

Abstract
The Hedgehog (HH) signaling pathway was discovered originally as a key pathway in embryonic patterning and development. Since its discovery, it has become increasingly clear that the HH pathway also plays important roles in a multitude of cancers. Therefore, HH signaling has emerged as a therapeutic target of interest for cancer therapy. In this review, we provide a brief overview of HH signaling and the key molecular players involved and offer an up-to-date summary of our current knowledge of endogenous and exogenous small molecules that modulate HH signaling. We discuss experiences and lessons learned from the decades-long efforts toward the development of cancer therapies targeting the HH pathway. Challenges to develop next-generation cancer therapies are highlighted.
Introduction
As with several signaling pathways, the HH pathway is essential for embryonic and stem cell programs, but pathway action is also linked to cancer, in particular, in maintaining tumor-initiating/stem cells (TISC) (Harris et al., 2012). Activating mutations in HH pathway components have been documented in medulloblastoma (MB) (Raffel et al., 1997), the most common childhood brain cancer; in basal cell carcinoma (BCC) (Xie et al., 1998), the most common cancer in the white population; and in rhabdomyosarcoma (RMS) (Tostar et al., 2006), the most common pediatric soft tissue cancer. In addition, modulation of the tumor microenvironment by HH signaling has been argued to play a broader role in several other cancers, including those of the breast (Mukherjee et al., 2006), lung (Watkins et al., 2003), liver (Huang et al., 2006), stomach (Berman et al., 2003), pancreas (Thayer et al., 2003), colon (Varnat et al., 2009), and prostate (Karhadkar et al., 2004). Not surprisingly given these early findings, HH signaling has emerged as an attractive target for targeted cancer therapy (Rubin and de Sauvage, 2006). Proof of principle has been demonstrated in the design of therapeutic approaches to modulate pathway activity in treating invasive BCC (Sekulic et al., 2012; Tang et al., 2012; Von Hoff et al., 2009). These initial findings raise optimism for extending the therapeutic range of HH pathway modulators.
Here, we provide an update on therapeutic development around the HH pathway with a focus on small-molecule regulators and cancer.
Overview of the HH Signaling Pathway
The hedgehog (hh) gene was discovered over 35 years ago in ground-breaking genetic screens examining segmental development in the fruit fly, Drosophila melanogaster (Nusslein-Vol-hard and Wieschaus, 1980). Evolutionary analysis reveals ancient components were likely repurposed and rearranged into a connected signaling pathway in conjunction with the emergence of multicellular metazoan life forms over a billion years ago (Hausmann et al., 2009; Ingham et al., 2011; Wilson and Chuang, 2010). In mammals, the core components of the HH pathway comprise: three HH ligands (Sonic hedgehog [SHH], Desert hedgehog [DHH], and Indian hedgehog [IHH]); a 12-pass transmembrane receptor Patched1 (PTCH1); a G-protein-coupled receptor-like seven-pass transmembrane protein Smoothened (SMO); and three transcription factors (GLI1, GLI2, and GLI3) named from the original association of one member (GLI1) with glioma (Ingham and McMahon, 2001). The primary cilium (PC), a subcellular membrane extension with a tubulin scaffold, provides a specific cellular compartment critical to the distribution and function of many of these pathway components (Figure 1) (Corbit et al., 2005; Haycraft et al., 2005; Rohatgi et al., 2007). There are likely diverse roles played by the PC, including the concentration of pathway components to effectively regulate the signaling response and regulation inherent in unique features of the organelle itself. For example, recent evidence indicates a distinct lipid membrane composition for the PC, critical for ciliary function and HH signaling (Chavez et al., 2015; Garcia-Gonzalo et al., 2015).
Figure 1. Schematic Illustrations of the Mammalian HH Signaling Pathway.
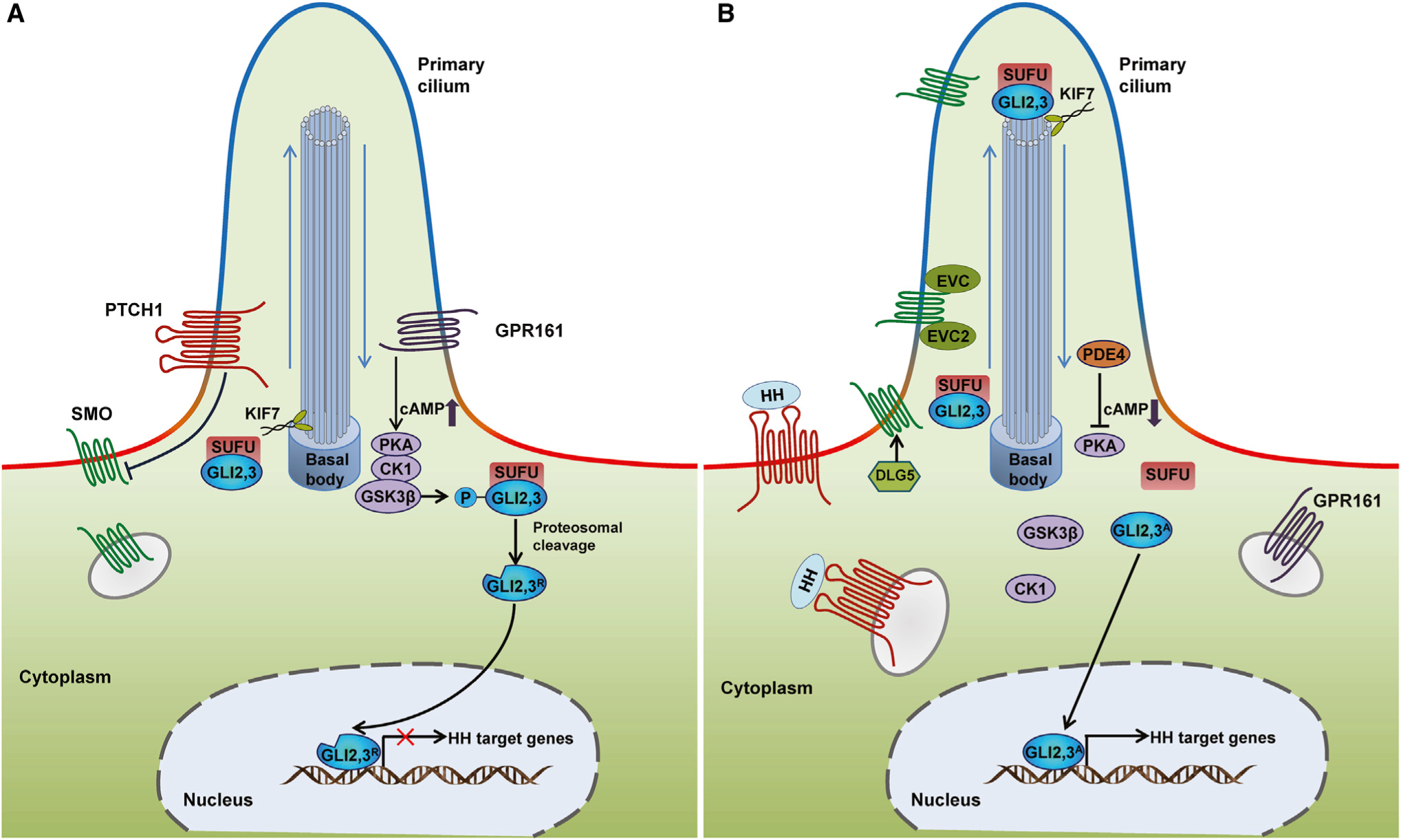
(A) In the absence of HH ligand, PTCH1 localizes at the base of the PC (a subcellular membrane extension with high levels of PI4P (blue) but low levels of PI(4,5)P2 (red)), and inhibits SMO accumulation in the PC and consequently SMO activity. The GLI transcription factors GLI2 and GLI3 are sequestered in the cytoplasm by SUFU and phosphorylated by PKA, CK1, and GSK3β. GPR161, a ciliary G-protein-coupled receptor localized at the base of the PC, can activate PKA through increasing the cAMP levels, promoting the phosphorylation of GLI2 and GLI3. Phosphorylated GLI2 and GLI3 are processed by the proteasome into repressor forms (GLI2R and GLI3R).
(B) Upon ligand binding, PTCH1 and GPR161 are displaced from the PC and SMO interacts with DLG5 and translocates into the PC. Within the PC, SMO forms a complex with EVC and EVC2 to transduce the HH signaling response. Activated SMO relieves SUFU-mediated suppression of GLI2 and GLI3 within the PC. GLI2 and GLI3 maintain their full-length status and bypass phosphorylation, as PKA activity is restrained by a decreased level of cAMP induced by the exit of GPR161 from PC and the degradation of cAMP by phosphodiesterase 4 (PDE4). The activator forms of GLI2 and GLI3 (GLI2A and GLI3A) translocate to the nucleus and induce the expression of HH target genes. Movement of GLI2 and GLI3 proteins within the PC occurs in conjunction with KIF7, a member of the kinesin family of anterograde motor proteins.
In the absence of an HH ligand, PTCH1 localizes to the PC, where it suppresses ciliary accumulation of SMO, a necessary step for pathway activation in mammals (but not in Drosophila) (Figure 1) (Rohatgi et al., 2007). Tuning PTCH1 ciliary intensity by truncating PTCH’s cytoplasmic tail by varying lengths leads to highly correlated SMO suppression, a strong indication of a critical role of ciliary PTCH1 in inhibiting SMO activity (Kim et al., 2015). Upon HH binding, PTCH1 exits the PC, thus relieving the inhibition of SMO and allowing the translocation of SMO into the PC (Rohatgi et al., 2007). The relationship between PTCH1 trafficking from the PC and SMO entry and pathway activation remains a mystery.
An attractive hypothesis based on PTCH1 homology with the resistance-nodulation-division (RND) family of bacterial membrane transporters posits a role for PTCH1 in the transport of endogenous small-molecule SMO modulator(s) (Sharpe et al., 2015b). However, a transport function of PTCH1 has not been demonstrated nor has a bona fide endogenous SMO activator/inhibitor been identified. Recent studies suggest cholesterol is an endogenous SMO activator (Byrne et al., 2016; Huang et al., 2016; Luchetti et al., 2016), while others argue that PTCH1 may mediate secretion of an endogenous SMO inhibitor (or inhibitors), possibly a sterol derivative (or derivatives) (Roberts et al., 2016; Sever et al., 2016). This topic is discussed further in the section on “Endogenous Chemical Modulation of SMO“.
Prior to entering into the PC, SMO interacts with Discs large, homolog 5 (DLG5) at the basal body; a necessary step for subsequent pathway activation (Chong et al., 2015). Within the cilia, SMO forms a complex with EVC (Ellis-van Creveld syndrome protein) and EVC2 to transduce the HH signal (Dorn et al., 2012; Yang et al., 2012). The activation of SMO triggers an intracellular signal transduction cascade that promotes trafficking and processing of GLI transcription factors in the PC, regulating their transcriptional activity (Liu et al., 2015; Shi et al., 2014).
The three mammalian GLI proteins share a similar DNA-binding domain comprising five tandem C2H2 zinc fingers and a C-terminal activation domain; however, only GLI2 and GLI3 contain N-terminal repressor domains (Aza-Blanc et al., 2000; Dai et al., 1999; Sasaki et al., 1999). GLI1 encodes a feedforward pathway activator amplifying the activation response (Lee et al., 1997; Park et al., 2000); Gli1 transcription is entirely dependent on active HH signaling (Lee et al., 1997; Ruiz i Altaba, 1998). GLI2 and GLI3 form full-length activators, or when truncated by processing, distinct repressor forms (Pan et al., 2006; Wang and Li, 2006). Among the targets of direct GLI action are cell fate determinants of tissue patterning, cell proliferation, and cell survival regulators, as well as a variety of HH pathway components that act in positive or negative feedback systems (Junker et al., 2014; Peterson et al., 2012; Vokes et al., 2008).
A number of studies have identified interacting proteins involved in GLI regulation of the HH pathway. Suppressor of fused (SUFU) and kinesin family member 7 (KIF7) are two core regulators of mammalian GLI proteins. SUFU negatively regulates the mammalian HH pathway by cytoplasmic retention of all three GLI proteins (Han et al., 2015; Kogerman et al., 1999; Stone et al., 1999). SUFU also interacts with GLI2 in the nucleus, inhibiting GLI2 activity (Han et al., 2015). SUFU co-occupies promoters of HH target genes with GLI proteins and p66β, a chromatin-remodeling factor, and Mycbp, a transcriptional regulatory factor (Han et al., 2015; Lin et al., 2014). KIF7 is a kinesin protein that acts in anterograde transport (from base to tip) in the PC (Cheung et al., 2009; Endoh-Yamagami et al., 2009; Liem et al., 2009; Tay et al., 2005). In addition, a set of kinases, including protein kinase A (PKA), casein kinase I (CK1), and glycogen synthase kinase 3β (GSK3β), regulate the state of GLI phosphorylation at the basal body of the PC (Barzi et al., 2010; Fumoto et al., 2006; Sillibourne et al., 2002).
In the unstimulated state, KIF7 localizes at the base of the PC and prevents GLI2 and GLI3 accumulation within the cilia (Endoh-Yamagami et al., 2009; Liem et al., 2009). GLI2 and GLI3 are phosphorylated by PKA, CK1, and GSK3β, and then processed by the proteasome into their respective repressor forms, GLI2R and GLI3R (Pan and Wang, 2007; Tempé et al., 2006; Wang and Li, 2006). In this, a cAMP boost modulated by GPR161, a ciliary G-protein-coupled receptor localized at the base of PC, may stimulate PKA activity (Mukhopadhyay et al., 2013). Upon pathway activation, KIF7 movement to the tip of the PC promotes activation of GLI2 and GLI3, most likely countering the action of SUFU (Endoh-Yamagami et al., 2009; Liem et al., 2009). PKA activity is restrained by decreasing cAMP levels due to the ciliary exit of GPR161 (Mukhopadhyay et al., 2013) and cAMP degradation by phosphodiesterase 4 (PDE4) (Williams et al., 2015). Above all, dissociation of GLI2 and GLI3 from SUFU results in fully activated GLI2 and GLI3 (GLI2A and GLI3A), which translocate to the nucleus and turn on their target genes. In normal HH signaling, most GLI-dependent activation of HH target genes is mediated by GLI2A (Bai et al., 2002).
Endogenous Chemical Modulation of SMO
The endogenous modulator(s) operating between PTCH1 and SMO remains the major mystery of the HH field. PTCH1 acts catalytically to suppress SMO, and despite early reports, to the contrary, it is now accepted that there is no physical interaction between PTCH1 and SMO (Taipale et al., 2002). Much of the attention has focused on steroidal metabolites.
SMO activation is impaired through a cholesterol deficiency, either by methyl-β-cyclodextrin (MβCD) stripping in cell culture, or by functionally perturbing the action of 7-dehydrocholesterol reductase (DHCR7) converting 7-dehydrocholesterol (7-DHC) to cholesterol; the genetic underpinning of Smith-Lemli-Opitz syndrome (SLOS) (Blassberg et al., 2016; Cooper et al., 2003). Sterol depletion also diminishes SMO accumulation on the PC (Huang et al., 2016). Provocatively, PTCH1 belongs to a diverse family of RND pump proteins whose bacterial role is to transport diverse molecules such as antibiotics and sterols across lipid membranes (Davies et al., 2000). PTCH1 also contains a putative sterol-sensing domain first identified in the context of sterol-sensing regulatory mechanisms governing sterol metabolism (Martin et al., 2001; Strutt et al., 2001).
From Oxysterols to Cholesterol: Endogenous SMO Agonists?
Oxysterols have been put forward as one class of endogenous SMO activators (Corcoran and Scott, 2006) although the most potent of these in SMO activation, 20(S)-hydroxycholesterol (20(S)-OHC), could not be identified in a cell line that responds to HH signals (Myers et al., 2013). SMO mutations that impair binding to 20(S)-OHC do not inhibit SMO activity in a manner that is tightly correlated to a loss of binding activity (Myers et al., 2013; Nachtergaele et al., 2013; Nedelcu et al., 2013). Further, 20(S)-OHC does not act synergistically with SHH ligand in pathway activation, as might be expected when these factors are present at sub-threshold levels (Huang et al., 2016; Nachtergaele et al., 2012). Taken together, it is unlikely that 20(S)-OHC is the endogenous SMO modulator.
New insights have come from a variety of studies, including crystal structure models of SMO action (Byrne et al., 2016; Huang et al., 2016; Jiang et al., 2016; Luchetti et al., 2016; Roberts et al., 2016; Sever et al., 2016). Unexpectedly, a cholesterol molecule binds the SMO cysteine rich domain (CRD) (Byrne et al., 2016). When delivered with MβCD, cholesterol activates SMO ciliary accumulation and SMO-directed signaling (Huang et al., 2016; Luchetti et al., 2016). Blockade of SMO ciliary accumulation and HH pathway activation by sterol depletion can be rescued by cholesterol. SMO lacking its CRD, or bearing mutations in key amino acids required for cholesterol binding, does not respond to cholesterol (Huang et al., 2016; Luchetti et al., 2016). Cholesterol binding to SMO is competed by both 20(S)-OHC and cyclopamine, consistent with a common binding site in the CRD (Byrne et al., 2016; Huang et al., 2016).
In contrast to 20(S)-OHC, cholesterol potentiates signaling evoked by the HH ligand (Huang et al., 2016; Luchetti et al., 2016). Interestingly, a mutant mouse Smo-G155F responds to cholesterol and the HH ligand but not 20(S)-OHC (Huang et al., 2016; Luchetti et al., 2016; Nachtergaele et al., 2012). Here, the conformations of SMO bound to cholesterol and 20(S)-OHC are distinct, consistent with these findings (Byrne et al., 2016; Luchetti et al., 2016). The current evidence supports cholesterol as a potential endogenous SMO activator, regulating activity in a distinct manner from 20(S)-OHC. Moreover, as the response of SMOΔCRD to HH ligand dampens but is not abolished (Aanstad et al., 2009; Huang et al., 2016; Luchetti et al., 2016; Myers et al., 2013; Nachtergaele et al., 2013; Nedelcu et al., 2013), the CRD, and cholesterol action, appear to be required for maximal levels of SMO activation.
It is not clear how a specificity for SMO would be created by this regulatory mechanism. Interestingly, low levels of cholesterol-dependent detergents permeabilize the plasma membrane but leave the ciliary membrane intact, suggesting a different sterol composition or accessibility in the PC membrane relative to the cell membrane that could play a role in modulating SMO activity (Ye et al., 2013).
A key factor enabling the discovery of the role of cholesterol in SMO regulation has been the application of MβCD to access the SMO CRD in the extracellular space. Previous studies probably missed the interactions here because cholesterol presentation was insufficient due to its hydrophobic nature and poor solubility. MβCD shields cholesterol from water, potentially enabling an effective concentration consistent with the rapid transfer of cholesterol to the SMO CRD. This begs the question of how cholesterol might be presented in vivo?
A Role for SMO Antagonists?
Data supporting an inhibitor as an endogenous SMO modulator also exist. There have been conflicting reports on whether sterol depletion blocks pathway activation mediated by SMOΔCRD (Huang et al., 2016; Luchetti et al., 2016; Myers et al., 2013). Deletion of the CRD results in high basal SMO activity, indicating this region can have an inhibitory role (Aanstad et al., 2009; Myers et al., 2013; Nachtergaele et al., 2013; Nedelcu et al., 2013), potentially binding an endogenous inhibitory molecule in the unstimulated state in vivo (Huang et al., 2016; Luchetti et al., 2016; Myers et al., 2013). MβCD removal of sterols actually enhances pathway activation by SAG1.5 (a potent SAG analog) (Sever et al., 2016), arguing for depletion of an inhibitory sterol in addition to the binding of a permissive sterol for pathway activation. Studying pathway activity in mosaic neuralized embryonic bodies, investigators have argued for a paracrine SMO inhibitory signal that is produced by PTCH1- and PTCH2-expressing cells and enhanced by the loss of DHCR7 (Roberts et al., 2016). Most recently, 3β,5α-dihydroxycholest-7-en-6-one (DHCEO), an endogenous derivative of the DHCR7 substrate 7-dehydrocholesterol (7-DHC) (Sever et al., 2016), and the most abundant oxysterol species in the brain of an SLOS animal model (Xu et al., 2012), was identified as an inhibitory molecule. Interestingly, DHCEO did not appear to bind to either the CRD or transmembrane (TM) domains of SMO and was not detected in cyclodextrin extracts (Sever et al., 2016); thus, the in vivo relevance of these findings is still in question.
Although the action of endogenous SMO modulators has not been resolved, examples exist of molecular classes whose members can have agonist or antagonist actions on SMO. One class are glucocorticoids identified in screens examining SMO ciliary accumulation and signaling activity (Rana et al., 2013; Wang et al., 2012b). One of these glucocorticoids, budesonide, is a pathway antagonist that binds the SMO CRD domain (Rana et al., 2013; Wang et al., 2012b). Similarly, certain oxysterol analogs compete against 20(S)-OHC for binding SMO CRD but inhibit SMO activity (Nedelcu et al., 2013).
CRD or TM: Which Sites Engage Endogenous Modulators?
Multiple sites on SMO bind natural or synthetic compounds, including the CRD, a transmembrane binding groove with the entry facing the CRD, and the cytoplasmic tail. Which of these engage endogenous molecules is an open question. Strikingly, small-molecule interactions might switch activity states. A mouse Smo mutant D477G/E522K that contains two point mutations, which block binding of an antagonist cyclopamine to the TM site but leave CRD binding unaffected, is activated by cyclopamine (Huang et al., 2016). SMOΔCRD activity in an SMO knockout cell line can be inhibited by overexpressing PTCH1 (Myers et al., 2013), indicating that PTCH1 can likely regulate via other sites on SMO. Mutations in the TM pocket that disrupt binding of synthetic modulators, including several drug-refractory mutations such as mouse Smo mutant D477G/E522K mentioned above, do not abolish HH and PTCH1 regulation of SMO, potentially pointing to distinct sites and mechanistic actions of synthetic and endogenous SMO modulatory factors.
While neither the CRD or the TM appears to be an exclusive site for mediating endogenous small-molecule regulation on SMO, allosteric interactions might communicate small-molecule interactions from one site to the other, as observed in crystal structures (Byrne et al., 2016). Previous analyses might be biased by the predictive separation of three domains based on their hypothetical location across the cellular membrane. In agreement with this view, SMO is no longer inhibited by PTCH1 when both CRD and the first two TM domains are replaced by corresponding portions of Frizzled5 (Fz5), an ortholog of SMO in the Wnt pathway (Murone et al., 1999). Future dissection of SMO domains guided by structural studies will provide a better understanding of structure, function, and regulatory interactions governing SMO activity.
PI4P, a New Player
The cytoplasmic tail of SMO appears to be another site involved in SMO’s endogenous modulation. Phosphatidylinositol 4-photosphate (PI4P) binds SMO’s cytoplasmic tail, promoting ciliary accumulation and pathway activation (Jiang et al., 2016). PTCH1 was found to sequester PI4P (Jiang et al., 2016). Further, HH pathway activation elevates the cellular PI4P level (Jiang et al., 2016). Intriguingly, PI4P is the major phosphoinositide in the primary cilium and appears to be required for normal HH signaling as knockout of inositol 5-phosphatase (INPP5E), which localizes in the PC and produces PI4P, decreased the cellular response to the HH signal (Chavez et al., 2015; Garcia-Gonzalo et al., 2015).
A possible mechanism of PTCH1 control of local membrane lipid composition important for SMO regulation is consistent with its reciprocal translocation with SMO in the PC and repellent localization in ciliary membrane domains of SMO and a PTCH1 mutant that retains in the PC upon HH stimulation (Kim et al., 2015). PTCH1 also localizes with a “skirt”-like distribution at the base of the PC (Rohatgi et al., 2007). Interestingly, single-molecule-resolution analysis of SMO trafficking in the PC suggests binding events at the ciliary base that are subject to PTCH1 regulation, which keep SMO out of the PC (Milenkovic et al., 2015; Ye et al., 2013). Further study of the mechanisms of ciliary compartmentalization of PTCH1, SMO, and these membrane lipids will likely provide important new insights into the PTCH1 endogenous regulation of SMO.
HH Signaling and Human Diseases
The HH signaling pathway controls numerous biological processes throughout embryonic development and exerts important regulatory functions in the adult. A variety of studies have linked deregulated HH signaling activity to a number of diseases: depressed pathway activity to tissue degeneration and excessive pathway activity to malignant tumorigenesis. Some, but not all, of these examples, correlate with normal HH signaling mediating tissue homeostasis through stem cell programs. For example, HH is required for neural stem cell (NSC) proliferation in the adult hippocampus (Machold et al., 2003), and inhibition of HH suppresses NSC proliferation both in vitro and in vivo (Lai et al., 2003). Likewise, cardiac progenitors directly respond to HH signals, and reduced HH signaling is reported to result in fewer myocardial progenitors in the mouse (Thomas et al., 2008). The importance of the HH pathway in normal progenitor/stem cell renewal is consistent with reports of HH pathway engagement in the regeneration of the bladder (Shin et al., 2011), prostate (Karhadkar et al., 2004), bone (Miyaji et al., 2003), tooth (Zhao et al., 2014), liver (Ochoa et al., 2010), and airways (Watkins et al., 2003).
Whereas controlled HH pathway activity is linked to tissue repair and tissue homeostasis, uncontrolled activation of the pathway promotes cancer (Table 1). The HH-cancer link was first evident with the discovery of PTCH1 heterozygosity in patients with Gorlin syndrome, characterized by the appearance of a distinct set of solid tumors: BCC, MB, and RMS (Hahn et al., 1996; Johnson et al., 1996). Tumorigenesis in these cases is ligand independent; pathway activation depends on mutations in HH pathway components in HH responsive cells (Figure 2A).
Table 1.
Human Cancers Associated with Deregulated HH Signaling
| Human Cancer | Type | Experimental Systems | Key References |
|---|---|---|---|
| Medulloblastoma (MB) | ligand-independent | mouse model; clinical trial | (Berman et al., 2002; Geoerger et al., 2012; Romer et al., 2004) |
| Basal cell carcinoma (BCC) | ligand-independent | mouse model; clinical trial | (Migden et al., 2015; Sekulic et al., 2012; Xie et al., 1998) |
| Rhabdomyosarcoma (RMS) | ligand-independent | mouse model; clinical trial | (Hahn et al., 1998; Kieran et al., 2013) |
| Pancreatic cancer | ligand-dependent | mouse model; clinical trial | (Ko et al., 2016; Olive et al., 2009) |
| Chronic myeloid leukemia (CML) | ligand-dependent | mouse model; clinical trial | (Dierks et al., 2008; Martinelli et al., 2015) |
| Multiple myeloma | ligand-dependent | mouse model; clinical trial | (Dierks et al., 2007; Khan et al., 2015) |
| Lymphoma | ligand-dependent | mouse model; clinical trial | (Dierks et al., 2007; Tibes and Mesa, 2014) |
| Breast cancer | ligand-dependent | human breast cancer cell line; clinical trial | (Chu et al., 2014; Zhang et al., 2009) |
| Prostate cancer | ligand-dependent | human prostate cancer cell line; clinical trial | (Antonarakis et al., 2013; Karhadkar et al., 2004) |
| Small cell lung cancer | ligand-dependent | human lung cancer cell line; clinical trial | (Belani et al., 2013; Watkins et al., 2003) |
| Colorectal cancer | ligand-dependent | human colorectal cancer cell line; clinical trial | (Berlin et al., 2013; Monzo et al., 2006) |
| Liver cancer | ligand-dependent | human hepatocellular carcinoma cell line | (Huang et al., 2006) |
| Stomach cancer | ligand-dependent | human gastric carcinoma cell line; clinical trial | (Berman et al., 2003; Cohen et al., 2013) |
Figure 2. Distinct Mechanisms of HH Pathway Activation in Cancer.
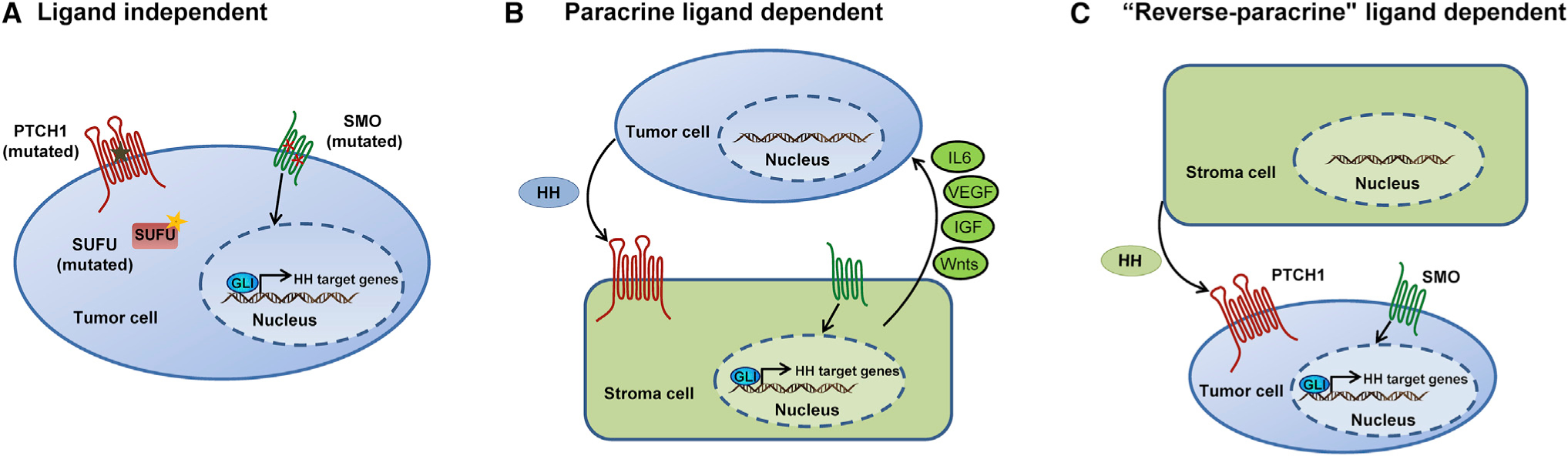
(A) Ligand-independent activation due to inactivating mutations in the negative regulators PTCH1 or SUFU, or activating mutations in the positive regulator SMO, or amplification of GLI activators.
(B and C) Ligand-dependent activation occurs through paracrine (B) or “reverse paracrine” mechanisms (C). (B) Paracrine signaling occurs when the tumor cells secrete the ligand and surrounding cells (e.g., stromal cells for epithelial tumors) respond and secrete tumor supporting signals. These include IL6, VEGF, IGF, and Wnts depending on the HH target tissue and tumor. (C) “Reverse paracrine” signaling occurs when tumor cells receive HH secreted by adjacent non-tumor cells (e.g., stromal cells) to promote tumor growth.
Almost all cases of BCC are linked to hyperactive HH signaling, most commonly through inactivating mutations in PTCH1 or activating mutations in SMO (Gailani et al., 1996; Reifenberger et al., 1998; Unden et al., 1996). Distinct forms of MB follow from activation of different signaling pathways; constitutively active HH signaling accounts for approximately 30% of patients with MB with mutations in PTCH1 and SUFU as the most common causes (Ellison et al., 2011; Kool et al., 2012). In line with these clinical observations, mice heterozygous for loss-of-function mutations in the Ptch1 gene develop MB frequently, and are more susceptible to UV-induced BCC (Aszterbaum et al., 1999). Further, Ptch1+−p53−]/− mice develop MB at a much high frequency, creating a useful experimental cancer model (Romer et al., 2004), complimenting approaches that conditionally remove Ptch1 activity (Adolphe et al., 2006) or conditionally switch on an oncogenic form of SMO (Mao et al., 2005).
HH pathway activation also promotes tumorigenesis through ligand-dependent mechanisms (Figures 2B and 2C). HH protein secreted by tumor cells induces pathway activation in adjacent stromal cells, which in turn promote the growth of the tumor through a number of paracrine signals, including interleukin-6 (IL6), vascular endothelial growth factor (VEGF), insulin-like growth factor (IGF), and Wnts (Figure 2B) (Mills et al., 2013; Pola et al., 2001; Yauch et al., 2008). This mechanism received support from titration studies with cyclopamine (Yauch et al., 2008), an HH pathway inhibitor frequently used in cancer studies (see later), that separated cyclopamine’s direct action in HH signaling from high-dose cytotoxic off-target effects that might have misled researchers to posit an autocrine mechanism of HH action (Berman et al., 2003; Karhadkar et al., 2004; Mukherjee et al., 2006; Thayer et al., 2003; Varnat et al., 2009; Watkins et al., 2003). In human xenograft mouse models of pancreatic and colorectal cancers, HH protein was produced by the tumor cells and HH target gene expression increased specifically in tumor-infiltrating stromal cells but not in tumor cells themselves (Yauch et al., 2008). Further, when GLI1 was ablated in mice, the growth of mutant KRAS-induced tumors was suppressed in a pancreatic microenvironment with reduced HH responsiveness (Mills et al., 2013). A reverse paracrine mechanism has also been invoked in lymphomas and multiple myelomas, where HH ligand produced by bone marrow stroma is thought to signal to neighboring tumor cells to activate the HH pathway (Figure 2C) (Dierks et al., 2007).
Strategies to Modulate HH Signaling
Given the importance of HH signaling in a multitude of cancers, it is not surprising that the HH pathway has attracted a great deal of interest as a therapeutic target. Several pathway components can be targeted pharmacologically (Figure 3). To date, a number of small molecules have been discovered as HH pathway antagonists (Table 2). Pathway agonists have also been identified, which could be useful for the development of regenerative therapies (Table 2).
Figure 3. Current Strategies for Modulating Activity of the HH Signaling Pathway.
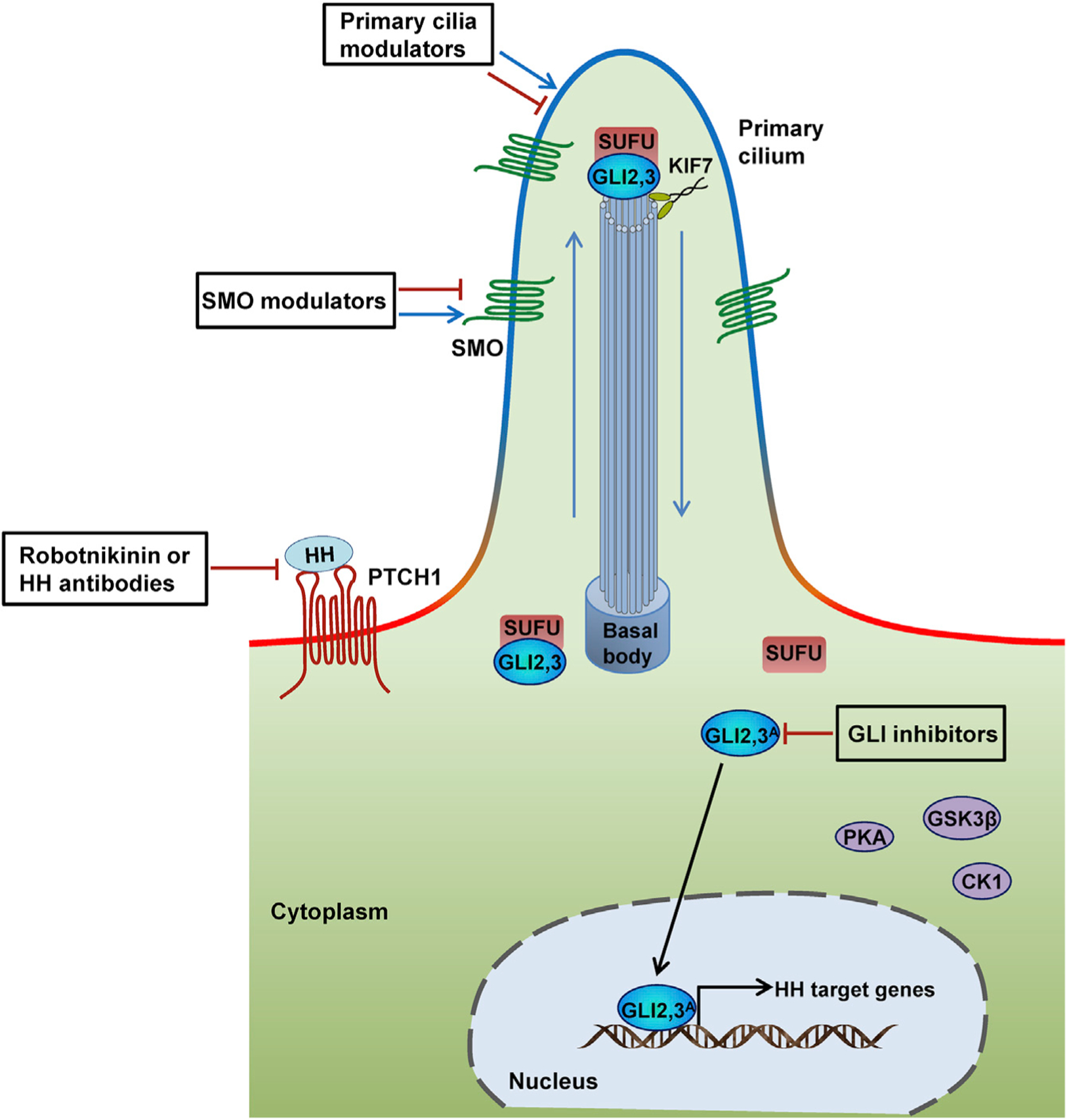
The HH signaling pathway can be modulated at many different levels, including interfering with the interaction of HH ligand and receptor through anti-HH antibodies or robotnikinin; modulating SMO activity; regulating ciliogenesis and the ciliary localization of pathway components; targeting GLI transcription factors directly or indirectly by modulating GLI interacting or regulatory factors.
Table 2.
A List of HH Pathway Modulators
| Pathway Modulators | Structure | Type | Functions | References |
|---|---|---|---|---|
| Upstream of SMO | ||||
| Robotnikinin | 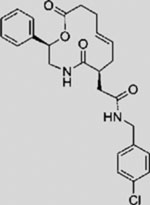 |
SMI | interferes with SHH protein | (Stanton et al., 2009) |
| CA1 | 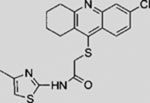 |
SMI | inhibits cilia biogenesis | (Wu et al., 2012) |
| CA2 | 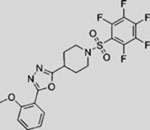 |
SMI | inhibits cilia biogenesis | (Wu et al., 2012) |
| RU-SKI 39 |  |
SMI | inhibits HH acyltransferase | (Petrova et al., 2013) |
| RU-SKI 41 |  |
SMI | inhibits HH acyltransferase | (Petrova et al., 2013) |
| RU-SKI 43 | 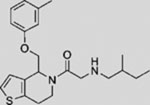 |
SMI | inhibits HH acyltransferase | (Petrova et al., 2013) |
| RU-SKI 50 | 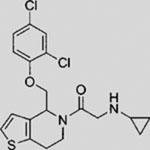 |
SMI | inhibits HH acyltransferase | (Petrova et al., 2013) |
| HH antibody 5E1 | NP | mAb | blocks SHH protein activity | (Ericson et al., 1996) |
| At SMO Level | ||||
| HH-Ag1.1 | 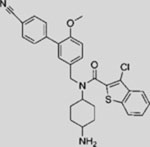 |
SMA | activates SMO | (Frank-Kamenetsky et al., 2002) |
| HH-Ag1.2 | 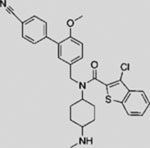 |
SMA | activates SMO | (Frank-Kamenetsky et al., 2002) |
| HH-Ag1.3 (SAG) | 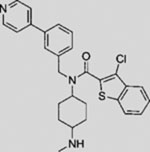 |
SMA | activates SMO | (Frank-Kamenetsky et al., 2002) |
| HH-Ag1.5 | 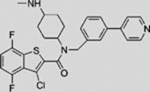 |
SMA | activates SMO | (Frank-Kamenetsky et al., 2002) |
| Purmorphamine |  |
SMA | activates SMO | (Sinha and Chen, 2006; Wu et al., 2002; Wu et al., 2004) |
| Oxysterols (e.g., 20(S)-OHC) | 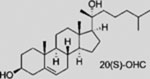 |
SMA | activates SMO | (Myers et al., 2013; Nachtergaele et al., 2013; Nedelcu et al., 2013) |
| Cholesterol | 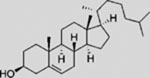 |
SMA | activates SMO | (Byrne et al., 2016; Huang et al., 2016; Luchetti et al., 2016) |
| PI4P | 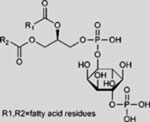 |
SMA | activates SMO | (Jiang et al., 2016) |
| DHCEO | 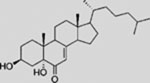 |
SMI | inhibits SMO | (Sever et al., 2016) |
| Fluocinolone acetonide | 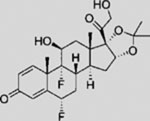 |
SMA | activates SMO ciliary translocation | (Wang et al., 2012b) |
| Triamcinolone Acetonide | 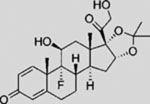 |
SMA | activates SMO ciliary translocation | (Wang et al., 2012b) |
| Budesonide | 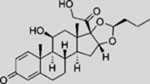 |
SMI | inhibits SMO ciliary translocation | (Wang et al., 2012b) |
| Ciclesonide | 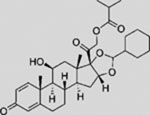 |
SMI | inhibits SMO ciliary translocation | (Wang et al., 2012b) |
| GSA-10 | 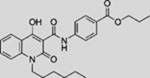 |
SMA | activates SMO | (Gorojankina et al., 2013) |
| SMANT | 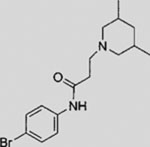 |
SMI | inhibits SMO | (Wang et al., 2012a) |
| DY131 | 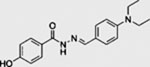 |
SMI | inhibits SMO | (Wang et al., 2012a) |
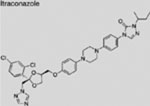
| Itraconazole | 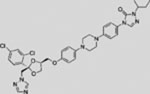 |
SMI | inhibits SMO | (Kim et al., 2010b) |
| ALLO-1 | 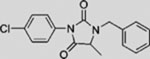 |
SMI | inhibits SMO | (Tao et al., 2011) |
| ALLO-2 | SMI | inhibits SMO | (Tao et al., 2011) | |
| Compound 5 | 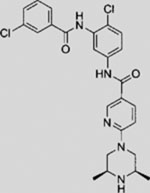 |
SMI | inhibits SMO | (Dijkgraaf et al., 2011) |
| Cyclopamine | 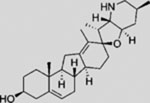 |
SMI | inhibits SMO | (Taipale et al., 2000) |
| KAAD-cyclopamine | 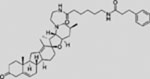 |
SMI | inhibits SMO | (Taipale et al., 2000) |
| Jervine | 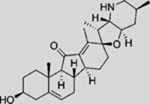 |
SMI | inhibits SMO | (Borzillo and Lippa, 2005) |
| Cur-61414 | 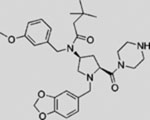 |
SMI | inhibits SMO | (Williams et al., 2003) |
| SANT-1 | SMI | inhibits SMO | (Chen et al., 2002b) | |
| SANT-2 | 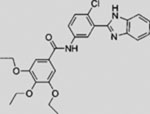 |
SMI | inhibits SMO | (Chen et al., 2002b) |
| SANT-3 | 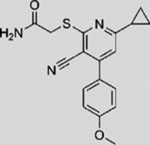 |
SMI | inhibits SMO | (Chen et al., 2002b) |
| SANT-4 | 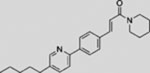 |
SMI | inhibits SMO | (Chen et al., 2002b) |
| Vismodegib (GDC-0449) | 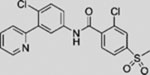 |
SMI | inhibits SMO | (Robarge et al., 2009) |
| Saridegib (IPI-926) | 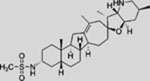 |
SMI | inhibits SMO | (Tremblay et al., 2009) |
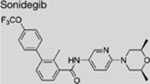
| Sonidegib (LDE225 | 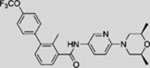 |
SMI | inhibits SMO | (Pan et al., 2010) |
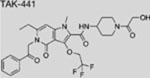
| TAK-441 | 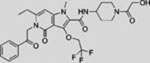 |
SMI | inhibits SMO | (Ohashi et al., 2012) |
| BMS-833923 | 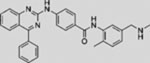 |
SMI | inhibits SMO | (Gendreau et al., 2009) |
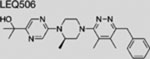
| LEQ506 | SMI | inhibits SMO | (Peukert et al., 2013) | |
| PF-04449913 | 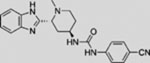 |
SMI | inhibits SMO | (Munchhof et al., 2011) |
| LY2940680 | 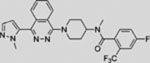 |
SMI | inhibits SMO | (Bender et al., 2011) |
| Vitamin D3 | 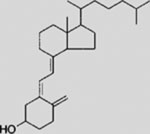 |
SMI | inhibits SMO | (Bijlsma et al., 2006) |
| BRD-6851 | 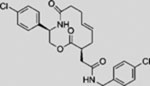 |
SMI | inhibits SMO | (Dockendorff et al., 2012) |
| SEN450 | 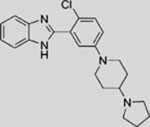 |
SMI | inhibits SMO | (Ferruzzi et al., 2012) |
| MRT-83 | SMI | inhibits SMO | (Roudaut et al., 2011) | |
| Downstream of SMO | ||||
| GANT58 | 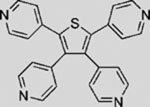 |
SMI | inhibits GLI1-mediated GLI luciferase | (Lauth et al., 2007) |
| GANT61 | 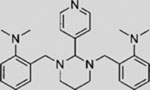 |
SMI | inhibits GLI1-mediated GLI luciferase | (Lauth et al., 2007) |
| Arcyriaflavin C | 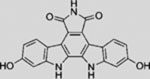 |
SMI | inhibits GLI1-mediated GLI luciferase | (Hosoya et al., 2008) |
| Physalins F | 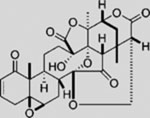 |
SMI | inhibits GLI1-mediated GLI luciferase | (Hosoya et al., 2008) |
| HPI1 | 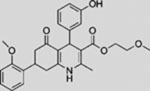 |
SMI | modulates GLI1 processing, activation, and/or trafficking | (Hyman et al., 2009) |
| HPI2 | 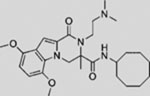 |
SMI | modulates GLI1 processing, activation, and/or trafficking | (Hyman et al., 2009) |
| HPI3 | 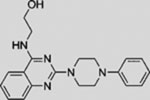 |
SMI | modulates GLI1 processing, activation, and/or trafficking | (Hyman et al., 2009) |
| HPI4 | 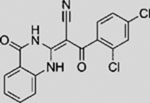 |
SMI | modulates GLI1 processing, activation, and/or trafficking | (Hyman et al., 2009) |
| Arsenic trioxide | As2O3 | SMI | inhibits GLI transcription factors | (Beauchamp et al., 2011; Kim et al., 2010a) |
| Glabrescione B | 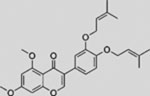 |
SMI | interferes with the binding of GLI1 and DNA | (Infante et al., 2015) |
SMI, small-molecule inhibitor; SMA, small-molecule activator; mAb, monoclonal antibody; NP, not provided.
Interfering with the Ligand and Its Binding to Receptor
Palmitoylation, catalyzed by an HH-directed acyltransferase (HHAT), is required for stability and normal activity of the HH ligand. Recently, Petrova et al. (2013) discovered that RU-SKI inhibits HH signaling blocking HHAT-mediated SHH palmitoylation. 5E1 (Ericson et al., 1996), an anti-SHH monoclonal antibody, and robotnikinin (Stanton et al., 2009), a small molecule that physically interacts with SHH, interfere with SHHPTCH1 interaction, thereby inhibiting HH signaling. Experiments in various developmental and cancer studies have confirmed the efficacy of 5E1 with sub-nanomolar inhibitory potency (Bailey et al., 2009; Wallace and Raff, 1999; Yauch et al., 2008). Although these molecules offer approaches for pathway intervention, their use to date has been limited to the laboratory. Further, their inhibitory functions would only limit ligand-dependent HH pathway actions.
Modulating SMO Activity
In addition to natural metabolites and putative endogenous modulators discussed above, a wide variety of SMO agonists and antagonists have been identified and optimized. The first small-molecule modulator of HH pathway activity was identified from the realization that mouse mutants lacking HH signaling showed facial features (cyclopia among these) observed in offspring of sheep ingesting corn lily plants (Veratrum californicum) (Bryden et al., 1971). Cyclopamine, a steroidal alkaloid produced by the plant, provided the connection (Cooper et al., 1998; Incardona et al., 1998; Keeler, 1970, 1973); cyclopamine binds to and inhibits SMO activity (Chen et al., 2002a).
SMO, like a wide range of other 7-pass membrane proteins, has emerged as the predominant “druggable” target from nearly two decades of unbiased drug discovery efforts that have focused on transcriptional output at the end of the pathway to identify any drug-susceptible step in the signal transduction chain (Frank-Kamenetsky et al., 2002). The “first-in-class” HH pathway regulator vismodegib (also known as GDC-0449), a specific and potent SMO antagonist, was approved by the United States Food and Drug Administration (FDA) in 2012 for the treatment of metastatic or locally advanced BCC (Dlugosz et al., 2012). In 2015, the FDA approved another SMO inhibitor, sonidegib (also known as LDE225), for treatment of locally advanced BCC (Burness, 2015).
Additional SMO inhibitors, including saridegib (Jimeno et al., 2013), BMS-833923 (Siu et al., 2009), and TAK-441 (Goldman et al., 2015), have given good clinical responses (Table 3). Interestingly, itraconazole, a systemic antifungal drug, emerged as an SMO inhibitor with a mechanism distinct from that of cyclopamine and other known SMO antagonists. Systemic administration of itraconazole suppressed HH pathway activity and the growth of MB in a mouse allograft model (Kim et al., 2010b). Moreover, itraconazole is reported to inhibit HH pathway activity from all known drug-resistant SMO mutants (Kim et al., 2013). Results from exploratory trials in human BCC have shown anti-HH pathway activity but marginal tumor shrinkage at best for itraconazole by itself, or in combination with arsenic trioxide (Ally et al., 2016; Kim et al., 2014a).
Table 3.
Summary of Selected Clinical Trials of HH Pathway Antagonists for Cancer Therapy
| Drug | Cancer Types | Combination Drugs | Phase | Clinicaltrials.gov Identifier | Current Status |
|---|---|---|---|---|---|
 |
BCC | 2 | NCT01201915 | recruiting | |
| BCC | 2 | NCT00833417 | completed | ||
| metastatic pancreatic cancer | administered with gemcitabine, nab-paclitaxel | 2 | NCT01088815 | active, not recruiting | |
| pancreatic ductal adenocarcinoma | 2 | NCT01096732 | terminated | ||
| recurrent or refractory MB | 2 | NCT00939484 | completed | ||
| advanced pancreatic cancer | administered with gemcitabine hydrochloride | 2 | NCT01195415 | active, not recruiting | |
| advanced or metastatic sarcoma | administered with RO4929097(a γ-secretase inhibitor) | 1b/2 | NCT01154452 | completed | |
| BCC | compared with placebo | 2 | NCT01543581 | completed | |
| BCC | 4 | NCT01160250 | approved for marketing | ||
| BCC; ovarian cancer; metastatic colorectal cancer | administered alone or with FOLFOX/FOLFIRI, bevacizumab | 2 | NCT00959647 | completed | |
| advanced or metastatic solid tumors | 1b | NCT00968981 | completed | ||
| advanced or metastatic solid tumors | 1 | NCT00607724 | completed | ||
| BCC | 2b | NCT01700049 | recruiting | ||
| BCC | 2 | NCT01835626 | recruiting | ||
| BCC | NP | NCT01631331 | active, not recruiting | ||
| BCC | compared with placebo | 2 | NCT02067104 | recruiting | |
| BCC | compared with placebo | 2 | NCT01898598 | recruiting | |
| BCC | administered with placebo in two vismodegib regimens | 2 | NCT01815840 | active, not recruiting | |
| BCC | compared with aminolevulinic acid 20% topical solution | 2 | NCT01556009 | recruiting | |
| refractory pontine glioma | 2 | NCT01774253 | terminated | ||
| recurrent MB | 1 | NCT00822458 | completed | ||
| MB | administered with cyclophosphamide or cisplatin or vincristine or pemetrexed or gemcitabine | 2 | NCT01878617 | recruiting | |
| MB | administered with temozolomide (compared with temozolomide alone) | 1/2 | NCT01601184 | recruiting | |
| AML | administered with decitabine and ribavirin | 2 | NCT02073838 | recruiting | |
| BCC | 4 | NCT02436408 | recruiting | ||
 |
prostate cancer | 1 | NCT02111187 | recruiting | |
| recurrent ovarian cancer | 1b | NCT02195973 | recruiting | ||
| multiple myeloma | administered with bortezomib | 2 | NCT02254551 | terminated | |
| advanced breast cancer | administered with docetaxel | 1b | NCT02027376 | active, not recruiting | |
| locally advanced or metastatic pancreatic cancer | administered with gemcitabine | 1b | NCT01487785 | completed | |
| relapsed or refractory acute leukemia | 2 | NCT01826214 | completed | ||
| recurrent or refractory MB | 1/2 | NCT01125800 | completed | ||
| MB; BCC | 1 | NCT01208831 | completed | ||
| BCC | 2 | NCT00961896 | completed | ||
| BCC | 2 | NCT01327053 | active, not recruiting | ||
| MB | 2 | NCT01708174 | active, not recruiting | ||
| MB; BCC | 1 | NCT00880308 | completed | ||
| chronic or accelerated phase myeloid leukemia | administered with nilotinib | 1b | NCT01456676 | completed | |
| myeloid malignancies | administered with azacitidine or decitabine | 1 | NCT02129101 | recruiting | |
| advanced solid tumors | administered with BKM120 | 1b | NCT01576666 | completed | |
| advanced or metastatic BCC | administered with buparlisib | NP | NCT02303041 | active, not recruiting | |
 |
advanced nonhematologic malignancies; BCC | 1 | NCT01204073 | recruiting | |
 |
advanced solid tumors | 1 | NCT01106508 | recruiting | |
 |
BCC | 2b | NCT02354261 | recruiting | |
| BCC | 0 | NCT02120677 | recruiting | ||
| BCC | 2 | NCT01108094 | completed | ||
 |
acute leukemia | 2 | NCT01841333 | recruiting | |
| myelodysplastic syndrome | administered with azacitidine | 1b/2 | NCT02367456 | recruiting | |
| AML | administered alone or with low-dose ARA-C or with daunorubicin and cytarabine | 1 | NCT02038777 | recruiting | |
| AML | administered with low-dose ARA-C or decitabine or daunorubicin or cytarabine | 1b/2 | NCT01546038 | recruiting | |
| solid tumors | 1 | NCT01286467 | completed | ||
| myelofibrosis | compared with placebo | 2 | NCT02226172 | recruiting | |
| hematologic malignancies | 1 | NCT00953758 | completed | ||
| MDS; CMML | 2 | NCT01842646 | active, not Recruiting | ||
 |
small cell lung cancer | administered with carboplatin and etoposide (compared with placebo with carboplatin and etoposide) | 1b/2 | NCT01722292 | terminated |
| neoplasm metastasis | 1 | NCT01919398 | active, not recruiting | ||
| advanced cancer | 1 | NCT01226485 | active, not recruiting | ||
 |
myelofibrosis | 2 | NCT01371617 | completed | |
| metastatic pancreatic cancer | administered with gemcitabine (compared with placebo with gemcitabine) | 1b/2 | NCT01130142 | completed | |
| advanced and/or metastatic solid tumors | 1 | NCT00761696 | completed | ||
| advanced pancreatic adenocarcinoma | administered with FOLFIRINOX | 1 | NCT01383538 | recruiting | |
| BCC; chondrosarcoma | NP | NCT01609179 | recruiting | ||
| recurrent head and neck cancer | administered with cetuximab | 1 | NCT01255800 | completed | |
| metastatic or locally advanced chondrosarcoma | compared with placebo | 2 | NCT01310816 | completed | |
 |
solid tumors | 1 | NCT01413906 | completed | |
| advanced or metastatic cancer | 1 | NCT00670189 | recruiting | ||
| CML | administered with dasatinib | 1/2 | NCT01218477 | completed | |
| small cell lung cancer | administered with carboplatin and etoposide | 1b | NCT00927875 | completed | |
| stomach neoplasms; esophageal neoplasms | administered with cisplatin and capecitabine | 1b | NCT00909402 | completed | |
| basal cell nevus syndrom | NP | NCT02100371 | active, not recruiting | ||
| multiple myeloma | administered alone or with lenalidomide and dexamethasossne or with bortezomib | 1b | NCT00884546 | completed | |
| leukemia | administered with dasatinib (compared with dasatinib alone) | 2 | NCT01357655 | completed | |
 |
BCC | administered with diclofenac (compared with vitamin D3 alone or diclofenac alone) | 2 | NCT01358045 | recruiting |
| Arsenic trioxide, As2O3 | BCC | NP | NCT01791894 | recruiting | |
| advanced neuroblastoma or other childhood solid tumors | 2 | NCT00024258 | completed | ||
| brain and CNS tumors | administered with temozolomide and radiation therapy | 1 | NCT00720564 | completed | |
| brain and CNS tumors | 1 | NCT00095771 | completed | ||
| brain cancer | 1 | NCT00185861 | completed |
NCT, national clinical trial; FOLFOX, folinic acid, 5-fluorouracil, and oxaliplatin; FOLFIRI, folinic acid, 5-fluorouracil, and irinotecan; FOLFIRINOX, 5-fluorouracil, leucovorin, irinotecan, and oxaliplatin; ARA-C, cytosine arabinoside; NP, not provided.
Given the normal role of HH signaling in stem/progenitor regulation, in building and maintaining tissue and organ systems, activation of the HH pathway could be beneficial in the appropriate clinical context; for example, in treating degenerative diseases. SMO agonists and antagonists are often structurally related (Nachtergaele et al., 2013; Wang et al., 2012b; Yang et al., 2009). Therefore, activators of the HH pathway, predominantly SMO agonists, have been developed in parallel with SMO antagonists. The first SMO agonist is HH-Ag1.1 identified from a screen of 140,000 synthetic compounds in C3H10T1/2 cells; HH-Ag1.2, 1.3, 1.4, and 1.5 are derivatives of HH-Ag1.1 (Frank-Kamenetsky et al., 2002). Chemical epistasis studies showed that these agonists act at the level of SMO, and binding assays demonstrated that HH-Ag1.5 is a direct ligand of SMO (Frank-Kamenetsky et al., 2002). SAG (also named HH-Ag1.3 in Frank-Kamenetsky et al., 2002) binds the heptahelical bundle of SMO, as demonstrated by the inhibitory interaction against BODIPY-cyclopamine (Chen et al., 2002b). Another screening effort in a library consisting of 50,000 heterocyclic compounds discovered purmorphamine, a 2,6,9-trisubstituted purine compound, which induces the differentiation of multipotent mesenchymal mouse progenitor fibroblast cells (C3H10T1/2) into an osteoblast lineage (Wu et al., 2002). Biochemical experiments revealed that purmorphamine is a small-molecule agonist of HH signaling, which also functions at the level of SMO (Sinha and Chen, 2006; Wu et al., 2004). Another agonist, GSA-10, binds SMO at a site distinct from cyclopamine, and promotes the differentiation of C3H10T1/2 cells into osteoblasts (Gorojankina et al., 2013).
In addition to screening, recent structural studies on SMO open the door to rational drug design (Byrne et al., 2016; Huang et al., 2016; Nachtergaele et al., 2013; Rana et al., 2013; Wang et al., 2013, 2014; Weierstall et al., 2014). The SMO protein consists of an extracellular CRD-containing N-terminal region, a seven-transmembrane helical (7TM) domain, and an intracellular C-terminal domain. The 7TM domain and the CRD domain are two well-characterized ligand binding sites (Byrne et al., 2016; Huang et al., 2016; Nachtergaele et al., 2013; Rana et al., 2013; Wang et al., 2013, 2014; Weierstall et al., 2014). Several SMO antagonists (vismodegib, LY2940680, cyclopamine, SANT-1, and AntaXV) and the agonist HH-Ag1.5 bind to the 7TM domain of SMO at different residues (Byrne et al., 2016; Wang et al., 2013, 2014; Weierstall et al., 2014). Armed with structural details of the 7TM domain in human SMO, researchers have mapped chemo-resistance-associated mutations of SMO from BCC patients (Atwood et al., 2015; Sharpe et al., 2015a). Several synthetic or natural steroidal compounds, including 20(S)-OHC (Nachtergaele et al., 2013), cholesterol (Byrne et al., 2016; Huang et al., 2016), and budesonide (Rana et al., 2013), bind the CRD in SMO structures. Structural insights obtained from these studies should help in the design of next-generation SMO modulators.
Regulating Ciliary Localization and Ciliogenesis
Given the PC’s central role in HH signaling transduction in vertebrates, the PC is an attractive focus for the discovery of new modulators of HH pathway components in this important organelle. Several PC-focused studies have separated SMO activity from PC association (Rohatgi et al., 2009; Wang et al., 2009; Wilson et al., 2009). Although all SMO agonists appear to induce SMO accumulation to the PC, some antagonists, notably cyclopamine, also induce SMO accumulation in the PC (Wang et al., 2009). Ciliary accumulation of SMO correlates with prolonged hypersensitivity to pathway stimulation, raising concerns about such compounds in treating cancers (Peluso et al., 2014; Wang et al., 2012a).
We (Wang et al., 2012a, 2012b) and others (Wu et al., 2012) have established high-content screening platforms that directly monitor SMO trafficking into the PC. Wu et al. (2012) identified ten small-molecule pathway inhibitors (SA1–10) that alter the ciliary localization of YFP-tagged SMO. SA8 and 9, unlike the other eight hits, promote endogenous SMO to accumulate in the PC, consistent with previous observations that different inhibitors induce different outcomes of SMO ciliary localization (Wu et al., 2012). A selective compound screen by our laboratory for inhibition of SHH-driven SMO ciliary accumulation identified new SMO antagonists with both conventional and novel mechanisms (Wang et al., 2012a). Notably, one of these, “SMANT”, was equally effective in inhibiting either wild-type SMO or a drug-refractory variant (Wang et al., 2012a).
A second screen identified a number of glucocorticoids as SMO synergists, promoting SMO ciliary translocation; many of the compounds are already FDA approved as anti-inflammatory drugs (Wang et al., 2012b). A subset of these compounds were identified in a screen imaging β-arrestin internalization, which is associated with SMO translocation to the PC (Wang et al., 2010). These compounds are unable to efficiently activate the pathway but rather prime cells for a more robust signaling response, inducing SMO accumulation in the PC (Wang et al., 2012b).
Interestingly, co-administration of some glucocorticoids with vismodegib blunts HH pathway inhibition, suggesting an unhelpful crosstalk where patients might receive glucocorticoids in conjunction with vismodegib-directed anti-BCC therapy. These observations also raise the question of whether glucocorticoid action may help treat HH-dependent degenerative disease. Structure-activity relationship studies have also identified glucocorticoid antagonists of SMO ciliary accumulation. One of these in clinical use, budesonide, inhibits the ciliary localization and signaling activity of normal SMO and drug-resistant mutant forms (Wang et al., 2012b), most likely through binding SMO’s CRD, and allosteric effects on SMO’s binding site for oxysterols (Rana et al., 2013). Together, these findings have shed new light on the pharmacological “space” for diverse modulators of SMO activity, and importantly, the studies provide new opportunities for treating refractory mutant forms of SMO that render FDA-approved drugs ineffective (Atwood et al., 2015; Buonamici et al., 2010; Dijkgraaf et al., 2011; Sharpe et al., 2015a; Yauch et al., 2009).
Targeting GLI Transcription Factors
Targeting GLI transcription factors, directly or indirectly, would in theory be more broadly applicable than using a diverse array of more upstream inhibitors. Further, transcriptional suppressors would circumvent problems with therapy-dependent accumulation of drug-resistant forms of SMO. GANT58 and GANT61 have been identified as selective inhibitors of GLI-mediated gene transactivation and HH-driven tumor growth in vivo (Lauth et al., 2007). GANT61 was demonstrated to block GLI1 binding to its target DNA (Lauth et al., 2007). Arcyriaflavin C and physalins F might indirectly attenuate GLI1 activity through PKC/MAPK pathway blockade (Hosoya et al., 2008), and HPI1–4 are four inhibitors that regulate processing, activation, or ciliary trafficking of GLI1/2 (Hyman et al., 2009).
Arsenic trioxide (ATO), used in the treatment of acute promyelocytic leukemia, has been shown to act as a GLI antagonist and inhibits the growth of MB allografts derived from Ptch1+/−p53−/− mice (Kim et al., 2010a) and xenografts of Ewing sarcoma (Beauchamp et al., 2011). In combination, ATO and itraconazole suppressed growth of MB and BCC in vivo, and prolonged the survival of a mouse model of MB-harboring cells producing Smo-D477G, a drug-resistant SMO variant (Kim et al., 2013). These studies focused on the active forms of GLI. A recent report suggested context-dependent arsenic attenuation of both GLI activator and repressor forms (Li et al., 2016). Pan inhibition may explain why arsenic can be carcinogenic where GLI repressor plays a dominant role (Fei et al., 2010). These findings, together with the discovery of the function of glucocorticoids and itraconazole, open new possibilities of drug repositioning in the treatment of HH-driven cancers with careful thought to applications and implications, and further optimization of treatment strategies.
Given that transcription factors do not generally make attractive targets for small-molecule drug discovery, an insight into how GLI proteins are themselves regulated by proteins that are more readily “druggable” is another potential approach to HH pathway modification. Several studies have found PI3K/mTOR signaling linked to GLI regulation (Buonamici et al., 2010; Riobo et al., 2006; Wang et al., 2012c). A combination of sonidegib and the PI3K inhibitor buparlisib (also known as NVP-BKM120), or the dual PI3K/mTOR inhibitor dactolisib (also known as NVP-BEZ235), markedly delayed MB relapse (Buonamici et al., 2010). Further, atypical protein kinase C ι/λ (aPKC-ι/λ) phosphorylates and activates GLI downstream of SMO, and inhibition of aPKC-ι/λ with a myristoylated peptide inhibitor (PSI) suppresses HH signaling in SMO-inhibitor-resistant BCC cells, suggesting aPKC-ι/λ may be a potential target for GLI regulation (Atwood et al., 2013). BRD4 occupies GLI1 and GLI2 promoters to regulate the transcriptional output of the HH pathway (Tang et al., 2014). The BRD4 inhibitor JQ1 effectively antagonizes HH-driven tumorigenesis, even in cells resistant to clinically available SMO inhibitors (Tang et al., 2014). In addition, recent studies argue that U0126 (an inhibitor of MEK) (Liu et al., 2014) and FN1–8 (a small molecule that disrupts GLI/TAF9 interaction) (Bosco-Clement et al., 2014) inhibited GLI activity and HH-mediated cancer growth. Rational drug design based on GLI structure and GLI partner interactions identified Glabrescione B, a small molecule that binds directly to GLI1 zinc fingers, interfering with zinc-finger-dependent DNA binding and transcriptional activity (Infante et al., 2015). Structural information for SUFU and GLI interactions could help in identifying drugs that enhance this interaction, thereby suppressing GLI’s transcriptional activity (Zhang et al., 2013).
Clinical Studies for Cancer Therapies
In addition to the previously discussed FDA-approved anti-HH pathway therapeutics, several SMO inhibitors have entered clinical trials for the treatment of a wide range of cancers (for a list of clinical trials, please refer to Table 3 and http://www.clinicaltrials.gov/). In this section, we review in more detail the path to approval for current drugs and their therapeutic targets, as well as recent progress on developing treatments for both ligand-independent cancers, including BCC and MB, and ligand-dependent cancers, showcasing leukemia and pancreatic cancer. Experience and lessons learned from these studies are valuable for continuous development toward more effective therapies.
Clinical Studies for BCC
Although surgical resection is a routine therapeutic approach for early-stage BCC, no effective treatment existed for locally advanced or metastatic BCC prior to the approval of vismodegib. Hyperactive HH signaling plays a central role in the tumorigenesis of sporadic BCC, predominantly by inactivating mutations in PTCH1 and SUFU and activating mutations in SMO (Reifenberger et al., 2005; Smith et al., 2014; Xie et al., 1998). Consequently, dozens of clinical trials for BCC treatment with HH pathway inhibitors have been conducted or are ongoing. Among these, the leading drug vismodegib obtained complete or partial responses in BCC patients at a metastatic or locally advanced stage in phase 1 and 2 trials (NCT00833417, NCT01543581, NCT00959647, NCT00968981 and NCT00607724). Promising results were observed early in a phase 1 clinical trial (Graham et al., 2011; LoRusso et al., 2011; Von Hoff et al., 2009). Its efficacy was further evaluated in a phase 2 clinical study in which 104 enrolled patients with BCC received oral vismodegib. The objective response rate was 30% for the metastatic BCC group and 43% for the locally advanced BCC group (Sekulic et al., 2012). Consequently, vismodegib was approved by the FDA in January 2012. To achieve more precise patient stratification, an ongoing phase 2b study of vismodegib therapy in various BCC subtypes aims to identify patients with BCC who are more likely to respond based on their pathological properties (NCT01700049). Several clinical trials to extend the use of vismodegib to surgical BCC are underway (NCT01835626, NCT01631331, NCT02067104, and NCT01898598). Moreover, three clinical trials are also being conducted to explore alternative vismodegib dosing regimens for BCC treatment to avoid chronic adverse events (NCT01201915, NCT01815840, and NCT01556009).
In a phase 1 study of sonidegib, 6 of 16 patients with BCC(37.5%) achievedpartial orcompleteresponse, witha strongassociation between tumor response and HH pathway activation monitored by gene expression (NCT00880308) (Rodon et al., 2014). A phase 2 study (NCT01327053) of 230 patients with either locally advanced BCC (n = 194) or metastatic BCC (n = 36) reported objective responses in two groups receiving different doses of sonidegib (Migden et al., 2015). Overall, 33 (42%) of 79 patients in the 200 mg/day group, and 49 (32%) of 151 patients in the 800 mg/day group, responded, supporting an antitumor activity for sonidegib in BCC (Migden et al., 2015). In addition, several other structurally distinct SMO inhibitors are also being evaluated in phase 1 or 2 clinical trials to treat locally advanced or metastatic BCC, including LEQ506 (NCT01106508), BMS-833923 (NCT00670189 and NCT02100371), IPI-926 (NCT01609179), LY2940680 (NCT01226485), and TAK-441 (NCT01204073) (Table 3).
Clinical studies for repositioning certain old drugs in BCC treatment have also been conducted. Vitamin D3, an essential nutrient for bone and the immune system, showed an inhibitory activity on SMO and the growth of murine BCC in preclinical studies (Bijlsma et al., 2006; Tang et al., 2011). In the hope of assessing a potential antitumor effect of this highly tolerable drug, a phase 2 clinical trial of vitamin D3 was performed on BCC patients; the results have not been reported (NCT01358045). Itraconazole was also evaluated for treating BCC (NCT02120677 and NCT02354261). Results from a previous exploratory phase 2 trial (NCT01108094) showed that as a single agent, itraconazole reduced tumor area by 24% in BCC patients, although larger trials of longer duration are needed for conclusive data (Kim et al., 2014a). Lastly, ATO is also being investigated in patients with recurrent BCC (NCT01791894).
Clinical Studies for MB
MB can be classified into four subtypes based on the causative genetic abnormalities: HH driven, Wnt driven, c-Myc driven, and a fourth group associated with isochromosome 17q (Kool et al., 2012; Northcott et al., 2011; Rusert et al., 2014). Each of these categories has distinct pathophysiological characteristics, and potentially also distinct tumor origins (Gibson et al., 2010; Schuller et al., 2008). A critical step for the strongest clinical trial is to identify the HH category subset of MB patients at the onset.
The first case report of MB treatment showed a reduction of symptoms upon administration of vismodegib (Rudin et al., 2009); however, a relapse was reported after several months of treatment (Yauch et al., 2009), triggering follow-up studies on acquired drug resistance (Dijkgraaf et al., 2011; Tao et al., 2011). In a phase 1 study among children with refractory or relapsed MB (NCT00822458), vismodegib induced antitumor responses (Gajjar et al., 2013a). Importantly, the permanent defects in bone growth observed in young mice after brief administration of an SMO antagonist (Kimura et al., 2008) were not observed in human patients (Gajjar et al., 2013a). In two phase 2 trials (NCT00939484 and NCT01239316), vismodegib exhibited activity against recurrent MB but only in the HH subtype as expected; prolonged disease stabilization occurred in 41% of these patients (Robinson et al., 2015). Patient recruiting is underway to examine vismodegib in combination with other drugs for treating MB (NCT01601184 and NCT01878617).
Amakye et al. (2012) discovered a five-gene signature (GLI1, SPHK1, SHROOM2, PDLIM3, and OTX2) as a preselection tool to identify HH subtype MB. This five-gene signature could robustly identify MB patients with active HH signaling in clinical trials of sonidegib where a strong association was observed between the tumor response and the HH pathway engagement (Shou et al., 2015). An analysis of sonidegib efficacy (NCT0088030) showed all responders fell into the HH subtype of MB patients through the five-gene signature examination. Similarly, in a phase 1/2 clinical trial of sonidegib enrolling pediatric patients with recurrent MB (NCT01125800), analysis of 14 available MB tumor samples using the five-gene HH signature assay showed selective allocation of two complete responders into the HH subtype (Geoerger et al., 2012).
Clinical Studies for Leukemia
The HH pathway plays a vital role in the maintenance and expansion of TISC in leukemia (Dierks et al., 2008; Zhao et al., 2009), although the analysis of SMO removal within hematopoietic stem cells argues against a critical role for HH signaling in normal blood cell programs (Gao et al., 2009; Hofmann et al., 2009). Inhibition of HH signaling induces cell-cycle arrest of human leukemia-initiating cells and abrogates their dormancy (Sadarangani et al., 2015). The absence of a normal role in blood cells opens a therapeutic window for selective targeting of the HH pathway in treating hematologic malignancy (Irvine and Copland, 2012; Jagani et al., 2010).
Clinical trials have been initiated to examine SMO inhibitors in treating leukemia. A phase 1 trial of PF-04449913 (NCT00953758) observed clinical activity in 23 (49%) of 47 patients across several types of hematologic disease, including acute myeloid leukemia (AML), chronic myeloid leukemia (CML), chronic myelomonocytic leukemia (CMML), myelodysplastic syndrome (MDS), and myelofibrosis (MF) (Martinelli et al., 2015). On the basis of these results, phase 2 studies are planned for PF-04449913 alone (NCT01842646 and NCT01841333) or in combination with chemotherapeutic agents (NCT01546038 and NCT02367456). Currently, sonidegib combined with azacitidine (a chemotherapy drug and DNA methyltransferase inhibitor, NCT02129101), and vismodegib combined with decitabine (a chemotherapy drug and DNA methyltransferase inhibitor) and ribavirin (an anti-viral drug used for leukemia treatment, NCT02073838), are being clinically investigated for treating multiple types of leukemia. Results from these trials are awaited to substantiate potential opportunities for anti-HH targeted therapies in the treatment of leukemia.
Clinical Studies for Pancreatic Cancer
Uncontrolled activation of HH signaling has been linked to the emergence of pancreatic adenocarcinoma (Lau et al., 2006). Initial evidence for a role of HH signaling in pancreatic cancer came from the finding that SHH is abnormally expressed in tumors, and that hyperactive HH signaling is essential for tumor formation (Berman et al., 2003; Thayer et al., 2003).
A study in a mouse model of pancreatic ductal adenocarcinoma demonstrated that IPI-926, an SMO inhibitor, enhanced delivery of gemcitabine, a chemotherapy drug, through an IPI-926 inhibition of stromal myofibroblasts (Olive et al., 2009). However, follow-up clinical trials have been disappointing. A phase 1b/2 clinical trial of this same combination (NCT01130142) was closed early because the primary endpoint of overall survival was not met. Another trial of IPI-926 in combination with FOLFIRINOX (a chemotherapy regimen for pancreatic cancer treatment, including 5-fluorouracil, leucovorin, irinotecan, oxaliplatin) for advanced pancreatic adenocarcinoma also failed (Ko et al., 2016). Furthermore, treatment using vismodegib plus gemcitabine in patients with metastatic pancreatic adenocarcinoma showed no improvement to gemcitabine alone (Catenacci et al., 2015; Kim et al., 2014b). The concept of enhancing drug delivery through HH-inhibitor mediated actions may require revision. Indeed, recent studies of tumor growth in pancreatic (Liu et al., 2016; Rhim et al., 2014) and bladder (Shin et al., 2014) cancer suggest stromal cells may actually restrain tumor growth. These studies highlight the importance of obtaining a clear mechanistic understanding of the complexity of cell interactions in a given tumor for the development of effective therapeutic strategies.
Emerging Drug Resistance in Cancer
Acquired drug resistance to SMO antagonists has been observed (Chang and Oro, 2012; Iarrobino et al., 2013; Yauch et al., 2009). Studies in mice and humans that develop drug-refractory tumors provided insights into mechanisms that will have to be taken into account in the design and development of next-generation therapies (Figure 4).
Figure 4. Mechanisms of Drug Resistance to HH Pathway Inhibitors and Potential Approaches to Overcome Tumor Resistance.
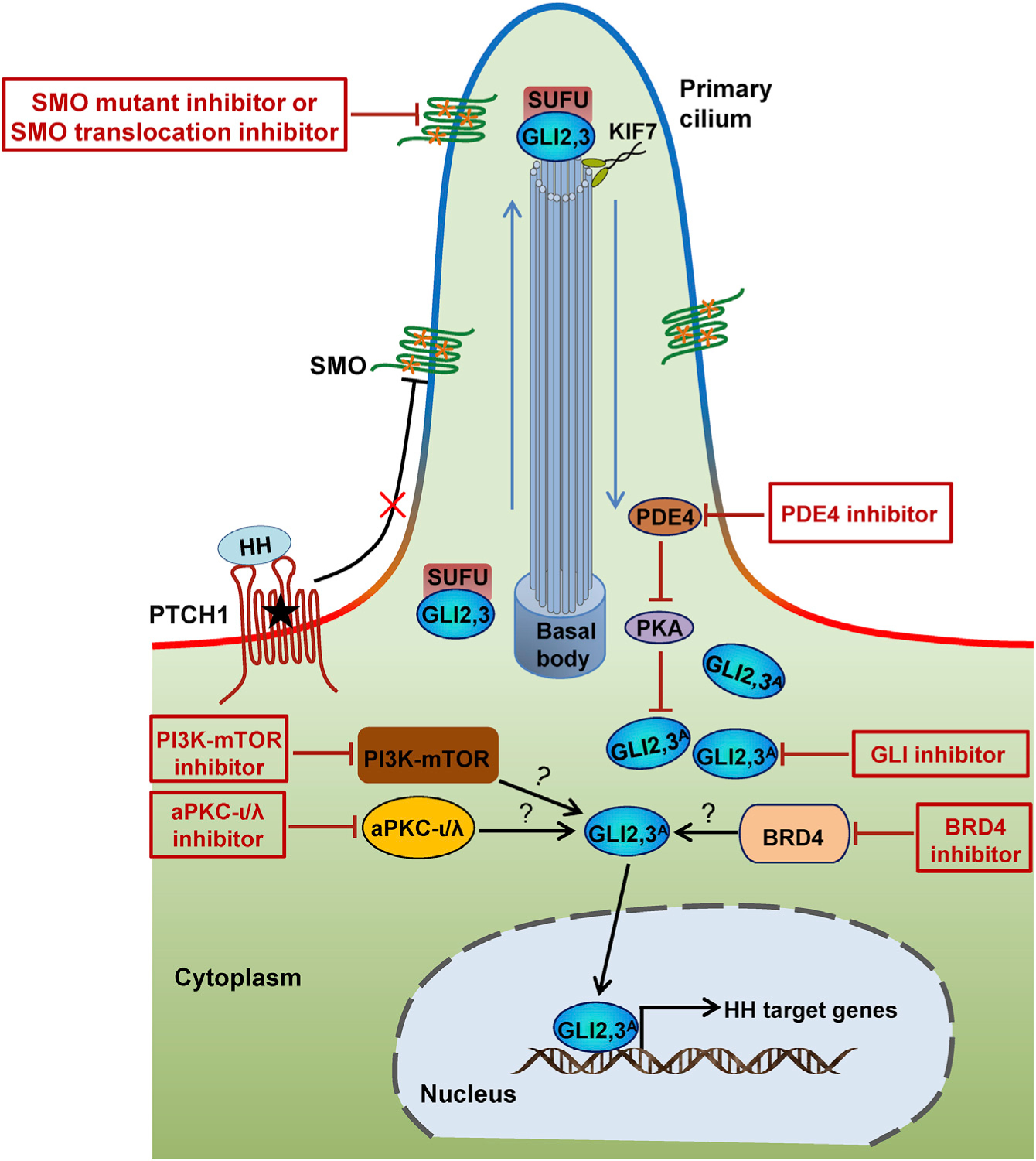
Drug resistance to SMO inhibitors is attributed in part to SMO mutations, the amplification of GLI transcription factors, upregulation of PI3K-mTOR signaling, aPKC-ι/λ activation, BRD4 activation, and PDE4 activation. Consequently, drugs targeting these components may circumvent the acquired resistance.
The first clinical identification of acquired resistance to vismodegib came from a metastatic MB patient who showed a dramatic response but a subsequent relapse (Rudin et al., 2009). A de novo D473H mutation in SMO was identified in the tumor; this missense mutation prevents vismodegib binding but leaves SMO’s signaling activity intact (Yauch et al., 2009). SMO mutations that block drug binding and/or confer constitutive activity, along with other genomic alterations that lead to SUFU loss of function or GLI2 gain of function, have been shown to be the primary causes of drug resistance through the genomic analysis of a large collection of human BCC samples refractory to vismodegib treatment (Atwood et al., 2015; Sharpe et al., 2015a). SMO mutations and GLI2 amplification were also identified in sonidegib-resistant tumors in a mouse study (Buonamici et al., 2010). Cross-resistance was observed for chemically distinct SMO inhibitors developed by different pharmaceutical companies in cell-culture experiments (Sharpe et al., 2015a) and, more importantly, an open clinical trial (Danial et al., 2016). Intra-tumor heterogeneity was also identified (Atwood et al., 2015; Sharpe et al., 2015a). These findings suggest that in order to circumvent drug resistance by SMO, a focus on downstream components of the SMO pathway and combination of therapies may be helpful.
Beyond the genetic alterations described above, drug resistance is also introduced by alterations outside the canonical HH pathway (Figure 4). By comparing the gene expression profiles in sonidegib-resistant and sonidegib-sensitive tumors, upregulation of PI3K-mTOR signaling was identified as a mechanism of resistance, which could be overcome combining sonidegib with buparlisib or dactolisib (Buonamici et al., 2010). Clinical trials (NCT01576666 and NCT02303041) are being conducted that will address this premise. Indeed, a phase 1b study of sonidegib in combination with buparlisib (NCT01576666) has provided promising results (Chu et al., 2014). In addition, laboratory studies in cell-culture and animal models suggest aPKC-ι/λ (Atwood et al., 2013), BRD4 (Tang et al., 2014), and PDE4 (Williams et al., 2015) as potential additional targets to overcome resistance to SMO inhibitors.
Conclusions and Perspectives
The latest surprises on potential endogenous SMO modulators reminded us that, even after decades of research, many things about the HH pathway are yet to be better understood. Considering these data, we suggest a model for SMO endogenous regulation by PTCH1 (Figure 5). In this model, multiple sites on SMO, including the CRD, the TM domains, and the cytoplasmic tail, are involved. Without HH ligand binding, PTCH1 controls the lipid composition of the ciliary membrane, possibly by retaining PI4P. Although a low level of SMO is constantly cycling in and out of the PC, its access to PI4P for activation is limited. Meanwhile, a potential antagonist bound to either the CRD, or part of the TM domains, or potentially both structures from the extracellular face, might contribute to suppressing SMO activation (Figure 5). This factor may normally be removed from the cell by PTCH1 in the absence of HH signals. Upon HH ligand binding, PTCH1-mediated secretion of the SMO antagonist may be blocked, thus allowing a possibly more ubiquitous agonist, such as cholesterol, or one also subject to PTCH1 regulation, to bind at similar SMO regulatory sites to those previously engaged by the PTCH1-dependent SMO antagonist. PTCH1 movement from the PC upon HH ligand binding may further reduce the access of this antagonist to the ciliary form of SMO. Further, a change in the local ciliary membrane composition to an elevated level of PI4P activates SMO (Figure 5). The PTCH1 “skirt” around the cilium base might also generate a possible inhibitory compartment there for SMO. This speculative model accounts for recent findings on cholesterol and PI4P regulation of HH signaling. The coordinated action of both endogenous agonist(s) and antagonist(s), engaging at multiple sites on SMO, unites seemingly conflicting lines of evidence. The insight that comes from a deeper mechanistic understanding of in vivo processes will likely provide valuable guidance toward developing more effective cancer therapies to treat HH-pathway-dependent tumors.
Figure 5. A Hypothetical Model for Endogenous SMO Modulation by PTCH1.
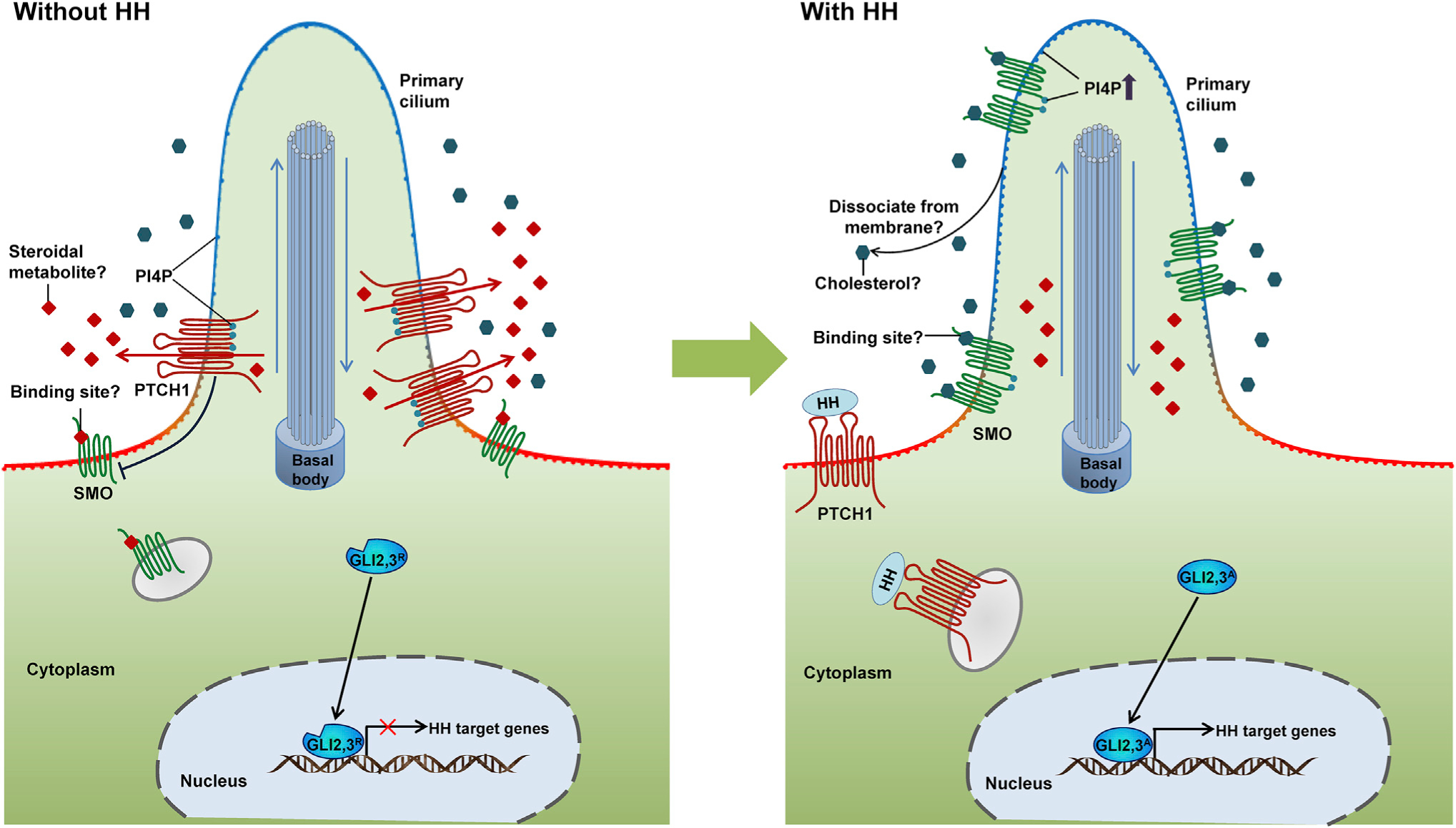
(Left) In the absence of HH ligand, PTCH1 suppresses SMO activity by retaining PI4P, limiting its access to SMO for activation, and potentially fluxing a steroidal antagonist (red diamonds) that binds SMO to its CRD and/or TM sites. (Right) When HH ligand binds PTCH1, its secretion of a potential antagonist might be blocked. PI4P is released from PTCH1 and enriched in the PC, which binds the SMO cytoplasmic tail and activates SMO by promoting phosphorylation and dimerization. Meanwhile, an ubiquitous agonist such as cholesterol or an agonist also subject to PTCH1 regulation might function synergistically in SMO activation via engaging the CRD and/or the TM domains.
Therapeutically, the HH pathway is an important target for both cancer therapy and regenerative medicine. Although the reactivation of normally quiescent HH signaling in the adult stage plays important roles in regeneration in multiple organs, including brain (Dellovade et al., 2006), heart (Wang et al., 2015), liver (Michelotti et al., 2013), lung (Peng et al., 2015), prostate (Karhadkar et al., 2004), and bladder (Shin et al., 2011), there have been no clinical efforts reported to utilize these constructive aspects of HH pathway action. Rather, the primary focus has been to target the HH pathway’s destructive action in promoting cancer. Indeed, the prominent role of HH signaling in stimulating or supporting cancer indicates that regenerative approaches will need to proceed with caution.
There is considerable scope for improving treatment for HH-pathway-dependent cancers, Currently, vismodegib and sonidegib, both of which are SMO inhibitors, are the only clinically approved treatments, and these are limited to locally advanced or metastatic BCC (Burness, 2015; Dlugosz et al., 2012). To date, all drugs being tested in clinical trials except ATO target SMO (Amakye et al., 2013). Drug resistance and cross-resistance accompany treatment with SMO antagonists (Atwood et al., 2015; Danial et al., 2016; Sharpe et al., 2015a). Intra-tumor heterogeneity has also been observed in drug-refractory patients (Atwood et al., 2015; Sharpe et al., 2015a). Circumventing drug resistance to current generation SMO inhibitors is critical. Furthermore, SMO inhibitors have limited effect in ligand-dependent cancers (Berlin et al., 2013; Kaye et al., 2012; Ko et al., 2016). This might reflect the higher complexity of these types of cancers at both molecular and cellular levels. Precise stratification of tumor subtypes to identify responsive patients will likely maximize the therapeutic opportunities.
As illustrated by studies on MB patients, approaches to stratifying patients to identify those most likely to respond will improve both the design and interpretation of clinical trials (Gajjar et al., 2013b; Geoerger et al., 2012). In addition, our understanding of tumorigenesis highlights an increasing complexity of signaling responses. Targeting other key pathways together with HH signaling may improve and extend current therapeutic options. Here, efforts to go beyond simple gene expression signatures to interpret active signaling processes from the data will be important.
ACKNOWLEDGMENTS
We are grateful for support from the National Basic Research Program of China (2015CB964800 and 2014CB964900), the National Natural Science Foundation of China (31571514), the Hundred Talents Program of Chinese Academy of Sciences, and State Key Laboratory of Stem Cell and Reproductive Biology. Work in A.P.M.’s laboratory is supported by a grant from the NIH (NS033642). A.P.M. has intellectual property around the HH pathway licensed to CURIS Inc., and through this, A.P.M. receives royalties in conjunction with the marketing of vismodegib.
REFERENCES
- Aanstad P, Santos N, Corbit KC, Scherz PJ, Trinh LA, Salvenmoser W, Huisken J, Reiter JF, and Stainier DYR (2009). The extracellular domain of smoothened regulates ciliary localization and is required for high-level Hh signaling. Curr. Biol 19, 1034–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adolphe C, Hetherington R, Ellis T, and Wainwright B (2006). Patched1 functions as a gatekeeper by promoting cell cycle progression. Cancer Res. 66, 2081–2088. [DOI] [PubMed] [Google Scholar]
- Ally MS, Ransohoff K, Sarin K, Atwood SX, Rezaee M, Bailey-Healy I, Kim J, Beachy PA, Chang AL, Oro A, et al. (2016). Effects of combined treatment with arsenic trioxide and itraconazole in patients with refractory metastatic basal cell carcinoma. JAMA Dermatol. 152, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amakye D, Robinson D, Rose K, Cho J, Ligon KL, Sharp T, Haider A, Bandaru R, Ando Y, Geoerger B, et al. (2012). Abstract 4818: the predictive value of a 5-gene signature as a patient pre-selection tool in medulloblastoma for Hedgehog pathway inhibitor therapy. Cancer Res. 72, 4818.22805309 [Google Scholar]
- Amakye D, Jagani Z, and Dorsch M (2013). Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat. Med 19, 1410–1422. [DOI] [PubMed] [Google Scholar]
- Antonarakis ES, Heath EI, Smith DC, Rathkopf D, Blackford AL, Danila DC, King S, Frost A, Ajiboye AS, and Zhao M (2013). Repurposing itraconazole as a treatment for advanced prostate cancer: a noncomparative randomized phase II trial in men with metastatic castration-resistant prostate cancer. Oncologist 18, 163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aszterbaum M, Epstein J, Oro A, Douglas V, LeBoit PE, Scott MP, and Epstein EH Jr. (1999). Ultraviolet and ionizing radiation enhance the growth of BCCs and trichoblastomas in patched heterozygous knockout mice. Nat. Med 5, 1285–1291. [DOI] [PubMed] [Google Scholar]
- Atwood SX, Li M, Lee A, Tang JY, and Oro AE (2013). GLI activation by atypical protein kinase C iota/lambda regulates the growth of basal cell carcinomas. Nature 494, 484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood SX, Sarin KY, Whitson RJ, Li JR, Kim G, Rezaee M, Ally MS, Kim J, Yao C, Chang AL, et al. (2015). Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell 27, 342–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aza-Blanc P, Lin HY, Ruiz i Altaba A, and Kornberg TB (2000). Expression of the vertebrate Gli proteins in Drosophila reveals a distribution of activator and repressor activities. Development 127, 4293–4301. [DOI] [PubMed] [Google Scholar]
- Bai CB, Auerbach W, Lee JS, Stephen D, and Joyner AL (2002). Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development 129, 4753–4761. [DOI] [PubMed] [Google Scholar]
- Bailey JM, Mohr AM, and Hollingsworth MA (2009). Sonic hedgehog paracrine signaling regulates metastasis and lymphangiogenesis in pancreatic cancer. Oncogene 28, 3513–3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzi M, Berenguer J, Menendez A, Alvarez-Rodriguez R, and Pons S (2010). Sonic-hedgehog-mediated proliferation requires the localization of PKA to the cilium base. J. Cell Sci 123, 62–69. [DOI] [PubMed] [Google Scholar]
- Beauchamp EM, Ringer L, Bulut G, Sajwan KP, Hall MD, Lee YC, Peaceman D, Ozdemirli M, Rodriguez O, Macdonald TJ, et al. (2011). Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J. Clin. Invest 121, 148–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belani CP, Dahlberg SE, Rudin CM, Fleisher M, Chen HX, Takebe N, Ramalingam SS, and Schiller JH (2013). Three-arm randomized phase II study of cisplatin and etoposide (CE) versus CE with either vismodegib (V) or cixutumumab (Cx) for patients with extensive stage-small cell lung cancer (ES-SCLC)(ECOG 1508). ASCO Annu. Meet. Proc 31, 7508. [Google Scholar]
- Bender MH, Hipskind PA, Capen AR, Cockman M, Credille KM, Gao H, Bastian JA, Clay JM, Lobb KL, and Sall DJ (2011). Identification and characterization of a novel smoothened antagonist for the treatment of cancer with deregulated hedgehog signaling. Cancer Res. 71, 2819. [Google Scholar]
- Berlin J, Bendell JC, Hart LL, Firdaus I, Gore I, Hermann RC, Mulcahy MF, Zalupski MM, Mackey HM, Yauch RL, et al. (2013). A randomized phase II trial of vismodegib versus placebo with FOLFOX or FOLFIRI and bevacizumab in patients with previously untreated metastatic colorectal cancer. Clin. Cancer Res 19, 258–267. [DOI] [PubMed] [Google Scholar]
- Berman DM, Karhadkar SS, Hallahan AR, Pritchard JI, Eberhart CG, Watkins DN, Chen JK, Cooper MK, Taipale J, Olson JM, et al. (2002). Medulloblastoma growth inhibition by hedgehog pathway blockade. Science 297, 1559–1561. [DOI] [PubMed] [Google Scholar]
- Berman DM, Karhadkar SS, Maitra A, Montes De Oca R, Gerstenblith MR, Briggs K, Parker AR, Shimada Y, Eshleman JR, Watkins DN, et al. (2003). Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 425, 846–851. [DOI] [PubMed] [Google Scholar]
- Bijlsma MF, Spek CA, Zivkovic D, van de Water S, Rezaee F, and Peppelenbosch MP (2006). Repression of smoothened by patched-dependent (Pro-)Vitamin D3 secretion. PLoS Biol. 4, 1397–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blassberg R, Macrae JI, Briscoe J, and Jacob J (2016). Reduced cholesterol levels impair Smoothened activation in Smith-Lemli-Opitz syndrome. Hum. Mol. Genet 25, 693–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borzillo GV, and Lippa B (2005). The Hedgehog signaling pathway as a target for anticancer drug discovery. Curr. Top. Med. Chem 5, 147–157. [DOI] [PubMed] [Google Scholar]
- Bosco-Clement G, Zhang F, Chen Z, Zhou H, Li H, Mikami I, Hirata T, Yagui-Beltran A, Lui N, and Do H (2014). Targeting Gli transcription activation by small molecule suppresses tumor growth. Oncogene 33, 2087–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryden MM, Evans HE, and Keeler RF (1971). Cyclopia in sheep caused by plant teratogens. J. Anat 110, 507. [PubMed] [Google Scholar]
- Buonamici S, Williams J, Morrissey M, Wang A, Guo R, Vattay A, Hsiao K, Yuan J, Green J, Ospina B, et al. (2010). Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci. Transl. Med 2, 3001599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burness CB (2015). Sonidegib: first global approval. Drugs 75, 1559–1566. [DOI] [PubMed] [Google Scholar]
- Byrne EF, Sircar R, Miller PS, Hedger G, Luchetti G, Nachtergaele S, Tully MD, Mydock-McGrane L, Covey DF, Rambo RP, et al. (2016). Structural basis of Smoothened regulation by its extracellular domains. Nature 535, 517–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catenacci DV, Junttila MR, Karrison T, Bahary N, Horiba MN, Nattam SR, Marsh R, Wallace J, Kozloff M, Rajdev L, et al. (2015). Randomized phase Ib/II study of gemcitabine plus placebo or vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. J. Clin. Oncol 33, 4284–4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang AL, and Oro AE (2012). Initial assessment of tumor regrowth after vismodegib in advanced basal cell carcinoma. Arch. Dermatol 148, 1324–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez M, Ena S, Van Sande J, de Kerchove d’Exaerde A, Schurmans S, and Schiffmann SN (2015). Modulation of ciliary phosphoinositide content regulates trafficking and sonic hedgehog signaling output. Dev. Cell 34, 338–350. [DOI] [PubMed] [Google Scholar]
- Chen JK, Taipale J, Cooper MK, and Beachy PA (2002a). Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 16, 2743–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JK, Taipale J, Young KE, Maiti T, and Beachy PA (2002b). Small molecule modulation of Smoothened activity. Proc. Natl. Acad. Sci. USA 99, 14071–14076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung HO, Zhang X, Ribeiro A, Mo R, Makino S, Puviindran V, Law KK, Briscoe J, and Hui CC (2009). The kinesin protein Kif7 is a critical regulator of Gli transcription factors in mammalian hedgehog signaling. Sci. Signal 2, 2000405. [DOI] [PubMed] [Google Scholar]
- Chong YC, Mann RK, Zhao C, Kato M, and Beachy PA (2015). Bifurcating action of Smoothened in Hedgehog signaling is mediated by Dlg5. Genes Dev. 29, 262–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu QS, Mahipal A, Schuler M, De Braud FGM, Dirix L, Rampersad A, Zhou J, Wu Y, Kalambakas S, and Wen PY (2014). 445ODOSE-ESCALATION study of sonidegib (LDE225) plus buparlisib (BKM120) in patients (PTS) with advanced solid tumors. Ann. Oncol 25, iv147–iv148. [Google Scholar]
- Cohen DJ, Christos PJ, Sparano JA, Kindler HL, Catenacci DVT, Bekaii-Saab TB, Tahiri S, Janjigian YY, Gibson MK, and Chan E (2013). A randomized phase II study of vismodegib (V), a hedgehog (HH) pathway inhibitor, combined with FOLFOX in patients (pts) with advanced gastric and gastroesophageal junction (GEJ) carcinoma: a New York Cancer Consortium led study. ASCO Annu. Meet. Proc 31, 67. [Google Scholar]
- Cooper MK, Porter JA, Young KE, and Beachy PA (1998). Teratogen-mediated inhibition of target tissue response to Shh signaling. Science 280, 1603–1607. [DOI] [PubMed] [Google Scholar]
- Cooper MK, Wassif CA, Krakowiak PA, Taipale J, Gong R, Kelley RI, Porter FD, and Beachy PA (2003). A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis. Nat. Genet 33, 508–513. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DYR, and Reiter JF (2005). Vertebrate Smoothened functions at the primary cilium. Nature 437, 1018–1021. [DOI] [PubMed] [Google Scholar]
- Corcoran RB, and Scott MP (2006). Oxysterols stimulate Sonic hedgehog signal transduction and proliferation of medulloblastoma cells. Proc. Natl. Acad. Sci. USA 103, 8408–8413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai P, Akimaru H, Tanaka Y, Maekawa T, Nakafuku M, and Ishii S (1999). Sonic hedgehog-induced activation of the Gli1 promoter is mediated by GLI3. J. Biol. Chem 274, 8143–8152. [DOI] [PubMed] [Google Scholar]
- Danial C, Sarin KY, Oro AE, and Chang AL (2016). An investigator-initiated open-label trial of sonidegib in advanced basal cell carcinoma patients resistant to vismodegib. Clin. Cancer Res 22, 1325–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JP, Chen FW, and Ioannou YA (2000). Transmembrane molecular pump activity of Niemann-Pick C1 protein. Science 290, 2295–2298. [DOI] [PubMed] [Google Scholar]
- Dellovade T, Romer JT, Curran T, and Rubin LL (2006). The hedgehog pathway and neurological disorders. Annu. Rev. Neurosci 29, 539–563. [DOI] [PubMed] [Google Scholar]
- Dierks C, Grbic J, Zirlik K, Beigi R, Englund NP, Guo GR, Veelken H, Engelhardt M, Mertelsmann R, Kelleher JF, et al. (2007). Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat. Med 13, 944–951. [DOI] [PubMed] [Google Scholar]
- Dierks C, Beigi R, Guo GR, Zirlik K, Stegert MR, Manley P, Trussell C, Schmitt-Graeff A, Landwerlin K, Veelken H, et al. (2008). Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell 14, 238–249. [DOI] [PubMed] [Google Scholar]
- Dijkgraaf GJ, Alicke B, Weinmann L, Januario T, West K, Modrusan Z, Burdick D, Goldsmith R, Robarge K, Sutherlin D, et al. (2011). Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 71, 435–444. [DOI] [PubMed] [Google Scholar]
- Dlugosz A, Agrawal S, and Kirkpatrick P (2012). Vismodegib. Nat. Rev. Drug Discov 11, 437–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dockendorff C, Nagiec MM, Weiwer M, Buhrlage S, Ting A, Nag PP, Germain A, Kim HJ, Youngsaye W, Scherer C, et al. (2012). Macrocyclic hedgehog pathway inhibitors: optimization of cellular activity and mode of action studies. ACS Med. Chem. Lett 3, 808–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn KV, Hughes CE, and Rohatgi R (2012). A smoothened-Evc2 complex transduces the hedgehog signal at primary cilia. Dev. Cell 23, 823–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison DW, Dalton J, Kocak M, Nicholson SL, Fraga C, Neale G, Kenney AM, Brat DJ, Perry A, Yong WH, et al. (2011). Medulloblastoma: clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol. 121, 381–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endoh-Yamagami S, Evangelista M, Wilson D, Wen X, Theunissen JW, Phamluong K, Davis M, Scales SJ, Solloway MJ, de Sauvage FJ, et al. (2009). The mammalian Cos2 homolog Kif7 plays an essential role in modulating Hh signal transduction during development. Curr. Biol 19, 1320–1326. [DOI] [PubMed] [Google Scholar]
- Ericson J, Morton S, Kawakami A, Roelink H, and Jessell TM (1996). Two critical periods of Sonic Hedgehog signaling required for the specification of motor neuron identity. Cell 87, 661–673. [DOI] [PubMed] [Google Scholar]
- Fei DL, Li H, Kozul CD, Black KE, Singh S, Gosse JA, DiRenzo J, Martin KA, Wang B, Hamilton JW, et al. (2010). Activation of Hedgehog signaling by the environmental toxicant arsenic may contribute to the etiology of arsenic-induced tumors. Cancer Res. 70, 1981–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferruzzi P, Mennillo F, De Rosa A, Giordano C, Rossi M, Benedetti G, Magrini R, Pericot Mohr G, Miragliotta V, Magnoni L, et al. (2012). In vitro and in vivo characterization of a novel Hedgehog signaling antagonist in human glioblastoma cell lines. Int. J. Cancer 131, 31. [DOI] [PubMed] [Google Scholar]
- Frank-Kamenetsky M, Zhang XM, Bottega S, Guicherit O, Wichterle H, Dudek H, Bumcrot D, Wang FY, Jones S, Shulok J, et al. (2002). Small-molecule modulators of Hedgehog signaling: identification and characterization of Smoothened agonists and antagonists. J. Biol 1, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumoto K, Hoogenraad CC, and Kikuchi A (2006). GSK-3beta-regulated interaction of BICD with dynein is involved in microtubule anchorage at centro-some. EMBO J. 25, 5670–5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gailani MR, Stahle-Backdahl M, Leffell DJ, Glynn M, Zaphiropoulos PG, Pressman C, Unden AB, Dean M, Brash DE, Bale AE, et al. (1996). The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat. Genet 14, 78–81. [DOI] [PubMed] [Google Scholar]
- Gajjar A, Stewart CF, Ellison DW, Kaste S, Kun LE, Packer RJ, Goldman S, Chintagumpala M, Wallace D, Takebe N, et al. (2013a). Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: a pediatric brain tumor consortium study. Clin. Cancer Res 19, 6305–6312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajjar AJ, Gururangan S, Qaddoumi IA, Packer R, Goldman S, Prados M, Desjardins A, Fouladi M, Takebe N, and Li S (2013b). A prospective phase II study to determine the efficacy of GDC 0449 (vismodegib) in adults with recurrent medulloblastoma (MB): a Pediatric Brain Tumor Consortium study (PBTC 25B). ASCO Annu. Meet. Proc 31, 2035. [Google Scholar]
- Gao J, Graves S, Koch U, Liu S, Jankovic V, Buonamici S, El Andaloussi A, Nimer SD, Kee BL, Taichman R, et al. (2009). Hedgehog signaling is dispensable for adult hematopoietic stem cell function. Cell Stem Cell 4, 548–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gonzalo FR, Phua SC, Roberson EC, Garcia G 3rd, Abedin M, Schurmans S, Inoue T, and Reiter JF (2015). Phosphoinositides regulate ciliary protein trafficking to modulate hedgehog signaling. Dev. Cell 34, 400–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendreau SB, Hawkins D, Ho CP, Lewin A, Lin T, Merchant A, Rowley RB, Wang Q, Matsui W, and Fargnoli J (2009). Abstract B192: preclinical characterization of BMS-833923 (XL139), a hedgehog (HH) pathway inhibitor in early clinical development. Mol. Cancer Ther 8, B192. [Google Scholar]
- Geoerger B, Aerts I, Casanova M, Chisholm JC, Hargrave DR, Leary S, Ashley DM, Bouffet E, MacDonald T, and Hurh E (2012). A phase I/II study of LDE225, a smoothened (Smo) antagonist, in pediatric patients with recurrent medulloblastoma (MB) or other solid tumors. ASCO Annu. Meet. Proc 30, 9519. [Google Scholar]
- Gibson P, Tong YA, Robinson G, Thompson MC, Currle DS, Eden C, Kranenburg TA, Hogg T, Poppleton H, Martin J, et al. (2010). Sub-types of medulloblastoma have distinct developmental origins. Nature 468, 1095–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman J, Eckhardt SG, Borad MJ, Curtis KK, Hidalgo M, Calvo E, Ryan DP, Wirth LJ, Parikh A, Partyka J, et al. (2015). Phase I dose-escalation trial of the oral investigational Hedgehog signaling pathway inhibitor TAK-441 in patients with advanced solid tumors. Clin. Cancer Res 21, 1002–1009. [DOI] [PubMed] [Google Scholar]
- Gorojankina T, Hoch L, Faure H, Roudaut H, Traiffort E, Schoenfelder A, Girard N, Mann A, Manetti F, Solinas A, et al. (2013). Discovery, molecular and pharmacological characterization of GSA-10, a novel small-molecule positive modulator of Smoothened. Mol. Pharmacol 83, 1020–1029. [DOI] [PubMed] [Google Scholar]
- Graham RA, Lum BL, Cheeti S, Jin JY, Jorga K, Von Hoff DD, Rudin CM, Reddy JC, Low JA, and Lorusso PM (2011). Pharmacokinetics of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with locally advanced or metastatic solid tumors: the role of alpha-1-acid glycoprotein binding. Clin. Cancer Res 17, 2512–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, Vorechovsky I, Holmberg E, Unden AB, Gillies S, et al. (1996). Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 85, 841–851. [DOI] [PubMed] [Google Scholar]
- Hahn H, Wojnowski L, Zimmer AM, Hall J, Miller G, and Zimmer A (1998). Rhabdomyosarcomas and radiation hypersensitivity in a mouse model of Gorlin syndrome. Nat. Med 4, 619–622. [DOI] [PubMed] [Google Scholar]
- Han Y, Shi Q, and Jiang J (2015). Multisite interaction with Sufu regulates Ci/Gli activity through distinct mechanisms in Hh signal transduction. Proc. Natl. Acad. Sci. USA 112, 6383–6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris PJ, Speranza G, and Dansky Ullmann C (2012). Targeting embryonic signaling pathways in cancer therapy. Expert Opin. Ther. Targets 16, 131–145. [DOI] [PubMed] [Google Scholar]
- Hausmann G, von Mering C, and Basler K (2009). The hedgehog signaling pathway: where did it come from? PLoS Biol. 7, e1000146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, and Yoder BK (2005). Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 1, e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann I, Stover EH, Cullen DE, Mao J, Morgan KJ, Lee BH, Kharas MG, Miller PG, Cornejo MG, Okabe R, et al. (2009). Hedgehog signaling is dispensable for adult murine hematopoietic stem cell function and hematopoiesis. Cell Stem Cell 4, 559–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoya T, Arai MA, Koyano T, Kowithayakorn T, and Ishibashi M (2008). Naturally occurring small-molecule inhibitors of hedgehog/GLI-mediated transcription. Chembiochem 9, 1082–1092. [DOI] [PubMed] [Google Scholar]
- Huang SH, He J, Zhang XL, Bian YH, Yang L, Xie GR, Zhang KF, Tang WD, Stelter AA, Wang Q, et al. (2006). Activation of the hedgehog pathway in human hepatocellular carcinomas. Carcinogenesis 27, 1334–1340. [DOI] [PubMed] [Google Scholar]
- Huang P, Nedelcu D, Watanabe M, Jao C, Kim Y, Liu J, and Salic A (2016). Cellular cholesterol directly activates smoothened in hedgehog signaling. Cell 166, 1176–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman JM, Firestone AJ, Heine VM, Zhao Y, Ocasio CA, Han K, Sun M, Rack PG, Sinha S, Wu JJ, et al. (2009). Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc. Natl. Acad. Sci. USA 106, 14132–14137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iarrobino A, Messina JL, Kudchadkar R, and Sondak VK (2013). Emergence of a squamous cell carcinoma phenotype following treatment of metastatic basal cell carcinoma with vismodegib. J. Am. Acad. Dermatol 69, 023. [DOI] [PubMed] [Google Scholar]
- Incardona JP, Gaffield W, Kapur RP, and Roelink H (1998). The teratogenic Veratrum alkaloid cyclopamine inhibits Sonic hedgehog signal transduction. Development 125, 3553–3562. [DOI] [PubMed] [Google Scholar]
- Infante P, Mori M, Alfonsi R, Ghirga F, Aiello F, Toscano S, Ingallina C, Siler M, Cucchi D, Po A, et al. (2015). Gli1/DNA interaction is a druggable target for Hedgehog-dependent tumors. EMBO J. 34, 200–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingham PW, and McMahon AP (2001). Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 15, 3059–3087. [DOI] [PubMed] [Google Scholar]
- Ingham PW, Nakano Y, and Seger C (2011). Mechanisms and functions of Hedgehog signalling across the metazoa. Nat. Rev. Genet 12, 393–406. [DOI] [PubMed] [Google Scholar]
- Irvine DA, and Copland M (2012). Targeting hedgehog in hematologic malignancy. Blood 119, 2196–2204. [DOI] [PubMed] [Google Scholar]
- Jagani Z, Dorsch M, and Warmuth M (2010). Hedgehog pathway activation in chronic myeloid leukemia. Cell Cycle 9, 3449–3456. [DOI] [PubMed] [Google Scholar]
- Jiang K, Liu Y, Fan J, Zhang J, Li X-A, Evers BM, Zhu H, and Jia J (2016). PI(4)P promotes phosphorylation and conformational change of smoothened through interaction with its C-terminal tail. PLoS Biol. 14, e1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimeno A, Weiss GJ, Miller WH Jr., Gettinger S, Eigl BJ, Chang AL, Dunbar J, Devens S, Faia K, Skliris G, et al. (2013). Phase I study of the Hedgehog pathway inhibitor IPI-926 in adult patients with solid tumors. Clin. Cancer Res 19, 2766–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM, Quinn AG, Myers RM, Cox DR, Epstein EH Jr., et al. (1996). Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 272, 1668–1671. [DOI] [PubMed] [Google Scholar]
- Junker JP, Peterson KA, Nishi Y, Mao J, McMahon AP, and van Oudenaarden A (2014). A predictive model of bifunctional transcription factor signaling during embryonic tissue patterning. Dev. Cell 31, 448–460. [DOI] [PubMed] [Google Scholar]
- Karhadkar SS, Bova GS, Abdallah N, Dhara S, Gardner D, Maitra A, Isaacs JT, Berman DM, and Beachy PA (2004). Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature 431, 707–712. [DOI] [PubMed] [Google Scholar]
- Kaye SB, Fehrenbacher L, Holloway R, Amit A, Karlan B, Slomovitz B, Sabbatini P, Fu L, Yauch RL, Chang I, et al. (2012). A phase II, randomized, placebo-controlled study of vismodegib as maintenance therapy in patients with ovarian cancer in second or third complete remission. Clin. Cancer Res 18, 6509–6518. [DOI] [PubMed] [Google Scholar]
- Keeler RF (1970). Teratogenic compounds of Veratrum californicum (Durand)X. Cyclopia in rabbits produced by cyclopamine. Teratology 3, 175–180. [DOI] [PubMed] [Google Scholar]
- Keeler RF (1973). Teratogenic compounds of Veratrum californicum (Durand). XIV. Limb deformities produced by cyclopamine. Proc. Soc. Exp. Biol. Med 142, 1287–1291. [DOI] [PubMed] [Google Scholar]
- Khan AA, Harrison CN, and McLornan DP (2015). Targeting of the Hedgehog pathway in myeloid malignancies: still a worthy chase? Br. J. Haematol 170, 323–335. [DOI] [PubMed] [Google Scholar]
- Kieran M, Geoerger B, Casanova M, Chisholm J, Aerts I, Bouffet E, Brandes AA, Leary SE, Sullivan M, and Bailey S (2013). A phase 1/2 safety and preliminary efficacy study of sonidegib (LDE225), a hedgehog pathway inhibitor, in pediatric and adult patients with relapsed or refractory medulloblastoma and other solid tumors. Neuro-Oncology 15 (Suppl 3 ), iii98–iii135. [Google Scholar]
- Kim J, Lee JJ, Gardner D, and Beachy PA (2010a). Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc. Natl. Acad. Sci. USA 107, 13432–13437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Tang JY, Gong R, Kim J, Lee JJ, Clemons KV, Chong CR, Chang KS, Fereshteh M, and Gardner D (2010b). Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell 17, 388–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Aftab BT, Tang JY, Kim D, Lee AH, Rezaee M, Chen B, King EM, Borodovsky A, Riggins GJ, et al. (2013). Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell 23, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DJ, Kim J, Spaunhurst K, Montoya J, Khodosh R, Chandra K, Fu T, Gilliam A, Molgo M, Beachy PA, et al. (2014a). Open-label, exploratory phase II trial of oral itraconazole for the treatment of basal cell carcinoma. J. Clin. Oncol 32, 745–751. [DOI] [PubMed] [Google Scholar]
- Kim EJ, Sahai V, Abel EV, Griffith KA, Greenson JK, Takebe N, Khan GN, Blau JL, Craig R, Balis UG, et al. (2014b). Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin. Cancer Res 20, 5937–5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Hsia EY, Brigui A, Plessis A, Beachy PA, and Zheng X (2015). The role of ciliary trafficking in Hedgehog receptor signaling. Sci. Signal 8, ra55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H, Ng JM, and Curran T (2008). Transient inhibition of the Hedgehog pathway in young mice causes permanent defects in bone structure. Cancer Cell 13, 249–260. [DOI] [PubMed] [Google Scholar]
- Ko AH, LoConte N, Tempero MA, Walker EJ, Kate Kelley R, Lewis S, Chang WC, Kantoff E, Vannier MW, Catenacci DV, et al. (2016). A phase I study of FOLFIRINOX plus IPI-926, a hedgehog pathway inhibitor, for advanced pancreatic adenocarcinoma. Pancreas 45, 370–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogerman P, Grimm T, Kogerman L, Krause D, Unden AB, Sandstedt B, Toftgard R, and Zaphiropoulos PG (1999). Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat. Cell Biol 1, 312–319. [DOI] [PubMed] [Google Scholar]
- Kool M, Korshunov A, Remke M, Jones DTW, Schlanstein M, Northcott PA, Cho Y-J, Koster J, Schouten-van Meeteren A, van Vuurden D, et al. (2012). Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 123, 473–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai K, Kaspar BK, Gage FH, and Schaffer DV (2003). Sonic hedgehog regulates adult neural progenitor proliferation in vitro and in vivo. Nat. Neurosci 6, 21–27. [DOI] [PubMed] [Google Scholar]
- Lau J, Kawahira H, and Hebrok M (2006). Hedgehog signaling in pancreas development and disease. Cell. Mol. Life Sci 63, 642–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauth M, Bergstrom A, Shimokawa T, and Toftgard R (2007). Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 104, 8455–8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Platt KA, Censullo P, and Altaba ARI (1997). Gli1 is a target of Sonic hedgehog that induces ventral neural tube development. Development 124, 2537–2552. [DOI] [PubMed] [Google Scholar]
- Li B, Giambelli C, Tang B, Winterbottom E, Long J, Jin K, Wang Z, Fei DL, Nguyen DM, Athar M, et al. (2016). Arsenic attenuates GLI signaling, increasing or decreasing its transcriptional program in a context-dependent manner. Mol. Pharmacol 89, 226–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liem KF Jr., He M, Ocbina PJ, and Anderson KV (2009). Mouse Kif7/Costal2 is a cilia-associated protein that regulates Sonic hedgehog signaling. Proc. Natl. Acad. Sci. USA 106, 13377–13382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Yao E, Wang K, Nozawa Y, Shimizu H, Johnson JR, Chen JN, Krogan NJ, and Chuang PT (2014). Regulation of Sufu activity by p66beta and Mycbp provides new insight into vertebrate Hedgehog signaling. Genes Dev. 28, 2547–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Li T, Reinhold M, and Naski M (2014). MEK1–RSK2 contributes to hedgehog signaling by stabilizing GLI2 transcription factor and inhibiting ubiquitination. Oncogene 33, 65–73. [DOI] [PubMed] [Google Scholar]
- Liu J, Zeng H, and Liu A (2015). The loss of Hh responsiveness by a nonciliary Gli2 variant. Development 142, 1651–1660. [DOI] [PubMed] [Google Scholar]
- Liu X, Pitarresi JR, Cuitino MC, Kladney RD, Woelke SA, Sizemore GM, Nayak SG, Egriboz O, Schweickert PG, Yu L, et al. (2016). Genetic ablation of Smoothened in pancreatic fibroblasts increases acinar-ductal metaplasia. Genes Dev. 30, 1943–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoRusso PM, Rudin CM, Reddy JC, Tibes R, Weiss GJ, Borad MJ, Hann CL, Brahmer JR, Chang I, Darbonne WC, et al. (2011). Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin. Cancer Res 17, 2502–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchetti G, Sircar R, Kong JH, Nachtergaele S, Sagner A, Byrne EF, Covey DF, Siebold C, and Rohatgi R (2016). Cholesterol activates the G-protein coupled receptor Smoothened to promote morphogenetic signaling. Elife 5, 20304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machold R, Hayashi S, Rutlin M, Muzumdar MD, Nery S, Corbin JG, Gritli-Linde A, Dellovade T, Porter JA, Rubin LL, et al. (2003). Sonic hedgehog is required for progenitor cell maintenance in telencephalic stem cell niches. Neuron 39, 937–950. [DOI] [PubMed] [Google Scholar]
- Mao J, Barrow J, McMahon J, Vaughan J, and McMahon AP (2005). An ES cell system for rapid, spatial and temporal analysis of gene function in vitro and in vivo. Nucleic Acids Res. 33, e155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin V, Carrillo G, Torroja C, and Guerrero I (2001). The sterol-sensing domain of Patched protein seems to control Smoothened activity through Patched vesicular trafficking. Curr. Biol 11, 601–607. [DOI] [PubMed] [Google Scholar]
- Martinelli G, Oehler VG, Papayannidis C, Courtney R, Shaik MN, Zhang X, O’Connell A, McLachlan KR, Zheng X, Radich J, et al. (2015). Treatment with PF-04449913, an oral smoothened antagonist, in patients with myeloid malignancies: a phase 1 safety and pharmacokinetics study. Lancet Haematol. 2, e339–e346. [DOI] [PubMed] [Google Scholar]
- Michelotti GA, Xie G, Swiderska M, Choi SS, Karaca G, Kruger L, Premont R, Yang L, Syn WK, Metzger D, et al. (2013). Smoothened is a master regulator of adult liver repair. J. Clin. Invest 123, 2380–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migden MR, Guminski A, Gutzmer R, Dirix L, Lewis KD, Combemale P, Herd RM, Kudchadkar R, Trefzer U, Gogov S, et al. (2015). Treatment with two different doses of sonidegib in patients with locally advanced or metastatic basal cell carcinoma (BOLT): a multicentre, randomised, double-blind phase 2 trial. Lancet Oncol. 16, 716–728. [DOI] [PubMed] [Google Scholar]
- Milenkovic L, Weiss LE, Yoon J, Roth TL, Su YS, Sahl SJ, Scott MP, and Moerner WE (2015). Single-molecule imaging of Hedgehog pathway protein Smoothened in primary cilia reveals binding events regulated by Patched1. Proc. Natl. Acad. Sci. USA 112, 8320–8325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills LD, Zhang Y, Marler RJ, Herreros-Villanueva M, Zhang L, Almada LL, Couch F, Wetmore C, Pasca di Magliano M, and Fernandez-Zapico ME (2013). Loss of the transcription factor GLI1 identifies a signaling network in the tumor microenvironment mediating KRAS oncogene-induced transformation. J. Biol. Chem 288, 11786–11794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyaji T, Nakase T, Iwasaki M, Kuriyama K, Tamai N, Higuchi C, Myoui A, Tomita T, and Yoshikawa H (2003). Expression and distribution of transcripts for sonic hedgehog in the early phase of fracture repair. Histochem. Cell Biol 119, 233–237. [DOI] [PubMed] [Google Scholar]
- Monzo M, Moreno I, Artells R, Ibeas R, Navarro A, Moreno J, Hernandez R, Granell M, and Pie J (2006). Sonic hedgehog mRNA expression by real-time quantitative PCR in normal and tumor tissues from colorectal cancer patients. Cancer Lett. 233, 117–123. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Frolova N, Sadlonova A, Novak Z, Steg A, Page GP, Welch DR, Lobo-Ruppert SM, Ruppert JM, Johnson MR, et al. (2006). Hedgehog signaling and response to cyclopamine differ in epithelial and stromal cells in benign breast and breast cancer. Cancer Biol. Ther 5, 674–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Wen X, Ratti N, Loktev A, Rangell L, Scales SJ, and Jackson PK (2013). The ciliary G-protein-coupled receptor Gpr161 negatively regulates the Sonic hedgehog pathway via cAMP signaling. Cell 152, 210–223. [DOI] [PubMed] [Google Scholar]
- Munchhof MJ, Li Q, Shavnya A, Borzillo GV, Boyden TL, Jones CS, LaGreca SD, Martinez-Alsina L, Patel N, Pelletier K, et al. (2011). Discovery of PF-04449913, a potent and orally bioavailable inhibitor of smoothened. ACS Med. Chem. Lett 3, 106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murone M, Rosenthal A, and de Sauvage FJ (1999). Sonic hedgehog signaling by the patched-smoothened receptor complex. Curr. Biol 9, 76–84. [DOI] [PubMed] [Google Scholar]
- Myers BR, Sever N, Chong YC, Kim J, Belani JD, Rychnovsky S, Bazan JF, and Beachy PA (2013). Hedgehog pathway modulation by multiple lipid binding sites on the smoothened effector of signal response. Dev. Cell 26, 346–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachtergaele S, Mydock LK, Krishnan K, Rammohan J, Schlesinger PH, Covey DF, and Rohatgi R (2012). Oxysterols are allosteric activators of the oncoprotein Smoothened. Nat. Chem. Biol 8, 211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachtergaele S, Whalen DM, Mydock LK, Zhao Z, Malinauskas T, Krishnan K, Ingham PW, Covey DF, Siebold C, and Rohatgi R (2013). Structure and function of the Smoothened extracellular domain in vertebrate Hedgehog signaling. Elife 29, 01340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedelcu D, Liu J, Xu Y, Jao C, and Salic A (2013). Oxysterol binding to the extracellular domain of Smoothened in Hedgehog signaling. Nat. Chem. Biol 9, 557–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S, Bouffet E, Clifford SC, Hawkins CE, French P, et al. (2011). Medulloblastoma comprises four distinct molecular variants. J. Clin. Oncol 29, 1408–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusslein-Volhard C, and Wieschaus E (1980). Mutations affecting segment number and polarity in Drosophila. Nature 287, 795–801. [DOI] [PubMed] [Google Scholar]
- Ochoa B, Syn WK, Delgado I, Karaca GF, Jung Y, Wang J, Zubiaga AM, Fresnedo O, Omenetti A, and Zdanowicz M (2010). Hedgehog signaling is critical for normal liver regeneration after partial hepatectomy in mice. Hepatology 51, 1712–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi T, Oguro Y, Tanaka T, Shiokawa Z, Tanaka Y, Shibata S, Sato Y, Yamakawa H, Hattori H, Yamamoto Y, et al. (2012). Discovery of the investigational drug TAK-441, a pyrrolo[3,2-c]pyridine derivative, as a highly potent and orally active hedgehog signaling inhibitor: modification of the core skeleton for improved solubility. Bioorg. Med. Chem 20, 5507–5517. [DOI] [PubMed] [Google Scholar]
- Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, et al. (2009). Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324, 1457–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, and Wang B (2007). A novel protein-processing domain in Gli2 and Gli3 differentially blocks complete protein degradation by the proteasome. J. Biol. Chem 282, 10846–10852. [DOI] [PubMed] [Google Scholar]
- Pan Y, Bai CB, Joyner AL, and Wang B (2006). Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell Biol 26, 3365–3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan S, Wu X, Jiang J, Gao W, Wan Y, Cheng D, Han D, Liu J, Englund NP, Wang Y, et al. (2010). Discovery of NVP-LDE225, a potent and selective smoothened antagonist. ACS Med. Chem. Lett 1, 130–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HL, Bai C, Platt KA, Matise MP, Beeghly A, Hui CC, Nakashima M, and Joyner AL (2000). Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development 127, 1593–1605. [DOI] [PubMed] [Google Scholar]
- Peluso MO, Campbell VT, Harari JA, Tibbitts TT, Proctor JL, White-bread N, Conley JM, White KF, Kutok JL, Read MA, et al. (2014). Impact of the Smoothened inhibitor, IPI-926, on smoothened ciliary localization and Hedgehog pathway activity. PLoS One 9, e90534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng T, Frank DB, Kadzik RS, Morley MP, Rathi KS, Wang T, Zhou S, Cheng L, Lu MM, and Morrisey EE (2015). Hedgehog actively maintains adult lung quiescence and regulates repair and regeneration. Nature 526, 578–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson KA, Nishi Y, Ma W, Vedenko A, Shokri L, Zhang X, McFarlane M, Baizabal JM, Junker JP, van Oudenaarden A, et al. (2012). Neural-specific Sox2 input and differential Gli-binding affinity provide context and positional information in Shh-directed neural patterning. Genes Dev. 26, 2802–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrova E, Rios-Esteves J, Ouerfelli O, Glickman JF, and Resh MD (2013). Inhibitors of hedgehog acyltransferase block sonic hedgehog signaling. Nat. Chem. Biol 9, 247–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peukert S, He F, Dai M, Zhang R, Sun Y, Miller-Moslin K, McEwan M, Lagu B, Wang K, Yusuff N, et al. (2013). Discovery of NVP-LEQ506, a second-generation inhibitor of smoothened. ChemMedChem 8, 1261–1265. [DOI] [PubMed] [Google Scholar]
- Pola R, Ling LE, Silver M, Corbley MJ, Kearney M, Blake Pepinsky R, Shapiro R, Taylor FR, Baker DP, Asahara T, et al. (2001). The morphogen Sonic hedgehog is an indirect angiogenic agent upregulating two families of angiogenic growth factors. Nat. Med 7, 706–711. [DOI] [PubMed] [Google Scholar]
- Raffel C, Jenkins RB, Frederick L, Hebrink D, Alderete B, Fults DW, and James CD (1997). Sporadic medulloblastomas contain PTCH mutations. Cancer Res. 57, 842–845. [PubMed] [Google Scholar]
- Rana R, Carroll CE, Lee H-J, Bao J, Marada S, Grace CRR, Guibao CD, Ogden SK, and Zheng JJ (2013). Structural insights into the role of the Smoothened cysteine-rich domain in Hedgehog signalling. Nat. Commun 4, 2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reifenberger J, Wolter M, Weber RG, Megahed M, Ruzicka T, Lichter P, and Reifenberger G (1998). Missense mutations in SMOH in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res. 58, 1798–1803. [PubMed] [Google Scholar]
- Reifenberger J, Wolter M, Knobbe CB, Kohler B, Schonicke A, Scharwachter C, Kumar K, Blaschke B, Ruzicka T, and Reifenberger G (2005). Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. Br. J. Dermatol 152, 43–51. [DOI] [PubMed] [Google Scholar]
- Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, and Tattersall IW (2014). Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 25, 735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riobo NA, Lu K, Ai X, Haines GM, and Emerson CP Jr. (2006). Phosphoinositide 3-kinase and Akt are essential for sonic hedgehog signaling. Proc. Natl. Acad. Sci. USA 103, 4505–4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robarge KD, Brunton SA, Castanedo GM, Cui Y, Dina MS, Goldsmith R, Gould SE, Guichert O, Gunzner JL, Halladay J, et al. (2009). GDC-0449-a potent inhibitor of the hedgehog pathway. Bioorg. Med. Chem. Lett 19, 5576–5581. [DOI] [PubMed] [Google Scholar]
- Roberts B, Casillas C, Alfaro AC, Jagers C, and Roelink H (2016). Patched1 and Patched2 inhibit Smoothened non-cell autonomously. Elife 23, 17634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson GW, Orr BA, Wu G, Gururangan S, Lin T, Qaddoumi I, Packer RJ, Goldman S, Prados MD, Desjardins A, et al. (2015). Vismodegib exerts targeted efficacy against recurrent sonic hedgehog-subgroup medulloblastoma: results from phase II pediatric brain tumor consortium studies PBTC-025B and PBTC-032. J. Clin. Oncol 33, 2646–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodon J, Tawbi HA, Thomas AL, Stoller RG, Turtschi CP, Baselga J, Sarantopoulos J, Mahalingam D, Shou Y, Moles MA, et al. (2014). A phase I, multicenter, open-label, first-in-human, dose-escalation study of the oral smoothened inhibitor Sonidegib (LDE225) in patients with advanced solid tumors. Clin. Cancer Res 20, 1900–1909. [DOI] [PubMed] [Google Scholar]
- Rohatgi R, Milenkovic L, and Scott MP (2007). Patched1 regulates Hedgehog signaling at the primary cilium. Science 317, 372–376. [DOI] [PubMed] [Google Scholar]
- Rohatgi R, Milenkovic L, Corcoran RB, and Scott MP (2009). Hedgehog signal transduction by Smoothened: pharmacologic evidence for a 2-step activation process. Proc. Natl. Acad. Sci. USA 106, 3196–3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romer JT, Kimura H, Magdaleno S, Sasai K, Fuller C, Baines H, Connelly M, Stewart CF, Gould S, Rubin LL, et al. (2004). Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/−)p53(−/−) mice. Cancer Cell 6, 229–240. [DOI] [PubMed] [Google Scholar]
- Roudaut H, Traiffort E, Gorojankina T, Vincent L, Faure H, Schoenfelder A, Mann A, Manetti F, Solinas A, Taddei M, et al. (2011). Identification and mechanism of action of the Acylguanidine MRT-83, a novel potent smoothened antagonist. Mol. Pharmacol 79, 453–460. [DOI] [PubMed] [Google Scholar]
- Rubin LL, and de Sauvage FJ (2006). Targeting the Hedgehog pathway in cancer. Nat. Rev. Drug Discov 5, 1026–1033. [DOI] [PubMed] [Google Scholar]
- Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, Holcomb T, Stinson J, Gould SE, Coleman B, et al. (2009). Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N. Engl. J. Med 361, 1173–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz i Altaba A (1998). Combinatorial Gli gene function in floor plate and neuronal inductions by Sonic hedgehog. Development 125, 2203–2212. [DOI] [PubMed] [Google Scholar]
- Rusert JM, Wu X, Eberhart CG, Taylor MD, and Wechsler-Reya RJ (2014). SnapShot: medulloblastoma. Cancer Cell 26, 940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadarangani A, Pineda G, Lennon KM, Chun H-J, Shih A, Schairer AE, Goff DJ, Prashad SL, Geron I, and Wall R (2015). GLI2 inhibition abrogates human leukemia stem cell dormancy. J. Transl. Med 13, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki H, Nishizaki Y, Hui CC, Nakafuku M, and Kondoh H (1999). Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development 126, 3915–3924. [DOI] [PubMed] [Google Scholar]
- Schuller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG, Huillard E, Sun T, Ligon AH, Qian Y, et al. (2008). Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell 14, 123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekulic A, Migden MR, Oro AE, Dirix L, Lewis KD, Hainsworth JD, Solomon JA, Yoo S, Arron ST, Friedlander PA, et al. (2012). Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med 366, 2171–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sever N, Mann RK, Xu L, Snell WJ, Hernandez-Lara CI, Porter NA, and Beachy PA (2016). Endogenous B-ring oxysterols inhibit the Hedgehog component Smoothened in a manner distinct from cyclopamine or side-chain oxysterols. Proc. Natl. Acad. Sci. USA 113, 5904–5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe HJ, Pau G, Dijkgraaf GJ, Basset-Seguin N, Modrusan Z, Januario T, Tsui V, Durham AB, Dlugosz AA, Haverty PM, et al. (2015a). Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell 27, 327–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe HJ, Wang W, Hannoush RN, and de Sauvage FJ (2015b). Regulation of the oncoprotein Smoothened by small molecules. Nat. Chem. Biol 11, 246–255. [DOI] [PubMed] [Google Scholar]
- Shi X, Zhang Z, Zhan X, Cao M, Satoh T, Akira S, Shpargel K, Magnuson T, Li Q, Wang R, et al. (2014). An epigenetic switch induced by Shh signalling regulates gene activation during development and medulloblastoma growth. Nat. Commun 5, 5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin K, Lee J, Guo N, Kim J, Lim A, Qu L, Mysorekar IU, and Beachy PA (2011). Hedgehog/Wnt feedback supports regenerative proliferation of epithelial stem cells in bladder. Nature 472, 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin K, Lim A, Zhao C, Sahoo D, Pan Y, Spiekerkoetter E, Liao JC, and Beachy PA (2014). Hedgehog signaling restrains bladder cancer progression by eliciting stromal production of urothelial differentiation factors. Cancer Cell 26, 521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shou Y, Robinson DM, Amakye DD, Rose KL, Cho YJ, Ligon KL, Sharp T, Haider AS, Bandaru R, Ando Y, et al. (2015). A five-gene hedgehog signature developed as a patient preselection tool for hedgehog inhibitor therapy in medulloblastoma. Clin. Cancer Res 21, 585–593. [DOI] [PubMed] [Google Scholar]
- Sillibourne JE, Milne DM, Takahashi M, Ono Y, and Meek DW (2002). Centrosomal anchoring of the protein kinase CK1delta mediated by attachment to the large, coiled-coil scaffolding protein CG-NAP/AKAP450. J. Mol. Biol 322, 785–797. [DOI] [PubMed] [Google Scholar]
- Sinha S, and Chen JK (2006). Purmorphamine activates the Hedgehog pathway by targeting Smoothened. Nat. Chem. Biol 2, 29–30. [DOI] [PubMed] [Google Scholar]
- Siu LL, Papadopoulos KP, Alberts SR, Taylor S, Patnaik A, Chen EX, Tolcher A, Vakkalaguada B, Lang L, Ahlers C, et al. (2009). Abstract A55: a first-in-human, phase I study of an oral hedgehog pathway antagonist, BMS-833923 (XL139), in subjects with advanced or metastatic solid tumors. Mol. Cancer Ther 8, A55. [Google Scholar]
- Smith MJ, Beetz C, Williams SG, Bhaskar SS, O’Sullivan J, Anderson B, Daly SB, Urquhart JE, Bholah Z, Oudit D, et al. (2014). Germline mutations in SUFU cause Gorlin syndrome-associated childhood medulloblastoma and redefine the risk associated with PTCH1 mutations. J. Clin. Oncol 32, 4155–4161. [DOI] [PubMed] [Google Scholar]
- Stanton BZ, Peng LF, Maloof N, Nakai K, Wang X, Duffner JL, Taveras KM, Hyman JM, Lee SW, Koehler AN, et al. (2009). A small molecule that binds Hedgehog and blocks its signaling in human cells. Nat. Chem. Biol 5, 154–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone DM, Murone M, Luoh S, Ye W, Armanini MP, Gurney A, Phillips H, Brush J, Goddard A, de Sauvage FJ, et al. (1999). Characterization of the human suppressor of fused, a negative regulator of the zinc-finger transcription factor Gli. J. Cell Sci 112, 4437–4448. [DOI] [PubMed] [Google Scholar]
- Strutt H, Thomas C, Nakano Y, Stark D, Neave B, Taylor AM, and Ingham PW (2001). Mutations in the sterol-sensing domain of Patched suggest a role for vesicular trafficking in Smoothened regulation. Curr. Biol 11, 608–613. [DOI] [PubMed] [Google Scholar]
- Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovic L, Scott MP, and Beachy PA (2000). Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 406, 1005–1009. [DOI] [PubMed] [Google Scholar]
- Taipale J, Cooper MK, Maiti T, and Beachy PA (2002). Patched acts catalytically to suppress the activity of Smoothened. Nature 418, 892–897. [DOI] [PubMed] [Google Scholar]
- Tang JY, Xiao TZ, Oda Y, Chang KS, Shpall E, Wu A, So PL, Hebert J, Bikle D, and Epstein EH Jr. (2011). Vitamin D3 inhibits hedgehog signaling and proliferation in murine basal cell carcinomas. Cancer Prev. Res 4, 744–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang JY, Mackay-Wiggan JM, Aszterbaum M, Yauch RL, Lindgren J, Chang K, Coppola C, Chanana AM, Marji J, Bickers DR, et al. (2012). Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. N. Engl. J. Med 366, 2180–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Gholamin S, Schubert S, Willardson MI, Lee A, Bandopadhayay P, Bergthold G, Masoud S, Nguyen B, Vue N, et al. (2014). Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat. Med 20, 732–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao H, Jin Q, Koo DI, Liao X, Englund NP, Wang Y, Ramamurthy A, Schultz PG, Dorsch M, Kelleher J, et al. (2011). Small molecule antagonists in distinct binding modes inhibit drug-resistant mutant of smoothened. Chem. Biol 18, 432–437. [DOI] [PubMed] [Google Scholar]
- Tay SY, Ingham PW, and Roy S (2005). A homologue of the Drosophila kinesin-like protein Costal2 regulates Hedgehog signal transduction in the vertebrate embryo. Development 132, 625–634. [DOI] [PubMed] [Google Scholar]
- Tempé D, Casas M, Karaz S, Blanchet-Tournier M-F, and Concordet J-P (2006). Multisite protein kinase A and glycogen synthase kinase 3β phosphorylation leads to Gli3 ubiquitination by SCFβTrCP. Mol. Cell. Biol 26, 4316–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, Qi YP, Gysin S, Fernandez-del Castillo C, Yajnik V, et al. (2003). Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 425, 851–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas NA, Koudijs M, van Eeden FJ, Joyner AL, and Yelon D (2008). Hedgehog signaling plays a cell-autonomous role in maximizing cardiac developmental potential. Development 135, 3789–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibes R, and Mesa RA (2014). Targeting hedgehog signaling in myelofibrosis and other hematologic malignancies. J. Hematol. Oncol 7, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tostar U, Malm CJ, Meis-Kindblom JM, Kindblom LG, Toftgard R, and Unden AB (2006). Deregulation of the hedgehog signalling pathway: a possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. J. Pathol 208, 17–25. [DOI] [PubMed] [Google Scholar]
- Tremblay MR, Lescarbeau A, Grogan MJ, Tan E, Lin G, Austad BC, Yu LC, Behnke ML, Nair SJ, Hagel M, et al. (2009). Discovery of a potent and orally active hedgehog pathway antagonist (IPI-926). J. Med. Chem 52, 4400–4418. [DOI] [PubMed] [Google Scholar]
- Unden AB, Holmberg E, Lundh-Rozell B, Stahle-Backdahl M, Zaphiropoulos PG, Toftgard R, and Vorechovsky I (1996). Mutations in the human homologue of Drosophila patched (PTCH) in basal cell carcinomas and the Gorlin syndrome: different in vivo mechanisms of PTCH inactivation. Cancer Res. 56, 4562–4565. [PubMed] [Google Scholar]
- Varnat F, Duquet A, Malerba M, Zbinden M, Mas C, Gervaz P, and Ruiz i Altaba A (2009). Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol. Med 1, 338–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vokes SA, Ji H, Wong WH, and McMahon AP (2008). A genome-scale analysis of the cis-regulatory circuitry underlying sonic hedgehog-mediated patterning of the mammalian limb. Genes Dev. 22, 2651–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Hoff DD, LoRusso PM, Rudin CM, Reddy JC, Yauch RL, Tibes R, Weiss GJ, Borad MJ, Hann CL, Brahmer JR, et al. (2009). Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N. Engl. J. Med 361, 1164–1172. [DOI] [PubMed] [Google Scholar]
- Wallace VA, and Raff MC (1999). A role for Sonic hedgehog in axon-toastrocyte signalling in the rodent optic nerve. Development 126, 2901–2909. [DOI] [PubMed] [Google Scholar]
- Wang B, and Li Y (2006). Evidence for the direct involvement of {beta}TrCP in Gli3 protein processing. Proc. Natl. Acad. Sci. USA 103, 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhou Z, Walsh CT, and McMahon AP (2009). Selective translocation of intracellular Smoothened to the primary cilium in response to Hedgehog pathway modulation. Proc. Natl. Acad. Sci. USA 106, 2623–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Lu J, Bond MC, Chen M, Ren XR, Lyerly HK, Barak LS, and Chen W (2010). Identification of select glucocorticoids as Smoothened agonists: potential utility for regenerative medicine. Proc. Natl. Acad. Sci. USA 107, 9323–9328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Arvanites AC, Davidow L, Blanchard J, Lam K, Yoo JW, Coy S, Rubin LL, and McMahon AP (2012a). Selective identification of hedgehog pathway antagonists by direct analysis of smoothened ciliary translocation. ACS Chem. Biol 7, 1040–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Davidow L, Arvanites AC, Blanchard J, Lam K, Xu K, Oza V, Yoo JW, Ng JM, Curran T, et al. (2012b). Glucocorticoid compounds modify smoothened localization and hedgehog pathway activity. Chem. Biol 19, 972–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ding Q, Yen CJ, Xia W, Izzo JG, Lang JY, Li CW, Hsu JL, Miller SA, Wang X, et al. (2012c). The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell 21, 374–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Wu H, Katritch V, Han GW, Huang X-P, Liu W, Siu FY, Roth BL, Cherezov V, and Stevens RC (2013). Structure of the human smoothened receptor bound to an antitumour agent. Nature 497, 338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Wu H, Evron T, Vardy E, Han GW, Huang X-P, Hufeisen SJ, Mangano TJ, Urban DJ, Katritch V, et al. (2014). Structural basis for Smoothened receptor modulation and chemoresistance to anticancer drugs. Nat. Commun 5, 4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Cao J, Dickson AL, and Poss KD (2015). Epicardial regeneration is guided by cardiac outflow tract and Hedgehog signalling. Nature 522, 226–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins DN, Berman DM, Burkholder SG, Wang B, Beachy PA, and Baylin SB (2003). Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 422, 313–317. [DOI] [PubMed] [Google Scholar]
- Weierstall U, James D, Wang C, White TA, Wang D, Liu W, Spence JCH, Bruce Doak R, Nelson G, Fromme P, et al. (2014). Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat. Commun 5, 3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JA, Guicherit OM, Zaharian BI, Xu Y, Chai L, Wichterle H, Kon C, Gatchalian C, Porter JA, Rubin LL, et al. (2003). Identification of a small molecule inhibitor of the hedgehog signaling pathway: effects on basal cell carcinoma-like lesions. Proc. Natl. Acad. Sci. USA 100, 4616–4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CH, Hempel JE, Hao J, Frist AY, Williams MM, Fleming JT, Sulikowski GA, Cooper MK, Chiang C, and Hong CC (2015). An in vivo chemical genetic screen identifies phosphodiesterase 4 as a pharmacological target for hedgehog signaling inhibition. Cell Rep. 11, 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CW, and Chuang PT (2010). Mechanism and evolution of cytosolic Hedgehog signal transduction. Development 137, 2079–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CW, Chen M-H, and Chuang P-T (2009). Smoothened adopts multiple active and inactive conformations capable of trafficking to the primary cilium. PLoS One 4, e5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Ding S, Ding Q, Gray NS, and Schultz PG (2002). A small molecule with osteogenesis-inducing activity in multipotent mesenchymal progenitor cells. J. Am. Chem. Soc 124, 14520–14521. [DOI] [PubMed] [Google Scholar]
- Wu X, Walker J, Zhang J, Ding S, and Schultz PG (2004). Purmorphamine induces osteogenesis by activation of the hedgehog signaling pathway. Chem. Biol 11, 1229–1238. [DOI] [PubMed] [Google Scholar]
- Wu VM, Chen SC, Arkin MR, and Reiter JF (2012). Small molecule inhibitors of Smoothened ciliary localization and ciliogenesis. Proc. Natl. Acad. Sci. USA 109, 13644–13649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Murone M, Luoh SM, Ryan A, Gu Q, Zhang C, Bonifas JM, Lam CW, Hynes M, Goddard A, et al. (1998). Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 391, 90–92. [DOI] [PubMed] [Google Scholar]
- Xu LB, Mirnics K, Bowman AB, Liu W, Da J, Porter NA, and Korade Z (2012). DHCEO accumulation is a critical mediator of pathophysiology in a Smith-Lemli-Opitz syndrome model. Neurobiol. Dis 45, 923–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Xiang J, Wang N, Zhao Y, Hyman J, Li S, Jiang J, Chen JK, Yang Z, and Lin S (2009). Converse conformational control of smoothened activity by structurally related small molecules. J. Biol. Chem 284, 20876–20884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CP, Chen WL, Chen YB, and Jiang J (2012). Smoothened transduces Hedgehog signal by forming a complex with Evc/Evc2. Cell Res. 22, 1593–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP, Marshall D, Fu L, Januario T, Kallop D, et al. (2008). A paracrine requirement for hedgehog signalling in cancer. Nature 455, 406–410. [DOI] [PubMed] [Google Scholar]
- Yauch RL, Dijkgraaf GJ, Alicke B, Januario T, Ahn CP, Holcomb T, Pujara K, Stinson J, Callahan CA, Tang T, et al. (2009). Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 326, 572–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F, Breslow DK, Koslover EF, Spakowitz AJ, Nelson WJ, and Nachury MV (2013). Single molecule imaging reveals a major role for diffusion in the exploration of ciliary space by signaling receptors. Elife 2, e00654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Harrington N, Moraes RC, Wu MF, Hilsenbeck SG, and Lewis MT (2009). Cyclopamine inhibition of human breast cancer cell growth independent of Smoothened (Smo). Breast Cancer Res. Treat 115, 505–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Fu L, Qi X, Zhang Z, Xia Y, Jia J, Jiang J, Zhao Y, and Wu G (2013). Structural insight into the mutual recognition and regulation between Suppressor of Fused and Gli/Ci. Nat. Commun 4, 2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J, Kwon HY, Kim J, Chute JP, Rizzieri D, et al. (2009). Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 458, 776–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Feng J, Seidel K, Shi S, Klein O, Sharpe P, and Chai Y (2014). Secretion of shh by a neurovascular bundle niche supports mesenchymal stem cell homeostasis in the adult mouse incisor. Cell Stem Cell 14, 160–173. [DOI] [PMC free article] [PubMed] [Google Scholar]