

SUMMARY
Emerging evidence suggests that the impact of dietary intake on human health and disease is linked to both the immune system and the microbiota. Yet, we lack an integrated mechanistic model for how these three complex systems relate, limiting our ability to understand and treat chronic and infectious disease. Here, we review recent findings at the interface of microbiology, immunology, and nutrition, with an emphasis on experimentally tractable models and hypothesis-driven mechanistic work. We outline emerging mechanistic concepts and generalizable approaches to bridge the gap between microbial ecology and molecular mechanism. These set the stage for a new era of precision human nutrition informed by a deep and comprehensive knowledge of the diverse cell types in and on the human body.
Keywords: microbiome, nutrition, immunology, metabolites, host receptors
eTOC BLURB
Alexander and Turnbaugh (XXX-YYY) review the current understanding of the mechanisms linking diet, the microbiome and immunity. In this context the authors propose general mechanistic concepts defining these interactions and outline important gaps in knowledge at the intersection of immunology and microbiota research.
INTRODUCTION
Dietary recommendations are in a constant state of flux. This is exemplified by the renewed debate on the negative health effects, or lack thereof, of red meat (Johnston et al., 2019). Searching for “anti-inflammatory diet” online yields a wealth of websites, books, and newspaper articles prompting us to eat less sugary processed foods and more vegetables, nuts, and fish. But what is it exactly in these foods that is pro- or anti- inflammatory? What components of diet alter immune function? How confident are scientists about the long-term health consequences of a diet? While these are all longstanding questions, they remain largely unaddressed due to gaps in our understanding of the complex inter-relationships between immunity, the trillions of microorganisms that colonize the human body (the human microbiota), and the pathogenesis and treatment of disease.
The pairwise interactions between dietary factors, the microbiota, and immunity have been extensively characterized. Host-associated microbes alter immunity through multiple mechanisms, discussed in-depth elsewhere (Blander et al., 2017; Honda and Littman, 2016; Skelly et al., 2019). Bacteria and fungi induce specific immune responses that can be either proor anti-inflammatory depending on the context. A seminal example of a member of the gut microbiota inducing a proinflammatory response is the induction of murine T helper 17 (Th17) cells (CD4+IL-17A+) by the adherence of segmented filamentous bacteria (SFB) to intestinal epithelial cells (Atarashi et al., 2015; Ivanov et al., 2009), leading to worsened disease in mouse models of autoimmunity (Bradley et al., 2017; Lee et al., 2011; Teng et al., 2016). In contrast, an example of a member of the gut microbiota inducing an anti-inflammatory response is the production of a zwitterionic peptide (polysaccharide A, PSA) by Bacteroides fragilis that induces regulatory T cells (CD4+Foxp3+), thereby rescuing mouse models of colitis (Round and Mazmanian, 2010). The role of diet in shaping the structure and function of the human microbiota has also been discussed extensively (Carmody and Turnbaugh, 2014; Kolodziejczyk et al., 2019). In both mice (Carmody et al., 2015) and humans (David et al., 2014), dietary perturbations can alter the gut microbiota within days, in some cases leading to changes to the gut microbiota that are transmissible across generations (Sonnenburg et al., 2016). Finally, the connection between diet and immune function is also well-established (Chandra, 1996). For example, vitamins A and D affect lymphocyte activation and proliferation, T helper cell differentiation, T cell migration, and isotype switching (Mora et al., 2008). Together, these studies provide strong support for the hypothesis that host phenotypes are driven in part by the net effects of the interactions between diet, the microbiome (defined herein as the aggregate genetic material and metabolic activity of the microbiota), and immunity. However, until recently, there have not been many studies investigating the mechanistic interplay between diet, microbiota, and immunity or the downstream consequences of these tripartite interactions for health and disease.
A major challenge to understanding the immunological consequences of diet is that the complexity of human diets makes it difficult to determine molecular mechanisms. Whole foods contain assorted macro- and micro-nutrients, among a vast “dark matter” of non-nutritive components and chemicals produced during food preparation (Carmody et al., 2019; Johnson et al., 2019; Wolf et al., 2019). We propose that the lack of an integrated mechanistic framework to understand the phenotypic consequences of diet, the microbiome, and immunity is a major reason for the underlying inconsistencies and controversial nature of dietary recommendations. More research is required to elucidate how individual food components (e.g., vitamins, antioxidants, and plant polysaccharides) impact immune-microbiome interactions at a cellular and molecular level. These data will help elucidate the origins of variable immune responses to dietary interventions (Zeevi et al., 2015) and their sensitivity to differences in host genotype, gut microbial community structure, and microbial metabolites (Poole et al., 2019).
Here we review recent findings that dissect these diet-microbiome-immune interactions at a mechanistic level, with a focus on immunological consequences. We classify these interactions into 6 general types of mechanisms. While we have binned studies into these themes, the themes and studies we highlight are by no means exhaustive and as future studies continue to shed light on these tripartite interactions, these classifications could and should be added to and refined. Our goal is to highlight how transitioning from observational studies in human cohorts to experimentally tractable models and controlled human interventions - while still considering the higher-level interactions between cell types and environmental factors -helps elucidate mechanisms of how the microbiota and dietary alterations impact immunity.
Getting in shape: diet-derived microbial metabolites sculpt immunity via host receptors
Perhaps the most well-studied mechanism through which diet and the microbiome conspire to alter host immunity is the recognition of diet-derived microbial metabolites by host signaling pathways (Figure 1). This includes the activation of G protein-coupled receptors (GPCRs) in both immune and non-immune cells by short-chain fatty acids (SCFAs) which are produced during the bacterial fermentation of oligo- and poly-saccharides, and bacterial amino acid metabolites. The microbially produced SCFAs propionate, acetate, and butyrate increase colonic CD4+Foxp3+ regulatory T cell (Treg) activity due to activation of the GPCR FFAR2 (GPCR43), protecting mice from colitis (Smith et al., 2013b). Activation of FFAR2 with an agonist, acetate, or to a lesser extent propionate, promotes type 3 innate lymphoid cell (ILC3) expansion. Propionate also increases interleukin 22 (IL-22)+ ILC3s, which are important for intestinal barrier maintenance (Chun et al., 2019). Mice with an Rorc-cre-mediated conditional deletion of Ffar2 (Ffar2ΔRorc) developed more severe colitis in the dextran sodium sulfate (DSS) mouse model of colitis compared to floxed controls. However, FFAR2 may not be the only pathway involved. While Ffar2ΔRorc mice have decreased IL-22+ ILC3 cells, they are not fully ablated. Treatment of Ffar2ΔRorc mice with acetate or propionate could help to determine the Ffar2-independent effects of SCFAs on ILC3s. Treatment of ILC3 cells with GPCR inhibitors (pertussis toxin and YM-254890) ablated IL-22 induction by the synthetic FFAR2 agonist but did not have as large an impact on propionate-mediated IL-22 induction. This suggests that IL-22 induction by the synthetic agonist is more dependent on GPCR signaling than propionate. Thus, different agonists for the same receptor can have disparate downstream effects on immunity which could be due to the differing affinities for the GPCR or the engagement of additional signaling pathways.
Figure 1. General types of interactions between diet, microbiota, and immune responses.
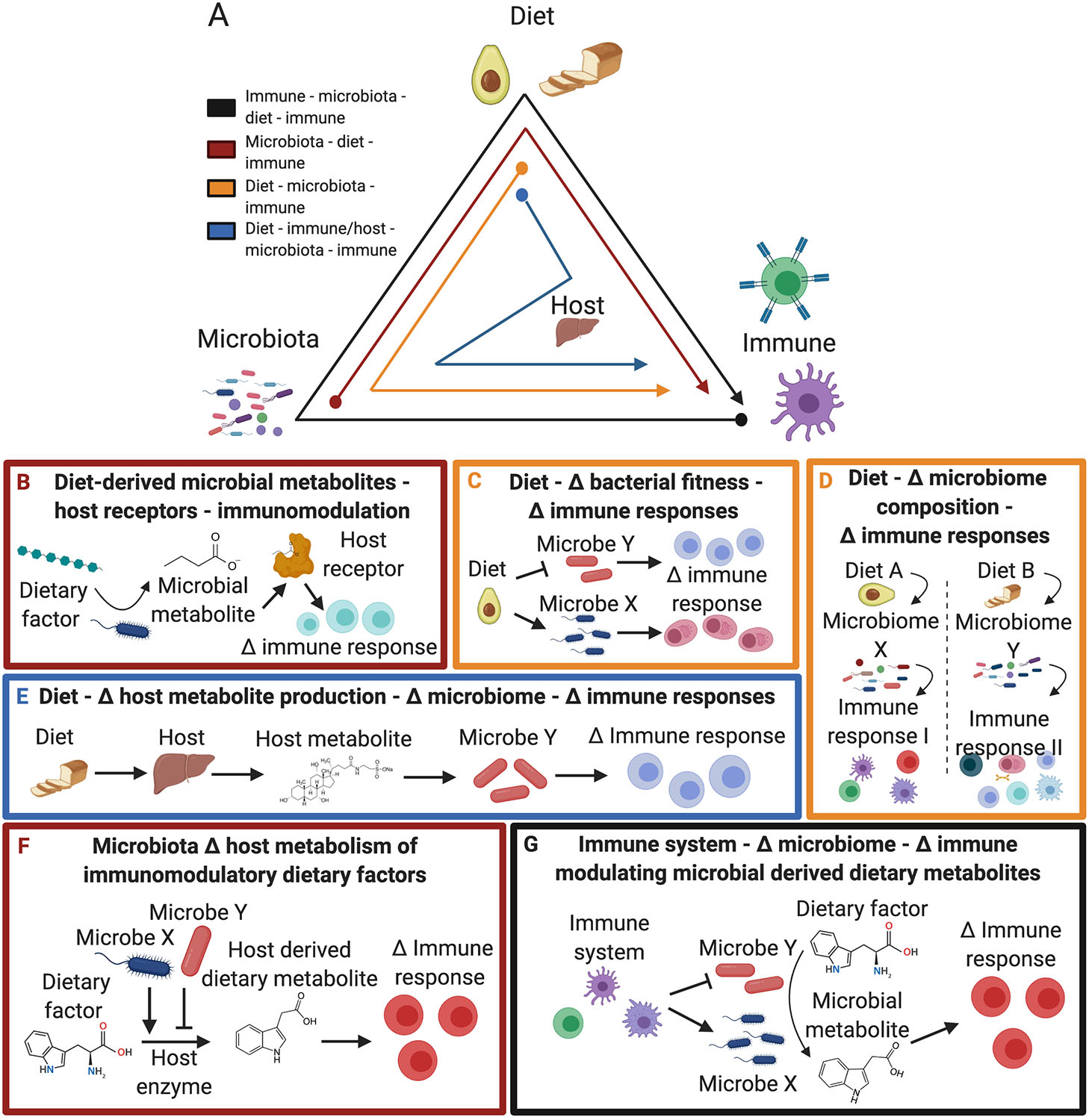
(A) (Black) The immune system shapes the microbiota, which alters the metabolism of immunomodulatory dietary factors. (Red) The microbiota metabolizes dietary substrates into immunomodulatory metabolites. (Orange) Diet influences the microbiota resulting in altered immune responses. (Blue) Dietary factors are metabolized by the host resulting in alterations to immunomodulatory microbes. (B-G) General mechanisms by which the diet, microbiome, and immune system interact to induce immunological changes: (B) Dietary derived microbial metabolites alter immune response via host receptor signaling; (C) Diet alters the fitness of immunomodulatory microbes; (D) Diet alters microbiome composition and activity, which modulates the immune response; (E) Diet alters host metabolite production which impacts immunomodulatory microbes; (F) Microbes shape host metabolism of immunomodulatory dietary factors; and (G) The immune system shapes the microbiome altering microbial metabolism of immunomodulatory dietary factors.
Another recent example of diet-dependent immunomodulatory microbial metabolites signaling via host receptors is microbial metabolism of fatty acids. The fatty acid linoleic acid can be metabolized by epoxide hydrolases encoded by the host, bacteria, and fungi into 12,13-diHOME (Levan et al., 2019). 12,13-diHOME is enriched in neonates at risk of childhood atopy and asthma corresponding to an enrichment of bacterial epoxide hydrolases including NP_814872 (Enterococcus faecalis), YP_003971091 (Bifidobacterium bifidum), and YP_003971333 (B. bifidum). Expression of these epoxide hydrolases in E. coli was sufficient to convert 12,13-EpOME to 12,13-diHOME. When mice challenged with cockroach antigen were treated with 12,13-diHOME they had increased pulmonary T cells, neutrophils, monocytes, and proinflammatory cytokines and decreased Treg cells, all hallmarks of allergic airway inflammation. In vitro administration of 12,13-diHOME to human dendritic cells decreased IL-10 secretion and activated PPARγ. PPARγ is a lipid-activated nuclear receptor that regulates inflammatory responses and the gut microbiota (Byndloss et al., 2017; Khare et al., 2015). However, although 12,13-diHOME is sufficient to induce PPARγ this does not rule other targets that could contribute to the mechanism of action of 12,13-diHOME. Additional studies are also warranted to determine the importance of variations in linoleic acid consumption and the primary microbial species, genes, and enzymes involved in its metabolism.
Microbial metabolism of tryptophan into aryl hydrocarbon receptor (AHR) ligands represents another example of diet-derived metabolites that signal via host receptors resulting in altered immune responses. Lactobacillus species metabolize tryptophan into indole metabolites such as indole-3-acetic acid (IAA), which act as AHR ligands and promote IL-22 production (Zelante et al., 2013). In a DSS model of colitis, intestinal inflammation was decreased when mice were inoculated with Lactobacillus species capable of metabolizing tryptophan into AHR ligands (L. murinus CNMC I-5020, L. taiwanensis CNCM I-5019, and L. reuteri CNMC I-5022) (Lamas et al., 2016). The role of AHR signaling in this protection was supported by findings that treatment of these mice with an AHR antagonist abrogated these effects. This example reveals that dietary amino acids such as tryptophan can be metabolized by Lactobacillus species, which signal via AHR resulting in immunomodulation. However, not all species of Lactobacillus promote AHR activity to the same extent; for example, L. murinus and L. taiwanensis induce AHR activity in vitro to a greater extent than L. reuteri. More research is needed to identify the factors that contribute to the differential AHR activation of Lactobacillus species or strains and their downstream consequences for intestinal inflammation and colitis severity. More broadly, it will be important to determine which other members of the gut microbiota produce AHR ligands and the relative contribution of dietary versus endogenous tryptophan in their biosynthesis.
They are what we eat: diet-dependent microbes feedback on immunity
The functional consequences of shifts in microbial communities are difficult to study, especially in humans, due to our lack of methods for precisely manipulating the human microbiome (Lam et al., 2019). One common approach is “microbiome transplantation experiments” where complex microbial communities are directly transferred between animals. An advantage of this approach is that it enables researchers to test hypotheses about the downstream effects of changes in the microbiome for immunity (Figure 2); however, its limitations have been the subject of debate (Walter et al., 2020). Here, we highlight studies that used this approach to test diet-microbiome-immune interactions. In our opinion, these studies represent a critical step towards reducing these complex systems to mechanisms and are complementary to the more reductionist models discussed in the prior section.
Figure 2. Diet alters the immunomodulatory potential of the gut microbiota.
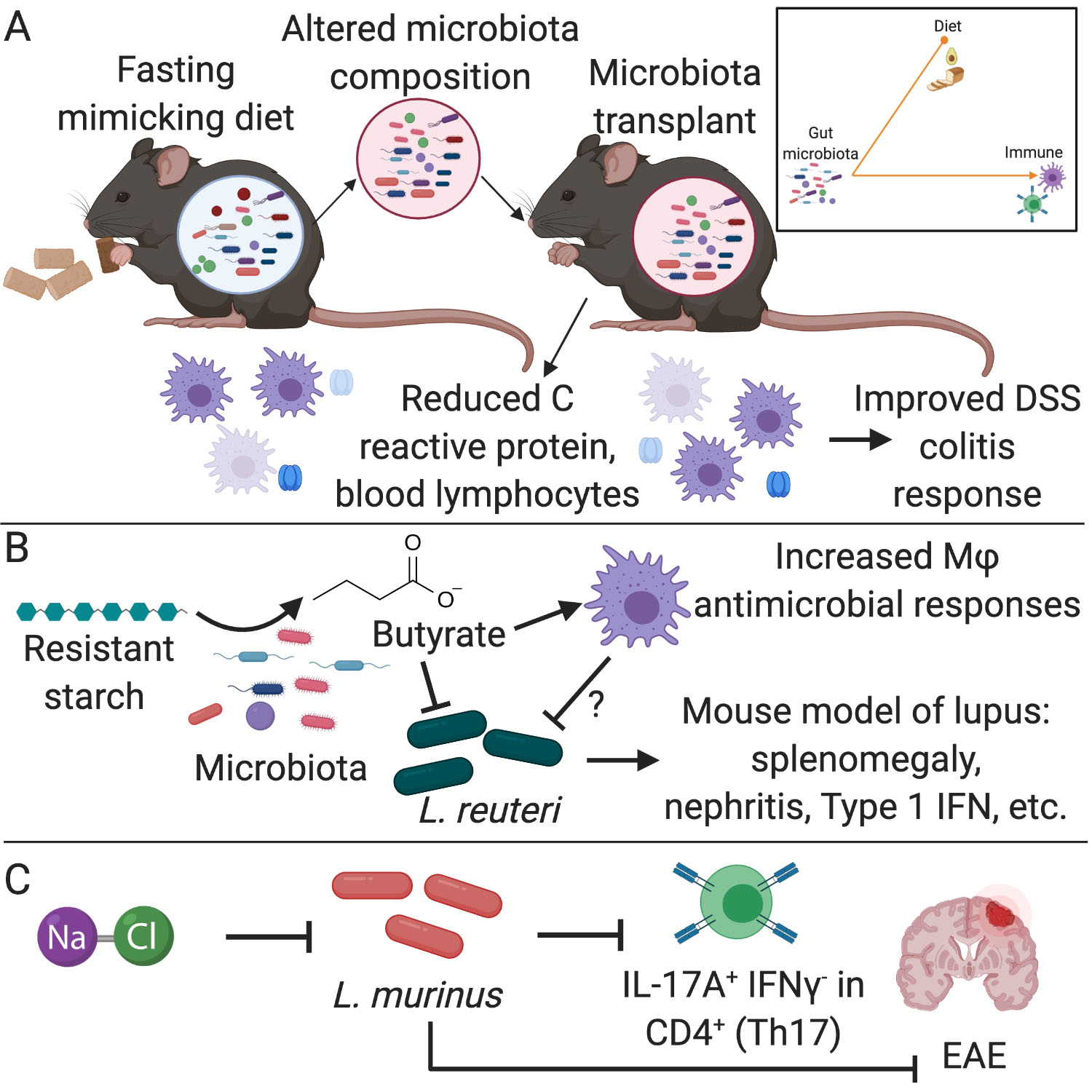
(A) Mice on a fasting mimicking diet have reduced blood lymphocytes and C reactive protein, which corresponds to alterations in the composition of the gut microbiota. Transplantation of this altered microbiota into germ-free (GF) donor mice phenocopies the donor mice and results in improved dextran sodium sulfate (DSS) colitis response (Rangan et al., 2019). (B) Resistant starch is metabolized by members of the gut microbiota into the short chain fatty acid (SCFA) butyrate which has direct effects on immune responses, such as the increased antimicrobial activity of macrophages (Schulthess et al., 2019). Additionally, butyrate can directly inhibit growth of L. reuteri, which has been shown to worsen lupus severity in a mouse model (Zegarra-Ruiz et al., 2019). (C) The growth of L. murinus is inhibited in vitro by increasing salt (NaCl) concentrations. Mice on a high salt diet supplemented with L. murinus had reduced Th17 levels and less severe disease in the experimental autoimmune encephalomyelitis (EAE) mouse model of multiple sclerosis (Wilck et al., 2017).
Multiple studies support the hypothesis that diet-induced shifts in the gut microbiome alter immune-related disease models (Figure 1c–d). For example, transplantation of the gut microbiota from intermittently fasted mice ameliorated the symptoms of experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis (Cignarella et al., 2018). Fecal transplants from mice fed a fasting mimicking diet led to reduced colitis severity in DSS treated mice (Rangan et al., 2019) (Figure 2a). While these studies provide support for a causal role of diet-induced shifts in the gut microbiota to protection from immune-driven disease models, the specific microbes and molecular mechanisms can be difficult to disentangle. Importantly, these findings do not imply that the microbiome is the only factor responsible for these complex phenotypes as there is a clear precedent for microbiome independent effects of diet on immune cells. For example, caloric restriction decreases circulating monocytes (Jordan et al., 2019) and fasting reduces B cells in Peyer’s Patches in both germ-free (GF) and specific pathogen free (SPF) mice (Nagai et al., 2019).
The contribution of microbiome signals versus the independent effects of diet on immunity is a central question. Recently, the relative impacts of these factors on intestinal Treg cell levels was investigated. GF mice have similar frequencies and numbers of small intestinal lamina propria Treg cells to SPF mice, suggesting a microbiome-independent accumulation of these cells (Kim et al., 2016; Weiss et al., 2012). When GF mice were sired from mice fed an antigen-free diet (AFD) and were raised on this diet, the levels of Treg cells (CD4+FoxP3+) in the small intestine significantly dropped compared to SPF and GF mice (Kim et al., 2016). This decrease in Treg cells was accompanied by an increased susceptibility to intestinal allergy in an ovalbumin (OVA) mouse model, where OVA-specific naïve OT-II CD4+ T cells were transferred into AFD mice challenged with OVA. These findings suggest that Treg cell induction in the small intestine is dependent on dietary antigens. To further understand the relative contributions of microbiota versus dietary antigen induction of Treg cells it would be important to test the effect of the AFD on SPF mice, although this diet could additionally alter the composition of the microbiota which could feedback on immunity. This caveat could be partially addressed with a microbiota transplant from SPF mice fed an AFD versus a control diet to GF mice to assess the effect of the alteration in microbiome composition on intestinal Treg cells.
Microbiome transplants have also been used to test whether alterations in the microbiota of undernourished children can recapitulate phenotypes associated with undernourishment, including immune deficiencies. Fecal transplants from twins with severe acute malnutrition (SAM) to GF mice resulted in decreased body weight relative to well-nourished twin controls (Smith et al., 2013a). The transplantation of immunoglobulin A (IgA)-coated microbes from undernourished children into GF mice resulted in a diet-dependent enteropathy phenotype (Kau et al., 2015) that was rescued by the addition of two bacterial species that were coated with IgA from the well-nourished co-twin. This observation that IgA may indicate bacteria that contribute to or prevent pathologies complicates our view of IgA immune targeting; IgA may play opposing roles depending on the dietary context. IgA targeting can decrease the colonization of bacteria but can also increase the colonization of the mucosal niche. In the context of undernourished children IgA may target pathology-inducing bacteria but in a healthy microbiota IgA may target pathology-preventing bacteria. More recently, two studies investigated the role of microbiota dynamics with paired metabolomic and proteomic analyses to characterize broad biological states of children with SAM transitioning to moderate acute malnutrition (MAM) (Gehrig et al., 2019; Raman et al., 2019). SAM children had higher protein levels of inflammatory markers such as IL-6, C-reactive protein, TNF-ɑ and other NF-κB agonists, but when these children were supplemented with a specific microbiota-directed complementary food - including peanut flour, chickpea flour, soy flour, and banana - these inflammatory markers were reduced. Consumption of this diet by mice colonized with microbiota derived from a donor with post-SAM MAM led to an increase in the size and number of submucosal lymphoid aggregates composed of B cells and T cells relative to GF controls. This difference between GF and colonized mice suggests that the dietary difference in lymphoid aggregates is dependent on microbial colonization, setting the stage for follow-up mechanistic studies. These studies also highlight the therapeutic potential of microbiota-directed nutritional interventions, motivating ongoing work in preclinical models and clinical studies to treat immune-related diseases.
An alternative to studying complex dietary shifts is to alter a single dietary component followed by paired analyses of the microbiota and immune responses. For example, mice fed a vitamin D deficient (D−) diet had lower frequencies of Treg cells (FoxP3+ and Rorγt+ FoxP3+) in the colon and more severe DSS colitis compared to a vitamin D containing (D+) diet (Cantorna et al., 2019). The microbiota transfer from D+ or D− mice to GF recipients phenocopied these results. B. thetaiotaomicron and the Clostridium cluster XIVa were decreased in D− mice corresponding to decreased Treg cells. SCFA production by these bacteria is implicated in Treg cell induction (Arpaia et al., 2013; Atarashi et al., 2013; Ohnmacht et al., 2015; Sefik et al., 2015; Smith et al., 2013b); therefore, the authors propose that decreased levels of these bacteria explain Treg cell decreases on a D− diet. Additional work is needed to test if these specific bacteria are necessary and sufficient for the phenotypic differences and if SCFAs are the primary mechanisms responsible for altered colitis severity and Treg cells. Similar microbiota transplantation studies have been performed to investigate the impact of emulsifiers on immune-microbiome interactions. Transplantation of the gut microbiotas of mice fed two commonly used emulsifiers (carboxymethylcellulose or polysorbate-80) recapitulated the worsened colitis severity and increased intestinal inflammation seen in donor mice (Chassaing et al., 2015). These findings suggest that multiple dietary perturbations can affect immune-microbiome interactions relevant to inflammatory bowel disease (IBD).
While microbiome transplants are a powerful research tool to test causal relationships between diet, the microbiome, and immunity, there are important caveats to consider in these studies. One issue is whether or not the altered microbial community structure initiated by diet is maintained in the recipient animals in the absence of the original selection pressure (Turnbaugh et al., 2009). Another challenge is the generalizability of these results given the logistical limitations of expanding the number of independent donor samples tested (Walter et al., 2020). A useful control is to include heat-killed samples to determine if the resulting phenotype requires viable cells or could instead be mediated by an immunomodulatory metabolite or protein. It can be incredibly difficult to elucidate the mechanisms through which diet shapes complex microbial communities let alone the numerous potential mechanisms through which an altered microbiome could shape immunity. More work is needed to explore systematic approaches to identify the active components of microbial communities, analogous to the activity-guided fractionation methods that are routinely used in natural products discovery.
The role of a dietary factor in shifting the microbiome and the associated functional consequences can be studied with the lack or supplementation of the factor. For example, mice fed a fiber-free diet have reduced colonic mucus layer thickness (Desai et al., 2016; Earle et al., 2015). The lack of complex dietary fibers results in the expansion of mucus degrading bacteria including Akkermansia muciniphila, Bacteroides caccae, Bacteroides ovatus, and Eubacterium rectale, in gnotobiotic mice colonized with a synthetic gut microbiota (comprised of 14 representative species from the 5 main phyla found in humans) (Desai et al., 2016). A strength of using a synthetic community is the availability of well annotated genomes and previous literature (Everard et al., 2013), which revealed a shift in the microbiome from fiber to carbohydrate active enzymes such as mucin O-glycan in mice fed a fiber-free diet (Desai et al., 2016). This corresponded to a decreased mucus layer in mice fed a fiber-free diet accompanied by increased protein levels of the inflammatory marker lipocalin 2 decreased and colon lengths. A key question is whether supplementation of a diet with purified dietary factors can alleviate phenotypes associated with a deficient diet. Supplementation of synthetically colonized mice on fiber-free diet with purified fibers (14 host indigestible fibers) did not mitigate mucus degradation seen in the fiber-free diet (Desai et al., 2016). However, a recent study found that supplementation of mice on a low fiber diet with Bifidobacterium longum and inulin prevented mucus deterioration (Schroeder et al., 2018). In a companion study, inulin supplementation to mice fed a high-fat diet (HFD) rescued colon shortening and restored production of the antimicrobial peptide, Reg3γ, and IL-22 levels (Zou et al., 2018). These protective effects of inulin were not observed in GF mice or Il22−/− mice, suggesting a dependence on the presence of an intact microbiota and IL-22 signaling (Zou et al., 2018). Together, these studies highlight strengths and drawbacks to synthetic microbiota studies. While these studies provide a strong tool to investigate alterations of a sequenced community, these communities may lack members of the microbiota that impact immune responses to dietary supplementation. Therefore, studies using both complex and synthetic communities can form a more complete picture of both the biology of how a dietary shift alters the metabolism of members of the microbiota as well as the broader question of the overall immune consequences of the diet mediated shifts in the gut microbiota.
An important component of adaptive immunity is the antigen-specific response. Members of the microbiota induce antigen-specific responses (Goto et al., 2014). Therefore, a question in the field is whether diet affects antigen expression in members of the microbiota thus impacting antigen-specific responses. Recently, antigen-specific responses to Bacteroides thetaiotaomicron were investigated (Wegorzewska et al., 2019). Using B. thetaiotaomicron antigen-specific T cells combined with dietary alterations, the authors observed that higher glycan levels and lower salt levels decreased B. thetaiotaomicron antigen expression in vitro. Dietary glucose decreased expression of the B. thetaiotaomicron antigen and decreased antigen-specific T cells in vivo suggesting that dietary factors can impact antigen dependent immunity.
Another intriguing hypothesis is that dietary components and downstream metabolites influence the composition of the microbiota and subsequent immune responses via direct effects on bacterial growth and viability (Carmody et al., 2019). For example, the SCFA butyrate inhibits Lactobacillus reuteri growth in vitro (Zegarra-Ruiz et al., 2019). Daily gavage of L. reuteri exacerbated a toll-like receptor 7 (TLR7)-dependent mouse model of lupus but a resistant starch diet ameliorated disease pathologies such as splenomegaly, type 1 IFN induction in the spleen and ileum, and nephritis (Figure 2b). These results suggest that the benefits of resistant starch in lupus may be mediated via butyrate production by other bacteria resulting in the decreased growth of L. reuteri. Dietary microbial metabolites can also alter host signaling pathways in ways that feed back on microbial fitness. For example, macrophages differentiated in the presence of butyrate have increased antimicrobial responses via histone deacetylase (HDAC) 3 inhibition (Schulthess et al., 2019), suggesting that butyrate may also act via host signaling pathways to repress the growth of L. reuteri (Figure 2b). Additional support for the growth inhibition hypothesis comes from studies of mice fed a high salt diet, which resulted in decreased levels of Lactobacillus murinus (Wilck et al., 2017). The growth of L. murinus is inhibited in vitro by increasing NaCl concentrations and supplementation of mice on a high salt diet with L. murinus reduced Th17 cells and symptoms in a mouse model of multiple sclerosis (EAE) (Figure 2c). However, the mechanisms of how high salt concentrations influence L. murinus growth in vivo remain unclear as other members of the microbiota are similarly affected by increasing salt concentrations.
Teaming up on immunity: diet and the microbiota affect host processes that influence immunity
Diet can shape the microbiota through indirect effects on host signaling (Figure 1e). For example, dietary fat intake induces host bile acid (BA) production (Carmody and Turnbaugh, 2014; David et al., 2014), affecting both gut microbial metabolism and immune cell function. Increased levels of Bilophila wadsworthia in mice fed a milk fat diet were attributed to the increased hepatic taurine conjugation of BAs (Devkota et al., 2012). Mice fed a low-fat diet supplemented with taurocholic acid had increased levels of B. wadsworthia, increased Th1 cell frequencies and numbers, and higher incidence of colitis in Il10−/− mice (Figure 3a). These findings are supported by data in human subjects which revealed that fecal BA concentrations, B. wadsworthia, and bacterial sulfite reductases are elevated in response to a high-fat animal-based diet relative to a plant-based diet (David et al., 2014). More recently, we showed that dietary increases in the host production of the ketone body β-hydroxybutyrate (βHB) leads to decreased intestinal levels of Bifidobacterium adolescentis and Th17 cells (Ang et al. 2020). More work is needed to explore the functional consequences of enteric ketone bodies for autoimmunity and host defense. Taken together, these studies highlight how changes in host metabolism in response to diet has broader impacts on the microbiome that feeds back on immunity.
Figure 3. Diet alters production of host and microbial metabolites feeding back on immune responses.
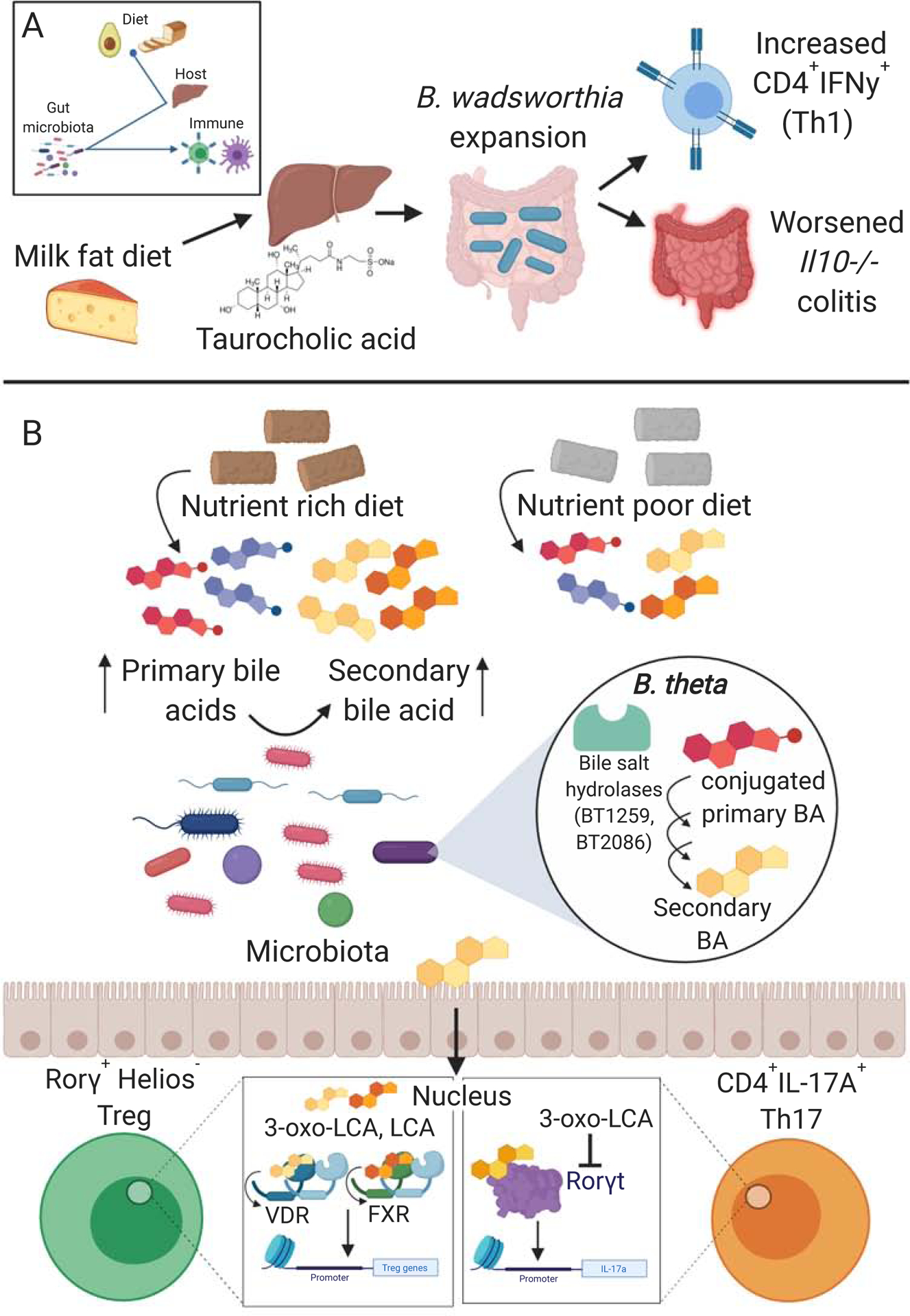
(A) Mice fed a milk fat diet had increased levels of hepatic taurine conjugation of bile acids, which was associated with a bloom in B. wadsworthia. Supplementation of mice on a low-fat diet with taurocholic acid resulted in increased levels of B. wadsworthia, Th1 cells, and higher incidence of colitis in Il10−/− mice (Devkota et al., 2012). (B) Mice on a nutrient rich diet compared to a nutrient poor (minimal) diet have different compositions of primary and secondary bile acid (BA) pools. Bile salt hydrolase enzymes along with other microbial enzymes are involved in the production of secondary bile acids from primary bile acids (Yao et al., 2018). Secondary bile acids can alter immune responses via their interactions with immune receptors such as the nuclear receptors vitamin D receptor (VDR) and farnesoid X receptor (FXR) (Song et al., 2020) and the Th17 cell master transcription factor Rorγt (Hang et al., 2019). For example, 3-oxo-lithocholic acid (3-oxo-LCA) directly binds and inhibits Rorγt resulting in decreased levels of CD4+IL-17A+ Th17 cells (Hang et al., 2019).
Diet can also influence the production of host metabolites that are further metabolized by the microbiota into immunomodulatory compounds. A diverse array of secondary BAs are produced following bacterial deconjugation and dehydroxylation of primary BAs produced by the host. Primary and secondary BAs have been studied for their relative impacts on immunity due to their ability to signal via GPCRs and nuclear receptors (Fiorucci et al., 2018). In a screen of 29 BA metabolites, two secondary BAs, 3-oxoLCA and isoalloLCA, had distinct effects on T cells, where 3-oxoLCA inhibited CD4+IL-17A+ Th17 cell differentiation and isoalloLCA promoted Treg cells in vitro and in vivo (Hang et al., 2019). Using retinoic acid receptor-related orphan receptor gamma (Rorγt) binding and luciferase activity assays, the authors provided evidence for an interaction between 3-oxoLCA and Rorγt, which results in the suppression of Rorγt activity. These data support a mechanistic model of a direct interaction between a secondary BA and an immune cell master transcription factor, which results in an altered immune response. However, it is unclear which microbial enzymes are responsible for the production of these BAs or if and how diet alters production of these compounds. In a subsequent study, a connection between diet, composition of BAs, and altered immunity was established. SPF mice fed a nutrient rich (NRD) or minimal diet (MD: a nutrient poor diet - no cholesterol, lower levels of nutrients, less diverse fatty acids, different amino acid compositions) had distinct Treg cell responses (Song et al., 2020). SPF mice on the NRD had higher levels of colonic Treg cell frequencies (Rorγ+Helios−) compared to SPF mice on a MD or GF mice on a rich diet. The higher frequencies of Treg cells were correlated with an altered colonic BA pool. Supplementation of the MD with combinations of secondary lithocholic/3-oxo-lithocholic acids was sufficient to restore Treg cell frequencies. The production of BA conjugates has been linked to bacterially encoded bile salt hydrolases (BSHs). For example, BSHs from Bacteroides thetaiotaomicron modulate the BA conjugate tauro-β-muricholic acid and downstream immune transcriptional pathways, as revealed by comparisons of gnotobiotic mice colonized with wild-type (wt) and isogenic BSH-deficient (BT2086) B. thetaiotaomicron (Yao et al., 2018). Consistent with these findings, monocolonization of BSH-deficient (BT1259 and BT2086) B. thetaiotaomicron led to decreased colonic Treg cell compared to colonized with wt B. thetaiotaomicron (Song et al., 2020). These data suggest that bacterial metabolism of BAs drives Treg cell accumulation. By analyzing transgenic mice deficient for 4 GPCRs and 5 combinations of nuclear receptors, the authors identified two nuclear receptors [vitamin D receptor (VDR) and to a lesser extent farnesoid X receptor (FXR)] that impact colonic Treg cells. More work is necessary to test the hypothesis that dietary shifts in BA levels and their subsequent bacterial metabolism and signaling through VDR shape Treg cells and downstream host phenotypes. Do GF VDR-deficient mice respond to purified BAs or the different strains of B. thetaiotaomicron? If supplementation of GF mice with combinations of secondary lithocholic/3-oxo-lithocholic acids rescues the Treg cell population as it did in SPF mice, then this would rule out the role of other diet- or microbiome-derived factors in BA-mediated Treg cell induction. Together, these studies establish that dietary and microbial factors influence the BA pool, which impacts immune responses such as Treg cell levels, in some cases through direct binding and inhibition of master transcription factors in the case of 3-oxoLCA and Rorγt or through signaling via host nuclear receptors (Figure 3b).
Members of the microbiota can alter host metabolism of immunomodulatory dietary factors (Figure 1f). Many dietary factors have reciprocal interactions with the microbiota, altering microbial community structure while being metabolized by microbial enzymes. For instance, the vitamin A metabolite retinoic acid (RA) increases FoxP3+ Treg cell (Elias et al., 2008; Xiao et al., 2008), inhibits Th17 cell polarization (Elias et al., 2008), and alters the gut microbiota (Hibberd et al., 2017). The microbiota can also suppress host metabolism of vitamin A into RA, having downstream effects on immunity (Grizotte-Lake et al., 2018). For instance, Clostridia suppress the expression of retinol dehydrogenase 7 (Rdh7) in intestinal epithelial cells (IECs) which is necessary for RA production. Deleting Rdh7 in IECs with Villin-Cre (RdhΔIEC) significantly decreases RA levels in the small intestine. This deletion also decreases IL-22 expression, IL-22+CD3+ and IL-22+CD3− cells (IL-22 producing ILC and T cells), and production of antimicrobial peptides (AMP) with a corresponding decrease in spatial segregation between the IECs and the gut microbiota. RdhΔIEC mice are protected from challenge with Salmonella typhimurium, suggesting that higher levels of IL-22 enhances the ability of S. typhimurium to colonize mice. IL-22 is important for barrier function and protection from other pathogens such as Citrobacter rodentium (Eidenschenk et al., 2014; Zheng et al., 2008); therefore, it is counterintuitive that IL-22 promotes S. typhimurium colonization. However, IL-22 has been shown to promote S. typhimurium by suppressing the growth of gut bacterial symbionts (Behnsen et al., 2014). This suggests that IL-22 may act as a rheostat where levels must be precisely tuned such that enteric symbionts flourish and provide colonization resistance to pathogens such as S. typhimurium. It remains unclear why the gut microbiota would be more sensitive to AMPs induced by IL-22 than S. typhimurium. Together, these findings reveal that the microbiota influences vitamin A metabolism by the host, which results in altered immunity (Figure 4a). Gnotobiotic mice colonized with a defined human gut bacterial community on a vitamin A deficient diet had compositional shifts and altered transcriptional activity (Hibberd et al., 2017). This effect could be due to direct effects of vitamin A on the microbiota or due to differential host responses. Further mechanistic studies are needed to tease apart the direct versus indirect effects of vitamin A on the structure and function of the gut microbiota. Together, these studies highlight the reciprocal interactions of vitamin A with the microbiota and immunity underscoring a central issue in studying the interplay between diet, microbiota and immunity, where feedback loops between these factors complicate our understanding of the overall role of each individual factor.
Figure 4. The gut microbiota alters the metabolism of immunomodulatory dietary factors and immune responses alter immunomodulatory bacteria.
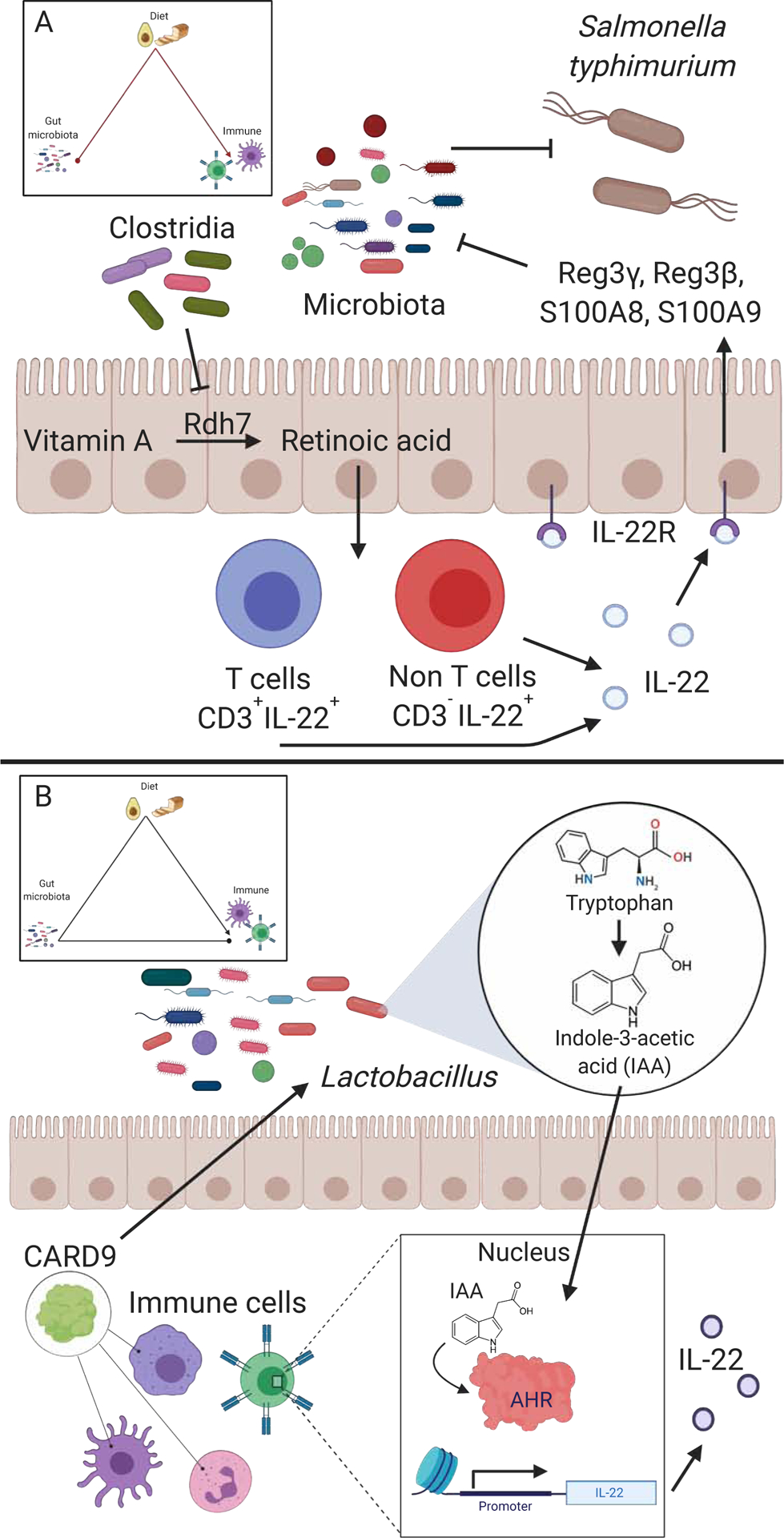
(A) Members of the Clostridia class inhibit the expression of retinol dehydrogenase 7 (Rdh7) in intestinal epithelial cells (IECs) (Grizotte-Lake et al., 2018). Rdh7 is responsible for metabolizing vitamin A into retinoic acid (RA). RA promotes the production of IL-22, which induces antimicrobial peptides (AMPs) -Reg3γ, Reg3B, S100A8, S100A9. These AMPs suppress the growth of gut microbiota. The inhibition of members of the gut microbiota by AMPs may contribute to the reduction of colonization resistance to Salmonella typhimurium. (B) Caspase Recruitment Domain-containing protein 9 (CARD9) is an important immune signalling protein involved in fungal immune responses. Card9−/− mice have decreased levels of the gut bacterium L. reuteri suggesting Card9 promotes L. reuteri colonization. L. reuteri metabolizes tryptophan into indole-3-acetic acid (IAA) which is an aryl hydrocarbon receptor (AHR) agonist (Lamas et al., 2016). AHR activation results in IL-22 production, among other things.
Regaining control: immune effects on microbial metabolism of dietary factors feeds back on immunity
Studies in transgenic mice have begun to reveal host immune factors that shape the gut microbiota and the subsequent immune response to dietary interventions (Figure 1g). There is a rich body of literature that has demonstrated an immunological influence on the microbiota (Hooper and Macpherson, 2010); including mucosal layers (Johansson et al., 2008; Van der Sluis et al., 2006), antimicrobial peptides (Brandl et al., 2007; Pütsep et al., 2000; Vaishnava et al., 2008), IgA production (Macpherson et al., 2000; Suzuki et al., 2004), T cell coordination of immune responses (Kubinak et al., 2015), inflammasome signaling (Elinav et al., 2011; Levy et al., 2015), and TLR5 signaling (Cullender et al., 2013; Fulde et al., 2018; Vijay-Kumar et al., 2010). However, few studies have evaluated whether or not modulations to the gut microbiota by the immune system can alter microbial metabolism of dietary products in a way that feeds back on immunity. One example of this comes from the Caspase Recruitment Domain-containing protein 9 (CARD9), which is a susceptibility gene for IBD in humans. IBD patients with the CARD9 risk allele have reduced AHR activation which correlates with a general decrease in IAA levels in IBD patients (Lamas et al., 2016). CARD9 plays an important role in immunity such as fungal immune responses as well as TLR signaling (Goodridge et al., 2009; Hara et al., 2008; Lanternier et al., 2015), and Card9−/− mice have impaired recovery from DSS colitis challenge (Lamas et al., 2016). Card9−/− mice have decreased levels of the gut bacterium L. reuteri, among other shifts in the gut microbiota. Transplantation of the Card9−/− microbiota to wt GF mice recapitulated the DSS colitis phenotype, potentially due to decreased metabolism of tryptophan by L. reuteri to AHR agonists, as discussed above. This example demonstrates that differences in a single immune signaling protein, CARD9, can lead to immunologically relevant shifts in the gut microbial community structure via alterations to the microbial metabolism of dietary products (Figure 4b).
Innate sensing of microbial signals has been tied to microbial community structure. When the ability to sense certain features of the microbiota, such as flagella in the case of TLR5, the composition of the microbiota is altered (Carvalho et al., 2012; Vijay-Kumar et al., 2010). Tlr5−/− mice on a HFD develop worsened metabolic syndrome compared to wt mice including increased immune infiltrates in the pancreas (Vijay-Kumar et al., 2010). The transfer of Tlr5−/− cecal content into GF mice was sufficient to recapitulate metabolic syndrome phenotypes compared to the transfer of wt content with no dietary intervention (Vijay-Kumar et al., 2010). Similarly, the deficiency of the TLR signaling adaptor protein MyD88 in T cells changes the composition of the microbiota via altered IgA targeting of the microbiota and metabolic syndrome developed when mice are fed a HFD (Kubinak et al., 2015; Petersen et al., 2019). This metabolic syndrome was abrogated when the mice are treated with antibiotics (Petersen et al., 2019). One of the central caveats with these studies is that the deletion of these key innate sensing pathways can have significant impacts on host signaling. Therefore, cell-specific deletion of these pathways such as the T cell specific Myd88−/− or follow-up studies which have specifically deleted Tlr5 from dendritic cells and intestinal epithelial cells (Chassaing et al., 2014) can begin to parse out which cells are involved in these phenotypes as well as decrease the overall impact of these deletions on other host processes. These studies provide evidence that innate shaping of the microbiota impacts metabolic syndrome phenotypes driven by a HFD.
Another example of immunological control of the microbiota and immunomodulatory diet-derived metabolites comes from studies of inflammasome mediated signals. The deletion of the Nlrp6 Asc-dependent inflammasome assembly alters the gut microbiota (Elinav et al., 2011; Levy et al., 2015) corresponding to decreased IL-18 levels, a major downstream product of inflammasome activation (Levy et al., 2015). The administration of IL-18 to Nlrp6−/− or Asc−/− mice altered the microbiota composition compared to wt controls. These findings suggest that shifts in the gut microbiota in mice deficient in Nlrp6 Asc-dependent inflammasome function is influenced by IL-18 production. However, these results do not fully establish that microbiota alterations in Nlrp6−/− mice are solely driven by decreased IL-18 and not other inflammasome dependent immune responses. A dual deletion of Il18 and Nlrp6 would further support this mechanism. Wt GF mice co-housed with Asc−/− SPF mice had lower IL-18 production than their wt cohoused counterparts suggesting that IL-18 is repressed by exposure to Asc−/− microbiota. Metabolite analysis revealed an enrichment for taurine and a depletion of spermidine and histamine from the cecal content of GF wt mice cohoused with Asc−/− SPF mice compared to the wt co-housed control. Taurine is an amino sulfonic acid and provides a substrate for BA formation and is modulated by members of the microbiota (Brestoff and Artis, 2013). Spermidine and histamine are amino acid metabolites produced by the microbiota and host (Levy et al., 2015). Taurine increased IL-18 levels while spermidine and histamine decreased IL-18 in cultured colon explants. Thus, alterations to the microbiota in Asc−/− mice contribute to differential levels of amino acid derivatives (taurine, spermidine, and histamine), which can feedback on immunity (IL-18). While there are other examples of host immune factors influencing intestinal microbial ecology, these studies provide examples of immune-mediated alterations to the microbiota impacting microbial metabolism of dietary derived immunomodulatory products.
CONCLUDING REMARKS
Observational human nutrition and microbiome studies have been invaluable in prioritizing dietary factors, immune cells, and microorganisms to study. Continued progress requires experimentally tractable systems that enable researchers to disentangle these tripartite interactions. These fundamental studies are complementary to ongoing observational and interventional studies in human cohorts, providing candidate biomarkers and therapeutic targets (Turnbaugh, 2020). More in-depth experiments dissecting pairwise interactions will also be critical, as these mechanistic insights set the stage for interdisciplinary cross-system studies.
Several broad patterns have emerged from the work to date. Diet shapes the gut microbiome through direct effects on bacterial growth and viability and more complex indirect effects mediated by diet-dependent changes to host cells. In turn, the microbiome plays a critical role in expanding our metabolism of dietary substrates, which has energetic implications as well as an important signaling role to immune cells through host receptors. We categorized these interactions into 6 general types of mechanisms (Figure 1b–g). However, given the novelty of this field of study, we believe these to be the tip of the iceberg and as more studies contribute insight into this area these mechanisms should be added to and refined. For instance, it is clear that the microbiota impacts the uptake of dietary factors via alterations to host signaling but the impact of this on immunity has not yet been fully investigated. For example, the microbiota affects expression of histone deacetylase 3 (HDAC3) which alters the expression of a lipid transporter CD36 (Kuang et al., 2019). While the link between microbiota and uptake of dietary factors is established, the impact on immunity remains unclear. Another example where pairwise interactions between diet-microbiota and microbiota-immune related disease have been established but the tripartite interactions have not yet been investigated comes from rheumatoid arthritis (RA). Strain-level variation in the Prevotella copri species is associated with high fiber intake (De Filippis et al., 2019) and this species is also linked to RA (Scher et al., 2013). However, it remains unclear if decreasing fiber intake would lower P. copri levels in RA patients or what the net consequences for immunity would be. This is a prime example of the broader trend where microbiome literature suggests that microbe X induces worsened mouse models of autoimmunity and is impacted by diet Y; however, the impact of the alteration to microbe X by diet Y on immunity remains untested.
A central question in the field is whether higher-level conclusions can be drawn about which microbiota members are involved in dietary immunomodulation. The historical focus on single culturable and genetically tractable model organisms may leave the impression that there are a small number of immunomodulatory members of the human microbiota. We propose that this observation is reflective of a bias in the existing literature, motivating more systematic efforts to determine which bacteria and other microorganisms interact with diet and host immunity (Geva-Zatorsky et al., 2017). Isolate-based screens also typically fail to capture genetic variations within a bacterial species (Bisanz et al., 2020); more work is needed to dissect the host-microbiota interactions at the strain-level. On a methodological level, multiple researchers have taken a “bottom-up” approach wherein individual dietary components, microbes, immune factors, or bacterial genes are studied in mechanistic detail. “Top-down” microbiome surveys provide valuable associations and causal relationships between complex microbial communities and host phenotypes. An important area for future work will be to bridge these two approaches by building more complex synthetic microbial communities and developing systematic approaches for identifying the dietary and microbial effectors of immune phenotypes.
Higher-order interactions are also beginning to be revealed, consistent with the hypothesis that there is extensive feedback between diet, the microbiome, and immunity. For example, the immune system can alter the abundance of bacteria, changing how dietary factors are metabolized by the microbiota, and then resulting in changes in immunity (Figure 1g). As with other complex systems, these interactions are likely dynamic and will be difficult to understand by only studying their component parts. While there are examples of microbiome-independent effects of diet on immunity, there is a substantial amount of literature that reveals a role of the microbiota in dietary immune modulation. Further, there are likely synergistic effects of diet and the microbiota on immunity, which is an important area of future investigation. Methods to tune each node in situ and in real-time are an essential step towards gaining more quantitative mechanistic insights.
To systematically address gaps in the field we must consider the current successful tools and approaches and propose novel techniques. Advances in nutrition, ecology, bacterial genetics, and chemical biology will provide important tools. There is a need to systematically pull apart and re-assemble dietary components to individually assess their impact on the microbiota and immunity. A seminal example of this is the defined perturbation in diet combined with combinatorial communities in gnotobiotic mice to model how changes in macronutrient intake alter the structure of the microbiota (Faith et al., 2011). Combining this approach with immune profiling could assess how these diet-mediated shifts in the microbiota impact immunity. Additionally, more subtle alterations to the diet could reveal the role of specific nutrients such as a single amino acid. These studies will be aided by efforts to build synthetic microbial communities that model complex interactions between 100s-1000s of microbial strains. By coupling these synthetic microbial communities to organoids and other in vitro or ex vivo models we can begin to more rapidly screen dietary components and microbiomes for their downstream effects on immunity. However, these approaches have several important limitations. As discussed earlier, while synthetic systems allow for better mechanistic investigations, we can miss important biology if members of the microbiota are excluded. Further, gnotobiotic mice and the currently available in vitro models are distinct from humans in numerous respects, motivating efforts to use model animals with a more “human-like” anatomy (e.g., pigs), to construct more sophisticated multicellular in vitro models, or to combine methods for generating “humanized” transgenic animals.
In parallel, a “synthetic nutrition” approach can be used in human cohorts to assess the causal role of specific dietary perturbations on the microbiome and immunity. A first step towards this goal was provided by a recent study wherein 34 subjects were sampled daily for 17 days, integrating 24 hour food records and metagenomics to track longitudinal relationships between food intake and the gut microbiota (Johnson et al., 2019). The design of this study would lend itself well to additional assessment of the immune system where subjects could be densely sampled and diet intake, microbiota composition, and immune profiling could be combined to longitudinally assess these tripartite relationships. Taking this a step further, randomized dietary interventions in humans could simultaneously characterize the microbiome and immunity.
Bacterial genetics and biochemistry will also be essential to understanding the role of the microbiome in nutrition and immunology. Our current knowledge base is built on a handful of model bacteria, resulting in a vast “dark matter” of unannotated genes, proteins, and metabolites. Multiple studies discussed here provide a strong proof-of-concept for the value of understanding bacterial genetics (Song et al., 2020; Yao et al., 2018). Combining these approaches with defined diet alterations will help to connect individual genes to diet-dependent immune phenotypes. The construction of both gain-of-function and loss-of-function libraries in diverse bacterial and fungal species will provide valuable resources, as demonstrated for B. thetaiotaomicron (Goodman et al., 2009). Chemical screens pairing diet-derived microbial metabolites to host receptors, as recently demonstrated for GPCRs (Chen et al., 2019; Colosimo et al., 2019), coupled to methods for activity-based protein profiling (Jariwala et al., 2019) will also be informative to identify the targets of diet-derived microbial metabolites and the microorganisms responsible for their production. Hypotheses developed through these reductionist studies can then be tested in humans and animal models to evaluate their physiological and clinical relevance.
In summary, the microbiome field is at an inflection point, where researchers are beginning to integrate findings from multiple reductionist studies, moving closer to the complexity revealed by surveys of the natural system. The studies discussed herein provide support for the feasibility and utility of this approach while also highlighting the gaps in our knowledge and limitations to currently available methods. We may have “miles to go before we sleep” but if the recent advances are any indication, we are well on our way.
Acknowledgments
We thank Kathy Lam, Djem Kissiov, and Qi Yan Ang, for critical feedback on the manuscript. Figures were created using BioRender.com. This project was supported by the National Institutes of Health (R01HL122593; R21CA227232; R01AR074500; 5T32AI060537, 1F32AI147456). PJT is a Chan Zuckerberg Biohub investigator and was a Nadia’s Gift Foundation Innovator supported, in part, by the Damon Runyon Cancer Research Foundation (DRR-42-16) and the Searle Scholars Program (SSP-2016-1352).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
Dr. Turnbaugh is on the scientific advisory boards for Kaleido, Pendulum, Seres, and SNIPRbiome; there is no direct overlap between the current study and these consulting duties. All other authors have no relevant declarations.
REFERENCES
- Ang QY, Alexander M, Newman JC, Tian Y, Cai J, Upadhyay V, Turnbaugh JA, Verdin E, Hall KD, Leibel LL, Ravussin E, Rosenbaum M, Patterson AD, and Turnbaugh PJ (2020). Ketogenesis alters the gut microbiome resulting in decreased intestinal Th17 levels. Cell, 10.1016/j.cell.2020.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, et al. (2013). Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504, 451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, et al. (2013). Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500, 232–236. [DOI] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, Narushima S, Suda W, Imaoka A, Setoyama H, Nagamori T, et al. (2015). Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell 163, 367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnsen J, Jellbauer S, Wong CP, Edwards RA, George MD, Ouyang W, and Raffatellu M (2014). The cytokine IL-22 promotes pathogen colonization by suppressing related commensal bacteria. Immunity 40, 262–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisanz JE, Soto-Perez P, Lam KN, Bess EN, and Haiser HJ (2020). A genomic toolkit for the mechanistic dissection of intractable human gut bacteria. Cell Host Microbe. 10.1016/j.chom.2020.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blander JM, Longman RS, Iliev ID, Sonnenberg GF, and Artis D (2017). Regulation of inflammation by microbiota interactions with the host. Nat. Immunol 18, 851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley CP, Teng F, Felix KM, Sano T, Naskar D, Block KE, Huang H, Knox KS, Littman DR, and Wu H-JJ (2017). Segmented filamentous bacteria provoke lung autoimmunity by inducing gut-lung axis Th17 cells expressing dual TCRs. Cell Host Microbe 22, 697–704.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandl K, Plitas G, Schnabl B, DeMatteo RP, and Pamer EG (2007). MyD88-mediated signals induce the bactericidal lectin RegIIIγ and protect mice against intestinal Listeria monocytogenes infection. J. Exp. Med 204, 1891–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brestoff JR, and Artis D (2013). Commensal bacteria at the interface of host metabolism and the immune system. Nat. Immunol 14, 676–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byndloss MX, Olsan EE, Rivera-Chávez F, Tiffany CR, Cevallos SA, Lokken KL, Torres TP, Byndloss AJ, Faber F, Gao Y, et al. (2017). Microbiota-activated PPAR-γ signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 357, 570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantorna MT, Lin Y-D, Arora J, Bora S, Tian Y, Nichols RG, and Patterson AD (2019). Vitamin D regulates the microbiota to control the numbers of RORγt/FoxP3 regulatory T cells in the colon. Frontiers in Immunology 10, 1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmody RN, and Turnbaugh PJ (2014). Host-microbial interactions in the metabolism of therapeutic and diet-derived xenobiotics. J. Clin. Invest 124, 4173–4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmody RN, Gerber GK, Luevano JM Jr, Gatti DM, Somes L, Svenson KL, and Turnbaugh PJ (2015). Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe 17, 72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmody RN, Bisanz JE, Bowen BP, Maurice CF, Lyalina S, Louie KB, Treen D, Chadaideh KS, Maini Rekdal V, Bess EN, et al. (2019). Cooking shapes the structure and function of the gut microbiome. Nat Microbiol 4, 2052–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho FA, Koren O, Goodrich JK, Johansson MEV, Nalbantoglu I, Aitken JD, Su Y, Chassaing B, Walters WA, González A, et al. (2012). Transient inability to manage proteobacteria promotes chronic gut inflammation in TLR5-deficient mice. Cell Host Microbe 12, 139–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra RK (1996). Nutrition, immunity and infection: From basic knowledge of dietary manipulation of immune responses to practical application of ameliorating suffering and improving survival. Proc. Natl. Acad. Sci. U. S. A 93, 14304–14307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassaing B, Ley RE, and Gewirtz AT (2014). Intestinal epithelial cell toll-like receptor 5 regulates the intestinal microbiota to prevent low-grade inflammation and metabolic syndrome in mice. Gastroenterology 147, 1363–1377.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassaing B, Koren O, Goodrich JK, Poole AC, Srinivasan S, Ley RE, and Gewirtz AT (2015). Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature 519, 92–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Nwe P-K, Yang Y, Rosen CE, Bielecka AA, Kuchroo M, Cline GW, Kruse AC, Ring AM, Crawford JM, et al. (2019). A forward chemical genetic screen reveals gut microbiota metabolites that modulate host physiology. Cell 177, 1217–1231.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun E, Lavoie S, Fonseca-Pereira D, Bae S, Michaud M, Hoveyda HR, Fraser GL, Gallini Comeau CA, Glickman JN, Fuller MH, et al. (2019). Metabolite-sensing receptor Ffar2 regulates colonic group 3 innate lymphoid cells and gut immunity. Immunity 51, 786–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cignarella F, Cantoni C, Ghezzi L, Salter A, Dorsett Y, Chen L, Phillips D, Weinstock GM, Fontana L, Cross AH, et al. (2018). Intermittent fasting confers protection in CNS autoimmunity by altering the gut microbiota. Cell Metab. 27, 1222–1235.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colosimo DA, Kohn JA, Luo PM, Piscotta FJ, Han SM, Pickard AJ, Rao A, Cross JR, Cohen LJ, and Brady SF (2019). Mapping interactions of microbial metabolites with human G-protein-coupled receptors. Cell Host Microbe 26, 273–282.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullender TC, Chassaing B, Janzon A, Kumar K, Muller CE, Werner JJ, Angenent LT, Bell ME, Hay AG, Peterson DA, et al. (2013). Innate and adaptive immunity interact to quench microbiome flagellar motility in the gut. Cell Host Microbe 14, 571–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, et al. (2014). Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Filippis F, Pasolli E, Tett A, Tarallo S, Naccarati A, De Angelis M, Neviani E, Cocolin L, Gobbetti M, Segata N, et al. (2019). Distinct genetic and functional traits of human intestinal Prevotella copri strains are associated with different habitual diets. Cell Host Microbe 25, 444–453.e3. [DOI] [PubMed] [Google Scholar]
- Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M, Pudlo NA, Kitamoto S, Terrapon N, Muller A, et al. (2016). A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell 167, 1339–1353.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A, Antonopoulos DA, Jabri B, and Chang EB (2012). Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature 487, 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earle KA, Billings G, Sigal M, Lichtman JS, Hansson GC, Elias JE, Amieva MR, Huang KC, and Sonnenburg JL (2015). Quantitative imaging of gut microbiota spatial organization. Cell Host Microbe 18, 478–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eidenschenk C, Rutz S, Liesenfeld O, and Ouyang W (2014). Role of IL-22 in microbial host defense. Curr. Top. Microbiol. Immunol 380, 213–236. [DOI] [PubMed] [Google Scholar]
- Elias KM, Laurence A, Davidson TS, Stephens G, Kanno Y, Shevach EM, and O’Shea JJ (2008). Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood 111, 1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, et al. (2011). NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, Guiot Y, Derrien M, Muccioli GG, Delzenne NM, et al. (2013). Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. U. S. A 110, 9066–9071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith JJ, McNulty NP, Rey FE, and Gordon JI (2011). Predicting a human gut microbiota’s response to diet in gnotobiotic mice. Science 333, 101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorucci S, Biagioli M, Zampella A, and Distrutti E (2018). Bile acids activated receptors regulate innate immunity. Front. Immunol 9, 1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulde M, Sommer F, Chassaing B, van Vorst K, Dupont A, Hensel M, Basic M, Klopfleisch R, Rosenstiel P, Bleich A, et al. (2018). Neonatal selection by Toll-like receptor 5 influences long-term gut microbiota composition. Nature 563, E25. [DOI] [PubMed] [Google Scholar]
- Gehrig JL, Venkatesh S, Chang H-W, Hibberd MC, Kung VL, Cheng J, Chen RY, Subramanian S, Cowardin CA, Meier MF, et al. (2019). Effects of microbiota-directed foods in gnotobiotic animals and undernourished children. Science 365, eaau4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geva-Zatorsky N, Sefik E, Kua L, Pasman L, Tan TG, Ortiz-Lopez A, Yanortsang TB, Yang L, Jupp R, Mathis D, et al. (2017). Mining the human gut microbiota for immunomodulatory organisms. Cell 168, 928–943.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman AL, McNulty NP, Zhao Y, Leip D, Mitra RD, Lozupone CA, Knight R, and Gordon JI (2009). Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe 6, 279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodridge HS, Shimada T, Wolf AJ, Hsu Y-MS, Becker CA, Lin X, and Underhill DM (2009). Differential use of CARD9 by dectin-1 in macrophages and dendritic cells. J. Immunol 182, 1146–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, Panea C, Nakato G, Cebula A, Lee C, Diez MG, Laufer TM, Ignatowicz L, and Ivanov II (2014). Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity 40, 594–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grizotte-Lake M, Zhong G, Duncan K, Kirkwood J, Iyer N, Smolenski I, Isoherranen N, and Vaishnava S (2018). Commensals suppress intestinal epithelial cell retinoic acid synthesis to regulate interleukin-22 activity and prevent microbial dysbiosis. Immunity 49, 1103–1115.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang S, Paik D, Yao L, Kim E, Jamma T, Lu J, Ha S, Nelson BN, Kelly SP, Wu L, et al. (2019). Bile acid metabolites control TH17 and Treg cell differentiation. Nature 576, 143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara H, Ishihara C, Takeuchi A, Xue L, Morris SW, Penninger JM, Yoshida H, and Saito T (2008). Cell type-specific regulation of ITAM-mediated NF-κB activation by the adaptors, CARMA1 and CARD9. The Journal of Immunology 181, 918–930. [DOI] [PubMed] [Google Scholar]
- Hibberd MC, Wu M, Rodionov DA, Li X, Cheng J, Griffin NW, Barratt MJ, Giannone RJ, Hettich RL, Osterman AL, et al. (2017). The effects of micronutrient deficiencies on bacterial species from the human gut microbiota. Sci. Transl. Med 9, eaal4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, and Littman DR (2016). The microbiota in adaptive immune homeostasis and disease. Nature 535, 75–84. [DOI] [PubMed] [Google Scholar]
- Hooper LV, and Macpherson AJ (2010). Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat. Rev. Immunol 10, 159–169. [DOI] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al. (2009). Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jariwala PB, Pellock SJ, Goldfarb D, Cloer EW, Artola M, Simpson JB, Bhatt AP, Walton WG, Roberts LR, Major MB, et al. (2019). Discovering the microbial enzymes driving drug toxicity with activity-based protein profiling. ACS Chem. Biol 15, 217–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson MEV, Phillipson M, Petersson J, Velcich A, Holm L, and Hansson GC (2008). The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc. Natl. Acad. Sci. U. S. A 105, 15064–15069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AJ, Vangay P, Al-Ghalith GA, Hillmann BM, Ward TL, Shields-Cutler RR, Kim AD, Shmagel AK, Syed AN, Personalized Microbiome Class Students, et al. (2019). Daily sampling reveals personalized diet-microbiome associations in humans. Cell Host Microbe 25, 789–802.e5. [DOI] [PubMed] [Google Scholar]
- Johnston BC, Zeraatkar D, Han MA, Vernooij RWM, Valli C, El Dib R, Marshall C, Stover PJ, Fairweather-Taitt S, Wójcik G, et al. (2019). Unprocessed red meat and processed meat consumption: dietary guideline recommendations from the nutritional recommendations (NutriRECS) consortium. Annals of Internal Medicine 171, 756. [DOI] [PubMed] [Google Scholar]
- Jordan S, Tung N, Casanova-Acebes M, Chang C, Cantoni C, Zhang D, Wirtz TH, Naik S, Rose SA, Brocker CN, et al. (2019). Dietary intake regulates the circulating inflammatory monocyte pool. Cell 178, 1102–1114.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kau AL, Planer JD, Liu J, Rao S, Yatsunenko T, Trehan I, Manary MJ, Liu T-C, Stappenbeck TS, Maleta KM, et al. (2015). Functional characterization of IgA-targeted bacterial taxa from undernourished Malawian children that produce diet-dependent enteropathy. Sci. Transl. Med 7, 276ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khare A, Chakraborty K, Raundhal M, Ray P, and Ray A (2015). Cutting edge: dual function of PPARγ in CD11c+ cells ensures immune tolerance in the airways. J. Immunol 195, 431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KS, Hong S-W, Han D, Yi J, Jung J, Yang B-G, Lee JY, Lee M, and Surh CD (2016). Dietary antigens limit mucosal immunity by inducing regulatory T cells in the small intestine. Science 351, 858–863. [DOI] [PubMed] [Google Scholar]
- Kolodziejczyk AA, Zheng D, and Elinav E (2019). Diet–microbiota interactions and personalized nutrition. Nat. Rev. Microbiol 17, 742–753. [DOI] [PubMed] [Google Scholar]
- Kuang Z, Wang Y, Li Y, Ye C, Ruhn KA, Behrendt CL, Olson EN, and Hooper LV (2019). The intestinal microbiota programs diurnal rhythms in host metabolism through histone deacetylase 3. Science 365, 1428–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubinak JL, Petersen C, Stephens WZ, Soto R, Bake E, O’Connell RM, and Round JL (2015). MyD88 signaling in T cells directs IgA-mediated control of the microbiota to promote health. Cell Host Microbe 17, 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam KN, Alexander M, and Turnbaugh PJ (2019). Precision medicine goes microscopic: engineering the microbiome to improve drug outcomes. Cell Host Microbe 26, 22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamas B, Richard ML, Leducq V, Pham H-P, Michel M-L, Da Costa G, Bridonneau C, Jegou S, Hoffmann TW, Natividad JM, et al. (2016). CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med 22, 598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanternier F, Mahdaviani SA, Barbati E, Chaussade H, Koumar Y, Levy R, Denis B, Brunel A-S, Martin S, Loop M, et al. (2015). Inherited CARD9 deficiency in otherwise healthy children and adults with Candida species–induced meningoencephalitis, colitis, or both. Journal of Allergy and Clinical Immunology 135, 1558–1568.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YK, Menezes JS, Umesaki Y, and Mazmanian SK (2011). Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. U. S. A 108 , 4615–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levan SR, Stamnes KA, Lin DL, Panzer AR, Fukui E, McCauley K, Fujimura KE, McKean M, Ownby DR, Zoratti EM, et al. (2019). Elevated faecal 12,13-diHOME concentration in neonates at high risk for asthma is produced by gut bacteria and impedes immune tolerance. Nature Microbiology 4, 1851–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy M, Thaiss CA, Zeevi D, Dohnalová L, Zilberman-Schapira G, Mahdi JA, David E, Savidor A, Korem T, Herzig Y, et al. (2015). Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell 163, 1428–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macpherson AJ, Gatto D, Sainsbury E, Harriman GR, Hengartner H, and Zinkernagel RM (2000). A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science 288, 2222–2226. [DOI] [PubMed] [Google Scholar]
- Mora JR, Iwata M, and von Andrian UH (2008). Vitamin effects on the immune system: vitamins A and D take centre stage. Nat. Rev. Immunol 8, 685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai M, Noguchi R, Takahashi D, Morikawa T, Koshida K, Komiyama S, Ishihara N, Yamada T, Kawamura YI, Muroi K, et al. (2019). Fasting-refeeding impacts immune cell dynamics and mucosal immune responses. Cell 178, 1072–1087.e14. [DOI] [PubMed] [Google Scholar]
- Ohnmacht C, Park J-H, Cording S, Wing JB, Atarashi K, Obata Y, Gaboriau-Routhiau V, Marques R, Dulauroy S, Fedoseeva M, et al. (2015). The microbiota regulates type 2 immunity through RORγt+ T cells. Science 349, 989–993. [DOI] [PubMed] [Google Scholar]
- Petersen C, Bell R, Klag KA, Lee SH, and Soto R (2019). T cell–mediated regulation of the microbiota protects against obesity. Science 365, eaat9351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole AC, Goodrich JK, Youngblut ND, Luque GG, Ruaud A, Sutter JL, Waters JL, Shi Q, El-Hadidi M, Johnson LM, et al. (2019). Human salivary amylase gene copy number impacts oral and gut microbiomes. Cell Host Microbe 25, 553–564.e7. [DOI] [PubMed] [Google Scholar]
- Pütsep K, Axelsson LG, Boman A, and Midtvedt T (2000). Germ-free and colonized mice generate the same products from enteric prodefensins. Journal of Biological Chemistry 275, 40478–40482. [DOI] [PubMed] [Google Scholar]
- Raman AS, Gehrig JL, Venkatesh S, Chang H-W, Hibberd MC, Subramanian S, Kang G, Bessong PO, Lima AAM, Kosek MN, et al. (2019). A sparse covarying unit that describes healthy and impaired human gut microbiota development. Science 365, eaau4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangan P, Choi I, Wei M, Navarrete G, Guen E, Brandhorst S, Enyati N, Pasia G, Maesincee D, Ocon V, et al. (2019). Fasting-mimicking diet modulates microbiota and promotes intestinal regeneration to reduce inflammatory bowel disease pathology. Cell Rep. 26, 2704–2719.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Round JL, and Mazmanian SK (2010). Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc. Natl. Acad. Sci. U. S. A 107, 12204–12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, Rostron T, Cerundolo V, Pamer EG, Abramson SB, et al. (2013). Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife 2, e01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder BO, Birchenough GMH, Ståhlman M, Arike L, Johansson MEV, Hansson GC, and Bäckhed F (2018). Bifidobacteria or fiber protects against diet-induced microbiota-mediated colonic mucus deterioration. Cell Host Microbe 23, 27–40.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulthess J, Pandey S, Capitani M, Rue-Albrecht KC, Arnold I, Franchini F, Chomka A, Ilott NE, Johnston DGW, Pires E, et al. (2019). The short chain fatty acid butyrate imprints an antimicrobial program in macrophages. Immunity 50, 432–445.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sefik E, Geva-Zatorsky N, Oh S, and Konnikova L (2015). Individual intestinal symbionts induce a distinct population of RORγ+ regulatory T cells. Science 349, 993–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelly AN, Sato Y, Kearney S, and Honda K (2019). Mining the microbiota for microbial and metabolite-based immunotherapies. Nat. Rev. Immunol 19, 305–323. [DOI] [PubMed] [Google Scholar]
- Smith MI, Yatsunenko T, Manary MJ, Trehan I, Mkakosya R, Cheng J, Kau AL, Rich SS, Concannon P, Mychaleckyj JC, et al. (2013a). Gut microbiomes of Malawian twin pairs discordant for kwashiorkor. Science 339, 548–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, Glickman JN, and Garrett WS (2013b). The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Sun X, Oh SF, Wu M, Zhang Y, Zheng W, Geva-Zatorsky N, Jupp R, Mathis D, Benoist C, et al. (2020). Microbial bile acid metabolites modulate gut RORγ+ regulatory T cell homeostasis. Nature 577, 410–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg ED, Smits SA, Tikhonov M, Higginbottom SK, Wingreen NS, and Sonnenburg JL (2016). Diet-induced extinctions in the gut microbiota compound over generations. Nature 529, 212–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Meek B, Doi Y, Muramatsu M, Chiba T, Honjo T, and Fagarasan S (2004). Aberrant expansion of segmented filamentous bacteria in IgA-deficient gut. Proc. Natl. Acad. Sci. U. S. A 101, 1981–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng F, Klinger CN, Felix KM, Bradley CP, Wu E, Tran NL, Umesaki Y, and Wu H-JJ (2016). Gut microbiota drive autoimmune arthritis by promoting differentiation and migration of Peyer’s Patch T Follicular Helper cells. Immunity 44, 875–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ (2020). Diet should be a tool for researchers, not a treatment. Nature 577, S23–S23. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, and Gordon JI (2009). The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med 1, 6ra14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, and Hooper LV (2008). Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc. Natl. Acad. Sci. U. S. A 105, 20858–20863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Sluis M, De Koning BAE, De Bruijn ACJM, Velcich A, Meijerink JPP, Van Goudoever JB, Büller HA, Dekker J, Van Seuningen I, Renes IB, et al. (2006). Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 131, 117–129. [DOI] [PubMed] [Google Scholar]
- Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, and Gewirtz AT (2010). Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 328, 228–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter J, Armet AM, Finlay BB, and Shanahan F (2020). Establishing or exaggerating causality for the gut microbiome: lessons from human microbiota-associated rodents. Cell 180, 221–232. [DOI] [PubMed] [Google Scholar]
- Wegorzewska MM, Glowacki RWP, Hsieh SA, Donermeyer DL, Hickey CA, Horvath SC, Martens EC, Stappenbeck TS, and Allen PM (2019). Diet modulates colonic T cell responses by regulating the expression of a Bacteroides thetaiotaomicron antigen. Sci Immunol 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JM, Bilate AM, Gobert M, Ding Y, Curotto de Lafaille MA, Parkhurst CN, Xiong H, Dolpady J, Frey AB, Ruocco MG, et al. (2012). Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. J. Exp. Med 209, 1723–1742, S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, Bartolomaeus H, Haase S, Mähler A, Balogh A, Markó L, et al. (2017). Salt-responsive gut commensal modulates TH17 axis and disease. Nature 551, 585–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf AR, Wesener DA, Cheng J, Houston-Ludlam AN, Beller ZW, Hibberd MC, Giannone RJ, Peters SL, Hettich RL, Leyn SA, et al. (2019). Bioremediation of a Common Product of Food Processing by a Human Gut Bacterium. Cell Host Microbe 26, 463–477.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao S, Jin H, Korn T, Liu SM, Oukka M, Lim B, and Kuchroo VK (2008). Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-β-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J. Immunol 181, 2277–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L, Seaton SC, Ndousse-Fetter S, Adhikari AA, DiBenedetto N, Mina AI, Banks AS, Bry L, and Sloan Devlin A (2018). A selective gut bacterial bile salt hydrolase alters host metabolism. eLife 7, e37182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeevi D, Korem T, Zmora N, Israeli D, Rothschild D, Weinberger A, Ben-Yacov O, Lador D, Avnit-Sagi T, Lotan-Pompan M, et al. (2015). Personalized nutrition by prediction of glycemic responses. Cell 163, 1079–1094. [DOI] [PubMed] [Google Scholar]
- Zegarra-Ruiz DF, El Beidaq A, Iñiguez AJ, Lubrano Di Ricco M, Manfredo Vieira S, Ruff WE, Mubiru D, Fine RL, Sterpka J, Greiling TM, et al. (2019). A diet-sensitive commensal lactobacillus strain mediates TLR7-dependent systemic autoimmunity. Cell Host Microbe 25, 113–127.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G, Zecchi R, D’Angelo C, Massi-Benedetti C, Fallarino F, et al. (2013). Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 39, 372–385. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, et al. (2008). Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med 14, 282–289. [DOI] [PubMed] [Google Scholar]
- Zou J, Chassaing B, Singh V, Pellizzon M, Ricci M, Fythe MD, Kumar MV, and Gewirtz AT (2018). Fiber-mediated nourishment of gut microbiota protects against diet-induced obesity by restoring IL-22-mediated colonic health. Cell Host Microbe 23, 41–53.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]