

Abstract
A novel severe acute respiratory syndrome coronavirus (SARS-CoV-2) has emerged as the causative agent behind the coronavirus disease 2019 (COVID-19) pandemic. Treatment efforts have been severely impeded due to the lack of specific effective antiviral drugs for the treatment of COVID-associated pathologies. In the present research endeavour the inhibitory prospects of cyanobacterial metabolites were assessed at the active binding pockets of the two vital SARS-CoV-2 proteases namely, main protease (Mpro) and the papain-like protease (PLpro) that proteolytically process viral polyproteins and facilitate viral replication, employing an in silico molecular interaction-based approach. It was evident from our analysis based on the binding energy scores that the metabolites cylindrospermopsin, deoxycylindrospermopsin, carrageenan, cryptophycin 52, eucapsitrione, tjipanazole, tolyporphin and apratoxin A exhibited promising inhibitory potential against the SARS-CoV-2 Mpro. The compounds cryptophycin 1, cryptophycin 52 and deoxycylindrospermopsin were observed to display encouraging binding energy scores with the PLpro of SARS-CoV-2. Subsequent estimation of physicochemical properties and potential toxicity of the metabolites followed by robust molecular dynamics simulations and analysis of MM-PBSA energy scoring function established deoxycylindrospermopsin as the most promising inhibitory candidate against both SARS-CoV-2 proteases. Present research findings bestow ample scopes to further exploit the potential of deoxycylindrospermopsin as a successful inhibitor of SARS-CoV-2 in vitro and in vivo and pave the foundation for the development of novel effective therapeutics against COVID-19.
Communicated by Ramaswamy H. Sarma
Keywords: SARS-CoV-2, cyanobacterial metabolites, molecular docking, drug-likeness, molecular dynamics simulations, MM-PBSA, deoxycylindrospermopsin
1. Introduction
In December 2019, the World Health Organization was alerted to the occurrence of a new strain of coronavirus which originated in Wuhan, China (Elfiky, 2020; Enayatkhani et al., 2020; Gupta et al., 2020; Huang et al., 2020; Pant et al., 2020; Xu et al., 2020). Since then, the virus has cataclysmically spread across the world and has infected more than 11 million patients effecting a death toll of around five hundred thousand (updated on 03 July, 2020) (https://www.worldometers.info/coronavirus/). The coronavirus disease (COVID-19), caused by the novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has been reported to be more fatal to elderly individuals and patients with comorbidities (Al-Khafaji et al., 2020; Elfiky, 2020). Though bats are believed to be the primary reservoirs for this novel coronavirus, intermediate hosts are suggested to be involved before its final spillover event into humans (Lu et al., 2020). Of the handful of coronaviruses that are known to infect humans, causing either mild illness or severe disease, SARS-CoV-2 is the seventh and is associated with symptoms including cough, fever and difficulty breathing which may progress to pneumonia and death if unattended (Andersen et al., 2020; Boopathi et al., 2020; Das et al., 2020; Elfiky, 2020; Lobo-Galo et al., 2020). Significant variability exists between the genome sequence of SARS-CoV-2 in comparison to SARS-CoV (approximately 21%), and MERS-CoV (approximately 50%), the causative agents of the SARS and MERS outbreaks that occurred in recent past (Lu et al., 2020). The SARS-CoV-2 genome encodes several structural and non-structural proteins (nsp) that play pivotal roles in attaching the virus to host cellular receptors, viral replication and subsequent infection (Gupta et al., 2020; Tai et al., 2020). The viral spike (S) protein is an important structural protein that facilitates viral attachment, fusion and subsequent entry into the human host via its interaction with the human (host) cellular receptor angiotensin converting enzyme II (ACE 2) (Abdelli et al., 2020; Sinha et al., 2020; Tai et al., 2020). After attachment to the ACE 2 receptor, the genome of SARS-CoV-2 is released into the cytoplasm and takes control of cellular apparatus to implement its own translation process (Hasan et al., 2020). The viral genome encodes several other vital structural proteins that include the envelop protein (a hydrophobic protein responsible for morphogenesis during assemblage of the viral genome), the membrane (M) protein (responsible for the maintenance of the shape of the viral envelope) and the nucleocapsid (N) protein (responsible for RNA binding and dimerization) (Andersen et al., 2020; Sarma et al., 2020; Wahedi et al., 2020). These structural proteins are produced via a complex proteolytic process involving two proteases namely the chymotrypsin-like cysteine protease (main protease [Mpro] or 3 C-like protease [3CLpro]) and the papain-like cysteine protease (PLpro) (Adeoye et al., 2020; Enmozhi et al., 2020; Islam et al., 2020; Joshi et al., 2020; Umesh et al., 2020). The vital role of the SARS-CoV Mpro and PLpro in proteolytic cleavage of the large viral polyprotein orf1ab and viral replication has established them as promising drug and vaccine targets in the areas of therapeutic research against COVID-19 (Aanouz et al., 2020; Elmezayen et al., 2020; Gyebi et al., 2020; Khan et al., 2020; 2020). The sequence of the Mpro/3CLpro has been conserved to up to 96.08% in SARS-CoV-2 (Prajapat et al., 2020; Ul Qamar et al., 2020). As such, several protease inhibitors including lopinavir and ritonavir and combinations thereof have been tried to inhibit SARS-CoV-2 protease function and arrest the infection and spread of the virus. However, lack of specific antiviral drugs targeted against SARS-CoV-2 proteases has complicated and impeded treatment efforts.
In recent decades, the global pharmaceutical industry has relied heavily on natural products from microbial sources for the development of novel and effective therapeutics. The present research employed a library of 23 chemically diverse biologically active metabolites (Figure 1(A,B)) from cyanobacteria to target the main protease (Mpro/3CLpro) as well as the papain-like protease of SARS-CoV-2 using a molecular docking approach. We undertook efforts to provide a comprehensive analysis of the physicochemical and drug-like features of the cyanobacterial metabolites that displayed encouraging inhibitory potential. Our results were further validated by robust molecular dynamics simulations and assessment of molecular mechanics-Poisson-Boltzmann surface area (MM-PBSA) binding free energy with a motive to profile the most promising inhibitory candidates against the SARS-CoV-2 Mpro and PLpro, targeted towards the development of effective therapeutics.
Figure 1.
The structure of the cyanobacterial metabolites investigated for their ability to interact with the Mpro and PLpro of SARS-CoV-2.


2. Materials and methods
2.1. Molecular docking of the cyanobacterial metabolites with SARS-CoV-2 mpro and PLpro
The high-resolution X-ray diffraction crystal structure of the Mpro (PDB ID: 6M03 Chain A; 2.0 Å resolution) and the PLpro (PDB ID: 6W9C Chain A; 2.7 Å resolution) of SARS-CoV-2 were downloaded from the PDB. The respective protein structures were prepared for docking analysis by removing the associated water and ligand molecules. Polar hydrogen atoms and Gasteiger charges were added to the structure using AutoDock tools. Ligands of interest were biologically active compounds isolated from cyanobacteria and were sourced from the NCBI PubChem and ZINC databases. Three dimensional structures of the respective compounds were prepared using the Open Babel server (O’Boyle et al., 2011) and the smiles structures were energy optimized using the PRODRG server (Schüttelkopf & Van Aalten, 2004). Gromos 96 forcefield was applied to the structures for energy minimization. The refined protein and ligand structures were subjected to molecular docking using AutoDock Vina software (Trott & Olson, 2010) in light of the existing information regarding the active binding pockets of the respective viral proteins (Báez-Santos et al., 2015; Jin et al., 2020). Molecular docking was performed employing a grid-based docking method opting a rigid protein receptor and flexible ligand docking protocol (Naidoo et al., 2020). Grid boxes of coordinates 30, 30 and 30 in x, y and z dimensions encompassing the active site residues were employed to carry out molecular docking at the binding pockets of the viral proteins (Naidoo et al., 2020). Protein-ligand complexes with the lowest binding energy scores and RMSD < 2.0 Å (Naidoo et al., 2020) were used for further interaction analysis using the PyMol software (version 1.7.4) and the Protein-Ligand Interaction Profiler (PLIP) web-server (Salentin et al., 2015). Binding affinities (ΔG) derived from AutoDock Vina were used to determine the inhibition constant (Ki) using the following equation:
| (1) |
where, ΔG1 represents the binding affinity of the ligand exposed to a conversion factor (4.184) to render kJ−1 mol−1, R is the gas constant (8.314 J mol−1 K−1) and T is the temperature of the system held at 298 K−1.
In order to corroborate the predicted potency of the concerned compounds, we determined the ligand efficiency (LE; Equation 2) and ligand lipophilic efficiency (LLE; Equation 3) as measures to quantify the effectiveness of the structural components of each compound to bind the concerned viral proteases (Hopkins et al., 2004; 2014, Shultz, 2013).
| (2) |
where, HA is the number of heavy atoms (non-hydrogen atoms) and pKi is the negative logarithm of the inhibition constant.
| (3) |
where, pKi is the negative logarithm of the inhibition constant and cLogP is the calculated partial coefficient.
2.2. Assessment of physicochemical features of the cyanobacterial metabolites
Cyanobacterial metabolites that displayed binding energy scores ≤ −7.5 kcal/mol with the respective viral proteins in AutoDock Vina were chosen for further estimation of physicochemical properties and drug-like features (Elfiky, 2020; Khan et al., 2020; Naidoo et al., 2020). The physicochemical properties were estimated employing the SwissADME server and were validated with the pkCSM server (Daina et al., 2017; Pires et al., 2015). SwissADME and FAF-Drugs4 (Lagorce et al., 2017) servers were used to detect the presence of pan-assay interference structures (PAINS) associated with structural alerts among the selected compounds. ProTox-II (Banerjee et al., 2018) and vNN-ADMET web servers (Schyman et al., 2017) were used to assess the toxicity indices like mutagenicity (AMES mutagenesis) and cytotoxicity of the compounds.
2.3. Molecular dynamics simulations
Molecular dynamics (MD) simulations offer a thorough validation of molecular docking results and allow reliable assessment of potential stability of a protein-ligand complex (Khan et al., 2020; Kumar et al., 2020). The ligands displaying the best interaction scores (in terms of binding energy values) with each SARS-CoV-2 Mpro and PLpro were subjected to MD simulation for a timescale of 120 nanoseconds (ns) using GROMACS software (version 2019) (Abraham et al., 2015). The MD simulations were performed as per the protocols opted by Khan et al. (2020). The equilibration steps were set with constant pressure and temperature (NPT) ensemble (Umesh et al., 2020) The MD simulations were carried out at a standard temperature of 300 K and pressure level of 1.013 bar (Umesh et al., 2020). The structural parameters pertaining to the MD simulations like the root mean square deviations (RMSD), the root mean square fluctuations (RMSF), the radius of gyration (RoG) and the hydrogen bonds of the respective protein-ligand complexes were estimated as a function of time (120 ns) to assess their conformational and structural stability (Khan et al., 2020).
2.4. Estimation of free energies of binding employing molecular mechanics-Poisson-Boltzmann surface area (MM-PBSA) method
Assessment of molecular mechanics-Poisson-Boltzmann surface area (MM-PBSA) based free energies of binding of protein-ligand complexes in combination with MD simulations provides apt estimate of the conformational stabilities of protein-ligand complexes (Bhardwaj et al., 2020). MM-PBSA based free energies of binding of the selected protein-ligand complexes were estimated employing the ‘g_mmpbsa’ script of GROMACS (Kumari et al., 2014). The total free energy of binding of a protein-ligand complex involves the potential energy, polar and non-polar solvation energies and was calculated as:
where ΔGbind signifies the total energy of binding of the receptor-ligand complex, ΔGreceptor and ΔGligand represent the energies of the free receptor and unbound ligand respectively (Bhardwaj et al., 2020).
3. Results and discussion
3.1. Molecular interactions of the cyanobacterial metabolites with SARS-CoV-2 Mpro
The Mpro (3CLpro) is encoded by the SARS-CoV-2 genome as an integral protease responsible for the cleavage of two polyproteins to form functional components (Andersen et al., 2020). The Mpro has been suggested as an important target candidate for antiviral drug development (Ul Qamar et al., 2020). It has thus become essential to discover and develop new drugs that specifically target Mpro and provide therapeutic relief against SARS-CoV-2. Cyanobacterial metabolites have been identified as promising therapeutics against a range of diseases and disorders owing to an array of biological activities (Tan & Phyo, 2020). In the present work, a collection of cyanobacterial metabolites that have displayed significant anticancer, antimicrobial and antiviral activities were assessed to explore their inhibitory prospects against the Mpro of SARS-CoV-2. Recent reports have suggested that the active site of the Mpro displays a stereoselective recognition towards non-covalent inhibitors (Jacobs et al., 2013; Karypidou et al., 2018). Our analyses thus focused on the identification of compounds that could potentially form strong non-covalent interactions with the catalytic dyad of the Mpro.
The detailed information regarding the binding energy scores of the 23 compounds with SARS-CoV-2 Mpro have been provided in Table 1 (compounds displaying binding energy scores ≤ −7.5 kcal/mol) and Table S1 [Supplementary material] (compounds displaying binding energy scores > −7.5 kcal/mol). It was evident from our analysis that out of the 23 compounds, cylindrospermopsin, deoxycylindrospermopsin, carrageenan, cryptophycin 52, eucapsitrione, tjipanazole, tolyporphin and apratoxin A displayed encouraging results with binding energy scores ≤ −7.5 kcal/mol (Table 1) (Elfiky, 2020; Khan et al., 2020; Naidoo et al., 2020). The cyanotoxin, deoxycylindrospermopsin originally isolated from Cylindrospermopsis sp. displayed the most promising binding efficiency (-8.6 ± 0.02 kcal/mol) and interacted with the important residues Thr26, Cys44, Phe140, Ser144, Cys145, His163 and Glu166 at the active binding pocket of Mpro (Table 1 and Figure 2). The compound cylindrospermopsin displayed a high binding energy value of −8.1 ± 0.03 kcal/mol and exhibited hydrophobic interactions with the residues Phe8 and Phe305 and formed hydrogen bonds with Gln110, Thr111, Gln127, Asn151, Asp153, Phe294, Asp295 and Arg298 at the active binding site of Mpro (Table 1).
Table 1.
Binding energy scores and interaction profile of the cyanobacterial metabolites with SARS-CoV-2 Mpro.
| Cyanobacterial compound | Predicted Binding Affinity (kcal/mol) | RMSD (Å) | Inhibition Constant (Ki) (μM) | Ligand Efficiency (LE) | Ligand Lipophilic Efficiency (LLE) | Amino acid interactions |
|---|---|---|---|---|---|---|
| Apratoxin A | −7.5 ± 0.02 | 0.02 | 3.16 | 0.13 | 1.21 | Met49,Asn142, Met165,Glu166, Gln189 |
| Carrageenan | −8.4 ± 0.02 | 0.01 | 0.22 | 0.18 | 12.79 | Asn119, Cys145, Gln189, Glu166, Thr26, Thr25, Gly143, His163, His41# |
| Cryptophycin 52 | −8.3 ± 0.01 | 0.01 | 0.82 | 0.18 | 1.25 | Thr25, Leu27, Glu166, Pro168,Gln189 |
| Cylindrospermopsin | −8.1 ± 0.03 | 0.01 | 1.15 | 0.29 | 7.27 | Phe8, Phe305,Gln110, Thr111, Gln127, Asn151, Asp153, Phe294, Asp295, Arg298 |
| Deoxycylindrospermopsin | −8.6 ± 0.02 | 0.02 | 0.49 | 0.32 | 6.86 | Glu166, Thr26, Cys44, Phe140, Ser144, Cys145, His163# |
| Eucapsitrione | −7.8 ± 0.03 | 0.01 | 1.90 | 0.29 | 8.29 | Val104, Arg298,Lys102, Gln110, Asp295 |
| Tjipanazole A1 | −8.0 ± 0.03 | 0.02 | 1.35 | 0.25 | 2.22 | Leu144, Asn142, His163, His164, Glu166 |
| Tolyporphin | −8.2 ± 0.02 | 0.02 | 1.00 | 0.25 | 3.74 | Val104, Ile106, Phe294, Phe305,Gln110, Asp153, Asp295, Arg298# |
Hydrophobic interactions are represented in italics, Hydrogen bonds are represented in bold and salt bridges are displayed with #.
Figure 2.

Mode of interaction of deoxycylindrospermopsin with SARS-CoV-2 Mpro. Cyan sphere represents SARS-CoV-2 Mpro. Orange sphere represents deoxycylindrospermopsin. Hydrophobic interactions have been represented as grey dashed lines and hydrogen bonds have been displayed as blue lines. Salt bridges have been represented as yellow dashed lines.
Similarly, carrageenan displayed an encouraging binding energy score (-8.4 ± 0.02 kcal/mol) and interacted with the two crucial residues of the catalytic dyad of Mpro namely, Cys145 through hydrogen bonding and His41 through a salt bridge with the carboxylate group (Table 1) (Jin et al., 2020). Interestingly, carrageenan did not form hydrophobic interactions with any of the residues at the binding site. The compound is not typically isolated from cyanobacteria but was included in this study after considering the multitude of medicinal uses that include antiviral activity against HIV-1 (Schaeffer & Krylov, 2000) and might be a promising avenue for the development of anti-coronavirus drugs.
The cryptophycins are a large class of peptolides with potent anticancer activity isolated from Nostoc sp. that has undergone clinical trials in cancer treatments. Derivatives of cryptophycin were used as conjugates for antibodies to bind to cysteine mutants that target HER2 and CD22 receptors in human breast cancer (Weiss et al., 2017). Of the two analogs investigated, cryptophycin 52 exhibited a better binding energy score (-8.3 ± 0.01 kcal/mol) (Table 1) relative to cryptophycin 1 (-7.0 ± 0.01 kcal/mol) (Table S1 [Supplementary material]). The compound cryptophycin 52 was noted to interact with the residues Thr25, Leu27, Glu166, Pro168 and Gln189 at the active binding pocket of Mpro (Table 1).
The tjipanazoles, isolated from Tolypothrix tjipanasensis are characteristic of carboxazoles exhibiting three benzenoid rings, a hexopyranose ring attached to the aromatic ring system and an indole type NH group (Bonjouklian et al., 1991). The compound formed hydrogen bonds with essential amino acids Asn142, Leu144, His163, His164 and Glu166 of Mpro and returned a binding energy score of −8.0 ± 0.03 kcal/mol (Table 1). Similarly, tolyporphin isolated as a resistance reversing agent from T. nodosa exhibited a binding energy score of −8.2 ± 0.02 kcal/mol and involved the residues Val104, Ile106, Gln110, Asp153, Phe294, Asp295, Arg298 and Phe305 in its interaction with Mpro (Table 1).
The apparent biosynthetic potential of cyanobacteria as a producer of diverse and biologically valuable chemicals is further highlighted by the apratoxins, a group of cyclodepsipeptides that contain polypeptide and polyketide fragments (Chen & Forsyth, 2003). The cytotoxicity of this group of compounds has established them as interesting targets for the development of anticancer drug leads (Luesch et al., 2001). In the current investigation, apratoxin A revealed a binding affinity of −7.5 ± 0.02 kcal/mol with the Mpro of SARS-CoV-2 forming interactions with the amino acid residues Met49, Asn142, Met165, Glu166 and Gln189 (Table 1).
Eucapsitrione is an anthraquinone derivative that was isolated as a potent anti-Mycobacterium tuberculosis agent from the cyanobacterium Eucapsis sp. (Sturdy et al., 2010). In the current study, the compound was observed to interact with the residues Lys102, Val104, Gln110, Asp295 and Arg298 at the active site of the Mpro and produced a binding energy score of −7.8 ± 0.01 kcal/mol (Table 1).
Thus, it was evident from the molecular docking results that the metabolites cylindrospermopsin, deoxycylindrospermopsin, carrageenan, cryptophycin 52, eucapsitrione, tjipanazole, tolyporphin and apratoxin A (Table 1) exhibited promising inhibitory prospects (binding energy scores ≤ −7.5 kcal/mol) (Elfiky, 2020; Khan et al., 2020; Naidoo et al., 2020) against SARS-CoV-2 Mpro and were accordingly selected for further analyses.
3.2. Molecular interactions of the cyanobacterial metabolites with SARS-CoV-2 PLpro
Similar to the Mpro, the PLpro is responsible for the cleavage of several polypeptides to produce functional viral components (Báez-Santos et al., 2015). The PLpro also plays an integral role in negating the effects of the host’s innate immune system response against viral infection (Báez-Santos et al., 2015). The human immune response is regulated by ubiquitin, a small protein and the ubiquitin-like modifier interferon stimulated gene 15 (ISG15) (Hu & Sun, 2016). The protease is capable of removing ubiquitin and ISG15 from cellular conjugates thereby, downregulating the differentiation of T-cells which essentially limits the response of the immune system to infections (Békés et al., 2015; Hu & Sun, 2016). The PLpro is thus an essential target for antiviral drugs (Báez-Santos et al., 2015). Present work involved a thorough molecular docking of the 23 selected biologically active cyanobacterial metabolites at the active binding pocket of the PLpro (Báez-Santos et al., 2015).
The binding energy scores of the 23 compounds with SARS-CoV-2 PLpro have been detailed in Table 2 (compounds displaying binding energy scores ≤ −7.5 kcal/mol) and Table S2 [Supplementary material] (compounds displaying binding energy scores > −7.5 kcal/mol). Of the 23 compounds, cryptophycin 1, cryptophycin 52 and deoxycylindrospermopsin showed promising inhibitory potential with binding energy scores ≤ −7.5 kcal/mol (Table 2) (Elfiky, 2020; Khan et al., 2020; Naidoo et al., 2020). The metabolites cryptophycin 1, cryptophycin 52 and deoxycylindrospermopsin displayed binding energy scores of −7.7 ± 0.05 kcal/mol, −7.6 ± 0.02 kcal/mol and −7.9 ± 0.04 kcal/mol respectively (Table 2). Deoxycylindrospermopsin seemed to be the most efficient and interacted with the amino acid residues Lys157, Gly163, Asp164, Arg166, Ala246, Tyr264, Thr301 and Asp302 at the active binding pocket of PLpro (Table 2 and Figure 3) (Adeoye et al., 2020). Based on the decent binding energy scores with SARS-CoV-2 PLpro these metabolites were selected for further analyses.
Table 2.
Binding energy scores and interaction profile of the cyanobacterial metabolites with SARS-CoV-2 PLpro.
| Cyanobacterial compound | Predicted Binding Affinity (kcal/mol) | RMSD (Å) | Inhibition Constant(Ki) (μM) | Ligand Efficiency (LE) | Ligand Lipophilic Efficiency (LLE) | Amino Acid Interactions |
|---|---|---|---|---|---|---|
| Cryptophycin 1 | −7.7 ± 0.05 | 0.01 | 2.25 | 0.17 | 1.00 | Tyr264, Tyr268,Glu167, Thr301 |
| Cryptophycin 52 | −7.6 ± 0.02 | 0.03 | 2.67 | 0.16 | 0.73 | Leu162,Asp164, Tyr171, Pro248,Arg166, Ser170, Tyr264 |
| Deoxycylindrospermopsin | −7.9 ± 0.04 | 0.02 | 1.60 | 0.29 | 6.34 | Tyr264,Lys157, Gly163, Asp164, Ala246, Thr301, Asp302, Arg166# |
Hydrophobic interactions are represented in italics, Hydrogen bonds are represented in bold and salt bridges are displayed with #.
Figure 3.

Mode of interaction of deoxycylindrospermopsin with SARS-CoV-2 PLpro. Blue sphere represents theSARS-CoV-2 PLpro. Orange sphere represents deoxycylindrospermopsin. Hydrophobic interactions have been represented as grey dashed lines and hydrogen bonds have been displayed as blue lines. Salt bridges have been represented as yellow dashed lines.
3.3. Predicted biological activity and efficiency of the cyanobacterial metabolites
The affinity of a proposed therapeutic candidate to a target receptor is a measure of the strength to which the concerned compound binds with the receptor. Binding affinity is an essential factor that determines the potency of a drug and is inversely proportional to the common measures of potency including the dissociation constant or the inhibition constant (Ki) (Hughes et al., 2011). Thus, the stronger the affinity of a compound, the lower the concentration required to produce the desired activity. A compound that exhibits a ki within micromolar range (1–40 μM) is considered a hit or lead compound and should be considered for further drug development (Hughes et al., 2011). The current study identified three compounds namely, carrageenan, cryptophycin 52 and deoxycylindrospermopsin that were predicted to be active within sub-nanomolar range against the viral proteins (Tables 1 and 2). However, the potency of a compound is not the only factor to consider when selecting a compound as a drug candidate. Several studies have resolved that the physicochemical features of a compound including molecular weight, lipophilicity, number of hydrogen bond donors and acceptors and non-hydrogen atom properties affect the solubility, membrane permeability, stability and toxicity of a compound (Daina et al., 2017; Hopkins et al., 2014). Ligand metrics were therefore designed to provide a measure of the biological potency of a compound after considering its physicochemical properties (Hopkins et al., 2014). For instance, the ligand efficiency (LE) metric deployed in the current study describes the potency of the compound per heavy atom (Hopkins et al., 2004). The compounds deoxycylindrospermopsin, carrageenan and cryptophycin 52 displayed LE values that fell below the threshold of 0.3 (Hopkins et al., 2004). In addition to the predicted biological activity of deoxycylindrospermopsin, the compound was predicted to be an efficient ligand. The lipophilic ligand efficiency (LLE) metric on the other hand, is a measure of the ability of the compound to bind specifically to the target receptor relative to its polar or hydrophobic state (log P) (Hopkins et al., 2014). The compounds carrageenan, eucapsitrione, cylindrospermopsin and deoxycylindrospermopsin exhibited LLE values greater than the proposed acceptable threshold (Leeson & Springthorpe, 2007; Ul Qamar et al., 2020).
3.4. Physicochemical properties and drug-like features of the cyanobacterial metabolites
The physicochemical properties of the most promising inhibitory candidates (binding energy score ≤ −7.5 kcal/mol) among the cyanobacterial metabolites were assessed in accordance with Lipinski’s rule of five which predicts the oral bioavailability and membrane permeability of a drug candidate in terms of the pharmacokinetic properties of a concerned compound (Table 3) (Lipinski et al., 2001). Lipinski's rule of five proposes that a drug candidate with logarithm of partial coefficient (log P) ≤ 5, molecular weight ≤ 500 Dalton, number of hydrogen bond acceptors ≤ 10 and number of hydrogen bond donors ≤ 5 exhibits decent gastrointestinal absorption and enhanced membrane permeability (Lipinski et al., 2001). Despite its promising inhibitory potential (Tables 1 and 2), carrageenan was noted to violate the Lipinski's rule of five (molecular weight > 500 Dalton) (Table 3). The compounds apratoxin A, cryptophycin 1, cryptophycin 52 and tolyporphin also showed violations owing to their higher molecular weights than the proposed threshold (Table 3). Interestingly, deoxycylindrospermopsin displayed decent physicochemical features which were observed to be in accordance with the Lipinski's rule of five (Table 3). The compounds apratoxin A, cryptophycin 1, cryptophycin 52 and tolyporphin were predicted to display poor solubility (Table 3), whereas, carrageenan, eucapsitrione and tjipanazole exhibited moderate solubility. Furthermore, apratoxin A, carrageenan and tolyporphin exhibited low bioavailability scores of 0.17 (Table 3) (Naidoo et al., 2020). However, deoxycylindrospermopsin was predicted to be highly soluble and showed decent bioavailability score of 0.55 (Table 3) (Naidoo et al., 2020). The solubility of a drug candidate is an essential pharmacokinetic index that defines how efficiently a drug will be metabolized (Naidoo et al., 2020). The cyanobacterial metabolites were predicted not to possess any structural alerts associated with toxic consequences on PAINS toxicophore estimation (Table 3) (Naidoo et al., 2020; Pouliot & Jeanmart, 2016). The compound tjipanazole A1 was predicted to be potentially mutagenic (Table 3). The cryptophycins have been reported to be associated with severe neurotoxicity in clinical trials as a result of their superior cytotoxic activity (Weiss et al., 2017). Although antibody conjugates of cryptophycin were deemed safer for anticancer therapy, in the interest of producing a rapid anti-coronavirus treatment, this might not be an appropriate choice (Weiss et al., 2017). Cylindrospermopsins are cyanotoxins produced by a variety of freshwater cyanobacteria (Berry et al., 2012). Despite the relatively non-toxic nature of deoxycylindrospermopsin, variants cylindrospermopsin and 7-epicylindrospermopsin have been found to accumulate in the liver of rats causing DNA fragmentation and display toxic effects (Solter & Beasley, 2013). Thus, a thorough analysis of the physicochemical features and toxicity assessment established deoxycylindrospermopsin as the most effective and safe candidate for further analyses as it fulfilled the rules of drug-likeness and was predicted to be non-toxic.
Table 3.
Physicochemical properties and drug-likeness of the most promising cyanobacterial metabolites.
| Cyanobacterial compound | Mol. wt. (Dalton) | Number of hydrogen bond acceptors | Number of hydrogen bond donors | Log P | Solubility | PAINS Alerts | Bioavailability | Mutagenicity (AMES mutagenesis) | Cytotoxicity |
|---|---|---|---|---|---|---|---|---|---|
| Apratoxin A | 840.12 | 9 | 2 | 4.38 | Poorly soluble | 0 alert | 0.17 | No | No |
| Carrageenan | 551.83 | 10 | 3 | −9.41 | Moderately Soluble | 0 alert | 0.17 | No | No |
| Cryptophycin 1 | 655.18 | 8 | 2 | 4.65 | Poorly soluble | 0 alert | 0.55 | No | No |
| Cryptophycin 52 | 699.20 | 8 | 2 | 4.84 | Poorly soluble | 0 alert | 0.55 | No | No |
| Cylindrospermopsin | 415.42 | 8 | 5 | −1.37 | Highly soluble | 0 alert | 0.55 | No | No |
| Deoxycylindrospermopsin | 399.42 | 7 | 4 | −0.55 | Highly soluble | 0 alert | 0.55 | No | No |
| Eucapsitrione | 358.30 | 6 | 3 | 2.57 | Moderately soluble | 0 alert | 0.55 | No | No |
| Tjipanazole A1 | 471.33 | 4 | 4 | 3.65 | Moderately soluble | 0 alert | 0.55 | Yes | No |
| Tolyporphin | 742.83 | 12 | 4 | 2.67 | Poorly soluble | 0 alert | 0.17 | No | No |
Log P-Logarithm of partial coefficient; PAINS-Pan-Assay Interference Structures.
3.5. Analysis of molecular dynamics simulations
The compound deoxycylindrospermopsin exhibited better binding potential with SARS-CoV-2 Mpro and PLpro in terms of binding energy in comparison to the other cyanobacterial metabolites (Tables 1 and 2 and Tables S1 and S2 [Supplementary material]). Furthermore, the physicochemical features and drug-like attributes of the compound was noted to be in accordance with the Lipinski’s rule of five and it was predicted to be non-toxic (Table 3). Accordingly, the complexes of deoxycylindrospermopsin with SARS-CoV-2 Mpro and PLpro proteins were selected for MD simulations for a timescale of 120 ns to validate the results of molecular docking and assess their conformational stability. A detailed RMSD analysis of the deoxycylindrospermopsin-SARS-CoV-2 Mpro complex revealed that it displayed fluctuations in RMSD values of Cα atoms until 94 ns and attained stability thereafter (Figure 4(A)). The average RMSD value of the complex was observed to be 1.4 Å. On the contrary, the free protein was found to display more fluctuations throughout the course of MD simulation displaying a higher average RMSD value of 1.9 Å (Figure 4(A)). Similar trend was noted from the RMSD analysis of the deoxycylindrospermopsin-SARS-CoV-2 PLpro complex which experienced fluctuations in RMSD values of Cα atoms until 92 ns and attained equilibrium after that and remained stable (Figure 4(B)). The average RMSD value of the complex was found to be 1.3 Å. However, the free protein was noted to display fluctuations throughout the timescale of 120 ns and returned a higher average RMSD value of 1.8 Å (Figure 4(B)). It has been suggested that lower RMSD values signify higher stability of a protein-ligand complex (Muralidharan et al., 2020). Thus, it was evident from our analysis that the binding of deoxycylindrospermopsin with SARS-CoV-2 Mpro and PLpro rendered considerable stability to the complexes. Our observations indicated towards the conformational stability and decent binding potential of deoxycylindrospermopsin with SARS-CoV-2 Mpro and PLpro (Khan et al., 2020).
Figure 4.

(A) RMSD analysis of the free receptor SARS-CoV-2 Mpro (green) and complex deoxycylindrospermopsin-CoV-2 Mpro (red) along a timescale of 120 ns (B) RMSD analysis of the free receptor SARS-CoV-2 PLpro (blue) and complex deoxycylindrospermopsin-SARS-CoV-2 PLpro (black) along a timescale of 120 ns.
Analysis of root mean square fluctuation (RMSF) of the residues of a protein-ligand complex is effective in addressing local changes along a protein chain and deciphering its conformational stability (Muralidharan et al., 2020). Extensive analysis of RMSF of the free proteins and the complexes revealed comparatively lower fluctuations among the complexes compared to the free proteins (Figures 5(A,B)), thus indicating higher stability of the protein-ligand complexes. The average RMSF values were 1.6 Å and 1.9 Å for the complexes deoxycylindrospermopsin-SARS-CoV-2 Mpro (Figure 5(A)) and deoxycylindrospermopsin-SARS-CoV-2 PLpro (Figure 5(B)) respectively. On the contrary the average RMSF values of the free receptors SARS-CoV-2 Mpro and PLpro were observed to be 2.2 Å (Figure 5(A)) and 2.6 Å (Figure 5(B)) respectively. The peaks in the RMSF plot indicate the residues which experience major fluctuations during the MD simulations (Muralidharan et al., 2020). It was evident from the RMSF analysis of the concerned protein-ligand complexes that the residues in the active binding pockets of the SARS-CoV-2 proteins that interacted with deoxycylindrospermopsin (Tables 1 and 2) were relatively stable (Figures 5(A,B)) throughout the course of the MD simulations of 120 ns and thus, reflected their conformation stability.
Figure 5.

(A) RMSF analysis of the free receptor SARS-CoV-2 Mpro (green) and complex deoxycylindrospermopsin-SARS-CoV-2 Mpro (red) along a timescale of 120 ns (B) RMSF analysis of the free receptor SARS-CoV-2 PLpro (blue) and complex deoxycylindrospermopsin-SARS-CoV-2 PLpro (black) along a timescale of 120 ns.
Intermolecular hydrogen bonds (H-bonds) impart stability to a protein-ligand complex and play a crucial role in molecular interaction (Khan et al., 2020; Kumar et al., 2020). Proper assessment of the formation and deformation of H-bonds is a vital step in analyzing MD simulations and inferring about the stability of a receptor-ligand complex (Khan et al., 2020). A detailed analysis of the hydrogen bonding trajectories associated with the MD simulations of the deoxycylindrospermopsin-CoV-2 Mpro and deoxycylindrospermopsin-CoV-2 PLpro complexes for a timescale of 120 ns revealed that both the complexes displayed a maximum number of 6 intermolecular hydrogen bonds (Figures 6(A,B)). The minimum number of hydrogen bonds for the complexes deoxycylindrospermopsin-CoV-2 Mpro and deoxycylindrospermopsin-CoV-2 PLpro were observed to be 2 and 1 respectively (Figures 6(A,B)). The average number of H-bonds was found to be 4 in both the cases (Figures 6(A,B)). It was evident that in the initial phases of MD simulations the number of H-bonds in both the complexes decreased, however, in the final stages the number of H-bonds increased to 6 in both the cases which implied the conformational stability of the complexes. It has been suggested that intermolecular H-bonds confer stability to a receptor-ligand complex (Kumar et al., 2020) and it was evident from our observations that the complexes of deoxycylindrospermopsin with SARS-CoV-2 Mpro and PLpro were considerably stable with 6 hydrogen bonds each at the end of MD simulations of 120 ns (Figure 6(A,B)).
Figure 6.

(A) Number of hydrogen bonds of the deoxycylindrospermopsin-SARS-CoV-2 Mpro complex (red) as a function of time during MD simulations of 120 ns. (B) Number of hydrogen bonds of the deoxycylindrospermopsin-SARS-CoV-2 PLpro complex (blue) as a function of time during MD simulations of 120 ns.
To further validate our observations, a detailed analysis of the interaction profiles of the concerned protein-ligand complexes after the MD simulations of 120 ns was performed. It was evident that the complexes deoxycylindrospermopsin-SARS-CoV-2 Mpro and deoxycylindrospermopsin-SARS-CoV-2 PLpro did not experience any change in interacting residues and pertaining hydrophobic and hydrogen bonds prior to and after the MD simulations of 120 ns (Figures 7(A,B) and Tables 1 and 2). Thus, the complexes were inferred to be conformationally stable (Khan et al., 2020) and correlated with the results of molecular docking.
Figure 7.
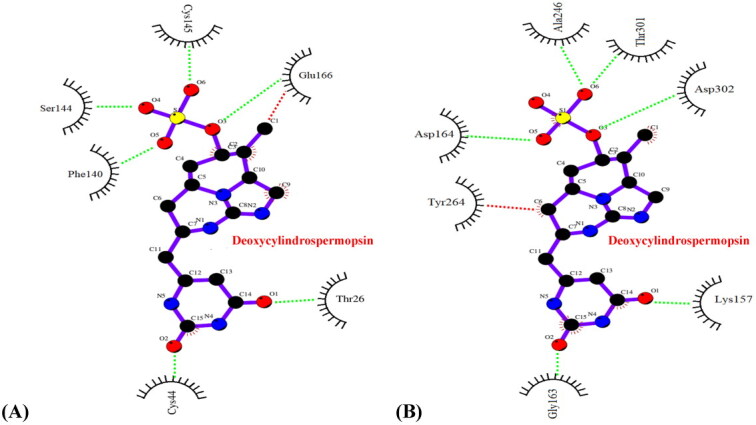
Interaction profile (Ligplot image) of (A) deoxycylindrospermopsin-SARS-CoV-2 Mpro complex after 120 ns MD simulations (B) deoxycylindrospermopsin-SARS-CoV-2 PLpro complex after 120 ns MD simulations. Hydrophobic and hydrogen bond interactions in the respective figures have been marked as red and green dashed lines respectively.
3.6. Analysis of MM-PBSA free energies of binding of deoxycylindrospermopsin with Mpro and PLpro and radius of gyration of the complexes
The deoxycylindrospermopsin-SARS-CoV-2 Mpro and deoxycylindrospermopsin-SARS-CoV-2 PLpro complexes were selected for further estimation of the free energies of binding using the molecular mechanics-Poisson-Boltzmann surface area (MM-PBSA) method. It has been suggested that MM-PBSA provides accurate estimates of free energies of binding of protein-ligand complexes and a more negative value indicates stronger binding (Gupta et al., 2020). The detailed information regarding the electrostatic energy, SASA (Solvent Accessible Surface Areas) energy, van der Waals energy, polar solvation energy and final binding energy from 20 ns to 120 ns with 20 ns interval has been detailed in Table 4. It was evident that all forms of energy, except the polar solvation energy, favourably contributed to the interaction deoxycylindrospermopsin with SARS-CoV-2 Mpro and SARS-CoV-2 PLpro (Table 4). The complexes deoxycylindrospermopsin-SARS-CoV-2 Mpro and deoxycylindrospermopsin-SARS-CoV-2 PLpro displayed final binding energy scores of −45.44 ± 1.66 kcal/mol and −41.39 ± 1.59 kcal/mol respectively (Table 4) which implied considerable conformational stability of the complexes after MD simulations of 120 ns.
Table 4.
Details of binding free energies (± standard deviation) for deoxycylindrospermopsin-SARS-CoV-2 Mpro and deoxycylindrospermopsin-SARS-CoV-2 PLpro complexes calculated using MM-PBSA method from 20 to 120 ns with 20 ns interval.
| Complexes | Time (ns) | van der Waals energy (kcal/mol) | SASA energy (kcal/mol) | Electrostatic energy (kcal/mol) | Polar solvation energy (kcal/mol) | Binding energy (kcal/mol) |
|---|---|---|---|---|---|---|
| deoxycylindrospermopsin- SARS-CoV-2 protease Mpro | 20 | −40.68 ± 2.43 | −3.77 ± 0.43 | −8.92 ± 0.41 | 16.96 ± 1.23 | −36.41 ± 2.04 |
| 40 | −42.90 ± 2.16 | −3.96 ± 0.35 | −9.24 ± 0.52 | 17.71 ± 1.34 | −38.39 ± 1.69 | |
| 60 | −44.33 ± 2.76 | −4.08 ± 0.49 | −9.53 ± 0.39 | 18.20 ± 1.10 | −39.74 ± 2.54 | |
| 80 | −46.04 ± 2.34 | −4.14 ± 0.46 | −9.74 ± 0.47 | 18.56 ± 0.87 | −41.36 ± 2.40 | |
| 100 | −49.73 ± 1.98 | −4.21 ± 0.32 | −9.84 ± 0.45 | 18.91 ± 0.73 | −44.87 ± 2.02 | |
| 120 | −50.38 ± 1.65 | −4.25 ± 0.26 | −9.98 ± 0.31 | 19.17 ± 0.56 | −45.44 ± 1.66 | |
| deoxycylindrospermopsin- SARS-CoV-2 protease PLpro | 20 | −38.29 ± 2.63 | −3.46 ± 0.34 | −8.20 ± 0.56 | 15.58 ± 1.34 | −34.37 ± 2.19 |
| 40 | −40.25 ± 2.45 | −3.64 ± 0.27 | −8.48 ± 0.61 | 16.27 ± 1.49 | −36.1 ± 1.84 | |
| 60 | −42.17 ± 2.58 | −3.75 ± 0.36 | −8.75 ± 0.43 | 16.72 ± 1.76 | −37.95 ± 1.61 | |
| 80 | −44.12 ± 2.32 | −3.89 ± 0.41 | −8.94 ± 0.52 | 17.05 ± 1.32 | −39.9 ± 1.93 | |
| 100 | −44.94 ± 2.15 | −3.97 ± 0.32 | −9.04 ± 0.41 | 17.37 ± 0.92 | −40.58 ± 1.96 | |
| 120 | −45.73 ± 1.79 | −4.10 ± 0.21 | −9.17 ± 0.36 | 17.61 ± 0.77 | −41.39 ± 1.59 |
Radius of gyration (RoG) confers meaningful information about the compactness associated with folding and unfolding of protein structures on binding with the ligands (Khan et al., 2020). Accordingly, RoG was calculated to assess the compactness and structural stability of the deoxycylindrospermopsin-SARS-CoV-2 Mpro and deoxycylindrospermopsin-SARS-CoV-2 PLpro complexes. It has been suggested that higher RoG values signify lower conformational stability and compactness due to unfolding while lower RoG values indicate higher compactness and stability due to folded state of a protein-ligand complex (Khan et al., 2020). The average RoG values of the deoxycylindrospermopsin-SARS-CoV-2 Mpro and deoxycylindrospermopsin-SARS-CoV-2 PLpro complexes were found to be 20.31 ± 0.2 Å and 21.43 ± 0.3 Å. Our findings revealed that the respective complexes were compact with significant conformational stability.
4. Conclusion
COVID-19 pandemic has shocked the world with its devastating impact on global public health. The numbers of infections and deaths have been escalating regularly with the rapid spread and transmission of the causative virus SARS-CoV-2. There has been a dire need to develop specific effective therapeutics and prophylactics against the menacing virus and combat COVID-19. The present research effort aimed at exploring the inhibitory prospects of 23 cyanobacterial metabolites against the SARS-CoV-2 Mpro and PLpro proteases that have been considered as promising drug targets. Our analysis revealed that the metabolites cylindrospermopsin, deoxycylindrospermopsin, carrageenan, cryptophycin 52, eucapsitrione, tjipanazole, tolyporphin and apratoxin A exhibited promising inhibitory potential against SARS-CoV-2 Mpro based on the binding energy scores of the interactions. The compounds cryptophycin 1, cryptophycin 52 and deoxycylindrospermopsin displayed significant inhibitory prospects against the PLpro of SARS-CoV-2, as evident from the binding energies. Subsequent analysis of physicochemical features, potential toxicity, molecular dynamics simulations and MM-PBSA energy scoring function established deoxycylindrospermopsin as the most promising common inhibitory candidate against both the SARS-CoV-2 proteases. Present findings promise to establish foundations to further validate the inhibitory potential of the compound deoxycylindrospermopsin in vitro and in vivo and deploy it as an effective inhibitor of SARS-CoV-2 Mpro and PLpro in the quest for effective drug development against COVID-19.
Supplementary Material
Acknowledgements
AR acknowledges Lovely Professional University, India for providing the necessary infrastructure facility. We also thank the anonymous reviewers for their suggestions which helped to improve the manuscript.
Funding Statement
The Mangosuthu University of Technology is acknowledged for financial support.
Disclosure statement
The authors wish to declare that there are no conflicts of interest.
References
- Aanouz, I., Belhassan, A., El Khatabi, K., Lakhlifi, T., El Idrissi, M., & Bouachrine, M. (2020). Moroccan medicinal plants as inhibitors of COVID-19: Computational investigations. Journal of Biomolecular Structure and Dynamics, 1–9. 10.1080/07391102.2020.1758790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelli, I., Hassani, F., Bekkel Brikci, S., & Ghalem, S. (2020). In silico study the inhibition of angiotensin converting enzyme 2 receptor of COVID-19 by Ammoides verticillata components harvested from Western Algeria. Journal of Biomolecular Structure and Dynamics, 1–14. 10.1080/07391102.2020.1763199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham, M. J., Murtola, T., Schulz, R., Páll, S., Smith, J. C., Hess, B., & Lindahl, E. (2015). GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX, 1–2, 19–25. 10.1016/j.softx.2015.06.001 [DOI] [Google Scholar]
- Adeoye, A. O., Oso, B. J., Olaoye, I. F., Tijjani, H., & Adebayo, A. I. (2020). Repurposing of chloroquine and some clinically approved antiviral drugs as effective therapeutics to prevent cellular entry and replication of coronavirus. Journal of Biomolecular Structure and Dynamics, 1–11. 10.1080/07391102.2020.1765876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Khafaji, K., Al-DuhaidahawiL, D., & Taskin Tok, T. (2020). Using integrated computational approaches to identify safe and rapid treatment for SARS-CoV-2. Journal of Biomolecular Structure and Dynamics, 1–9. 10.1080/07391102.2020.1764392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen, K. G., Rambaut, A., Lipkin, W. I., Holmes, E. C., & Garry, R. F. (2020). The proximal origin of SARS-CoV-2. Nature Medicine, 26(4), 450–452. 10.1038/s41591-020-0820-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Báez-Santos, Y. M., St. John, S. E., & Mesecar, A. D. (2015). The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antiviral Research, 115, 21–38. 10.1016/j.antiviral.2014.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee, P., Eckert, A. O., Schrey, A. K., & Preissner, R. (2018). ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Research, 46(W1), W257–263. 10.1093/nar/gky318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Békés, M., Rut, W., Kasperkiewicz, P., Mulder, M. P. C., Ovaa, H., Drag, M., Lima, C. D., & Huang, T. T. (2015). SARS hCoV papain-like protease is a unique Lys48 linkage-specific di-distributive deubiquitinating enzyme. The Biochemical Journal, 468(2), 215–226. 10.1042/BJ20141170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry, J. P., Gibbs, P. D. L., Schmale, M. C., & Saker, M. L. (2012). Toxicity of cylindrospermopsin, and other apparent metabolites from Cylindrospermopsis raciborskii and Aphanizomenon ovalisporum, to the zebrafish (Danio rerio) embryo. Toxicon: Official Journal of the International Society on Toxinology, 53(2), 289–299. 10.1016/j.toxicon.2008.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj, V. K., Singh, R., Sharma, J., Rajendran, V., Purohit, R., & Kumar, S. (2020). Identification of bioactive molecules from Tea plant as SARS-CoV-2 main protease inhibitors. Journal of Biomolecular Structure and Dynamics, 1–10. 10.1080/07391102.2020.1766572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonjouklian, R., Smitka, T. A., Doolin, L. E., Molloy, R. M., Debono, M., Shaffer, S. A., Moore, R. E., Stewart, J. B., & Patterson, G. M. L. (1991). Tjipanazoles, new antifungal agents from the blue-green alga Tolypothrix tjipanasensis. Tetrahedron Letter, 47(37), 7739–7750. 10.1016/S0040-4020(01)81932-3 [DOI] [Google Scholar]
- Boopathi, S., Poma, A. B., & Kolandaivel, P. (2020). Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. Journal of Biomolecular Structure and Dynamics, 1–10. 10.1080/07391102.2020.1758788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J., & Forsyth, C. J. (2003). Total synthesis of apratoxin A. Journal of the American Chemical Society, 125(29), 8734–8735. 10.1021/ja036050w [DOI] [PubMed] [Google Scholar]
- Daina, A., Michielin, O., & Zoete, V. (2017). SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules . Scientific Reports, 7, 42717. 10.1038/srep42717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das, S., Sarmah, S., Lyndem, S., & Singha Roy, A. (2020). An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. Journal of Biomolecular Structure and Dynamics, 1–11. 10.1080/07391102.2020.1763201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky, A. A. (2020). Natural products may interfere with SARS-CoV-2 attachment to the host cell. Journal of Biomolecular Structure and Dynamics, 1–10. 10.1080/07391102.2020.1761881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmezayen, A. D., Al-Obaidi, A., Şahin, A. T., & Yelekçi, K. (2020). Drug repurposing for coronavirus (COVID-19): In silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. Journal of Biomolecular Structure and Dynamics, 1–12. 10.1080/07391102.2020.1758791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enayatkhani, M., Hasaniazad, M., Faezi, S., Guklani, H., Davoodian, P., Ahmadi, N., Einakian, M. A., Karmostaji A., Ahmadi, K. (2020). Reverse vaccinology approach to design a novel multi-epitope vaccine candidate against COVID-19: An in silico study. Journal of Biomolecular Structure and Dynamics, 1–16. 10.1080/07391102.2020.1756411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enmozhi, S. K., Raja, K., Sebastine, I., & Joseph, J. (2020). Andrographolide as a potential inhibitor of SARS-CoV-2 main protease: An in silico approach. Journal of Biomolecular Structure and Dynamics, 1–7. 10.1080/07391102.2020.1760136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, M. K., Vemula, S., Donde, R., Gouda, G., Behera, L., & Vadde, R. (2020). In-silico approaches to detect inhibitors of the human severe acute respiratory syndrome coronavirus envelope protein ion channel. Journal of Biomolecular Structure and Dynamics, 1–11. 10.1080/07391102.2020.1751300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyebi, G. A., Ogunro, O. B., Adegunloye, A. P., Ogunyemi, O. M., & Afolabi, S. O. (2020). Potential inhibitors of coronavirus 3-chymotrypsin-like protease (3CLpro): An in silico screening of alkaloids and terpenoids from African medicinal plants. Journal of Biomolecular Structure and Dynamics, 1–13. 10.1080/07391102.2020.1764868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan, A., Paray, B. A., Hussain, A., Qadir, F. A., Attar, F., Aziz, F. M., Sharifi, M., Derakhshankhah, H., Rasti, B., Mehrabi, M., Shahpasand, K., Saboury, A. A., & Shahpasand, K. (2020). A review on the cleavage priming of the spike protein on coronavirus by angiotensin converting enzyme-2 and furin. Journal of Biomolecular Structure and Dynamics, 1–9. 10.1080/07391102.2020.1754293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins, A. L., Groom, C. R., & Alex, A. (2004). Ligand efficiency: A useful metric for lead selection. Drug Discovery Today, 9(10), 430–431. 10.1016/S1359-6446(04)03069-7 [DOI] [PubMed] [Google Scholar]
- Hopkins, A. L., Keserü, G. M., Leeson, P. D., Rees, D. C., & Reynolds, C. H. (2014). The role of ligand efficiency metrics in drug discovery. Nature Reviews Drug Discovery, 13(2), 105–121. 10.1038/nrd4163 [DOI] [PubMed] [Google Scholar]
- Hu, H., & Sun, S. C. (2016). Ubiquitin signaling in immune responses. Cell Research, 26(4), 457–483. 10.1038/cr.2016.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, C., Wang, Y., Li, X., Ren, L., Zhao, J., Hu, Y., Zhang, L., Fan, G., Xu, J., Gu, X., Cheng, Z., Yu, T., Xia, J., Wei, Y., Wu, W., Xie, X., Yin, W., Li, H., Liu, M., … Cao, B. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. The Lancet, 395(10223), 497–506. 10.1016/S0140-6736(20)30183-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, J., Rees, S., Kalindjian, S., & Philpott, K. (2011). Principles of early drug discovery. British Journal of Pharmacology, 162(6), 1239–1249. 10.1111/j.1476-5381.2010.01127.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam, R., Parves, R., Paul, A. S., Uddin, N., Rahman, M. S., Mamun, A. A., Hossain, M. N., Ali, M. A., & Halim, M. A. (2020). A molecular modeling approach to identify effective antiviral phytochemicals against the main protease of SARS-CoV-2. Journal of Biomolecular Structure and Dynamics, 1–12. 10.1080/07391102.2020.1761883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs, J., Grum-Tokars, V., Zhou, Y., Turlington, M., Saldanha, S. A., Chase, P., Eggler, A., Dawson, E. S., Baez-Santos, Y. M., Tomar, S., Mielech, A. M., Baker, S. C., Lindsley, C. W., Hodder, P., Mesecar, A., & Stauffer, S. R. (2013). Discovery, synthesis, and structure-based optimization of a series of N-(tert-butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. Journal of Medicinal Chemistry, 56(2), 534–546. 10.1021/jm301580n [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, Z., Du, X., Xu, Y., Deng, Y., Liu, M., Zhao, Y., Zhang, B., Li, X., Zhang, L., Peng, C., Duan, Y., Yu, J., Wang, L., Yang, K., Liu, F., Jiang, R., Yang, X., You, T., Liu, X., … Yang, H. (2020). Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature, 582(7811), 289–293. 10.1038/s41586-020-2223-y [DOI] [PubMed] [Google Scholar]
- Joshi, R. S., Jagdale, S. S., Bansode, S. B., Shankar, S. S., Tellis, M. B., Pandya, V. K., Chugh, A., Giri, A. P., & Kulkarni, M. J. (2020). Discovery of potential multi-target directed ligands by targeting host-specific SARS-CoV-2 structurally conserved main protease. Journal of Biomolecular Structure and Dynamics, 1–16. 10.1080/07391102.2020.1760137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karypidou, K., Ribone, S. R., Quevedo, M. A., Persoons, L., Pannecouque, C., Helsen, C., Claessens, F., & Dehaen, W. (2018). Synthesis, biological evaluation and molecular modeling of a novel series of fused 1,2,3-triazoles as potential anti-coronavirus agents . Bioorganic & Medicinal Chemistry Letters, 28(21), 3472–3476. 10.1016/j.bmcl.2018.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, M. T., Ali, A., Wang, Q., Irfan, M., Khan, A., Zeb, M. T., Zhang, Y. J., Chinnasamy, S., & Wei, D. Q. (2020). Marine natural compounds as potent inhibitors against the main protease of SARS-CoV-2. A molecular dynamic study. Journal of Biomolecular Structure and Dynamics, 1–11. 10.1080/07391102.2020.1769733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, R. J., Jha, R. K., Amera, G., Jain, M., Singh, E., Pathak, A., Singh, R. P., Muthukumaran, J., & Singh, A. K. (2020). Targeting SARS-Cov-2: A systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 2'-O-ribose methyltransferase. Journal of Biomolecular Structure and Dynamics, 1–14. 10.1080/07391102.2020.1753577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, S. A., Zia, K., Ashraf, S., Uddin, R., & Ul-Haq, Z. (2020). Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. Journal of Biomolecular Structure and Dynamics, 1–10. 10.1080/07391102.2020.1751298 [DOI] [PubMed] [Google Scholar]
- Kumar, D., Kumari, K., Jayaraj, A., Kumar, V., Kumar, R. V., Dass, S. K., Chandra, R., & Singh, P. (2020). Understanding the binding affinity of noscapines with protease of SARS-CoV-2 for COVID-19 using MD simulations at different temperatures. Journal of Biomolecular Structure and Dynamics, 1–14. 10.1080/07391102.2020.1752310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari, R., Kumar, R., Open Source Drug Discovery Consortium, & Lynn, A. (2014). g_mmpbsa-A GROMACS tool for high-throughput MM-PBSA calculations. Journal of Chemical Information and Modeling, 54(7), 1951–1962., 10.1021/ci500020m [DOI] [PubMed] [Google Scholar]
- Lagorce, D., Bouslama, L., Becot, J., Miteva, M. A., & Villoutreix, B. O. (2017). FAF-Drugs4: Free ADME-tox filtering computations for chemical biology and early stages drug discovery. Bioinformatics (Oxford, England), 33(22), 3658–3660. 10.1093/bioinformatics/btx491 [DOI] [PubMed] [Google Scholar]
- Leeson, P. D., & Springthorpe, B. (2007). The influence of drug-like concepts on decision-making in medicinal chemistry. Nature Reviews. Drug Discovery, 6(11), 881–890. 10.1038/nrd2445 [DOI] [PubMed] [Google Scholar]
- Lipinski, C. A., Lombardo, F., Dominy, B. W., & Feeney, P. J. (2001). Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews, 46(1–3), 3–26. 10.1016/s0169-409x(00)00129-0 [DOI] [PubMed] [Google Scholar]
- Lobo-Galo, N., Terrazas-López, M., Martínez-Martínez, A., & Díaz-Sánchez, Á. G. (2020). FDA-approved thiol reacting drugs that potentially bind into the SARS-CoV-2 main protease, essential for viral replication. Journal of Biomolecular Structure and Dynamics, 1–9. 10.1080/07391102.2020.1764393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, R., Zhao, X., Li, J., Niu, P., Yang, B., Wu, H., Wang, W., Song, H., Huang, B., Zhu, N., Bi, Y., Ma, X., Zhan, F., Wang, L., Hu, T., Zhou, H., Hu, Z., Zhou, W., Zhao, L., … Tan, W. (2020). Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. The Lancet, 395(10224), 565–574. 10.1016/S0140-6736(20)30251-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luesch, H., Yoshida, W. Y., Moore, R. E., Paul, V. J., & Corbett, T. H. (2001). Total structure determination of apratoxin A, a potent novel cytotoxin from the marine cyanobacterium Lyngbya majuscula. Journal of the American Chemical Society, 123(23), 5418–5423. 10.1021/ja010453j [DOI] [PubMed] [Google Scholar]
- Muralidharan, N., Sakthivel, R., Velmurugan, D., & Gromiha, M. M. (2020). Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 protease against COVID-19. Journal of Biomolecular Structure and Dynamics, 1–6. 10.1080/07391102.2020.1752802 [DOI] [PubMed] [Google Scholar]
- Naidoo, D., Roy, A., Slavětínská, L. P., Chukwujekwu, J. C., Gupta, S., & Van Staden, J. (2020). New role for crinamine as a potent, safe and selective inhibitor of human monoamine oxidase B: In vitro and in silico pharmacology and modeling. Journal of Ethnopharmacology, 248, 112305. 10.1016/j.jep.2019.112305 [DOI] [PubMed] [Google Scholar]
- O’Boyle, N. M., Banck, M., James, C. A., Morley, C., Vandermeersch, T., & Hutchison, G. R. (2011). Open babel: An open chemical toolbox. Journal of Cheminformatics, 3, 33. 10.1186/1758-2946-3-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pant, S., Singh, M., Ravichandiran, V., Murty, U. S. N., & Srivastava, H. K. (2020). Peptide like and small molecule inhibitors against Covid-19. Journal of Biomolecular Structure and Dynamics, 1–10. 10.1080/07391102.2020.1757510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires, D. E., Blundell, T. L., & Ascher, D. B. (2015). pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using Graph-Based Signatures. Journal of Medicinal Chemistry, 58(9), 4066–4072. 10.1021/acs.jmedchem.5b00104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouliot, M., & Jeanmart, S. (2016). Pan Assay Interference Compounds (PAINS) and other promiscuous compounds in antifungal research: Mini perspective. Journal of Medicinal Chemistry, 59(2), 497–503. 10.1021/acs.jmedchem.5b00361 [DOI] [PubMed] [Google Scholar]
- Prajapat, M., Sarma, P., Shekhar, N., Avti, P., Sinha, S., Kaur, H., Kumar, S., Bhattacharyya, A., Kumar, H., Bansal, S., & Medhi, B. (2020). Drug targets for corona virus: A systematic review. Indian Journal of Pharmacology, 52(1), 56–85. 10.4103/ijp.IJP_115_20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salentin, S., Schreiber, S., Haupt, V. J., Adasme, M. F., & Schroeder, M. (2015). PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Research, 43(W1), W443–447. 10.1093/nar/gkv315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarma, P., Sekhar, N., Prajapat, M., Avti, P., Kaur, H., Kumar, S., Singh, S., Kumar, H., Prakash, A., Dhibar, D. P., & Medhi, B. (2020). In-silico homology assisted identification of inhibitor of RNA binding against 2019-nCoV N-protein (N terminal domain). Journal of Biomolecular Structure and Dynamics, 1–9. 10.1080/07391102.2020.1753580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer, D. J., & Krylov, V. S. (2000). Anti-HIV activity of extracts and compounds from algae and cyanobacteria. Ecotoxicology and Environmental Safety, 45(3), 208–227. 10.1006/eesa.1999.1862 [DOI] [PubMed] [Google Scholar]
- Schüttelkopf, A. W., & Van Aalten, D. M. (2004). PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallographica. Section D, Biological Crystallography, 60(Pt 8), 1355–1363. 10.1107/S0907444904011679 [DOI] [PubMed] [Google Scholar]
- Schyman, P., Liu, R., Desai, V., & Wallqvist, A. (2017). vNN web server for ADMET predictions. Frontiers in Pharmacology, 8, 889. 10.3389/fphar.2017.00889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shultz, M. D. (2013). The thermodynamic basis for the use of lipophilic efficiency (LipE) in enthalpic optimizations. Bioorganic and Medicinal Chemistry Letters, 23(21), 5992–6000. 10.1016/j.bmcl.2013.08.030 [DOI] [PubMed] [Google Scholar]
- Sinha, S. K., Shakya, A., Prasad, S. K., Singh, S., Gurav, N. S., Prasad, R. S., & Gurav, S. S. (2020). An in-silico evaluation of different Saikosaponins for their potency against SARS-CoV-2 using NSP15 and fusion spike glycoprotein as targets. Journal of Biomolecular Structure and Dynamics, 1–12. 10.1080/07391102.2020.1762741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solter, P. F., & Beasley, V. R. (2013). Safety assessment including current and emerging issues in toxicologic pathology. In Haschek, W. M., Rousseauz, C. G., Wallig, M. A. (Eds.), Haschek and Rousseaux’s handbook of toxicologic pathology. Academic Press. 10.1016/C2010-1-67850-9 [DOI] [Google Scholar]
- Sturdy, M., Krunic, A., Cho, S., Franzblau, S., & Orjala, J. (2010). Eucapsitrione, an anti-Mycobacterium tuberculosis anthraquinone derivative from the cultured freshwater cyanobacterium Eucapsis sp. Journal of Natural Products, 73(8), 1441–1443. 10.1021/np100299v [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai, W., He, L., Zhang, X., Pu, J., Voronin, D., Jiang, S., Zhou, Y., & Du, L. (2020). Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cellular & Molecular Immunology, 17(6), 613–618. 10.1038/s41423-020-0400-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, L. T., & Phyo, M. Y. (2020). Marine cyanobacteria: A source of lead compounds and their clinically-relevant molecular targets. Molecules, 25(9), 2197. 10.3390/molecules25092197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott, O., & Olson, A. J. (2010). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31(2), 455–461. 10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ul Qamar, M. T., Alqahtani, S. M., Alamri, M. A., & Chen, L. L. (2020). Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. Journal of Pharmaceutical Analysis. 10.1016/j.jpha.2020.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umesh, K. D., Selvaraj, C., Singh, S. K., & Dubey, V. K. (2020). Identification of new anti-nCoV drug chemical compounds from Indian spices exploiting SARS-CoV-2 main protease as target. Journal of Biomolecular Structure and Dynamics, 1–9. 10.1080/07391102.2020.1763202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahedi, H. M., Ahmad, S., & Abbasi, S. W. (2020). Stilbene based natural compounds as promising drug candidates against COVID-19. Journal of Biomolecular Structure and Dynamics, 1–10. 10.1080/07391102.2020.1762743 [DOI] [PubMed] [Google Scholar]
- Weiss, C., Figueras, E., Borbely, A., & Sewald, N. (2017). Cryptophycins: Cytotoxic cyclodepsipeptides with potential for tumor targeting. Journal of Peptide Science: An Official Publication of the European Peptide Society, 23(7-8), 514–531. 10.1002/psc.3015 [DOI] [PubMed] [Google Scholar]
- Xu, X., Chen, P., Wang, J., Feng, J., Zhou, H., Li, X., Zhong, W., & Hao, P. (2020). Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Science China Life Sciences, 63(3), 457–460. 10.1007/s11427-020-1637-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.