

Abstract
Considering lack of validated therapeutic drugs or vaccines against contagious SARS-CoV2, various efforts have been focused on repurposing of existing drugs or identifying new agents. In an attempt to identify new and potential SARS-CoV2 inhibitors targeting specific enzyme of the pathogen, a few induced fit models of SARS-CoV2 main protease (Mpro) including N-aryl amide and aryl sulfonamide based fragments were subjected to a multi-step in silico strategy. Sub-structure query of co-crystallographic fragments provided numerous ZINC15 driven commercially available compounds that entered molecular docking stage to find binding interactions/modes inside Mpro active site. Docking results were reevaluated through time dependent stability of top-ranked ligand-protease complexes by molecular dynamics (MD) simulations within 50 ns. Relative contribution of interacted residues in binding to the most probable binding pose was estimated through amino acid decomposition analysis in B3LYP level of theory with Def2-TZVPP split basis set. In confirmation of docking results, MD simulations revealed less perceptible torsional distortions (more stable binding mode) in binding of ZINC_252512772 (ΔGb −9.18 kcal/mol) into Mpro active site. H-bond interactions and hydrophobic contacts were determinant forces in binding interactions of in silico hit. Quantum chemical calculations confirmed MD results and proved the pivotal role of a conserved residue (Glu166) in making permanent hydrogen bond (98% of MD simulations time) with ZINC_252512772. Drug-like physicochemical properties as well as desirable target binding interactions nominated ZINC_252512772 as a desirable in silico hit for further development toward SARS-CoV2 inhibitors.
Highlights
A few N-aryl amide/aryl sulfonamide based fragments were subjected to a multi-step in silico strategy to afford potential SARS-CoV2 Mpro inhibitors.
MD simulations revealed less perceptible torsional distortions (more stable binding mode) in binding of ZINC_252512772 (ΔGb -9.18 kcal/mol) into Mpro active site.
H-bond interactions and hydrophobic contacts were determinant forces in binding interactions of in silico hit.
Quantum chemical calculations confirmed MD results and proved pivotal role of a conserved residue (Glu166) in making permanent hydrogen bond (98% of MD simulations time) with ZINC_252512772.
Communicated by Ramaswamy H. Sarma
Keywords: Covid-19, main protease, molecular dynamics, in silico hit
1. Introduction
Coronaviruses (CoVs) are enveloped viruses with positive single-stranded RNA genomes infecting both animals and human (Fung & Liu, 2019). CoVs are caused of several diseases from common cold to more severe highly pathogenic ones such as SARS (Severe acute respiratory syndrome), MERS (Middle-east respiratory syndrome) and Coronavirus disease 2019 (Covid-19) with higher mortalities (Cui et al., 2019). A novel coronavirus SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2) is responsible for Covid-19 that first appeared in Wuhan region of China within December 2019 (Zhu et al., 2020). Due to high transmission rate, SARS-CoV-2 spread quickly in multiple countries and the WHO declared an outbreak of COVID-19 a pandemic on March 2020 (https://www.who.int/emergencies/diseases/novel-coronavirus-2019; Gorbalenya et al., 2020). At the time of writing present manuscript, more than 200 countries were affected with more than 2600000 confirmed cases and about 180000 deaths making it a serious threat to global public health (https://www.who.int/emergencies/diseases/novel-coronavirus-2019).
Similar to other members of beta-coronaviruses (SARS-CoV and MERS-CoV), SARS-CoV-2 cause viral pneumonia by attacking lower respiratory system, entry into alveolar epithelial cells, rapid replication and triggering a strong immune response leading to pulmonary tissue damage (Villar et al., 2019). More vulnerable population that must be majorly managed on prevention of SARS-CoV2 infection, are the people with decreased immune function such as the elderly and individuals suffering from basic diseases (Chen et al., 2020).
Given the urgency of highly contagious SARS-CoV2 and considering the lack of validated therapeutic drugs or vaccines against this pathogen, several research groups rely on repurposing FDA approved drugs with proven effectiveness against similar infectious diseases (Boopathi et al., 2020; Liu et al., 2020). Numerous drugs, such as ribavirin, lopinavir and interferon have been tried against SARS or MERS, although the efficacies of some drugs remained controversial (Zumla et al., 2016). A few reports indicated that broad-spectrum antiviral drug arbidol that has been used against influenza outbreaks, could enter the clinical trials (Li & De Clercq, 2020). Optimistic results on the application of chloroquine and remdesivir against clinical isolate of 2019-nCoV have been attained (Wang et al., 2020) and remdesivir is also currently under clinical investigations against Ebola virus infection (Mulangu et al., 2019). Barcitinib was proposed as a potential anti-Covid-19 agents on the basis of its anti-inflammatory effect and possibly reducing viral entry (Richardson et al., 2020). In an open-label control study, Cai et al. demonstrated that favipiravir could control Covid-19 progression and viral clearance (Cai et al., 2020). Another open-label non-randomized clinical trial survey indicated that viral load reduction/disappearance in hydroxychloroquine administered Covid-19 patients were reinforced by azithromycin (Gautret et al., 2020). In addition to synthetic drugs (Adeoye et al., 2020; Lobo-Galo et al., 2020), traditional Chinese medicines (Ren et al., 2020; Tahir et al., 2019) and other medicinal plants (Aanouz et al., 2020; Enmozhi et al., 2020; Islam et al., 2020; Umesh et al., 2020) also draw attention in treatment of Covid-19.
Elucidation of SARS-CoV2 pathogenic mechanisms with the aim of identifying potential drug targets, contribute significantly to successful drug repurposing purposes and also design and development of novel therapeutic agents with probably less side effects (Al-Khafaji et al., 2020). Human CoVs are characterized by structural proteins such as spike protein (Protein S) and also nonstructural proteins including 3-chymotrypsin-like cysteine protease (3CLpro), papain-like protease (PLpro) and RNA dependent RNA polymerase (RdRp) (Ibrahim et al., 2019). During viral infection progression, both structural and nonstructural proteins play a key part, proposing that targeting them could be a promising strategy for controlling COVID-19 infection (Duan et al., 2020). It should be noted that some of the considered proteins were previously proven to be drug discovery targets in the case of SARS-CoV and MERS-CoV (ul Qamar et al., 2020) and may be looked upon as druggable targets in the case of SARS-CoV2 due to the sequence similarity with SARS-CoV (ul Qamar et al., 2020) and also similar binding sites (Joshi et al., 2020; Liu et al., 2020). Hoffmann et al. conducted an interesting research on TMPRSS2 (host-cell produced transmembrane protease serine 2) that facilitates binding of spike protein to angiotensin-converting enzyme 2 (ACE2) on the host cell surface membrane. It was demonstrated that SARS-CoV-2 uses the SARS-CoV ACE2 and TMPRSS2 for host cell entry and clinically proven TMPRSS2 inhibitor camostat mesylate could block SARS-CoV-2 entry (Hoffmann et al., 2020).
Along with conducting in vitro and clinical assessments with the aim of successful drug repurposing, few evaluations were also dedicated to in silico development or computational molecular modeling of new phytochemicals or synthetic compounds (Elfiky, 2020d; Elfiky & Azzam, 2020; Gyebi et al., 2020; Wahedi et al., 2020) or FDA approved drugs against different SARS-CoV2 targets such as RdRp (Elfiky, 2020a, 2020b, 2020c), 3CLpro (Khan, Jha, et al. 2020a; Khan, Zia, et al. 2020b; Muralidharan et al., 2020; Pant et al., 2020), helicase (Beck et al., 2020), hydrolase (Elmezayen et al., 2020), nucleocapsid protein (Sarma et al., 2020), envelope protein (Gupta et al., 2020), cell-surface Heat Shock Protein A5, ACE2 and spike protein (Abdelli et al., 2020; Ibrahim et al., 2020; Sinha et al., 2020). Moreover, some studies focused on the design of a vaccine and a preventative peptidomimetic SARS-CoV2 antagonist (Enayatkhani et al., 2020; Hasan et al., 2020; Robson, 2020). SARS-CoV2 protease enzymes are of significant interest in targeting and modeling studies since they have determinant role in cleaving viral polyproteins into effector proteins, deubiquitinating certain host cell proteins and immune suppression (Baez-Santos et al., 2015; Liu et al., 2020).
Given the vital role of SARS-CoV2 protease in viral infection, current study focused on the identification of in silico hit compounds as potential SARS-CoV2 inhibitors via multi-step modeling strategy comprising docking, molecular dynamics (MD) simulations and ab initio based amino acid decomposition analysis of a ZINC retrieved chemical entities (Sterling & Irwin, 2015). Our structure based simulations run on high-resolution 3D structures of SARS-CoV2 main protease (Mpro) in complex with several fragment ligands (PDB IDs 5R7Y, 5R7Z, 5R80, 5R81, 5R82, 5R83 and 5R84) which were recently unveiled by Fearon et al. (Fearon et al., 2020). To get more information, hierarchical view of the multi-step simulations is depicted in Figure 1.
Figure 1.

Hierarchical view of the multi-step simulations to identify hit compounds as potential SARS-CoV2 inhibitors; Covid-19 related 3 D Mpro structures were screened and subjected to AutoDock4.2 validation step to afford three holo structures with PDB IDs 5R80, 5R81 & 5R84 (Fearon et al., 2020). Corresponding co-crystallographic fragment ligands were subjected to substructure query in Zinc15 database with the criteria set on all subsets including purchasable compounds (Sterling & Irwin, 2015). 295 compounds were selected and entered into molecular docking study to attain three top-ranked analogues with highest binding affinities to related protease targets. Time dependent stability of top-ranked ligand-protease complexes were checked by MD simulations within 50 ns and the cycle might be repeated for next top-ranked candidates if non-desirable results were achieved. Subsequent amino acid decomposition analysis was performed on in silico hit compounds to acquire ligand-residue intermolecular binding energies by functional B3LYP in association with split valence basis set using polarization functions (Def2-TZVPP). Schematic 3 D representation of fragment-enzyme complexes were generated by NGL which is a WebGL based 3 D viewer incorporated into PDB (Rose et al., 2018).
2. Materials and methods
2.1. Target dataset
3D structures of SARS-CoV2 Mpro in complex with several fragment ligands (5R7Y, 5R7Z, 5R80, 5R81, 5R82, 5R83 and 5R84) were retrieved from the Protein Data Bank (www.rcsb.org) (Fearon et al., 2020). Co-crystallographic dimethyl sulfoxide and water molecules were all removed from the original PDB files and non-polar hydrogens merged. Acquired files were converted to pdbqt format after assigning Kollman charges and solvation parameters via AutoDock Tools program (ADT) (Morris et al., 2009; Sanner, 1999).
2.2. Ligand dataset
Internal validation of AutoDock4.2 software was done by seven co-crystallographic ligands (5R7Y, 5R7Z, 5R80, 5R81, 5R82, 5R83 and 5R84) using their interacted conformations as starting point. Detailed characteristics of cognate ligands are summarized in internal validation section. All archived ligands that were retrieved in MOL format from Zinc15 database (Sterling & Irwin, 2015) were finally converted to pdbqt using ADT subsequent to the assignment of Gasteiger charges and solvation parameters (Morris et al., 2009; Sanner, 1999). Physicochemical descriptors and ADME parameters were computed by web-based SwissADME program (Daina et al., 2017).
2.3. Molecular docking
Lamarckian genetic algorithm (LGA) incorporated into AutoDock4.2 package was used to run ligand flexible docking simulations on Zinc driven small molecules (Morris et al., 2009; Sanner, 1999). All the processing steps and docking parameters were arranged according to previous protocols (Alikhani et al., 2018). Post-docking representation and analysis of ligand-enzyme interactions were performed by the aid of the protein-ligand interaction profiler (PLIP) web server (Salentin et al., 2015). Besides protein-ligand interaction profiler (PLIP) package, discovery studio visualizer (ViewerLite) 3.5 was also used to graphically represent the molecules.
2.4. MD Simulation study
The all-atom MD simulations were performed using GROMACS 4.6.5 simulation package (Hess et al., 2008) with standard AMBER99SB-ILDN force fields (Lindorff‐Larsen et al., 2010). The compatible topologies of the ligands were prepared using the General Amber Force Field (GAFF) (Wang et al., 2004) and ANTECHAMBER suite of programs (Case et al, 2005), applying the semiempirical AM1BCC charges (Jakalian et al., 2002). MD simulations were performed according to previous studies (Ebadi et al., 2013; Razzaghi-Asl et al., 2018). Briefly, two 10000 and 5000 steps steepest descent and conjugated gradient minimizations were performed to remove potential clashes between water, ions, and ligand-enzyme in the simulation box. Following initial optimization, the system was equilibrated at 300 K during 500 ps NVT and 1000 ps NPT ensembles. 50 ns MD production was performed in NPT ensemble without any restraint. The temperature and pressure were controlled using the modified V-rescale thermostat from Berendsen (Berendsen et al., 1984) with 0.1 ps time constant and Parrinello-Rahman barostat (Parrinello & Rahman, 1981) with 2 ps time constant for coupling respectively.
2.5. Amino acid decomposition analysis
Analyzing of intermolecular binding energy components was performed via constructing a chemical environment including key interacted SARS-CoV2 residues around a docked molecule. N-terminals of interacted amino acids were acetylated while C-terminals were methyl amidated without disturbing original conformational and configurational features. Polar hydrogen bonds were optimized using B3LYP/Def2-TZVPP method through heavy atom fixing (HAF) approximation (constrained optimizations) (Fogarasi et al., 1992). All the ligand-residue binding energies were estimated by the same method and basis set. The whole calculations were performed with the ORCA ab initio quantum chemistry package (Neese, 2012).
3. Results
3.1. Molecular docking
To check the validity of LGA in predicting binding modes within SARS-CoV2 Mpro binding site, candidate co-crystalized ligands were extracted from original complex structures and subjected to re-docking into the corresponding induced fit models (5R7Y, 5R7Z, 5R80, 5R81, 5R82, 5R83 and 5R84). For this purpose, root-mean-square deviation (RMSD) of the Cartesian coordinates of re-docked co-crystallographic ligand atoms were used as validation criteria (Vyas et al., 2008). On the basis of re-docking results, adaptable predictability levels (≤ 2 Å) could be achieved with 100 independent GA runs and 2.5 × 107 maximum number of evaluations for complexes 5R80, 5R81 and 5R84 (Table 1).
Table 1.
AutoDock4.2 validation results for different 3 D holo SARS-CoV2 protease-ligand complexes retrieved from PDB.
| No. | PDB code | Resolution (Ǻ) | Co-crystallographic ligand | Top-ranked population (out of 50) | ΔGb (kcal/mol) | RMSD from reference (Å) |
|---|---|---|---|---|---|---|
| 1 | 5R7Y | 1.65 | 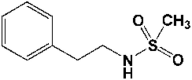 |
23 | −5.82 | 6.42 |
| 2 | 5R7Z | 1.59 | 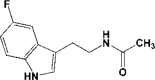 |
33 | −6.05 | 4.17 |
| 3 | 5R80 | 1.93 | 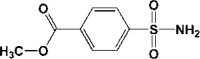 |
49 | −5.02 | 1.99 |
| 4 | 5R81 | 1.95 | 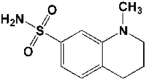 |
49 | −6.82 | 1.58 |
| 5 | 5R82 | 1.31 | 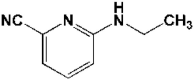 |
45 | −5.16 | 7.25 |
| 6 | 5R83a | 1.58 | 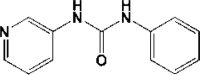 |
50 | −6.94 | 6.93 |
| 7 | 5R84 | 1.83 | 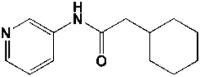 |
32 | −7.21 | 0.40 |
Drug-enzyme inhibition constants (Ki) were estimated via calculation of free binding energies (equation 1) which was incorporated into the force-field of AutoDock4 as a dependent variable (equation 2):
| (1) |
| (2) |
In equation 1, ΔGb is indicative of free binding energy (kcal/mol), R is the gas constant (Cal.K−1.mol−1) and T represents temperature in kelvin (298.15 K). A number 2.71828 is indicates Napier's constant. In equation 2, ΔGTorsional is a term which shows loss of torsional free energy upon binding to enzyme binding site. EvdW and EH-bond and EElectrostatic define hydrophobic interaction energy (van der Waals energy), hydrogen bond interaction energy and electrostatic interaction energies. EDesolvation represents desolvation energy or ‘hydrophobicity’ for drug target interaction. To explain more, EDesolvation represents an energy required to replace ligand-water bonds with that of ligand target ones.
Corresponding co-crystallographic fragments (PDB IDs 5R80, 5R81 & 5R84) were subjected to substructure query in Zinc15 database with a criteria set on ‘all subsets’ to afford 295 purchasable compounds for docking simulations (Sterling & Irwin, 2015). Achieved docked poses were ranked to attain best and worst conformations associated with their binding affinities (Table 2).
Table 2.
AutoDock4.2 results for top-ranked Zinc15 driven compounds with highest binding affinities to SARS-CoV2 protease; within each structure, primary fragment used as a criteria for substructure query is depicted by dashed rectangle/ellipsoid frames.
| No. | PDB code | Top-ranked compound (Best conformation) | Top-ranked population (out of 50) | ΔGb (kcal/mol) |
||
|---|---|---|---|---|---|---|
| Best conformation | Co-crystallographic conformation | Worst conformation | ||||
| 1 | 5R80 |
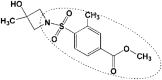 ZINC_170619726 ZINC_170619726
|
33 | −7.14 | −5.02 | −6.52 ZINC_74941908 |
| 2 | 5R81 |
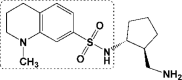 ZINC_108126011 ZINC_108126011
|
16 | −9.39 | −6.82 | −6.52 ZINC_74941908 |
| 3 | 5R84 |
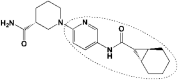 ZINC_252512772 ZINC_252512772
|
20 | −9.18 | −7.21 | −6.77 ZINC_70069 |
To characterize binding modes of screened compounds inside the active site of SARS-CoV2 Mpro, all intermolecular interactions of ZINC_170619726, ZINC_108126011 and ZINC_252512772 with key residues, schematic representation of ligand binding poses are depicted in Figure 2. It should be noted that for salt bridge interactions, the distance is reported between charge centers.
Figure 2.

Schematic 3 D representation of binding poses of ZINC_170619726, ZINC_108126011 and ZINC_252512772 inside binding site of SARS-CoV2 protease with PDB accession codes a) 5R80, b) 5R81 & c) 5R84, respectively; Yellow spheres within ZINC_170619726 conformation indicates charge centers of salt bridge between SARS-CoV2 and ligand.
3.2. MD Simulations
All systems reached steady-state after a few ps from the beginning of MD simulation. The convergence of energy and temperature was evaluated by calculating mean and RSD during the 50 ns MD simulation. The temperature of the holo 5R84, ZINC_170619726, ZINC_108126011 and ZINC_252512772 systems were stable at 300 K (300.0 (RSD: 0.38) in four systems). The energy of holo 5R84, Zinc_170619726, ZINC_108126011, and ZINC_252512772 systems reached −173007 (RSD: 0.16%), −168959 (RSD: 0.16), −170846 (0.16) and −173011 (0.16) kcal/mol, respectively and were stable during 50 ns MD simulation.
The stability of SARS-CoV2 Mpro during MD simulation was evaluated through monitoring the RMSD matrix, the radius of gyration, and intra-enzyme hydrogen bonds. The 3 D structure of SARS-CoV2 Mpro was stable during the 50 ns MD simulation. The RMSD of protein in crystallographic structure (5R84) increased to 0.12 nm in 20 ps at the beginning of production run. The RMSD was stable during 50 ns MD simulation (0.19 ± 0.014 nm (RSD: 7.7%)). The radius of gyration (Rg) was constant during 50 ns MD simulation (2.21 ± 0.009 nm). The mean number of intra-hydrogen bonds in SARS-CoV2 Mpro for crystallographic structure (5R84) was calculated 231.7 ± 7.3.
Figure 3 shows the RMSD matrix and radius of gyration of three potential inhibitors of SARS-CoV2 Mpro found via virtual screening. The maximum RMSD changes in the RMSD matrix of three systems were less than 0.3 nm indicating the convergence of SARS-CoV2 Mpro to equilibrium structure.
Figure 3.

RMSD matrix and radius of gyration plots of Zinc_170619726, ZINC_108126011 (PDB 5R80), and ZINC_252512772 (PDB 5R84) in complex with SARS-CoV2 Mpro.
The mean number of intra-hydrogen bonds in SARS-CoV2 Mpro for Zinc_170619726, ZINC_108126011, and ZINC_252512772 systems were calculated as 227.3 ± 6.9, 226.0 ± 7.2 and 225.6 ± 6.9 in each frame of trajectories, respectively.
MD simulation of holo structure of SARS-CoV2 Mpro revealed that the Mpro formed stable complex with Z31792168 (Figure 4). Z31792168 was stable in the active site of SARS-CoV2 Mpro. Main conformational changes about 9 ns (red line in the RMSD matrix) was due to the rotation of cyclohexane ring and did not change the orientation of Z31792168 in the active site.
Figure 4.

The orientation of Z31792168 in holo structure of SARS-CoV2 Mpro (PDB 5R84) after 6, 10 and 40 ns of MD simulations. Cys145, Glu166, and Thr190 were depicted as blue sticks to represent the dimensions of the active site for better comparison.
The stability of Zinc_170619726 in complex with SARS-CoV2 Mpro was evaluated in terms of RMSD matrix and clustering analysis. The conformational changes in Zinc_170619726 during the 50 ns MD simulation led to 14 clusters by considering 2 nm cutoff and using gromos method. The size of the top three clusters was obtained as 83.8, 6.5, and 4.8% of the total population, respectively. Conformational changes of Zinc_170619726 in the active site of SARS-CoV2 Mpro are depicted in Figure 5. The red color in the margin of the RMSD matrix before 7 ns indicated the major conformational change in comparison to the initial structure. In predicted docking pose, the hydroxyl moiety of Zinc_170619726 formed a hydrogen bond with residue Thr190. But during the first 7 ns of MD simulations, it began to turn and orient toward Glu166. Zinc_170619726 formed an average hydrogen bond of 0.6 ± 0.7 per ns indicating that the hydrogen bonds formed between Zinc_170619726 and SARS-CoV2 Mpro were not relatively stable. No hydrogen bond with Glu166 was detected by analyzing the MD trajectories. The sulfone group of Zinc_170619726 formed a hydrogen bond with Gln192 in 37% of MD simulation time.
Figure 5.

The orientation of Zinc_170619726 (PDB 5R80) in the active site of SARS-CoV2 Mpro for the most populated cluster (83.8%), after 4, and 40 ns of MD simulations. Cys145, Glu166, and Thr190 were depicted as blue sticks to represent the dimensions of the active site for better comparison.
Figure 6 shows that ZINC_108126011 experienced a high level of conformational variation. The clustering of trajectories during the 50 ns MD simulation resulted in 109 clusters. The size of the most populated cluster was 31.9% of the total population. At the time of 8 ns of MD simulation, ZINC_108126011 leaped out of the binding site of SARS-CoV2 Mpro. After a short period (< 4 ns), ZINC_108126011 bound almost in the same pose as the initial docking structure. But in the newly formed complex, quaternary amine was more exposed to the bulk water. The amine group of ZINC_108126011 formed a salt bridge with Glu166 in 31.9% of trajectories. During this time, ZINC_108126011 formed a stable hydrogen bond with Asn142 indicating a different induced fit in the structure of SARS-CoV2 Mpro. In the last 10 ns of MD simulation, the amino moiety was only exposed to the bulk water.
Figure 6.

The orientation of ZINC_108126011 (PDB 5R81) in the active site of SARS-CoV2 Mpro for the most populated cluster (31.9%), after 8, and 16 ns of MD simulations. Cys145, Glu166, and Thr190 were depicted as blue sticks to represent the dimensions of the active site for better comparison.
Conformational changes of ZINC_252512772 in the initial predicted binding mode were less than Zinc_170619726 and ZINC_108126011 (Figure 7). The maximum change of the RMSD matrix was 0.34 nm. ZINC_252512772 formed a permanent hydrogen bond with Glu166 during 98% of MD simulations time.
Figure 7.

The orientation of ZINC_252512772 (PDB 5R84) in the active site of SARS-CoV2 Mpro for the most populated cluster (93.3%), after 8, and 22 ns of MD simulations. Cys145, Glu166, and Thr190 were depicted as blue sticks to represent the dimensions of the active site for better comparison.
The short range electrostatic and vdW interactions between studied ligands and the active site of SARS-CoV2 Mpro is reported in Table 3.
Table 3.
MD estimated van der Waals and electrostatic energies of 5r80 (Zinc_170619726), 5r81 (ZINC_108126011) and 5r84 (ZINC_252512772).
| Compound | Electrostatic (kcal/mol) | vdW (kcal/mol) |
|---|---|---|
| 5r84 (holo structure) | −11.3 (±3.7) | −32.2 (±3.2) |
| 5r80 | −11.7 (±5.0) | −32.0 (±2.4) |
| 5r81 | −6.6 (±3.6) | 32.6 (±3.6) |
| 5r84 | −16.9 (±3.8) | −41.4 (±2.4) |
3.3. Intermolecular binding energy components
On the basis of molecular docking and MD simulations results, ZINC_252512772 exhibited superior binding features and less conformational variations during 50 ns inside active site of SARS-CoV2 Mpro. In order to determine the relative contribution of each interacted Mpro residue in binding to ZINC_252512772, we were convinced to estimate the binding energies of the most probable binding pose of ZINC_252512772 with key residues surrounding active site in B3LYP level of theory with Def2-TZVPP split basis set (Table 4). It should be noted that Cys145 and Thr190 were also taken into consideration as the interacted environment due to MD outputs. All the ligand-residue binding interaction energies (ΔEb) were calculated through equation 3:
| (3) |
Table 4.
Estimated binding energies of ZINC_252512772 inside the binding site of SARS-CoV2 Mpro (PDB code 5R84) in B3LYP level of theory with the Def2-TZVPP as split basis set.
| Residue | Binding energy (kcal/mol) |
|---|---|
| His41 | 2.51 |
| Pro52 | 0.13 |
| Phe140 | −0.08 |
| Asn142 | −3.16 |
| Cys145 | −0.35 |
| Met165 | 5.27 |
| Glu166 | −31.66 |
| Arg188 | 1.98 |
| Gln189 | 0.90 |
| Thr190 | −0.09 |
ELR is indicative of residue ligand interaction energy while ER and EL stand for electronic energies for residues and ligand, respectively.
3.4. Pharmacokinetics prediction
In silico prediction of pharmacokinetics profile was performed through SwissADME calculator (Table 5).
Table 5.
Estimated physicochemical and ADME properties of selected in silico hits (ZINC_170619726, ZINC_108126011 and ZINC_252512772) predicted by swiss ADME.
| Compound | Physicochemical properties |
||||||
|---|---|---|---|---|---|---|---|
| MW | RTBs | HBA | HBD | TPSA (Å2) | logS / Class | Consensus Log Po/w | |
| ZINC_170619726 | 299.34 | 4 | 6 | 1 | 92.29 | −2.05 | 1.10 |
| ZINC_108126011 | 325.47 | 4 | 5 | 2 | 83.81 | −2.55 | 1.26 |
|
ZINC_252512772 |
355.46 |
5 |
3 |
2 |
88.32 |
−3.43 |
2.12 |
| Pharmacokinetics |
|||||||
| Compound |
GIA |
BBB permeant |
P-gp substrate |
CYP1A2 inhibitor |
CYP2C9 inhibitor |
CYP2D6 inhibitor |
CYP3A4 inhibitor |
| ZINC_170619726 | High | No | No | No | No | No | No |
| ZINC_108126011 | High | No | Yes | No | No | No | No |
| ZINC_252512772 | High | No | Yes | No | No | Yes | No |
4. Discussion
4.1. Analysis of binding affinity/mode
Growing fragments into drug-like structures via adding layers of rings, substituents and linkers has been regarded as a key approach for drug design (Jacquemard & Kellenberger, 2019). Desirable fragment libraries are appropriate starting points for developing privileged medicinal structures provided that further interactions to hotspots inside a target binding region can be reached upon molecular extension. Analysis of docked poses exhibited that ZINC_170619726, ZINC_108126011 and ZINC_252512772 were located in the binding site of SARS-CoV2 protease (Figure 8).
Figure 8.

Cluster of Zinc driven docked poses (ZINC_170619726, ZINC_108126011 and ZINC_252512772), co-crystallographic fragments (thick stick) and top-ranked poses (thick stick) within binding site residues of SARS-CoV2 protease binding site; PDB IDs a) 5R80, b) 5R81 & c) 5R84.
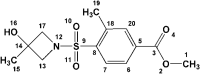
Assessed compounds participated in H-bond interaction and hydrophobic contacts with 13 hydrophilic and hydrophobic residues of SARS-CoV2 protease. In addition to H-bond interactions and hydrophobic contacts, Zinc_170619726 built salt bridge via carboxylate moiety with His41 imidazole nitrogens (depicted with yellow spheres as charge centers in Figure 2 and the reported distance is between charge centers). His41 is one of the catalytic dyad residues building an active site of the enzyme (Das et al., 2020; Yang et al., 2003). Comparison of interaction models indicated that Zinc_170619726 tended to made more hydrophilic bonds (Table 6) while hydrophobic contacts were dominant for ZINC_252512772 through involvement of cyclic carbon atoms (Table 7). Although more diverse residues were interacted, Zinc_170619726 was associated with lower free binding energy with regard to other three molecules.
Table 6.
Estimated binding interactions of Zinc_170619726 inside the binding site of SARS-CoV2 protease (Chain A; PDB 5R80).
 | ||||||
|---|---|---|---|---|---|---|
| No. | Residue | Interaction type | Distancea (nm) | Hydrogen bond donor angle (°) | Ligand atom/group | Protein atom |
| 1 | Met165 | Hydrophobic | 3.47 | – | C19 | CB (Side chain) |
| 2 | Glu166 | Hydrophobic | 3.59 | – | C7 | CB (Side chain) |
| 3 | Leu167 | Hydrophobic | 3.66 | – | C13 | CD2 (Side chain) |
| 4 | Pro168 | Hydrophobic | 3.97 | – | C13 | CG (Side chain) |
| 5 | Glu166 | Hydrogen bond | 2.88 | 105.98 | O4 | NH (Backbone) |
| 6 | Arg188 | Hydrogen bond | 1.74 | 122.07 | O16H | O (Backbone) |
| 7 | Thr190 | Hydrogen bond | 2.17 | 144.22 | O16 | NH (Backbone) |
| 8 | Gln192 | Hydrogen bond | 2.01 | 131.95 | O16 | NE2H (Side chain) |
| 9 | His163 | Salt bridge | 5.10 | – | Carboxylate | Imidazole Ns |
aFor hydrophobic interactions distance is reported between interacting carbon atoms; For H-bonds distance is reported between hydrogen and acceptor atom; For salt bridges distance is reported between charge centers.
bFor Hydrogen bonds donor angle is reported as the angle between donor, acceptor and hydrogen atoms.
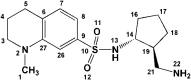
Table 7.
Estimated binding interactions of ZINC_252512772 inside the binding site of SARS-CoV2 protease (Chain A; PDB 5R84).
 | ||||||
|---|---|---|---|---|---|---|
| No. | Residue | Interaction type | Distance a (nm) | Hydrogen bond donor angle (°) | Ligand atom/group | Protein atom |
| 1 | His41 | Hydrophobic | 3.10 | – | C5 | CB (Side chain) |
| 2 | Pro52 | Hydrophobic | 3.01 | – | C4 | CG (Side chain) |
| 3 | Phe140 | Hydrophobic | 3.75 | – | C19 | CB (Side chain) |
| 4 | Met165 | Hydrophobic | 3.85 | – | C16 | CB (Side chain) |
| 5 | Glu166 | Hydrophobic | 3.28 | – | C19 | CB (Side chain) |
| 6 | Asn142 | Hydrogen bond | 1.95 | 135.79 | O25 | NH (Backbone) |
| 7 | Glu166 | Hydrogen bond | 2.16 | 114.39 | N24H | OE1 (Side chain) |
| 8 | Glu166 | Hydrogen bond | 1.96 | 165.50 | N13 | NH (Backbone) |
| 9 | Arg188 | Hydrogen bond | 3.60 | 104.22 | N10H | O (Backbone) |
| 10 | Gln189 | Hydrogen bond | 3.56 | 106.91 | N10 | NE2H (Side chain) |
aFor hydrophobic interactions distance is reported between interacting carbon atoms; For H-bonds distance is reported between hydrogen and acceptor atom; For salt bridges distance is reported between charge centers.
bFor Hydrogen bonds donor angle is reported as the angle between donor, acceptor and hydrogen atoms.
Met165 and Glu166 were found to form H-bond and hydrophobic interactions with all of the three molecules (Tables 6–8) and hence may be regarded as key residues in maintaining relevant complexes (Kumar et al., 2020). Perpendicular orientation of pyridyl ring and piperidinyl ring of ZINC_252512772 in the active site of SARS-CoV2 protease provided two hydrogen bonds with side chain carboxylate oxygen and backbone NH of Glu166 (Figure 2c). Similar H-bond pattern could be detected for ZINC_170619726 between ester carbonyl oxygen and Glu166 backbone NH (Figure 2a). Comparable hydrogen bond interaction was recorded between Glu166 carboxylate and amino methyl substituent of ZINC_108126011.
Table 8.
Estimated binding interactions of ZINC_108126011 inside the binding site of SARS-CoV2 protease (Chain A; PDB 5R81).
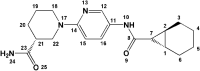 | ||||||
|---|---|---|---|---|---|---|
| No. | Residue | Interaction type | Distancea (nm) | Hydrogen bond donor angle (°) | Ligand atom/group | Protein atom |
| 1 | Met165 | Hydrophobic | 3.60 | – | C6 | CB (Side chain) |
| 2 | Pro168 | Hydrophobic | 3.63 | – | C17 | CB (Side chain) |
| 3 | Pro168 | Hydrophobic | 3.45 | – | C16 | CD2 (Side chain) |
| 4 | Gln189 | Hydrophobic | 3.75 | – | C26 | CB (Side chain) |
| 5 | Glu166 | Hydrogen bond | 1.68 | 154.57 | N22 | OE1H (Side chain) |
| 6 | Glu166 | Hydrogen bond | 1.91 | 162.59 | N13H | O (Backbone) |
| 7 | Leu167 | Hydrogen bond | 3.60 | 106.70 | N22H | O (Backbone) |
aFor hydrophobic interactions distance is reported between interacting carbon atoms; For H-bonds distance is reported between hydrogen and acceptor atom; For salt bridges distance is reported between charge centers.
bFor Hydrogen bonds donor angle is reported as the angle between donor, acceptor and hydrogen atoms.
Docking results proposed that N-aryl amide derivative (ZINC_252512772) could bind to SARS-CoV2 protease with high binding energy (ΔGb −9.18 kcal/mol) and hence might be a good anti-coronavirus candidate for further development strategies. Most of the established interactions were hydrophobic in nature (Table 7) and probably Glu166 was the most significant residue that participated in three key interactions with C19, pyridyl nitrogen and terminal amide nitrogen of ZINC_252512772. Similar results could be found on drug repurposing studies of an anti-asthmatic drug montelukast which showed highly binding affinity to SARS-CoV2 Mpro through hydrophobic contacts with Thr24, Leu27, His41, Phe140, Cys145, His163, Met165, Pro168 and His172 (Wu et al., 2020).
In the case of sulfonamide derivatives (ZINC_170619726 and ZINC_108126011), it seemed that non-rigidified sulfonamide (ZINC_108126011) could bind more tightly to the SARS-CoV2 binding site. Attachment of a primary amine into cyclopentyl ring of ZINC_108126011 provided a good HBD (hydrogen bond donor) and at the same time HBA (hydrogen bond acceptor) site to interact through H-bonding with Glu166 and Leu167. Similar H-bond interaction could not be recorded for ZINC_170619726 due to the lack of hydrogen on sulfonamide nitrogen and also its non-protruding orientation. Moreover, none of the sulfonamide oxygens formed H-bonds with SARS-CoV2 residues.
4.2. MD Analysis
Enzymes are flexible macromolecules that can adopt a limited induced-fit models in order to achieve probably best steric and electronic complementary with their ligands. To evaluate the stability of predicted SARS-CoV2 Mpro complexes with regard to the dynamic characteristics of the enzyme, 50 ns MD simulations of three systems (ZINC_170619726, ZINC_108126011 and ZINC_252512772) were conducted in explicit water. The obtained results of three studied systems were compared to the result of 50 ns MD simulation of crystallographic holo structure (PDB ID: 5R84).
The evaluation of temperature and energy during MD simulations indicated that the systems reached to the steady-state and conservation of energy law was satisfied within four systems.
A comparison of MD results indicated that density of polar surface area in the structure of SARS-CoV2 Mpro inhibitor is the dominant factor in stability of ligand-enzyme complex. Distribution of polar surfaces (O and N atoms) in the structure of ZINC_252512772 is more than ZINC_170619726 and ZINC_108126011. The active site of SARS-CoV2 Mpro is exposed to the surface of enzyme and polar hydroxyl and quaternary amine in the structures of ZINC_170619726 and ZINC_108126011 tended to be more exposed to the bulk water than interacting with enzyme active site. This structural feature has been seen in the structure of other known protease inhibitors especially in -navir and -previr classes (Lv et al., 2015). Appropriate but not the most potent interaction with ionized carboxylate of Glu166 eased the binding and also increased the stability of the ligand-enzyme complex.
Comparing the RMSF of three systems during the 50 ns MD simulations showed that ZINC_252512772 formed tight binding with SARS-CoV2 Mpro. The RMSF in residue Glu166 indicated a significant change among three systems. The RMSF of Glu166 in ZINC_252512772, ZINC_108126011 and Zinc_170619726 was 0.08, 0.13 and 0.12 nm respectively. ZINC_108126011 with quaternary amine formed less favorable interactions with Glu166. In comparison, ZINC_252512772, which interacted with Glu166 via its primary amid moiety, had the most binding energy. Interestingly, ZINC_108126011 and Zinc_170619726 had more constructive interactions with hydrophobic residues (His41, Pro52, and Phe140). In this regard, the most noticeable change in the binding pose of Zinc_170619726 was the protruding of methyl benzoate moiety into the SARS-CoV2 Mpro active site (Figure 4). Similar shift in binding pose could be detected for ZINC_108126011 in a way that tetrahydroquinoline ring permeated into the hydrophobic pocket and made the sulfonamide and quaternary amine more water exposable (Figure 5).
The electrostatic interaction decreased in the following order: 5r84 > 5r80 ∼ 5r84 (holo structure) > 5r81. The vdW interaction of 5r84 with the active site of SARS-CoV2 Mpro was more than 5r81, 5r84 (holo structure) and 5r80.
According to obtained results, ZINC_252512772 showed better steric and electronic complementary fitness with the SARS-CoV2 Mpro active site in comparison to ZINC_108126011 and Zinc_170619726.
4.3. Amino acid decomposition analysis
MD results confirmed that within 50 ns simulations, less conformational changes were met in binding of ZINC_252512772 with regard to ZINC_170619726 and ZINC_108126011. More stable ZINC_252512772-Mpro complex convinced us to determine relative contribution of each interacted Mpro residue in binding to the most probable conformation of the corresponding ligand. On the basis of achieved results (Table 4), five residues (Phe140, Asn142, Cys145, Glu166 & Thr190) were associated with attractive binding forces while His41, Pro52, Met165, Arg188 and Gln189 were associated with repulsive binding forces.
Probably the most favorable result could be achieved in the case of Glu166 which is one of the catalytic dyad residues. Quantum chemical calculations confirmed MD results and indicated pivotal role of Glu166 in making permanent hydrogen bond (98% of MD simulations time) with ZINC_252512772 (ΔEb −31.66 kcal/mol). Our calculations showed that Glu166 was responsible for tight binding of ZINC_252512772 into the Mpro active site. Interaction of Glu166 with a few synthetic and phytochemical agents were previously reported (ul Qamar et al., 2020) and moreover functional role of conserved residue Glu166 within substrate-binding site of SARS-CoV was pointed before (Lin et al., 2004).
Some hydrophobic contacts were associated with lower binding energies (Phe140 − 0.08 kcal/mol) in B3LYP/Def2-TZVPP level of calculation. Second-ranked interacting residues were found to be Asn142 with −3.16 kcal/mol supported binding energy. Modeling studies showed a hydrogen bond between Asn142 backbone NH and terminal amide oxygen of ZINC_252512772 and estimated binding energy for MD simulated complex might be attributed to the stability of this key interaction. Despite prediction of H-bond interaction between Arg188 backbone oxygen and internal amide NH of ZINC_252512772 by molecular docking, QM calculations did not dedicate attractive binding force to the corresponding interaction (1.98 kcal/mol). According to this, we were prompted to estimate the binding energy to Arg188 in docked pose of ZINC_252512772 which was found to be −0.98 kcal/mol. Different binding energies confirmed MD results and indicated that predicted hydrogen bond between Arg188 backbone oxygen and internal amide NH of ZINC_252512772 might not be a stable interaction since considerable torsional deviation occurred within the most probable binding pose of the ligand during MD time.
4.4. In silico ADMET prediction
Physiochemical properties have substantial effects on the behavior of compounds within a living system and therefore it is significant to predict ADMET properties through hit identification. Results of estimated parameters indicated that ZINC_170619726, ZINC_108126011 and ZINC_252512772 met the criteria for drug-likeness and followed Lipinski’s rule of five.
5. Conclusion
A new pathogen (SARS-CoV2) that first emerged in Wuhan region of China in December 2019 spread quickly into all countries of the world. Given an urgent need toward therapeutic drugs or vaccines against the pathogen, seeking for new privileged anti-SARS-CoV2 molecules with good synthetic accessibility or commercial availability is now an important attitude for structure-based hit/lead generation. In the current study potential SARS-CoV2 inhibitors were proposed through in silico analysis of ZINC15 driven commercially available compounds against Mpro as the specific enzyme of pathogen with determinant role in maturation of viral nonstructural proteins (Nsps) and hence viral life cycle. For this purpose, a few co-crystallographic ligands of SARS-CoV2 Mpro with N-aryl amide and aryl sulfonamide based fragments were used to identify new and potential enzyme inhibitors via screening ZINC driven commercially available developed structures. Molecular docking simulations proposed ZINC_108126011 and ZINC_252512772 as superior Mpro binders with free binding energies comparable to a few re-purposed drugs. H-bond interactions and hydrophobic contacts were dominant attractive forces in binding to target. MD simulations on top-ranked docked poses demonstrated less conformational changes (more stable binding mode) for ZINC_252512772 within Mpro active site during 50 ns trajectory. Per-residue binding energy analysis of ZINC_252512772-Mpro complex determined relative contribution of individual interacted residues in binding to the selected hit. Quantum chemical calculations confirmed MD results and proved the pivotal role of Glu166 in making permanent hydrogen bond (98% of MD simulations time) with ZINC_252512772. Obtained results showed that ZINC_252512772 might be a desirable in silico hit for further development of potent SARS-CoV2 inhibitors. Although additional effort remains to be made for in vitro, in vivo and clinical validations, it is hoped that proposed compound could serve as appropriate hit molecules for further development of anti-Covid-19 agents.
Glossary
Abbreviations
- ACE2
Angiotensin-converting enzyme 2
- ADT
AutoDock Tools
- ADMET
Adsorption Distribution Metabolism Excretion Toxicity
- 3-CLpro
3-chymotrypsin-like cysteine protease
- Covid-19
Coronavirus disease 2019
- Def2-TZVPP
Valence triple-zeta with two sets of polarization functions
- FDA
Food and Drug Administration
- HAF
Heavy Atom Fixation
- LGA
Lamarckian Genetic Algorithm
- MD
Molecular Dynamics
- MERS
Middle East Respiratory Syndrome
- Mpro
Main protease
- PDB
Protein Data Bank
- PLIP
Protein Ligand Interaction profiler
- PLpro
Papin-Like protease
- QM
Quantum Mechanical
- RMSD
Root Mean Square Deviation
- SARS
Severe Acute Respiratory Syndrome
- TMPRSS2
Transmembrane Serine Protease 2
Funding Statement
This work was supported by the Ardabil University of Medical Sciences under Grant no. IR.ARUMS.REC.1399.002.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Aanouz, I., Belhassan, A., El-Khatabi, K., Lakhlifi, T., El-Ldrissi, M., & Bouachrine, M. (2020). Moroccan Medicinal plants as inhibitors against SARS-CoV-2 main protease: Computational investigations. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1758790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelli, I., Hassani, F., Brikci, S. B., & Ghalem, S. (2020). In silico study the inhibition of angiotensin converting enzyme 2 receptor of COVID-19 by Ammoides verticillata components harvested from Western Algeria. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1763199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adeoye, A. O., Oso, B. J., Olaoye, I. F., Tijjani, H., & Adebayo, A. I. (2020). Repurposing of chloroquine and some clinically approved antiviral drugs as effective therapeutics to prevent cellular entry and replication of coronavirus. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1765876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alikhani, R., Razzaghi-Asl, N., Ramazani, A., & Hosseinzadeh, Z. (2018). Insights into the structural/conformational requirements of cytotoxic oxadiazoles as potential chemotherapeutic target binding agents. Journal of Molecular Structure, 1164, 9–22. 10.1016/j.molstruc.2018.03.034 [DOI] [Google Scholar]
- Al-Khafaji, K., Al-Duhaidahawi, D., & Tok, T. T. (2020). Using integrated computational approaches to identify safe and rapid treatment for SARS-CoV-2. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1764392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baez-Santos, Y. M., St John, S. E., & Mesecar, A. D. (2015). The SARS coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antiviral Research, 115, 21–38. 10.1016/j.antiviral.2014.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck, B. R., Shin, B., Choi, Y., Park, S., & Kang, K. (2020). Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Computational and Structural Biotechnology Journal, 18, 784–790. 10.1016/j.csbj.2020.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., DiNola, A., & Haak, J. R. (1984). Molecular dynamics with coupling to an external bath. The Journal of Chemical Physics, 81(8), 3684–3690. 10.1063/1.448118 [DOI] [Google Scholar]
- Boopathi, S., Poma, A. B., & Kolandaivel, P. (2020). Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1758788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, Q., Yang, M., Liu, D., Chen, J., Shu, D., Xia, J., Liao, X., Gu, Y., Cai, Q., Yang, Y., Shen, C., Li, X., Peng, L., Huang, D., Zhang, J., Zhang, S., Wang, F., Liu, J., Chen, L., Chen, S., … Liu, L. (2020). Experimental treatment with favipiravir for Covid-19: An open-label control study. Engineering, . 10.1016/j.eng.2020.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case, D. A., Cheatham, T. E., Darden, T., Gohlke, H., Luo, R., Merz, K. M., Jr., Onufriev, A., Simmerling, C., Wang, B., & Woods, R. J. (2005). The Amber biomolecular simulation programs. Journal of Computational Chemistry, 26(16), 1668–1688. 10.1002/jcc.20290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, N., Zhou, M., Dong, X., Qu, J., Gong, F., Han, Y., Qiu, Y., Wang, J., Liu, Y., Wei, Y., Xia, J., Yu, T., Zhang, X., & Zhang, L. (2020). Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet (London, England), 395(10223), 507–513. 10.1016/S0140-6736(20)30211-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, J., Li, F., & Shi, Z. L. (2019). Origin and evolution of pathogenic coronaviruses. Nature Reviews. Microbiology, 17(3), 181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina, A., Michielin, O., & Zoete, V. (2017). SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific Reports, 7, 42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das, S., Sarmah, S., Lyndem, S., & Roy, A. S. (2020). An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1763201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan, Y., Zhu, H. L., & Zhou, C. (2020). Advance of promising targets and agents against COVID-19 in China . Drug Discov. Today, 25(5), 810–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebadi, A., Razzaghi-Asl, N., Khoshneviszadeh, M., & Miri, R. (2013). Comparative amino acid decomposition analysis of potent type I p38α inhibitors. Daru : Journal of Faculty of Pharmacy, Tehran University of Medical Sciences, 21(1), 41. 10.1186/2008-2231-21-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky, A. A. (2020. a). Anti-HCV, nucleotide inhibitors, repurposing against COVID-19. Life Sciences, 248, 117477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky, A. A. (2020. b). Natural products may interfere with SARS-CoV-2 attachment to the host cell. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1761881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky, A. A. (2020. c). Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): A molecular docking study. Life Sciences, 253, 117592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky, A. A. (2020. d). SARS-CoV-2 RNA dependent RNA polymerase (RdRp) targeting: An in silico perspective. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1761882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky, A. A., & Azzam, E. B. (2020). Novel guanosine derivatives against MERS CoV polymerase: An in silico perspective. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1758789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmezayen, A. D., Al-Obaidi, A., Şahin, A. T., & Yelekçi, K. (2020). Drug repurposing for coronavirus (COVID-19): in silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1758791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enayatkhani, M., Hasaniazad, M., Faezi, S., Guklani, H., Davoodian, P., Ahmadi, N., Einakian, M. A., Karmostaji, A., & Ahmadi, K. (2020). Reverse vaccinology approach to design a novel multi-epitope vaccine candidate against COVID-19: An in silico study. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1756411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enmozhi, S. K., Raja, K., Sebastine, I., & Joseph, J. (2020). Andrographolide as a potential inhibitor of SARS-CoV-2 main protease: An in silico approach. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1760136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon, D., Powell, A. J., Douangamath, A., Owen, C. D., Wild, C., Krojer, T., Lukacik, P., Strain-Damerell, C. M., Walsh, M. A., & von Delft, F. (2020). PanDDA analysis of COVID-19 main protease against the DSI-poised fragment library. 10.2210/pdb5R7Y/pdb, , , , , , [DOI]
- Fogarasi, G., Zhou, X., Taylor, P. W., & Pulay, P. (1992). The calculation of ab initio molecular geometries: Efficient optimization by natural internal coordinates and empirical correction by offwet forces. Journal of the American Chemical Society, 114(21), 8191–8201. 10.1021/ja00047a032 [DOI] [Google Scholar]
- Fung, T. S., & Liu, D. X. (2019). Human coronavirus: Host-pathogen interaction . Annual Review of Microbiology, 73, 529–557. [DOI] [PubMed] [Google Scholar]
- Gautret, P., Lagier, J.-C., Parola, P., Hoang, V. T., Meddeb, L., Mailhe, M., Doudier, B., Courjon, J., Giordanengo, V., Vieira, V. E., Tissot Dupont, H., Honoré, S., Colson, P., Chabrière, E., La Scola, B., Rolain, J.-M., Brouqui, P., & Raoult, D. (2020). Hydroxychloroquine and azithromycin as a treatment of COVID-19: Results of an open-label non-randomized clinical trial. International Journal of Antimicrobial Agents, 105949. 10.1016/j.ijantimicag.2020.105949 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Gorbalenya, A. E., Baker, S. C., Baric, R. S., de Groot, R. J., Drosten, C., Gulyaeva, A. A., Haagmans, B. L., Lauber, C., Leontovich, A. M., Neuman, B. W., Penzar, D., Perlman, S., Poon, L. L. M., Samborskiy, D. V., Sidorov, I. A., Sola, I., & Ziebuhr, J. (2020). The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nature Microbiology, 5, 536–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, M. K., Vemula, S., Donde, R., Gouda, G., Behera, L., & Vadde, R. (2020). In-silico approaches to detect inhibitors of the human severe acute respiratory syndrome coronavirus envelope protein ion channel. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1751300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyebi, G. A., Ogunro, O. B., Adegunloye, A. P., Ogunyemi, O. M., & Afolabi, S. O. (2020). Potential inhibitors of coronavirus 3-chymotrypsin-like protease (3CLpro): An in silico screening of alkaloids and terpenoids from African medicinal plants. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1764868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan, A., Paray, B. A., Hussain, A., Qadir, F. A., Attar, F., Aziz, F. M., Sharifi, M., Derakhshankhah, H., Rasti, B., Mehrabi, M., Shahpasand, K., Saboury, A. A., & Falahati, M. (2020). A review on the cleavage priming of the spike protein on coronavirus by angiotensin-converting enzyme-2 and furin. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1754293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess, B., Kutzner, C., Van Der Spoel, D., & Lindahl, E. (2008). GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. Journal of Chemical Theory and Computation, 4(3), 435–447. [DOI] [PubMed] [Google Scholar]
- Hoffmann, M., Kleine-Weber, H., Schroeder, S., Kruger, N., Herrler, T., Erichsen, S., Schiergens, T. S., Herrler, G., Wu, N.-H., Nitsche, A., Muller, M. A., Drosten, C., & Pohlmann, S. (2020). SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell, 181(2), 271–280. https://www.who.int/emergencies/diseases/novel-coronavirus-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim, I. M., Abdelmalek, D. H., & Elfiky, A. A. (2019). GRP78: A cell’s response to stress. Life Sciences, 226, 156–163. 10.1016/j.lfs.2019.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim, I. M., Abdelmalek, D. H., Elshahat, M. E., & Elfiky, A. A. (2020). COVID-19 spike-host cell receptor GRP78 binding site prediction. The Journal of Infection, 80(5), 554–562. 10.1016/j.jinf.2020.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam, R., Parves, M. R., Paul, A. S., Uddin, N., Rahman, M., S., Al Mamun, A., Hossain, M. N., Ali, M. A., & Halim, M. A. (2020). A molecular modeling approach to identify effective antiviral phytochemicals against the main protease of SARS-CoV-2. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1761883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemard, C., & Kellenberger, E. (2019). A bright future for fragment-based drug discovery: What does it hold? Expert Opinion on Drug Discovery, 14(5), 413–416. 10.1080/17460441.2019.1583643 [DOI] [PubMed] [Google Scholar]
- Jakalian, A., Jack, D. B., & Bayly, C. I. (2002). Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. Journal of Computational Chemistry, 23(16), 1623–1641. [DOI] [PubMed] [Google Scholar]
- Joshi, R. S., Jagdale, S. S., Bansode, S. B., Shankar, S. S., Tellis, M. B., Pandya, V. K., Chugh, A., Giri, A. P., & Kulkarni, M. J. (2020). Discovery of potential multi-target-directed ligands by targeting host-specific SARS-CoV-2 structurally conserved main protease. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1760137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, R. J., Jha, R. K., Amera, G. M., Jain, M., Singh, E., Pathak, A., Singh, R. P., Muthukumaran, J., & Singh, A. K. (2020. a). Targeting SARS-CoV-2: A systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 2′-O-ribose methyltransferase. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1753577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, S. A., Zia, K., Ashraf, S., Uddin, R., & Ul-Haq, Z. (2020. b). Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1751298. [DOI] [PubMed] [Google Scholar]
- Kumar, D., Kumari, K., Jayaraj, A., Kumar, V., Kumar, R. V., Dass, S. K., Chandra, R., & Singh, P. (2020). Understanding the binding affinity of noscapines with protease of SARS-CoV-2 for COVID-19 using MD simulations at different temperatures. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1752310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, G., & De Clercq, E. (2020). Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nature Reviews. Drug Discovery, 19(3), 149–150. [DOI] [PubMed] [Google Scholar]
- Lin, C.-W., Tsai, C.-H., Tsai, F.-J., Chen, P.-J., Lai, C.-C., Wan, L., Chiu, H.-H., & Lin, K.-H. (2004). Characterization of trans- and cis-cleavage activity of the SARS coronavirus 3CLpro protease: Basis for the in vitro screening of anti-SARS drugs. FEBS Letters, 574(1-3), 131–137. 10.1016/j.febslet.2004.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindorff‐Larsen, K., Piana, S., Palmo, K., Maragakis, P., Klepeis, J. L., Dror, R. O., & Shaw, D. E. (2010). Improved side-chain torsion potentials for the Amber ff99SB protein force field . Proteins, 78(8), 1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, C., Zhou, Q., Li, Y., Garner, L. V., Watkins, S. P., Carter, L. J., Smoot, J., Gregg, A. C., Daniels, A. D., Jervey, S., & Albaiu, D. (2020). Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Central Science, 6(3), 315–331. 10.1021/acscentsci.0c00272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo-Galo, N., Terrazas-López, M., Martínez-Martínez, A., & Díaz-Sánchez, Á. G. (2020). FDA-approved thiol-reacting drugs that potentially bind into the SARS-CoV-2 main protease, essential for viral replication. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1764393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv, Z., Chu, Y., & Wang, Y. (2015). HIV protease inhibitors: A review of molecular selectivity and toxicity. HIV/AIDS (Auckland, N.Z.).), 7, 95–104. 10.2147/HIV.S79956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, G., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., & Olson, A. J. (2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry, 30(16), 2785–2791. 10.1002/jcc.21256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulangu, S., Dodd, L. E., Davey, R. T., Jr, Tshiani, Mbaya, O., Proschan, M., Mukadi, D., Lusakibanza Manzo, M., Nzolo, D., Tshomba Oloma, A., Ibanda, A., Ali, R., Coulibaly, S., Levine, A. C., Grais, R., Diaz, J., Lane, H. C., Muyembe-Tamfum, J. J., Sivahera, B., … Nordwall, J, PALM Consortium Study Team (2019). A randomized, controlled trial of Ebola virus disease therapeutics. The New England Journal of Medicine, 381(24), 2293–2303. PALM Writing Group [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidharan, N., Sakthivel, R., Velmurugan, D., & Gromiha, M. M. (2020). Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 protease against COVID-19. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1752802. [DOI] [PubMed] [Google Scholar]
- Neese, F. (2012). The ORCA program system. WIREs Computational Molecular Science, 2(1), 73–78. 10.1002/wcms.81 [DOI] [Google Scholar]
- Pant, S., Singh, M., Ravichandiran, V., Murty, U. S. N., & Srivastava, H. K. (2020). Peptide-like and small-molecule inhibitors against Covid-19. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1757510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrinello, M., & Rahman, A. (1981). Polymorphic transitions in single crystals: A new molecular dynamics method. Journal of Applied Physics, 52(12), 7182–7190. 10.1063/1.328693 [DOI] [Google Scholar]
- Razzaghi-Asl, N., Karimi, A., & Ebadi, A. (2018). The potential of natural product vs neurodegenerative disorders: In silico study of artoflavanocoumarin as BACE-1 inhibitor. Computational Biology and Chemistry, 77, 307–317. [DOI] [PubMed] [Google Scholar]
- Ren, J. l., Zhang, A. H., & Wang, X. J. (2020). Traditional Chinese medicine for COVID-19 treatment. Pharmacological Research, 155, 104743. 10.1016/j.phrs.2020.104743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson, P., Griffin, I., Tucker, C., Smith, D., Oechsle, O., Phelan, A., Rawling, M., Savory, E., & Stebbing, J. (2020). Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet (London, England), 395(10223), e30–e31. 10.1016/S0140-6736(20)30304-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson, B. (2020). Computers and viral diseases. Preliminary bioinformatics studies on the design of a synthetic vaccine and a preventative peptidomimetic antagonist against the SARS-CoV-2 (2019-nCoV, COVID-19) coronavirus. Computers in Biology and Medicine, 119, 103670. 10.1016/j.compbiomed.2020.103670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose, A. S., Bradley, A. R., Valasatava, Y., Duarte, J. M., Prlić, A., & Rose, P. W. (2018). NGL viewer: Web-based molecular graphics for large complexes. Bioinformatics (Oxford, England), 34(21), 3755–3758. 10.1093/bioinformatics/bty419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salentin, S., Schreiber, S., Haupt, V. J., Adasme, M. F., & Schroeder, M. (2015). PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Research, 43(W1), W443–W447. 10.1093/nar/gkv315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanner, M. (1999). Python: a programming language for software integration and development. Journal of Molecular Graphics & Modelling, 17(1), 57–61. [PubMed] [Google Scholar]
- Sarma, P., Shekhar, N., Prajapat, M., Avti, P., Kaur, H., Kumar, S., Singh, S., Kumar, H., Prakash, A., Dhibar, D. P., & Medhi, B. (2020). In-silico homology assisted identification of inhibitor of RNA binding against 2019-nCoV N-protein (N terminal domain). Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1753580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha, S. K., Shakya, A., Prasad, S. K., Singh, S., Gurav, N. S., Prasad, R. S., & Gurav, S. S. (2020). An in-silico evaluation of different Saikosaponins for their potency against SARS-CoV-2 using NSP15 and fusion spike glycoprotein as targets. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1762741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterling, T., & Irwin, J. J. (2015). ZINC 15 - Ligand discovery for everyone. Journal of Chemical Information and Modeling, 55(11), 2324–2337. 10.1021/acs.jcim.5b00559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahir, U. Q. M., Maryam, A., Muneer, I., Xing, F., Ashfaq, U. A., Khan, F. A., Anwar, F., Geesi, M. H., Khalid, R. R., Rauf, S. A., & Siddiqi, A. R. (2019). Computational screening of medicinal plant phytochemicals to discover potent pan-serotype inhibitors against dengue virus. Scientific Reports, 9(1), 1433. 10.1038/s41598-018-38450-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ul Qamar, M. T., Alqahtani, S. M., Alamri, M. A., & Chen, L. L. (2020). Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. Journal of Pharmaceutical Analysis, . 10.1016/j.jpha.2020.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umesh, K., D., Selvaraj, C., Singh, S. K., & Dubey, V. K. (2020). Identification of new anti-nCoV drug chemical compounds from Indian spices exploiting SARS-CoV-2 main protease as target. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1763202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villar, J., Zhang, H., & Slutsky, A. S. (2019). Lung repair and regeneration in ARDS: Role of PECAM1 and Wnt signaling. Chest, 155(3), 587–594. 10.1016/j.chest.2018.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas, A., Jain, A., & Gupta, A. (2008). Virtual screening: A fast tool for drug design. Scientia Pharmaceutica, 76(3), 333–360. 10.3797/scipharm.0803-03 [DOI] [Google Scholar]
- Wahedi, H. M., Ahmad, S., & Abbasi, S. W. (2020). Stilbene-based natural compounds as promising drug candidates against COVID-19. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1762743. [DOI] [PubMed] [Google Scholar]
- Wang, M., Cao, R., Zhang, L., Yang, X., Liu, J., Xu, M., Shi, Z., Hu, Z., Zhong, W., & Xiao, G. (2020). Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Research, 30(3), 269–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A., & Case, D. A. (2004). Development and testing of a general amber force field. Journal of Computational Chemistry, 25(9), 1157–1174. 10.1002/jcc.20035 [DOI] [PubMed] [Google Scholar]
- Wu, C., Liu, Y., Yang, Y., Zhang, P., Zhong, W., Wang, Y., Wang, Q., Xu, Y., Li, M., Li, X., Zheng, M., Chen, L., & Li, H. (2020). Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharmaceutica Sinica B, 10(5), 766–788. 10.1016/j.apsb.2020.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H., Yang, M., Ding, Y., Liu, Y., Lou, Z., Zhou, Z., Sun, L., Mo, L., Ye, S., Pang, H., Gao, G. F., Anand, K., Bartlam, M., Hilgenfeld, R., & Rao, Z. (2003). The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proceedings of the National Academy of Sciences, 100(23), 13190–13195. 10.1073/pnas.1835675100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, N., Zhang, D., Wang, W., Li, X., Yang, B., Song, J., Zhao, X., Huang, B., Shi, W., Lu, R., Niu, P., Zhan, F., Ma, X., Wang, D., Xu, W., Wu, G., Gao, G. F., & Tan, W, China Novel Coronavirus Investigating and Research Team. (2020). A novel coronavirus from patients with pneumonia in China, 2019. The New England Journal of Medicine, 382(8), 727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumla, A., Chan, J. F., Azhar, E. I., Hui, D. S., & Yuen, K. Y. (2016). Coronaviruses - drug discovery and therapeutic options. Nature Reviews. Drug Discovery, 15(5), 327–347. [DOI] [PMC free article] [PubMed] [Google Scholar]