

Abstract
The coronavirus disease-2019 caused by a novel SARS CoV-2 virus has emerged as a global threat. Still, no drugs are available for its treatment. The main protease is the most conserved structure responsible for the posttranslational processing of non-structural polyproteins of this virus. Therefore, it can be the potential target for drug discovery against SARS CoV-2. Twenty-one thousand two hundred and seven chemical compounds used for sequential virtual screening studies including coronavirus screening compounds (Life Chemical database) and antiviral compounds (Asinex database). The Schrodinger suite 2019 employed for high throughput screening, molecular docking and MM-GBSA through the Glide module. Subsequently, 23 compounds were selected in the phase first selection criteria for re-docking with AutoDock and iDock followed by ADMET prediction. The drug-likeness predicted through Lipinski’s rule of five, Veber’s rule and Muegge’s rule. Finally, three ligands were selected for molecular dynamics simulation studies over 150 ns against the main protease of the SARS CoV-2. They showed promising docking scores on Glide, iDock and AutoDock Vina algorithms (ligand F2679-0163: −10.75, −10.29 and −9.2; ligand F6355-0442: −9.38, −8.61 and −7.6; ligand 8250: −9.795, −7.94 and −7.5), respectively. The RMSD parameter remained stable at 2.5 Å for all the three ligands for 150 ns. The high RMSF fluctuations, RoG of around 22 Å and the binding free energy were favorable in each case. The hydrogen bond interactions of 8250, F6355-0442 and F2679-0163 were six, five and three, respectively. These compounds can be further explored for in vitro experimental validation against SARS-CoV-2.
Communicated by Ramaswamy H. Sarma
Keywords: Main protease, COVID-19, SARS CoV-2, docking, molecular dynamics simulation
1. Introduction
The COVID-19 has been an extremely contagious and pathogenic viral infection first observed in the Wuhan city of China (Singhal, 2020). The causative agent was found to be a novel virus belonging to the coronavirus family, particularly from the beta subfamily (Cascella et al., 2020). The virus has been designated as SARS CoV-2, as it causes severe acute respiratory syndrome and pneumonia (Pal et al., 2020). The earlier viruses from the same family such as SARS and MERS have shown 10% to 36% mortality (da Costa et al., 2020; Paules et al., 2020) which was much higher than 2–4% by SARS CoV-2 (Roussel et al., 2020), but attack rate of this virus is said to be very high, and therefore, it has been declared as a global pandemic by World Health Organization (Zarocostas, 2020).
The coronaviruses are positive-stranded spherical enveloped RNA viruses that infect a variety of animals and human beings (Fehr & Perlman, 2015; Ye et al., 2020). They measure from 60 to 80 nm with 26 to 32 kb genome size (Lu et al., 2020). The majority of the portion of RNA encodes RNA dependent RNA polymerase and two overlapping non-structural proteins (NSPs) called polyproteins (pp1a and pp1ab; Elfiky, 2020a, 2020b; Gao et al., 2020), the remaining genomic portion codes for four structural proteins. The functional polypeptides are produced by the proteolysis of these polyproteins (Krichel et al., 2020). The structural proteins are namely Spike (S), Envelope (E), Membrane (M) and Nucleocapsid (N; Hasan et al., 2020; McBride et al., 2014; Yadav et al., 2020). The latest SARS CoV-2 virus is a beta virus (Xie & Chen, 2020). It has been predicted that spike protein interacts with the ACE2 receptors on the human cells (Ortega et al., 2020), and it takeovers the host machinery for synthesis and replication of new viruses (Xu et al., 2011).
The highly conserved main proteases (Mpro) named as 3-C-like protease (3CLpro) is predominantly responsible for the posttranslational processing of various viral proteins (Boopathi et al., 2020; Chen et al., 2020; Stobart et al., 2013). However, papain-like protease (PLpro) also cleaves the large polyproteins into NSPs (Báez-Santos et al., 2015). These NSP play an essential role in viral genomic expression and replication (Snijder et al., 2016). Both of them have been used as targets for potential drug therapy for SARS CoV-2 infections (Lindner et al., 2005; Wu et al., 2020).
Some compounds have been found as a potential treatment against SARS CoV-2 in many studies (Caly et al., 2020; Elmezayen et al., 2020; Hendaus, 2020; Zhou et al., 2020). There are studies in which antiviral drugs such as Anti-HIV, Anti-HCV, Anti-HBV and drugs for other infectious diseases have been suggested as therapies (Barlow et al., 2020; Bhatnagar et al., 2020; Cherian et al., 2020; Fan et al., 2020; Sayad et al., 2020). Some of them have suggested that combinations of drugs may provide rapid viral clearance than the single agent alone (Cao et al., 2020; Sheahan et al., 2020). Additionally, many studies have ventured into finding the treatment in the phytochemicals as well (Elfiky, 2020a, 2020b; Islam et al., 2020). Several studies have shown the drugs that can be suitably explored as inhibitors of Mpro as well (Aanouz et al., 2020; Bhardwaj et al., 2020; Das et al., 2020; Enmozhi et al., 2020; Gupta et al., 2020; Havranek & Islam, 2020; Kumar et al., 2020; Umesh et al., 2020). Even after so many studies, there is no approved drug yet against this virus. Therefore, there is an urgent need to conduct basic research to identify the drugs against the SARS CoV-2. Therefore, this study was conducted with the newly categorized coronavirus screening compounds by the Life Chemicals database and other antiviral drug-like chemical compounds to find the potential treatment through molecular docking, molecular dynamics simulations (MDS) and ADME/T (absorption, distribution, metabolism, elimination and toxicity [ADME/T]) studies.
2. Material and methods
2.1. Chemical compounds library preparation and target selection
Figure 1 shows the virtual screening flow of the study, wherein three separate databases were used for the computational screening of 21,207 ligands against the target protein of SARS CoV-2. One was a very new group placed in the Life Chemical database (https://lifechemicals.com/) as coronavirus screening (Anti-nCoV2) compound library (Lib-A), and second, from the same database was taken as Antiviral drugs (Lib-B). The third library of chemical compounds with antiviral properties was taken from the Asinex database (Lib-C; http://www.asinex.com/antiviral/). Out of the total 21,207 compounds, the Lib-A had 2327 Anti-nCoV2 screening ligands, Lib-B had 10,158 compounds with antiviral activity and Lib-C had 8722 chemical moieties with antiviral properties.
Figure 1.

Virtual screening flow of the study.
The main protease target protein of SARS CoV-2 (PBD IDs: 7BRP) was used in this study as it is the recently released crystal structure and is highly conserved in nature. The three-dimensional structure of the target protein was retrieved from the protein data bank (https://www.rcsb.org/structure/7BRP). This protein structure was selected because of its high resolution (1.8 Å) and recognized by the X-ray diffraction technique. The three-dimensional structure of Mpro and its interactive co-crystallized ligand site on its structure is represented in Figure 2(A–C).
Figure 2.

(A) Three-dimensional structure of main protease of SARS CoV-2 (PDB ID: 7BRP) Chains A and B associated with co-crystal ligand (a). (B) Three-dimensional structure of Chain A associated with co-crystal ligand (a). (C) Three-dimensional structure of Chain A after protein preparation.
2.2. Protein preparation and ligand preparation
The target protein was prepared before starting the docking processes. It was done with the help of Schrodinger’s protein preparation wizard tool (Madhavi Sastry et al., 2013). The three-dimensional structure of the target protein (PDB IDs: 7BRP) was prepared. This was done by correcting bonds, removing unrelated chemical complexes, eliminating water molecules from het groups, creating zero-order bonds to metal atoms, the addition of hydrogen bonds, conversion of selenomethionine to methionine, filling in missing side chains and generating het states utilizing EpikPh 7 to +2. The OPLS3e was used for the optimization of protein HBs through overlying and minimizing hydrogen utilization. The molecular modeling package of the ligand preparation module in OPLS3e was used to prepare ligands with appropriate parameters such as optimization, determination of promoters, tautomers, ionization state at pH 7.0, ring confirmation, two dimensional to three-dimensional conversion and correction of partial atomic charges (Madhavi Sastry et al., 2013).
2.3. Active site prediction and receptor grid generation
One of the most crucial aspect in designing a drug via computational docking or digital screening was recognizing the suitable active site for binding the ligand molecules. The grid generation was the next step with the selection of co-crystal ligand of the target protein (Figure 2). The OPLS3e force field was used to generate partial cut off charge of 0.25 Å and mounting of the protein atoms through default parameters of 1 Å radii of Van der Waal’s scaling factor. The dimensions of the grid box and receptor setup were x = 20 Å, y = 20 Å, z = 20 Å and x = 10 Å, y = 10 Å, z = 10 Å during docking study, respectively, with a grid space of 1 Å (Halgren et al., 2004).
2.4. Molecular docking
2.4.1. Glide
The sequential molecular docking study of the selected ligands (Lib-A: 2327 Anti-nCoV2 screening compounds from Life Chemicals, Lib-B: 10,158 antiviral drugs like compounds from Life Chemicals, and Lib-C: 8722 antiviral compounds from Asinex database) was done against the main protease of SARS CoV-2. High throughput virtual screening of the selected chemical compounds from Lib-A, B and C was performed against target protein using Schrodinger’s Ligand docking module with a flexible docking parameter. The flow of the docking study has been described in Figure 1 wherein 10% of the ligands (300 Ligands from Lib-A, 1000 from Lib-B and 870 Ligands from Lib-C) were selected for the standard precision (SP) docking. After that, 10% of the compounds (30 from Lib-A, 100 from Lib-B and 87 from Lib-C) from SP docking underwent extra precision (XP) docking (Kesharwani et al., 2016). Subsequently, to get more insights into these docking results, the iDock and AutoDock Vina docking program were also employed. Thus, compounds with docking scores more negative than −8 were selected for docking with iDock and AutoDock Vina (see 2.7 Lead compounds selection criteria). The molecular mechanics generalized born surface area (MM-GBSA) methods were used for binding free energy (ΔG bind) calculation of these selected ligand molecules.
2.4.2. AutoDock Vina and iDock
The AutoDock Vina (Trott & Olson, 2009) and iDock (Li et al., 2012) programs were used for further validating the findings of Glide docking analysis of selected 23 ligands (8 from Lib-A, six from Lib-B and nine from Lib-C). All the ligands were converted into ‘pdbqt’ format using the AutoDock tools. The dimensions of the grid box and receptor setup were similar to the Glide docking grid box edges.
2.5. MM-GBSA and binding free energies
The OPLS 3e Force field of Schrodinger suite module was used for prime MM/GBSA calculation with ΔG bind between protein and ligand complexes (Genheden & Ryde, 2015). The prime MM-GBSA strategy was used to calculate the ΔG bind of every selected ligand. Thus, the top three compounds extracted after the docking process underwent the ΔG scores. Therefore, the final prioritization of the optimized lead compound was based on docking scores, ADME/T studies and ΔG bind, which were further followed up by the molecular dynamic studies.
2.6. ADME/T screening
The drug likeliness properties of the final 23 compounds from Lib-A, B and C, were calculated by using SwissADME (Daina et al., 2017) and admetSAR-2.0 online tools (http://lmmd.ecust.edu.cn/admetsar2/). The predicted result consists of physiochemical properties, lipophilicity, water-solubility, pharmacokinetics, drug-likeness and toxicity studies.
The physicochemical properties include molecular weight (MW), number of the rotatable bonds (NRB), hydrogen bond acceptor (HBA), hydrogen bond donor (HBD), molar refractivity (MR) and topological surface area (TSA). The other two significant determinant are lipophilicity and solubility that are monitored for favorable drug development. The preceding parameter is predicted by using an average of five logarithmic predictions of the octanol-water coefficient (Po/w). They include consensus of values based on atomistic and knowledge Log Po/w (XLOGP3), physics Log Po/w (iLOGP), atomistic Log Po/w (WLOGP), topological Log Po/w (MLOGP) and hybrid fragmental/topological Log Po/w (SILICOS-IT). The latter property is also based on logarithmic solubility (Log S) predicted values scale. The solubility characteristic of the compounds as per this scale is defined as insoluble if more negative than −10. The solubility ranges from poorly soluble to highly soluble corresponding to the value of −10 to greater than zero, respectively. The values of the poorly soluble compounds lie in between −10 and − 6. The higher than −6 and less than −4 is classified as moderately soluble. The soluble compounds are in between −4 and −2. The values between −2 and 0 are very soluble, while higher than zero are highly soluble.
The estimation of different pharmacokinetic properties such as ADME/T along with drug likeliness was done for these compounds by applying three established methods such as Lipinski’s rule of 5 (Lipinski, 2004), Veber’s rule and Muegge’s rule (Muegge, 2003).
The Lipinski’s rule defines an orally active drug, which confirms to the number of hydrogen bonds acceptor ≤ 10, HBD ≤ 5, MW < 500 Da and LogP (The logarithm of Octanol-water partition coefficient) ≤ 5. However, Veber’s rule takes two parameters to determine the good oral bioavailability in the majority of the compounds, such as < 10 rotatable bonds (ROTB) and polar surface area (PSA) less than 140 Å. Moreover, according to the Muegge’s rule, for a drug like chemical compound to become a successful drug molecule, it has to pass a pharmacophore point filter such as MW between 200 and 600 Da, XLogP −2 to 5, TPSA less than 150, number of rings less than 7, number of carbon less than 4, number of heteroatoms less than 1, number of rotatable bonds less than 15, HBA less than 10, HBD less than 5.
The prediction of toxicity was made by a new online admetSAR-2.0 webserver. The parameters such as human ether-a-go-go-related gene inhibition, AMES toxicity, acute oral toxicity (c) and carcinogenicity (Class-3) were determined. The US EPA based criteria were used to categorize predicted LD50 values into four classes. The Class I comprises compounds with LD50 lower than or equal to 50 mg/kg. Class II includes the compounds with LD50 values higher than 50 mg/kg but lower than 500 mg/kg. The Class III compounds have values more than 500 mg/kg but lower than 5000 mg/kg. The compounds with values higher than 5000 mg/kg are classified as Class IV. However, the rat carcinogenicity is classified as per the Carcinogenicity Potency Database of TD 50 values as ‘Danger’, Warning’ and ‘Non-required’. The ’Danger’ compounds have TD50 value of ≤ 10 mg/kg body wt./day. The ’Warning’ compounds have TD50 > 10 mg/kg body wt./day. However, non-carcinogenic compounds are assigned as ’Non-required’.
2.7. Lead compounds selection criteria
The selection of the final lead compounds was made with two sets of selection criteria extends over two phases of the process based on criteria such as the protein-ligand binding affinity, binding free energy, drug likeliness properties, physiochemical interactions between ligand and the target protein. The Glide docking score less than −8 was taken for the selection of ligands in the phase one- selection criteria. The further selection of the potential compounds in the second phase involves consideration of four parameters. These were docking score lower than or equal to the co-crystal ligand docking scores with respective docking tools, binding fee energy less than −60 (kcal/mol), ability to interact with a minimum of four amino acids implicated in positioning the acceptor substrate and drug likeliness properties through Lipinski’s rule of five, Veber’s rules criteria, Muegge’s rules and toxicities study. These filtering criteria would lead us to conclude the potential compounds for the MDSs and drug designing against SARS CoV-2.
2.8. Molecular dynamics simulations
The MDS studies were performed to analyze the stability of ligand-protein interactions with respect to the physical transition of the structural aspect of macromolecules to the functional relevance of the complex. Additionally, it demonstrates strength, pattern, dynamic conformational changes and intermolecular properties of the interactions. The selected complex then underwent MDS for 150 ns using AMBER 18 software used for the MDSs of ligand-protein complexes. It evaluates the root mean square deviation (RMSD), root mean square fluctuation (RMSF), intermolecular hydrogen bonds interactions, binding free energy and radius of gyration (RoG) for the elucidation of conformational, structural and compactness of the protein-ligand (Nandekar et al., 2013).
The Leap module of the software created the molecular receptor topology. The ligand-protein complex obtained from molecular docking was the starting structure of each MD simulations. Subsequently, followed established protocol such as neutralization and submerging complex into the water molecule rectangular box (TIP3P) where the protein atoms were 10 Å away from the nearest edge of the box. The minimization of the solvent system was achieved by freezing the protein and removing the bad contact with restraint on the heavy atoms, first through 2500 steepest descent method and then conjugation gradient for 2000 further steps. The system was gradually heated from 0 to 300 K temperature at 1 atm pressure (NPT conditions). It was achieved for 50 ps followed by 50ps of density equilibrium and 1 ns of constant equilibrium to exchange potential and kinetic energies. Afterward, the temperature was kept constant by using the Berendsen algorithm.
The MDS was run for 150 ns to evaluate the stability of the docked ligand-receptor complexes preceded by the system equilibration for 500ps on the canonical NPT ensemble. The covalently bound hydrogen atoms were constrained by the SHAKE algorithm with 2 fs time and temperature control by Langevin dynamics. Finally, the recording was made for every 5ps using the particle-mesh Ewald summation method for treating electrostatic interaction. The CPPTRAJ module of the Amber18 software was used to analyze the MD trajectories for every 20 ns. The MM-GBSA binding free energy was calculated by inbuilt Amber Tools. The RMSD, RMSF and RoG values were recorded for 150 ns.
3. Result and discussion
3.1. Reliability of protein structure and grid generation
The process of drug designing necessitates the accuracy of the quality and reliability of the three-dimensional structure of the target protein. That can be judged by using the PROCHECK server to develop Ramachandran plot, which displays allowed, and the disallowed regions regarding backbone dihedrals of protein residues. The essential condition of being a model of good quality having more than 90% of residues in favored regions. This determines a good worth of stereo-chemical quality of three-dimensional protein structure with minimum steric interaction concerning the forbidden psi and phi angles. Figure 3 and Table 1 show that 91.2% residues lie in the most favored region, and only 0.6% lie in the disallowed region of the three-dimensional structure of target protein (PDB ID: 7BRP). The active site was predicted after the selection of the co-crystal ligand of the target protein. The receptor grid of the protein was done similarly to the co-crystal ligands of the protein. Their identification helps in designing potent protein inhibitors through their binding sites.
Figure 3.

Ramachandran plots of 7brp proteins structure describe favored and the disallowed.
Table 1.
Ramachandran plot statistics showing residues present in favored and disallowed regions of protein structure of main protease from SARS CoV-2.
| Properties | Residues | Percentage (%) |
|---|---|---|
| The most favored regions | 476 | 91.2% |
| Additional allowed regions | 43 | 8.2% |
| Generously allowed regions | 0 | 0.0% |
| Disallowed regions | 3 | 0.6% |
| Number of non-glycine and non-proline residues | 522 | 100.0% |
| The end residues (excluding Glycine and Proline) | 4 | |
| Number of glycine residues (shown as triangles) | 50 | |
| Glycine | 26 | |
| Total number of residues | 602 | 100.0% |
3.2. Virtual screening and molecular docking, binding free energy calculation
Identification of active site residues and location of the target structures help in designing potent drug molecules via ligand-protein docking. The active site was predicted after the selection of the three-dimensional structure of the target protein. Twenty-one thousand two hundred seven compounds were screened and docked against the target protein (Figure 4(A)). After virtual screening and molecular docking, 23 compounds were finalized (eight from Lib-A, six from Lib-B and nine from Lib-C) in first phase selection criteria based on their Glide docking scores (less than −8 docking score) with the target proteins. These 23 ligand molecules were selected for ADMET prediction, ΔG bind calculation and docking with AutoDock Vina and iDock. Three ligand molecules (two compounds from Lib-B and one compound from Lib-C) were found to be potential hit candidates for further exploration after the second phase (see lead compounds selection criteria in the Material and Methods section). The comparative docking score of these three compounds shown in Figure 4(B) with respect to the reference ligands.
Figure 4.

(A) Glide docking scores of selected 23 compounds from Lib-A, Lib-B and Lib-C. (B) Comparative docking score of best three ligands with respect to reference ligand molecule (co-crystal ligand).
3.2.1. Docking score (glide, iDock and AutoDock vina) and binding free energy (ΔG bind) calculation by prime MM-GBSA of top eight compounds from Library-A
Table 2 (A-1 to A-8) shows the two-dimensional structures, free binding energy, docking scores, number of hydrogen bonds and interactive residues of best eight ligand molecules from Library-A. These eight ligand molecules had less than −8 docking scores with glide docking in phase one. Their free binding energies and docking scores, such as Glide, iDock and AutoDock Vina scores were recorded.
Table 2.
Two-dimensional structure, free binding energy, docking scores, number of hydrogen bond and interactive residues of best 23 ligands (from Lib-A: A-1 to A-8; Lib-B: B-9 to B-14 and Lib-C: C-15 to C-23) with target protein structure of SARS CoV-2.
| S/No | Life chemicals ID | 2D structures | ΔD structures ID | Docking
score (kcal/mol) |
NHB | H-bond interactive residues Chain A | ||
|---|---|---|---|---|---|---|---|---|
| Glide | iDock | AutoDock Vina | ||||||
| A-1 | F0015-0201 | 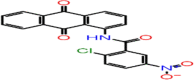 |
–60.87 | –8.817 | –10.46 | –8.5 | 3 | GLU166 GLY143 CYS145 |
| A-2 | F0265-1326 | 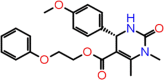 |
–64.79 | –8.799 | –7.68 | –7.00 | 4 | LEU141 GLY143 CYS145 GLU166 |
| A-3 | F1641-0167 |  |
–51.52 | –8.446 | –8.35 | –7.3 | 3 | GLY143 SER144 CYS145 |
| A-4 | F1011-1885 |  |
–51.51 | –8.31 | –8.08 | –7.6 | 5 | HIS164 LEU141 GLY143 SER144 CYS145 |
| A-5 | F0886-0045 | 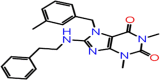 |
–67.95 | –8.148 | –8.70 | –6.5 | 4 | GLY143 SER144 CYS145 HIS41 |
| A-6 | F2711-0202 | 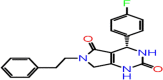 |
–56.93 | –8.113 | –8.63 | –7.6 | 4 | LEU141 GLY143 CYS145 GLU166 |
| A-7 | F1057-0056 | 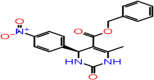 |
–61.60 | –8.093 | –8.16 | –7.1 | 4 | LEU141 GLY143 CYS145 GLU166 |
| A-8 | F0452-4293 | 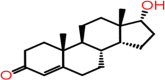 |
–44.90 | –8.008 | –8.24 | –7.3 | 4 | GLY143 SER144 CYS145 ARG188 |
| B-9 | F2679-0163 | 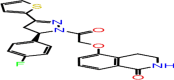 |
–61.37 | –10.75 | –10.29 | –9.2 | 4 | LEU141 GLY143 CYS145 GLU166 |
| B-10 | F6355-0442 | 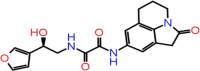 |
–59.75 | –9.38 | –8.61 | –7.6 | 4 | GLU166(x2) CYS145 GLY143 |
| B-11 | F0648-0053 |  |
–68.78 | –8.99 | –9.09 | –8.5 | 4 | GLN192 GLU166 CYS145 GLY143 |
| B-12 | F0648-0756 |  |
–63.03 | –8.584 | –8.81 | –8.1 | 4 | GLY143 SER144 CYS145 GLU166 |
| B-13 | F0612-0047 | 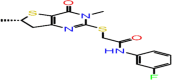 |
–56.87 | –8.56 | –7.93 | –6.8 | 4 | GLY143 SER144 CYS145 GLU166 |
| B-14 | F2644-0465 | 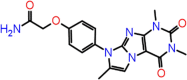 |
–62.56 | –8.46 | –8.50 | –7.6 | 5 | THR190 GLN192 GLY143 SER144 CYS145 |
| C-15 | 8250 | 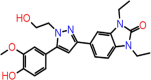 |
–63.70 | –9.795 | –7.94 | –7.5 | 4 | THR190 GLY43 SER144 CYS145 |
| C-16 | 7017 | 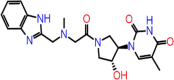 |
–55.29 | –8.986 | –9.32 | –8.3 | 5 | GLU166(x2) GLN189 CYS145 SER144 |
| C-17 | 5804 | 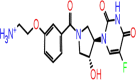 |
–58.71 | –8.672 | –8.37 | –7.4 | 7 | THR190 GLU166 LEU141 GLY143 CYS145 THR26(x2) |
| C-18 | 8396 | 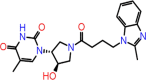 |
–63.62 | –8.402 | –9.12 | –7.8 | 6 | THR26(x2) CYS145 GLY143 LEU141 GLU166 |
| C-19 | 5510 | 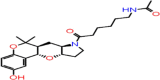 |
–65.92 | –8.4 | –8.61 | –7.2 | 5 | GLU166(x2) HIE41 ASN142 THR26 |
| C-20 | 728 | 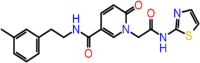 |
–58.57 | –8.388 | –8.49 | –7.5 | 3 | GLY143 CYS145 THR26 |
| C-21 | 7018 | 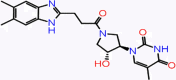 |
–48.74 | –8.369 | –8.86 | –7.9 | 4 | ARG188 CYS145 SER144 GLY143 |
| C-22 | 2676 | 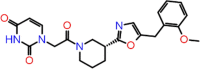 |
–64.03 | –8.358 | –8.71 | –8.1 | 3 | CYS145 SER144 GLY143 |
| C-23 | 8489 | 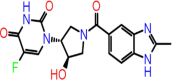 |
–62.32 | –8.212 | –9.67 | –8.1 | 5 | LEU141 CYS145 THR26(x2) GLU166 |
The range of ΔG bind lies between −60.87 and −67.88 of the most notable top eight compounds. The docking scores of these eight compounds are between −8.0 and −8.81 with Glide; −7.68 to −10.46 with iDock and −6.5 to −8.5 with AutoDock Vina methodology. The F0015-0201 compound had good docking scores of −8.817, −10.46 and −8.5 with Glide, iDock and AutoDock Vina, respectively, but it was not proceeded for further MDS study because of unfavorable AMES toxicity results. The number of hydrogen bonds made and interactions with amino acid residues are shown in Table 2. Out of the total eight compounds, one interacted with five hydrogen bonds, five of them formed four hydrogen bonds, and the rest two compounds interaction with three hydrogen bonds. All hydrogen bond interactive amino acids are HIS164, LEU141, GLY143, SER144, CYS145, GLU166, ARG188, but interestingly, all compounds interaction was shown mainly with two common amino acids GLY143, CYS145.
3.2.2. Docking score (glide, iDock and AutoDock vina) and binding free energy (ΔG bind) calculation by prime MM-GBSA of top-six compounds from Library-B
Table 2 (B-9 to B-14) shows two-dimensional structures, binding free energy, docking score, hydrogen bond and hydrogen bond interactive residues of best six compounds selected from Lib-B. Their range of ΔG bind lies between −56.87 and −68.78 of most notable top six compounds. The docking scores of these six compounds are between −8.46 and −10.75 with Glide, −8.46 to −10.75 with iDock and 6.8 to −9.2 with AutoDock Vina. Programs.
One compound forms five hydrogen bond interactions while the other five compounds have shown four hydrogen bond interactions with the target protein. All hydrogen bond interactive amino acids are LEU141, GLU166, THR190, GLN192, GLY143, SER144 and CYS145. All six compounds have shown interaction with two common residues GLY143, CYS145, similar to Lib-A. Finally, two compounds were selected for further MDS from Lib-B as they pass all the selection criteria (see lead compounds selection criteria in the Material and Methods section).
The first compound (F2679-0163) has a good docking score of −10.75, −10.29 and 9.2 with Glide, iDock and AutoDock Vina, respectively. Its binding free energy is −61.37 and forms four hydrogen bond interactions with LEU141, GLY143, CYS145 and GLU166 (Figure 5). Docking scores of second compounds with Glide, iDock, AutoDock Vina are −9.38, −8.61 and 7.6, respectively, and four hydrogen bond interactions with GLU166(x2), CYS145, GLY143. The ΔG bind of the second compounds is −59.75 (≈ −60.00). Both compounds have shown good docking scores with all docking methods and more negative than crystal ligand Glide docking scores.
Figure 5.

Binding mode and chemical interactions of best three lead molecules with residues.
3.2.3. Docking score (glide, iDock and AutoDock vina) and binding free energy (ΔG bind) calculation by prime MM-GBSA of top nine compounds from Library-C
Table 2 (C-15 to C-23) elaborates on the best nine compounds from Lib-C (Asinex Antiviral compound database) concerning the two-dimensional structure, ΔG bind, docking scores, number of hydrogen bonds and hydrogen bond associated amino acids.
The ΔG bind lies between −48.74 and −65.92 of the most notable top nine compounds from Lib-C. The docking score of these nine compounds is between −8.212 and −9.795 with Glide, −7.94 to −9.67 with iDock and −7.2 to −8.3 with Auto Dock Vina. Among all nine compounds, one compound formed seven hydrogen bond interactions, one-formed six, the other three formed five, two of them formed four and rest two compounds formed three hydrogen bond interactions with the target protein. All hydrogen bond interactive amino acids are HIS164, LEU141, GLY143, SER144, CYS145, GLU166 and ARG188. Among these residues, most of the compounds interact with three amino acid residues, including CYS145, SER144 and GLY143. One compound was passed all the selection criteria (see lead compounds selection criteria in the Material and Methods section) and selected for further MDS from Lib-C among all the best nine compounds.
One compound stands out with its parameter in this database, which can be worthy of further development of drugs against the Mpro of SARS CoV-2. The ΔG bind of this compound (Asinex ID: 8250) is −63.70 and Glide, iDock and AutoDock Vina docking scores are of −9.795, 7.94 and 7.5, respectively. Four hydrogen bond interactions formed with THR190, GLY43, SER144 and CYS145 (Figure 5).
3.3. Physiochemical properties, ADME/T and drug likeness properties
In silico tools such as SwissADME and admetSAR −2.0 webserver can lead to early predictions of Physiochemical properties, ADME/T and drug-likeness properties. The oral bioavailability of the possible active compounds was calculated through Lipinski’s rule of five and Veber’s rule. While the Muegge’s rule determined the possibility of a compound to become a successful drug molecule by the pharmacophore point calculation. All the compounds followed the Lipinski, Veber’s and Muegge’s rule, and their values are shown in Table 3. Therefore, final compounds were predicted to have good bioavailability and satisfied the drug likeliness parameters according to these rules. Moreover, 20 compounds show high human intestinal absorption except three compounds (F0648-0053, F0648-0756 and 5804) which have low absorption.
Table 3.
The physiochemical, lipophilicity, water solubility, pharmacokinetics, drug likeliness and toxicity predictions of the best 23 antiviral small molecules selected from Lib-A (A-1 to A-8), Lib-B (B-9 to B-14) and Lib-C (C-15 to C-23).
| S/N | Ligand ID | SwissADME
tools |
admetSAR -
2.0 |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Physiochemical
properties |
Lipophilicity |
Water
solubility |
Pharmacokinetics |
Drug
likeness |
Toxicity
prediction |
||||||||||||
| MW | NRB | HBA | HBD | MR | TPSA | Consensus Log Po/w | Class | GI absorption | Lipinski | Veber | Muegge | Human ether-a-go-go-related gene inhibition | AMES toxicity | Acute oral toxicity (c) | Carcinogenicity (three-class) | ||
| A-1 | F0015-0201 | 406.78 | 4 | 5 | 1 | 107.8 | 109.06 | 3.17 | Moderately | High | 0 | 0 | 0 | Weak inhibitor | AMES toxic | III | Non-required |
| A-2 | F0265-1326 | 396.44 | 8 | 5 | 1 | 115.18 | 77.1 | 2.68 | Soluble | High | 0 | 0 | 0 | Weak inhibitor | Non-AMES toxic | III | Non-required |
| A-3 | F1641-0167 | 317.34 | 4 | 3 | 0 | 95.38 | 46.61 | 3.16 | Moderately | High | 0 | 0 | 0 | Weak inhibitor | Non-AMES toxic | III | Non-required |
| A-4 | F1011-1885 | 386.79 | 5 | 4 | 3 | 109.74 | 116.05 | 1.79 | Soluble | High | 0 | 0 | 0 | Weak inhibitor | AMES toxic | III | Non-required |
| A-5 | F0886-0045 | 403.48 | 6 | 3 | 1 | 120.09 | 73.85 | 3.15 | Moderately | High | 0 | 0 | 0 | Strong inhibitor | Non-AMES toxic | III | Non-required |
| A-6 | F2711-0202 | 351.37 | 4 | 3 | 2 | 105.83 | 61.44 | 2.23 | Soluble | High | 0 | 0 | 0 | Strong inhibitor | Non-AMES toxic | III | Non-required |
| A-7 | F1057-0056 | 367.36 | 6 | 5 | 2 | 106.28 | 113.25 | 1.71 | Soluble | High | 0 | 0 | 0 | Weak inhibitor | AMES toxic | III | Non-required |
| A-8 | F0452-4293 | 288.42 | 0 | 2 | 1 | 85.36 | 37.3 | 3.46 | Soluble | Soluble | 0 | 0 | 0 | Weak inhibitor | Non-AMES toxic | III | Warning |
| B-9 | F2679-0163 | 449.5 | 6 | 5 | 1 | 130.64 | 99.24 | 3.77 | Moderately | 0 | 0 | 0 | Weak inhibitor | Non-AMES toxic | III | Non-required | |
| B-10 | F6355-0442 | 369.37 | 7 | 5 | 3 | 99.14 | 111.88 | 0.79 | Soluble | High | 0 | 0 | 0 | Weak inhibitor | Non-AMES toxic | III | Non-required |
| B-11 | F0648-0053 | 492.51 | 8 | 7 | 3 | 128.48 | 169.29 | 2.11 | Soluble | Low | 1 | 1 | 1 | Weak inhibitor | Non-AMES toxic | III | Non-required |
| B-12 | F0648-0756 | 459.48 | 8 | 7 | 3 | 113.24 | 191.96 | 1.93 | Soluble | Low | 0 | 1 | 1 | Weak inhibitor | Non-AMES toxic | III | Non-required |
| B-13 | F0612-0047 | 365.45 | 5 | 4 | 1 | 94.65 | 114.59 | 2.81 | Soluble | High | 0 | 0 | 0 | Weak inhibitor | Non-AMES toxic | III | Non-required |
| B-14 | F2644-0465 | 382.37 | 4 | 5 | 1 | 102.24 | 118.55 | 0.79 | Soluble | High | 0 | 0 | 0 | Weak inhibitor | Non-AMES toxic | III | Non-required |
| C-15 | 8250 | 422.48 | 7 | 5 | 2 | 120.74 | 94.44 | 2.66 | Soluble | High | 0 | 0 | 0 | Weak inhibitor | Non-AMES toxic | III | Non-required |
| C-16 | 7017 | 412.44 | 6 | 6 | 3 | 114.29 | 127.32 | 0.23 | Soluble | High | 0 | 0 | 0 | Weak inhibitor | Non-AMES toxic | III | Non-carcinogens |
| C-17 | 5804 | 379.36 | 6 | 6 | 3 | 97.8 | 132.27 | –0.85 | Very Soluble | Low | 0 | 0 | 0 | Weak inhibitor | Non-AMES toxic | III | Non-carcinogens |
| C-18 | 8396 | 411.45 | 6 | 5 | 2 | 116.3 | 113.22 | 1.06 | Soluble | High | 0 | 0 | 0 | Weak inhibitor | Non-AMES toxic | III | Non-required |
| C-19 | 5510 | 430.54 | 8 | 5 | 2 | 121.63 | 88.1 | 2.62 | Soluble | High | 0 | 0 | 0 | Weak inhibitor | Non-AMES toxic | III | Non-required |
| C-20 | 728 | 396.46 | 9 | 4 | 2 | 108.73 | 121.33 | 2.31 | Soluble | High | 0 | 0 | 0 | Weak inhibitor | Non-AMES toxic | III | Non-required |
| C-21 | 7018 | 411.45 | 5 | 5 | 3 | 116.52 | 124.08 | 1.3 | Soluble | High | 0 | 0 | 0 | Weak inhibitor | Non-AMES toxic | III | Non-required |
| C-22 | 2676 | 424.45 | 7 | 6 | 1 | 116.82 | 110.43 | 1.75 | Soluble | High | 0 | 0 | 0 | Weak inhibitor | Non-AMES toxic | III | Non-required |
| C-23 | 8489 | 373.34 | 3 | 6 | 3 | 97.16 | 124.08 | 0.6 | Soluble | High | 0 | 0 | 0 | Weak inhibitor | Non-AMES toxic | III | Non-required |
Log S scale predicted the solubility level of 23 compounds, out of which, one compound is very soluble, four compounds are moderately soluble, and remaining18 compounds are soluble. Out of 23 compounds, three compounds (F0015-0201, F1011-1885 and F1057-0056), have AMES toxic nature, and one (F0452-4293) has carcinogenic nature based on the toxicity screening results of ligands. The rest 19 compounds were relatively safe (Table 3).
3.4. Molecular dynamics simulations
The compounds that qualify two-phase lead optimization screening selection criteria were taken for the molecular dynamics studies. Out of the 23 compounds, three compounds (F2679-0163, F6355-0442 and 8250) fulfilled all the parameters further needed for molecular dynamic simulations. The parameters explored for analyses such as RMSD, RMSF, inter-molecular hydrogen bonding (H bonding), RoG and binding free energy through MDS.
3.4.1. RMSD analysis
The RMSD assess the structural stability of the protein-ligand complexes within a particular period. The behavior of protein-ligand complexes was calculated as a function of time performed over 150 ns by MDS. The RMSD values of protein-ligand are depicted in Figure 6(A). The ligand F6355-0442 is gradually reached the stabilized value of 2.5 Å in 50 ns and remained at the same value for 150 ns. However, it showed slightly non-significant fluctuation around 90 and 130 ns. Moreover, the RMSD graph for ligand F2679-0163 showed a similar pattern to maintain the stabilization. However, the ligand 8250 reached the average value of 2.5 Å only in 10 ns and remained there for 140 ns. Additionally, it showed little higher values at the end of the plot for the last 10 ns. Thus, all three compounds were stabilized for the majority of the MDS time duration.
Figure 6.

(A) Molecular dynamics simulation (MDS) results of three ligand-protein complex for 150 ns. (B) Mean square fluctuation (RMSF) results of three ligand-protein complex for 150 ns.
3.4.2. RMSF analysis
The RMSF determines the deviation of the particle from their original position in the macromolecule three-dimensional structure. It details the conformational flexibility of the protein structure by identifying the flexible and rigid structures of the proteins. We find the high fluctuations in the loop of three protein-ligand complexes (Mpro-F2679-0163, Mpro-8250 and Mpro-F6355-0442) depicting their accommodating nature of the binding site of the proteins (Figure 6(B)). It measures the deviation of the particle from their original positions in the rigid, flexible regions.
3.4.3. Intermolecular hydrogen bonding
The number of hydrogen bonds formed between ligand and protein measures the binding affinity of the ligand. Therefore, more number of a hydrogen bonds between the protein and ligands depict stronger binding affinity. Therefore, MDS analysis takes into consideration the formation and deformation of hydrogen bonds. Our study observed a maximum of six hydrogen bonds at the end of 150 ns simulation in the case of ligand F6355-0442. The Mpro-8250 and Mpro- F2679-0163 show five and three HB, respectively (Figure 7(A)).
Figure 7.

(A) Number of Intermolecular hydrogen bonds between the ligands and amino acid residues of the target protein for 150 ns. (B) Radius of gyration (RoG) results of the ligand-protein complex for 150 ns.
3.4.4. RoG analysis
The folding and compactness of the proteins-ligand complexes can be judged with the help of RoG. It is an important method of revealing the influence of ligand molecules on the three-dimensional conformational structural changes after the interaction of the ligand with it. The high value of RoG depicts the sustenance of loose packing and folding behavior of the protein after the interaction with the ligands. All three ligand-protein complexes show an average high value of around 22 Å throughout the 150 ns (Figure 7(B)). They are in its native structure as there was not much variation observed throughout the 150 ns in the RoG graph except in case of ligand 8250 where values went even higher gradually starting from 110 to 150 ns. However, there were few minor variations in RoG due to conformational changes in the secondary structure of protein during the MDS process. The RoG graph of all three complexes (Figure 7(B)) shows that ligand remains tightly bound to the active site of the protein.
3.4.5. MM-GBSA calculations
The average binding free energy (ΔG) of these three compounds was calculated for every 20 ns by the MM-GBSA approach up to 150 ns. Table 4 shows the change of ΔG values at each data point along with their standard deviations. The minimum values of ΔG for ligands F2679-0163, F6355-0442 and 8250 were −22.90 ± 2.97 Kcal/mol (90 ns), −24.78 ± 3.38 Kcal/mol and (150 ns) −35.94 ± 3.05 (10 ns) and maximum values were −38.99 ± 2.73 Kcal/mol (30 ns), −30.61 ± 2.47 Kcal/mol (90 ns) and −42.44 ± 3.35 Kcal/mol (110 ns), respectively. Therefore, the order of binding free was in a favorable range that can be appreciated graphically in Figure 8.
Table 4.
Average binding free energy (ΔG) with standard deviation (SD) values for three ligand-protein complexes at 20 nanosecond (ns) intervals.
| Average
binding free energy (ΔG) (Kcal/mol)/standard
deviation (SD) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Time | 10 ns | 30 ns | 50 ns | 70 ns | 90 ns | 110 ns | 130 ns | 150 ns |
| Ligand-protein complex | ||||||||
| 7brp_0163 | –37.50 ± 5.58 | –38.99 ± 2.73 | –33.05 ± 3.25 | –34.77 ± 3.20 | –27.99 ± 3.33 | –22.90 ± 2.97 | –23.78 ± 3.89 | –25.22 ± 3.09 |
| 7brp_0442 | –29.24 ± 3.75 | –27.98 ± 2.71 | –30.46 ± 3.32 | –30.04 ± 2.56 | –30.61 ± 2.47 | –30.22 ± 2.09 | –30.13 ± 2.33 | –24.78 ± 3.38 |
| 7brp_8250 | –35.94 ± 3.05 | –42.38 ± 2.87 | –42.01 ± 3.79 | –40.93 ± 3.17 | –41.32 ± 2.92 | –42.44 ± 3.35 | –41.76 ± 3.49 | –41.24 ± 3.54 |
Figure 8.

Binding free energy between protein-ligand (F2679-0163, F6355-0442 and 8250) complexes at every 20 nanoseconds for 150 ns.
4. Conclusions
The study has used the main protease of SARS CoV-2 as it is the recently released crystal structure (PDB ID: 7brp) and is highly conserved, predominantly responsible for posttranslational processing of various viral proteins. This study was conducted with 21,207 compounds from Life Chemicals and Asinex database to find the potential drug molecules through computational drug designing techniques. The molecular docking results showed a good binding affinity with the target protein of SARS CoV-2. The docking and MDSs studies have delineated three chemical compounds, which can be the potential drug molecules against the main protease of SARS-CoV-2. Two final compounds (Compound id: F2679-0163, F6355-0442) are from the Life Chemicals library, and the third chemical compounds library having an antiviral property is from the Asinex database (Compound ID: 8250). All three ligand-protein complexes have favorable parameter values in RMSD, RMSF, RoG, intermolecular hydrogen bonding and binding free energy for 150 ns. These compounds (F2679-0163, F6355-0442 and 8250) can be further explored for in vitro experimental validation against SARS-CoV-2.
Acknowledgments
Thanks to the Department of Pharmacology, AIIMS, Rishikesh for providing the facility. Special thanks to Dr. Prajwal Nandekar, Dr. Vinod and Schrödinger's team for their corporation.
Glossary
Abbreviations
- CoV
coronavirus
- COVID–19
coronavirus disease 2019
- FDA
food and drug administration
- HB
hydrogen bond
- MDS
molecular dynamics simulation
- MERS
middle east respiratory syndrome
- PDB
Protein Data Bank
- SARS
severe acute respiratory syndrome
- SP
standard precision
- WHO
World Health Organization
- XP
extra precision
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Aanouz, I., Belhassan, A., El-Khatabi, K., Lakhlifi, T., El-Ldrissi, M., & Bouachrine, M. (2020). Moroccan medicinal plants as inhibitors against SARS-CoV-2 main protease: Computational investigations. Journal of Biomolecular Structure and Dynamics, 38, 1–9. 10.1080/07391102.2020.1758790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Báez-Santos, Y. M., St. John, S. E., & Mesecar, A. D. (2015). The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antiviral Research, 115, 21–38. 10.1016/j.antiviral.2014.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow, A., Landolf, K. M., Barlow, B., Yeung, S. Y. A., Heavner, J. J., Claassen, C. W., & Heavner, M. S. (2020). Review of Emerging Pharmacotherapy for the Treatment of Coronavirus Disease 2019. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy, 40(5), 416–437. 10.1002/phar.2398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj, V. K., Singh, R., Sharma, J., Rajendran, V., Purohit, R., & Kumar, S. (2020). Identification of bioactive molecules from tea plant as SARS-CoV-2 main protease inhibitors. Journal of Biomolecular Structure and Dynamics, 38, 1–10. 10.1080/07391102.2020.1766572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar, T., Murhekar, M. V., Soneja, M., Gupta, N., Giri, S., Wig, N., & Gangakhedkar, R. (2020). Lopinavir/ritonavir combination therapy amongst symptomatic coronavirus disease 2019 patients in India: Protocol for restricted public health emergency use. The Indian Journal of Medical Research, 151(2&3), 184–189. 10.4103/ijmr.IJMR_502_20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boopathi, S., Poma, A. B., & Kolandaivel, P. (2020). Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. Journal of Biomolecular Structure and Dynamics, 38, 1–14. 10.1080/07391102.2020.1758788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caly, L., Druce, J. D., Catton, M. G., Jans, D. A., & Wagstaff, K. M. (2020). The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antiviral Research, 178, 104787. 10.1016/j.antiviral.2020.104787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, B., Wang, Y., Wen, D., Liu, W., Wang, J., Fan, G., Ruan, L., Song, B., Cai, Y., Wei, M., Li, X., Xia, J., Chen, N., Xiang, J., Yu, T., Bai, T., Xie, X., Zhang, L., Li, C., … Wang, C. (2020). A trial of lopinavir-ritonavir in adults hospitalized with severe covid-19. The New England Journal of Medicine, 382(19), 1787–1799. 10.1056/NEJMoa2001282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascella, M., Rajnik, M., Cuomo, A., Dulebohn, S. C., & Di Napoli, R. (2020). Features, evaluation and treatment coronavirus (COVID-19). StatPearls. http://www.ncbi.nlm.nih.gov/pubmed/32150360 [PubMed] [Google Scholar]
- Chen, Y. W., Yiu, C.-P B., & Wong, K.-Y. (2020). Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CL pro) structure: Virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Research, 9, 129. 10.12688/f1000research.22457.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherian, S. S., Agrawa, M., Basu, A., Abraham, P., Gangakhedkar, R. R., & Bhargava, B. (2020). Perspectives for repurposing drugs for the coronavirus disease 2019. Indian Journal of Medical Research, 151(2 & 3), 160–171. 10.4103/ijmr.IJMR_585_20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Costa, V. G., Moreli, M. L., & Saivish, M. V. (2020). The emergence of SARS, MERS and novel SARS-2 coronaviruses in the 21st century. Archives of Virology, 165(7), 1517–1526. 10.1007/s00705-020-04628-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina, A., Michielin, O., & Zoete, V. (2017). SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific Reports, 7(1), 1–13. 10.1038/srep42717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das, S., Sarmah, S., Lyndem, S., & Roy, A. S. (2020). An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. Journal of Biomolecular Structure and Dynamics, 38, 1–11. 10.1080/07391102.2020.1763201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky, A. A. (2020. a). Natural products may interfere with SARS-CoV-2 attachment to the host cell. Journal of Biomolecular Structure and Dynamics, 38, 1–10. 10.1080/07391102.2020.1761881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky, A. A. (2020. b). SARS-CoV-2 RNA dependent RNA polymerase (RdRp) targeting: An in silico perspective. Journal of Biomolecular Structure & Dynamics, 38, 1–15. 10.1080/07391102.2020.1761882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmezayen, A. D., Al-Obaidi, A., Şahin, A. T., & Yelekçi, K. (2020). Drug repurposing for coronavirus (COVID-19): In silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. Journal of Biomolecular Structure and Dynamics, 38, 1–13. 10.1080/07391102.2020.1758791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enmozhi, S. K., Raja, K., Sebastine, I., & Joseph, J. (2020). Andrographolide as a potential inhibitor of SARS-CoV-2 main protease: An in silico approach. Journal of Biomolecular Structure and Dynamics, 38, 1–10. 10.1080/07391102.2020.1760136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, H. H., Wang, L.-Q., Liu, W.-L., An, X.-P., Liu, Z.-D., He, X.-Q., Song, L.-H., & Tong, Y.-G. (2020). Repurposing of clinically approved drugs for treatment of coronavirus disease 2019 in a 2019-novel coronavirus-related coronavirus model. Chinese Medical Journal, 133(9), 1051–1056. 10.1097/CM9.0000000000000797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehr, A. R., & Perlman, S. (2015). Coronaviruses: An overview of their replication and pathogenesis’, In HelenaJane Maier et al. (Eds.), Coronaviruses: Methods and Protocols, 1–23. New York: Springer Science. 10.1007/978-1-4939-2438-7_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, Y., Yan, L., Huang, Y., Liu, F., Zhao, Y., Cao, L., Wang, T., Sun, Q., Ming, Z., Zhang, L., Ge, J., Zheng, L., Zhang, Y., Wang, H., Zhu, Y., Zhu, C., Hu, T., Hua, T., Zhang, B., … Rao, Z. (2020). Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science (New York, N.Y.), 368(6492), 779–782. 10.1126/science.abb7498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genheden, S., & Ryde, U. (2015). The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opinion on Drug Discovery, 10(5), 449–461. 10.1517/17460441.2015.1032936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, S., Singh, A. K., Kushwaha, P. P., Prajapati, K. S., Shuaib, M., Senapati, S., & Kumar, S. (2020). Identification of potential natural inhibitors of SARS-CoV2 main protease by molecular docking and simulation studies. Journal of Biomolecular Structure and Dynamics, 38, 1–19. 10.1080/07391102.2020.1776157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halgren, T. A., Murphy, R. B., Friesner, R. A., Beard, H. S., Frye, L. L., Pollard, W. T., & Banks, J. L. (2004). Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening . Journal of Medicinal Chemistry, 47(7), 1750–1759. 10.1021/jm030644s [DOI] [PubMed] [Google Scholar]
- Hasan, A., Paray, B. A., Hussain, A., Qadir, F. A., Attar, F., Aziz, F. M., Sharifi, M., Derakhshankhah, H., Rasti, B., Mehrabi, M., & Shahpasand, K. (2020). A review on the cleavage priming of the spike protein on coronavirus by angiotensin-converting enzyme-2 and furin. Journal of Biomolecular Structure and Dynamics, 38, 1–9. 10.1080/07391102.2020.1754293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havranek, B., & Islam, S. M. (2020). An in silico approach for identification of novel inhibitors as potential therapeutics targeting COVID-19 main protease. Journal of Biomolecular Structure & Dynamics, 38, 1–12. 10.1080/07391102.2020.1776158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendaus, M. A. (2020). Remdesivir in the treatment of coronavirus disease 2019 (COVID-19): A simplified summary. Journal of Biomolecular Structure and Dynamics, 38, 1–6. 10.1080/07391102.2020.1767691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam, R., Parves, M. R., Paul, A. S., Uddin, N., Rahman, M. S., Al Mamun, A., Hossain, M. N., Ali, M. A., & Halim, M. A. (2020). A molecular modeling approach to identify effective antiviral phytochemicals against the main protease of SARS-CoV-2. Journal of Biomolecular Structure & Dynamics, 38, 1–20. 10.1080/07391102.2020.1761883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesharwani, S. S., Nandekar, P. P., Pragyan, P., Rathod, V., & Sangamwar, A. T. (2016). Characterization of differences in substrate specificity among CYP1A1, CYP1A2 and CYP1B1: An integrated approach employing molecular docking and molecular dynamics simulations. Journal of Molecular Recognition: JMR, 29(8), 370–390. 10.1002/jmr.2537 [DOI] [PubMed] [Google Scholar]
- Krichel, B., Falke, S., Hilgenfeld, R., Redecke, L., & Uetrecht, C. (2020). Processing of the SARS-CoV pp1a/ab nsp7-10 region. The Biochemical Journal, 477(5), 1009–1019. 10.1042/BCJ20200029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, A., Choudhir, G., Shukla, S. K., Sharma, M., Tyagi, P., Bhushan, A., & Rathore, M. (2020). Identification of phytochemical inhibitors against main protease of COVID-19 using molecular modeling approaches. Journal of Biomolecular Structure and Dynamics, 38, 1–11. 10.1080/07391102.2020.1772112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H., Leung, K. S., Wong, M. H. (2012). Idock: A multithreaded virtual screening tool for flexible ligand docking [Paper presentation]. IEEE Symposium on Computational Intelligence and Computational Biology, CIBCB 2012 (pp. 77–84). IEEE. 10.1109/CIBCB.2012.6217214 [DOI] [Google Scholar]
- Lindner, H. A., Fotouhi-Ardakani, N., Lytvyn, V., Lachance, P., Sulea, T., & Ménard, R. (2005). The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. Journal of Virology, 79(24), 15199–15208. 10.1128/JVI.79.24.15199-15208.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski, C. A. (2004). Lead- and drug-like compounds: The rule-of-five revolution. Drug Discovery Today: Technologies, 1(4), 337–341. 10.1016/j.ddtec.2004.11.007 [DOI] [PubMed] [Google Scholar]
- Lu, R., Zhao, X., Li, J., Niu, P., Yang, B., Wu, H., Wang, W., Song, H., Huang, B., Zhu, N., Bi, Y., Ma, X., Zhan, F., Wang, L., Hu, T., Zhou, H., Hu, Z., Zhou, W., Zhao, L., … Tan, W. (2020). Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. The Lancet, 395(10224), 565–574. 10.1016/S0140-6736(20)30251-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavi Sastry, G., Adzhigirey, M., Day, T., Annabhimoju, R., & Sherman, W. (2013). Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. Journal of Computer-Aided Molecular Design, 27(3), 221–234. 10.1007/s10822-013-9644-8 [DOI] [PubMed] [Google Scholar]
- McBride, R., van Zyl, M., & Fielding, B. C. (2014). The coronavirus nucleocapsid is a multifunctional protein. Viruses, 6(8), 2991–3018. 10.3390/v6082991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muegge, I. (2003). Selection criteria for drug-like compounds. Medicinal Research Reviews, 23(3), 302–321. 10.1002/med.10041 [DOI] [PubMed] [Google Scholar]
- Nandekar, P. P., Tumbi, K. M., Bansal, N., Rathod, V. P., Labhsetwar, L. B., Soumya, N., Singh, S., & Sangamwar, A. T. (2013). Chem-bioinformatics and in vitro approaches for candidate optimization: A case study of NSC745689 as a promising antitumor agent. Medicinal Chemistry Research, 22(8), 3728–3742. 10.1007/s00044-012-0364-8 [DOI] [Google Scholar]
- Ortega, J. T., Serrano, M. L., Pujol, F. H., & Rangel, H. R. (2020). Role of changes in SARS-CoV-2 spike protein in the interaction with the human ACE2 receptor: An in silico analysis. EXCLI Journal, 19, 410–417. 10.17179/excli2020-1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal, M., Berhanu, G., Desalegn, C., & Kandi, V. (2020). Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2): An update. Cureus, 12(3), e7423. 10.7759/cureus.7423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paules, C. I., Marston, H. D., & Fauci, A. S. (2020). Coronavirus infections-more than just the common cold. JAMA, 323(8), 707–708. 10.1001/jama.2020.0757 [DOI] [PubMed] [Google Scholar]
- Roussel, Y., Giraud-Gatineau, A., Jimeno, M.-T., Rolain, J.-M., Zandotti, C., Colson, P., & Raoult, D. (2020). SARS-CoV-2: Fear versus data. International Journal of Antimicrobial Agents, 55(5), 105947. 10.1016/j.ijantimicag.2020.105947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayad, B., Sobhani, M., & Khodarahmi, R. (2020). Sofosbuvir as repurposed antiviral drug against COVID-19: Why were we convinced to evaluate the drug in a registered/approved clinical trial? Archives of Medical Research. 10.1016/j.arcmed.2020.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheahan, T. P., Sims, A. C., Leist, S. R., Schäfer, A., Won, J., Brown, A. J., Montgomery, S. A., Hogg, A., Babusis, D., Clarke, M. O., Spahn, J. E., Bauer, L., Sellers, S., Porter, D., Feng, J. Y., Cihlar, T., Jordan, R., Denison, M. R., & Baric, R. S. (2020). Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nature Communications, 11(1), 222. 10.1038/s41467-019-13940-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal, T. (2020). A review of coronavirus disease-2019 (COVID-19). Indian Journal of Pediatrics, 87(4), 281–286. 10.1007/s12098-020-03263-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder, E. J., Decroly, E., & Ziebuhr, J. (2016). The nonstructural proteins directing coronavirus RNA synthesis and processing. In Ziebuhr J (Ed.), Advances in virus research, 59–126. Academic Press Inc. 10.1016/bs.aivir.2016.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stobart, C. C., Sexton, N. R., Munjal, H., Lu, X., Molland, K. L., Tomar, S., Mesecar, A. D., & Denison, M. R. (2013). Chimeric exchange of coronavirus nsp5 proteases (3CLpro) identifies common and divergent regulatory determinants of protease activity. Journal of Virology, 87(23), 12611–12618. 10.1128/JVI.02050-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott, O., & Olson, A. J. (2009). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31(2), 455–461. 10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umesh, U., Kundu, D., Selvaraj, C., Singh, S. K., & Dubey, V. K. (2020). Identification of new anti-nCoV drug chemical compounds from Indian spices exploiting SARS-CoV-2 main protease as target. Journal of Biomolecular Structure and Dynamics, 38, 1–9. 10.1080/07391102.2020.1763202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, C., Liu, Y., Yang, Y., Zhang, P., Zhong, W., Wang, Y., Wang, Q., Xu, Y., Li, M., Li, X., Zheng, M., Chen, L., & Li, H. (2020). Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharmaceutica Sinica B, 10(5), 766–788. 10.1016/j.apsb.2020.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, M., & Chen, Q. (2020). Insight into 2019 novel coronavirus — An updated interim review and lessons from SARS-CoV and MERS-CoV. International Journal of Infectious Diseases: IJID: Official Publication of the International Society for Infectious Diseases, 94, 119–124. 10.1016/j.ijid.2020.03.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, L. H., Huang, M., Fang, S. G., & Liu, D. X. (2011). Coronavirus infection induces DNA replication stress partly through interaction of its nonstructural protein 13 with the p125 subunit of DNA polymerase δ. The Journal of Biological Chemistry, 286(45), 39546–39559. 10.1074/jbc.M111.242206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav, R., Imran, M., Dhamija, P., Suchal, K., & Handua, S. (2020). Virtual screening and dynamics of potential inhibitors targeting RNA binding domain of nucleocapsid phosphoprotein from SARS-CoV-2. Journal of Biomolecular Structure & Dynamics, 38, 1–16. 10.1080/07391102.2020.1778536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, Z.-W., Yuan, S., Yuen, K.-S., Fung, S.-Y., Chan, C.-P., & Jin, D.-Y. (2020). Zoonotic origins of human coronaviruses. International Journal of Biological Sciences, 16(10), 1686–1697. 10.7150/ijbs.45472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarocostas, J. (2020). What next for the coronavirus response? The Lancet (London, England), 395(10222), 401. 10.1016/S0140-6736(20)30292-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Y., Hou, Y., Shen, J., Huang, Y., Martin, W., & Cheng, F. (2020). Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV. Cell Discovery, 6(1), 2. 10.1038/s41421-020-0153-3 [DOI] [PMC free article] [PubMed] [Google Scholar]