

Abstract
The COVID-19 pandemic has been responsible for several deaths worldwide. The causative agent behind this disease is the Severe Acute Respiratory Syndrome – novel Coronavirus 2 (SARS-CoV-2). SARS-CoV-2 belongs to the category of RNA viruses. The main protease, responsible for the cleavage of the viral polyprotein is considered as one of the hot targets for treating COVID-19. Earlier reports suggest the use of HIV anti-viral drugs for targeting the main protease of SARS-CoV, which caused SARS in the year 2002–2003. Hence, drug repurposing approach may prove to be useful in targeting the main protease of SARS-CoV-2. The high-resolution crystal structure of the main protease of SARS-CoV-2 (PDB ID: 6LU7) was used as the target. The Food and Drug Administration approved and SWEETLEAD database of drug molecules were screened. The apo form of the main protease was simulated for a cumulative of 150 ns and 10 μs open-source simulation data was used, to obtain conformations for ensemble docking. The representative structures for docking were selected using RMSD-based clustering and Markov State Modeling analysis. This ensemble docking approach for the main protease helped in exploring the conformational variation in the drug-binding site of the main protease leading to the efficient binding of more relevant drug molecules. The drugs obtained as top hits from the ensemble docking possessed anti-bacterial and anti-viral properties. This in silico ensemble docking approach would support the identification of potential candidates for repurposing against COVID-19.
Communicated by Ramaswamy H. Sarma
Keywords: COVID-19, SARS-CoV-2, main protease, repurposing, cryptic pockets, 3CLpro
Introduction
The COVID-19 pandemic caused by the SARS-CoV-2 is known to spread quite rapidly. The first incidence of this disease was found in Wuhan, China in December 2019. The World Health Organization (WHO) has reported over 2.8 million affected individuals and 1.9 million deaths by the end of April 2020. In a very short period COVID-19 has spread all over the globe. The SARS-CoV-2, has an RNA genome of around 30 K nucleotides. This genome is known to code for the entire viral proteome (Xu et.al., 2005). The entire coding RNA is divided into three regions, nonstructural protein (nsp), structural protein and accessory protein-coding region (Andersen et al., 2020; Kim et al., 2020; Wang & Chiou, 2020). The nonstructural protein region consisting of ORF1a and ORF1b, that codes for the polyprotein pp1a and pp1b. These polyproteins are further cleaved to form the 16 nsp. The structural protein region codes for the Spike(S)-glycoprotein, envelope protein, membrane protein and nucleoprotein (Kim et al., 2020). These proteins are responsible for the viral replication, viral functioning and viral–host interaction. Hence, therapeutic studies targeting these proteins have gained importance in the drug industry (Li & De Clercq, 2020). Few of these major proteins which have been considered as potential drug targets based on the earlier therapeutics developed against old coronaviruses include, viral proteases, RNA dependent RNA polymerase and viral surface Spike protein (Nadeem et al., 2020; Prajapat et al., 2020). The need of the hour lies in the development of fast therapeutics for combating the SARS-CoV-2. Drug repurposing is one such strategy that is being extensively used worldwide to design vaccines against this coronavirus (Asai et al., 2020; Cherian et al., 2020; Pawar, 2020; Tu et al., 2020). To understand how similar is the SARS-CoV-2 in comparison to the earlier coronaviruses sequence comparison studies have been performed to understand the variation in the sequence of these potential target proteins (Bauer et al., 2020; Forster et al., 2020; Li et al., 2020; Stefanelli et al., 2020; Wang et al., 2020; Wu et al., 2020; Yadav et al., 2020). These studies would help in repurposing and repositioning the drugs that have been used earlier in order to target this novel coronavirus. Drug repurposing studies are being performed through experimental as well as computational techniques (Adeoye et al., 2020; Ahmad et al., 2020; Amin & Abbas, 2020; Anwar et al., 2020; Baron et al., 2020; Beura & Chetti, 2020; Boopathi et al., 2020; Borkotoky & Banerjee, 2020; Chandra et al., 2020; Chen et al., 2020; de Oliveira et al., 2020; Elfiky, 2020a, 2020b; Elmezayen et al., 2020; Fan et al., 2020; Khan et al., 2020b; Kruse, 2020; Liu et al., 2020; Muralidharan et al., 2020; Phadke & Saunik, 2020; Quimque et al., 2020; Rosa & Santos, 2020; Serafin et al., 2020; Shah et al., 2020). All the three, drug targets mentioned have been extensively studied using drug repurposing. Main protease, RNA dependent RNA polymerase and spike protein have been screened for small molecules from the Food and Drug Administration (FDA) approved database as well as naturally occurring compounds (Aanouz et al., 2020; Abdelli et al., 2020; Al-Khafaji et al., 2020; Arya & Dwivedi, 2020; Babadaei, Hasan, Bloukh et al., 2020; Babadaei, Hasan, Vahdani et al., 2020; Basit et al., 2020; Bhardwaj et al., 2020; Caly et al., 2020; Choudhury, 2020; Das et al., 2020;Elfiky, 2020c, 2020d; Elfiky & Azzam, 2020; Enmozhi et al., 2020; Gao et al., 2020; Gupta et al., 2020a, 2020b; Gyebi et al., 2020; Hasan et al., 2020; Hendaus, 2020; Islam et al., 2020; Joshi et al., 2020; Khan et al., 2020a, 2020c; Kumar et al., 2020a, 2020b, 2020c, 2020d; Lobo-Galo et al., 2020; Mahanta et al., 2020; Mittal et al., 2020; Pant et al., 2020; Sarma et al., 2020; Sinha et al., 2020; Sk et al., 2020; Smith & Smith, 2020; Umesh et al., 2020; Wahedi et al., 2020). The viral main protease also known as 3Chymotrypsin-like protease (3CLpro) or main protease is formed by the autocleavage of the polyprotein and further responsible for cleavage of various other nsp (Kim et al., 2020). 3CLpro is the nsp5 amongst the 16 nsp. The high-resolution crystal structure of 3CLpro was elucidated in February 2020 (Jin et al., 2020). Before the availability of this structure, modeling studies were performed for the SARS-CoV-2 3CLpro and it was found to be similar to the SARS-CoV and MERS main protease (Phadke & Saunik, 2020). Drug repurposing studies on this model using in silico approaches revealed the role of previously used antivirals as a potential drug for COVID19 (Phadke & Saunik, 2020). The inhibitor interactions with 3CLpro have been well explored using electron density maps, the residues crucial to initiate the inhibitory effect upon interacting with the drugs have been identified in these experimental studies (Fearon et al., 2020). There are reports on protein–protein interaction networks, where the entire viral proteome has been studied to find its interactions with the human proteins (Gordon et al., 2020). This study reveals the role of 3CLpro in obstructing the inflammatory and interferon pathway in humans by inhibiting the nuclear transport of epigenetic regulatory proteins (Gordon et al., 2020). There are experimental studies that reveal the inhibitor binding site of the 3CLpro is divided into various subsites (Zhang et al., 2020). These studies also suggest that interaction with the residues in these sites would help in designing SARS-CoV-2 3CLpro inhibitors (Zhang et al., 2020). The crucial residues HIS 41 and CYS 145 form the catalytic dyad which is responsible to form strong interactions with the inhibitor. In the case of the inhibitor N3, it was observed to form a covalent bond with CYS 145. The residues surrounding this catalytic dyad are known to form the complete active site of 3CLpro. This active site consists of subsites S1 and S2 (Figure 1) (Zhang et al., 2020). The S1 subsite consists of the residues PHE 140, GLY 143, CYS 145, HIS 163, GLU 166 and HIS 172. The S2 subsite consists of the residues THR 25, HIS 41, MET 49, MET 165 and GLN 189 (Zhang et al., 2020). A Pan DDA analysis of different inhibitor molecules of 3CLpro also suggests that the inhibitory effect is observed when the molecule forms strong bonded or nonbonded interactions with these critical residues of the 3CLpro active site (Fearon et al., 2020).
Figure 1.

Inhibitor binding site as seen in 6LU7. N3 inhibitor (represented in stick), Subsite S1 (F140, G143, C145, H163, E166, H172) and Subsite S2 (T25, H41, M49, M165, Q189).
This current article describes the scope of repurposing drugs for COVID-19 considering the conformational variation in the inhibitory binding site of 3CLpro of SARS-CoV-2. The dynamic states of the protein than remain unexplored in experimental techniques such as X-ray crystallography were witnessed using molecular dynamics simulations. These findings further helped in identifying even better potential candidates for drug repurposing. The crystal structure with PDB ID 6LU7 was observed to have the covalently bound inhibitor N3 in its active site (Figure 1) (Jin et al., 2020). This structure has been widely used by the research community for performing molecular docking and simulation studies (Hall & Ji, 2020; Kandeel & Al-Nazawi, 2020; Komatsu et al., 2020; Ortega et al., 2020). The 3D coordinates for the apo form of 3CLpro were obtained from the PDB ID 6LU7 (Jin et al., 2020). This SARS-CoV-2 drug target was screened for probable drug candidates against the FDA approved and SWEETLEAD drug database (Centre for Drug Evaluation and Research (US), 2004; Novick et al., 2013). Two approaches were used for drug repurposing studies against the drug target 3CLpro (Figure 2). The first approach involved docking of the FDA approved and SWEETLEAD drug database against the crystal structure 6LU7 (Figure 2(A)). This approach has been referred to as ‘Direct Docking’ further. The second approach involved docking of the two mentioned databases against an ensemble of structures obtained from molecular dynamics (MD) simulations of the apo form of 3CLpro (Figure 2(B)). This approach has been referred to as ‘Ensemble docking’ further. MD simulations were performed for a cumulative of 150 ns for the apo 3CLpro. An open-source 10 μs MD simulation data of 3CLpro dimer was obtained from the simulations performed on the MDGRAPE-4A supercomputing cluster located at RIKEN BDR, Japan (Komatsu et al., 2020). An ensemble generation to perform ensemble docking was obtained using Root Mean Square Deviation (RMSD) based clustering and Markov State Modelling (MSM) analysis. A total of 16 conformations of 3CLpro were obtained from RMSD-based clustering and MSM analysis. Further, these 16 conformations were docked against the FDA approved and SWEETLEAD drug database (Figure 2(B)). The top-ranked drugs obtained from both these approaches belonged to the class of anti-bacterial and anti-viral drugs. The docking scores obtained for the ensemble docking revealed to be better than those obtained through direct docking. The interaction energies observed for the ensemble docked compounds were significantly better as compared to those seen through direct docking. The conformation variation in the drug-binding site of 3CLpro was observed, as a greater number of target protein residues interacted with the drug molecule. This lacked when the drugs were docked directly on to the crystal structure. More number of interactions between the drug molecule and the 3CLpro inferred that the drug-binding pockets were found to be more accessible in case of the ensemble docking. The findings obtained through the direct docking and ensemble docking of 3CLpro of SARS-CoV-2 have been discussed further in this article. These observations may prove to be useful in silico approaches in designing/repurposing drugs against COVID-19.
Figure 2.

Two approaches used for the drug repurposing and docking studies viz. Direct docking (A) and Ensemble docking (B) (ref.41: Komutsu et.al., 2020).
Methodology
The high-resolution crystal structure of 3CLpro was retrieved from the Protein Data Bank with PDB ID: 6LU7 (Jin et al., 2020). This PDB file was cleaned by removing the ligand coordinates. This PDB in the apo form was further considered for the molecular dynamics simulations and docking studies. The detailed protocol followed has been explained in Figure 2. Direct docking was performed on the 3CLpro protein against the FDA approved drug database and the SWEETLEAD database. This docking was performed using DOCK 6 (Allen et al., 2015). The receptor preparation was done using UCSF Chimera and further the active site pocket identification and docking was performed using DOCK 6 (Figure 2(A)) (Allen et al., 2015; Pettersen et al., 2004). In the case of ensemble docking, the coordinates for 3CLpro were obtained from molecular dynamics simulations (Figure 2(B)). The simulations were performed using the AMBER 16 simulation package (Case et al., 2016). The AMBER FF14SB force field was used for generation of the parameters. The system was neutralized by Na + ions and solvated using the TIP3P water model. The minimization was performed for 20,000 steps using the steepest descent and the conjugate gradient method. The system was gradually heated to 300 K using the Langevin thermostat. The SHAKE algorithm was employed for dealing with the hydrogen restraints. The equilibration was performed for 1 ns at NPT with temperature being 300 K and pressure being 1 atm. The production run was performed for 50 ns using the NPT ensemble. Three parallel runs of 50 ns each were performed based on the explained MD protocol, hence, a cumulative of 150 ns data was generated. A 10 µs simulation data on the dimer of 3CLpro was obtained MDGRAPE-4A, at RIKEN BDR, Japan (Komatsu et al., 2020). However, only monomer data was used from this 10 µs as the simulations performed in-house belonged to the monomer. A cumulative data of 10.15 µs was subjected to RMSD based clustering and Markov State Modelling (MSM) analysis. The RMSD cut-off of 1.7 Å was used for the clustering using the dbscan method of cpptraj module of AmberTools 17 (Ester et al., 1966). A total of 12 representative conformations were obtained through RMSD-based clustering. The MSM analysis was performed using the PyEmma software (Scherer et al., 2015). The backbone dihedral angles were used as the collective variable for performing the MSM analysis. The complete details of the MSM analysis performed in order to obtain the different states of 3CLpro have been given in the Supporting Information as SI1. MSM analysis has been widely used for significant sampling of the MD simulation data (Chodera & Noé, 2014; Chodera et al., 2007; Jani et al., 2019; Prinz et al., 2011; Sirur et al., 2016). A similar methodology for identifying significant states from the simulation data was used in one of the earlier works reported by our group (Jani et al., 2019). A total of 4 representative conformations for 3CLpro were obtained from the MSM analysis. Hence, a total of 16 conformations exploring the ensemble of 3CLpro were considered for the ensemble docking approach described in Figure 2(B). The FDA approved and SWEETLEAD drug databases were screened against these 16 ensemble structures (as shown in Supporting Information Figures S1 and S2). The details of choosing top scored ligands from ensemble docking has been discussed in details in the ‘Results & Discussion’ section (Supporting Information Figures S1 and S2). The interaction energies for the docked complexes were calculated using the Prodigy-LIG server (Vangone et al., 2019). Prodigy-LIG predicts the interaction energy between protein–ligand complexes using aa contact-based prediction method. It takes into account the inhibition constant obtained from the available protein–ligand complexes. A HADDOCK refinement method is employed, to predict the intermolecular energies based on the number of atomic contacts formed between the protein and the ligand (Vangone et al., 2019).
Results and discussion
Direct docking
The direct docking of the 3CLpro against the FDA approved drug database was performed using DOCK6 (Allen et al., 2015). The drugs were screened and ranked based on their grid score. The grid scores quantify the strength of binding of any small molecule owing to the nonbonded interactions it forms with the active site of the receptor molecule. Hence, a better grid score indicated a better binding of that small molecule to the receptor. The grid scores are energy values which are obtained using force field equation given below,
Where, E is the grid score and is the van der Waals contribution and is the electrostatic contribution. Each of these terms in double summation over ligand atoms i and receptor atoms j.
A more negative value of E, indicates better stability of docking obtained by that particular molecule. The top ranked drug would be the one which would have the most negative value of the grid score (E). An E value greater than zero indicates unfavorable binding of the molecule to the receptor which may have resulted due to steric clashes between the ligand molecule and the amino acid residues of the receptor molecule. Only 9% of the drugs were observed to have grid scores above zero while screening the FDA approved. The drugs with the top ranked grid scores have been discussed below.
The drugs that ranked in the top five were ceftazidime (PubChem CID: 5481173), quetiapine (PubChem CID: 5002), cabergoline (PubChem CID: 54746), enoxolone (PubChem CID: 10114) and apremilast (PubChem CID: 11561674) in the order of decreasing rank. These drugs and their current usage for treating different diseases have been listed in Table 1.
Table 1.
Top five ranked drugs from the FDA approved database obtained through direct docking of 3CLpro of SARS-CoV-2.
| PubChem CID | Name of the drug | Earlier purpose | Structure |
|---|---|---|---|
| 5481173 | Ceftazidime | Antibacterial, used in pneumonia | 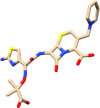 |
| 5002 | Quetiapine | Bipolar disorder, used as a treatment against Schizophrenia |  |
| 54746 | Cabergoline | Dopamine agonists, used in Parkinson’s disease |  |
| 1011 | Enoxolone | Consists of Glycyrrhetinic acid, a plant derivative, used as anti-allergitic, anti-bacterial and anti-viral |  |
| 11561674 | Apremilast | Psoriasis | 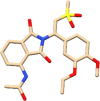 |
The information on the current usage of these drugs was obtained from PubChem. The grid scores obtained for these drugs have been shown in Figure 3(A). The top-ranked drug ceftazidime is known to be used as an antibacterial in respiratory tract infections. Enoxolone, which ranked fourth as per the grid score has glycyrrhetinic acid as a subcomponent, which is a known plant derivative and is also known to possess anti-bacterial and anti-viral properties. The drugs, quetiapine and cabergoline, that ranked as second and third, respectively, are known to be effective in treating neurological diseases such as bipolar disorder and Parkison’s disease. Apremilast, which ranked as fifth is known to be used in treating psoriasis. The goal of docking against the FDA approved drug database was to find previously known drugs that would be effective in treating the symptoms of the current disease in investigation. The results obtained from direct docking infer that out of the top five docked drugs, two of them appear to carry antibacterial and antiviral properties which were also found to be specific to the respiratory tract infections.
Figure 3.

The top five ranked drugs from the FDA approved (A) and SWEETLEAD (B) drug database obtained through direct docking approach.
The direct docking of 3CLpro was performed against the SWEETLEAD drug database using DOCK 6. The SWEETLEAD drug hosts around more than 10 K drug molecules which include the approved drugs, rejected drugs and molecules isolated from traditional medicinal herbs (Novick et al., 2013). One of the docking studies by Smith and Smith on the SARS-CoV-2 spike protein mentioned the use of SWEETLEAD for identifying few molecules with potential inhibitory activity against the viral spike protein (Smith & Smith, 2020). The docking of 3CLpro against this database led to the identification of a few small molecules which may prove to be potential candidates for drug development against the COVID-19 disease (Smith & Smith, 2020). In the case, of SWEETLEAD drug database only 17% of the drugs were observed to have grid score above zero, the top ranked drugs have been described further. The drugs that ranked in the top five were dibekacin (PubChem CID: 470999), micronomicin (PubChem CID: 3037206), catalposide (PubChem CID: 93039), dihydro-alpha-ergocryptine (PubChem CID: 114948) and itopride (PubChem CID: 3792) in the decreasing order of their ranks. The details about the current usage of these drugs for treating various diseases and their respective grid scores have been given in Table 2.
Table 2.
Top five ranked drugs from the SWEETLEAD database obtained through direct docking of 3CLpro of SARS-CoV-2.
| PubChem CID | Name of the drug | Earlier purpose | Structure |
|---|---|---|---|
| 470999 | Dibekacin | Aminoglycoside Antibiotic | 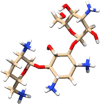 |
| 3037206 | Micronomicin | Aminoglycoside Antibiotic |  |
| 93039 | Catalposide | Plant derivative, anti-inflammatory effect | 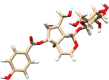 |
| 114948 | Dihydro-alpha-ergocryptine | Parkinson’s diseases |  |
| 3792 | Itopride | Functional dyspepsia, gastrointestinal drug, Blocks dopamine receptor |  |
The grid scores of each of these drugs has been shown in Figure 3(B). The drugs that were observed to be ranked in the top two were dibekacin and micronomicin which belonged to the class of aminoglycoside antibiotics. Catalposide which ranked third is known to be a plant derivative and is known to be used as an anti-inflammatory agent. The remaining two drugs dihydro-alpha-ergocryptine and itopride are used for treating Parkinson’s disease and gastrointestinal ailments, respectively. Two of the drugs amongst the top five ranked drugs are known to have antibacterial activity viz. dibekacin and micronomicin.
Ensemble generation
The molecular dynamics simulations of the apo 3CLpro were performed to obtain an ensemble of 3CLpro conformations capturing the protein dynamics. An additional open source simulation data of 10 µs was used for ensemble generation (Komatsu et al., 2020). The conformational variation in the 3CLpro was measured by calculating the backbone RMSD of the monomers against the 6LU7. Figure 4 shows the backbone RMSD of the 3CLpro against 6LU7 for each of the monomers. It was observed that the RMSD values for 10 μs open source simulation ranged between 1 and 4 Å (Figure 4(A)). In case of one of the monomers the RMSD ranged around 3.5–4 Å in the last 3 μs, whereas in the intial 6.5 μs it ranged around 1–2 Å. In case of the other monomer, the initial 2 μs had an RMSD within the range of 1–2 Å. The remaining 8 μs had the RMSD values ranging within 2–3 Å. Figure 4(B) represents the backbone RMSD values for the three replicates of 50 ns simulations performed for the monomer 3CLpro system. It was observed that for all the three replicates the RMSD values ranged between 1 and 2 Å throughout the simulations. The different ranges of RMSD suggest that different conformations of the 3CLpro were explored in all these simulations. In order to generate different representatives of these ensembles clustering was performed using two different approaches. The first method employed was RMSD-based clustering. The simulation data of 10.15 µs was clustered based on the all-atom RMSD of all the residues of the 3CLpro system. The reference used was the 6LU7 structure of 3CLpro. The cpptraj module of AMBERTOOLS 17 was used for performing this clustering using the dbscan method (Ester et al., 1996). The RMSD cut-off used here was of 1.7 Å for the in-house as well as the open-source simulation data. A total of three clusters were obtained from the in-house simulation data and nine clusters were obtained from the open-source simulation data. The representatives of these 12 clusters were considered for further docking studies. Figure 5 shows the RMSD values of these cluster representatives against the experimental structure of 3CLpro, i.e. PDB ID: 6LU7. The structures represented in blue were the ones that were obtained from the RMSD based clustering. Ten of the representative structures from the ensemble had the RMSD value below 3 Å. In order, to have more significant conformations from the ensemble a second approach of MSM analysis was performed on the entire simulation data. The collective variable used for the MSM analysis was the backbone torsion angle. Based on this CV, four significant states were obtained for the entire simulation data (Supporting Information SI 1). The RMSD values of these four representative structures obtained from MSM analysis have been given in the Figure 5. The representative structures shown in purple were obtained from the MSM analysis (Figure 5). All the four representative structures had an RMSD of less than 3 Å, whereas three of them showed an RMSD below 2 Å. These 16 structures represented the ensemble covered by the protein throughout the simulations. The varying RMSD values infer that the dynamics of the protein helped in surfacing out conformations that differ from the experimentally derived static conformation of the protein. The flexibility of the protein was captured in these representative structures which further helped in the docking of a few other small molecules. Considering these ensemble structures, helped to explore a wider range of drug molecules that would bind to the target protein, in this case the 3CLpro protein. There are studies where the role of molecular dynamics in exploring the different conformations of the binding site also referred to as cryptic pockets help in computer-aided drug discovery (Kuzmanic et al., 2020). Hence, the identification of different states of 3CLpro through MD simulations helped in visiting different conformations of the binding site. The information on varying conformations of the binding site may also lead to the identification of more significant drug molecules, further increasing the scope of therapeutics through drug repurposing.
Figure 4.

(A) Backbone RMSD of the two monomers against 6LU7 (open source 10 μs simulation data). (B) Backbone RMSD of the monomers against 6LU7 for three replicates of 50 ns simulation data).
Figure 5.

Root Mean Square deviation (RMSD) values against 6LU7 for the 16 ensemble representatives (C1 to C16) of 3CLpro obtained through MD simulations.
Ensemble docking
A total of 16 ensemble representatives of 3CLpro, as explained in the subsection ‘Ensemble Generation’ were selected from the MD simulation data using RMSD-based clustering and MSM analysis. Sixteen independent molecular docking were performed on each of these 16 ensemble representatives for screening each of the two databases separately, using the identical protocol as explained in the ‘Methodology’ section of the article. Hence, a total of 32 independent docking studies were performed for screening the FDA approved and the SWEETLEAD drug database against these 16 representative structures of 3CLpro. The Supporting Information Figure S1 depicts the grid scores obtained from each of the 16 independent docking exercises. In the Supporting Information Figure S1, the X-axis represents the drug molecules from the FDA drug database and the Y-axis represents the grid scores obtained through DOCK 6. The top ranked drug is at position 1 of the X-axis obtained from C1 to C16 independent docking exercises. These top ranked drugs have been shown in Figure 6(A) of the manuscript and also in Table 3. The top ranked drugs from the SWEETLEAD database also have been identified using the similar procedure. Figure 6 explains the name of the top-ranked drugs against these ensemble representatives and their corresponding grid scores obtained from DOCK 6. Around, 3–10% of the drugs from either of database were observed to have a grid score above zero in these 16 ensembles. These top ranked drugs obtained for each of the 16 ensembles has been discussed below. A schematic representation of the 16 independent docking leading to the identification of 16 top ranked drugs from the FDA approved drug database has been given in Supporting Information Figure S2. The Supporting Information Figure S2 depicts the top ranked drug from the FDA database for every ensemble representative. The docking performed considering the C1 ensemble representative as receptor, resulted in Hesperidin as the top ranked drug. Similarly, in every individual docking exercise, the drug that ranked first has been shown in Table 3 and Figure 6(A) of the manuscript. Table 4 and Figure 6(B) show the top ranked drugs obtained through individual docking exercise while screening the SWEETLEAD drug database. The details about these drugs obtained from FDA approved and SWEETLEAD drug database mentioning their earlier purpose and their chemical structure has been described in Tables 3 and 4, respectively.
Figure 6.

The top ranked drugs from the FDA approved (A) and SWEETLEAD (B) drug database obtained through ensemble docking approach.
Table 3.
Top ranked drugs of the FDA approved drug database obtained through 16 independent molecular docking of 3CLpro ensemble representatives of SARS-CoV-2.
| 3CLpro Ensemble representative | Name of the drug (PubChem CID) | Earlier purpose | Structure |
|---|---|---|---|
| RMSD-based clustering | |||
| C1 | Hesperidin (3594) | Bioflavonoid, anti-oxidant, anti-inflammatory |  |
| C2 | Etoposide (36462) | Chemotherapy drug, used in lung cancer too |  |
| C3 | Pranlukast (4887) | Anti-asthamatic, reduces bronchospasm |  |
| C4 | Azelnidipine (65948) | Treats hypertension, calcium channel blocker |  |
| C5 | Epicatechin gallate (107905) | Flavonoid, treated for pre-diabetes |  |
| C6 | Brinzolamide (68844) | Ocular Hypertension | 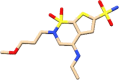 |
| C7 | Ceftiofur (6328657) | Anti-bacterial, veterinary drug | 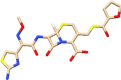 |
| C8 | Artesunate (6917864) | Treats malaria, combination therapy mefloquine | 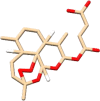 |
| C9 | Ivermectin (6321424) | Treats parasitic infections | 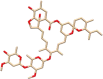 |
| C10 | Peimine (131900) | Anti-inflammatory |  |
| C11 | Empagliflozin (11949646) | Treats Type2-Diabetes | 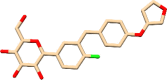 |
| C12 | Agenerase/Amprenavir (65016) | Antiviral, inhibits HIV protease |  |
| Markov state modelling (MSM) Analysis | |||
| C13 | Cefazedone (71736) | Antibacterial |  |
| C14 | Indinavir (5362440) | Antiviral, inhibits HIV protease | 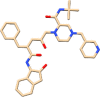 |
| C15 | Ceftin (6321416) | Antibacterial, used against pneumonia | 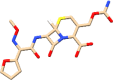 |
| C16 | Ceftizoxime (6533629) | Antibiotic, used against life threating bacterial infections |  |
Table 4.
Top ranked drugs of the SWEETLEAD drug database obtained through 16 independent molecular docking of 3CLpro ensemble representatives of SARS-CoV-2.
| 3CLpro Ensemble representative | Name of the drug (PubChem CID) | Earlier purpose | Structure |
|---|---|---|---|
| RMSD-based clustering | |||
| C1 | Tobramycin (36294) | Antibiotic, antibacterial activity |  |
| C2 | Lanreotide acetate (71349) | Used to treat Acromegaly, inhibits the growth hormone |  |
| C3 | Lenapenem (216262) | carbapenem antibiotic with bactericidal activity, penicillin binding protein |  |
| C4 | Neomycin (8378) | Aminoglycoside antibiotic |  |
| C5 | Riboflavin tetrabutyrate (92140) | One component of the multi-vitamin drugs | 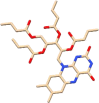 |
| C6 | Sennosides (5199) | Stimulant laxative |  |
| C7 | Gentamicin (3467) | Antibacterial, used against pneumonia | 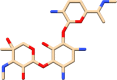 |
| C8 | Terlipressin (72081) | Vasoactive drug, used to manage low blood pressure | 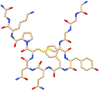 |
| C9 | Ribostamycin (33042) | Aminoglycoside-aminocyclitol antibiotic | 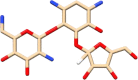 |
| C10 | Tobramycin (36294) | Antibiotic, antibacterial activity | 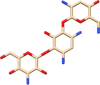 |
| C11 | Neomycin (8378) | Aminoglycoside antibiotic | 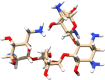 |
| C12 | Neomycin (8378) | Aminoglycoside antibiotic |  |
| Markov State Modelling (MSM) Analysis | |||
| C13 | Lypressin (644076) | Used against diabetes insipidus |  |
| C14 | Amikacin (37768) | Antibiotic, Multi-drug resistant tuberculosis | 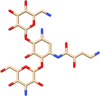 |
| C15 | Vasopressin tannate (8230) | Antidiuretic drug, used against diabetes insipidus | 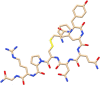 |
| C16 | Netilmicin (441306) | Antibiotic, treatment against severe bacterial infections | 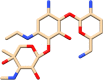 |
Figure 6(A) explains the screening of the FDA approved drug database against the ensemble representative structures. The drug with the lowest value of the grid score was indinavir (PubChem CID: 5362440), which is a known HIV protease inhibitor which was obtained for the C14 ensemble representative. This was followed by ceftin (PubChem CID: 6321416) which a cephalosporin-derivative and is used as an antibiotic to fight bacterial infection (C15 ensemble representative). The third lowest value of the grid score was observed for the drug ivermectin (PubChem CID: 6321424), which is used in treating head lice, and is known to possess anti-parasitic property (C9 ensemble representative). One of the in vitro drug repurposing studies, approves the use of ivermectin as a repurposed drug against COVID-19 (Caly et al., 2020). However, three more drugs that belong to the group of cephalosporin-derivatives viz. ceftiofur (PubChem CID: 6328657), cefazedone (PubChem CID: 71736) and ceftizoxime (PubChem CID: 6533629) were also ranked through ensemble docking. All these drugs are known to possess anti-bacterial property and are used to treat severe bacterial infections. The top-ranked drug obtained through direct docking, ceftazidime, is also a cephalosporin derivative (Figure 3(A)). Amprenavir (PubChem CID: 65016), which is also a known HIV protease inhibitor ranked as the top hit for the C12 ensemble representative. However, the value of the grid score was comparatively higher than the top hit drug molecules for the other ensemble structures of 3CLpro.
Figure 6(B) lists the top-ranked drugs obtained on screening the SWEETLEAD drug database using the ensemble docking approach. Amongst, the 16 representative structures used for docking, three structures (C4, C11 and C12) were observed to show the anti-bacterial drug, Neomycin (PubChem CID: 8378) as the top ranked molecule. The drug with the lowest value of the grid score was neomycin, in comparison to the rest the of the top ranked drugs for each of the 3CLpro ensemble representative. The drug with the second-lowest value of grid score was vasopressin tannate (PubChem CID: 8230) which is known to be used as an anti-diuretic drug to treat diabetes insipidus (C15). The drug with the third-lowest value of grid score was again neomycin for the ensemble representative C11. However, amikacin which is also known for its antibacterial property was the one with fourth-lowest value of grid score (top ranked for C14). Ten of the 16 ensemble structures screened drug molecules possessing anti-bacterial activity viz. tobramycin (PubChem CID: 36294), lenapenem (PubChem CID: 216262), neomycin, gentamicin (PubChem CID: 3467), ribostamycin (PubChem CID: 33042), amikacin (PubChem CID: 37768) and netilmicin (PubChem CID: 441306) as top-ranked. Neomycin and tobramycin appeared as the top ranked for three and two of the 16 representative structures, respectively.
Drug–3CLpro interactions
Interaction energies
The interaction energies between the top-ranked docked ligands and 3CLpro were calculated using the Prodigy-LIG server (Vangone et al., 2019). This interaction energy obtained from Prodigy-LIG server, is calculated based on the number of contacts formed by the atoms of the ligands with the residues of the protein. Lower the value of the interaction energy better would be the binding between the protein and ligand molecule. Figure 7(A) depicts the interaction energies for the top ranked drugs docked drugs obtained on screening the FDA approved drug database. It was observed that the interaction energies for the top-ranked drugs through ensemble docking (yellow) were significantly better from the top five ranked drugs obtained through direct docking. A difference of 3–6 kcal/mole was observed between the drugs obtained through the two docking approaches, the ensemble docking showing better interaction energies. The drug with the best interaction energy was ivermectin, which was also one of the top ranked drugs w.r.t the grid score obtained from DOCK 6. Figure 7(B) depicts the interaction energies for the top hit drugs obtained on screening the SWEETLEAD drug database. All the drugs obtained through direct and ensemble docking approaches were observed to have interaction energies in the range of −4 to −6 kcal/mole.
Figure 7.

Interaction energies obtained for the docked complexes obtained through direct (blue) and ensemble (yellow) docking of FDA approved (A) and SWEETLEAD (B) drug database.
As the interaction energy calculation depends on the number of contacts formed by the ligand with the residues of the protein, it may be inferred that in case of the FDA approved drugs the top-ranked drugs obtained through ensemble docking showed better interactions with the residues of 3CLpro as compared to those obtained through direct docking. Amongst the ensemble docked FDA approved drugs, ivermectin would have formed a greater number of contacts with the residues of 3CLpro as compared to other drugs. In the case of the SWEETLEAD drugs, all the top-ranked drugs from either of the docking approach showed similar values of interaction energies. This may infer that most of these drugs formed similar number of contacts with the residues of 3CLpro.
Crucial residues: direct docking
The active site of the main protease shows the presence of a few polar residues viz. histidine, asparagine, glutamate and glutamine. These residues were observed to be involved in hydrogen bonding and hydrophobic interactions with the drug molecules. The LigPlus and PLIP were used to calculate the various interactions between the drug molecule and the 3CLpro (Laskowski & Swindells, 2011; Salentin et al., 2015). Figure S3 Supporting Information shows the residues of 3CLpro present in the vicinity of the FDA approved ligand molecules and the residues that are involved in hydrogen bonding. The drug ceftazidime which showed the lowest value of grid score was observed to form hydrogen bonds with GLY 143 and HIS 164 (Figure 8). Ceftazidime was also involved in forming hydrophobic interactions with GLU 166 and π-π interactions with HIS 41 (Figure 8). Quetiapine formed hydrogen bonds with GLY 143 and THR 26. It showed hydrophobic interactions with ASP 187 and GLU 189 and π–π interactions with HIS 41. Cabergoline had no hydrogen bonding interactions, however, it showed hydrophobic interactions with PHE 140, GLU 166 and GLN 189. π–π interaction with HIS 41 was observed for cabergoline too. Enoxolone was observed to form hydrogen bonds with GLU 166 and GLU 192. It was also involved in hydrophobic interactions with THR 25, ASN 142, MET 165 and GLU 189. Apremilast was observed to form hydrogen bonding interactions with THR 26, GLY 143, ASN 142 and SER 144 and hydrophobic interactions with ASN 142 and MET 165. It was observed that HIS 41, GLY 143, ASN 142 and GLU 166 were involved in interacting with the three of the drug molecules amongst the top five ranked drugs. However, π-π interactions with HIS 41 were observed in the top three molecules. The region around CYS 145 of the main protease is known to interact with human proteins viz. human deacetylase 2 (HDAC2) tRNA-methyl transferase 1 (TRMT1) (Gordon et al., 2020). Both these proteins are known epigenetic regulators and their nuclear localization is blocked by this viral protease (Gordon et al., 2020). The docking studies performed here revealed that the residues ASN142 and GLY143, neighboring to the CYS145 were known to be involved in interacting with the drug molecules. This may suggest their crucial role in inhibiting the protein–protein interaction between the main protease and the human epigenetic regulatory proteins.
Figure 8.

Ceftazidime forming hydrogen bonds with G143 and H164, π-π and hydrophobic interactions with H41 and E166, respectively.
Figure S4 Supporting Information shows the residues of 3CLpro present in the vicinity of the SWEETLEAD drug molecules and the residues that are involved in hydrogen bonding. The top-ranked drug, dibekacin was observed to form five hydrogen bonds with THR 25, THR 26, SER 46, CYS 145, GLU 166 and GLN 189 (Figure 9). Micronomicin, which ranked second in terms of the grid score was observed to form hydrogen bonds with THR 24, SER 46 and ASN 142. The next drug in the top five ranked drugs was catalposide which showed hydrogen bonding interactions with THR 24, THR 26, GLY 143 and GLU 166. It also showed π- π interactions with HIS 41 and hydrophobic interactions with MET 165 and GLN 189. Dihydro-alpha ergocryptine, which ranked fourth in terms of the grid score was observed to form hydrogen bonds with ASN 142 and GLY 143. It was also involved in hydrophobic interactions with GLU 166. Itopride which ranked last amongst the top five drug molecules did not show any significant interactions with the residues of the protein.
Figure 9.

Dibekacin forming hydrogen bonds with T25, T26, S46, C145, E166 and Q189.
Crucial residues: ensemble docking
Experimental studies performed to elucidate the crystal structure of 3CLpro suggests that the inhibitor binding site of this protein is divided into subsites (Zhang et al., 2020). The S1 subsite consists of PHE 140, ASN 142, GLU 166, HIS 163 and HIS 172. Whereas, the S2 subsite consists of the hydrophobic pocket made by the residues viz. HIS 41, MET 49, TYR 54 and MET 165. The CYS 145 is involved in covalent bond with the inhibitor N3 (Figure 1) (Jin et al., 2020; Zhang et al., 2020). It was observed that most of these residues which play a crucial role in interacting with the inhibitor showed similar results for the drugs obtained through direct and ensemble docking. However, obtaining the same drug as the top-ranked in case of different ensemble representative states with varying interactions suggests conformational variability in the inhibitor binding site of the 3CLpro.
The residues interacting with the top-ranked drugs from the FDA approved and the SWEETLEAD database for all the 16 representative clusters have been shown in Supporting Information Figures S5 and S6, respectively. The ensemble docking of FDA approved drug database had the three drugs indinavir, ceftin and ivermectin as top ranked drugs for three of the 3CLpro representatives. These three had the lowest value of the grid scores in comparison to the other top-ranked drugs from the remaining 13 3CLpro representative. Indinavir, ceftin and ivermectin were observed to show interactions with THR 24, LEU 27, VAL 42, THR 45, ARG 60, LYS 61, ASN 142, GLU 166 and GLN 189 (Figure 10). Figure S5 Supporting Information depicts the hydrogen bonding interactions of all the top-ranked drug molecules of the 16 representative structures. It was observed that apart from the residues mentioned above HIS 41, PHE 140, HIS163 and GLN 192 were also responsible for forming hydrogen bonding interactions with the other top-ranked drug molecules. The ensemble docking of SWEETLEAD drug database had the three drugs neomycin, vasopressin tannate and amikacin as top-ranked in five of the 3CLpro representatives. The grid scores for these three drug molecules had the lowest values in comparison to the remaining top-ranked drugs in the remaining 11 3CLpro representatives. Neomycin was observed to be the top-ranked drug in the case of three ensemble structures and in comparison, to the other top-ranked drugs had the first and third lowest value of the grid score. The conformational variability in the ensemble structures was visible on observing the number of interactions of the drug, neomycin in the three different 3CLpro states that were captured (Figure 11). GLU 166 was involved in the formation of strong hydrogen bonding with the atoms of the neomycin in all the three states. GLU 166 formed 3 (Figure 11(A)), 1 (Figure 11(B)) and 2 (Figure 11(C)) hydrogen bonds in the three representative structures of the ensemble. The other residues involved in hydrogen bonding were THR 24, SER 46, HIS 164, MET 165, PRO 168 and GLN 189. Vasopressin tannate, was the drug with second lowest value of grid score amongst the remaining top-ranked drug molecules. It was observed to form hydrogen bonds with THR 24, THR 25, HIS 41 and SER 46 residues of the 3CLpro. Amikacin, the drug obtained with the fourth-lowest value of grid score formed hydrogen bonds with CYS 145, ASN 142 and MET 165.
Figure 10.

Hydrogen bonding interactions of indinavir (A), ceftin (B) and ivermectin (C) with the residues of 3CLpro.
Figure 11.

Hydrogen bonding of neomycin with residues of 3CLpro captured in three different states of during ensemble generation.
Conclusion
The high throughput docking and ensemble docking studies of 3C-like protease reveal few potential drugs that can be considered for repurposing. The docking against the FDA approved and the SWEETLEAD drug database helped to enlist few antibacterial and antiviral drugs that may be used as candidates for repurposing studies against 3C-like protease. Indinavir, ivermectin, cephalosporin-derivatives, neomycin and amprenavir were few of the drugs which may prove to be effective against the symptoms seen in COVID-19. As, the earlier purpose of indinavir and amprenavir states inhibition of the HIV protease. Similarly, ivermectin, cephalosporin-derivatives and neomycin are used against treating anti-parasitic and anti-bacterial infections especially respiratory tract infections. In support of these findings, these drugs were also observed to show better docking scores in comparison to other drugs. The ensemble docking approach helped to explore the conformational variability of the inhibitor binding site of 3C-like protease. The conformations captured through effective sampling methods like Markov State Modelling analysis reveal a more accessible region for inhibitors to bind to the 3C-like protease. The drugs with anti-viral and anti-bacterial properties that were not ranked at the top by direct docking were identified through ensemble docking. The grid scores for the drugs when docked against the ensemble structures of 3CLpro were observed to be better in comparison to direct docking. Inferring, the conformational flexibility of the 3CLpro to accommodate potential drug molecules. The drug residue interactions also complement the role of crucial residues that were earlier defined by the electron-density studies of 3C-like protease and its inhibitors (Fearon et al., 2020). The ensemble docking approach coupled with a strong sampling technique would help to explore the more accessible conformations of the drug target which would further help in designing a better drug as an inhibitor.
Supplementary Material
Acknowledgements
The authors would like to acknowledge the PARAM supercomputing facility and the Bioinformatics Resources and Applications Facility (BRAF) at Centre for Development of Advanced Computing (C-DAC), Pune for providing the computing infrastructure. The authors would like to acknowledge SR Rajesh Kumar, Saurabh Patil and Akash Khade for their timely administrative support and services.
Funding Statement
The authors would like to acknowledge the National Supercomputing Mission (NSM), Ministry of Electronics and Information Technology (MeitY), Government of India for funding this work.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Aanouz, I., Belhassan, A., El-Khatabi, K., Lakhlifi, T., El-Ldrissi, M., & Bouachrine, M. (2020). Moroccan Medicinal plants as inhibitors against SARS-CoV-2 main protease: Computational investigations. Journal of Biomolecular Structure and Dynamics, 6, 1–9. 10.1080/07391102.2020.1758790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelli, I., Hassani, F., Brikci, B., & Ghalem, S. (2020). S. In silico study the inhibition of angiotensin converting enzyme 2 receptor of COVID-19 by Ammoides verticillata components harvested from Western Algeria. Journal of Biomolecular Structure and Dynamics, 14, 1–14. 10.1080/07391102.2020.1763199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adeoye, A. O., Oso, B. J., Olaoye, I. F., Tijjani, H., & Adebayo, A. I. (2020). Repurposing of chloroquine and some clinically approved antiviral drugs as effective therapeutics to prevent cellular entry and replication of coronavirus. Journal of Biomolecular Structure & Dynamics. 10.1080/07391102.2020.1765876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad, S., Abbasi, H. W., Shahid, S., Gul, S., & Abbasi, S. W. (2020). Molecular docking, simulation and MM-PBSA studies of nigella sativa compounds: A computational quest to identify potential natural antiviral for COVID-19 treatment. Journal of Biomolecular Structure and Dynamics, 1–16. 10.1080/07391102.2020.1775129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Khafaji, K., Al-Duhaidahawi, D., & Tok, T. (2020). Using integrated computational approaches to identify safe and rapid treatment for SARS-CoV-2. Journal of Biomolecular Structure and Dynamics, 15, 1–9. 10.1080/07391102.2020.1764392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen, W. J., Balius, T. E., Mukherjee, S., Brozell, S. R., Moustakas, D. T., Lang, P. T., Case, D. A., Kuntz, I. D., & Rizzo, R. C. (2015). DOCK 6: Impact of new features and current docking performance. Journal of Computational Chemistry, 36(15), 1132–1156. 10.1002/jcc.23905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin, M., & Abbas, G. (2020). Docking study of Chloroquine and Hydroxychloroquine interaction with SARS-CoV-2 spike glycoprotein-An in silico insight into the comparative efficacy of repurposing antiviral drugs. Journal of Biomolecular Structure and Dynamics, 29, 1–11. 10.1080/07391102.2020.1775703 [DOI] [PubMed] [Google Scholar]
- Andersen, K. G., Rambaut, A., Lipkin, W. I., Holmes, E. C., & Garry, R. F. (2020). The proximal origin of SARS-CoV-2. Nature Medicine, 26(4), 450–452. 10.1038/s41591-020-0820-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anwar, F., Altayb, H. N., Al-Abbasi, F. A., Al-Malki, A. L., Kamal, M. A. & Kumar, V. (2020). Antiviral effects of probiotic metabolites on COVID-19. Journal of Biomolecular Structure and Dynamics, 9, 1–10. 10.1080/07391102.2020.1775123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arya, A., & Dwivedi, V. D. (2020). Synergistic effect of vitamin D and remdesivir can fight C OVID-19. Journal of Biomolecular Structure and Dynamics, 9, 1–2. 10.1080/07391102.2020.1773929 [DOI] [PubMed] [Google Scholar]
- Asai, A., Konno, M., Ozaki, M., Otsuka, C., Vecchione, A., Arai, T., Kitagawa, T., Ofusa, K., Yabumoto, M., Hirotsu, T., Taniguchi, M., Eguchi, H., Doki, Y., & Ishii, H. (2020). COVID-19 drug discovery using intensive approaches. International Journal of Molecular Sciences, 21(8), 2839. 10.3390/ijms, 21082839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babadaei, M. M. N., Hasan, A., Bloukh, S. H., Edis, Z., Sharifi, M., Kachooei, E., & Falahati, M. (2020). The expression level of angiotensin-converting enzyme 2 determines the severity of COVID-19: Lung and heart tissue as targets. Journal of Biomolecular Structure and Dynamics, 1–7. 10.1080/07391102.2020.1767211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babadaei, M. M. N., Hasan, A., Vahdani, Y., Bloukh, S. H., Sharifi, M., Kachooei, E., Haghighat, S., & Falahati, M. (2020). Development of remdesivir repositioning as a nucleotide analog against COVID-19 RNA dependent RNA polymerase. Journal of Biomolecular Structure and Dynamics, 1–9. 10.1080/07391102.2020.1767210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron, S. A., Devaux, C., Colson, P., Raoult, D., & Rolain, J. M. (2020). Teicoplanin: An alternative drug for the treatment of COVID-19? International Journal of Antimicrobial Agents, 55(4), 105944. 10.1016/j.ijantimicag.2020.105944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basit, A., Ali, T., & Rehman, S. U. (2020). Truncated human angiotensin converting enzyme 2; a potential inhibitor of SARS-CoV-2 spike glycoprotein and potent COVID-19 therapeutic agent. Journal of Biomolecular Structure and Dynamics, 1–10. 10.1080/07391102.2020.1768150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer, D. C., Tay, A. P., Wilson, L. O. W., Reti, D., Hosking, C., McAuley, A. J., Pharo, E., Todd, S., Stevens, V., Neave, M. J., Tachedjian, M., Drew, T. W., & Vasan, S. S. (2020). Supporting pandemic response using genomics and bioinformatics: A case study on the emergent SARS-CoV-2 outbreak. Transboundary and Emerging Diseases, 67(4), 1453–1462. 10.1111/tbed.13588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beura, S., & Chetti, P. (2020). In-silico strategies for probing chloroquine based inhibitors against SARS-CoV-2. Journal of Biomolecular Structure and Dynamics, 8, 1–13. 10.1080/07391102.2020.1772111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj, V. K., Singh, R., Sharma, J., Rajendran, V., Purohit, R., & Kumar, S. (2020). Identification of bioactive molecules from tea plant as SARS-CoV-2 main protease inhibitors. Journal of Biomolecular Structure and Dynamics, 1–10. 10.1080/07391102.2020.1766572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boopathi, S., Poma, A. B., & Kolandaivel, P. (2020). Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. Journal of Biomolecular Structure and Dynamics, 30, 1–10. 10.1080/07391102.2020.1758788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkotoky, S., & Banerjee, M. (2020). A computational prediction of SARS-CoV-2 structural protein inhibitors from Azadirachta indica (Neem). Journal of Biomolecular Structure and Dynamics, 28, 1–17. 10.1080/07391102.2020.1774419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caly, L., Druce, J. D., Catton, M. G., Jans, D. A., & Wagstaff, K. M. (2020). The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antiviral Research, 178, 104787. 10.1016/j.antiviral.2020.104787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case, D. A., Betz, R. M., Cerutti, D. S, Cheatham, T. E., Darden, T. A., Duke, R. E., Giese, T. J., Gohlke, H., Goetz, A. W., Homeyer, N., Izadi, S., Janowski, P., Kaus, J., Kovalenko, A., Lee, T. S., LeGrand, S., Li, P., Lin, C., Luchko, T., … Kollman, P. A. (2016). AMBER 2016. University of California. [Google Scholar]
- Center for Drug Evaluation and Research (U.S.). (2004). Drugs@FDA. U.S. Food and Drug Administration, Center for Drug and Evaluation Research. [Google Scholar]
- Chandra, A., Gurjar, V., Qamar, I., & Singh, N. (2020). Identification of potential inhibitors of SARS-COV-2 endoribonuclease (EndoU) from FDA approved drugs: A drug repurposing approach to find therapeutics for COVID-19. Journal of Biomolecular Structure and Dynamics, 9, 1–11. 10.1080/07391102.2020.1775127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y. W., Yiu, C. B., & Wong, K. Y. (2020). Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CL pro) structure: Virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Research, 9, 129. 10.12688/f1000research, 22457.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherian, S. S., Agrawa, M., Basu, A., Abraham, P., Gangakhedkar, R. R., & Bhargava, B. (2020). Perspectives for repurposing drugs for the coronavirus disease 2019. Indian Journal of Medical Research, 151(2), 160–171. 10.4103/ijmr.IJMR_585_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodera, J. D., & Noé, F. (2014). Markov state models of biomolecular conformational dynamics . Current Opinion in Structural Biology, 25, 135–144. 10.1016/j.sbi.2014.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodera, J. D., Singhal, N., Pande, V. S., Dill, K. A., & Swope, W. C. (2007). Automatic discovery of metastable states for the construction of Markov models of macromolecular conformational dynamics. The Journal of Chemical Physics, 126(15), 155101–17461665. 10.1063/1.2714538 [DOI] [PubMed] [Google Scholar]
- Choudhury, C. (2020). Fragment tailoring strategy to design novel chemical entities as potential binders of novel corona virus main protease. Journal of Biomolecular Structure and Dynamics, 1, 1–14. 10.1080/07391102.2020.1771424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das, S., Sarmah, S., Lyndem, S., & Roy, S. (2020). A. An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. Journal of Biomolecular Structure and Dynamics, 13, 1–11. 10.1080/07391102.2020.1763201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira, O. V., Rocha, G. B., Paluch, A. S., & Costa, L. T. (2020). Repurposing approved drugs as inhibitors of SARS-CoV-2? S-protein from molecular modeling and virtual screening. Journal of Biomolecular Structure and Dynamics, 2, 1–10. 10.1080/07391102.2020.1772885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky, A. A. (2020. a). Anti-HCV, nucleotide inhibitors, repurposing against COVID-19. Life Sciences, 248, 117477. 10.1016/j.lfs.2020.117477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky, A. A. (2020. b). Natural products may interfere with SARS-CoV-2 attachment to the host cell. Journal of Biomolecular Structure and Dynamics, 5, 1–10. 10.1080/07391102.2020.1761881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky, A. A. (2020. c). Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): A molecular docking study. Life Sciences, 253, 117592. 10.1016/j.lfs.2020.117592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky, A. A. (2020. d). SARS-CoV-2 RNA dependent RNA polymerase (RdRp) targeting: An in silicoperspective. Journal of Biomolecular Structure and Dynamics, 6, 1–9. 10.1080/07391102.2020.1761882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky, A. A., & Azzam, E. B. (2020). Novel guanosine derivatives against MERS CoV polymerase: An in silico perspective. Journal of Biomolecular Structure and Dynamics, 27, 1–9. 10.1080/07391102.2020.1758789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmezayen, A. D., Al-Obaidi, A., Sahin, A. T., & Yelekçi, K. (2020). Drug repurposing for coronavirus (COVID-19): In silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. Journal of Biomolecular Structure and Dynamics, 26, 1–13. 10.1080/07391102.2020.1758791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enmozhi, S. K., Raja, K., Sebastine, I., & Joseph, J. (2020). Andrographolide as a potential inhibitor of SARS-CoV-2 main protease: An in silico approach. Journal of Biomolecular Structure and Dynamics, 5, 1–7. 10.1080/07391102.2020.1760136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ester, M., Kriegel, H., Sander, J., & Xu, X. (1996). A density-based algorithm for discovering clusters in large spatial databases with noise. Proceedings of the Second International Conference on Knowledge Discovery and Data Mining (KDD-96), ACM-Digital Library (pp. 226–231). [Google Scholar]
- Fan, H. H., Wang, L. Q., Liu, W. L., An, X. P., Liu, Z. D., He, X. Q., Song, L. H., & Tong, Y. G. (2020). Repurposing of clinically approved drugs for treatment of coronavirus disease 2019 in a 2019-novel coronavirus (2019-nCoV) related coronavirus model. Chinese Medical Journal, 133(9):1051–1056. 10.1097/CM9.0000000000000797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon, D., Owen, C. D., Douangamath, A., Lukacik, P., Powell, A. J., Strain-Damerell, C. M., Resnick, E., Krojer, T., Gehrtz, P., Wild, C., Aimon, A., Brandao-Neto, J., Carbery, A., Dunnett, L., Skyner, R., Snee, M., London, N., Walsh, M. A., & von Delft, F. (2020). PanDDA analysis group deposition of SARS-CoV-2 mainprotease fragment screen. RCSB-PDB. [Google Scholar]
- Forster, P., Forster, L., Renfrew, C., & Forster, M. (2020). Phylogenetic network analysis of SARS-CoV-2 genomes. Proceedings of the National Academy of Sciences of the United States of America, 8, 202004999. 10.1073/pnas.2004999117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, J., Tian, Z., & Yang, X. (2020). Breakthrough: Chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Bioscience Trends, 14(1), 72–73. 10.5582/bst.2020.01047 [DOI] [PubMed] [Google Scholar]
- Gordon, D. E., Jang, G. M., Bouhaddou, M., Xu, J., Obernier, K., White, K. M., O’Meara, M. J., Rezelj, V. V., Guo, J. Z., Swaney, D. L. and Tummino, T. A. (2020). A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 10.1038/s41586-020-2286-9 [DOI] [PMC free article] [PubMed]
- Gupta, M. K., Vemula, S., Donde, R., Gouda, G., Behera, L., & Vadde, R. (2020. a). In-silico approaches to detect inhibitors of the human severe acute respiratory syndrome coronavirus envelope protein ion channel. Journal of Biomolecular Structure and Dynamics, 15, 1–11. 10.1080/07391102.2020.1751300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, S., Singh, A. K., Kushwaha, P. P., Prajapati, K. S., Shuaib, M., Senapati, S., & Kumar, S. (2020. b). Identification of potential natural inhibitors of SARS-CoV2 main protease by molecular docking and simulation studies. Journal of Biomolecular Structure and Dynamics, 1, 1–19. 10.1080/07391102.2020.1776157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyebi, G. A., Ogunro, O. B., Adegunloye, A. P., Ogunyemi, O. M., & Afolabi, S. O. (2020). Potential inhibitors of coronavirus 3-chymotrypsin-like protease (3CLpro): An in silico screening of alkaloids and terpenoids from African medicinal plants. Journal of Biomolecular Structure and Dynamics, 18, 1–13. 10.1080/07391102.2020.1764868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, D. C., Jr., & Ji, H. F. (2020). A search for medications to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2 spike glycoprotein and 3CL protease. Travel Medicine and Infectious Disease, 35, 101646. 10.1016/j.tmaid.2020.101646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan, A., Paray, B. A., Hussain, A., Qadir, F. A., Attar, F., Aziz, F. M., Sharifi, M., Derakhshankhah, H., Rasti, B., Mehrabi, M., Shahpasand, K., Saboury, A. A., & Falahati, M. (2020). A review on the cleavage priming of the spike protein on coronavirus by angiotensin-converting enzyme-2 and furin. Journal of Biomolecular Structure and Dynamics, 22, 1–9. 10.1080/07391102.2020.1754293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendaus, M. A. (2020). Remdesivir in the treatment of coronavirus disease 2019 (COVID-19): A simplified summary. Journal of Biomolecular Structure and Dynamics, 1–6. 10.1080/07391102.2020.1767691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam, R., Parves, M. R., Paul, A. S., Uddin, N., Rahman, M. S., Mamun, A. A., Hossain, M. N., Ali, M. A., & Halim, M. A. A. (2020). Molecular modeling approach to identify effective antiviral phytochemicals against the main protease of SARS-CoV-2. Journal of Biomolecular Structure and Dynamics, 12, 1–12. 10.1080/07391102.2020.1761883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jani, V., Sonavane, U., & Joshi, R. (2019). Detecting early stage structural changes in wild type, pathogenic and non-pathogenic prion variants using Markov state model. RSC Advances, 9(25), 14567–14579. 10.1039/C9RA01507H [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, Z., Du, X., Xu, Y., Deng, Y., Liu, M., Zhao, Y., Zhang, B., Li, X., Zhang, L., Peng, C., Duan, Y., Yu, J., Wang, L., Yang, K., Liu, F., Jiang, R., Yang, X., You, T., Liu, X., … Yang, H. (2020). Structure of M(pro) from COVID-19 virus and discovery of its inhibitors. Nature, 598, 289–293 10.1038/s41586-020-2223-y [DOI] [PubMed] [Google Scholar]
- Joshi, R. S., Jagdale, S. S., Bansode, S. B., Shankar, S. S., Tellis, M. B., Pandya, V. K., Chugh, A., Giri, A. P., & Kulkarni, M. J. (2020). Discovery of potential multi-target-directed ligands by targeting host-specific SARS-CoV-2 structurally conserved main protease. Journal of Biomolecular Structure and Dynamics, 5, 1–16. 10.1080/07391102.2020.1760137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandeel, M., & Al-Nazawi, M. (2020). Virtual screening and repurposing of FDA approved drugs against COVID-19 main protease. Life Sciences, 251, 117627. 10.1016/j.lfs.2020.117627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, M. T., A. A., Wang, Q., Irfan, M., Khan, A., Zeb, M. T., Zhang, Y. J., Chinnasamy, S., & Wei, D. Q. (2020. a). Marine natural compounds as potents inhibitors against the main protease of SARS-CoV-2-a molecular dynamic study. Journal of Biomolecular Structure and Dynamics, 1, 1–11. 10.1080/07391102.2020.1769733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, R. J., Jha, R. K., Amera, G. M., Jain, M., Singh, E., Pathak, A., Singh, R. P., Muthukumaran, J., & Singh, A. K. (2020. b). Targeting SARS-CoV-2: A systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 2'-O-ribose methyltransferase. Journal of Biomolecular Structure and Dynamics, 1–14. 10.1080/07391102.2020.1753577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, S. A., Ashraf, Z. K., Uddin, S., & Ul-Haq, R. (2020. c). Z. Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. Journal of Biomolecular Structure and Dynamics, 13, 1–10. 10.1080/07391102.2020.1751298 [DOI] [PubMed] [Google Scholar]
- Kim, D., Lee, J. Y., Yang, J. S., Kim, J. W., Kim, V. N., & Chang, H. (2020). The architecture of SARS-CoV-2 transcriptome. Cell, (20), 30402–30406. 10.1016/j.cell.2020.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu, T. S., Koyama, Y. M., Okimoto, N., Morimoto, G., Ohno, Y., & Taiji, M. (2020). COVID-19 related trajectory data of 10 microseconds all atom molecular dynamics simulation of SARS-CoV-2 dimeric main protease. Mendeley Data, v1. 10.17632/vpps4vhryg.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse, R. L. (2020). Therapeutic strategies in an outbreak scenario to treat the novel coronavirus originating in Wuhan, China. F1000Research, 9, 72. 10.12688/f1000research.22211.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, A., Choudhir, G., Shukla, S. K., Sharma, M., Tyagi, P., Bhushan, A., & Rathore, M. (2020. a). Identification of phytochemical inhibitors against main protease of COVID-19 using molecular modeling approaches. Journal of Biomolecular Structure and Dynamics, 4, 1–11. 10.1080/07391102.2020.1772112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, D., Kumari, K., Jayaraj, A., Kumar, V., Kumar, R. V., Dass, S. K., Chandra, R., & Singh, P. (2020. b). Understanding the binding affinity of noscapines with protease of SARS-CoV-2 for COVID-19 using MD simulations at different temperatures. Journal of Biomolecular Structure and Dynamics, 4, 1–14. 10.1080/07391102.2020.1752310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, V., Dhanjal, J. K., Bhargava, P., Kaul, A., Wang, J., Zhang, H., Kaul, S. C., Wadhwa, R., & Sundar, D. (2020. c). Withanone and withaferin-A are predicted to interact with transmembrane protease serine 2 (TMPRSS2) and block entry of SARS-CoV-2 into cells. Journal of Biomolecular Structure and Dynamics, 29, 1–27. 10.1080/07391102.2020.1775704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, V., Dhanjal, J. K., Kaul, S. C., Wadhwa, R., & Sundar, D. (2020. d). Withanone and caffeic acid phenethyl ester are predicted to interact with main protease (Mpro) of SARS-CoV-2 and inhibit its activity. Journal of Biomolecular Structure and Dynamics, 1, 1–13. 10.1080/07391102.2020.1772108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmanic, A., Bowman, G. R., Juarez-Jimenez, J., Michel, J., & Gervasio, F. L. (2020). Investigating cryptic binding sites by molecular dynamics simulations . Accounts of Chemical Research, 53(3), 654–661. 10.1021/acs.accounts.9b00613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski, R. A., & Swindells, M. B. (2011). LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. Journal of Chemical Information and Modeling, 51(10), 2778–2786. 10.1021/ci200227u [DOI] [PubMed] [Google Scholar]
- Li, C., Yang, Y., & Ren, L. (2020). Genetic evolution analysis of 2019 novel coronavirus and coronavirus from other species. Infection, Genetics and Evolution : Journal of Molecular Epidemiology and Evolutionary Genetics in Infectious Diseases, 82, 104285. 10.1016/j.meegid.2020.104285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, G., & De Clercq, E. (2020). Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nature Reviews Drug Discovery, 19(3), 149–150. 10.1038/d41573-020-00016-0 [DOI] [PubMed] [Google Scholar]
- Liu, S., Zheng, Q., & Wang, Z. (2020). Potential covalent drugs targeting the main protease of the SARS-CoV-2 coronavirus. Bioinformatics (Oxford, England), 36(11), 3295–3298. 10.1093/bioinformatics/btaa224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo-Galo, N., Terrazas-López, M., Martínez-Martínez, A., & Díaz-Sánchez, Á. G. (2020). FDA-approved thiol-reacting drugs that potentially bind into the SARS-CoV-2 main protease, essential for viral replication. Journal of Biomolecular Structure and Dynamics, 14, 1–9. 10.1080/07391102.2020.1764393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahanta, S., Chowdhury, P., Gogoi, N., Goswami, N., Borah, D., Kumar, R., Chetia, D., Borah, P., Buragohain, A. K., & Gogoi, B. (2020). Potential anti-viral activity of approved repurposed drug against main protease of SARS-CoV-2: An in silico based approach. Journal of Biomolecular Structure and Dynamics, 25, 1–10. 10.1080/07391102.2020.1768902 [DOI] [PubMed] [Google Scholar]
- Mittal, L., Kumari, A., Srivastava, M., Singh, M., & Asthana, S. (2020). Identification of potential molecules against COVID-19 main protease through structure-guided virtual screening approach. Journal of Biomolecular Structure and Dynamics, 20, 1–19. 10.1080/07391102.2020.1768151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidharan, N., Sakthivel, R., Velmurugan, D., & Gromiha, M. M. (2020). Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 protease against COVID-19. Journal of Biomolecular Structure and Dynamics, 16, 1–6. 10.1080/07391102.2020.1752802 [DOI] [PubMed] [Google Scholar]
- Nadeem, M. S., Zamzami, M. A., Choudhry, H., Murtaza, B. N., Kazmi, I., Ahmad, H., & Shakoori, A. R. (2020). Origin, potential therapeutic targets and treatment for coronavirus disease (COVID-19). Pathogens, 9(4), 307. 10.3390/pathogens9040307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick, P. A., Ortiz, O. F., Poelman, J., Abdulhay, A. Y., & Pande, V. S. (2013). SWEETLEAD: An in silico database of approved drugs, regulated chemicals, and herbal isolates for computer-aided drug discovery. PLoS One, 8(11), e79568. 10.1371/journal.pone.0079568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega, J. T., Serrano, M. L., Pujol, F. H., & Rangel, H. R. (2020). Unrevealing sequence and structural features of novel coronavirus using in silico approaches: The main protease as molecular target. EXCLI Journal, 19, 400–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pant, S., Singh, M., Ravichandiran, V., Murty, U. S. N., & Srivastava, H. K. (2020). Peptide-like and small-molecule inhibitors against Covid-19. Journal of Biomolecular Structure and Dynamics, 6, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawar, A. Y. (2020). Combating Devastating COVID -19 by Drug Repurposing. International Journal of Antimicrobial Agents, 105984. 10.1016/j.ijantimicag.2020.105984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., & Ferrin, T. E. (2004). UCSF Chimera-a visualization system for exploratory research and analysis. Journal of Computational Chemistry, 25(13), 1605–1612. 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- Phadke, M., & Saunik, S. (2020). COVID-19 treatment by repurposing drugs until the vaccine is in sight. Drug Development Research. 10.1002/ddr.21666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prajapat, M., Sarma, P., Shekhar, N., Avti, P., Sinha, S., Kaur, H., Kumar, S., Bhattacharyya, A., Kumar, H., Bansal, S., & Medhi, B. (2020). Drug targets for corona virus: A systematic review. Indian Journal of Pharmacology, 52(1), 56–65. 10.4103/ijp.IJP_115_20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz, J. H., Wu, H., Sarich, M., Keller, B., Senne, M., Held, M., Chodera, J. D., Schütte, C., & Noé, F. (2011). Markov models of molecular kinetics: Generation and validation. The Journal of Chemical Physics, 134(17), 174105. 10.1063/1.3565032 [DOI] [PubMed] [Google Scholar]
- Quimque, M. T. J., Notarte, K. I. R., Fernandez, R. A. T., Mendoza, M. A. O., Liman, R. A. D., Lim, J. A. K., Pilapil, L. A. E., Ong, J. K. H., Pastrana, A. M., Khan, A., Wei, D. Q., & Macabeo, A. (2020). Virtual screening-driven drug discovery of SARS-CoV2 enzyme inhibitors targeting viral attachment, replication, post-translational modification and host immunity evasion infection mechanisms. Journal of Biomolecular Structure and Dynamics, 1, 1–23. 10.1080/07391102.2020.1776639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa, S. G. V., & Santos, W. C. (2020). Clinical trials on drug repositioning for COVID-19 treatment. Revista Panamericana de Salud Pública, 44, e40. 10.26633/RPSP.2020.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salentin, S., Schreiber, S., Haupt, V. J., Adasme, M. F., & Schroeder, M. (2015). PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Research, 43(W1), W443–W447. 10.1093/nar/gkv315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarma, P., Shekhar, N., Prajapat, M., Avti, P., Kaur, H., Kumar, S., Singh, S., Kumar, H., Prakash, A., Dhibar, D. P., & Medhi, B. (2020). In-silico homology assisted identification of inhibitor of RNA binding against 2019-nCoV N-protein (N terminal domain). Journal of Biomolecular Structure and Dynamics, 18, 1–9. 10.1080/07391102.2020.1753580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer, M. K., Trendelkamp-Schroer, B., Paul, F., Pérez-Hernández, G., Hoffmann, M., Plattner, N., Wehmeyer, C., Prinz, J. H., & Noé, F. (2015). PyEMMA 2: A software package for estimation, validation, and analysis of Markov models. Journal of Chemical Theory and Computation, 11(11), 5525–5542. 10.1021/acs.jctc.5b00743 [DOI] [PubMed] [Google Scholar]
- Serafin, M. B., Bottega, A., Foletto, V. S., da Rosa, T. F., Hörner, A., & Hörner, R. (2020). Drug repositioning is an alternative for the treatment of coronavirus COVID-19. International Journal of Antimicrobial Agents, 55(6), 105969. 10.1016/j.ijantimicag.2020.105969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah, B., Modi, P., & Sagar, S. R. (2020). In silico studies on therapeutic agents for COVID-19: Drug repurposing approach. Life Sciences, 252, 117652. 10.1016/j.lfs.2020.117652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha, S. K., Shakya, A., Prasad, S. K., Singh, S., Gurav, N. S., Prasad, R. S., & Gurav, S. S. (2020). An in-silico evaluation of different Saikosaponins for their potency against SARS-CoV-2 using NSP15 and fusion spike glycoprotein as targets. Journal of Biomolecular Structure and Dynamics, 13, 1–12. 10.1080/07391102.2020.1762741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirur, A., De Sancho, D., & Best, R. B. (2016). Markov state models of protein misfolding. The Journal of Chemical Physics, 144(7), 075101. 10.1063/1.4941579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sk, M. F., Roy, R., Jonniya, N. A., Poddar, S., & Kar, P. (2020). Elucidating biophysical basis of binding of inhibitors to SARS-CoV-2 main protease by using molecular dynamics simulations and free energy calculations. Journal of Biomolecular Structure and Dynamics, 1, 1–13. 10.1080/07391102.2020.1768149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, M., & Smith, J. C. (2020). Repurposing therapeutics for COVID-19: Supercomputer-based docking to the SARS-CoV-2 viral spike protein and viral spike protein-human ACE2 interface. ChemRxiv. Preprint. 10.26434/chemrxiv.11871402.v4 [DOI] [Google Scholar]
- Stefanelli, P., Faggioni, G., Lo Presti, A., Fiore, S., Marchi, A., Benedetti, E., Fabiani, C., Anselmo, A., Ciammaruconi, A., Fortunato, A., De Santis, R., Fillo, S., Capobianchi, M. R., Gismondo, M. R., Ciervo, A., Rezza, G., Castrucci, M. R., Lista, F., & On Behalf of ISS COVID-19 Study Group. (2020). Whole genome and phylogenetic analysis of two SARS-CoV-2 strains isolated in Italy in January and February 2020: Additional clues on multiple introductions and further circulation in Europe. Euro Surveill, 25(13). 10.2807/1560-7917.ES.2020.25.13.2000305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu, Y. F., Chien, C. S., Yarmishyn, A. A., Lin, Y. Y., Luo, Y. H., Lin, Y. T., Lai, W. Y., Yang, D. M., Chou, S. J., Yang, Y. P., Wang, M. L., & Chiou, S. H. (2020). A review of SARS-CoV-2 and the ongoing clinical trials. International Journal of Molecular Sciences, 21(7), 2657. 10.3390/ijms, 21072657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umesh, Kundu, D., Selvaraj, C., Singh, S. K., & Dubey, V. K. (2020). Identification of new anti-nCoV drug chemical compounds from Indian spices exploiting SARS-CoV-2 main protease as target. Journal of Biomolecular Structure and Dynamics, 2, 1–9. 10.1080/07391102.2020.1763202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vangone, A., Schaarschmidt, J., Koukos, P., Geng, C., Citro, N., Trellet, M. E., Xue, L. C., & Bonvin, A. (2019). Large-scale prediction of binding affinity in protein-small ligand complexes: The PRODIGY-LIG web server. Bioinformatics (Oxford, England), 35(9), 1585–1587. 10.1093/bioinformatics/bty816 [DOI] [PubMed] [Google Scholar]
- Wahedi, H. M., Ahmad, S., & Abbasi, S. W. (2020). Stilbene-based natural compounds as promising drug candidates against COVID-19. Journal of Biomolecular Structure and Dynamics, 12, 1–10. 10.1080/07391102.2020.1762743 [DOI] [PubMed] [Google Scholar]
- Wang, C., Liu, Z., Chen, Z., Huang, X., Xu, M., He, T., & Zhang, Z. (2020). The establishment of reference sequence for SARS-CoV-2 and variation analysis. Journal of Medical Virology, 92(6), 667–674. 10.1002/jmv.25762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. WHO. https://covid19.who.int/
- Wu, F., Zhao, S., Yu, B., Chen, Y. M., Wang, W., Song, Z. G., Hu, Y., Tao, Z. W., Tian, J. H., Pei, Y. Y., Yuan, M. L., Zhang, Y. L., Dai, F. H., Liu, Y., Wang, Q. M., Zheng, J. J., Xu, L., Holmes, E. C., & Zhang, Y. Z. (2020). Author correction: A new coronavirus associated with human respiratory disease in China. Nature, 580(7803), E7. 10.1038/s41586-020-2202-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, D., Zhang, Z., Jin, L., Chu, F., Mao, Y., Wang, H., Liu, M., Wang, M., Zhang, L., Gao, G. F., & Wang, F. S. (2005). The genetic sequence, origin, and diagnosis of SARS-CoV-2. European Journal of Clinical Microbiology & Infectious Diseases, 24(3), 165–171. 10.1007/s10096-020-03899-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav, P. D., Potdar, V. A., Choudhary, M. L., Nyayanit, D. A., Agrawal, M., Jadhav, S. M., Majumdar, T. D., Shete-Aich, A., Basu, A., Abraham, P., & Cherian, S. S. (2020). Full-genome sequences of the first two SARS-CoV-2 viruses from India. Indian Journal of Medical Research, 151(2), 200-209. 10.4103/ijmr.IJMR_663_20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L., Lin, D., Sun, X., Curth, U., Drosten, C., Sauerhering, L., Becker, S., Rox, K., & Hilgenfeld, R. (2020). Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors. Science, 368(6489), 409–412. 10.1126/science.abb3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.