

Abstract
We report here a rare case of Bardet–Biedl syndrome (BBS). A 7-year-old boy was diagnosed to have BBS based on the clinical features: retinitis pigmentosa sine pigmento, obesity, postaxial polydactyly, syndactyly, and hypogenitalism. It was associated with mild hepatomegaly with deranged liver function test and mild renal involvement radiologically, high-arched palate, and low intelligence quotient. The patient was prescribed proper refractive correction and subjected to multidisciplinary management. BBS has ocular and systemic manifestations, requiring a multidisciplinary approach to treatment.
Keywords: Bardet–Biedl syndrome, hypogonadism, polydactyly, retinitis pigmentosa sine pigmento
Introduction
BBS is an autosomal recessive condition characterized mainly by rod-cone dystrophy, postaxial polydactyly, central obesity, mental retardation, hypogonadism, and renal dysfunction. The most common gene is BBS1 located at chromosome 11. Retinal dystrophy is the most important clinical feature of the disorder. Attenuated vessels, mottling, granularity of the retinal pigment epithelial, bone spicule formation, and waxy pallor of the disc are typical fundus findings in RP. BBS is the most commonly associated RP sine pigmento, present in our case. BBS is a chronic disease without any specific cure.
Case Report
An 8-year-old boy presented with his parents with chief complaint of diminution of vision in both eyes since birth. He was also not able to see properly at night, as conveyed by parents. He had full-term normal vaginal delivery by a nonconsanguineous parentage. His birth weight was 3 kg with no history of neonatal intensive care unit admission postbirth. He had one brother and two sisters, but with no such similar complaints. He had low intelligence and poor school performance as compared to children of his own age, as told by his parents. On ocular examination, his visual acuity was finger count at 1 M in both the eyes with accurate projection of rays in all the four quadrants. His intraocular pressure was 16 mmHg by noncontact tonometer in both the eyes. Cycloplegic refraction by 2% homatropine revealed power of −5.50 Dsp/−1.50 DCy @180 degree in the right eye and − 6.50 DSp/−0.75 DCy @10 degree in the left eye with best-corrected visual acuity of 6/36 and 6/24 on Snellen's chart in the right and left eyes, respectively. Adnexa were normal. Pupils were 3 mm in size and briskly reactive to light with no afferent pupillary defect. The patient was orthophoric with a full range of ocular movements. Fundus examination by indirect ophthalmoscopy revealed cup-to-disc ratio of 0.4, with disc pallor and temporal crescent in both the eyes. There was generalized chorioretinal atrophy without any pigmentation in both the eyes [Figure 1], suggesting retinitis pigmentosa (RP) sine pigmento.
Figure 1.
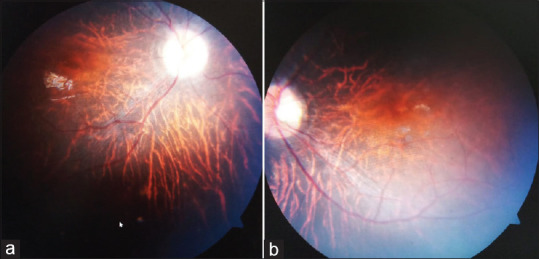
Colored photo of both eyes (a and b) fundus examination revealed cup-to-disc ratio of 0.4, with disc pallor and temporal crescent in both the eyes. There was generalized chorioretinal atrophy and without any pigmentation
Visual field examination was not possible due to low understanding of the patient. Detailed systemic examination revealed postaxial polydactyly in the left hand [Figure 2a] and in the bilateral feet [Figure 2b], high-arched palate [Figure 2c], syndactyly of the second and third toe, micropenis [Figure 2d], and truncal obesity [Figure 2e].
Figure 2.

Postaxial polydactyly in the left hand (a) and in the bilateral feet (b), high-arched palate (c), syndactyly of the second and third toe, micropenis (d), and truncal obesity (e)
Detailed ENT examination revealed high-arched palate and bilateral tonsillar hypertrophy. There was no associated sensorineural hearing loss. Electrocardiogram and echocardiography revealed normal heart both structurally and functionally. Ultrasonography of the abdomen demonstrated mild hepatomegaly with small hemangioma, bilateral small renal concretions, and right renal calculus. There was no associated dental malocclusion. Detailed serological investigations revealed mildly deranged liver function test and normal Renal function test (RFT). There was a gross reduction in luteinizing hormone and follicle-stimulating hormone signifying hypogonadism.
Based on the ocular and systemic findings, the diagnosis of Bardet–Biedl syndrome (BBS) with associated RP sine pigmento and myopia was made. The patient was prescribed proper refractive spectacles. He was subjected to multidisciplinary management. The parents were explained about the nature of the disease and poor visual prognosis, and a course of Vitamin A (15,000 IU of retinol palmitate) was prescribed for 30 days.
Discussion
BBS was described by Bardet–Bidel in 1920. BBS is an autosomal recessive condition characterized mainly by rod-cone dystrophy, postaxial polydactyly, central obesity, mental retardation, hypogonadism, and renal dysfunction. In this type of inheritance, both parents will be carriers, meaning they have one gene for the syndrome paired with one normal gene. Each of their children then has a 25% chance (or 1 chance in 4) of inheriting the two Bardet–Biedl genes (one from each parent) needed to cause the disorder. Carriers are unaffected because they have only one copy of the gene. Beales et al. have given a diagnostic criterion for BBS: the presence of either four primary features or a combination of three primary and two secondary features.[1] The primary features are rod-cone dystrophy, polydactyly, obesity, learning disabilities, hypogenitalism in males, and renal anomalies. Secondary features are speech disorder, cataracts, astigmatism, strabismus, syndactyly, brachydactyly, developmental delay, polyuria, and polydipsia. Additional secondary features are hypodontia/small tooth roots, dental crowding, high-arched palate, mild spasticity, diabetes mellitus, left ventricular hypertrophy/congenital heart disease, hepatic fibrosis, ataxia, poor coordination, and imbalance. BBS patients may have learning disabilities and short-term memory deficit but gifted by excellent long-term memory. These patients perform much better at performance rather than verbal skills.
BBS is genetically heterogeneous with 6 loci mapped till date. The most common gene is BBS1 located at chromosome 11.[2] Retinal dystrophy is the most important clinical feature of the disorder. BBS has previously been described as a rod-cone, cone-rod, and choroidal or global retinal dystrophy.[3] It has also been reported that patients with difficulty to classify early photoreceptor dysfunction later on progress to a classic stage of rod degeneration.[4] Attenuated vessels, mottling, granularity of the retinal pigment epithelial, bone spicule formation, and waxy pallor of the disc are typical fundus findings in RP. BBS is the most commonly associated RP sine pigmento, present in our case. Obesity is the second major clinical feature. Truncal obesity is mostly found. Limb abnormalities are the third major clinical feature, with polydactyly being most common. Brachydactyly and rarely syndactyly can also be found. Syndactyly is less commonly seen but is usually partial and confined to the feet,[1] also presented in our case between the second and third toe in both the feet. A minority of patients are mentally retarded. Male hypogenitalism and complex female genitourinary malformations are also associated. Renal dysfunction is a major cause of morbidity and mortality. The diagnosis is essentially based on clinical features. BBS is distinguished from much rarer Laurence–Moon–Biedl syndrome, in which retinal pigmentary degeneration, mental retardation, and hypogonadism occur but without polydactyly.
Our patient demonstrated several classic clinical features as RP sine pigmento, central obesity, postaxial polydactyly, syndactyly in the feet, and hypogenitalism. There was associated high-arched palate and myopia.
BBS is a chronic disease without any specific cure. Parents require proper counseling with proper advice regarding the life-threatening systemic complications of the syndrome. A course of Vitamin A may be given for nyctalopia. Speech therapy and proper rehabilitation may be necessary for some children affected by this syndrome. It is a chronic disease without any cure. Time to time symptomatic treatment is required by multidisciplinary approach. The pediatrician should be involved in the overall care of the patient and should refer the patient to appropriate subspecialties when necessary.
Conclusion
BBS is a rare syndrome requiring early diagnosis and expert multidisciplinary management as it may be associated with life-threatening complications. Parents require proper counseling with proper advice regarding the life-threatening systemic complications of the syndrome. This case report may add in the knowledge for the understanding of this syndrome.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient has given his consent for his images and other clinical information to be reported in the journal. The patient understands that name and initials will not be published and due efforts will be made to conceal identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
The authors declare that there are no conflicts of interests of this paper.
References
- 1.Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: Results of a population survey. J Med Genet. 1999;36:437–46. [PMC free article] [PubMed] [Google Scholar]
- 2.Mykytyn K, Nishimura DY, Searby CC, Shastri M, Yen HJ, Beck JS, et al. Identification of the gene (BBS1) most commonly involved in Bardet-Biedl syndrome, a complex human obesity syndrome. Nat Genet. 2002;31:435–8. doi: 10.1038/ng935. [DOI] [PubMed] [Google Scholar]
- 3.Riise R, Andréasson S, Tornqvist K. Full-field electroretinograms in individuals with the Laurence-Mood-Bardet-Biedl syndrome. Acta Ophthalmol Scand. 1996;74:618–20. doi: 10.1111/j.1600-0420.1996.tb00747.x. [DOI] [PubMed] [Google Scholar]
- 4.Azari AA, Aleman TS, Cideciyan AV, Schwartz SB, Windsor EA, Sumaroka A, et al. Retinal disease expression in Bardet-Biedl syndrome-1 (BBS1) is a spectrum from maculopathy to retina-wide degeneration. Invest Ophthalmol Vis Sci. 2006;47:5004–10. doi: 10.1167/iovs.06-0517. [DOI] [PubMed] [Google Scholar]