

Abstract
Despite the intensive research efforts towards antiviral drug against COVID-19, no potential drug or vaccines has not yet discovered. Initially, the binding site of COVID-19 main protease was predicted which located between regions 2 and 3. Structure-based virtual screening was performed through a hierarchal mode of elimination technique after generating a grid box. This led to the identification of five top hit molecules that were selected on the basis of docking score and visualization of non-bonding interactions. The docking results revealed that the hydrogen bonding and hydrophobic interactions are the major contributing factors in the stabilization of complexes. The docking scores were found between −7.524 and −6.711 kcal/mol indicating strong ligand-protein interactions. Amino acid residues Phe140, Leu141, Gly143, Asn142, Thr26, Glu166 and Thr190 (hydrogen bonding interactions) and Phe140, Cys145, Cys44, Met49, Leu167, Pro168, Met165, Val42, Leu27 and Ala191 (hydrophobic interactions) formed the binding pocket of COVID-19 main protease. From identified hits, ZINC13144609 and ZINC01581128 were selected for atomistic MD simulation and density functional theory calculations. MD simulation results confirm that the protein interacting with both hit molecules is stabilized in the chosen POPC lipid bilayer membrane. The presence of lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) in the hydrophobic region of the hit molecules leads to favorable ligand-protein contacts. The calculated pharmacokinetic descriptors were found to be in their acceptable range and therefore confirming their drug-like properties. Hence, the present investigation can serve as the basis for designing and developing COVID-19 inhibitors.
Communicated by Ramaswamy H. Sarma
Keywords: COVID-19, molecular dynamics, virtual screening, synthetic feasibility, quantum mechanical calculations
Graphical abstract

1. Introduction
In the twenty-first century, the novel coronavirus 2019 is a major cause of disaster due to a lack of vaccine, drugs to treatment. The pandemic has been born from the family of coronavirus and can cause illness such as elevated body temperature, common cold, pneumonia, bronchiolitis, rhinitis, pharyngitis, sinusitis, Middle East respiratory syndrome (MERS) and the severe acute respiratory syndrome (SARS) (Amanat & Krammer, 2020). In 2003, SARS had killed 774 people, whereas MERS killed 858 people between 2012 and 2019 (Boopathi et al., 2020). In 2019, a new virus identified in china namely novel coronavirus disease 2019 (COVID-19) (Boopathi et al., 2020).
Coronavirus has a single-stranded RNA genome (Yang et al., 2006), pleomorphic or spherical shape, and bears club-shaped projections of glycoproteins on its surface (80–120 nm diameter) (Guo et al., 2010; Belouzard et al., 2012). The RNA genome is made up of ∼30,000 nucleotides and covered by enveloped structure. It encodes four structural proteins, nucleocapsid (N) protein, membrane (M) protein, spike (S) protein, and envelop (E) protein and several non-structural proteins (nsp) (Boopathi et al., 2020). COVID-19 packages its genome in a nucleocapsid (N) protein and forms a ribonucleoprotein (RNP) complex. The RNP is essential for viral replication, transcription, and assembly. Several studies suggested that the modulation of N protein oligomerization by small molecules is a possible antiviral drug development strategy (Chang et al., 2016; Zumla et al., 2016).
The membrane (M) protein found in the surface of the virus and supposed to be the central organizer for the COVID-19 assembly (Sarma et al., 2020). The spike (S) protein integrated throughout the viral surface and involves the attachment with the host cell surface receptors and facilitates viral entry into the host cell by fusion between the viral and host cell membranes (Kirchdoerfer et al., 2016). The S protein is composed of three identical chains with 1273 amino acid residue each and characterized by two well-defined protein domain regions: S1 and S2 subunits. These S1 and S2 subunits are involved in cell recognition and the fusion of viral and cellular membranes respectively (Walls et al., 2020). The envelop (E) protein, a small component of the virus particle, is a small membrane protein made up of ∼76–109 amino acid residues and involves in host cell interaction and virus assembly (Boopathi et al., 2020; Gupta et al., 2020).
The structural spike (S) protein of Coronavirus binds with the angiotensin-converting enzyme 2 (ACE2) receptors that are present on the surface of many human cells, including lungs (Hasan et al., 2020). The S protein is associated with proteolytic cleavages by host proteases, in two domain regions located at the boundary between the S1 and S2 subunits. In a later stage happens the cleavage of the S2 subunit to release the fusion peptide (Boopathi et al., 2020; Lin et al., 2020). This process will trigger the activation of the membrane fusion mechanism (Tahir Khan et al., 2020). Therefore, inhibition of the activation of membrane fusion could be a possible target for antiviral drug development against COVID-19. Coronavirus entry, replication, transcription, and formation of RNP complex are depicted in Figure 1.
Figure 1.

A schematic representation of the possible mechanism of COVID-19 entry, replication, transcription and formation of RNP complex.
Furthermore, endosome opens to release COVID-19 to the cytoplasm and translation of viral polymerase protein takes place (Li, 2016). The positive RNA genome is translated to generate replicase proteins that use the genome as a template to generated full-length negative-sense RNAs, which subsequently serve as templates in generating addition full-length genomes (Masters, 2006; Van Hemert et al., 2008). The nucleocapsids are formed from the encapsidation of replicated genomes. The structural proteins S, M, and E are synthesized in the cytoplasm and further translation results in the transfer to endoplasmic reticulum-Golgi intermediate compartment (ERGIC) (Masters, 2006). This ERGIC membrane coalesces with the nucleocapsids to self-assembly into new virions. Finally, new virions are exported from infected cells by exocytosis, so that can affect other cells.
The ongoing COVID-19 threat that emerged in China has rapidly spread to the whole world and has been declared as a global public health emergency by the World Health Organization (WHO) (Muralidharan et al., 2020). Many efforts have been directed to develop potential drugs or vaccine but no drug or vaccine has not launched. Therefore, all nations implementing appropriate preventive and control measures only. So, it is an urgent need to develop effective and potential antiviral-COVID-19 candidates for reducing disease severity, and transmission to block the COVID-19 outbreaks.
To accomplish the objective of the study, we have carried out the structure modeling of the crystal structure of COVID-19. The prepared protein structure was used to perform high-throughput virtual screening (HTVS) of chemical libraries. The novel hit molecules identified from docking study were selected based on the docking score, binding energy calculations, and their other interactions with amino acid residues. Also, we performed molecular dynamics simulations and molecular orbital analysis for the best two hit molecules. Pharmacokinetic parameters and bioavailability score were also assessed.
2. Materials and methodology
The entire computational investigation was performed on Windows 7 (64-bit) operating systems with 4 GB RAM and 2.66 GHz IntelVR CoreTM 2 Quad Q8400 processor except for molecular dynamics simulations. Molecular dynamics simulations were performed on Ubuntu 14.04.5 version in the linux environment with 4 GB RAM by Desmond.
2.1. Target protein preparation
The three-dimensional crystal structure of the COVID-19 main protease (PDB-ID: 6Y84) was retrieved from the Protein Data Bank (http://www.rscb.org/pdb). The target protein has a resolution of 1.39 Å and this indicates its good quality as a resolution of 2.0 Å is the recommended maximum for a good structural protein for docking (Hajduk et al., 2005). The retrieved protein was pre-processed using the protein preparation wizard by adding missing loops and atoms, adding missing hydrogens and assigning bond orders. Further, we removed non-essential water molecules and hetero atoms, before restrained minimization using OPLS-2005.
2.2. Binding site identification
The objective is to bind the ligands from the molecular docking to the binding site of the COVID-19 main protease protein. SiteMap tool of Maestro was employed to recognize potential binding pocket by joining together ‘sitepoints’ that most probably subsidize to tie protein-protein or protein-ligand binding (Halgren, 2007, 2009). The overall site score describes the rank of computed binding pocket using a linear combination of various terms.
2.3. Receptor grid generation
The active site of COVID-19 main protease protein was located in region 3 site and the receptor grid files were generated using Grid Generation panel of Glide module. This establishes two cubical boxes having a common centroid to organize the calculations: a larger enclosing and a smaller binding box (Athar et al., 2016).
2.4. Ligand preparation and virtual screening
Structure-based virtual screening is of major importance for the computational drug discovery process to speed up drug development. This approach emerged as an important tool in identifying small drug-like molecules through various computational tools, by using knowledge about the target protein, its constitutional information, or by known active compounds for target protein (Kar & Roy, 2013). In this process of virtual screening, the selected dataset consists of all compound libraries of the ZINC database in SDF format (Rollinger et al., 2008). The complete virtual screening was performed against the above said databases by the Glide module of Maestro. These database molecules were docked into the binding site of the protein utilizing HTVS protocol to calculate ligand–protein binding affinities. Molecules filtered from HTVS were subjected to SP docking which can dock few tens to millions of ligands with high accuracy. A further precise amount of molecule was then subjected to glide XP docking where further elimination of false positives is carried out by more extensive sampling and advanced scoring resulting in higher enrichment. Based on their Glide Gscore, the top five hit molecules were selected for further investigation.
2.5. Binding energy calculation
Molecular mechanics-generalized born surface area (MM-GBSA) method was used for the calculation of binding free energy for the docked complexes (Su et al., 2015). This method used the following equation:
Where solv and SA represent the salvation energy and surface area associated energy and E represents the energy minimized states of ligand-protein used in the calculations. For this calculation, the top-scored ligand–protein complexes were selected with default parameters such as OPLS-2005 force field and VSGB solvent model.
2.6. Molecular dynamics simulation
To analyze the structural stability of the COVID-19 main protease protein–ligand complexes, molecular dynamics simulations were carried out by using Desmond in the presence of the POPC bilayer membrane (Shekhar et al., 2019). The protein–ligand complex was pre-processed using the protein preparation wizard panel to add missing atoms and refinement of side chains. The solvated model of a complex was prepared by selecting POPC (300 K) as a membrane model. The solvated system was in an orthorhombic box to provide SPC solvent model and the system was neutralized using Na+ and Cl− ions. The salt concentration was set as 0.15 M to maintain physiological conditions. To minimize the short range bad contacts, energy minimization was carried out using the hybrid method steepest decent and the limited-memory Broyden–Fletcher–Goldfarb–Shanno (LBFGS) algorithms (Guo et al., 2010) for 2000 iterations. Finally, these systems were subjected to 50 ns MD simulation production runs at 300 K temperature and 1 bar as pressure. The model was relaxed before the production system run because it makes a series of predefined minimizations and MD executions. The MD simulations were analyzed in terms of RMSD and RMSF to estimate the variations in the position of the systems in terms of simulation time. Protein–ligand contacts and torsion profiles were also examined during the simulation.
2.7. Prediction of bioavailability and synthetic feasibility
In silico bioavailability and the synthetic feasibility of the top-scored molecules were evaluated through the Swiss ADME tool. Consideration of synthetic practicability produces a number, between 1 – for simply synthesized compounds, and 10 – for compounds that are challenging to synthesize.
2.8. Pharmacokinetic and bioavailability calculations
The assessment of pharmacokinetic parameters of top-scored molecules obtained from virtual screening protocol is believed to be an essential step of the computational drug discovery process. It is known that the major concern for the failure of drug candidates in clinical trials is poor pharmacokinetics (Venugopal et al., 2020). Hence, the evaluation of pharmacokinetic properties in previous stages of drug discovery reduces the chance of failure. With this objective, the pharmacokinetic properties of the top-scored molecules of COVID-19 obtained from the present investigation were predicted using the Qikprop module (Kumar et al., 2018).
2.9. Density functional theory calculations
Single point energy calculations using density functional theory were performed using Jaguar (Bochevarov et al., 2013) to explain ligand-bound protein in electronic level. The structures were optimized using hybrid functional B3LYP parameters with 6-31 G** basis set. Various properties such as electrostatic potential, average local ionization energy, gas-phase energy, canonical orbital, etc. were calculated. The positions of lowest unoccupied molecular orbital (LUMO)–highest occupied molecular orbital (HOMO) orbitals of selected hit molecules were analyzed to evaluate the binding pattern at the quantum level.
3. Results and discussion
Present computational work aimed to search novel, potential inhibitors of COVID-19, and characterize the binding pattern to identify the structural features through structure-based screening and molecular dynamics simulation studies. Subsequent in silico bioavailability and synthetic feasibility was also evaluated to strengthen the scope of work.
3.1. Analysis of active site identification
The localization and identification of the binding site are necessary as the function and structure of the protein are related to specific interaction with binding site functionalities before structure-based drug design. For predicting the binding site, a comprehensive search was done by SiteMap. It indicates the locations of hydrogen-bonded, van der Waals, and hydrophobic regions in the representative COVID-19 proteins. The calculated structural features of binding sites have shown in Table 1. The druggability of binding sites was determined which is given in the terms of site score. In all possible binding sites, site_1 has 0.961 site score along with 274.743 as volume. Same as the site score of other site_2, site_3, site_4, and site_5 were found to be 0.946, 0.837, 0.591, and 0.571 respectively. Based on the results, site_1 has the potential to mechanistically justify the binding pattern and answer for the protein–ligand interactions. Therefore, site_1 was considered as an appropriate binding site to perform virtual screening
Table 1.
Summary of structural features of the binding site predicted by SiteMap.
| Sitemap_site | Site_1 | Site_2 | Site_3 | Site_4 | Site_5 |
|---|---|---|---|---|---|
| SiteScore | 0.961 | 0.946 | 0.837 | 0.591 | 0.571 |
| Dscore | 0.986 | 0.993 | 0.720 | 0.547 | 0.407 |
| Volume | 274.743 | 299.782 | 160.181 | 50.764 | 55.909 |
| Exposure | 0.605 | 0.741 | 0.557 | 0.652 | 0.667 |
| Enclosure | 0.642 | 0.588 | 0.745 | 0.574 | 0.616 |
| Contact | 0.851 | 0.777 | 1.089 | 0.922 | 0.873 |
| Don/acc | 1.056 | 0.983 | 0.790 | 1.748 | 0.767 |
COVID-19 main protease protein composed of three regions of 306 amino acid residues. The first (blue), second (green), and third (orange) regions contain 8–101, 102–184, and 201–305 residues, respectively. The potential binding site lies between region 2 and 3 that is connected through a long loop (Figure 2).
Figure 2.

Predicted binding pocket where virtual screening was performed.
3.2. Virtual screening and interaction analysis of complex
Small molecules of the ZINC database have been used for structure-based screening of COVID-19 main protease protein. The active site with the best site score (top-ranked potential receptor binding cavity) was taken as a prerequisite for receptor grid generation. The executed approach of virtual screening was the hierarchal mode of elimination technique i.e. HTVS followed by SP and further the XP docking. Glide XP combines accurately energy-based scoring terms and thorough sampling that resulted in compounds with docking scores between −7.524 and −6.711 kcal/mol indicating strong ligand-protein interactions. Final shortlisting of hit molecules was based on visual observation of the major amino acid residues involved in the binding that included hydrogen bonding with Glu166, Phe140, Gly143, Leu141, Asn142, Thr26 and Thr190. From these observations, we selected the top five compounds, the docking score, binding free energies of hit molecules are provided in Table 2.
Table 2.
Docking score and binding free energies of selected five-hit molecules.
| Ligand No. | Docking score | dG Bind | dG Bind Coulomb | dG Bind Lipo | dG Bind Hbond | dG Bind vdW |
|---|---|---|---|---|---|---|
| ZINC13144609 | −7.524 | −38.461 | −1.285 | −18.163 | −1.478 | −33.972 |
| ZINC01581128 | −7.429 | −69.390 | −70.280 | −34.362 | −2.880 | −35.765 |
| ZINC06062920 | −6.994 | −62.967 | −54.608 | −34.027 | −2.650 | −40.284 |
| ZINC03022586 | −6.947 | −68.341 | 7.655 | −24.258 | −1.830 | −37.597 |
| ZINC00134422 | −6.711 | −61.889 | −23.221 | −22.414 | −1.492 | −33.074 |
The top hit molecule ZINC13144609 (1,4-bis(1H-benzo[d]imidazol-2-yl)butane-1,2,3,4-tetraol) showed hydrogen bonding with amino acid residue Glu166, Leu141 and Asn142 with a docking score of −7.524 kcal/mol. The selected five potential hit molecules in the binding site of protease protein, interacting with amino acid residues Phe140, Gly143, Thr26, Thr190, Glu166, Pro168, Met165 and Leu141 with a docking score of −7.524 and −6.711 kcal/mol. The number of hydrogen bond along with residue and other interactions is tabulated in Table 3. All hit molecules showed hydrogen–bonding interactions with main amino acid residues Phe140, Leu141, Gly143, Asn142, Thr26, Glu166 and Thr190 with a well-fitted pose within the binding site in the hydrophobic pocket formed by Phe140, Cys145, Cys44, Met49, Leu167, Pro168, Met165, Val42, Leu27 and Ala191. The binding pattern within the binding site pocket of all top-scored hit molecules was quite same and additional π-π interactions contributed for the binding affinity of the molecules.
Table 3.
Top five hit molecules with their number of interacting hydrogen bonds, residues and other interactions.
| Ligand No. | Number of
hydrogen bond |
H-bond
interaction |
Other interactions | ||
|---|---|---|---|---|---|
| Backbone | Sidechain | Backbone | Sidechain | ||
| ZINC13144609 | 3 | 3 | Glu166, Leu141 | Glu166, Asn142 | – |
| ZINC01581128 | 5 | 1 | Gly143, Leu141, Phe140, Glu166 | Glu166 | Π–Π stacking |
| ZINC06062920 | 4 | 1 | Gly143, Phe140, Glu166 | Glu166 | Π–Π stacking |
| ZINC03022586 | 5 | 1 | Thr26, Gly143, Thr190, Glu166 | Gln192 | – |
| ZINC00134422 | 4 | – | Thr26, Gly143, Glu166 | – | – |
The docking pattern of the docked hit molecule ZINC13144609 and ZINC01581128 is represented in Figures 3 and 4. The docking orientation of the remaining hit molecules ZINC06062920, ZINC03022586 and ZINC00134422 are depicted in Figures S1–S3.
Figure 3.

COVID-19 main protease protein and ZINC13144609 binding interaction diagram.
Figure 4.

COVID-19 main protease protein and ZINC01581128 binding interaction diagram.
3.3. Molecular dynamics simulation analysis
A molecular dynamics simulation time step involves a computationally intensive force calculation for each atom of a chemical system, followed by a less expensive integration step that advances the positions of the atoms according to classical laws of motion. It enables the atomic-level characterization of numerous biomolecular processes such as investigation of the stability of protein–ligand interactions associated with activation and deactivation of various molecular pathways. The atomistic MD simulation was performed to obtain insights into the stability behavior of top hit molecules ZINC13144609 and ZINC01581128 at the binding pocket of 6Y84. The complex was embedded within the POPC bilayer chosen with default parameters and simulated for 50 ns. Each complex was soaked in an orthorhombic box with SPC water molecules. To maintain a neutral system, Na+ and Cl− ions were added with 0.15 M salt concentration. The trajectories generated were analyzed by plotting the RMSD, RMSF of each frame as a function of simulation time.
For hit molecule ZINC13144609, the RMSD value of the protein Cα was found to increase up to a value of 2.6 Å with respect to its starting coordinate (t = 0) for first 10 ns and stabilize around an average value of 2.347 Å for rest of the MD trajectories which indicates no change in the protein backbone. Further, the RMSF of the backbone at the binding site of 6Y84 is found to be in the range of 1.0 Å to 2.573 Å which suggests lower degree of flexibility in binding region. From the above observation, it is proved that the ligand movement was stable during the simulation. The ligand–protein contacts are illustrated in Figure 5. It is found that the hydrogen bonds with Glu166 and hydrophobic interactions with Pro168, Leu167, Met 49, His41are major contributing factor for stabilizing hit molecule ZINC13144609 at the binding site which is in accordance with our docking result. His41 found to exhibit π-π stacking with the benzene ring for 61% of the trajectory. Among the six-hydrogen bond predicted by docking, only four are found to be preserved during the simulation.
Figure 5.

RMSD, RMSF and residue interaction plots of 6Y84 protein complexed with ZINC13144609.
For hit molecule ZINC01581128, the RMSD and RNSF plots are given in Figure 6. The analysis of 6Y84 protein-ZINC01581128 complex showed low RMSD value, 2.586 Å indicating the stability of the atomistic molecular system. The plot showed that the system attained stability after 20 ns of simulations. The RMSF value is found to be in the range of 1.2–3.0 Å which indicates low fluctuations in the binding site. The hydrogen-bonding interactions with Gly143, Phe140 and Glu166 were retained throughout the simulations. It suggests the importance of these interactions in stabilizing the simulated complex.
Figure 6.

The protein RMSD, RMSF and protein–ligand contacts diagram of complex 6Y84 with ZINC01581128.
From the above results, it is clear that the protein interacting with both hit molecules is stabilized in the chosen POPC bilayer membrane. The top hit molecules are stabilized through, hydrogen bonding, hydrophobic and π–π interactions with various residues indicate its better stabilization in 6Y84 protein.
3.4. Pharmacokinetic parameters and bioavailability prediction
In the drug discovery process, the molecules under consideration need to possess good pharmacokinetic profiles as well as potency to confirm their bioavailability and effectiveness. The various physicochemical and therapeutic significant descriptors were calculated and documented in Tables 4 and 5, respectively.
Table 4.
Summary of physicochemical properties of top five hit molecules.
| Ligand no. | MW | Dipole | donorHB | accptHB | Volume | Rotor | PSA | Rule of five |
|---|---|---|---|---|---|---|---|---|
| ZINC13144609 | 354.365 | 9.895 | 6 | 9.8 | 1067.801 | 9 | 131.487 | 1 |
| ZINC01581128 | 411.46 | 4.735 | 5 | 7.7 | 1241.312 | 13 | 148.557 | 1 |
| ZINC06062920 | 411.46 | 7.793 | 5 | 7.95 | 1242.611 | 12 | 159.536 | 1 |
| ZINC03022586 | 426.458 | 4.967 | 4 | 14.4 | 1215.403 | 10 | 178.68 | 0 |
| ZINC00134422 | 304.259 | 3.251 | 2 | 9.4 | 913.267 | 6 | 167.007 | 0 |
| Rangea | 130.0–725.0 | 1.0–12.5 | 1.0–6.0 | 2.0–20.0 | 500.0–2000.0 | 0–15 | 7.0–200 | Max 4 |
aRange: for 95% oral drugs.
Table 5.
Therapeutic significant properties of top five hit molecules.
| Ligand No. | CNS | QPlogPo/w | QPlogS | QPlogBB | QPPMDCK | Metab | QPlogKhsa |
| ZINC13144609 | −2 | 0.421 | −2.727 | −2.005 | 44.317 | 4 | −0.693 |
| ZINC01581128 | −2 | 0.689 | −1.188 | −1.896 | 1.152 | 9 | −0.232 |
| ZINC06062920 | −2 | 0.457 | −1.421 | −2.09 | 0.729 | 8 | −0.23 |
| ZINC03022586 | −2 | −1.301 | −2.892 | −3.745 | 1.987 | 2 | −1.097 |
| ZINC00134422 | −2 | −0.8 | −2.191 | −2.579 | 5.219 | 2 | −0.799 |
| Rangea | −2 to +2 | −2 to 6.5 | −6.5 to 0.5 | −3.0 to 1.2 | <25 p >500 g | 1–8 | −1.5 to 1.5 |
aRange: for 95% oral drugs.
All the calculated physicochemical and therapeutic significant properties were found to be in their permissible range and therefore confirming their drug-likeness.
The bioavailability of top scored hit molecules was determined through SwissADME tool. The bioavailability radar chart of ZINC13144609 and ZINC01581128 is illustrated in Figure 7. The pink area shown in Figure 7 represents most favorable area for each property like INSATU (unsaturation), INSOLU (insolubility), FLEX (rotatable bonds), LIPO (lipophilicity), SIZE (molecular weight) and POLAR (polar surface area). The predicted bioavailability score is tabulated in Table 6.
Figure 7.

The bioavailability radar chart for top hit molecule (A) ZINC13144609 (B) ZINC01581128.
Table 6.
Synthetic accessibility and bioavailability score of top screened molecules.
| Ligand No. | Hit structures | Bioavailability score | Synthetic accessibility score |
|---|---|---|---|
| ZINC13144609 1,4-bis(1H-benzo[d]imidazol-2-yl)butane-1,2,3,4-tetraol |
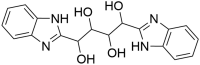 |
0.55 | 3.79 |
| ZINC01581128 3-((7,10-dihydroxy-2-(2-((2-hydroxyethyl) ammonio)ethyl)-6-oxo-2,6-dihydrodibenzo [cd,g]indazol-5-yl) amino)propan-1-aminium |
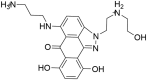 |
0.55 | 3.26 |
| ZINC06062920 3-((6-hydroxy-2-(2-((2-hydroxyethyl)ammonio)ethyl)-7,10-dioxo-1,2,6,7,10,10b-hexahydrodibenzo[cd,g]indazol-5-yl)amino)propan-1-aminium |
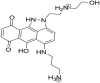 |
0.55 | 3.57 |
| ZINC03022586 N2,N7-bis(2-hydroxyethyl)-9H-fluorene-2,7-disulfonamide |
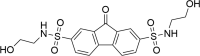 |
0.55 | 3.19 |
| ZINC00134422 2-(2-hydroxyethyl)-6-propylpyrrolo[3,4-f]isoindole-1,3,5,7(2H,6H)-tetraone |
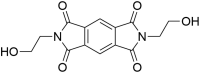 |
0.55 | 2.15 |
3.5. Estimation of synthetic feasibility
The synthetic feasibility of top scored molecules is shown in Table 6. The synthetic feasibility score is in the range of 1 for simply synthesizable and 10 for difficult to synthesize. All top hit molecules showed synthetic feasibility score around 3.0 that indicates all are easily synthesizable. Furthermore, we forward to plan future synthesis with the best plausible route.
3.6. Quantum mechanical calculations
The hit molecule ZINC13144609 and ZINC01581128 were chosen for energy calculations. The HOMO–LUMO energy gap represents the chemical reactivity of molecules. The electronic properties of screened hit molecules were shown in Table 7.
Table 7.
Summary of calculated electronic properties of hit molecules.
| Electronic properties | ZINC13144609 | ZINC01581128 |
|---|---|---|
| Number of canonical orbitals | 478 | 581 |
| Gas phase energy | −1216.649911 | −1390.606133 |
| HOMO | −0.21684 | −0.34043 |
| LUMO | −0.022889 | −0.23834 |
| Electrostatic potential mean (kcal/mol) | −0.17 | 116.53 |
| Average local ionization energy mean (kcal/mol) | 261.64 | 400.01 |
The position of HOMO-LUMO orbitals of ZINC13144609 (Figure 8) suggests that the hydrophobic region of molecule alters the topology of HOMO-LUMO orbitals and also energies are localized. HOMO and LUMO covers mainly benzimidazole ring of hit molecule. The ZINC01581128 molecule found to have LUMO orbital over hydrophobic region and HOMO orbital mainly cover polar region (Figure 9). Therefore, the presence of HOMO and LUMO orbitals near the hydrophobic part of the hit molecules is important to from stable ligand–protein complex.
Figure 8.

Surface maps for DFT results for ZINC13144609: (A) HOMO map, (B) LUMO map, (C) Interaction strength map and (D) Electrostatic potential map.
Figure 9.

Surface maps for DFT results for ZINC01581128: (A) HOMO map, (B) LUMO map, (C) Interaction strength map and (D) Electrostatic potential map.
4. Conclusions
In the present investigation, structure-based virtual screening, DFT calculation, MM/GBSA, pharmacokinetic parameters calculation, and molecular dynamics simulation studies in search of the antiviral drug against COVID-19 were performed. The COVID-19 main protease protein has made up of three regions of 306 amino acid residues. The predicted binding site lies between regions 2 and 3. Virtual screening was executed through a hierarchal mode of elimination technique in the prerequisite binding site. Further, the top five hit molecules were shortlisted based on docking score and non-bonding interactions. The reliability of docking analysis was confirmed by the low RMSD value of docked ligand and protein. The highest scoring hit molecule ZINC13144609 (−7.524 kcal/mol) exhibited six hydrogen bonds with amino acid residues Glu166, Leu141 and Asn142. The second hit molecule ZINC01581128 showed a docking score −7.429 kcal/mol with six hydrogen bonding (Gly143, Leu141, Phe140 and Glu166) and Π–Π stacking interactions. The docking study suggested that the polar part of the molecules forms hydrogen bonding network. It is evident from the MD simulation study that the ligand–protein complexes have equilibrated and changes were perfectly acceptable for small, globular proteins. The LUMO–HOMO orbital study described the interaction pattern of hits with the protein at the quantum level. The pharmacokinetic properties calculation of top scored hits obtained from present work ensured their safe administration in the human body. Lastly, all top-scored hits can be easily synthesizable based on the least synthetic accessibility score. Therefore, investigated hits could be potent and efficacious drugs targeting COVID-19 main protease protein for the COVID-19 treatment. Experimentally, the need to validate the molecular modeling results reported here with in vitro and/or in vivo inhibition evaluation is acknowledged but due to lack of funding, work is limited. Therefore, investigated inhibitors could be potent and efficacious drugs targeting COVID-19 main protease protein for the COVID-19 treatment.
Supplementary Material
Acknowledgments
The authors are thankful to Dr. Sourav Kalra, Centre for Human Genetics & Molecular Medicine, Central University of Punjab for providing computational facilities to accomplish this research work.
Glossary
Abbreviations
- MERS
Middle East respiratory syndrome
- SARS
Severe acute respiratory syndrome
- LUMO
Lowest unoccupied molecular orbital
- HOMO
Highest occupied molecular orbital
- HTVS
High throughput virtual screening
- SP
Standard precision
- XP
Extra precision
- MM-GBSA
Molecular mechanics-generalized born surface area
- LBFGS
Limited-memory Broyden-Fletcher-Goldfarb-Shanno
- RMSF
Root mean square fluctuation
- RMSD
Root mean square deviation
- POPC
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
Disclosure statement
No competing interests exist.
References
- Amanat, F., & Krammer, F. (2020). SARS-CoV-2 vaccines: Status Report. Immunity, 52(4), 583–589. 10.1016/j.immuni.2020.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athar, M., Lone, M. Y., Khedkar, V. M., & Jha, P. C. (2016). Pharmacophore model prediction, 3D-QSAR and molecular docking studies on vinyl sulfones targeting Nrf2-mediated gene transcription intended for anti-Parkinson drug design. Journal of Biomolecular Structure & Dynamics, 34(6), 1282–1297. 10.1080/07391102.2015.1077343 [DOI] [PubMed] [Google Scholar]
- Belouzard, S., Millet, J. K., Licitra, B. N., & Whittaker, G. R. (2012). Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses, 4(6), 1011–1033. 10.3390/v4061011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochevarov, A. D., Harder, E., Hughes, T. F., Greenwood, J. R., Braden, D. A., Philipp, D. M., Rinaldo, D., Halls, M. D., Zhang, J., & Friesner, R. A. (2013). Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. International Journal of Quantum Chemistry, 113(18), 2110–2142. 10.1002/qua.24481 [DOI] [Google Scholar]
- Boopathi, S., Poma, A. B., & Kolandaivel, P. (2020). Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. Journal of Biomolecular Structure and Dynamics, 1–10. 10.1080/07391102.2020.1758788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, C., Lo, S.-C., Wang, Y.-S., & Hou, M.-H. (2016). Recent insights into the development of therapeutics against coronavirus diseases by targeting N protein. Drug Discovery Today, 21(4), 562–572. 10.1016/j.drudis.2015.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, Z., Mohanty, U., Noehre, J., Sawyer, T. K., Sherman, W., & Krilov, G. (2010). Probing the alpha-helical structural stability of stapled p53 peptides: molecular dynamics simulations and analysis. Chemical Biology & Drug Design, 75(4), 348–359. 10.1111/j.1747-0285.2010.00951.x [DOI] [PubMed] [Google Scholar]
- Gupta, M. K., Vemula, S., Donde, R., Gouda, G., Behera, L., & Vadde, R. (2020). In-silico approaches to detect inhibitors of the human severe acute respiratory syndrome coronavirus envelope protein ion channel. Journal of Biomolecular Structure and Dynamics, 1–11. 10.1080/07391102.2020.1751300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajduk, P. J., Huth, J. R., & Tse, C. (2005). Predicting protein druggability. Drug Discovery Today, 10(23-24), 1675–1682. 10.1016/S1359-6446(05)03624-X [DOI] [PubMed] [Google Scholar]
- Halgren, T. A. (2009). Identifying and characterizing binding sites and assessing druggability. Journal of Chemical Information and Modeling, 49(2), 377–389. 10.1021/ci800324m [DOI] [PubMed] [Google Scholar]
- Halgren, T. (2007). New method for fast and accurate binding-site identification and analysis. Chemical Biology & Drug Design, 69(2), 146–148. 10.1111/j.1747-0285.2007.00483.x [DOI] [PubMed] [Google Scholar]
- Hasan, A., Paray, B. A., Hussain, A., Qadir, F. A., Attar, F., Aziz, F. M., Sharifi, M., Derakhshankhah, H., Rasti, B., Mehrabi, M., & Shahpasand, K. Saboury, A. A, & Falahati, M. (2020). A review on the cleavage priming of the spike protein on coronavirus by angiotensin-converting enzyme-2 and furin. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1754293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar, S., & Roy, K. (2013). How far can virtual screening take us in drug discovery? Expert Opinion on Drug Discovery, 8(3), 245–261. 10.1517/17460441.2013.761204 [DOI] [PubMed] [Google Scholar]
- Kirchdoerfer, R. N., Cottrell, C. A., Wang, N., Pallesen, J., Yassine, H. M., Turner, H. L., Corbett, K. S., Graham, B. S., McLellan, J. S., & Ward, A. B. (2016). Pre-fusion structure of a human coronavirus spike protein. Nature, 531(7592), 118–121. 10.1038/nature17200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, N., Mishra, S. S., Sharma, C. S., Singh, H. P., & Kalra, S. (2018). In silico binding mechanism prediction of benzimidazole based corticotropin releasing factor-1 receptor antagonists by quantitative structure activity relationship, molecular docking and pharmacokinetic parameters calculation. Journal of Biomolecular Structure & Dynamics, 36(7), 1691–1712. PMC][ 10.1080/07391102.2017.1332688] [28521603] [DOI] [PubMed] [Google Scholar]
- Li, F. (2016). Structure, Function, and Evolution of Coronavirus Spike Proteins. Annual Review of Virology, 3(1), 237–261. 10.1146/annurev-virology-110615-042301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, S.-M., Lin, S.-C., Hsu, J.-N., Chang, C.-K., Chien, C.-M., Wang, Y.-S., Wu, H.-Y., Jeng, U.-S., Kehn-Hall, K., & Hou, M.-H. (2020). Structure-based stabilization of non-native protein-protein interactions of coronavirus nucleocapsid proteins in antiviral drug design. Journal of Medicinal Chemistry, 63(6), 3131–3141. 10.1021/acs.jmedchem.9b01913 [DOI] [PubMed] [Google Scholar]
- Masters, P. S. (2006). The molecular biology of coronaviruses. Advances in Virus Research, 66, 193–292. 10.1016/s0065-3527(06)66005-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidharan, N., Sakthivel, R., Velmurugan, D., & Gromiha, M. M. (2020). Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 protease against COVID-19. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1752802 [DOI] [PubMed] [Google Scholar]
- Rollinger, J. M., Stuppner, H., & Langer, T. (2008). Virtual screening for the discovery of bioactive natural products. Natural Compounds as Drugs, I, 211–249. 10.1007/978-3-7643-8117-2_6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarma, P., Shekhar, N., Prajapat, M., Avti, P., Kaur, H., Kumar, S., Singh, S., Kumar, H., & Prakash, A., Dhibar, D. P.. & Medhi, B. (2020) In-silico homology assisted identification of inhibitor of RNA binding against 2019-nCoV N-protein (N terminal domain). Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1753580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar, M. S., Venkatachalam, T., Sharma, C. S., Pratap Singh, H., Kalra, S., & Kumar, N. (2019). Computational investigation of binding mechanism of substituted pyrazinones targeting corticotropin releasing factor-1 receptor deliberated for anti-depressant drug design. Journal of Biomolecular Structure & Dynamics, 37(12), 3226–3244. 10.1080/07391102.2018.1513379 [DOI] [PubMed] [Google Scholar]
- Su, P.-C., Tsai, C.-C., Mehboob, S., Hevener, K. E., & Johnson, M. E. (2015). Comparison of radii sets, entropy, QM methods, and sampling on MM-PBSA, MM-GBSA, and QM/MM-GBSA ligand binding energies of F. tularensis enoyl-ACP reductase (FabI)). Journal of Computational Chemistry, 36(25), 1859–1873. 10.1002/jcc.24011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahir Khan, M., Ali, A., Wang, Muhammad Irfan, Q., Khan, A., Zeb, M. T., Zhang, Y.-J., Chinnasamy, S., & Wei, D.-Q. (2020). Marine natural compounds as potents inhibitors against the main protease of SARS-CoV-2. A molecular dynamic study. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2020.1769733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hemert, M. J., van den Worm, S. H. E., Knoops, K., Mommaas, A. M., Gorbalenya, A. E., & Snijder, E. J. (2008). SARS-coronavirus replication/transcription complexes are membrane-protected and need a host factor for activity in vitro. PLoS Pathogens, 4(5), e1000054. 10.1371/journal.ppat.1000054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venugopal, P. P., Das, B. K., Soorya, E., & Chakraborty, D. (2020). Effect of hydrophobic and hydrogen bonding interactions on the potency of ß‐alanine analogs of G‐protein coupled glucagon receptor inhibitors. Proteins: Structure, Function, and Bioinformatics, 88(2), 327–344. 10.1002/prot.25807 [DOI] [PubMed] [Google Scholar]
- Walls, A. C., Park, Y.-J., Tortorici, M. A., Wall, A., McGuire, A. T., & Veesler, D. (2020). Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell, 181(2), 281–292.e6. 10.1016/j.cell.2020.02.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H., Bartlam, M., & Rao, Z. (2006). Drug design targeting the main protease, the Achilles' heel of coronaviruses . Current Pharmaceutical Design, 12(35), 4573–4590. 10.2174/138161206779010369 [DOI] [PubMed] [Google Scholar]
- Zumla, A., Chan, J. F. W., Azhar, E. I., Hui, D. S. C., & Yuen, K.-Y. (2016). Coronaviruses – drug discovery and therapeutic options. Nature Reviews. Drug Discovery, 15(5), 327–347. 10.1038/nrd.2015.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.