipidemia and hepatic steatosis. J Clin Invest. 2009;119:315–322. doi: 10.1172/JCI37432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Braccini L, Ciraolo E, Campa CC, Perino A, Longo DL, Tibolla G, et al. PI3K-C2γ is a Rab5 effector selectively controlling endosomal Akt2 activation downstream of insulin signalling. Nat Commun. 2015;6:7400. doi: 10.1038/ncomms8400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168:960–976. doi: 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. 2020;21:183–203. doi: 10.1038/s41580-019-0199-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boden G, Cheung P, Kresge K, Homko C, Powers B, Ferrer L. Insulin resistance is associated with diminished endoplasmic reticulum stress responses in adipose tissue of healthy and diabetic subjects. Diabetes. 2014;63:2977–2983. doi: 10.2337/db14-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ghosh A, Abdo S, Zhao S, Wu CH, Shi Y, Lo CS, et al. Insulin inhibits Nrf2 gene expression via heterogeneous nuclear ribonucleoprotein F/K in diabetic mice. Endocrinology. 2017;158:903–919. doi: 10.1210/en.2016-1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yamamoto M, Kensler TW, Motohashi H. The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol Rev. 2018;98:1169–1203. doi: 10.1152/physrev.00023.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tan Y, Ichikawa T, Li J, Si Q, Yang H, Chen X, et al. Diabetic downregulation of Nrf2 activity via ERK contributes to oxidative stress-induced insulin resistance in cardiac cells in vitro and in vivo. Diabetes. 2011;60:625–633. doi: 10.2337/db10-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Othman EM, Leyh A, Stopper H. Insulin mediated DNA damage in mammalian colon cells and human lymphocytes in vitro. Mutat Res.2013;745–746:34–9. [DOI] [PubMed]
- 55.Bucris E, Beck A, Boura-Halfon S, Isaac R, Vinik Y, Rosenzweig T, et al. Prolonged insulin treatment sensitizes apoptosis pathways in pancreatic beta cells. J Endocrinol. 2016;230:291–307. doi: 10.1530/JOE-15-0505. [DOI] [PubMed] [Google Scholar]
- 56.Chow HM, Shi M, Cheng A, Gao Y, Chen G, Song X, et al. Age-related hyperinsulinemia leads to insulin resistance in neurons and cell-cycle-induced senescence. Nat Neurosci. 2019;22:1806–1819. doi: 10.1038/s41593-019-0505-1. [DOI] [PubMed] [Google Scholar]
- 57.Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, et al. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature. 1997;389:994–999. doi: 10.1038/40194. [DOI] [PubMed] [Google Scholar]
- 58.Clancy DJ, Gems D, Harshman LG, Oldham S, Stocker H, Hafen E, et al. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science. 2001;292:104–106. doi: 10.1126/science.1057991. [DOI] [PubMed] [Google Scholar]
- 59.Kim SS, Lee CH. Growth signaling and longevity in mouse models. In BMB Reports. 2019;52:70–85. doi: 10.5483/BMBRep.2019.52.1.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Previs SF, Withers DJ, Ren JM, White MF, Shulman GI. Contrasting effects of IRS-1 versus IRS-2 gene disruption on carbohydrate and lipid metabolism in vivo. J Biol Chem. 2000;275:38990–38994. doi: 10.1074/jbc.M006490200. [DOI] [PubMed] [Google Scholar]
- 61.Selman C, Partridge L, Withers DJ. Replication of extended lifespan phenotype in mice with deletion of insulin receptor substrate 1. PLoS One. 2011;6:e16144. doi: 10.1371/journal.pone.0016144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bluher M, Kahn BB, Kahn CR. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science. 2003;299:572–574. doi: 10.1126/science.1078223. [DOI] [PubMed] [Google Scholar]
- 63.Templeman NM, Flibotte S, Chik JHL, Sinha S, Lim GE, Foster LJ, et al. Reduced circulating insulin enhances insulin sensitivity in old mice and extends lifespan. Cell Rep. 2017;20:451–463. doi: 10.1016/j.celrep.2017.06.048. [DOI] [PubMed] [Google Scholar]
- 64.Barzilai N, Huffman DM, Muzumdar RH, Bartke A. The critical role of metabolic pathways in aging. Diabetes. 2012;61:1315–1322. doi: 10.2337/db11-1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kennedy BK, Lamming DW. The mechanistic target of rapamycin: the grand ConducTOR of metabolism and aging. Cell Metab. 2016;23:990–1003. doi: 10.1016/j.cmet.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guillen C, Benito M. mTORC1 overactivation as a key aging factor in the progression to type 2 diabetes mellitus. Front Endocrinol (Lausanne) 2018;9:621. doi: 10.3389/fendo.2018.00621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Deelen J, van den Akker EB, Trompet S, van HD, Mooijaart SP, Slagboom PE et al Employing biomarkers of healthy ageing for leveraging genetic studies into human longevity. Exp Gerontol2016;82:166–174. [DOI] [PubMed]
- 68.Boucher J, Kleinridders A, Kahn CR. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol. 2014;6. [DOI] [PMC free article] [PubMed]
- 69.Corbit KC, Camporez JPG, Tran JL, Wilson CG, Lowe DA, Nordstrom SM, et al. Adipocyte JAK2 mediates growth hormone-induced hepatic insulin resistance. JCI Insight. 2017;2:e91001. doi: 10.1172/jci.insight.91001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Milner KL, Jenkins AB, Trenell M, Tid-Ang J, Samocha-Bonet D, Weltman M, et al. Eradicating hepatitis C virus ameliorates insulin resistance without change in adipose depots. J Viral Hepat. 2014;21:325–332. doi: 10.1111/jvh.12143. [DOI] [PubMed] [Google Scholar]
- 71.Del PS, Leonetti F, Simonson DC, Sheehan P, Matsuda M, DeFronzo RA. Effect of sustained physiologic hyperinsulinaemia and hyperglycaemia on insulin secretion and insulin sensitivity in man. Diabetologia. 1994;37:1025–1035. doi: 10.1007/BF00400466. [DOI] [PubMed] [Google Scholar]
- 72.Kobayashi M, Olefsky JM. Effects of streptozotocin-induced diabetes on insulin binding, glucose transport, and intracellular glucose metabolism in isolated rat adipocytes. Diabetes. 1979;28:87–95. doi: 10.2337/diab.28.2.87. [DOI] [PubMed] [Google Scholar]
- 73.Czech MP. Insulin action and resistance in obesity and type 2 diabetes. Nat Med. 2017;23:804–814. doi: 10.1038/nm.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim MK, Reaven GM, Chen YD, Kim E, Kim SH. Hyperinsulinemia in individuals with obesity: role of insulin clearance. Obesity (Silver Spring).2015;23:2430–4. [DOI] [PMC free article] [PubMed]
- 75.Montagnani M, Chen H, Barr VA, Quon MJ. Insulin-stimulated activation of eNOS is independent of Ca2+ but requires phosphorylation by Akt at Ser1179. J Biol Chem. 2001;276:30392–30398. doi: 10.1074/jbc.M103702200. [DOI] [PubMed] [Google Scholar]
- 76.King GL, Park K, Li Q. Selective insulin resistance and the development of cardiovascular diseases in diabetes: the 2015 Edwin Bierman Award Lecture. Diabetes. 2016;65:1462–1471. doi: 10.2337/db16-0152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gutierrez A, Contreras C, Sanchez A, Prieto D. Role of phosphatidylinositol 3-kinase (PI3K), mitogen-activated protein kinase (MAPK), and protein kinase C (PKC) in calcium signaling pathways linked to the α1-adrenoceptor in resistance arteries. Front Physiol. 2019;10:55. doi: 10.3389/fphys.2019.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pinho JF, Medeiros MA, Capettini LS, Rezende BA, Campos PP, Andrade SP, et al. Phosphatidylinositol 3-kinase-delta up-regulates L-type Ca2+ currents and increases vascular contractility in a mouse model of type 1 diabetes. Br J Pharmacol. 2010;161:1458–1471. doi: 10.1111/j.1476-5381.2010.00955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Montagnani M, Golovchenko I, Kim I, Koh GY, Goalstone ML, Mundhekar AN, et al. Inhibition of phosphatidylinositol 3-kinase enhances mitogenic actions of insulin in endothelial cells. J Biol Chem. 2002;277:1794–1799. doi: 10.1074/jbc.M103728200. [DOI] [PubMed] [Google Scholar]
- 80.Madonna R, Massaro M, De CR. Insulin potentiates cytokine-induced VCAM-1 expression in human endothelial cells. Biochim Biophys Acta. 2008;1782:511–516. doi: 10.1016/j.bbadis.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 81.Aroor AR, Jia G, Sowers JR. Cellular mechanisms underlying obesity-induced arterial stiffness. Am J Physiol Regul Integr Comp Physiol. 2018;314:R387–R398. doi: 10.1152/ajpregu.00235.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Brillante DG, O’Sullivan AJ, Howes LG. Arterial stiffness in insulin resistance: the role of nitric oxide and angiotensin II receptors. Vasc Health Risk Manag. 2009;5:73–78. [PMC free article] [PubMed] [Google Scholar]
- 83.Muniyappa R, Yavuz S. Metabolic actions of angiotensin II and insulin: a microvascular endothelial balancing act. Mol Cell Endocrinol. 2013;378:59–69. doi: 10.1016/j.mce.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Berendji-Grun D, Kolb-Bachofen V, Kroncke KD. Nitric oxide inhibits endothelial IL-1β-induced ICAM-1 gene expression at the transcriptional level decreasing Sp1 and AP-1 activity. Mol Med. 2001;7:748–754. [PMC free article] [PubMed] [Google Scholar]
- 85.Horita S, Nakamura M, Suzuki M, Satoh N, Suzuki A, Seki G. Selective insulin resistance in the kidney. Biomed Res Int. 2016;2016:5825170. doi: 10.1155/2016/5825170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tanaka M. Improving obesity and blood pressure. Hypertens Res. 2020;43:79–89. doi: 10.1038/s41440-019-0348-x. [DOI] [PubMed] [Google Scholar]
- 87.da Silva AA, do Carmo JM, Li X, Wang Z, Mouton AJ, Hall JE. Role of hyperinsulinemia and insulin resistance in hypertension: metabolic syndrome revisited. Can J Cardiol2020;36:671–682. [DOI] [PMC free article] [PubMed]
- 88.Wang F, Han L, Hu D. Fasting insulin, insulin resistance and risk of hypertension in the general population: a meta-analysis. Clin Chim Acta. 2017;464:57–63. doi: 10.1016/j.cca.2016.11.009. [DOI] [PubMed] [Google Scholar]
- 89.Stout RW. Insulin and atheroma. 20-yr perspective. Diabetes Care. 1990;13:631–654. doi: 10.2337/diacare.13.6.631. [DOI] [PubMed] [Google Scholar]
- 90.Qiu G, Ho AC, Yu W, Hill JS. Suppression of endothelial or lipoprotein lipase in THP-1 macrophages attenuates proinflammatory cytokine secretion. J Lipid Res. 2007;48:385–394. doi: 10.1194/jlr.M600304-JLR200. [DOI] [PubMed] [Google Scholar]
- 91.Takahashi M, Yagyu H, Tazoe F, Nagashima S, Ohshiro T, Okada K, et al. Macrophage lipoprotein lipase modulates the development of atherosclerosis but not adiposity. J Lipid Res. 2013;54:1124–1134. doi: 10.1194/jlr.M035568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.He PP, OuYang XP, Li Y, Lv YC, Wang ZB, Yao F, et al. MicroRNA-590 inhibits lipoprotein lipase expression and prevents atherosclerosis in apoE knockout mice. PLoS One. 2015;10:e0138788. doi: 10.1371/journal.pone.0138788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Behr SR, Kraemer FB. Insulin deficiency decreases lipoprotein lipase secretion by murine macrophages. Diabetes. 1988;37:1076–1081. doi: 10.2337/diab.37.8.1076. [DOI] [PubMed] [Google Scholar]
- 94.Stout RW, Vallance-Owen J. Insulin and atheroma. Lancet. 1969;1:1078–1080. doi: 10.1016/s0140-6736(69)91711-5. [DOI] [PubMed] [Google Scholar]
- 95.Adeva-Andany MM, Martinez-Rodriguez J, Gonzalez-Lucan M, Fernandez-Fernandez C, Castro-Quintela E. Insulin resistance is a cardiovascular risk factor in humans. Diabetes Metab Syndr. 2019;13:1449–1455. doi: 10.1016/j.dsx.2019.02.023. [DOI] [PubMed] [Google Scholar]
- 96.Paneni F, Luscher TF. Cardiovascular protection in the treatment of type 2 diabetes: a review of clinical trial results across drug classes. Am J Med. 2017;130:S18–S29. doi: 10.1016/j.amjmed.2017.04.008. [DOI] [PubMed] [Google Scholar]
- 97.United Kingdom Prospective Diabetes Study (UKPDS). 13: Relative efficacy of randomly allocated diet, sulphonylurea, insulin, or metformin in patients with newly diagnosed non-insulin dependent diabetes followed for three years. BMJ.1995;310:83–8. [PMC free article] [PubMed]
- 98.Herman ME, O’Keefe JH, Bell DSH, Schwartz SS. Insulin therapy increases cardiovascular risk in type 2 diabetes. Prog Cardiovasc Dis. 2017;60:422–434. doi: 10.1016/j.pcad.2017.09.001. [DOI] [PubMed] [Google Scholar]
- 99.Han Y, Xie H, Liu Y, Gao P, Yang X, Shen Z. Effect of metformin on all-cause and cardiovascular mortality in patients with coronary artery diseases: a systematic review and an updated meta-analysis. Cardiovasc Diabetol. 2019;18:96. doi: 10.1186/s12933-019-0900-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Luo F, Das A, Chen J, Wu P, Li X, Fang Z. Metformin in patients with and without diabetes: a paradigm shift in cardiovascular disease management. Cardiovasc Diabetol. 2019;18:54. doi: 10.1186/s12933-019-0860-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bailey CJ. Metformin: historical overview. Diabetologia. 2017;60:1566–1576. doi: 10.1007/s00125-017-4318-z. [DOI] [PubMed] [Google Scholar]
- 102.Rensing KL, Reuwer AQ, Arsenault BJ, von der Thusen JH, Hoekstra JB, Kastelein JJ, et al. Reducing cardiovascular disease risk in patients with type 2 diabetes and concomitant macrovascular disease: can insulin be too much of a good thing? Diabetes Obes Metab. 2011;13:1073–1087. doi: 10.1111/j.1463-1326.2011.01468.x. [DOI] [PubMed] [Google Scholar]
- 103.Dailey G, Wang E. A review of cardiovascular outcomes in the treatment of people with type 2 diabetes. Diabetes Ther. 2014;5:385–402. doi: 10.1007/s13300-014-0091-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mogensen UM, Andersson C, Fosbol EL, Schramm TK, Vaag A, Scheller NM, et al. Sulfonylurea in combination with insulin is associated with increased mortality compared with a combination of insulin and metformin in a retrospective Danish nationwide study. Diabetologia. 2015;58:50–58. doi: 10.1007/s00125-014-3372-z. [DOI] [PubMed] [Google Scholar]
- 105.Price HI, Agnew MD, Gamble JM. Comparative cardiovascular morbidity and mortality in patients taking different insulin regimens for type 2 diabetes: a systematic review. BMJ Open. 2015;5:e006341. doi: 10.1136/bmjopen-2014-006341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Stoekenbroek RM, Rensing KL, Bernelot Moens SJ, Nieuwdorp M, DeVries JH, Zwinderman AH, et al. High daily insulin exposure in patients with type 2 diabetes is associated with increased risk of cardiovascular events. Atherosclerosis. 2015;240:318–323. doi: 10.1016/j.atherosclerosis.2015.03.040. [DOI] [PubMed] [Google Scholar]
- 107.Seong JM, Choi NK, Shin JY, Chang Y, Kim YJ, Lee J, et al. Differential cardiovascular outcomes after dipeptidyl peptidase-4 inhibitor, sulfonylurea, and pioglitazone therapy, all in combination with metformin, for type 2 diabetes: a population-based cohort study. PLoS One. 2015;10:e0124287. doi: 10.1371/journal.pone.0124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Standl E, Schnell O, McGuire DK. Heart failure considerations of antihyperglycemic medications for type 2 diabetes. Circ Res. 2016;118:1830–1843. doi: 10.1161/CIRCRESAHA.116.306924. [DOI] [PubMed] [Google Scholar]
- 109.Inoue T, Maeda Y, Sonoda N, Sasaki S, Kabemura T, Kobayashi K, et al. Hyperinsulinemia and sulfonylurea use are independently associated with left ventricular diastolic dysfunction in patients with type 2 diabetes mellitus with suboptimal blood glucose control. BMJ Open Diabetes Res Care. 2016;4:e000223. doi: 10.1136/bmjdrc-2016-000223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Patti G, Lucerna M, Cavallari I, Ricottini E, Renda G, Pecen L, et al. Insulin-requiring versus noninsulin-requiring diabetes and thromboembolic risk in patients with atrial fibrillation: PREFER in AF. J Am Coll Cardiol. 2017;69:409–419. doi: 10.1016/j.jacc.2016.10.069. [DOI] [PubMed] [Google Scholar]
- 111.Holden SE, Jenkins-Jones S, Morgan CL, Schernthaner G, Currie CJ. Glucose-lowering with exogenous insulin monotherapy in type 2 diabetes: dose association with all-cause mortality, cardiovascular events and cancer. Diabetes Obes Metab. 2015;17:350–362. doi: 10.1111/dom.12412. [DOI] [PubMed] [Google Scholar]
- 112.Roumie CL, Greevy RA, Grijalva CG, Hung AM, Liu X, Murff HJ, et al. Association between intensification of metformin treatment with insulin vs sulfonylureas and cardiovascular events and all-cause mortality among patients with diabetes. JAMA. 2014;311:2288–2296. doi: 10.1001/jama.2014.4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nystrom T, Bodegard J, Nathanson D, Thuresson M, Norhammar A, Eriksson JW. Second line initiation of insulin compared with DPP-4 inhibitors after metformin monotherapy is associated with increased risk of all-cause mortality, cardiovascular events, and severe hypoglycemia. Diabetes Res Clin Pract. 2017;123:199–208. doi: 10.1016/j.diabres.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 114.Jil M, Rajnikant M, Richard D, Iskandar I. The effects of dual-therapy intensification with insulin or dipeptidylpeptidase-4 inhibitor on cardiovascular events and all-cause mortality in patients with type 2 diabetes: a retrospective cohort study. Diab Vasc Dis Res. 2017;14:295–303. doi: 10.1177/1479164116687102. [DOI] [PubMed] [Google Scholar]
- 115.Mellbin LG, Malmberg K, Norhammar A, Wedel H, Ryden L. Prognostic implications of glucose-lowering treatment in patients with acute myocardial infarction and diabetes: experiences from an extended follow-up of the Diabetes Mellitus Insulin-Glucose Infusion in Acute Myocardial Infarction (DIGAMI) 2 Study. Diabetologia. 2011;54:1308–1317. doi: 10.1007/s00125-011-2084-x. [DOI] [PubMed] [Google Scholar]
- 116.Gamble JM, Chibrikov E, Twells LK, Midodzi WK, Young SW, MacDonald D, et al. Association of insulin dosage with mortality or major adverse cardiovascular events: a retrospective cohort study. Lancet Diabetes Endocrinol. 2017;5:43–52. doi: 10.1016/S2213-8587(16)30316-3. [DOI] [PubMed] [Google Scholar]
- 117.Gamble JM, Simpson SH, Eurich DT, Majumdar SR, Johnson JA. Insulin use and increased risk of mortality in type 2 diabetes: a cohort study. Diabetes Obes Metab. 2010;12:47–53. doi: 10.1111/j.1463-1326.2009.01125.x. [DOI] [PubMed] [Google Scholar]
- 118.Anselmino M, Ohrvik J, Malmberg K, Standl E, Ryden L. Glucose lowering treatment in patients with coronary artery disease is prognostically important not only in established but also in newly detected diabetes mellitus: a report from the Euro Heart Survey on Diabetes and the Heart. Eur Heart J. 2008;29:177–184. doi: 10.1093/eurheartj/ehm519. [DOI] [PubMed] [Google Scholar]
- 119.Ekstrom N, Svensson AM, Miftaraj M, Franzen S, Zethelius B, Eliasson B, et al. Cardiovascular safety of glucose-lowering agents as add-on medication to metformin treatment in type 2 diabetes: report from the Swedish National Diabetes Register. Diabetes Obes Metab. 2016;18:990–998. doi: 10.1111/dom.12704. [DOI] [PubMed] [Google Scholar]
- 120.Currie CJ, Poole CD, Evans M, Peters JR, Morgan CL. Mortality and other important diabetes-related outcomes with insulin vs other antihyperglycemic therapies in type 2 diabetes. J Clin Endocrinol Metab. 2013;98:668–677. doi: 10.1210/jc.2012-3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Saleh N, Petursson P, Lagerqvist B, Skuladottir H, Svensson A, Eliasson B, et al. Long-term mortality in patients with type 2 diabetes undergoing coronary angiography: the impact of glucose-lowering treatment. Diabetologia. 2012;55:2109–2117. doi: 10.1007/s00125-012-2565-6. [DOI] [PubMed] [Google Scholar]
- 122.Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet.1998;352:837–53. [PubMed]
- 123.Gerstein HC, Bosch J, Dagenais GR, Diaz R, Jung H, Maggioni AP, et al. Basal insulin and cardiovascular and other outcomes in dysglycemia. N Engl J Med. 2012;367:319–328. doi: 10.1056/NEJMoa1203858. [DOI] [PubMed] [Google Scholar]
- 124.Abitbol A, Brown RE, Jiandani D, Sauriol L, Aronson R. Real-world health outcomes of insulin glargine 300 U/mL vs insulin glargine 100 U/mL in adults with type 1 and type 2 diabetes in the Canadian LMC Diabetes Patient Registry: the REALITY Study. Can J Diabetes. 2019;43:504–509. doi: 10.1016/j.jcjd.2019.04.012. [DOI] [PubMed] [Google Scholar]
- 125.Gupta S, Wang H, Skolnik N, Tong L, Liebert RM, Lee LK, et al. Treatment dosing patterns and clinical outcomes for patients with type 2 diabetes starting or switching to treatment with insulin glargine (300 units per milliliter) in a real-world setting: a retrospective observational study. Adv Ther. 2018;35:43–55. doi: 10.1007/s12325-017-0651-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Siegmund T, Tentolouris N, Knudsen ST, Lapolla A, Prager R, Phan TM, et al. A European, multicentre, retrospective, non-interventional study (EU-TREAT) of the effectiveness of insulin degludec after switching basal insulin in a population with type 1 or type 2 diabetes. Diabetes Obes Metab. 2018;20:689–697. doi: 10.1111/dom.13149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yaghootkar H, Scott RA, White CC, Zhang W, Speliotes E, Munroe PB, et al. Genetic evidence for a normal-weight “metabolically obese” phenotype linking insulin resistance, hypertension, coronary artery disease, and type 2 diabetes. Diabetes. 2014;63:4369–4377. doi: 10.2337/db14-0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhao JV, Luo S, Schooling CM. Sex-specific Mendelian randomization study of genetically predicted insulin and cardiovascular events in the UK Biobank. Commun Biol. 2019;2:332. doi: 10.1038/s42003-019-0579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Corkey BE. Diabetes: have we got it all wrong? Insulin hypersecretion and food additives: cause of obesity and diabetes? Diabetes Care. 2012;35:2432–2437. doi: 10.2337/dc12-0825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lean MEJ, Leslie WS, Barnes AC, Brosnahan N, Thom G, McCombie L, et al. Durability of a primary care-led weight-management intervention for remission of type 2 diabetes: 2-year results of the DiRECT open-label, cluster-randomised trial. Lancet Diabetes Endocrinol. 2019;7:344–355. doi: 10.1016/S2213-8587(19)30068-3. [DOI] [PubMed] [Google Scholar]
- 131.Kempf K, Altpeter B, Berger J, Reuss O, Fuchs M, Schneider M, et al. Efficacy of the telemedical lifestyle intervention program TeLiPro in advanced stages of type 2 diabetes: a randomized controlled trial. Diabetes Care. 2017;40:863–871. doi: 10.2337/dc17-0303. [DOI] [PubMed] [Google Scholar]
- 132.Mattson MP, Longo VD, Harvie M. Impact of intermittent fasting on health and disease processes. Ageing Res Rev. 2017;39:46–58. doi: 10.1016/j.arr.2016.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kurauti MA, Freitas-Dias R, Ferreira SM, Vettorazzi JF, Nardelli TR, Araujo HN, et al. Acute exercise improves insulin clearance and increases the expression of insulin-degrading enzyme in the liver and skeletal muscle of Swiss mice. PLoS One. 2016;11:e0160239. doi: 10.1371/journal.pone.0160239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pareek M, Schauer PR, Kaplan LM, Leiter LA, Rubino F, Bhatt DL. Metabolic surgery: weight loss, diabetes, and beyond. J Am Coll Cardiol. 2018;71:670–687. doi: 10.1016/j.jacc.2017.12.014. [DOI] [PubMed] [Google Scholar]
- 135.Purnell JQ, Johnson GS, Wahed AS, Dalla MC, Piccinini F, Cobelli C, et al. Prospective evaluation of insulin and incretin dynamics in obese adults with and without diabetes for 2 years after Roux-en-Y gastric bypass. Diabetologia. 2018;61:1142–1154. doi: 10.1007/s00125-018-4553-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Thomas DD, Corkey BE, Istfan NW, Apovian CM. Hyperinsulinemia: an early indicator of metabolic dysfunction. J Endocr Soc. 2019;3:1727–1747. doi: 10.1210/js.2019-00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Davies MJ, D’Alessio DA, Fradkin J, Kernan WN, Mathieu C, Mingrone G, et al. Management of hyperglycemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD) Diabetes Care. 2018;41:2669–2701. doi: 10.2337/dci18-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Supplementary Table 1. List of all regression confounders for the creation of adjusted hazard ratios.
Data Availability Statement
Data for this review were identified by searches from MEDLINE, PubMed, and references from relevant articles using the search terms “hyperinsulinaemia,” “insulin and longevity or Nrf2 or autophagy,” “insulin treatment and type 2 diabetes,” and “insulin signaling pathways,” In order to limit the number of references, more recently published papers referring to several previously published articles were cited, if possible. Only articles published in English were selected.