

Abstract
Björklund P, Pacak K, Crona J (Uppsala University, Uppsala, Sweden; Section on Medical Neuroendocrinology, Program in Reproductive and Adult Endocrinology, Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), National Institutes of Health (NIH), Bethesda, MD, USA). Precision medicine in pheochromocytoma and paraganglioma: current and future concepts.
Pheochromocytoma and paraganglioma (PPGL) are rare diseases but are also amongst the most characterized tumour types. Hence, patients with PPGL have greatly benefited from precision medicine for more than two decades. According to current molecular biology and genetics-based taxonomy, PPGL can be divided into three different clusters characterized by: Krebs cycle reprogramming with oncometabolite accumulation or depletion (group 1a); activation of the (pseudo) hypoxia signalling pathway with increased tumour cell proliferation, invasiveness and migration (group 1b); and aberrant kinase signalling causing a pro-mitogenic and anti-apoptotic state (group 2). Categorization into these clusters is highly dependent on mutation subtypes. At least 12 different syndromes with distinct genetic causes, phenotypes and outcomes have been described. Genetic screening tests have a documented benefit, as different PPGL syndromes require specific approaches for optimal diagnosis and localization of various syndrome-related tumours. Genotype-tailored treatment options, follow-up and preventive care are being investigated. Future new developments in precision medicine for PPGL will mainly focus on further identification of driver mechanisms behind both disease initiation and malignant progression. Identification of novel druggable targets and prospective validation of treatment options are eagerly awaited. To achieve these goals, we predict that collaborative large-scale studies will be needed: Pheochromocytoma may provide an example for developing precision medicine in orphan diseases that could ultimately aid in similar efforts for other rare conditions.
Keywords: cancer metabolism, neuroendocrine tumours, paraganglioma, personalized medicine, pheochromocytoma, precision medicine
Management of pheochromocytoma and paraganglioma: current state of the art
Pheochromocytoma (PCC) and paraganglioma [(PGL) together referred to herein as PPGL] arise from neuroendocrine cells in the adrenal medulla as well as autonomous sympathetic and parasympathetic ganglia. According to the 2004 World Health Organization classification, PPGLs arising from the adrenal medulla should be termed PCCs, whereas tumours originating from autonomic ganglia are PGLs [1].
Diagnosis
The gold standard for diagnosing PPGL is through the detection of excess catecholamine and/or dopamine production. Analysis of free metanephrines and metoxytyramine in plasma is currently the method of choice to both detect and exclude the disease [2–4]. In addition, 24-h urine collections may be used. To obtain optimal specificity of plasma free metanephrine analysis, the patient should rest in the supine position for 20–30 min before blood test from a peripheral vein [5]. The most common causes of false-positive results are medications, such as tricyclic antidepressants, that give rise to an increase in catecholamine release [5, 6]. There are no definitive pathological or molecular criteria for malignant PPGL. Instead, malignant cases are classified by the presence of distant metastasis that is defined by the presence of PPGL tumour cells in nonchromaffin organs [7]. Metastatic disease occurs in 10–20% of PCCs and in 15–35% of abdominal PGLs [8–10].
Staging
Recent guidelines recommend the use of computed tomography (CT) for the localization of PCCs that occur in patients without a known genetic cause [11–14]. Magnetic resonance imaging (MRI) should be used in patients with metastatic PPGL or for localizing head and neck PGL [15]. Molecular imaging may allow for increased sensitivity in the detection of metastatic lesions [14]. Metaiodobenzylguanidine (MIBG)-based scintigraphy has long been used in the management of these patients and was recently found to have diagnostic value. Positron emission tomography (PET)/CT with 18F-dihydroxyphenylalanine, 18F-fluorodeoxyglucose (FDG) and 68 Ga-DOTATATE tracers have a role in selected patients [16, 17]. However, the sensitivity of PET/CT is suboptimal, especially for metastatic PPGL. Currently, the highest sensitivity for staging of PCC and PGL tumours can be achieved through a combination of anatomical and functional imaging [15].
Treatment
Cure of localized PPGL can be achieved through complete surgical resection [18, 19]. Adequate preoperative preparation is required to optimize the patient for surgical intervention, and to reduce the risk of potentially life-threatening intra and postoperative complications as a result of haemodynamic instability [20, 21]. The choice of surgical technique is dependent on both tumour location and size. In most cases, adrenal PCCs may be resected laparoscopically with our without adrenal-sparing technique [22, 23]. The larger abdominal PGLs and PCCs can be resected using open approaches.
According to a recent study, the 1-year progression-free survival of those with metastatic PPGL was 46% at diagnosis for therapy-naive patients with metastases [24]. Taking into account the relatively long survival of these patients, a ‘wait and see’ approach may therefore sometimes be advocated. The rationale for surgery would be to reduce the cumulative toxic side effects of both medical and radiation treatment. Surgical resection and/or locoregional therapy can also be considered in patients with metastatic PPGL in order to provide palliate relief or to allow long-term remission [25]. Combination chemotherapy with CVD regimen (cyclophosphamide, dacarbazine and vincristine) as well as 131I-labelled MIBG are the most validated systemic strategies for patients with unresectable tumours [26]. Positive effects of radiolabelled somatostatin compounds (177Lu-DOTATATE) have also been described [27, 28].
Precision medicine in PPGL
The concept of precision medicine, where the care of the individual patient is tailored by taking personal variability into account, has a long tradition in the management of PPGL. The individual’s genomic sequence is the most important factor for patient stratification. A total of 12 different syndromes with an increased risk of PPGL have been described to date (Table 1) [9, 18]. These syndromes have been linked to specific loci: SDHA [29], SDHB [30], SDHC [31], SDHD [32], SDHAF2 [33], FH [34], VHL [35], EPAS1 [36], RET [37], NF1 [38], TMEM127 [39] and MAX (Fig. 1) [40]. Genetic screening is currently performed in patients with a high probability of an underlying genetic disorder, i.e. those presenting at a young age, and/or with multifocal and/or metastatic tumours. The diagnostic procedure utilizes a stepwise approach with prioritization of the genes to be analysed. If a genetic syndrome can be established by genetic diagnosis, patient management can be individually tailored based on the specific characteristics of the syndrome.
Table 1.
Syndromes with increased risk of PPGL
| Syndrome | Gene | Location | Transmission | Original reference |
|---|---|---|---|---|
| PGL1 | SDHD | 11q23 | AD, paternal | Baysal et al. (32) |
| PGL2 | SDHAF2 | 11q12 | AD, paternal | Hao et al. (33) |
| PGL3 | SDHC | 1q23.3 | AD | Niemann et al. (31) |
| PGL4 | SDHB | 1p36.13 | AD | Astuti et al. (30) |
| PGL5 | SDHA | 5p15 | NA | Burnichon et al. (29) |
| FH | FH | 1q42.1 | NA | Letouze et al. (34) |
| Von Hippel Lindau | VHL | 3p25.3 | AD | Latif et al. (35) |
| Paraganglioma-Polycytemia disease | EPAS1 | 2p21 | NA | Zhuang et al. (36) |
| Neurofibromatosis type 1 | NF1 | 17q11.2 | AD | Wallace et al. (38) |
| MEN2 | RET | 10q11.21 | AD | Mulligan et al. (37) |
| TMEM127 | TMEM127 | 2q11.2 | AD | Qin et al. (39) |
| MAX | MAX | 14q23.3 | AD, paternal | Comino-Mendez et al. (40) |
PGL, paraganglioma; PPGL, pheochromocytoma and paraganglioma; FH, fumarate hydratase; MEN2, multiple endocrine neoplasia type 2; TMEM127, transmembrane protein 127; MAX, myc-associated factor X; AD, autosomal dominant; VHL, Von Hippel Lindau syndrome; NF1, Neurofibromatosis type 1; NA, not available.
Fig. 1.
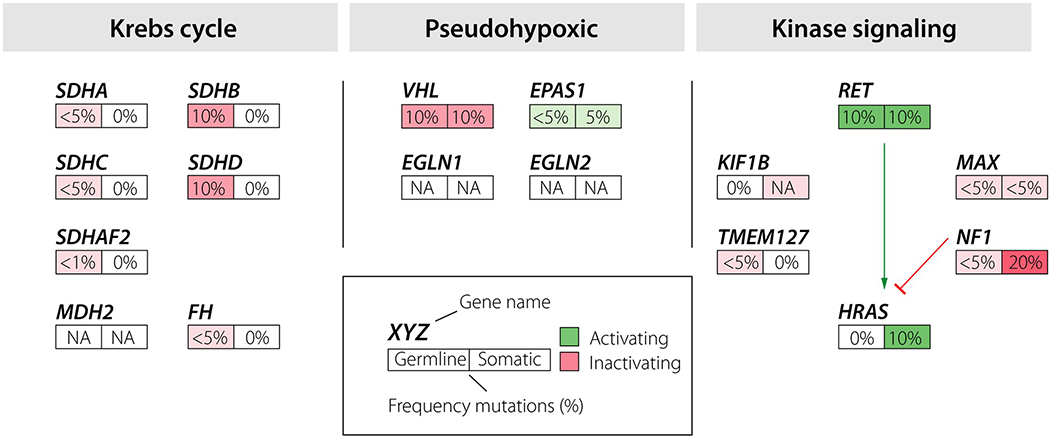
Description of genes involved in pheochromocytoma and paraganglioma tumourigenesis grouped in molecular clusters, and the estimated overall frequency of pathogenic mutations in these diseases.
This may provide not only information on the future risk for disease, but also empirical knowledge of the clinical and molecular characteristics of the particular PPGL (Fig. 2) [41]. These phenotype differences for PPGLs manifest as different hormonal profiles, anatomical localizations and risks of metastatic disease. Genotype-tailored selection of molecular imaging tracers has been shown to increase the sensitivity of such investigations; 18F-FDG is preferred in patients with SDHx (SDHA-D and SDHAF2) -related disease to detect primary and metastatic lesions [12, 42]. It has been hypothesized that, among PPGL patients with metastatic disease, some could respond differently to systemic treatment; however, this remains to be confirmed in prospective randomized trials.
Fig. 2.
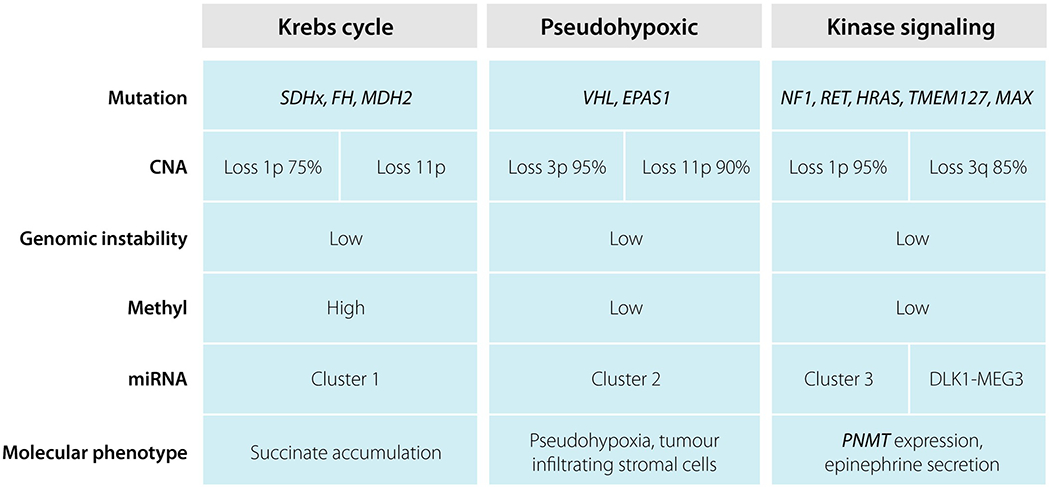
Summary of the genetic and molecular characteristics of pheochromocytoma and paraganglioma in the three different clusters.
Krebs cycle cluster and familial PGLs
Truncating mutations in proteins constituting the Krebs cycle cause familial PGL syndromes types 1–5, as well as fumarate hydratase-associated PPGL (Fig. 3a). These diseases are transmitted in an autosomal dominant fashion. PGL types 1–3 (SDHD, SDHAF2 and SDHC) are mainly characterized by localized parasympathetic PGLs. By contrast, familial PGL type 4 (SDHB) is associated with significant morbidity and increased mortality due to increased risk of metastatic PPGL, as well as other cancers such as renal cell carcinoma. Familial PGL type 5 (SDHA) is associated with PPGL but a clear phenotype remains to be discovered. It was previously shown that inactivating mutations in the FH gene cause dominant hereditary leiomyomatosis and renal cell cancer. Such mutations were recently associated with PPGL with an increased frequency of metastatic disease. It was recently suggested that constitutional mutations in MDH2, which encodes a third enzyme involved in the Krebs cycle, may be involved in PPGL tumourigenesis [43].
Fig. 3.
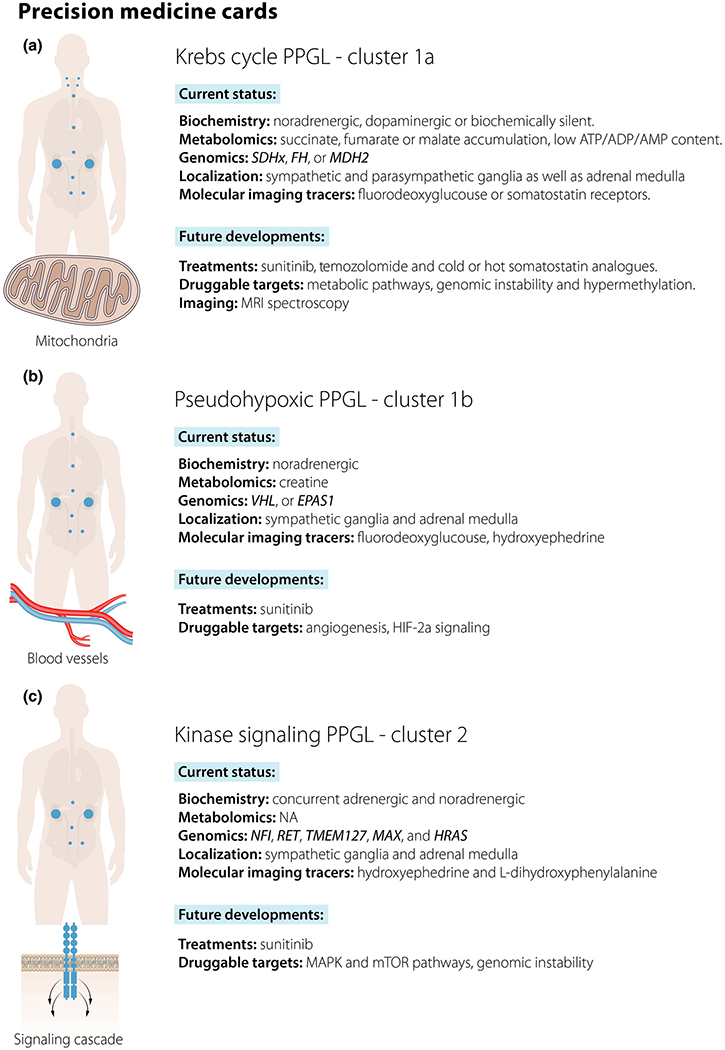
(a–c) Current and future perspectives of precision medicine in pheochromocytoma and paraganglioma stratified by molecular subgroup.
Familial PGL type 1
Familial PGL type 1 is caused by loss of function mutations in the SDHD gene and shows near complete penetrance for parasympathetic PGL tumours that are most commonly located in the head and neck region [44–46]. Unilateral PCC is reported to occur in about 50% of patients [46, 47]. Metastatic tumours are rare. SDHD-related PPGLs are heterogeneous with regard to hormone production; head and neck PGLs are often biochemically silent or produce either dopamine or methoxytyramine, whereas PCCs show norepinephrine secretion [48]. Additional pathologies have been reported, including renal cell carcinoma, gastrointestinal stromal cell tumours (GIST) and pituitary adenomas [49, 50]. It was recently suggested that mortality is not increased in carriers of SDHD mutations compared to a normal population [51].
Familial PGL type 2
Familial PGL type 2 is caused by loss of function mutations in the SDHAF2 gene and has so far been detected in a limited number of families [33, 52–54]. All reported cases have presented with parasympathetic head and neck PGL without signs of metastatic disease.
Familial PGL type 3
Familial PGL type 3 is caused by loss of function mutations in the SDHC gene and is mainly characterized by parasympathetic PGLs located in the head and neck region with a low risk of metastases [55–57].
Familial PGL type 4
Familial PGL type 4 is caused by loss of function mutations in SDHB and is associated with sympathetic PGL with an increased risk of malignancy. The high frequency of malignant PPGLs results in increased patient morbidity and mortality [47, 58, 59]. PCC and head and neck PGL are less commonly reported. Tumours secrete norepinephrine and/or dopamine. Patients with PGL type 4 show an increased risk of developing renal cell carcinoma as well as GIST [44]. Mutations in SDHx genes have also been associated with pituitary adenomas [49, 60].
Familial PGL type 5
Familial PGL type 5 is caused by truncating mutations in the SDHA gene. These mutations have been associated with both PCC and PGL as well as GIST. It is thought that carriers of SDHA mutations have a low penetrance for the development of PPGL tumours, and concomitant presentation with GIST is infrequently observed [29, 61, 62].
Fumarate hydratase
Constitutional loss of function mutations in the fumarate hydratase (FH) gene were recently described in patients with PPGL [34, 63, 64]. The phenotype is characterized by multiple primary tumours as well as metastatic disease [64]. It was previously demonstrated that germline FH mutations cause autosomal dominant hereditary leiomyomatosis and renal cell cancer [65].
Molecular genetics of familial PGLs
The SDHx genes encode different subunits of the succinate dehydrogenase complex that catalyses reactions in the tricarboxylic acid cycle (oxidation of succinate to fumarate) and in the respiratory electron transfer chain (complex II). Loss of function mutations in these genes causes an accumulation of succinate that inhibits EGLN1-3 enzyme activity (Fig. 2) [66]. Inhibition of this enzyme family causes decreased hydroxylation and subsequent ubiqutination of hypoxia-inducible factor (HIF) transcription factors, resulting in a pseudohypoxic state similar to that seen in VHL- and EPAS1-mutated tumours. It has recently been suggested that accumulation of succinate in SDHx-mutated tumours results in a hypermethylator phenotype through inhibition of histone and DNA methylases [34]. Tumours with FH mutations have increased levels of fumarate, and similar molecular consequences to those of SDHx-mutated tumours [63, 64].
The pseudohypoxic cluster: von Hippel–Lindau and PGL–polycythemia syndromes
Mutations in genes associated with regulation of cellular oxygen sensation and response to hypoxia have been shown to give rise to PPGL (Fig. 3b). The von Hippel–Lindau (VHL) disease confers susceptibility to multiple tumours including PPGL. Gain of function mutations in the EPAS1 gene causes PGL–somatostatinoma–polycythemia syndrome [67].
VHL syndrome
VHL syndrome is caused by loss of function mutations in the tumour suppressor gene VHL with an incidence of 1/36 000 [68]. It is characterized by an increased risk of retinal haemangioma, central nervous system haemangioblastoma and PPGL. Pancreatic neuroendocrine tumours, renal clear cell carcinoma and lymphatic sac tumours occur but at lower frequencies. Patients with C598T mutation present with Chuvash polycythemia [69]. The penetrance of PPGL is 10–25% and about 50% of patients with PCC have bilateral disease [18, 70–72]. VHL-related PPGLs predominantly produce norepinephrine and less than 5% of patients develop metastatic disease [70–73].
PGL–polycythemia syndrome
Gain of function mutations within the EPAS1 gene that encodes the hypoxia-inducible factor-2alpha (HIF2A) protein is associated with autosomal dominant polycythemia [74]. Activating amino acid transitions at HIF2A hydroxylation sites was recently described as a cause of PPGL, polycythemia and somatostatinoma. Ocular manifestations have also been reported [75]. Germline mutations at these codons have been considered embryologically lethal and the mutations are absent in constitutional DNA in syndrome carriers [76]. The presence of the somatic mutations in different tissues indicates mosaic carrier status in these patients [34]. To determine the tissue distribution of somatic EPAS1 mutations, deep sequencing of peripheral blood and/or buccal swabs should be performed [77]. The gender distribution for PGL–polycythemia syndrome shows a strong female bias; the cause of this trend remains to be determined [67].
Molecular genetics of VHL and PGL–polycythemia syndromes
Mutations in VHL and EPAS1 cause a cellular pseudohypoxic state through the stabilization of HIF transcription factor proteins (Fig. 2) [78]. Activated HIF alters transcription of target genes that results in increased angiogenesis as well as cellular proliferation and reduced apoptosis. Loss of function mutations in the VHL gene results in reduced ubiqutination of HIF transcription factors with subsequent reduction of degradation by the proteasome. This causes HIF accumulation and nuclear translocation [79]. Gain of function mutations at EPAS1 hydroxylation sites causes reduced VHL protein binding that diminishes HIF2A ubiqutination enabling HIF escape from degradation [36, 67, 80, 81]. EPAS1-mutated tumours have been shown to have similar transcriptomic and metabolomic signatures compared to PPGL tumours with VHL mutations [36].
Mutations in EGLN1 and EGLN2, genes that encode the proteins responsible for prolyl hydroxylation targeting HIF for degradation, may also confer susceptibility to PPGL [82, 83].
These tumours predominantly secrete norepinephrine, due to low expression of phenylethanolamine N-methyltransferase (PNMT), and have been linked to hypermethylation of the PNMT gene promoter [84]. Or: These tumours predominantly secrete norepinephrine due to low expression of phenylethanolamine N-methyltransferase (PNMT), which has been linked to hypermethylation of the PNMT gene promoter [84].
Kinase signalling cluster
Mutated genes classified as cluster 2 PPGL are tightly linked to regulation of signalling in either RAS/RAF/MAPK or (mTOR) pathways (Fig. 3c). This cluster comprises the majority of PCCs, and shows a wide molecular spectrum. The associated syndromes show a different range of diseases involving the nervous system [neurofibromatosis type 1 (NF1)] and endocrine glands [multiple endocrine neoplasia type 2 (MEN2)], as well as exclusive association with PPGL in those with TMEM127and MAX mutations. Somatic HRAS mutations are common in PPGL, but no cases with constitutional HRAS mutation and PPGL have been presented.
NF1
NF1, also known as von Recklinghausen’s disease [85], is caused by loss of function mutations in the neurofibromin 1 (NF1) gene with an incidence of 1 : 2500–3000. The clinical phenotype is characterized by fibromatous skin lesions, lichen eye nodules, optic gliomas and café au lait spots [86]. NF1 syndrome has a relatively low penetrance for PCC of about 5% and is mainly associated with unilateral PCC with mixed epinephrine/norepinephrine production [87].
MEN2
MEN2 is caused by gain of function mutations within the RET (rearranged during transfection) gene that encodes a tyrosine kinase receptor [37, 88, 89]. MEN2 is characterized by susceptibility to multiple endocrine neoplasms: medullary thyroid carcinoma, PCC and parathyroid adenomas. Generally, bilateral PCCs occur in about 50% of probands [90]. Mutations in the cysteine-rich extracellular domains located in exons 10–11 underlie a majority of MEN2 cases. Disease-causing variants within noncysteine regions located in exons 13–16 are less common and characterized by pronounced interpatient phenotypic heterogeneity [91]. Among carriers of noncysteine mutations, only a minority develops PCC [91]. MEN2-associated PCCs typically produce a mixture of epinephrine/norepinephrine and rarely metastasize [9, 10, 18, 92–94].
TMEM127
Loss of function mutations in the TMEM127 (transmembrane protein 127) gene causes susceptibility to PCC, and less frequently abdominal PGL [39, 95, 96]. Association with renal cell carcinoma has been described but remains to be validated [97]. Results from a large Brazilian family showed 30% penetrance for PPGL with median onset at 43 years of age [98]. Bilateral PCCs are common. Mixed epinephrine/norepinephrine secretion is observed in TMEM127-associated PPGLs.
MAX
Loss of function mutations in the MAX (myc-associated factor X) gene has been shown to cause hereditary PCC and less commonly PGL [40, 99]. PCCs are commonly bilateral and show an intermediate biochemical profile with moderately elevated levels of epinephrine [99]. No additional phenotypic associations have been reported.
Molecular genetics of PPGL within the kinase signalling cluster
Cluster 2 tumours are characterized by an increased activity in the mitogenic signalling pathways ERK/MAPK and PI3K/AKT/mTOR (Fig. 2). Both these pathways are frequently altered in a wide variety of human cancers. However, heterogeneity within this cluster occurs and the tumours can be further subclassified according to their transcriptome and miRNAome profiles [84, 100].
It has been suggested that tumours with mutations in NF1, RET and HRAS overlap with regard to their mechanisms of tumourigenesis, that diverge on RAS-mediated signalling [84]. Mutations in NF1 GTPas domain result in reduced inhibition of RAS intrinsic activity, whilst ligand or mutant-dependent activation of RET results in activation of RAS through downstream signalling [101–103]. Somatic mutations in the NF1 gene have been described as frequent events in patients with sporadic PCC and were associated with activation of the ERK/MAPK signalling pathway [73, 99, 104, 105]. Somatic gain of function mutations in H-RAS has been described in a significant proportion of PPGLs [84, 106–109]. Activating mutations in RET occurs at phosphorylation sites causing intrinsic activation resulting in downstream activation of RAS/RAF/MAPK and PI3K/AKT signalling pathways [110, 111].
Loss of function mutations in TMEM127 results in reduced inhibition of the mTOR pathway in an RAS/RAF/MAPK- and PI3K/AKT-independent manner [39]. The definitive role of the TMEM127 protein in PPGL tumourigenesis remains to be identified.
Mutated MAX causes deregulation of the MYC–MAX–MXD1 pathway that leads to altered transcription and signalling in the NRAS–PIK3CA–AKT1–mTOR pathway [40]. It has been suggested that MAX-mutated tumours have a unique transcriptomic signature [100]. The fact that MAX-mutated tumours are distinct from those with NF1/RET/HRAS mutations is also supported by their intermediate expression of PNMT, and subsequent lower production of epinephrine.
The genetic landscape of PPGL
Genome-wide characterization is a valuable tool not only for identification of genetic factors driving tumourigenesis, but also to provide information about the degree and spectrum of genetic instability. It has consistently been found that PPGLs are characterized by a relatively low degree of genetic instability both at the nucleotide and chromosomal level [84, 100, 112–114]. Furthermore, the observed mutational spectrum was consistent with ageing, and there was limited evidence of previous or ongoing mutagenic processes such as smoking or defects in cellular chromatin maintenance.
Somatic mutations may also provide ancestral information through chronological and spatial classification. Such characterization may allow for better understanding of the dynamic life of tumours from initiation, through acquisition of malignant capabilities, to metastatic dissemination [115]. In order to transform cells in the adrenal medulla or autonomous ganglia to initiate a tumour, it is likely that both nucleotide mutations and copy number alterations are needed (Fig. 4) [114, 116]. This hypothesis is supported by the classification of somatic mutations in driver genes and specific somatic copy number alterations as both obligate and early events [114]. VHL-mutated PPGLs show loss of 3p and 11p, whereas those with NF1 mutations show loss of 17q. Cluster 2 tumours display loss of 1p and 3q with high frequencies [84, 116]. The pathogenic mechanisms of 11p inactivation in VHL tumours and losses of 1p and 3q in cluster 2 PPGL remain to be determined [117, 118].
Fig. 4.
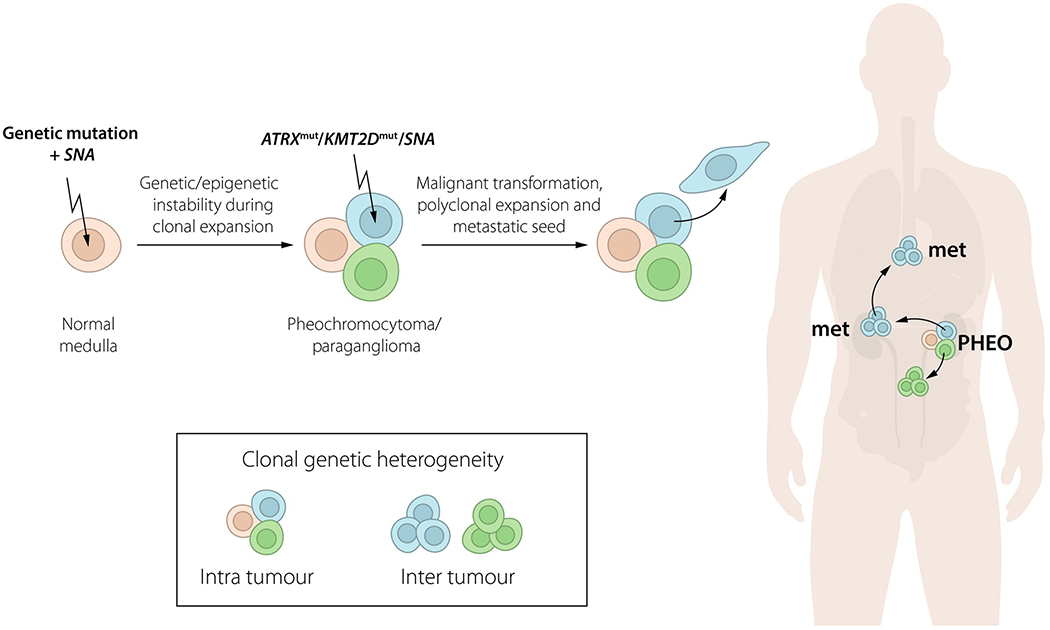
Proposed evolution of pheochromocytoma and paraganglioma from tumour initiation to metastatic seed. Genetic heterogeneity may occur within tumours as well as between different tumour lesions. SNA, somatic copy number alterations.
Identifying the molecular mechanisms involved in malignant transformation of benign tumours can be regarded as the ‘holy grail’ of PPGL translational research. Novel findings indicate that somatic ATRX mutations occur in a subset of PPGLs that display a high frequency of metastatic disease [84, 100, 112]. Extrapolating findings from other neuroendocrine tumours, inactivation of ATRX is most probably a secondary event that promotes malignancy through establishing the alternative lengthening of telomeres phenotype [119, 120]. Somatic mutations in the TERT promoter (SDHx-deficient PPGL), as well as in the chromatin modifier KMT2D, have also been identified but remain to be validated by independent observers [113, 121]. It has also been demonstrated that metastases accumulate chromosomal aberrations compared to the paired primary tumours [114]. Elucidating whether these somatic changes are drivers of malignant transformation remains a priority.
Future concepts for precision medicine
Molecular characterization
In this review, we describe 13 loci (SDHA-D, SDHAF2, VHL, EPAS1, NF1, RET, HRAS, MAX and TMEM127) that have been suggested to cause PPGL. ATRX, KMT2D and TERT promoter have also been identified with recurrent mutations but their contribution to the disease remains to be identified. Despite investigation with exome sequencing, there is still no identified disease-causing mechanism for a large proportion of PPGLs [84, 100]. Recurrent copy number alterations as well as miRNA and methylation patterns have been identified, but the particular mechanisms in which these events are involved in PPGL tumourigenesis remain to be clarified [84].
It is known from previous studies that the miRNAome of PPGL clusters in a similar manner to the transcriptome [122]. Further investigation of the PPGL miRNAome revealed a distinct subset of PPGLs with copy number neutral LOH of chromosome 14, that was subsequently linked to deregulation of the DLK1-MEG3 miRNA [84]. These cases were all characterized by aberrant kinase signalling, but no disease-causing mutations were detected by exome sequencing.
Deregulation of epigenetic maintenance was recently found to occur as a consequence of succinate accumulation [34, 123]. Whether specific epimutations can be directly linked to tumour formation or malignancy remains to be determined. Of particular interest for PPGLs was the recent identification of epimutated SDHC as a cause of GIST [124].
Up to 10% of patients with nonsyndromic presentation are carriers of germline mutations. Hence, a prioritization approach in diagnostic genetic screening may miss patients with an underlying genetic cause [125–127]. The limited use of genetic screening in the clinical setting is mainly due to the shortcomings of current methodologies. This includes poor cost effectiveness and long analysis times especially for extensive analyses such as genes of interest in PPGL. The advent of next-generation sequencing methods has the potential to decrease the cost of sequencing and enable all patients with PPGLs to be screened for all relevant loci [128]. Much work is needed to design such a comprehensive PPGL screening test: there are at least 20 relevant genes that span more than 200 exons and the assay should be able to detect nucleotide substations as well as gross deletions [128, 129].
Phenotypic characterization of known syndromes
A correct genetic diagnosis may predict PPGL tumour characteristics, as well as the risk of other diseases associated with the specific syndrome. Pioneering work was performed with the traditional tumour susceptibility syndromes VHL, MEN and NF1, where appropriate surveillance may facilitate early diagnosis and treatment. A similar approach might soon be used for familial PGL syndromes where patients with SDHB mutations have been most commonly studied. SDHB carriers have a substantially increased risk of malignant tumours as well as higher mortality, which motivates extensive follow-up [47]. Recently, another group has been recognized with polycythemia PGL syndrome, where patients have an increased risk of developing duodenal somatostatinoma in addition to PPGL [67]. Improved guidelines for the management of patients with familial PPGL diseases are eagerly awaited.
Diagnostics and localization
The detection of excess hormone production underlies diagnosis in PPGL, and the hormone profile of individual tumours depends on mutation subgroup (Table 2). Tumours with mutations in FH, VHL, EPAS1 and succinate dehydrogenase complex genes predominantly produce norepinephrine with low levels of epinephrine. Tumours with RET, NF1, TMEM127, MAX and HRAS mutations show increased levels of both epinephrine and norepinephrine [130, 131]. The molecular rationale for this phenomenon may be promoter methylation, and subsequent decreased transcription of PNMT, which is responsible for conversion of norepinephrine to epinephrine [84]. Cluster 1a tumours may secrete dopamine or may be biochemically silent [132].
Table 2.
Clinical characteristics of mutation subtypes.
| Genes | Biochemistry | Radiology | Molecular imaging |
|---|---|---|---|
| SDHx, FH | NE, MT, dopamine | MRI | 18F-FDG or 68Ga-Som PET |
| VHL, EPAS1 | NE | CT | 18F-DOPA or 11C-HED PET |
| RET | NE, E | CT | 18F-DOPA or 11C-HED PET |
NE, norepinephrine; E, epinephrine; FDG, fluorodeoxyglucose; Som, somatostatin receptor; DOPA, L-dihydroxyphenylalanine; HED, hydroxyephedrine; CT, computed tomography; MRI, magnetic resonance imaging; PET, positron emission tomographyMT, methoxytyramine.
Gene mutation status can also guide in the selection of appropriate imaging tests with SDHx tumours having high uptake of FDG as well with somatostatin-labelled PET tracers [16, 42]. Introduction of tumour- and/or genotype-specific radiopharmaceuticals will help to assess the degree of, for example, hypoxia, apoptosis, angiogenesis and invasiveness. Utilization of H-HRMAS NMR may help to identify specific mutations even without genetic testing [133–135]. Similarly MRI spectroscopy with hyperpolarization has the potential for in vivo molecular metabolomics profiling [136].
Treatment
With the discovery of different mechanisms driving PPGL formation in the three clusters, it was hypothesized that mutation subtypes may be used as biomarkers to predict sensitivity to systemic therapy. Patients with pro-angiogenic PPGLs could benefit from antiangiogenic therapy, whereas those with PPGLs in which the kinase signalling cascades are activated could respond to inhibitors of these particular pathways [137, 138]. Within the ongoing Phase II studies of systemic therapy in metastatic PPGL, these hypotheses have a potential to be tested.
Genetic instability could be a potential druggable target in PPGL patients despite the low overall degree of genetic fragmentation [84, 100, 112]. The identification of deregulated telomere maintenance, especially in the alternative lengthening of telomeres (ALT) context, may have therapeutic implications as such tumours have been shown to respond to ATR inhibition [112, 121, 139, 140]. In the rare cases that have increased genetic instability, it is probable that the number of immunoreactive neo-antigens will be increased, thus conferring susceptibility to immunotherapy [84, 141, 142].
It has been demonstrated that succinate dehydrogenase-deficient PPGLs exhibit a hypermethylator phenotype, and related tumours (GIST) have shown epimutations in SDHC [124]. This implies that agents modulating the epigenome could be effective in the treatment of these tumours.
It was recently suggested that SDHx tumours had a relatively high response to temozolomide [138]. The explanation for the increased sensitivity to this alkalyting agent was the observed hypermethylation of the MGMT promoter [143]. In addition, SDHx-mutated tumours show increased expression of somatostatin receptors that could enable the use of somatostatin receptor-directed diagnostics and treatment [144]. The skewed metabolism in SDHx-deficient PPGLs may also render these tumours vulnerable to antimetabolite therapy.
Tumour heterogeneity
A complication in the development of genetic biomarkers is genetic heterogeneity within and between PPGL lesions [84, 100, 114, 145]. Preliminary findings indicate that genetic divergence is considerable between clones observed within primary and metastatic lesions of the same patient (Fig. 4). Therefore, analyses from primary tumours should be extrapolated to metastases with caution. Before the introduction of genetic biomarkers, their chronological position in PPGL tumour evolution needs to be determined. Technologies such as liquid biopsies and/or circulating tumour cells may provide a way to circumvent the problem of spatial heterogeneity but remain to be evaluated in PPGLs [146, 147].
Concluding remarks
Despite the orphan status of PPGL, considerable progress has been made in our understanding of the molecular biology underlying the diseases. This has been translated into improved diagnostics and allowed for identification of an increasing number of syndrome carriers. By contrast, the treatment of PPGL has changed marginally during the last decades and particularly the care of patients with metastatic disease has an urgent need for improvement. It remains to be proven whether translation of experimental data can provide new treatment options or serve as predictive factors for existing therapies. Developing and introducing new treatments may be the biggest challenge for the PPGL community, and the ultimate success will rely on strong cross-cultural collaboration between physicians, researchers and patients worldwide.
Footnotes
Conflict of interest statement
No conflicts of interest to declare.
References
- 1.Ronald ADR, Philipp UH, Charis E. Pathology and Genetics of Tumours of Endocrine Organs (IARC/World Health Organization Classification of Tumours; ). 2004. [Google Scholar]
- 2.Weise M, Merke DP, Pacak K, Walther MM, Eisenhofer G. Utility of plasma free metanephrines for detecting childhood pheochromocytoma. J Clin Endocrinol Metab 2002; 87: 1955–60. [DOI] [PubMed] [Google Scholar]
- 3.Lenders JW, Pacak K, Walther MM et al. Biochemical diagnosis of pheochromocytoma: which test is best? JAMA 2002; 287: 1427–34. [DOI] [PubMed] [Google Scholar]
- 4.Eisenhofer G, Lenders JW, Linehan WM, Walther MM, Goldstein DS, Keiser HR. Plasma normetanephrine and metanephrine for detecting pheochromocytoma in von Hippel-Lindau disease and multiple endocrine neoplasia type 2. N Engl J Med 1999; 340: 1872–9. [DOI] [PubMed] [Google Scholar]
- 5.Lenders JW, Willemsen JJ, Eisenhofer G et al. Is supine rest necessary before blood sampling for plasma metanephrines? Clin Chem 2007; 53: 352–4. [DOI] [PubMed] [Google Scholar]
- 6.Neary NM, King KS, Pacak K. Drugs and pheochromocytoma–don’t be fooled by every elevated metanephrine. N Engl J Med 2011; 364: 2268–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeLellis R, Lloyd R, Heitz P, Eng C. Pathology and Genetics of Tumours of Endocrine Organs (IARC/World Health Organization Classification of Tumours). Lyon: IARC Press, 2004. [Google Scholar]
- 8.O’Riordain DS, Young WF Jr, Grant CS, Carney JA, van Heerden JA. Clinical spectrum and outcome of functional extraadrenal paraganglioma. World J Surg 1996; 20: 916–21; discussion 22. [DOI] [PubMed] [Google Scholar]
- 9.Amar L, Bertherat J, Baudin E et al. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol 2005; 23: 8812–8. [DOI] [PubMed] [Google Scholar]
- 10.King KS, Prodanov T, Kantorovich V et al. Metastatic pheochromocytoma/paraganglioma related to primary tumor development in childhood or adolescence: significant link to SDHB mutations. J Clin Oncol 2011; 29: 4137–42. Epub 2011 Oct 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lumachi F, Tregnaghi A, Zucchetta P. Cristina Marzola M, Cecchin D, Grassetto G, Bui F. Sensitivity and positive predictive value of CT, MRI and 123I-MIBG scintigraphy in localizing pheochromocytomas: a prospective study. Nucl Med Commun 2006; 27: 583–7. [DOI] [PubMed] [Google Scholar]
- 12.Timmers HJ, Kozupa A, Chen CC et al. Superiority of fluorodeoxyglucose positron emission tomography to other functional imaging techniques in the evaluation of metastatic SDHB-associated pheochromocytoma and paraganglioma. J Clin Oncol 2007; 21: 3888–95. [DOI] [PubMed] [Google Scholar]
- 13.Zelinka T, Timmers HJ, Kozupa A et al. Role of positron emission tomography and bone scintigraphy in the evaluation of bone involvement in metastatic pheochromocytoma and paraganglioma: specific implications for succinate dehydrogenase enzyme subunit B gene mutations. Endocr Relat Cancer 2008; 15: 311–23. [DOI] [PubMed] [Google Scholar]
- 14.Lenders JW, Duh QY, Eisenhofer G et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2014; 99: 1915–42. [DOI] [PubMed] [Google Scholar]
- 15.Gimenez-Roqueplo AP, Caumont-Prim A, Houzard C et al. Imaging work-up for screening of paraganglioma and pheochromocytoma in SDHx mutation carriers: a multicenter prospective study from the PGL.EVA investigators. J Clin Endocrinol Metab 2012; 15: 15. [DOI] [PubMed] [Google Scholar]
- 16.Janssen I, Blanchet EM, Adams K et al. Superiority of [68 Ga]-DOTATATE PET/CT to other functional imaging modalities in the localization of SDHB-associated metastatic pheochromocytoma and paraganglioma. Clin Cancer Res 2015; 21: 3888–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Timmers HJ, Chen CC, Carrasquillo JA et al. Comparison of 18F-fluoro-L-DOPA, 18F-fluoro-deoxyglucose, and 18F-fluorodopamine PET and 123I-MIBG scintigraphy in the localization of pheochromocytoma and paraganglioma. J Clin Endocrinol Metab 2009; 94: 4757–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mannelli M, Castellano M, Schiavi F et al. Clinically guided genetic screening in a large cohort of italian patients with pheochromocytomas and/or functional or nonfunctional paragangliomas. J Clin Endocrinol Metab 2009; 94: 1541–7. [DOI] [PubMed] [Google Scholar]
- 19.Amar L, Servais A, Gimenez-Roqueplo AP, Zinzindohoue F, Chatellier G, Plouin PF. Year of diagnosis, features at presentation, and risk of recurrence in patients with pheochromocytoma or secreting paraganglioma. J Clin Endocrinol Metab 2005; 90: 2110–6. Epub 005 Jan 11. [DOI] [PubMed] [Google Scholar]
- 20.Plouin PF, Duclos JM, Soppelsa F, Boublil G, Chatellier G. Factors associated with perioperative morbidity and mortality in patients with pheochromocytoma: analysis of 165 operations at a single center. J Clin Endocrinol Metab 2001; 86: 1480–6. [DOI] [PubMed] [Google Scholar]
- 21.Goldstein RE, O’Neill JA Jr, Holcomb GW III et al. Clinical experience over 48 years with pheochromocytoma. Ann Surg 1999; 229: 755–64; discussion 64-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen WT, Grogan R, Vriens M, Clark OH, Duh QY. One hundred two patients with pheochromocytoma treated at a single institution since the introduction of laparoscopic adrenalectomy. Arch Surg 2010; 145: 893–7. [DOI] [PubMed] [Google Scholar]
- 23.Agarwal G, Sadacharan D, Aggarwal V et al. Surgical management of organ-contained unilateral pheochromocytoma: comparative outcomes of laparoscopic and conventional open surgical procedures in a large single-institution series. Langenbecks Arch Surg 2012; 397: 1109–16. [DOI] [PubMed] [Google Scholar]
- 24.Hescot S, Leboulleux S, Amar L et al. One-year progression-free survival of therapy-naive patients with malignant pheochromocytoma and paraganglioma. J Clin Endocrinol Metab 2013; 24: 24. [DOI] [PubMed] [Google Scholar]
- 25.Mannelli M, Dralle H, Lenders JW. Perioperative Management of Pheochromocytoma/Paraganglioma: is There a State of the Art? Horm Metab Res 2012; 19: 19. [DOI] [PubMed] [Google Scholar]
- 26.Plouin PF, Fitzgerald P, Rich T et al. Metastatic pheochromocytoma and paraganglioma: focus on therapeutics. Horm Metab Res 2012; 44: 390–9. Epub 2012 Feb 7. [DOI] [PubMed] [Google Scholar]
- 27.Forrer F, Riedweg I, Maecke HR, Mueller-Brand J. Radiolabeled DOTATOC in patients with advanced paraganglioma and pheochromocytoma. Q J Nucl Med Mol Imaging 2008; 52: 334–40. Epub 2008 May 16. [PubMed] [Google Scholar]
- 28.Van Essen M, Krenning EP, De Jong M, Valkema R, Kwekkeboom DJ. Peptide Receptor Radionuclide Therapy with radiolabelled somatostatin analogues in patients with somatostatin receptor positive tumours. Acta Oncol 2007; 46: 723–34. [DOI] [PubMed] [Google Scholar]
- 29.Burnichon N, Briere JJ, Libe R et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet 2010; 19: 3011–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Astuti D, Latif F, Dallol A et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet 2001; 69: 49–54. Epub 2001 Jun 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Niemann S, Muller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet 2000; 26: 268–70. [DOI] [PubMed] [Google Scholar]
- 32.Baysal BE, Ferrell RE, Willett-Brozick JE et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000; 287: 848–51. [DOI] [PubMed] [Google Scholar]
- 33.Hao HX, Khalimonchuk O, Schraders M et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science 2009; 325: 1139–42. Epub 2009 Jul 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Letouze E, Martinelli C, Loriot C et al. SDH Mutations Establish a Hypermethylator Phenotype in Paraganglioma. Cancer Cell 2013; 21: 00183–9. [DOI] [PubMed] [Google Scholar]
- 35.Latif F, Tory K, Gnarra J et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993; 260: 1317–20. [DOI] [PubMed] [Google Scholar]
- 36.Zhuang Z, Yang C, Lorenzo F et al. Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia. N Engl J Med 2012; 367: 922–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mulligan LM, Kwok JB, Healey CS et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature 1993; 363: 458–60. [DOI] [PubMed] [Google Scholar]
- 38.Wallace MR, Marchuk DA, Andersen LB et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science 1990; 249: 181–6. [DOI] [PubMed] [Google Scholar]
- 39.Qin Y, Yao L, King EE et al. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet 2010; 42: 229–33. Epub 2010 Feb 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Comino-Mendez I, Gracia-Aznarez FJ, Schiavi F et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet 2011; 43: 663–7. [DOI] [PubMed] [Google Scholar]
- 41.Dahia PL. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat Rev Cancer 2014; 14: 108–19. [DOI] [PubMed] [Google Scholar]
- 42.Timmers HJ, Chen CC, Carrasquillo JA et al. Staging and functional characterization of pheochromocytoma and paraganglioma by 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography. J Natl Cancer Inst 2012; 104: 700–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cascon A, Comino-Mendez I, Curras-Freixes M et al. Whole-exome sequencing identifies MDH2 as a new familial paraganglioma gene. J Natl Cancer Inst 2015; 107: pii: djv053. [DOI] [PubMed] [Google Scholar]
- 44.van der Mey AG, Maaswinkel-Mooy PD, Cornelisse CJ, Schmidt PH, van de Kamp JJ. Genomic imprinting in hereditary glomus tumours: evidence for new genetic theory. Lancet 1989; 2: 1291–4. [DOI] [PubMed] [Google Scholar]
- 45.Burnichon N, Rohmer V, Amar L et al. The succinate dehydrogenase genetic testing in a large prospective series of patients with paragangliomas. J Clin Endocrinol Metab 2009; 94: 2817–27. Epub 009 May 19. [DOI] [PubMed] [Google Scholar]
- 46.Neumann HP, Pawlu C, Peczkowska M et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 2004; 292: 943–51. [DOI] [PubMed] [Google Scholar]
- 47.van Hulsteijn LT, Dekkers OM, Hes FJ, Smit JW, Corssmit EP. Risk of malignant paraganglioma in SDHB-mutation and SDHD-mutation carriers: a systematic review and meta-analysis. J Med Genet 2012; 25: 25. [DOI] [PubMed] [Google Scholar]
- 48.Havekes B, van der Klaauw AA, Weiss MM et al. Pheochromocytomas and extra-adrenal paragangliomas detected by screening in patients with SDHD-associated head-and-neck paragangliomas. Endocr Relat Cancer 2009; 16: 527–36. [DOI] [PubMed] [Google Scholar]
- 49.Xekouki P, Szarek E, Bullova P et al. Pituitary adenoma with paraganglioma/pheochromocytoma (3PAs) and succinate dehydrogenase defects in human and mice. J Clin Endocrinol Metab 2015; 100: E710–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pasini B, McWhinney SR, Bei T et al. Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet 2008; 16: 79–88. [DOI] [PubMed] [Google Scholar]
- 51.van Hulsteijn LT, Heesterman B, Jansen JC et al. No evidence for increased mortality in SDHD variant carriers compared with the general population. Eur J Hum Genet 2015; 23: 1713–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Baars F, Cremers C, van den Broek P, Geerts S, Veldman J. Genetic aspects of nonchromaffin paraganglioma. Hum Genet 1982; 60: 305–9. [DOI] [PubMed] [Google Scholar]
- 53.Bayley JP, Kunst HP, Cascon A et al. SDHAF2 mutations in familial and sporadic paraganglioma and phaeochromocytoma. Lancet Oncol 2010; 11: 366–72. Epub 2010 Jan 11. [DOI] [PubMed] [Google Scholar]
- 54.Casey R, Garrahy A, Tuthill A et al. Universal genetic screening uncovers a novel presentation of an SDHAF2 mutation. J Clin Endocrinol Metab 2014; 99: E1392–6. [DOI] [PubMed] [Google Scholar]
- 55.Niemann S, Steinberger D, Muller U. PGL3, a third, not maternally imprinted locus in autosomal dominant paraganglioma. Neurogenetics 1999; 2: 167–70. [DOI] [PubMed] [Google Scholar]
- 56.Hensen EF, van Duinen N, Jansen JC et al. High prevalence of founder mutations of the succinate dehydrogenase genes in the Netherlands. Clin Genet 2012; 81: 284–8. [DOI] [PubMed] [Google Scholar]
- 57.Mannelli M, Ercolino T, Giache V, Simi L, Cirami C, Parenti G. Genetic screening for pheochromocytoma: should SDHC gene analysis be included? J Med Genet 2007; 44: 586–7. Epub 2007 Jun 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gimenez-Roqueplo AP, Favier J, Rustin P et al. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res 2003; 63: 5615–21. [PubMed] [Google Scholar]
- 59.Bogdasarian R, Lotz P. Multiple simultaneous paragangliomas of the head and neck in association with multiple retroperitoneal pheochromocytomas. Otolaryngol Head Neck Surg 1979; 87: 648–52. [DOI] [PubMed] [Google Scholar]
- 60.Denes J, Swords F, Rattenberry E et al. Heterogeneous genetic background of the association of pheochromocytoma/paraganglioma and pituitary adenoma: results from a large patient cohort. J Clin Endocrinol Metab 2015; 100: E531–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Korpershoek E, Favier J, Gaal J et al. SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J Clin Endocrinol Metab 2011; 96: E1472–6. [DOI] [PubMed] [Google Scholar]
- 62.Pantaleo MA, Astolfi A, Indio V et al. SDHA loss-of-function mutations in KIT-PDGFRA wild-type gastrointestinal stromal tumors identified by massively parallel sequencing. J Natl Cancer Inst 2011; 103: 983–7. [DOI] [PubMed] [Google Scholar]
- 63.Clark GR, Sciacovelli M, Gaude E et al. Germline FH mutations presenting with pheochromocytoma. J Clin Endocrinol Metab 2014; 99: E2046–50. [DOI] [PubMed] [Google Scholar]
- 64.Castro-Vega LJ, Buffet A, De Cubas AA et al. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum Mol Genet 2014; 23: 2440–6. [DOI] [PubMed] [Google Scholar]
- 65.Tomlinson IP, Alam NA, Rowan AJ et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 2002; 30: 406–10. [DOI] [PubMed] [Google Scholar]
- 66.Selak MA, Armour SM, MacKenzie ED et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005; 7: 77–85. [DOI] [PubMed] [Google Scholar]
- 67.Pacak K, Jochmanova I, Prodanov T et al. New Syndrome of Paraganglioma and Somatostatinoma Associated With Polycythemia. J Clin Oncol 2013; 18: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Maher ER, Iselius L, Yates JR et al. Von Hippel-Lindau disease: a genetic study. J Med Genet 1991; 28: 443–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ang SO, Chen H, Hirota K et al. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat Genet 2002; 32: 614–21. [DOI] [PubMed] [Google Scholar]
- 70.Richard S, Beigelman C, Duclos JM et al. Pheochromocytoma as the first manifestation of von Hippel-Lindau disease. Surgery 1994; 116: 1076–81. [PubMed] [Google Scholar]
- 71.Baghai M, Thompson GB, Young WF Jr, Grant CS, Michels VV, van Heerden JA. Pheochromocytomas and paragangliomas in von Hippel-Lindau disease: a role for laparoscopic and cortical-sparing surgery. Arch Surg 2002; 137: 682–8; discussion 8-9. [DOI] [PubMed] [Google Scholar]
- 72.Neumann HP, Berger DP, Sigmund G et al. Pheochromocytomas, multiple endocrine neoplasia type 2, and von Hippel-Lindau disease. N Engl J Med 1993; 329: 1531–8. [DOI] [PubMed] [Google Scholar]
- 73.Burnichon N, Vescovo L, Amar L et al. Integrative genomic analysis reveals somatic mutations in pheochromocytoma and paraganglioma. Hum Mol Genet 2011; 20: 3974–85. [DOI] [PubMed] [Google Scholar]
- 74.Percy MJ, Furlow PW, Lucas GS et al. A gain-of-function mutation in the HIF2A gene in familial erythrocytosis. N Engl J Med 2008; 358: 162–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pacak K, Chew EY, Pappo AS et al. Ocular manifestations of hypoxia-inducible factor-2alpha paraganglioma-somatostatinoma-polycythemia syndrome. Ophthalmology 2014; 121: 2291–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lorenzo FR, Yang C, Ng Tang Fui M et al. A novel EPAS1/HIF2A germline mutation in a congenital polycythemia with paraganglioma. J Mol Med (Berl) 2013; 91: 507–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Buffet A, Smati S, Mansuy L et al. Mosaicism in HIF2A-related polycythemia-paraganglioma syndrome. J Clin Endocrinol Metab 2014; 99: E369–73. [DOI] [PubMed] [Google Scholar]
- 78.Favier J, Gimenez-Roqueplo AP. Pheochromocytomas: the (pseudo)-hypoxia hypothesis. Best Pract Res Clin Endocrinol Metab 2010; 24: 957–68. [DOI] [PubMed] [Google Scholar]
- 79.Berra E, Richard DE, Gothie E, Pouyssegur J. HIF-1-dependent transcriptional activity is required for oxygen-mediated HIF-1alpha degradation. FEBS Lett 2001; 491: 85–90. [DOI] [PubMed] [Google Scholar]
- 80.Lorenzo FR, Yang C, Ng Tang Fui M et al. A novel EPAS1/HIF2A germline mutation in a congenital polycythemia with paraganglioma. J Mol Med 2012; 23: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Comino-Mendez I, de Cubas AA, Bernal C et al. Tumoral EPAS1 (HIF2A) mutations explain sporadic pheochromocytoma and paraganglioma in the absence of erythrocytosis. Hum Mol Genet 2013; 26: 26. [DOI] [PubMed] [Google Scholar]
- 82.Ladroue C, Carcenac R, Leporrier M et al. PHD2 mutation and congenital erythrocytosis with paraganglioma. N Engl J Med 2008; 359: 2685–92. [DOI] [PubMed] [Google Scholar]
- 83.Yang C, Zhuang Z, Fliedner SM et al. Germ-line PHD1 and PHD2 mutations detected in patients with pheochromocytoma/paraganglioma-polycythemia. J Mol Med (Berl) 2015; 93:93–104. [DOI] [PubMed] [Google Scholar]
- 84.Castro-Vega LJ, Letouze E, Burnichon N et al. Multi-omics analysis defines core genomic alterations in pheochromocytomas and paragangliomas. Nat Commun 2015; 6: 6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Crowe F, Schull W, Neel J. A Clinical, Pathologicala and Genetic Study of Multiple Neurofibromatosis. Springfield, IL: Charles C Thomas, 1956. [Google Scholar]
- 86.McGaughran JM, Harris DI, Donnai D et al. A clinical study of type 1 neurofibromatosis in north west England. J Med Genet 1999; 36: 197–203. [PMC free article] [PubMed] [Google Scholar]
- 87.Walther MM, Herring J, Enquist E, Keiser HR, Linehan WM. von Recklinghausen’s disease and pheochromocytomas. J Urol 1999; 162: 1582–6. [PubMed] [Google Scholar]
- 88.Donis-Keller H, Dou S, Chi D et al. Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Hum Mol Genet 1993; 2: 851–6. [DOI] [PubMed] [Google Scholar]
- 89.Sipple J. The association of pheochromocytoma with carcinoma of the thyroid gland. Am J Med 1961; 31: 163–6. [Google Scholar]
- 90.Ponder BA. The phenotypes associated with ret mutations in the multiple endocrine neoplasia type 2 syndrome. Cancer Res 1999; 59: 1736s–41s; discussion 42s. [PubMed] [Google Scholar]
- 91.Mukherjee S, Zakalik D. RET codon 804 mutations in multiple endocrine neoplasia 2: genotype-phenotype correlations and implications in clinical management. Clin Genet 2011; 79: 1–16. [DOI] [PubMed] [Google Scholar]
- 92.Lairmore TC, Ball DW, Baylin SB, Wells SA Jr. Management of pheochromocytomas in patients with multiple endocrine neoplasia type 2 syndromes. Ann Surg 1993; 217: 595–601; discussion -3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Modigliani E, Vasen HM, Raue K et al. Pheochromocytoma in multiple endocrine neoplasia type 2: European study. The Euromen Study Group. J Intern Med 1995; 238: 363–7. [DOI] [PubMed] [Google Scholar]
- 94.Welander J, Soderkvist P, Gimm O. Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocr Relat Cancer 2011; 18: R253–76. [DOI] [PubMed] [Google Scholar]
- 95.Yao L, Schiavi F, Cascon A et al. Spectrum and prevalence of FP/TMEM127 gene mutations in pheochromocytomas and paragangliomas. JAMA 2010; 304: 2611–9. [DOI] [PubMed] [Google Scholar]
- 96.Abermil N, Guillaud-Bataille M, Burnichon N et al. TMEM127 screening in a large cohort of patients with pheochromocytoma and/or paraganglioma. J Clin Endocrinol Metab 2012; 97: E805–9. Epub 2012 Mar 14. [DOI] [PubMed] [Google Scholar]
- 97.Qin Y, Deng Y, Ricketts CJ et al. The tumor susceptibility gene TMEM127 is mutated in renal cell carcinomas and modulates endolysosomal function. Hum Mol Genet 2014; 23: 2428–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Toledo SP, Lourenco DM Jr, Sekiya T et al. Penetrance and clinical features of pheochromocytoma in a six-generation family carrying a germline TMEM127 mutation. J Clin Endocrinol Metab 2015; 100: E308–18. [DOI] [PubMed] [Google Scholar]
- 99.Burnichon N, Cascon A, Schiavi F et al. MAX mutations cause hereditary and sporadic pheochromocytoma and paraganglioma. Clin Cancer Res 2012; 27: 27. [DOI] [PubMed] [Google Scholar]
- 100.Flynn A, Benn D, Clifton-Bligh R et al. The genomic landscape of phaeochromocytoma. J Pathol 2015; 236: 78–89. [DOI] [PubMed] [Google Scholar]
- 101.Martin GA, Viskochil D, Bollag G et al. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell 1990; 63: 843–9. [DOI] [PubMed] [Google Scholar]
- 102.Califano D, Rizzo C, D’Alessio A et al. Signaling through Ras is essential for ret oncogene-induced cell differentiation in PC12 cells. J Biol Chem 2000; 275: 19297–305. [DOI] [PubMed] [Google Scholar]
- 103.Le LQ, Parada LF. Tumor microenvironment and neurofibromatosis type I: connecting the GAPs. Oncogene 2007; 26: 4609–16. Epub 2007 Feb 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Welander J, Larsson C, Backdahl M et al. Integrative genomics reveals frequent somatic NF1 mutations in sporadic pheochromocytomas. Hum Mol Genet 2012; 24: 24. [DOI] [PubMed] [Google Scholar]
- 105.Welander J, Andreasson A, Juhlin CC et al. Rare germline mutations identified by targeted next-generation sequencing of susceptibility genes in pheochromocytoma and paraganglioma. J Clin Endocrinol Metab 2014; 99: E1352–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Crona J, Delgado Verdugo A, Maharjan R et al. Somatic mutations in H-RAS in sporadic pheochromocytoma and paraganglioma identified by exome sequencing. J Clin Endocrinol Metab 2013; 98: E1266–71. [DOI] [PubMed] [Google Scholar]
- 107.Luchetti A, Walsh D, Rodger F et al. Profiling of somatic mutations in phaeochromocytoma and paraganglioma by targeted next generation sequencing analysis. Int J Endocrinol 2015; 2015: 138573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Oudijk L, deKrijger RR, Rapa I et al. H-RAS mutations are restricted to sporadic pheochromocytomas lacking specific clinical or pathological features: data from a multi-institutional series. J Clin Endocrinol Metab 2014; 99: E1376–80. [DOI] [PubMed] [Google Scholar]
- 109.Crona J, Nordling M, Maharjan R et al. Integrative genetic characterization and phenotype correlations in pheochromocytoma and paraganglioma tumours. PLoS ONE 2014; 9: e86756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Besset V, Scott RP, Ibanez CF. Signaling complexes and protein-protein interactions involved in the activation of the Ras and phosphatidylinositol 3-kinase pathways by the c-Ret receptor tyrosine kinase. J Biol Chem 2000; 275: 39159–66. [DOI] [PubMed] [Google Scholar]
- 111.Adari H, Lowy DR, Willumsen BM, Der CJ, McCormick F. Guanosine triphosphatase activating protein (GAP) interacts with the p21 ras effector binding domain. Science 1988; 240: 518–21. [DOI] [PubMed] [Google Scholar]
- 112.Fishbein L, Khare S, Wubbenhorst B et al. Whole-exome sequencing identifies somatic ATRX mutations in pheochromocytomas and paragangliomas. Nat Commun 2015; 6: 6140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Juhlin CC, Stenman A, Haglund F et al. Whole-exome sequencing defines the mutational landscape of pheochromocytoma and identifies KMT2D as a recurrently mutated gene. Genes Chromosom Cancer 2015; 54: 542–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Crona J, Backman S, Maharjan R et al. Spatio-temporal heterogeneity characterizes the genetic landscape of pheochromocytoma and defines early events in tumourigenesis. Clin Cancer Res 2015; 21: 4451–60. [DOI] [PubMed] [Google Scholar]
- 115.Nik-Zainal S, Van Loo P, Wedge DC et al. The life history of 21 breast cancers. Cell 2012; 149: 994–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Korpershoek E, Petri BJ, Post E et al. Adrenal medullary hyperplasia is a precursor lesion for pheochromocytoma in MEN2 syndrome. Neoplasia 2014; 16: 868–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kugelberg J, Welander J, Schiavi F et al. Role of SDHAF2 and SDHD in von Hippel-Lindau associated pheochromocytomas. World J Surg 2014; 38: 724–32. [DOI] [PubMed] [Google Scholar]
- 118.Aarts M, Dannenberg H, deLeeuw RJ et al. Microarray-based CGH of sporadic and syndrome-related pheochromocytomas using a 0.1-0.2 Mb bacterial artificial chromosome array spanning chromosome arm 1p. Genes Chromosom Cancer 2006; 45: 83–93. [DOI] [PubMed] [Google Scholar]
- 119.Marinoni I, Kurrer AS, Vassella E et al. Loss of DAXX and ATRX are associated with chromosome instability and reduced survival of patients with pancreatic neuroendocrine tumors. Gastroenterology 2014; 146(453–60): e5. [DOI] [PubMed] [Google Scholar]
- 120.de Wilde RF, Heaphy CM, Maitra A et al. Loss of ATRX or DAXX expression and concomitant acquisition of the alternative lengthening of telomeres phenotype are late events in a small subset of MEN-1 syndrome pancreatic neuroendocrine tumors. Mod Pathol 2012; 25: 1033–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Papathomas T, Oudijk L, Zwarthoff EC et al. TERT promoter mutations in tumors originating from the adrenal gland and extra-adrenal paraganglia. Endocr Relat Cancer 2014; 21: 653–61. [DOI] [PubMed] [Google Scholar]
- 122.de Cubas AA, Leandro-Garcia LJ, Schiavi F et al. Integrative analysis of miRNA and mRNA expression profiles in pheochromocytoma and paraganglioma identifies genotype-specific markers and potentially regulated pathways. Endocr Relat Cancer 2013; 20: 477–93. [DOI] [PubMed] [Google Scholar]
- 123.de Cubas AA, Korpershoek E, Inglada-Perez L et al. DNA methylation profiling in pheochromocytoma and paraganglioma reveals diagnostic and prognostic markers. Clin Cancer Res 2015; 21: 3020–30. [DOI] [PubMed] [Google Scholar]
- 124.Killian JK, Miettinen M, Walker RL et al. Recurrent epimutation of SDHC in gastrointestinal stromal tumors. Sci Transl Med 2014; 6: 268ra177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Brito JP, Asi N, Bancos I et al. Testing for germline mutations in sporadic pheochromocytoma/paraganglioma: a systematic review. Clin Endocrinol (Oxf) 2014; 82: 338–45. [DOI] [PubMed] [Google Scholar]
- 126.Neumann HP, Bausch B, McWhinney SR et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 2002; 346: 1459–66. [DOI] [PubMed] [Google Scholar]
- 127.Muth A, Abel F, Jansson S, Nilsson O, Ahlman H, Wangberg B. Prevalence of germline mutations in patients with pheochromocytoma or abdominal paraganglioma and sporadic presentation: a population-based study in Western sweden. World J Surg 2012; 36: 1389–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Toledo RA, Dahia PL. Next-generation sequencing for the diagnosis of hereditary pheochromocytoma and paraganglioma syndromes. Curr Opin Endocrinol Diabetes Obes 2015; 22: 169–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Crona J, Ljungstrom V, Welin S, Walz MK, Hellman P, Bjorklund P. Bioinformatic challenges in clinical diagnostic application of targeted next generation sequencing: experience from pheochromocytoma. PLoS ONE 2015; 10: e0133210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Eisenhofer G, Walther MM, Huynh TT et al. Pheochromocytomas in von Hippel-Lindau syndrome and multiple endocrine neoplasia type 2 display distinct biochemical and clinical phenotypes. J Clin Endocrinol Metab 2001; 86: 1999–2008. [DOI] [PubMed] [Google Scholar]
- 131.Eisenhofer G, Huynh TT, Pacak K et al. Distinct gene expression profiles in norepinephrine- and epinephrine-producing hereditary and sporadic pheochromocytomas: activation of hypoxia-driven angiogenic pathways in von Hippel-Lindau syndrome. Endocr Relat Cancer 2004; 11: 897–911. [DOI] [PubMed] [Google Scholar]
- 132.Proye C, Fossati P, Fontaine P et al. Dopamine-secreting pheochromocytoma: an unrecognized entity? Classification of pheochromocytomas according to their type of secretion. Surgery 1986; 100: 1154–62. [PubMed] [Google Scholar]
- 133.Richter S, Peitzsch M, Rapizzi E et al. Krebs cycle metabolite profiling for identification and stratification of pheochromocytomas/paragangliomas due to succinate dehydrogenase deficiency. J Clin Endocrinol Metab 2014; 99: 3903–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rao JU, Engelke UF, Sweep FC et al. Genotype-specific differences in the tumor metabolite profile of pheochromocytoma and paraganglioma using untargeted and targeted metabolomics. J Clin Endocrinol Metab 2015; 100: E214–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rao JU, Engelke UF, Rodenburg RJ et al. Genotype-specific abnormalities in mitochondrial function associate with distinct profiles of energy metabolism and catecholamine content in pheochromocytoma and paraganglioma. Clin Cancer Res 2013; 19: 3787–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bhattacharya P, Chekmenev EY, Perman WH et al. Towards hyperpolarized (13)C-succinate imaging of brain cancer. J Magn Reson 2007; 186: 150–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nolting S, Grossman AB. Signaling pathways in pheochromocytomas and paragangliomas: prospects for future therapies. Endocr Pathol 2012; 23: 21–33. [DOI] [PubMed] [Google Scholar]
- 138.Favier J, Igaz P, Burnichon N et al. Rationale for antiangiogenic therapy in pheochromocytoma and paraganglioma. Endocr Pathol 2012; 23: 34–42. [DOI] [PubMed] [Google Scholar]
- 139.Liu T, Brown TC, Juhlin CC et al. The activating TERT promoter mutation C228T is recurrent in subsets of adrenal tumors. Endocr Relat Cancer 2014; 21: 427–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Flynn RL, Cox KE, Jeitany M et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 2015; 347: 273–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Snyder A, Makarov V, Merghoub T et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014; 371: 2189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rizvi NA, Hellmann MD, Snyder A et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015; 348: 124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hegi ME, Diserens AC, Gorlia T et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 2005; 352: 997–1003. [DOI] [PubMed] [Google Scholar]
- 144.Elston MS, Meyer-Rochow GY, Conaglen HM et al. Increased SSTR2A and SSTR3 expression in succinate dehydrogenase-deficient pheochromocytomas and paragangliomas. Hum Pathol 2014; 46: 390–6. [DOI] [PubMed] [Google Scholar]
- 145.Korpershoek E, Stobbe CK, van Nederveen FH, de Krijger RR, Dinjens WN. Intra-tumoral molecular heterogeneity in benign and malignant pheochromocytomas and extra-adrenal sympathetic paragangliomas. Endocr Relat Cancer 2010; 17: 653–62. [DOI] [PubMed] [Google Scholar]
- 146.Khan MS, Kirkwood A, Tsigani T et al. Circulating tumor cells as prognostic markers in neuroendocrine tumors. J Clin Oncol 2013; 31: 365–72. [DOI] [PubMed] [Google Scholar]
- 147.Bettegowda C, Sausen M, Leary RJ et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 2014; 6: 224ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]