

Abstract
The sharp spurt in positive cases of novel coronavirus-19 (SARS-CoV-2) worldwide has created a big threat to human. In view to expedite new drug leads for COVID-19, Main Proteases (Mpro) of novel Coronavirus (SARS‐CoV‐2) has emerged as a crucial target for this virus. Nitric oxide (NO) inhibits the replication cycle of SARS-CoV. Inhalation of nitric oxide is used in the treatment of severe acute respiratory syndrome. Herein, we evaluated the phenyl furoxan, a well-known exogenous NO donor to identify the possible potent inhibitors through in silico studies such as molecular docking as per target analysis for candidates bound to substrate binding pocket of SARS-COV-2 Mpro. Molecular dynamics (MD) simulations of most stable docked complexes (Mpro-22 and Mpro-26) helped to confirm the notable conformational stability of these docked complexes under dynamic state. Furthermore, Molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) calculations revealed energetic contributions of key residues of Mpro in binding with potent furoxan derivatives 22, 26. In the present study to validate the molecular docking, MD simulation and MM-PBSA results, crystal structure of Mpro bound to experimentally known inhibitor X77 was used as control and the obtained results are presented herein. We envisaged that spiro-isoquinolino-piperidine-furoxan moieties can be used as effective ligand for SARS-CoV-2 Mpro inhibition due to the presence of key isoquinolino-piperidine skeleton with additional NO effect.
Communicated by Ramaswamy H. Sarma
Keywords: COVID-19, SARS-CoV-2 Mpro inhibition, nitric oxide, furoxan, in silico analysis
Introduction
Novel Coronavirus (SARS-CoV-2) causing COVID-19 disease was originated from Hubei province of China, and has been exponentially increasing in whole world with United States of America, China, Europe, India and Middle East countries becoming the greatest concern. It is one of three Coronaviruses which have been associated with pneumonia, including severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle-East respiratory syndrome coronavirus (MERS-CoV). The World Health Organization announced that the outbreak of this deadly and fast-spreading infectious pneumonia constitutes a global health emergency (WHO, 2019-nCoV Situation Report, 2020). As on 22nd June 2020, the total numbers of cases around the world were recorded to be 9,072,803 with more than 471,178 deaths (worldometers.info/coronavirus).
WHO has been declared COVID-19 as a global pandemic disease but currently there is no effective treatment available and we only depend on preventive and supportive therapies (Cheng, 2019). Researchers around the globe are desperately working to discover either effective vaccines or antiviral drugs, however these experimental methods for drug discovery are expensive and time-consuming. In silico based screening methods offer easy way, fast and low-cost techniques for novel testable hypotheses to discover potential lead drug candidate (Cheng, 2019). There are many clinical trials in progress by pharmaceutical companies to afford the potential agents but very limited data with respect to in vitro and in vivo activities of the lead drugs is available in literatures which are in clinical studies for treatment of COVID-19 (Lu, 2020).
Nitric oxide (NO) is involved in a broad range of processes and act as an important signaling molecule. Thus, the designing of NO-donors/hybrid molecules with NO releasing group may be beneficial for a variety of disorders, such as: cardiovascular, inflammatory, bacterial, fungal, parasitic, ocular diseases and cancer, arthritis, asthma, cerebral ischemia, Parkinson’s disease, neurodegenerative diseases, seizures and viral (Serafim et al., 2012). The well know NO donor, furoxan is 1,2,5-oxadiazole-2-oxide that could release NO in the presence of thiols as well as by enzymatic action in tissue with complex process that probably involves generation more than one redox form of NO (Gasco et al., 2004).
Nitric oxide (NO) is an antiviral effector of the immune system and inhibits the replication of a variety of viruses (Saura et al., 1999). NO can inhibit the replication of viruses that encode cysteine proteases, such as members of the Picornavirus family and the Coronavirus family (Mannick, 1995; Reiss & Komatsu, 1998). Endogenously generated NO can inhibit the replication cycle of SARS-CoV and hence exhibit the desired antiviral effect (Åkerström et al., 2009). NO inhibits the replication of SARS-CoV by dual mechanisms. First, NO or its derivatives cause a reduction in the palmitoylation of nascently expressed spike (S) protein which affects the fusion between the S protein and its cognate receptor i.e. angiotensin converting enzyme 2 (ACE-2). Second, NO or its derivatives cause a reduction in viral RNA production in the early steps of viral replication, and this could possibly be due to an effect on one or both of the cysteine proteases encoded in Orf1a of SARS-CoV (Åkerström et al., 2009).
In-vitro studies have identified significant inhibition activity of NO against SARS-coronavirus infection by S-Nitroso-N-acetylpenicillamine, a nitric oxide donor (Keyaerts et al., 2004). In patients with severe acute respiratory syndrome (SARS), inhalation of nitric oxide (NO) has improved arterial oxygenation and enabled the reduction of inspired oxygen therapy and airway pressure (Chen et al., 2004). Currently, clinical trial testing of inducible nitric oxide (iNO) is underway in COVID-19 infected patients complicated with Acute respiratory distress syndrome (ARDS) [NCT04306393 and NCT04305457] (clinicaltrials.gov).
Theophylline derivative and pyrimidone derivative were identified as inhibitor of RNA binding agent against 2019-nCoV N protein (N terminal domain) by in-silico studies (Sarma et al., 2020). Many natural compounds were studied for their properties to block the activity of the angiotensin converting enzyme 2 (ACE-2) as a receptor for SARS-CoV-2 and as a potential therapeutic target of the COVID-19 virus using in silico study (Abdelli et al., 2020). The extensive research is required by using computer simulations such as (A) Exploration of the energetic binding affinity of SARS-CoV-2 Mpro with new inhibitors based on free energy calculations. (B) Investigation of the structural properties, flexibility, conformational changes of the SARS-CoV-2 Mpro, and study interaction pattern between virus and membrane, virus and inhibitor. C) Monitoring of the thermodynamics (virus-water interaction) properties of SARS-CoV-2 Mpro in the presence/absence of the antiviral inhibitor (Boopathi et al., 2020). Thus computational study could be an effective and swift approach on discovering novel uses for existing medications or approved antiviral drugs to inhibit SARS-CoV-2 Mpro (Al-Khafaji et al., 2020; Khan et al., 2020a, 2020b; Lobo-Galo et al., 2020; Muralidharan et al., 2020; Pant et al., 2020) or SARS-CoV-2 RdRp (Elfiky, 2020a). The researchers have also studied the potential application of natural origin phytochemicals and bioactive derivatives with an anti-viral properties as inhibitors of SARS-CoV-2 Mpro using molecular docking (Aanouz et al., 2020; Das et al., 2020; Elfiky, 2020b; Gyebi et al., 2020; Islam et al., 2020; Joshi et al., 2020; Sinha et al., 2020). The advent of high resolution experimental structure of the target, the predictions of virtual screening studies and binding energy calculations are generally more accurate. The high resolution experimental structure of the target, the main protease of SARS-CoV-2 (Jin et al., 2020; Zhang et al., 2020), was utilized in the current study as the target for molecular docking study.
Keeping in view the importance of early screening of NO donor hybrids for effective anti-viral attributes, the present study focus on evaluation of phenyl furoxan as potential drug candidates by using molecular docking, MD simulation and MM-PBSA approach. The predictions of this study will provide information that can be utilized further for development of efficient drugs by testing it in vitro, in vivo and through clinical trials to control SARS-CoV-2.
Experimental
Molecular docking
Preparation and selection of target
The crystal structure of the SARS-CoV-2 Mpro bound to potent broad-spectrum non-covalent inhibitor X77 (PDB ID: 6W63) was selected for the docking analysis (www.rcsb.org; www.pdb.org). The protein structure was selected on the basis of resolution and method of determination. The selected protein structure was analyzed for the Ramachandran plot using V life MDS 4.6 (www.vlifesciences.com). Selected protein structure of SARS-CoV-2 Mpro was refined for removal of native ligand structure X77 keeping its native conformation intact (Pettersen et al., 2004). The refined protein structure was utilized further for docking analysis.
Preparation of ligand structures
Molecular structures of the selected furoxan derivatives was developed using 2D molecular builder in V life MDS and further converted in to 3D geometry and optimized via application of merck molecular force field (MMFF) to get optimized ligand structures (vlifesciences.com). These optimized ligand structures were utilized for docking analysis.
Redocking analysis
The redocking protocol is utilized for the validation of the docking protocol. The redocking was carried out via docking of co crystallized ligand X77 in SARS-CoV-2 Mpro. The RMSD was determined between docked ligand X77 and co crystallized ligand X77. The RMSD value less than 1.0 Å was considered to be satisfactory and which will ascertain that the binding mode of the selected furoxan derivative will be similar to that of co crystallized ligand X77.
Docking analysis
Docking simulations was performed to predict the possible binding mode of the selected furoxan derivatives. The Grip based docking analysis followed by redocking using AutoDock Vina (Trott & Olson, 2010) which uses Lamarckian Genetic Algorithm (LGA) was performed keeping ligand structures (Table 1) in the flexible conformations. The rotational angle between the two conformations was kept at 10° and total number of rotation to 30 in grip based docking. The best docking pose was selected on the basis of the docking score, binding energy and types of interactions.
Table 1.
Docking result of selected NO donor furoxan.
| Sr. No | Structure & Name | Binding Affinity (kcal/mol) | Docking Score | Interactions |
|||
|---|---|---|---|---|---|---|---|
| H bond | Aromatic | Charge | Hydrophobic | ||||
| 1 |
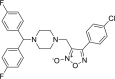 3-({4-[bis(4-fluorophenyl)methyl] piperazin-1- yl}methyl)-4-(4-chloro phenyl)-1,2,5-oxadiazol-2-ium-2-olate 3-({4-[bis(4-fluorophenyl)methyl] piperazin-1- yl}methyl)-4-(4-chloro phenyl)-1,2,5-oxadiazol-2-ium-2-olate |
−7.1 | −58.33 | HIS41 | GLU166 | LEU141 ASN142 MET165 GLU166 | |
| 2 |
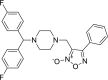 3-({4-[bis(4-fluorophenyl)methyl] piperazin-1- yl}methyl)-4-phenyl-1,2,5-oxadiazol-2-ium-2-olate 3-({4-[bis(4-fluorophenyl)methyl] piperazin-1- yl}methyl)-4-phenyl-1,2,5-oxadiazol-2-ium-2-olate |
−6.7 | −66.94 | SER144 | HIS41 | HIS41 | ASN142 MET165 |
| 3 |
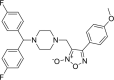 3-({4-[bis(4-fluorophenyl)methyl] piperazin-1-yl}methyl)-4- (4-methoxyphenyl)-1,2,5-oxadiazol-2-ium-2-olate 3-({4-[bis(4-fluorophenyl)methyl] piperazin-1-yl}methyl)-4- (4-methoxyphenyl)-1,2,5-oxadiazol-2-ium-2-olate |
−8.1 | −67.36 | HIS41 | GLU166 | ASN142 MET165 GLU166 | |
| 4 |
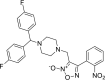 3-({4-[bis(4-fluorophenyl)methyl] piperazin-1-yl}methyl)-4- (2-nitro phenyl)-1,2,5-oxadiazol-2-ium-2-olate 3-({4-[bis(4-fluorophenyl)methyl] piperazin-1-yl}methyl)-4- (2-nitro phenyl)-1,2,5-oxadiazol-2-ium-2-olate |
−8.4 | −46.14 | GLN189 | MET49 GLN189 | ||
| 5 |
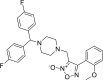 3-({4-[bis(4-fluorophenyl)methyl] piperazin-1-yl}methyl)-4- (2-methyl phenyl)-1,2,5-oxadiazol-2-ium-2-olate 3-({4-[bis(4-fluorophenyl)methyl] piperazin-1-yl}methyl)-4- (2-methyl phenyl)-1,2,5-oxadiazol-2-ium-2-olate |
−7.6 | −67.09 | ASN142 | HIS41 | GLU166 | ASN142 MET165 GLU166 |
| 6 |
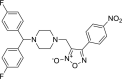 3-({4-[bis(4-fluorophenyl)methyl] piperazin-1-yl}methyl)-4- (4-nitro phenyl)-1,2,5-oxadiazol-2-ium-2-olate amine 3-({4-[bis(4-fluorophenyl)methyl] piperazin-1-yl}methyl)-4- (4-nitro phenyl)-1,2,5-oxadiazol-2-ium-2-olate amine |
−7.9 | −63.99 | HIS41 | MET49 ASN142 GLU166 | ||
| 7 |
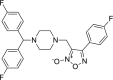 3-({4-[bis(4-fluorophenyl)methyl] piperazin-1-yl}methyl)-4- (4-fluoro phenyl)-1,2,5-oxadiazol-2-ium-2-olate 3-({4-[bis(4-fluorophenyl)methyl] piperazin-1-yl}methyl)-4- (4-fluoro phenyl)-1,2,5-oxadiazol-2-ium-2-olate |
−7.2 | −70.55 | SER144 | HIS41 HIS163 | MET165 GLU166 LEU167 PRO168 | |
| 8 |
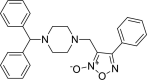 3-{[4-(diphenylmethyl)piperazin-1-yl] methyl}-4-phenyl-1,2,5-oxa diazol-2-ium-2-olate 3-{[4-(diphenylmethyl)piperazin-1-yl] methyl}-4-phenyl-1,2,5-oxa diazol-2-ium-2-olate |
−7.5 | −76.22 | HIS41 | GLU166 | MET165 GLU166 | |
| 9 |
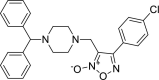 3-{[4-(diphenylmethyl)piperazin-1-yl]methyl}- 4-(4-chlorophenyl)-1,2,5 -oxadiazol-2-ium-2-olate 3-{[4-(diphenylmethyl)piperazin-1-yl]methyl}- 4-(4-chlorophenyl)-1,2,5 -oxadiazol-2-ium-2-olate |
−8.5 | −66.26 | CYS145 | HIS41 HIS163 | GLU166 | LEU141 ASN142 GLU166 |
| 10 |
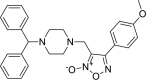 3-{[4-(diphenylmethyl)piperazin-1-yl]methyl}- 4-(4-methoxyphenyl)-1,2,5-oxadiazol-2-ium-2-olate 3-{[4-(diphenylmethyl)piperazin-1-yl]methyl}- 4-(4-methoxyphenyl)-1,2,5-oxadiazol-2-ium-2-olate |
−7.1 | −70.45 | GLU166 | HIS41 | HIS41 | HIS41 ASN142 HIS164 MET165 GLU166 |
| 11 |
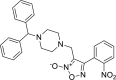 3-{[4-(diphenylmethyl)piperazin-1-yl]methyl}- 4-(2-nitrophenyl)-1,2,5-oxadiazol-2-ium-2-olate 3-{[4-(diphenylmethyl)piperazin-1-yl]methyl}- 4-(2-nitrophenyl)-1,2,5-oxadiazol-2-ium-2-olate |
−7.7 | −64.58 | GLU166 CYS145 | GLU166 | LEU141 ASN142 GLU166 | |
| 12 |
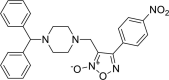 3-{[4-(diphenylmethyl)piperazin-1-yl]methyl}- 4-(4-nitrophenyl)-1,2,5-oxadiazol-2-ium-2-olate 3-{[4-(diphenylmethyl)piperazin-1-yl]methyl}- 4-(4-nitrophenyl)-1,2,5-oxadiazol-2-ium-2-olate |
−7.9 | −69.80 | CYS145 | HIS163 | GLU166 | LEU141 ASN142 GLU166 |
| 13 |
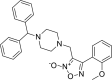 3-{[4-(diphenylmethyl)piperazin-1-yl]methyl}- 4-(2-methoxyphenyl)-1,2,5-oxadiazol-2-ium-2-olate 3-{[4-(diphenylmethyl)piperazin-1-yl]methyl}- 4-(2-methoxyphenyl)-1,2,5-oxadiazol-2-ium-2-olate |
−8.4 | −47.40 | HIS41 | HIS41 | THR25 LEU27 HIS41 VAL42 CYS44 | |
| 14 |
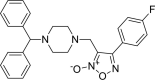 3-{[4-(diphenylmethyl)piperazin-1-yl]methyl}- 4-(4-fluorophenyl)-1,2,5-oxadiazol-2-ium-2-olate 3-{[4-(diphenylmethyl)piperazin-1-yl]methyl}- 4-(4-fluorophenyl)-1,2,5-oxadiazol-2-ium-2-olate |
−8.4 | −57.37 | CYS145 | HIS41 HIS163 | GLU166 | ASN142 MET165 GLU166 |
| 15 |
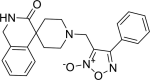 3-({3-oxo-2,3-dihydro-1H-spiro [isoquinoline-4,4′-piperidine]-1′-yl} methyl)-4-phenyl-1,2,5-oxadiazol-2-ium-2-olate 3-({3-oxo-2,3-dihydro-1H-spiro [isoquinoline-4,4′-piperidine]-1′-yl} methyl)-4-phenyl-1,2,5-oxadiazol-2-ium-2-olate |
−8.0 | −40.26 | GLU166 | HIS41 | GLU166 PRO168 | |
| 16 |
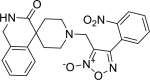 4-(2-nitrophenyl)-3-({3-oxo-2,3-di hydro-1H- spiro[isoquinoline-4,4′-piperidine]-1′-yl} methyl)-1,2,5-oxadiazol-2-ium-2-olate 4-(2-nitrophenyl)-3-({3-oxo-2,3-di hydro-1H- spiro[isoquinoline-4,4′-piperidine]-1′-yl} methyl)-1,2,5-oxadiazol-2-ium-2-olate |
−7.5 | −57.77 | SER144 | HIS41 | MET165 GLU166 | |
| 17 |
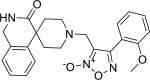 4-(2-methoxyphenyl)-3-({3-oxo-2,3-dihydro-1H- spiro[isoquinoline-4,4′-piperidine]-1′-yl}methyl)-1, 2,5-oxadiazol-2-ium-2-olate 4-(2-methoxyphenyl)-3-({3-oxo-2,3-dihydro-1H- spiro[isoquinoline-4,4′-piperidine]-1′-yl}methyl)-1, 2,5-oxadiazol-2-ium-2-olate |
−7.6 | −75.44 | GLY143 CYS145 GLN189 | HIS41 | MET49 ASN142 CYC145 HIS165 ARG188 GLN189 | |
| 18 |
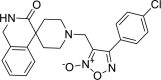 4-(2-methoxyphenyl)-3-({3-oxo-2,3-dihydro-1H- spiro[isoquinoline-4,4′-piperidine]-1′-yl}methyl)-1, 2,5-oxadiazol-2-ium-2-olate 4-(2-methoxyphenyl)-3-({3-oxo-2,3-dihydro-1H- spiro[isoquinoline-4,4′-piperidine]-1′-yl}methyl)-1, 2,5-oxadiazol-2-ium-2-olate |
−8.3 | −62.94 | HIS41 | MET49 MET165 GLU166 ARG188 GLN189 | ||
| 19 |
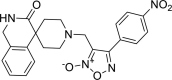 4-(4-nitrophenyl)-3-({3-oxo-2,3-dihydro-1H-spiro [isoquinoline-4,4′-piperidine]-1′-yl}methyl)-1,2, 5-oxadiazol-2-ium-2-olate 4-(4-nitrophenyl)-3-({3-oxo-2,3-dihydro-1H-spiro [isoquinoline-4,4′-piperidine]-1′-yl}methyl)-1,2, 5-oxadiazol-2-ium-2-olate |
−8.8 | −43.83 | GLN192 | HIS41 | MET49 | |
| 20 |
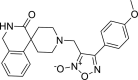 4-(4-methoxyphenyl)-3-({3-oxo-2,3-dihydro- 1H-spiro[isoquinoline-4,4′-piperidine]-1′-yl}methyl)-1, 2,5-oxadiazol-2-ium-2-olate 4-(4-methoxyphenyl)-3-({3-oxo-2,3-dihydro- 1H-spiro[isoquinoline-4,4′-piperidine]-1′-yl}methyl)-1, 2,5-oxadiazol-2-ium-2-olate |
−8.4 | −88.64 | GLY143, SER144 | HIS41 | HIS41, ASN142, MET165, GLU166, PRO168. ASP187, ARG188 | |
| 21 |
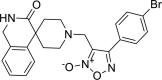 4-(4-bromophenyl)-3-({3-oxo-2,3-dihydro-1H- spiro[isoquinoline-4,4′-piperidine]-1′-yl}methyl)-1, 2,5-oxadiazol-2-ium-2-olate 4-(4-bromophenyl)-3-({3-oxo-2,3-dihydro-1H- spiro[isoquinoline-4,4′-piperidine]-1′-yl}methyl)-1, 2,5-oxadiazol-2-ium-2-olate |
−8.5 | −66.78 | GLN189 | HIS41 | CYS44 MET149 MET165 GLU166 ARG188 GLN189 | |
| 22 |
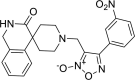 4-(3-nitrophenyl)-3-({3-oxo-2,3-di hydro-1H- spiro[isoquinoline-4,4′-piperidine]-1′-yl}methyl)-1, 2,5-oxadiazol-2-ium-2-olate 4-(3-nitrophenyl)-3-({3-oxo-2,3-di hydro-1H- spiro[isoquinoline-4,4′-piperidine]-1′-yl}methyl)-1, 2,5-oxadiazol-2-ium-2-olate |
−9.6 | −90.93 | CYS145 SER144 | HIS41 HIS163 | HIS41, CYS44, MET49, ASN142, GLY143, CYS145 MET165 GLU166 | |
| 23 |
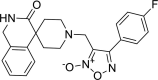 4-(4-flurophenyl)-3-({3-oxo-2,3-dihydro- 1H-spiro[isoquinoline-4,4′-piperidine]-1′-yl}methyl)-1, 2,5-oxadiazol-2-ium-2-olate 4-(4-flurophenyl)-3-({3-oxo-2,3-dihydro- 1H-spiro[isoquinoline-4,4′-piperidine]-1′-yl}methyl)-1, 2,5-oxadiazol-2-ium-2-olate |
−8.3 | −89.44 | GLN189, GLY143. | HIS41 HIS163 | MET49, ASN142, MET165, ARG188 GLN189 | |
| 24 |
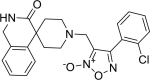 4-(2-chlorophenyl)-3-({3-oxo-2,3-dihydro- 1H-spiro[isoquinoline-4,4′-piperidine]-1′-yl} methyl)-1,2,5-oxadiazol-2-ium-2-olate 4-(2-chlorophenyl)-3-({3-oxo-2,3-dihydro- 1H-spiro[isoquinoline-4,4′-piperidine]-1′-yl} methyl)-1,2,5-oxadiazol-2-ium-2-olate |
−8.1 | −84.66 | SER144 | HIS163 | CYS44, MET49, ASN142, CYS145, MET165. | |
| 25 |
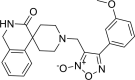 4-(3-methoxyphenyl)-3-({3-oxo-2,3-dihydro-1H- spiro[isoquinoline-4,4′-piperidine]-1′-yl} methyl)-1,2,5-oxadiazol-2-ium-2-olate 4-(3-methoxyphenyl)-3-({3-oxo-2,3-dihydro-1H- spiro[isoquinoline-4,4′-piperidine]-1′-yl} methyl)-1,2,5-oxadiazol-2-ium-2-olate |
−7.5 | −80.77 | GLY143 | HIS41 HIS163 | LEU141 ASN142 MET165 GLU166 ASP187 ARG188 | |
| 26 |
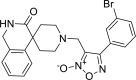 4-(3-bromophenyl)-3-({3-oxo-2,3-dihydro- 1H-spiro[isoquinoline-4,4′-piperidine]-1′-yl} methyl)-1,2,5-oxadiazol-2-ium-2-olate 4-(3-bromophenyl)-3-({3-oxo-2,3-dihydro- 1H-spiro[isoquinoline-4,4′-piperidine]-1′-yl} methyl)-1,2,5-oxadiazol-2-ium-2-olate |
−9.8 | −90.91 | CYS145 SER144. | HIS41 HIS163. | HIS41, CYS44, MET49, ASN142, GLY143, CYS145 MET165 GLU166 | |
| 27 |
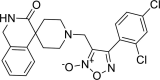 4-(2,4-dichlorophenyl)-3-({3-oxo-2,3-dihydro-1H- spiro[isoquinoline-4,4′-piperidine]-1′-yl}methyl)-1, 2,5-oxadiazol-2-ium-2-olate 4-(2,4-dichlorophenyl)-3-({3-oxo-2,3-dihydro-1H- spiro[isoquinoline-4,4′-piperidine]-1′-yl}methyl)-1, 2,5-oxadiazol-2-ium-2-olate |
−8.0 | −61.69 | HIS41 | MST19 MET165 GLU166 ARG188 GLN189 | ||
| 28 |
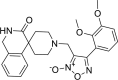 4-(2,3-dimethoxyphenyl)-3-({3-oxo-2,3-dihydro- 1H-spiro[isoquinoline-4,4′-piperidine]-1′-yl}methyl)-1, 2,5-oxadiazol-2-ium-2-olate 4-(2,3-dimethoxyphenyl)-3-({3-oxo-2,3-dihydro- 1H-spiro[isoquinoline-4,4′-piperidine]-1′-yl}methyl)-1, 2,5-oxadiazol-2-ium-2-olate |
−8.0 | −78.90 | GLY143 | HIS41 | HIS41 CYS44 MET49 ASN142 MET165 GLU166 | |
Molecular dynamics (MD) simulation study
The MD simulations study of Mpro-X77 (Control) and most stable docked complexes obtained as per binding energy and docking scores i.e. Mpro-22 and Mpro-26 was performed by using GROMACS 5.0.5 (www.gromacs.org) program (Abraham et al., 2015) on Linux operating system to examine the structural behavior in terms of stability of Mpro-22 and Mpro-26 complexes in comparison to control under dynamic state. The receptor and ligand molecules from control and docked complexes were separated, and their topology files were built separately. The topology file of receptor Mpro was built using optimized potentials for liquid simulations of all atoms (OPLS-AA) force field (Kaminski et al., 2001). Whereas, topology files of ligand X77 (control), 22 and 26 were generated using an automated online PRODRG server (Van Aalten et al., 1996). For dynamics study the simulation parameters were kept uniform and constant for all three complexes. Initially the solvation of all the complexes was carried out in the system of a cubic box with a size of 1.0 nm by using SPC (simple point-charge) water molecules to provide an aqueous environment. The systems were then neutralized by adding three Na+ ions. The periodic boundary conditions (PBC) were applied in all directions. Energy minimization of the solvated complexes was performed with the steepest descent (SD) method for 50,000 steps at 300 K to remove bad contacts until a tolerance of 1000 kJ/mol was achieved. The linear constraint solver (LINCS) algorithm was used to constrain all bond lengths (Hess et al., 1997). A cutoff of 1.4 nm for the van der Waals interaction and 1.2 nm for electrostatic interactions were used for the simulation of all three complexes. The energy minimization of complexes was then followed by equilibration in two phases. In the first phase of equilibration, an NVT (constant number of particles, volume, and temperature) ensemble with a constant temperature of 300 K was used with a coupling constant of 0.1 picosecond for 500 picosecond, whereas in second phase an NPT (constant number of particles, pressure, and temperature) ensemble with a constant pressure of 1 bar was used with a coupling constant of 2 picosecond for 1 ns equilibration period. For both ensembles of equilibration, the coupling scheme of V-rescale was used and Particle Mesh Ewald (PME) algorithm (Essmann et al., 1995) was used to calculate long-range electrostatic interactions. After equilibration, the production MD run was performed for 100 ns to carry out structural analysis on all complexes. All resulting trajectories obtained from 100 ns MD simulations were analyzed using GROMACS utilities (Abraham et al., 2015). The root mean square deviation (RMSD) was calculated over the 100 ns production simulation for all back-bone atoms of each complex. Similarly, root mean square fluctuations (RMSF), radius of gyration (Rg) relative to the initial structure and H bonds were also calculated for both the complexes and control to assess the stability under dynamic state. MD simulations for all complexes were performed in replicates to check the consistency and reproducibility of the results.
Binding free energy calculation by MM-PBSA
The binding free-energy (ΔGbind) calculations for trajectories obtained from 100 ns MD simulation of complexes viz; Control, Mpro-22 and Mpro-26 were performed using g_mmpbsa tool which implements the Molecular Mechanics Poisson-Boltzmann Surface Area (MM-PBSA) to estimate interaction free energies (Kumari et al., 2014). g_mmpbsa tool represents the ΔGbind of receptor with ligand (inhibitor) in the solvent by using following equations:
where, Gcomplex corresponds to total free energy of the complex, Greceptor and Gligand are the total energies of the only receptor and ligand respectively.
where, ΔGMM indicates the average molecular mechanics potential energy in vacuum and ΔGsolv represents the free energy of solvation.
whereas, ΔGvdw corresponds to Van der Waals energy and ΔGelec represents electrostatic energy.
To calculate binding free energies, the VDW energy, electrostatic energy, molecular mechanics energy, polar solvation energy, and solvent-accessible surface area (SASA) energy were calculated for the trajectories obtained from 100 ns MD simulation of all three complexes. The contribution of each residue in binding free energy was calculated by MmPbSaDecomp.py python script (Kumari et al., 2014). This has been helpful in determining the crucial residues and their thermodynamic state involved in interaction with the SARS-CoV-2 Mpro – inhibitor complexes. The binding energy calculated using g_mmpbsa shows an apparent correlation of 0.80-0.85 with the experimental binding free energy (Kumari et al., 2014).
Results and discussion
Multiple small molecules have been reported to exhibit potent SARS-CoV Mpro inhibition activity (Ghosh et al., 2020). Moreover, Andrographolide (Enmozhi et al., 2020) and resveratrol (Wahedi et al., 2020) were found to be potential inhibitors of SARS-CoV-2 main protease. The 28 different nitric oxide donor furoxan derivatives coupled with benzhydrylpiperazine and spiro-isoquinolino-piperidine from our previous research work (Prabhuling et al., 2020; Pudukulatham et al., 2016) were selected for the current in silico analysis.
Development of small molecules with selective action on the Mpro of the COVID-19 is challenging task as protease inhibition is often correlated with the peptide like structures. The development of imidazole carboxamide derivatives as potent SARS-CoV-2 Mpro inhibitors open the avenue for discovery of small peptide or the peptide like structures as an attractive approach for investigation of potent inhibitors against SARS-CoV-2 Mpro. As selected furoxan derivatives have peptide like structures and due to their structural resemblance with reported imidazole carboxamide (X77) they are virtually analyzed against SARS-CoV-2 Mpro to explore their binding potential.
Molecular docking
The results of the predicted pharmacokinetics properties of the analyzed furoxan derivatives are displayed in Figures 1–7 and Table 1. The molecular docking revealed that the spiro-isoquinolino-piperidine class of molecules have more strong binding potentials than benzhydrylpiperazine class of molecules. The docking analysis indicated that the spiro-isoquinolino-piperidine ring is responsible for the anchoring of the molecule in the binding pocket in such a way that other parts of molecule will be adopting the perpendicular orientation in the bind pocket which is responsible for the key interactions in the binding pocket of SARS-CoV-2 Mpro. In case of the benzhydrylpiperazine class of molecules the planner ring orientation of the piperazine pushing interactive phenyl ring in the lower plane and two phenyl ring in the co planner conformation which might be responsible for steric repulsion and subsequently misfitting in the binding pocket of SARS-CoV-2 Mpro.
Figure 1.

Ramachandran plot of SARS-CoV-2 Mpro (PDB ID: 6W63).
Figure 2.

Binding pose of co-crystallized ligand X77 and 22 with crystal structure of SARS-CoV-2 Mpro (PDB ID: 6W63) (UCSF chimera).
Figure 3.

Binding pose for 22 with crystal structure of SARS-CoV-2 Mpro (PDB ID: 6W63) (Generated using Biopredicta Module of Vlife MDS 4.6).
Figure 4.

Binding pose for 26 with crystal structure of SARS-CoV-2 Mpro (PDB ID: 6W63) (Generated using Biopredicta Module of Vlife MDS 4.6).
Figure 5.

Binding pose for 23 with crystal structure of SARS-CoV-2 Mpro (PDB ID: 6W63) (Generated using Biopredicta Module of Vlife MDS 4.6).
Figure 6.

Binding pose for 20 with crystal structure of SARS-CoV-2 Mpro (PDB ID: 6W63) (Generated using Biopredicta Module of Vlife MDS 4.6).
Figure 7.

Binding pose for 24 with crystal structure of SARS-CoV-2 Mpro (PDB ID: 6W63) (Generated using Biopredicta Module of Vlife MDS 4.6).
The crystal structure of the SARS-CoV-2 Mpro bound to potent broad-spectrum non-covalent inhibitor X77 (PDB ID: 6W63) was selected for the docking analysis. The resolution of the selected SARS-CoV-2 Mpro was found to be 2.10 Å and as it is X-ray determined structure, which was found to be suitable for docking process. Ramachandran plot of the selected protein structure was determined to ascertain the structural geometry and to validate the selected protein structure. The Core region of the protein was found to be contain 84.16%, allowed region was found to contain 11.22% of amino acids, which indicated selected protein structure is in good shape and having excellent structural geometry (Figure 1).
Redocking analysis was performed to validate the docking protocol. The RMSD between docked non-covalent inhibitor X77 and co-crystalized X77 was found to be 0.11 Å which validated the docking protocol. Results of the redocking process indicated that using applied docking protocol to the selected furoxan derivatives will give similar type of binding potential to that of co-crystalized X77 (Figure 2).
Docking analysis of the studied furoxan derivatives were found to have similar binding ability to SARS-CoV-2 Mpro as compared to reported inhibitors. Derivative 4-(3-nitrophenyl)-3-({3-oxo-2,3-dihydro-1H-spiro[isoquinoline-4,4′-piperidine]-1′-yl}methyl)-1,2,5-oxadiazol-2-ium-2-olate (22) have binding affinity of −9.6 kcal/mol, and docking score of −90.93, with important hydrogen binding interactions with CYS145 (2.208 A0) and SER144 (2.164 A0) and aromatic interactions with HIS41 (5.281 A0) and HIS 163 (4.628 A0) as shown in Figure 3.
Derivative 4-(3-bromophenyl)-3-({3-oxo-2,3-dihydro-1H-spiro[isoquinoline-4,4′-piperidine]-1′-yl}methyl)-1,2,5-oxadiazol-2-ium-2-olate (26) shown binding affinity of −9.8 kcal/mol and docking score of −90.91 as compared to 22. Hydrogen binding interactions with CYS145 (2.153 A0) and SER144 (1.823 A0) and aromatic interactions with HIS41 (5.175 A0) and HIS 163(4.760 A0) as shown in Figure 4.
Derivative 4-(4-fluorophenyl)-3-({3-oxo-2,3-dihydro-1H-spiro[isoquinoline-4,4′-piperidine]-1′-yl}methyl)-1,2,5-oxadiazol-2-ium-2-olate (23) was shown binding affinity of −8.3 kcal/mol and docking score of −89.44. Important hydrogen binding interactions was observed with GLN189 (1.926 A0), GLY143 (1.565 A0) and aromatic interactions with HIS41(4.521 A0) and HIS 163(4.994 A0) as shown in Figure 5.
Derivative 4-(4-methoxyphenyl)-3-({3-oxo-2,3-dihydro-1H-spiro[isoquinoline-4,4′-piperidine]-1′-yl}methyl)-1,2,5-oxadiazol-2-ium-2-olate (20) displays binding affinity of −8.4 kcal/mol and docking score of −88.64. Hydrogen bond interactions with GLY 143 (1.507 A0), SER144 (2.533 A0) and aromatic interactions with HIS41 (4.323 A0) as shown in Figure 6.
4-(2-chlorophenyl)-3-({3-oxo-2,3-dihydro-1H-spiro[isoquinoline-4,4′-piperidine]-1′-yl}methyl)-1,2,5-oxadiazol-2-ium-2-olate (24) showed binding affinity of −8.1 kcal/mol and docking score −84.66, along with interaction with SER144 (2.364 A0) via formation of hydrogen bond and with HIS163 (4.797 A0) via formation of aromatic interaction as shown in Figure 7.
Molecular dynamics simulation study
The MD simulation study of most stable docked complexes viz: Mpro-22 and Mpro-26 helped us to confirm the stable binding ability of furoxan derivatives 22 and 26 towards Mpro receptor structure and compare it with experimentally known inhibitor of Mpro i.e. X77. Thus, MD simulation study provided a clear picture of the overall stability of Mpro-22 and Mpro-26 systems in comparison with Mpro-X77 (Control) (Parulekar & Sonawane, 2018a, 2018b). All systems are subjected to 100 ns MD simulations and their stability in dynamic state was investigated by using GROMACS utilities for calculating RMSD, RMSF, Rg and H bonds (Abraham et al., 2015). The RMSD is a significant parameter used to analyze the stability as per output obtained from MD trajectories which is used to verify the convergence of simulations. RMSD of the protein backbone atoms are plotted as a function of time to check the stability of each complex throughout the simulation. From Figure 8A it is clear that control system reached stable equilibrium after 10 ns whereas, both Mpro-22 and Mpro-26 systems reached equilibrium after 15 ns. The average RMSDs for all simulated systems were calculated and shown in Table 2. These average RMSDs vary between 0.18 nm to 0.28 nm for all systems (Table 2). The RMSD values of Control and Mpro-22 complex were found to be in the range of 0.1 nm to 0.2 nm and remained stable over entire simulation time after reaching equilibrium (Figure 8A). The RMSD values of Mpro-26 complexes are in the range of 0.2 nm to 0.3 nm with slight higher deviation and remained stable throughout 100 ns simulation after attaining equilibrium (Figure 8A). The RMSF parameter in MD simulation is used as a mean describing flexibility differences among residues. The higher value of RMSF generally indicates more flexibility, whereas the low RMSF value indicates limited movements of residues during simulation in relation to its average position. Hence, we analyzed fluctuations of Cα atoms from both docked complexes along with control to study the flexibility of the residues from respective structures in presence of furoxan derivatives 22, 26 and X77 (control). The RMSF values of both the docked complexes along with control are shown in Figure-8B. These RMSF results clearly depicts that residues of Mpro in the presence of 22 and 26 showed similar type of behavior as in presence of X77 (control) [Figure 8B]. Precisely, the active site residues of Mpro i.e. CYS145, SER144, HIS41, HIS163 and GLU166 involved in binding with 22 and 26 showed significantly less fluctuations indicating the stable hydrogen and aromatics interactions of these residues with both the inhibitors as observed for X77 inhibitor used as control (Table 1, Figure 8B). We also determined Rg and calculated average values as reported in Table 2 to understand the level of compactness in the structure of Mpro in presence of furoxan derivatives 22, 26 and X77 (control). Interestingly, the Rg values of both the complexes and control were found to be in the range of 2.2 nm to 2.3 nm and were consistent over entire 100 ns simulation time scale (Figure 8C). Thus, this suggests the similarity in compactness of receptor protein Mpro in the presence of 22, 26 and X77 (control) [Figure 8C]. Additionally, the effective binding ability of furoxan derivatives 22, 26 with SARS-CoV-2 Mpro protein was illustrated by H bonds analysis (Figure 9). Both inhibitors 22, 26 and more prominently 26 showed strong and consistent hydrogen bonding with Mpro in comparison to X77 (control) over entire 100 ns simulation run (Figure 9). Altogether, MD simulation results for both the complexes were in accordance with control system which significantly suggests the effective binding ability of inhibitors 22, 26 as like X77 (control) towards SARS-CoV-2 Mpro receptor structure (Figure 8A–C and Figure 9).
Figure 8.

Analysis of MD simulation trajectories for control and docked complexes in terms of root mean square deviations (RMSD), root mean square fluctuations (RMSF), and radius of gyration (Rg). (A) Backbone RMSDs of Mpro-X77 (control), Mpro-22 and Mpro-26 systems for 100 ns simulation time. (B) RMSF plot of Cα atoms from Mpro receptor structure in presence of X77 (control), 22 and 26 inhibitor molecules (C) Rg of Mpro receptor protein in presence of X77 (control), 22 and 26 inhibitor molecules for 100 ns showing similarity in compactness of SARS-CoV-2 Mpro protein.
Table 2.
The average RMSD and Rg for all the 100 ns simulated systems.
| System | Backbone RMSD (nm) with S.D. | Radius of gyration Rg (nm) with S.D. |
|---|---|---|
| Control | 0.18 nm (0.02) | 2.20 nm (0.01) |
| Mpro-22 | 0.20 nm (0.02) | 2.24 nm (0.01) |
| Mpro-26 | 0.28 nm (0.03) | 2.25 nm (0.02) |
*S.D.= standard deviation.
Figure 9.

The comparative hydrogen bonds analysis of control and both the docked complexes (Mpro-22 and Mpro-26) over 100 ns simulation run.
Binding free energy calculations and residue contributions study by MM-PBSA
The binding free energy calculations on the basis of different energetic terms for control, Mpro-22 and Mpro-26 complexes confirmed the binding efficiency of X77 (control) and furoxan derivatives 22, 26 at the known inhibitor binding site region of SARS-CoV-2 Mpro structure (Table 3). As seen from Table 3 control showed involvement of higher VDW energies for stabilizing the Mpro-X77 complex whereas, Mpro-22 and Mpro-26 complexes showed significant involvement of higher electrostatic energies for binding thus suggesting prominent role of hydrogen bonds in stabilizing both the docked complexes (Table 3, Figure 9). The control system seems to be more stabilized by non-bonded contacts in comparison to hydrogen bonds which could be the reason for higher VDW energies and lower electrostatic energies observed for control (Table 3, Figure 9). Similarly, polar salvation energy values were also obtained higher for Mpro-22 and Mpro-26 complexes in comparison to control (Table 3). The estimated binding energies (ΔGbind) for control was obtained as −130.014 ± 9.977 whereas, ΔGbind for Mpro-22 and Mpro-26 was observed to be −183.860 ± 9.106 and −171.972 ± 8.757 respectively (Table 3). Hence, on comparative account 22, 26 showed more effective binding than X77 (control) towards SARS-CoV-2 Mpro receptor on different energetic terms (Table 3).
Table 3.
Binding energy components comparison for SARS-CoV-2 Mpro protein in complex with X77 (control) and most stable inhibitor molecules 22, 26 as obtained from in silico investigation.
| Complex | ΔEvdwa | ΔEelec | ΔEMM | ΔGpolar | ΔGnon-polar | ΔGbinding |
|---|---|---|---|---|---|---|
| Control | −168.312 ± 13.357 | −7.801 ± 4.448 | −176.113 ± 11.457 | 61.034 ± 4.082 | −14.934 ± 0.054 | −130.014 ± 9.977 |
| Mpro-22 | −123.792 ± 7.327 | −151.609 ± 9.399 | −275.402 ± 15.326 | 102.911 ± 7.036 | −11.370 ± 0.993 | −183.860 ± 9.106 |
| Mpro-26 | −113.942 ± 7.049 | −245.111 ± 13.287 | −359.053 ± 19.781 | 219.180 ± 13.912 | −12.099 ± 0.602 | −171.972 ± 8.757 |
aΔEvdw, ΔEelec, ΔEMM, ΔGpolar and ΔGnon-polar are binding energy components of van der Waals, electrostatic, molecular mechanics, polar and non-polar solvation (SASA) energies, respectively. ΔGbinding is the total binding energy. The unit of energy is kJ/mol.
Furthermore, individual energy decomposition of all residues from SARS-CoV-2 Mpro receptor structure was calculated to investigate the key residue of Mpro involved in binding with X77 (control) and 22, 26 (Figure 10A–C). The results obtained from the calculation of energetic contribution of individual residues clearly confirm the effective binding ability of 22, 26 in comparison to X77 (control) towards Mpro (Figure 10A–C). These results are totally in accordance with the results obtained for the binding free energy calculations of all three complexes on the basis of different energetic terms (Table 3, Figure 10A–C). As seen from Figure 10A–C less residue wise energy contribution is observed for control whereas, significant high values (especially electrostatics and polar solvation) are observed for complexes Mpro-22 and Mpro-26 thus reflecting the strong binding nature of 22, 26 towards SARS-CoV-2 Mpro structure. Residue wise decomposition results confirms that the interacting residues from binding pocket of SARS-CoV-2 Mpro as obtained from docking analysis (Table 1) also shows significantly higher energetic contribution in binding with potent furoxan derivatives 22, 26 in comparison to control (Figure 10A–C).
Figure 10.

The energetic contribution of individual residues from all three complexes to binding energy in kJ/mol for 100 ns MD simulation, (A) all residue contribution of the Mpro-X77 (control) complex in the binding energy (kJ/mol). (B) all residue contribution of the Mpro-22 complex in the binding energy (kJ/mol) and (C) all residue contribution of the Mpro-26 complex in the binding energy (kJ/mol).
Conclusion
Thus, overall in silico analysis performed in the current study reveal the effective binding affinity of investigated spiro-isoquinolino-piperidine-furoxan derivatives in comparison to the experimentally known inhibitor of Mpro i.e. X77 (control) and also they were found to be superior to the corresponding benzhydrylpiperazine derivatives. Further modification of these spiro-isoquinolino-piperidine-furoxan derivatives can lead to discovery of potent SARS-CoV-2 Mpro inhibitors having the advantage of exogenous NO release feature. Although the molecules have shown prominent results in the in silico analysis, the wet lab experiment are required to validate the results. Present results can act as starting point for development of potent selective SARS-CoV-2 Mpro inhibitors.
Acknowledgements
The authors are thankful to V life Sciences, Pune for providing trial version of software for research work. The authors are thankful to the Institute of research and consulting studies at King Khalid University for funding this research through grant number 3-N-20/21 and the support of Research center for advanced materials Science is highly acknowledged.
Disclosure statement
There is no conflict of interest.
References
- Aanouz, I., Belhassan, A., El Khatabi, K., Lakhlifi, T., El Idrissi, M., & Bouachrine, M. (2020). Moroccan medicinal plants as inhibitors against SARS-CoV-2 main protease: Computational investigations. Journal of Biomolecular Structure and Dynamics, 1–9. 10.1080/07391102.2020.1758790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelli, I., Hassani, F., Bekkel Brikci, S., & Ghalem, S. (2020). In silico study the inhibition of Angiotensin converting enzyme 2 receptor of COVID-19 by Ammoides verticillata components harvested from western Algeria. Journal of Biomolecular Structure and Dynamics, 1–17. 10.1080/07391102.2020.1763199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham, M. J., Murtola, T., Schulz, R., Páll, S., Smith, J. C., Hess, B., & Lindahl, E. (2015). GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX, 1-2, 19–25. 10.1016/j.softx.2015.06.001 [DOI] [Google Scholar]
- Åkerström, S., Mousavi-Jazi, M., Klingström, J., Leijon, M., Lundkvist, Å., & Mirazimi, A. (2005). Nitric oxide inhibits the replication cycle of severe acute respiratory syndrome coronavirus. Journal of Virology, 79(3), 1966–1969. 10.1128/JVI.79.3.1966-1969.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Åkerström, S., Gunalan, V., Keng, C. T., Tan, Y. J., & Mirazimi, A. (2009). Dual effect of nitric oxide on SARS-CoV replication: Viral RNA production and palmitoylation of the S protein are affected. Virology, 395(1), 1–9. 10.1016/j.virol.2009.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Khafaji, K., Al-DuhaidahawiL, D., & Taskin Tok, T. (2020). Using Integrated Computational Approaches to Identify Safe and Rapid Treatment for SARS-CoV-2. Journal of Biomolecular Structure and Dynamics, 1–11. 10.1080/07391102.2020.1764392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boopathi, S., Poma, A. B., & Kolandaivel, P. (2020). Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. Journal of Biomolecular Structure and Dynamics, 1–14. 10.1080/07391102.2020.1758788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, L., Liu, P., Gao, H., Sun, B., Chao, D., Wang, F., Zhu, Y., Hedenstierna, G., & Wang, C. G. (2004). Inhalation of nitric oxide in the treatment of severe acute respiratory syndrome: A rescue trial in Beijing. Clinical Infectious Diseases: An Official Publication of the Infectious Diseases Society of America, 39(10), 1531–1535. 10.1086/425357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, F. (2019). In silico oncology drug repositioning and polypharmacology. In Cancer Bioinformatics. Humana Press. 1878, 243–261 10.1007/978-1-4939-8868-6_15 [DOI] [PubMed] [Google Scholar]
- Das, S., Sarmah, S., Lyndem, S., & Singha Roy, A. (2020). An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. Journal of Biomolecular Structure and Dynamics, 1–18. 10.1080/07391102.2020.1763201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky, A. A. (2020. a). SARS-CoV-2 RNA dependent RNA polymerase (RdRp) targeting: An in silico perspective. Journal of Biomolecular Structure and Dynamics, 1–9. 10.1080/07391102.2020.1761882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky, A. A. (2020. b). Natural products may interfere with SARSCoV-2 attachment to the host cell. Journal of Biomolecular Structure and Dynamics. 10.1080/07391102.2020.1761881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enmozhi, S. K., Raja, K., Sebastine, I., & Joseph, J. (2020). Andrographolide as a potential inhibitor of SARS-CoV-2 main protease: An in silico approach. Journal of Biomolecular Structure and Dynamics, 1–7. 10.1080/07391102.2020.1760136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essmann, U., Perera, L., Berkowitz, M. L., Darden, T., Lee, H., & Pedersen, L. G. (1995). A smooth particle mesh Ewald method. The Journal of Chemical Physics, 103(19), 8577–8593. 10.1063/1.470117 [DOI] [Google Scholar]
- Gasco, A., Fruttero, R., Sorba, G., Di Stilo, A., & Calvino, R. (2004). NO donors: Focus on furoxan derivatives. Pure and Applied Chemistry, 76(5), 973–981. 10.1351/pac200476050973 [DOI] [Google Scholar]
- Ghosh, A. K., Brindisi, M., Shahabi, D., Chapman, M. E., & Mesecar, A. D. (2020). Drug Development and Medicinal Chemistry Efforts Toward SARS‐Coronavirus and Covid‐19 Therapeutics. ChemMedChem., 15(11), 907–932. 10.1002/cmdc.202000223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyebi, G. A., Ogunro, O. B., Adegunloye, A. P., Ogunyemi, O. M., & Afolabi, S. O. (2020). Potential Inhibitors of Coronavirus 3-Chymotrypsin-Like Protease (3CLpro): An in silico screening of Alkaloids and Terpenoids from African medicinal plants. Journal of Biomolecular Structure and Dynamics, 1–19. 10.1080/07391102.2020.1764868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess, B., Bekker, H., Berendsen, H. J., & Fraaije, J. G. (1997). LINCS: A linear constraint solver for molecular simulations. Journal of Computational Chemistry, 18(12), 1463–1472. [DOI] [Google Scholar]
- Islam, R., Parves, R., Paul, A. S., Uddin, N., Rahman, M. S., Mamun, A. A., Hossain, M. N., Ali, M. A., & Halim, M. A. (2020). A Molecular Modeling Approach to Identify Effective Antiviral Phytochemicals against the Main Protease of SARS-CoV-2. Journal of Biomolecular Structure and Dynamics, 1–20. 10.1080/07391102.2020.1761883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, Z., Du, X., Xu, Y., Deng, Y., Liu, M., Zhao, Y., Zhang, B., Li, X., Zhang, L., Peng, C., Duan, Y., Yu, J., Wang, L., Yang, K., Liu, F., Jiang, R., Yang, X., You, T., Liu, X., … Yang, H. (2020). Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature, 582(7811), 289–293. (2019). 10.1038/s41586-020-2223-y [DOI] [PubMed] [Google Scholar]
- Joshi, R. S., Jagdale, S. S., Bansode, S. B., Shankar, S. S., Tellis, M. B., Pandya, V. K., Chugh, A., Giri, A. P., & Kulkarni, M. J. (2020). Discovery of potential multi-target-directed ligands by targeting host-specific SARS-CoV-2 structurally conserved main protease. Journal of Biomolecular Structure and Dynamics, 1–16. 10.1080/07391102.2020.1760137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminski, G. A., Friesner, R. A., Tirado-Rives, J., & Jorgensen, W. L. (2001). Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. The Journal of Physical Chemistry B, 105(28), 6474–6487. 10.1021/jp003919d [DOI] [Google Scholar]
- Keyaerts, E., Vijgen, L., Chen, L., Maes, P., Hedenstierna, G., & Van Ranst, M. (2004). Inhibition of SARS-coronavirus infection in vitro by S-nitroso-N-acetylpenicillamine, a nitric oxide donor compound. International Journal of Infectious Diseases : IJID : official Publication of the International Society for Infectious Diseases, 8(4), 223–226. 10.1016/j.ijid.2004.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, R. J., Jha, R. K., Amera, G., Jain, M., Singh, E., Pathak, A., Singh, R. P., Muthukumaran, J., & Singh, A. K. (2020. a). Targeting SARS-CoV-2: A systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 2'- O-ribose methyltransferase. Journal of Biomolecular Structure and Dynamics, 1–14. 10.1080/07391102.2020.1753577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, S. A., Zia, K., Ashraf, S., Uddin, R., & Ul-Haq, Z. (2020. b). Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. Journal of Biomolecular Structure and Dynamics, 1–10. 10.1080/07391102.2020.1751298. [DOI] [PubMed] [Google Scholar]
- Kumari, R., Kumar, R., & Lynn, A. (2014). g_mmpbsa-a GROMACS tool for high-throughput MM-PBSA calculations. Journal of Chemical Information and Modeling, 54(7), 1951–1962. 10.1021/ci500020m [DOI] [PubMed] [Google Scholar]
- Lobo-Galo, N., Terrazas-López, M., Martínez-Martínez, A., & Díaz-Sánchez, Á. G. (2020). FDA-approved thiol-reacting drugs that potentially bind into the SARS-CoV-2 main protease, essential for viral replication. Journal of Biomolecular Structure and Dynamics, 1–12. 10.1080/07391102.2020.1764393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, H. (2020). Drug treatment options for the 2019-new coronavirus (2019-nCoV). Bioscience Trends, 14(1), 69–71. 10.5582/bst.2020.01020 [DOI] [PubMed] [Google Scholar]
- Mannick, J. B. (1995). The antiviral role of nitric oxide. Research in Immunology, 146(9), 693–697. 10.1016/0923-2494(96)84920-0 [DOI] [PubMed] [Google Scholar]
- Muralidharan, N., Sakthivel, R., Velmurugan, D., & Gromiha, M. M. (2020). Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 protease against COVID-19. Journal of Biomolecular Structure and Dynamics, 1–6. 10.1080/07391102.2020.1752802. [DOI] [PubMed] [Google Scholar]
- Pant, S., Singh, M., Ravichandiran, V., Murty, U. S. N., & Srivastava, H. K. (2020). Peptidelike and small-molecule inhibitors against Covid-19. Journal of Biomolecular Structure and Dynamics, 1–10. 10.1080/07391102.2020.1757510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parulekar, R. S., & Sonawane, K. D. (2018. a). Molecular modeling studies to explore the binding affinity of virtually screened inhibitor toward different aminoglycoside kinases from diverse MDR strains. Journal of Cellular Biochemistry, 119(3), 2679–2695. 10.1002/jcb.26435 [DOI] [PubMed] [Google Scholar]
- Parulekar, R. S., & Sonawane, K. D. (2018. b). Insights into the antibiotic resistance and inhibition mechanism of aminoglycoside phosphotransferase from Bacillus cereus: In silico and in vitro perspective. Journal of Cellular Biochemistry, 119(11), 9444–9461. 10.1002/jcb.27261 [DOI] [PubMed] [Google Scholar]
- Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., & Ferrin, T. E. (2004). UCSF Chimera-a visualization system for exploratory research and analysis. Journal of Computational Chemistry, 25(13), 1605–1612. 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- Prabhuling, S., Tamboli, Y., Choudhari, P. B., Bhatia, M. S., Mohanta, T. K., Al-Harrasi, A., & Pudukulathan, Z. K. (2020). Synthesis and Modeling Studies of Furoxan Coupled Spiro-Isoquinolino Piperidine Derivatives as NO Releasing PDE 5 Inhibitors. Biomedicines, 8(5), 121. 10.3390/biomedicines8050121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pudukulatham, Z., Zhang, F. X., Gadotti, V. M., M’Dahoma, S., Swami, P., Tamboli, Y., & Zamponi, G. W. (2016). Synthesis and characterization of a disubstitutedpiperazine derivative with T-type channel blocking action and analgesic properties. Molecular Pain, 12, 174480691664167. 10.1177/1744806916641678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss, C. S., & Komatsu, T. (1998). Does nitric oxide play a critical role in viral infections? Journal of Virology, 72(6), 4547–4551. 10.1128/JVI.72.6.4547-4551.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarma, P., Sekhar, N., Prajapat, M., Avti, P., Kaur, H., Kumar, S., Singh, S., Kumar, H., Prakash, A., & Dhibar, D. P. (2020). In-silico homology assisted identification of inhibitor of RNA binding against 2019-nCoV N-protein (N terminal domain). Journal of Biomolecular Structure and Dynamics (Just-Accepted), 1–11. 10.1080/07391102.2020.1753580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura, M., Zaragoza, C., McMillan, A., Quick, R. A., Hohenadl, C., Lowenstein, J. M., & Lowenstein, C. J. (1999). An antiviral mechanism of nitric oxide: Inhibition of a viral protease. Immunity, 10(1), 21–28. 10.1016/S1074-7613(00)80003-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafim, R. A. M., Primi, M. C., Trossini, G. H. G., & Ferreira, E. I. (2012). Nitric oxide: State of the art in drug design. Current Medicinal Chemistry, 19(3), 386–405. 10.2174/092986712803414321 [DOI] [PubMed] [Google Scholar]
- Sinha, S. K., Shakya, A., Prasad, S. K., Singh, S., Gurav, N. S., Prasad, R. S., & Gurav, S. S. (2020). An in-silico evaluation of different Saikosaponins for their potency against SARS-CoV-2 using NSP15 and fusion spike glycoprotein as targets. Journal of Biomolecular Structure and Dynamics, 1–13. 10.1080/07391102.2020.1762741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott, O., & Olson, A. J. (2010). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31(2), 455–461. 10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Aalten, D. M., Bywater, R., Findlay, J. B., Hendlich, M., Hooft, R. W., & Vriend, G. (1996). PRODRG, a program for generating molecular topologies and unique molecular descriptors from coordinates of small molecules. Journal of Computer-Aided Molecular Design, 10(3), 255–262. 10.1007/BF00355047 [DOI] [PubMed] [Google Scholar]
- Wahedi, H. M., Ahmad, S., & Abbasi, S. W. (2020). Stilbene-based natural compounds as promising drug candidates against COVID-19. Journal of Biomolecular Structure and Dynamics, 1–16. 10.1080/07391102.2020.1762743 [DOI] [PubMed] [Google Scholar]
- www.rcsb.org
- www.vlifesciences.com
- Zhang, L., Lin, D., Sun, X., Curth, U., Drosten, C., Sauerhering, L., Becker, S., Rox, K., & Hilgenfeld, R. (2020). Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science, 368(6489), 409–412. 10.1126/science.abb3405 [DOI] [PMC free article] [PubMed] [Google Scholar]